

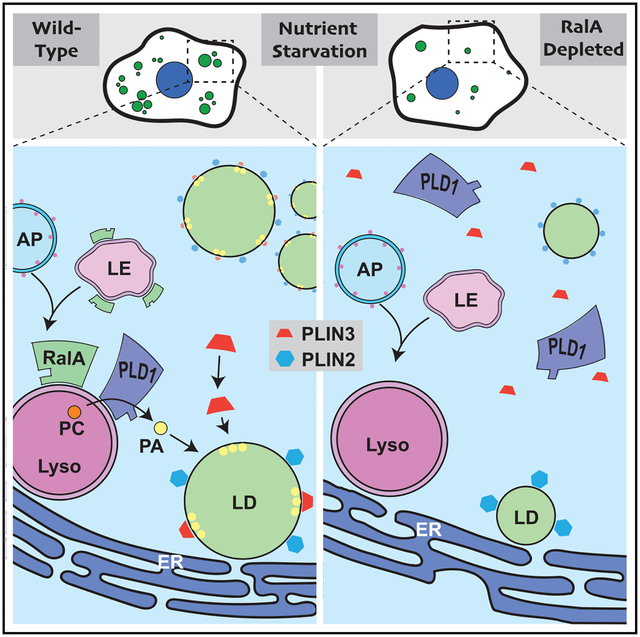
SUMMARY
Lipid droplets (LDs) are dynamic organelles that undergo dynamic changes in response to changing cellular conditions. During nutrient depletion, LD numbers increase to protect cells against toxic fatty acids generated through autophagy and provide fuel for beta-oxidation. However, the precise mechanisms through which these changes are regulated have remained unclear. Here, we show that the small GTPase RalA acts downstream of autophagy to directly facilitate LD growth during nutrient depletion. Mechanistically, RalA performs this function through phospholipase D1 (PLD1), an enzyme that converts phosphatidylcholine (PC) to phosphatidic acid (PA) and that is recruited to lysosomes during nutrient stress in a RalA-dependent fashion. RalA inhibition prevents recruitment of the LD-associated protein perilipin 3, which is required for LD growth. Our data support a model in which RalA recruits PLD1 to lysosomes during nutrient deprivation to promote the localized production of PA and the recruitment of perilipin 3 to expanding LDs.
Graphical abstract
In brief
Lipid droplets (LDs) are dynamic metabolic hubs that undergo changes in size, number, and abundance in response to cellular nutritional cues. Hussain et al. identify the small GTPase RalA and its interacting partner PLD1 as important regulators of LD accumulation in response to nutrient starvation.
INTRODUCTION
Cells maintain homeostasis during nutrient scarcity with a coordinated effort between organelles. Nutrient-sensing pathways initiate autophagy, a lysosomal-dependent cellular recycling process (Kroemer et al., 2010). Among the building blocks generated by autophagy are fatty acids (FAs). FAs are toxic if left in the cytosol and are quickly shuttled into lipid droplets (LDs), organelles that specialize in neutral lipid storage (Listenberger et al., 2003; Nguyen et al., 2017; Rambold et al., 2015). This process leads to a dramatic accumulation of cellular LDs (Nguyen et al., 2017; Rambold et al., 2015). Concurrently, nutrient scarcity shifts the mitochondrial networks toward an elongated morphology and induces closer spatial proximity between mitochondria and LDs (Nguyen et al., 2017; Rambold et al., 2011). These changes are required for the efficient flux of FAs into the mitochondria and for β-oxidation and ATP production. The ultimate goal of this dynamic response is to shift the cell’s reliance from glucose to FAs for energy production.
RalA and RalB proteins are members of the Ras family of small GTPases (Chardin and Tavitian, 1986). Ral proteins carry out their cellular functions by engaging with an overlapping set of effector proteins and are involved in shared processes including vesicular trafficking, membrane dynamics, and cell migration (Gentry et al., 2014; Yan and Theodorescu, 2018). Ral proteins mediate the response to various cellular conditions and extracellular stimuli. RalA is activated upon amino acid and glucose stimulation and is necessary for mTORC1 signaling during nutrient-replete conditions (Maehama et al., 2008). RalA is necessary for insulin-stimulated GLUT4 translocation to the plasma membrane (PM) and glucose uptake in adipocytes (Chen et al., 2007; Skorobogatko et al., 2018). RalB triggers innate immunity signaling through Sec5-TBK1 in response to infection (Simicek et al., 2013). Interestingly, RalB was found to mediate the cellular response to nutrient scarcity by initiating autophagosome formation (Bodemann et al., 2011; Simicek et al., 2013). There is currently no identified role for RalA in the cellular starvation response. Thus, we investigated whether RalA was involved in dynamic processes during nutrient starvation.
Here we report that RalA is essential for nutrient-starvation-induced LD accumulation. Acting downstream of autophagy, RalA recruits phospholipase D1 (PLD1), an enzyme that produces phosphatidic acid (PA), to the lysosome, which forms close contacts with LDs and the endoplasmic reticulum (ER), the site of LD biogenesis. The localized production of PA then supports the recruitment of the LD-associated protein perilipin 3 (PLIN3) and the subsequent expansion of the LD pool.
RESULTS
RalA is required for starvation-induced LD accumulation
To determine if RalA plays a role in starvation-induced LD formation, we cultured wild-type (WT) and RalA knockout (KO) mouse embryonic fibroblasts (MEFs) (Figure 1A) in Hank’s balanced salt solution (HBSS) for 4 h and compared LD levels. Unlike complete DMEM, HBSS has decreased glucose (25 mM versus 5.5 mM) and lacks amino acids and serum. This commonly used in vitro model for nutrient stress exposes cells to near-physiological concentrations of glucose and concentrations of amino acids comparable to those measured in certain tumor microenvironments (Kamphorst et al., 2015; Sullivan et al., 2019). Consistent with previous reports (Rambold et al., 2015), WT MEFs exhibit a significant increase in LDs upon nutrient starvation (Figures 1B and 1C). Surprisingly, RalA KO MEFs fail to accumulate LDs to the same extent as WT MEFs and have significantly fewer LDs by 4 h of HBSS starvation (Figures 1B and 1C). Similar results are observed in RalA knockdown HeLa cells (Figures 1E and 1F). Importantly, LD accumulation during nutrient starvation is rescued in RalA KO MEFs that stably express exogenous RalA at levels comparable to those of WT MEFs (Figures 1G–1I). These data indicate that RalA is necessary for starvation-induced LD accumulation.
Figure 1. RalA is required for starvation-induced lipid droplet accumulation.

(A) Western blot confirming knockout (KO) of RalA protein expression in MEFs.
(B) Representative images of LDs in WT and RalA KO MEFs starved in HBSS media for 4 h. LDs in green (Bodipy 493/503) and nuclei in blue (DAPI). Scale bars, 20 μm.
(C) Quantification of LD area per cell from (B). At least 100 cells were analyzed per time point. n = 3. mean ± SEM. **p < 0.01.
(D) Western blot confirming knockdown of RalA protein expression in HeLa cells.
(E) Representative images of LDs in scramble control and RalA knockdown HeLa cells. Scale bars, 20 μm.
(F) Quantification of LD area per cell from (E). At least 100 cells were analyzed per time point. n = 3. mean ± SEM. *p < 0.05.
(G) Western blot confirming expression of myc-RalA in RalA KO MEFs.
(H) Representative images of LDs in RalA KO MEFs expressing myc-RalA. Scale bars, 20 μm.
(I) Quantification of LD area per cell from (H). At least 100 cells were analyzed per time point. n = 3. mean ± SEM. **p < 0.01.
(J) Representative images of LDs in MEFs stimulated with 200 μM oleic acid in DMEM for 4 h. Scale bars, 20 μm.
(K) Quantification of LD area per cell from (J). At least 100 cells were analyzed for each time point. n = 3. mean ± SEM. ***p < 0.001.
See also Figure S1.
In both MEFs and HeLa cells, the LD content in unstarved cells is comparable between WT and RalA-depleted cells (Figures 1C and 1F), suggesting that cells lacking RalA are able to form LDs under nutrient-replete conditions. Because LD formation can be induced by exogenous FA supplementation (Listenberger et al., 2003; de Vries et al., 1997), we stimulated MEFs and HeLa cells with oleic acid for 4 h and monitored LD levels. Interestingly, MEFs and HeLa cells lacking RalA accumulate LDs at levels comparable to those of WT cells, indicating that RalA is not necessary for oleic-acid-stimulated LD formation (Figures 1J, 1K, and S1). From these data, we conclude that RalA regulation of LD accumulation is not universal but limited to specific conditions, including nutrient starvation.
Triacylglyceride levels increase in a RalA-dependent manner during nutrient starvation
The primary function of LDs is storage of triacylglycerides (TAGs) and cholesteryl esters (CEs). Considering the substantial difference in LD levels between cells lacking RalA and control cells, we sought to understand if these changes could be detected at the level of different lipid species. Therefore, we performed liquid chromatography-mass spectrometry (LC-MS) to measure the lipid composition of WT and RalA KO MEFs during nutrient starvation. WT MEFs exhibit a significant increase in the total amount of TGs measured compared with a minor increase in RalA KO MEFs. (Figure 2A). This effect is also observed at the level of individual TG species (Figure 2B). The levels of other lipid classes measured are not significantly altered over the course of nutrient starvation nor are they RalA dependent (Figures 2A and S2). Together, these results are consistent with LD imaging data and support the requirement for RalA in starvation-induced LD growth.
Figure 2. Analysis of lipid composition in nutrient-starved MEFs.
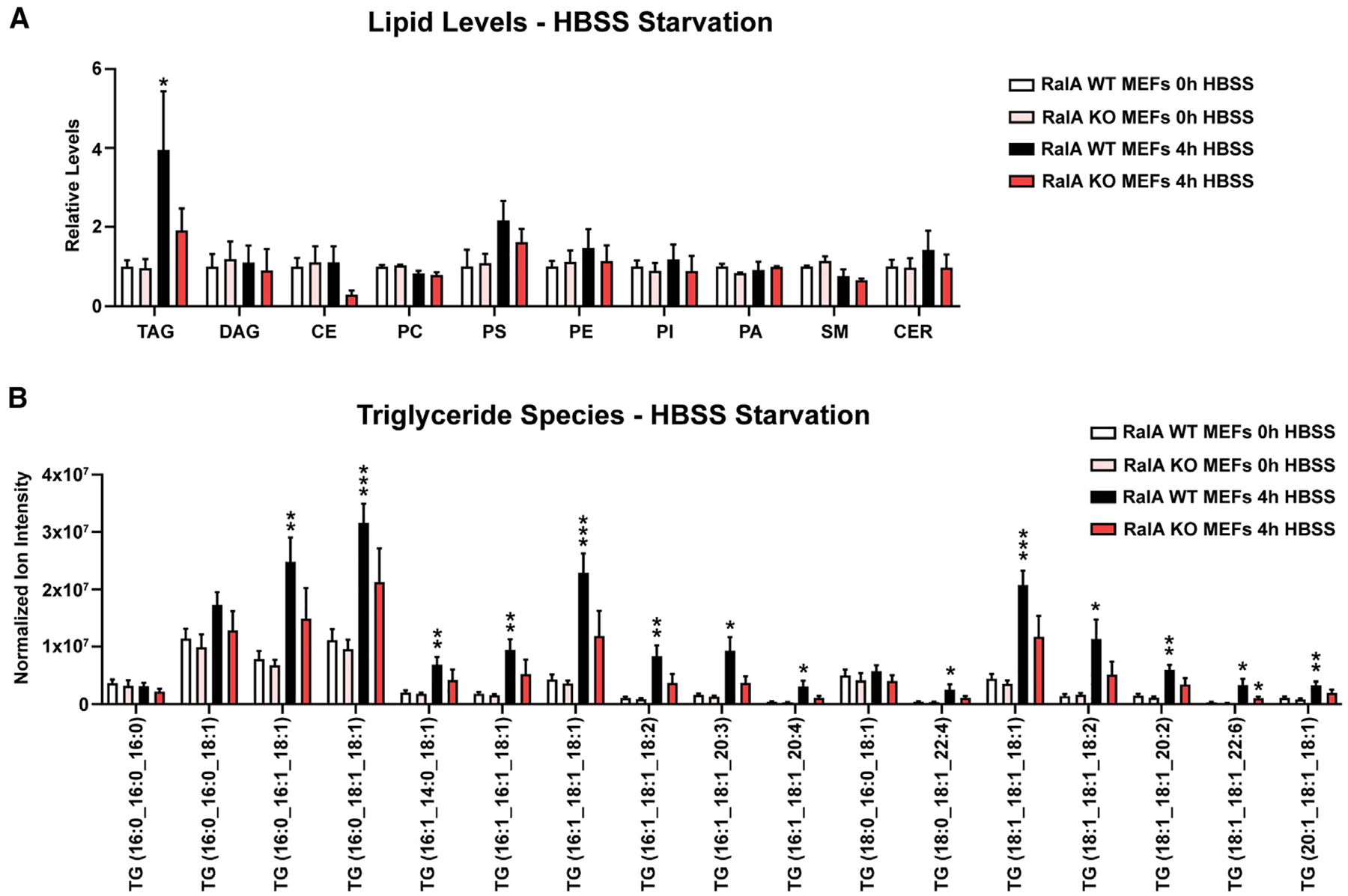
MEFs were incubated in either DMEM (0 h) or HBSS (4 h) for 4 h; lipids were extracted by a modified Folch method and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
(A) Relative levels of various lipids in MEFs. Raw values were normalized to cell counts taken prior to lipid extraction. n = 5. Two-way ANOVA using Tukey’s multiple comparisons test was done for each set of individual lipid type. mean ± SEM. *p < 0.05.
(B) Normalized ion intensity values for the most-abundant individual TAG species detected. Raw values were normalized to cell counts taken prior to lipid extraction. Separate multiple t tests were used to compare 0-h and 4-h WT MEFs and 0-h and 4-h RalA KO MEFs. n = 5. mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figure S2.
RalA acts downstream of autophagy in starvation-induced LD accumulation
Starvation-induced LD formation requires autophagy as a source of FAs (Rambold et al., 2015). Because RalB regulates starvation-induced autophagy induction, we next sought to determine whether RalA functions in autophagy (Bodemann et al., 2011). As such, we repeated the HBSS starvation in the presence of the pharmacological inhibitor chloroquine. Autophagy inhibition prevents LD accumulation in WT MEFs but does not lead to further defects in RalA KO MEFs (Figures 3A and 3B). Similar results are observed when we used the autophagy inhibitor Bafilomycin A1 (Figures S3A and S3B). As a complementary approach, we genetically blocked autophagy initiation by knocking down ATG5, a key protein required for autophagosome biogenesis (Mizushima et al., 2001), in WT and RalA KO MEFs (Figures 3C and 3D). Similar to drug inhibition, knockdown of ATG5 prevents LD accumulation during HBSS starvation in WT MEFs and has no effect on LD composition in RalA KO MEFs (Figures 3E and 3F). These results indicate that autophagy is necessary for RalA-dependent LD accumulation and consistent with RalA and autophagy functioning in the same pathway during nutrient starvation.
Figure 3. RalA facilitates LD accumulation downstream of autophagy.
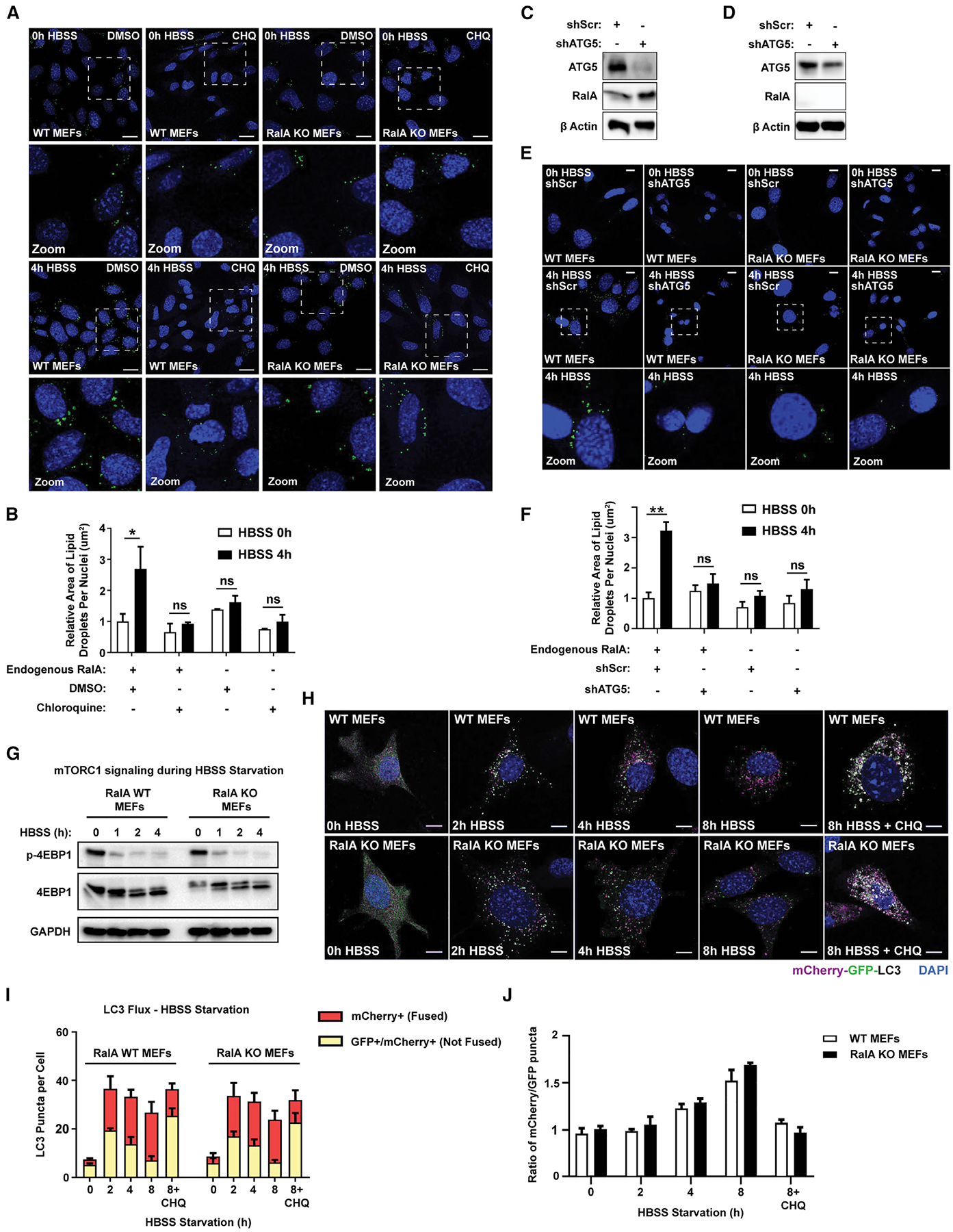
(A) Representative images of LDs in WT and RalA KO MEFs incubated in DMEM or HBSS media ± 100 uM chloroquine for 4 h. Scale bars, 20 μm.
(B) Quantification of LD area per cell from (A). At least 100 cells were analyzed per time point. n = 3. mean ± SEM. *p < 0.05.
(C) Western blot confirming knockdown of ATG5 protein expression in WT MEFs.
(D) Western blot confirming knockdown of ATG5 protein expression in RalA KO MEFs.
(E) Representative images of LDs (green) in WT and RalA KO MEFs expressing either shScramble or shATG5 constructs during HBSS starvation. Scale bars, 20 μm.
(F) Quantification of LD area per cell from (E). At least 100 cells were analyzed per time point. n = 3. mean ± SEM. **p < 0.01.
(G) Western blot analysis of phospho-4E-BP1 (Thr37/46), total 4E-BP1, and GAPDH protein levels in MEFs starved in HBSS for the indicated time points.
(H) Representative images of mCherry-EGFP-LC3B expression in MEFs starved in HBSS ± 100 uM chloroquine for the indicated time points. mCherry-only LC3B punctae indicate autophagosomes fused with lysosomes. mCherry-EGFP-expressing LC3B punctae indicate autophagosomes not fused with lysosomes. Scale bars, 20 μm.
(I) Quantification of mCherry-EGFP-LC3B punctae from (H). Approximately 35–40 punctae were analyzed per cell, and approximately 50 cells were analyzed per time point. n = 3. mean ± SEM.
(J) Ratio of mCherry+/GFP+ puncta calculated from (H).
See also Figure S3.
We next sought to determine whether RalA acts upstream or downstream of autophagy. We began by assessing whether the nutrient-sensing pathway upstream of autophagy is intact. mTORC1 signaling negatively regulates autophagy and is inhibited during nutrient starvation to allow for autophagy initiation (Jung et al., 2009; Noda and Ohsumi, 1998). We hypothesized that RalA-deficient cells may be unable to accumulate LDs due to dysregulation of mTORC1 signaling during nutrient starvation. We monitored phosphorylation of the mTORC1 target 4E-BP1 during HBSS starvation and observed a decrease in phosphorylated 4E-BP1 starting at 1 h of HBSS starvation in both WT and RalA KO MEFs (Figure 3G). This result indicates that mTORC1 signaling is inhibited by starvation independently of RalA and the defect in LD induction in RalA-deficient cells is not due to a failure in nutrient sensing.
Next, we sought to determine whether the loss of RalA prevents cells from efficiently undergoing autophagy. We measured autophagic flux in WT and RalA KO MEFs by using an mCherry-GFP-LC3 reporter (Pankiv et al., 2007). mCherry fluorescence is insensitive to lysosomal pH and does not degrade upon autophagosomal fusion to lysosomes. GFP, however, is sensitive to lysosomal pH and degrades once fusion has occurred. We starved WT and RalA KO MEFs stably expressing mCherry-GFP-LC3 and measured the rate of autophagosomal fusion with lysosomes (indicated by decrease in GFP punctae) over 8 h of starvation. We observe no impairment in the rate of autophagy in RalA KO MEFs compared with WT MEFs during starvation (Figures 3H–3J). Similar results are observed when comparing LC3 lipidation by western blot (Figure S3C). Collectively, these results suggest that autophagy is intact in RalA-deficient cells and that RalA acts downstream of autophagy to promote LD accumulation in low-nutrient conditions.
PLIN3 recruitment to LDs during nutrient starvation is dependent on RalA
With autophagy unaffected, we reasoned two ways that the loss of RalA could result in impaired LD accumulation. RalA could either promote LD growth or prevent LD breakdown. LDs are broken down by two mechanisms, namely, lipolysis and lipophagy (Singh et al., 2009; Zechner et al., 2017; Zimmermann et al., 2004). Lipolysis involves the enzymatic release of FAs stored in LDs by the sequential action of cytoplasmic lipases. Lipophagy is a selective form of autophagy that targets the bulk degradation of LDs through autophagosomal engulfment followed by fusion with lysosomes. Unlike lipophagy, lipolysis allows for fine-tuned release and use of FAs from LDs for energy production. Different nutrient-deprivation conditions (e.g., serum starved, glucose starved, and amino acid starved) trigger specific cellular responses for LD breakdown (Zechner et al., 2017). In the case of HBSS starvation, cells use lipolysis but not lipophagy (Rambold et al., 2015). FAs released by lipolysis are trafficked into the mitochondria, which form close contact sites with LDs (Nguyen et al., 2017; Rambold et al., 2015).
To determine whether RalA-dependent LD accumulation is due to the negative regulation of lipolysis, we compared the rates of FA transfer from LDs to the mitochondria in starved WT and RalA KO MEFs. We performed a pulse-chase assay (Figure S4A) to visualize FA movement between LDs and mitochondria by using BODIPY 558/568 (Red C12), a fluorescent FA analog validated to track FA movement (Rambold et al., 2015; Wang et al., 2010). At the beginning of the chase (0 h), Red C12 is highly colocalized with LDs (Figures S4B and S4D). By 24 h, Red C12 has relocalized from the LDs to the mitochondria in a significant number of cells (Figures S4B and S4D). Importantly, the rate of Red C12 movement is comparable between WT and RalA KO MEFs (Figures S4C and S4E), suggesting that FA release from LDs is not altered by the loss of RalA. Collectively, these data are consistent with a model in which RalA is acting downstream of autophagy but upstream of FA mobilization in starvation-induced LD accumulation.
We next investigated whether RalA directly impacts LD growth during nutrient starvation. As a starting point, we examined Perilipin localization following starvation. Perilipins (1–5) are LD-associated proteins involved in LD stabilization and growth (Itabe et al., 2017). We focused on perilipin 2 (PLIN2) and PLIN3 because they are expressed in most cell types (Brasaemle et al., 1997; Robenek et al., 2005; Sztalryd et al., 2006; Wolins et al., 2001). Furthermore, PLIN3 translocates to LDs during growth-stimulating conditions and is thought to aid in LD formation (Bulankina et al., 2009; Nose et al., 2013; Wolins et al., 2001, 2005). In both WT and RalA KO MEFs, PLIN2 forms ring-like structures around LDs under basal and starved conditions (Figures 4A and 4B). PLIN3, however, exhibits a dramatic shift in localization upon starvation from a dispersed cytoplasmic stain to focused punctae overlapping LDs (Figures 4C and 4D). Notably, PLIN3 does not relocalize to LDs in RalA KO MEFs (Figures 4C and 4D). This effect is not due to differences in the amount of PLIN3 protein during nutrient starvation (Figure 4E). These results indicate that RalA is required for starvation-induced recruitment of PLIN3 to LDs. Loss of PLIN3 protein expression reduces LDs and TAGs in a variety of cells (Bell et al., 2008; Buers et al., 2009; Bulankina et al., 2009; Gu et al., 2010; Nose et al., 2013). We next asked whether the loss of PLIN3 is sufficient to impair LD accumulation during nutrient stress. We knocked down PLIN3 in WT and RalA KO MEFs and subjected these cells to HBSS starvation (Figure 4F). Consistent with our hypothesis, knockdown of PLIN3 significantly impairs LD accumulation in WT MEFs during nutrient starvation compared to scramble control (Figures 4G and 4H). In contrast, PLIN3 knockdown has no additional effect on LDs in RalA KO MEFs, consistent with a model in which RalA and PLIN3 regulate LD expansion as part of the same pathway. Taken together, these data support a model in which RalA promotes LD growth during nutrient starvation by influencing PLIN3 recruitment to LDs.
Figure 4. PLIN3 recruitment to LDs during nutrient starvation is dependent on RalA.

(A) Representative images of PLIN2 and LDs in MEFs during 0 h and 4 h of HBSS starvation. PLIN2 in magenta (antibody), LDs in green (Bodipy 493/503), and nuclei in blue (DAPI). Scale bar, 20 μm.
(B) Relative colocalization of PLIN2 and LDs during 0 h and 4 h of HBSS starvation performed by Pearson’s correlation coefficient. Each point on the graph represents a Pearson’s coefficient value for one cell. At least 25 cells were analyzed per time point. n = 2. mean ± SEM.
(C) Representative images of PLIN3 and LDs in MEFs during 0 h and 4 h of HBSS starvation. PLIN3 in magenta (antibody), LDs in green (Bodipy 493/503), and nuclei in blue (DAPI). Scale bar, 20 μm.
(D) Relative colocalization of Perilipin 3 and LDs during 0 h and 4 h of HBSS starvation performed by Pearson’s correlation coefficient. Each point on the graph represents a Pearson’s coefficient value for one cell. At least 75 cells were analyzed per time point. n = 3. mean ± SEM. ***p < 0.001.
(E) Western blot analysis of PLIN3 protein levels in MEFs incubated in HBSS media for 0 or 4 h.
(F) Western blot confirming knockdown of PLIN3 protein expression in MEFs.
(G) Representative images of PLIN3 (magenta) and LDs (green) in WT and RalA KO MEFs expressing either shScramble or shPLIN3 constructs during 0 h and 4 h of HBSS starvation. Scale bar, 20 μm.
(H) Quantification of LD area per cell from (G). At least 75 cells were analyzed for each time point. n = 3. mean ± SEM. **p < 0.01.
See also Figure S4.
PLD1 is required for RalA-dependent starvation-induced LD accumulation
We next aimed to understand the molecular mechanisms through which RalA regulates PLIN3 recruitment and LD accumulation. RalA fulfills cellular functions by engaging downstream effectors and non-effector-interacting proteins (Cantor et al., 1995; Gentry et al., 2014; Moskalenko et al., 2003; Ohta et al., 1999; Vitale et al., 2005). RalA cycles between GTP and GDP-bound states, and nucleotide binding dictates RalA effector binding (Gentry et al., 2014). Under nutrient-replete conditions, intracellular levels of GTP are several-fold higher than GDP, but the GTP/GDP ratio decreases upon nutrient starvation (Rudoni et al., 2001). We hypothesized that RalA is acting in a nucleotide-dependent fashion to promote starvation-induced LD accumulation. However, we do not detect significant changes in RalA-nucleotide-bound status during nutrient starvation (Figures S5A and S5B), which is in agreement with a previous study (Bodemann et al., 2011). This finding is in contrast to RalB, which shifts toward a GTP-bound state during nutrient starvation (Bodemann et al., 2011). Instead of total cellular RalA activity, it is possible that only a subset of RalA in specific cellular compartments is acting to promote LD accumulation. A recent study showed that RalB activity is increased specifically at autophagosomes upon starvation (Singh et al., 2019). Thus, we stably expressed either a GTPase-deficient RalA Q72L (GTP bound) or the dominant-negative RalA S28N mutant (GDP bound) in RalA KO MEFs (Figure S5C). Both mutants rescue LD accumulation in RalA KO MEFs to levels similar to those of WT MEFs (Figures S5D and S5E). Collectively, these data indicate that RalA acts in a nucleotide-independent manner to facilitate starvation-induced LD accumulation. Further strengthening this conclusion is the fact that knocking out RalBP1, a well-known RalA effector, had no effect on LD accumulation during nutrient starvation (Figures S5F–S5H).
We next focused on downstream RalA-interacting proteins that bind RalA outside its nucleotide-binding domain. We first investigated phospholipase D (PLD), an enzyme that catalyzes the conversion of phosphatidylcholine into PA and choline. Mammalian cells encode two forms of PLD, namely, PLD1 and PLD2. PLD1 and PA have been linked to LD formation and growth (Andersson et al., 2006; Fei et al., 2011; Marchesan et al., 2003; Pagac et al., 2016). Knockdown of PLD1 in NIH 3T3 cells results in significantly fewer cellular LDs than that of control cells (Andersson et al., 2006). Additionally, PLD1 can translocate to LDs (Nakamura et al., 2005). PLD2 has not been shown to localize to LDs or influence LD formation. Based on this finding, we hypothesized that RalA is acting through PLD1 to facilitate LD accumulation. To test this hypothesis, we treated scramble and RalA KD HeLa cells with FIPI (5-Fluoro-2-indolyl des-chlorohalopemide), a potent PLD inhibitor in either DMEM or HBSS for 4 h (Su et al., 2009). FIPI treatment significantly reduces LD accumulation after 4 h of starvation in scramble HeLa cells but has no effect on RalA KD HeLa cells (Figures 5A and 5B). Similar results are observed in MEFs (Figures S5I and S5J). This result suggests that PLD1 and/or PLD2 and RalA function together to facilitate LD growth. Additionally, FIPI inhibits PLD by binding to the catalytic site, suggesting that the effects of PLD inhibition are directly related to its enzymatic activity, the production of PA.
Figure 5. PLD1 is required for RalA-dependent starvation-induced LD accumulation.
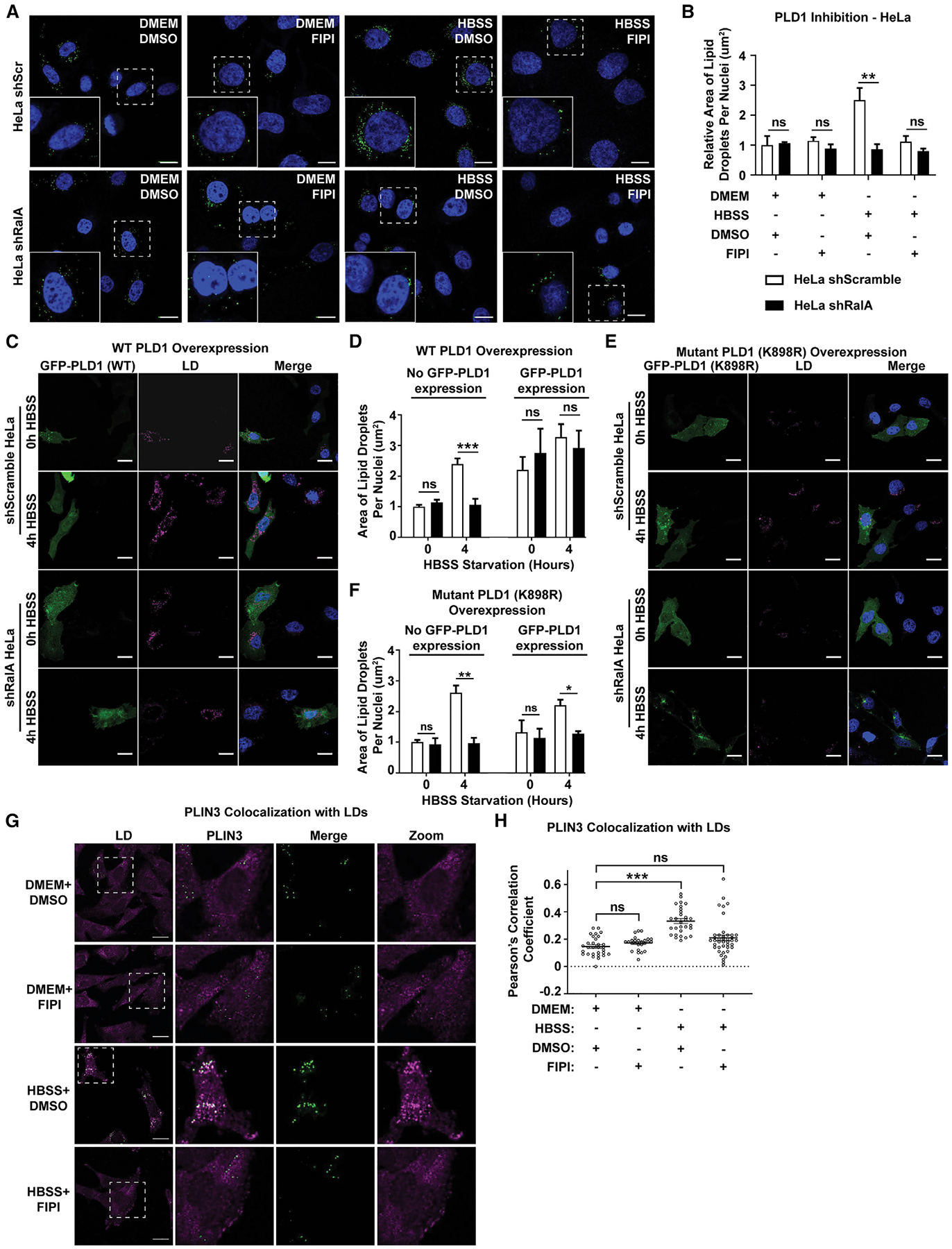
(A) Representative images of LDs in scramble and RalA KD HeLa cells incubated in DMEM or HBSS media ± 750 nM FIPI for 4 h. Scale bars, 20 μm.
(B) Quantification of LD area per cell from (A). At least 100 cells were analyzed for each time point. n = 3. mean ± SEM. **p < 0.01.
(C and E). Representative images of LDs in scramble and RalA KD HeLa cells expressing GFP-PLD1 (C) or GFP-PLD1 K898R (E). Cells were starved in HBSS for the indicated time points. LDs (magenta) labeled with LipidTOX deep red. Scale bars, 20 μm.
(D and F) Quantification of LD area per cell from (C) and (E). At least 40 cells were analyzed per time point. n = 3. mean ± SEM. *p < 0.05, **p < 0.01,***p < 0.001.
(G) Representative images of PLIN3 (magenta) and LDs (green) in MEFs in DMEM or HBSS media ± 750 nM FIPI for 4 h. Scale bar, 20 μm.
(H) Relative colocalization of PLIN3 and LDs from (G) performed by Pearson’s correlation coefficient. Each point on the graph represents a Pearson’s coefficient value for one cell. At least 30 cells were analyzed per time point. n = 2. mean ± SEM. ***p < 0.001.
See also Figure S5.
To gain further insight into whether PLD1 acts upstream or downstream of RalA, we transiently transfected GFP-PLD1 into shRalA HeLa cells and asked whether overexpression of PLD1 rescues LD accumulation during starvation. We took advantage of the fact that transient transfection efficiently delivers GFP-PLD1 to some but not all HeLa cells and compared LD accumulation in non-transfected cells (cells with no GFP expression) to cells transfected with GFP-PLD1 (cells with GFP expression). Remarkably, overexpression of GFP-PLD1 in shRalA HeLa rescues LD accumulation to levels comparable to control (cells with no GFP expression) (Figures 5C and 5D). The ability of cells to overcome the loss of RalA with the addition of PLD1 suggests that PLD1 acts downstream of RalA. To further characterize the role of PLD1 activity in LD accumulation, we used a catalytically inactive PLD1 that has a lysine-to-arginine point mutation at amino acid 898 (K898R) (Sung et al., 1997). Interestingly, transfection of the catalytically inactive GFP-PLD1 (K898R) fails to rescue LD accumulation during starvation in shRalA HeLa cells (Figures 5E and 5F). This finding complements the results from the FIPI experiments and further implicates PLD1 activity, the production of PA, in LD accumulation.
As RalA is required for PLIN3 recruitment to LDs, we sought to determine whether PLD1 and PA similarly impact PLIN3 recruitment to LDs during nutrient stress. PA is a bioactive molecule involved in lipid biogenesis, signaling, and protein recruitment. Due to its cone-shape structure, PA introduces a negative curvature on membranes that can facilitate membrane protein association. A study using artificial phospholipid monolayers showed increased PLIN3 insertion into monolayers enriched with PA (Mirheydari et al., 2016). Based on this study, we hypothesized that PLD1-dependent PA production is necessary for proper PLIN3 recruitment to budding or fully formed LDs and subsequent growth of LDs during nutrient starvation. To test this hypothesis, we measured PLIN3 localization upon inhibition of PLD1 activity by using FIPI during HBSS starvation. Similar to the previous experiment, PLIN3 relocalizes to LDs upon HBSS starvation in control WT MEFs treated with DMSO (Figures 5G and 5H). However, PLIN3 fails to relocalize to LDs in MEFs treated with FIPI (Figures 5G and 5H). This result is consistent with the hypothesis that PLIN3 recruitment during HBSS starvation is dependent on PA production.
RalA recruits PLD1 to lysosomes during nutrient stress
RalA and PLD1 are membrane-associated proteins (Han et al.,2002;Kinsella et al.,1991). RalA is localized to the PM, endosomal system, lysosomes, and mitochondria under different cellular conditions (Chen et al., 2006; Corrotte et al., 2010; Kashatus et al., 2011; Neyraud et al., 2012; Shipitsin and Feig, 2004). Like RalA, PLD1 is found in a diversity of organelles including the PM, Golgi apparatus, nucleus, ER, endosomes, and lysosomes (Brown et al., 1998; Corrotte et al., 2010; Du et al., 2003; Freyberg et al., 2001; Toda et al., 1999). Additionally, both PLD1 and RalA can localize to LDs in certain cells (Bersuker et al., 2018; Liu et al., 2004; Nakamura et al., 2005). We monitored the localization of fluorescently tagged RalA and PLD1. Interestingly, RalA and PLD1 colocalize under nutrient-replete conditions and their colocalization is not significantly altered upon starvation (Figure S6A). To our surprise, neither RalA nor PLD1 are detected at LDs (Figure S6B). However, we observe significant redistribution of both proteins to internal membranes upon starvation. Given that autophagy is induced during nutrient stress, we sought to determine if RalA relocalizes to autophagosomes or the endolysosomal system. We observe minimal RalA association with autophagosomes or early endosomes (Figures S6C and S6D). However, RalA clearly localizes to late endosomal/lysosomal vesicles labeled with GFP-Rab7 under both fed and starved conditions (Figure 6A). Additionally, RalA forms ring-like structures around Limp2-positive lysosomes under starved, but not fed, conditions (Figures 6A and 6B). Consistent with RalA and PLD1 functioning together during nutrient stress, we also observe that a subset of GFP-PLD1 becomes enriched at lysosomes during starved, but not fed, conditions (Figures 6C and 6D). Furthermore, the redistribution of PLD1 to the lysosome is dependent on RalA (Figures 6C and 6D). Collectively, these data support a model in which RalA recruits PLD1 to lysosomes upon nutrient starvation.
Figure 6. RalA recruits PLD1 to lysosomes during HBSS starvation.
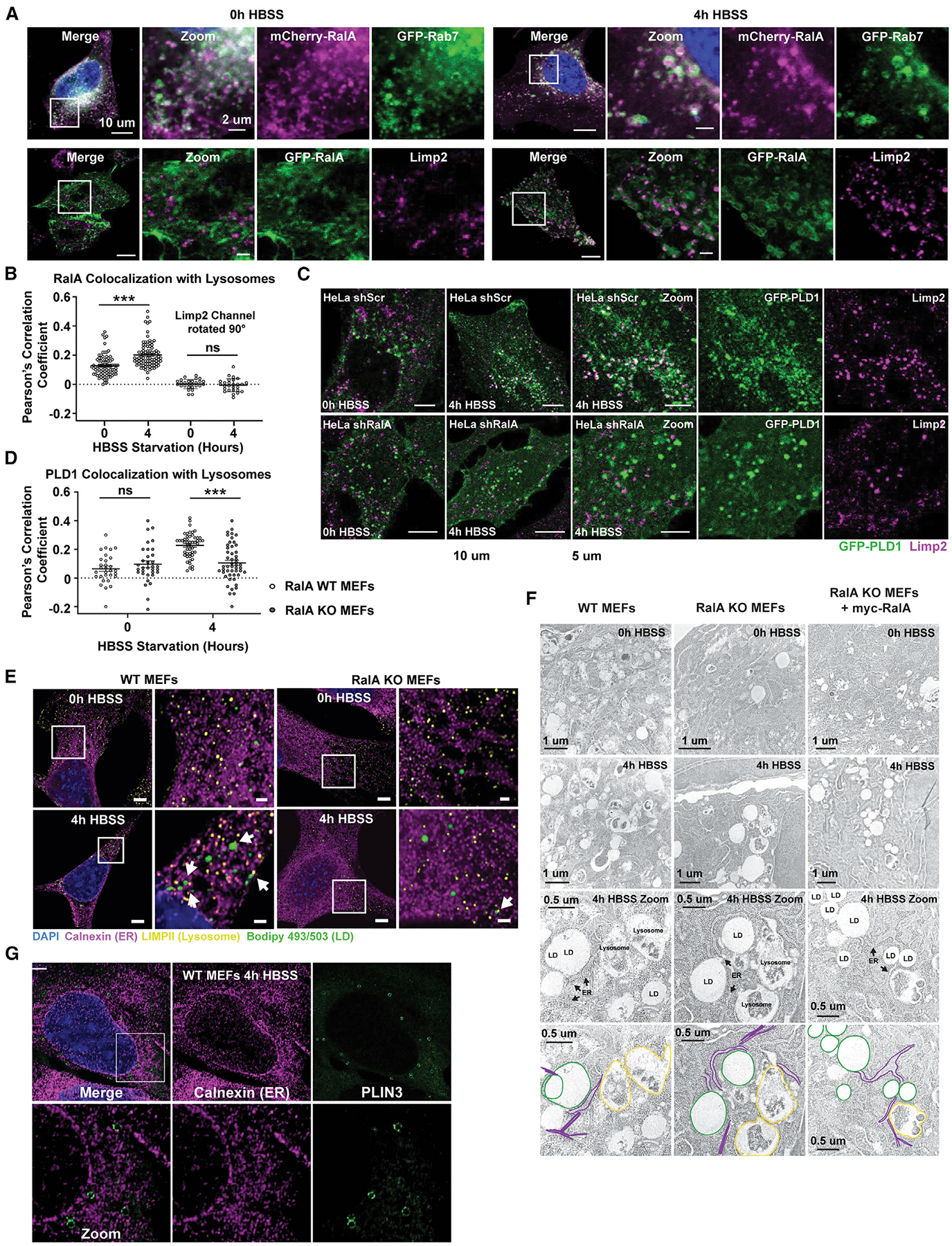
(A) Representative images of fluorescently tagged RalA localization to late endosomes (GFP-Rab7) and lysosomes (Limp2 ab) in HeLa cells. Cells were starved in HBSS for the indicated time points. Scale bar, 10 μm in merge panels and 2 μm in zoom panels.
(B) Quantification of relative colocalization of GFP-RalA and Limp2 from (A) performed by Pearson’s correlation coefficient. Each point on the graph represents a Pearson’s coefficient value from one cell. At least 50 cells were analyzed per time point. n = 3. mean ± SEM. ***p < 0.001.
(C) Representative images of lysosomes (magenta) and GFP-PLD1 (green) in scramble and RalA KD HeLa cells starved in HBSS. Scale bar, 10 μm for merge panels and 5 μm for zoom panels.
(D) Quantification of relative colocalization from (C) performed by Pearson’s correlation coefficient. Each point on the graph represents a Pearson’s coefficient value for one cell. At least 50 cells were analyzed per time point. n = 3. mean ± SEM. ***p < 0.001.
(E) Airyscan images of LDs, lysosomes, and ER localization in WT and RalA KO MEFs. LDs in green (Bodipy 493/503). Lysosomes in yellow (Limp2 ab). ER in magenta (calnexin ab). Cells were starved in HBSS for the indicated time points. White arrows point to instances of close proximity of organelles. Scale bar, 5 μm for merge images and 2 μm for zoom images.
(F) Electron micrographs of WT, RalA KO, and RalA KO+myc-RalA MEFs during 0 h and 4 h of HBSS starvation. LDs, lysosomes, and the ER are annotated and outlined.
(G) Airyscan images of PLIN3 (green) and calnexin (magenta) in WT MEFs after 4 h of HBSS starvation. Scale bar, 5 μm for merge and 2 μm for zoom.
See also Figure S6.
The localization of RalA and PLD1 to lysosomes but not LDs during HBSS starvation raises the question of how lysosomal RalA and PLD1 can influence LD accumulation and PLIN3 recruitment. We envision two plausible ways, both involving organelle contacts. First, there may be direct contacts between lysosomes and LDs, which has been reported in mammalian and yeast cells (Drizyte-Miller et al., 2020; Hariri et al., 2019; Kaushik and Cuervo, 2015; Wang et al., 2014; van Zutphen et al., 2014). Contact between lysosomes and LDs could allow for PA generation at the lysosome and subsequent transfer to LDs to promote PLIN3 recruitment. Recent studies have highlighted stable LD-lysosomal contact sites involved in lipid and protein transfer (Kaushik and Cuervo, 2015; Schroeder et al., 2015; Schulze et al., 2020). Second, both lysosomes and LDs may be connected by a third organelle, the ER. The ER is the site of LD biogenesis and makes membrane contacts with both lysosomes and LDs (Henne et al., 2015; Jacquier et al., 2011; Pan et al., 2000; Schuldiner and Bohnert, 2017; Szymanski et al., 2007; Wilfling et al., 2013). Lysosomal PA may traffic to the ER through ER-lysosome contacts, where it can promote LD biogenesis through PLIN3 recruitment to sites of LD biogenesis. Consistent with both of these models, high-resolution airyscan confocal analysis of ER, lysosomes, and LDs reveals direct lysosome-LD contacts following HBSS starvation, as well as multiple ER-lysosome and ER-LD contacts (Figure 6E). Notably, we still observe these contacts in RalA KO MEFs, suggesting that the defect in LD growth is due to the loss of PLD1 recruitment, rather than a loss of organelle contact. Importantly, we also observe close proximity between these organelles by electron microscopy (Figure 6F). These findings are in agreement with a recent study in yeast showing that ER-vacuole contacts become sites for LD biogenesis during nutrient stress (Hariri et al., 2018). Given the close proximity of the ER to the lysosomes and LDs, we hypothesized that PLIN3 redistribution during HBSS starvation in WT MEFs occurs at sites of LD biogenesis on the ER. PLIN3 has previously been shown to be recruited to nascent LDs emerging from the ER (Kassan et al., 2013; Skinner et al., 2009). Consistent with this, high-resolution confocal microscopy reveals PLIN3 ring-like structures forming adjacent to and budding from the ER in WT MEFs upon HBSS starvation (Figure 6G). Combined, these results suggest that specific organelle contacts between lysosomes, ER, and LDs are critical for RalA-induced LD accumulation.
Enrichment and depletion of PA at lysosomes impact LD accumulation and PLIN3 redistribution
The model that PLD1-derived PA generated at the lysosome traffics to LDs to support PLIN3 recruitment and LD expansion predicts that independently increasing PA levels at either lysosomes or LDs should rescue starvation-induced LD growth in the absence of RalA. To test this prediction, we initially targeted PLD1 directly to LDs by fusing it in frame with the C terminus of PLIN2 (GFP-PLIN2-PLD1) and stably introducing it into WT and RalA KO MEFs (Figure S7A). Although both GFP-PLIN2-PLD1 and the control GFP-PLIN2 localize to LDs (Figure S7B), only GFP-PLIN2-PLD1 expression is able to rescue HBSS-induced LD growth in RalA KO cells (Figures S7C, S7D, and S7E). Importantly, GFP-PLIN2-PLD1 expression also rescues recruitment of PLIN3 to LDs in RalA KO MEFs, indicating that increasing the levels of PA at LDs is sufficient to promote PLIN3 recruitment and LD growth during nutrient stress (Figure S7F). Next, to test whether recruitment of PLD1 to lysosomes independently of RalA is sufficient to rescue the loss of PLIN3 recruitment and LD growth, we fused PLD1 to the C terminus of TMEM192, a lysosomal transmembrane protein with both N and C termini facing the cytosol, and confirmed lysosomal localization in both HeLa cells and MEFs (Figures 7A, 7B, and S7G). Expression of lysosome-targeted PLD1 (mCherry-TMEM192-PLD1), but not the mCherry-TMEM192 control, rescues both LD growth and PLIN3 recruitment in RalA KO MEFs (Figures 7C, 7E, and 7G). These data demonstrate that the function of RalA in starvation-induced LD growth can be replaced with a lysosome-targeted PLD1 and thus further predict that decreasing the levels of PA at the lysosome independent of RalA or PLD1 manipulation should recapitulate the effects of RalA or PLD1 inhibition. To test this prediction, we fused the PA phosphatase LIPIN1 to the C terminus of TMEM192 (mCherry-TMEM192-LIPIN1) and stably expressed it in WT and RalA KO MEFs (Figures 7B and S7G). Consistent with our model, expression of mCherry-TMEM192-LIPIN1, but not the control mCherry-TMEM192, impairs both LD accumulation and PLIN3 recruitment in WT MEFs during HBSS starvation (Figures 7C, 7D, and 7F).
Figure 7. Enrichment and depletion of PA at lysosomes impact LD accumulation and PLIN3 redistribution.
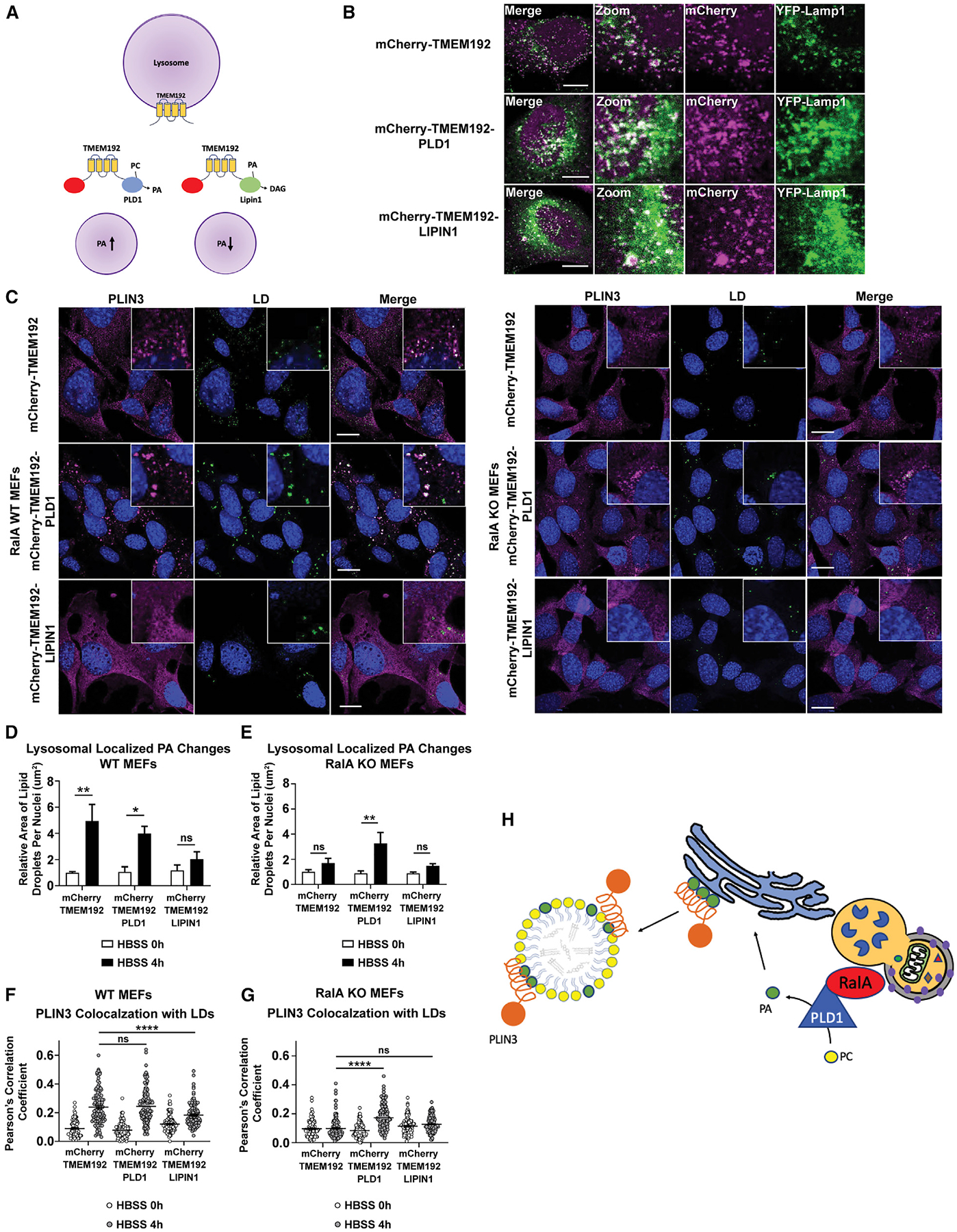
(A) Schematic of lysosomal-localized mCherryTMEM192 with PLD1 or LIPIN1 fused to the C terminus.
(B) Localization of mCherryTMEM192, mCherryTMEM192-PLD1, and mCherry-TMEM192-LIPIN1 to lysosomes (green, Limp2 ab) in HeLa cells. Scale bar, 10 μm.
(C) Representative images of PLIN3 (magenta) and LDs (green) in WT and RalA KO MEFs expressing either mCherryTMEM192, mCherryTMEM192-PLD1, or mCherryTMEM192-LIPIN1 during 4 h of HBSS starvation. Scale bar, 20 μm.
(D and E) Quantification of LD area per cell in WT MEFs (D) and RalA KO MEFs (E) expressing the lysosomal-targeted fusion proteins. At least 100 cells were analyzed per time point. n = 4. mean ± SEM. *p < 0.05, **p < 0.01.
(F and G) Quantification of relative colocalization between PLIN3 and LDs in WT MEFs (F) and RalA KO MEFs
(G) during 4 h of HBSS starvation. Colocalization performed by Pearson’s correlation coefficient. Each point on the graph represents a Pearson’s coefficient value for one cell. At least 50 cells were analyzed per time point. n = 3. mean ± SEM. ****p < 0.0001.
(H) Schematic summarizing the model for RalA-dependent-starvation-induced LD accumulation.
Collectively, the data from these experiments demonstrate that manipulation of PA at the lysosome is sufficient to impact both LD growth and PLIN3 recruitment during starvation and is consistent with the model that RalA and PLD1 relocalize to lysosomes during nutrient-stress-induced autophagy for which PLD1 converts PC to PA. This PA then traffics to the ER where it supports LD growth through the recruitment of PLIN3 to budding or newly formed LDs (Figure 7H).
DISCUSSION
How cells respond to fluctuations in nutrients has implications for both normal and pathological physiology. LDs have emerged as important metabolic hubs and have an important function during nutrient starvation in providing FAs for energy while buffering lipotoxic effects (Nguyen et al., 2017; Listenberger et al., 2003; Rambold et al., 2015). Here, we describe a pathway through which RalA and PLD1 regulate FA storage during cellular starvation. Our data indicate that RalA acts downstream of autophagy to directly facilitate LD growth under nutrient stress through the recruitment of PLD1 to lysosomes and the localized production of PA. Although, how this PA is transported to sites of LD biogenesis remains to be determined, we demonstrate that lysosomal-generated PA facilitates recruitment of PLIN3 and subsequent LD expansion.
RalA recruitment of PLD1 to lysosomes is consistent with its function in directing proteins to distinct cellular compartments. During mitosis, RalA recruits RalBP1 to mitochondria to promote mitochondrial fission (Kashatus et al., 2011). Similarly, RalA recruits Filamin to the cell surface to induce filopodia formation (Ohta et al., 1999). Although RalA can precipitate PLD1 activity in a nucleotide-independent manner (Jiang et al., 1995), whether lysosomal recruitment of PLD1 by RalA is through direct binding remains unclear. Intriguingly, we observe only a subpopulation of PLD1 relocalized to lysosomes. Both RalA and PLD1 share overlapping localizations under certain conditions. A subpopulation of RalA and PLD1 are localized at endosomes, and it is plausible that only this endosomal pool is relocalizing to lysosomes. Consistent with this possibility, we detect RalA on late endosomes. Other studies report that PLD1 partially relocalizes from the endosomal compartment to LC3 puncta and lysosomes in HeLa cells during nutrient deprivation (Dall’Armi et al., 2010). Interestingly, in HEK293 cells, PLD1 translocates to lysosomes during amino acid stimulation in a manner dependent on Vps34, a protein required for autophagy (Yoon et al., 2011). It is likely that PLD1 movement in the cell is regulated by multiple proteins and cellular conditions, adding spatial and temporal control of its cellular functions.
Further studies are needed to fully understand how RalA and PLD1 cooperate to facilitate LD accumulation during nutrient deprivation. We demonstrate that PLD1’s ability to rescue starvation-induced LD accumulation in RalA-deficient cells depends on its enzymatic activity. The enzymatic product PA is a multi-functional lipid involved in intracellular trafficking, lipid signaling, and protein recruitment (Athenstaedt and Daum, 1999; Tanguy et al., 2019). Evidence has emerged of PA involvement in LD formation and growth. Elevated PA levels in yeast cells lead to LD coalescence and subsequent formation of “supersized” LDs (Fei et al., 2011). Additionally, PA and PLD1 are required for LD formation in a microsome-based system (Marchesan et al., 2003). Given its unique anionic, conical shape, PA is proposed to be involved in LD formation at the ER and scission of LDs from the ER (Skinner et al., 2009). Others have reported increased PLD1 activity during HBSS starvation (Dall’Armi et al., 2010). An increase in PLD1 activity might suggest increased cellular PA levels; however, we did not observe significant changes in total PA levels in MEFs following starvation. Because PA is a precursor to downstream metabolites such as diacylglyceride (DAG), it may be quickly converted upon synthesis during nutrient starvation, explaining the lack in detectable changes. Alternatively, increased PA at the lysosome may be offset by the loss of PA production from other sites. A number of PLD1-interacting proteins such as Arf, PKC, and Rho increase PLD1 activity upon binding (Brown et al., 1993; Frohman, 2015; Kuribara et al., 1995; Singer et al., 1996). Although RalA interacts with PLD1, this interaction has no effect on PLD1 activity (Luo et al., 1998). We speculate that the RalA-PLD1 axis is facilitating LD accumulation without enhancing its ability to generate PA. Instead, it is the localized production of PA at lysosomes that leads to LD accumulation. This is consistent with the lysosome releasing PC, a substrate for PLD1, from recycled membranes. Remarkably, by forcing PLD1 to the lysosome in MEFs lacking RalA, LD accumulation as well as PLIN3 relocalization to LDs during HBSS starvation is rescued. This result also confirms that the RalA-PLD1 interaction is not necessary for PLD1-dependent PA production. However, this finding does not rule out the involvement of other proteins, such as the PLD1-activating proteins mentioned above.
Although the precise mechanism of PA involvement remains to be fully elucidated, gaining a better understanding of the cellular distribution of PA during nutrient starvation would prove useful. PA can potentially be used for TAG production and packaged into LDs for insertion into the ER bilayer to facilitate LD formation or for insertion into the LD monolayer to facilitate protein recruitment, among other possibilities. Our data are consistent with PLD1-produced PA being used in any of these ways. PLIN3 rapidly localizes to growing LDs upon FA-stimulated conditions (Bulankina et al., 2009; Kassan et al., 2013; Nose et al., 2013). PA and DAG are proposed to recruit PLIN3 onto LD monolayers as well as LD formation sites on the ER (Mirheydari et al., 2016; Skinner et al., 2009). Upon nutrient starvation, we observe a remarkable RalA-dependent relocalization of PLIN3 from the cytosol to LDs. These data support a model in which RalA function during LD formation is to promote PLIN3 recruitment through compartmentalization of PA production. PA produced at the lysosome instead of the ER raises the question of how it is transferred between these organelles. Close inspection by EM and high-resolution confocal microscopy shows clear examples of LDs, lysosomes, and ER in close proximity to each other. These observations suggest that PA transfer is through membrane contact sites (MCSs) and not vesicular trafficking. It is unlikely that PA is entering the ER by passive diffusion. A more likely scenario is that a lipid transfer protein (LTP) shuttles PA from the lysosome to the ER. Interest in ER-endolysosomal MCSs has grown in recent years. A variety of LTPs are enriched at ER-endolysosomal MCSs (Alpy et al., 2013; Höglinger et al., 2019; Kvam and Goldfarb, 2004; Lim et al., 2019; Ouimet et al., 2016; Rocha et al., 2009). Although a specific PA LTP at the ER has yet to be reported, PA LTPs have been identified in other organelles, such as the mitochondria (Connerth et al., 2012). The cellular coordination in forming spatial localization of organelles during HBSS starvation is unclear, although RalA does not appear to be involved in this process, as we observe the ER, LD, and lysosomes in close proximity regardless of RalA expression.
Several questions are raised by these findings. How does RalA sense starvation and through what molecular mechanism? Although current evidence does not support RalA nucleotide binding in this regulation, RalA contains a number of post-translational modification sites that may be playing a role (Neyraud et al., 2012; Sablina et al., 2007; Wu et al., 2005). Phosphorylation at serine 194 relocalizes RalA from the PM to the mitochondria (Kashatus et al., 2011). Additionally, phosphorylation of RalB at serine 198 relocalizes RalB from the PM to the perinuclear region (Martin et al., 2012). Furthermore, non-degradative mono-ubiquitination on lysine residues can impact Ral localization (Neyraud et al., 2012). We speculate RalA phosphorylation and/or ubiquitination following nutrient starvation drives its movement to lysosomes. We recently reported that RalA accumulates at depolarized mitochondria in a clathrin-meditated-endocytosis-dependent manner (Pollock et al., 2019). It is possible that RalA relocalization to lysosomes during nutrient stress is also reliant on the endocytic pathway. A key function of the endocytic pathway is internalization of nutrients found outside the cell. Imported nutrients are sorted into the early endosome, a subset of which mature into late endosomes and eventually fuse with lysosomes. Consistent with this idea and in agreement with other reported studies, we detect RalA on late endosomes under fed conditions and on lysosomes under both fed and starved conditions. These data point to a mechanism by which RalA can localize to lysosomes, but further exploration of the upstream signaling and RalA trafficking in the cell is needed to fully understand the RalA response to nutrient starvation.
In summary, we report that RalA and PLD1 function to facilitate LD accumulation under nutrient stress. Cellular regulation of energy stored as TAGs is critical. Exceeding the physiological capacity of TAG storage is linked to diseases such as obesity, diabetes, and cancer. Several studies in recent years report altered LD content in different types of malignancies and metabolic disorders. Additionally, dysfunction of LD-related processes like lipolysis and lipophagy lead to physiological conditions such as neutral lipid storage disease, fatty liver disease, and atherosclerosis. As our knowledge of LD biology expands, new therapies and preventative measures for diseases will arise. Further characterization of the mechanism by which RalA and PLD1 regulate LDs will contribute to these efforts.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David Kashatus (kashatus@virginia.edu).
Materials availability
All material generated in this study is available upon request by the lead contact, David Kashatus (kashatus@virginia.edu).
Data and code availability
All data reported in this paper will be shared upon request by the lead contact. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
HeLa cells were obtained from ATCC. Mouse Embryonic Fibroblasts (MEFs) were obtained from Christopher Marshall (Peschard et al., 2012). Sex of HeLa cells is female. Sex of MEF cells used in this study has not been determined. Cells were kept at 37°C with 5% CO2. Both HeLa and MEFs were maintained in Dulbecco’s Modified Eagle Medium (DMEM - GIBCO) supplemented with 10% Fetal Bovine Serum (FBS – GIBCO) and 1% Penicillin-Streptomycin (GIBCO). For all nutrient starvation studies performed, cells were replenished with fresh DMEM 1 hour prior to starvation in Hank’s Balanced Salt Solution (HBSS – GIBCO) supplemented with 800 μM MgCl2 and 1.8 mM CaCl2. HeLa cells, and all of the stable genetic variants of those HeLa cells, are periodically authenticated using short tandem repeat (STR) profiling. MEF cell lines are regularly authenticated through PCR and immunoblot to confirm expression or deletion of genes.
METHOD DETAILS
Chemicals
The following chemicals were used at the indicated concentrations: 100 μM Chloroquine (MP Biomedicals), 200 nM Bafilomycin A1 (Sigma-Aldrich), 750 nM FIPI (Calbiochem), 1 ug/ml BODIPY 493/503 (Invitrogen) 1 μM BODIPY 558/568 C12 (Invitrogen), 1:250 LipidTOX Deep Red (Invitrogen). All inhibitor concentrations were used at sub-lethal doses.
Western Blotting
Cells were lysed in RIPA buffer containing protease inhibitors. Protein concentration was determined by Bio-Rad Protein Assay (Bio-Rad). Equivalent protein was loaded and resolved by SDS-PAGE. Gels were transferred onto PVDF membranes (Millipore) and immunoblotted using the following antibodies: anti-RalA (BD Transduction Laboratories), anti-GAPDH (Cell Signaling Technology - CST), anti-β-Actin (CST), anti-β -Tubulin (CST), anti-4E-BP1 (CST), anti-phospho-4E-BP1 (Thr37/46 – CST), anti-LC3B (CST), anti-Perilipin 3/Tip47 (Santa Cruz), anti-RalBP1 (Santa Cruz), anti-Vinculin (CST). Goat α-rabbit and goat α-mouse HRP secondary antibodies (Jackson ImmunoResearch Laboratories, Inc) were used for detection. Western blot images were taken on a ChemiDoc (Bio-Rad) using chemiluminescence detection with WesternBright ECL (Advansta). Where applicable, band pixel intensities were quantified using ImageJ.
Immunofluorescence
Cells were seeded at 30% confluency onto glass coverslips (Fisher Scientific). After 48 hours, cells were treated with HBSS/Oleic Acid/inhibitors as described elsewhere in methods. After treatment, cells were fixed with 3% formaldehyde in PBS for 10 min, washed 3x in PBS and permeabilized in microscopy buffer for 10 min. Microscopy buffer contains 0.1% Saponin (Sigma-Aldrich), 1% BSA (Fisher Scientific) in 1x PBS. Cells were then blocked in either 5% normal goat or 5% normal donkey serum (Cell Signaling Technology) in microscopy buffer for 20 mins. After blocking, cells were incubated for 1 hr in one of the following antibodies diluted in microscopy buffer: anti-Perilipin2/ADRP (Santa Cruz), anti-Perilipin 3-Tip47 (Santa Cruz), anti-Limp2 (Gift from John D. Castle – University of Virginia), anti-Calnexin (Gift from John D. Castle). Cells were then washed 3x in PBS and incubated in one of the following secondaries diluted in microscopy buffer for 30 mins: Alexa Fluor 488 (Invitrogen), Alexa Fluor 568 (Invitrogen) Alexa Fluor 647 (Invitrogen). Lastly, cells were washed 3x in PBS and mounted in DAPI (4′,6-diamidino-2-phenylindole) solution (Cell Signaling Technology) to visualize nuclei. Images were taken on Zeiss LSM 700, 710, and 880 confocal microscopes (UVA Advanced Microscopy Facility). Airyscan images were taken using Zeiss LSM 880 (UVA Advanced Microscopy Facility).
For lipid droplet imaging, cells were seeded and fixed in formaldehyde as described above. Next, cell were incubated in 1 mg/ml BODIPY 493/503 (Invitrogen), 1:200 LipidTOX Deep Red (Invitrogen), or 100 μM Monodansylpentane (Abcepta) in PBS for 30 min. In immunofluorescence studies with co-staining of LDs, 1 mg/ml BODIPY 493/503 was added to the secondary antibody step. After staining, cells are washed 3x in PBS before mounting onto slides.
Image Analysis
Zeiss Zen, ImageJ and Fiji software were used to process and analyze images captured by confocal microscopy. ImageJ was used to process electron micrographs. LD quantitation was done on Image/Fiji. Uniform threshold function was applied to LD images and used to generate a mask on LDs. Analyze particles function was used to generate total area of LDs. Fiji coloc2 function was used to generate Pearson’s correlation coefficient’s for colocalization studies. ImageJ was used to quantitate mCherry and GFP LC3B punctae and ratios.
Transfection and Constructs
For transient transfections, cells were seeded at 30% confluency onto glass coverslips (VWR). After 24 hours, cells were transfected with the indicated constructs using FuGENE 6 (Promega). 500 ng of the following fluorescent constructs were used per coverslip: pEGFP-RalA, mCherry-RalA, pEGFP-PLD1, pEGFP-PLD1 (K898R), pEx-YFP-LC3, pEGFP-Rab7, pEGFP-EEA1. pEGFP-PLD1 and mutant pEGFP-PLD1 (K898R) were gifts from the Michael A. Frohman lab (Stony Brook University). pEGFP-Rab7 was a gift from the James E. Casanova lab (UVA). 24–48 hours post-transfection, cells were fixed in 3% formaldehyde in PBS for 5 mins, washed in PBS and mounted on glass slides.
All stable cell lines were generated using Calcium-phosphate transfection using the retroviral packaging vector, pCL-10A1 (Novus Biologicals) followed by antibiotic selection. The following constructs were used to generate stable cell lines: pBabe puro mCherry-EGFP-LC3B, pWZL blasti mito-YFP, pbabe puro myc-RalA, pbabe puro myc-RalA S28N, pbabe puro myc-RalA Q72L, pbabe puro GFP-PLIN2, pbabe puro GFP-PLIN2-PLD1, pbabe neo mCherryTmem192, pbabe neo mCherryTMEM192-PLD1, pbabe neo mCherryTMEM192-LIPIN1. Site directed mutagenesis was used generate pbabe puro myc-RalA S28N and pbabe puro myc-RalA Q72L from pbabe puro myc-RalA (WT).
Knockdowns and Knockouts
For stable knockdown of RalA, short hairpin RNA (shRNA) target sequence (5′- AAGACAGGTTTCTGTAGAAGA-3′) was used as reported previously (Lim et al., 2005). For stable knockdown of PLIN3, shRNA target sequence (5′- GAATGAGACATGTGTTTAA –3′) was used as reported previously (Gu et al., 2019; Sztalryd et al., 2006). For stable knockdown of ATG5, shRNA target sequence (5′- CCTTGGAACATCACAGTACAT –3′) was used as reported previously (Hollomon et al., 2013). All shRNA constructs were made in a pSuperior-Retro vector backbone and stable cell lines expressing constructs were generated as describe above.
Knockout of RalBP1 was done by CRISPR. The guide RNAs (sgRNA) target sequences are listed below. sgRNA sequences were cloned into lentiCRISPR v2 vector (addgene #52961). psPAX2 (Addgene #12260) and pCMV-VSV-G (Addgene #8454) was used to package shRNA constructs and generate lentiviral particles via calcium-phosphate transfection.
sgRalBP1–1 (exon 2): 5′-CACCGTCACTAGGGCTGCTGCTCGG-3′
sgRalBP1–2 (exon 3): 5′- CACCGCAGCTGATGTTGTTAAACAG-3′
sgRalBP1–3 (exon 4): 5′-CACCGAAAAGCAGCCTATGACCGAG –3′
Fatty Acid Pulse Chase Assay
Fluorescent fatty acid pulse chase experiments were performed as described previously with slight modifications (Rambold et al., 2015). MEFs were seeded at 30% confluency onto glass coverslips. After 48 hours, cells were incubated in DMEM containing 1 μM BODIPY 558/568 C12 (Red C12 – Invitrogen) overnight. Cells were washed in DMEM to remove Red C12 and incubated in DMEM for 1 hour. Cells were then chased in HBSS for the indicated time points. At each time point, cells were washed and fixed in 3% formaldehyde before mounting onto glass slides in DAPI so006Cution (Cell Signaling Technology). For LD co-staining experiments, cells were incubated with 1 mg/ml BODIPY 493/503 (Invitrogen) for 30 mins after fixation to visualize LDs. For mitochondria co-staining experiments, MEFs stably expressing Mito-YFP to visualize the mitochondria were used.
Oleic Acid Stimulation
Oleic acid was conjugated to fatty acid-free BSA (Fisher Scientific) at an approximately 6:1 molar ratio. Oleic acid was solubilized in 70% EtOH and heated at 70°C for 15 min. Fatty acid-free BSA was dissolved in DMEM at 37°C for 30 min with vortexing. Heated EtOH containing oleic acid was slowly added to BSA in DMEM and mixed thoroughly. Cells were seeded onto coverslips as described above. DMEM was replenished on cells 1 hour prior to oleic acid stimulation. Cells were treated with a final concentration of 200 μM oleic acid. LDs were stained and imaged as described above.
Ral GTP Pulldown
Generation of RalBP1-RBD Sepharose beads were done as described previously (Pollock et al., 2019). Briefly, BL21 Escherichia coli cells expressing pGEX-KG-RalBD were induced with Isopropyl β- d-1-thiogalactopyranoside (IPTG) and rocked at 37°C for 3 hours. Bacteria pellets were lysed in PBST containing inhibitors and sonicated. Supernatant containing GST-RalBP1-RBD was collected and rocked overnight with Gluthathione Sepharose 4B (Sigma-Aldrich) beads overnight at 4°C. Beads were washed and resuspended in lysis buffer. To measure RalA GTP status, cells were incubated in HBSS starvation media for 0, 1, 2, and 4 hours. At each endpoint, cells were washed in ice-cold PBS, scraped and pelleted. Cells were lysed in GTP lysis buffer (1% NP-40, 50 mM Tris pH 7.5, 200 mM NaCl, 10 mM MgCl2, 1% aprotinin, 1 mM PMSF, 0.5 mM DTT), spun down and supernatant was collected. Protein concentration was determined by Bradford Assay (Bio-Rad). 500 ug of protein was incubated with GST-RalBP1-RBD beads for 45 minutes at 4°C. Total RalA and GTP-RalA levels were detected by SDS-PAGE and immunoblotting.
Lipid Extraction
A modified Folch method (CHCl3:MeOH:H2O/2:1:2; 0.1N HCl) was used to extract lipids from RalA MEF cell pellets. Antioxidant BHT (butylated hydroxy toluene) was added at 50 μg/mL during extraction. CHCl3 (2 mL), and MeOH (1 mL) were added with 1.5 mL of ddH2O containing resuspended MEFs in a two-dram vial. 1N HCl (500 μL) was added last to bring the final concentration of acid to 0.1 N. Samples were vortexed, incubated on ice for 20 min, and centrifuged at 2,000 × g for 5 min at 4°C. The organic layer was transferred and aqueous layer extracted with addition of 1.5 mL of 2:1 CHCl3:MeOH solution. The extracted organic layers were combined and dried down under nitrogen stream. Samples were resuspended in 240 μL of 1:1 MeOH:isopropanol and stored at −80°C until further analysis.
LC-MS/MS Analysis of Lipid Extracts
The lipid samples were analyzed by LC-MS/MS. A Dionex Ultimate 3000 RS UHPLC system was used with an analytical column (Kinetex® 1.7 μm C18 100Å, Phenomenex, LC column 100 × 2.1 mm) and reverse phase LC solvents (C: ACN:H2O/50:50, 10 mM NH4HCO2, 1% formic acid; D: ACN:IPA:H2O/10:88:2, 10 mM NH4HCO2, 1% formic acid) with the following gradient: Flowrate 0.25 mL/min, 0 min 65% C, 4 min 40% C, 12 min 15% C, 21 min 0% C, 24 min, 0% C, 24.1 min 100% C, 27 min 0% C, 30 min 100% C, 33 min 0% C, 35 min 65% C. The eluted lipids were ionized by electrospray using a HESI-II probe into an Orbitrap Q-Exactive Plus mass spectrometer (Thermo Scientific). Data acquisition was performed using a Top10 data-dependent (ddMS2) global analysis method that consisted of one full MS1 scan (250–1,200 m/z) followed by 10 MS2 scans of the most abundant ions recorded in the MS1 scan. Lipid identifications and peak alignments were performed using LipidSearch™ software (version 4.0) while quantitative analysis of the aligned intensities was exported and analyzed using Prism GraphPad (version 7.03). Positive and negative ion annotations for each sample were combined and aligned within a chromatographic time window to allow greater confidence in lipid identifications using appropriate MS2 product ions and neutral losses from the compiled dataset in LipidSearch™ analysis software.
Lipid Species Identification from LC-MS/MS Analysis
Data analysis was performed using the LipidSearch™ software package, which used MS1 monoisotopic precursor ions for the in-silico database search followed by the product ions search from MS2 spectra (search parameters: Target database: Q Exactive, SearchType: product, ExpType: LC-MS, Precursor tolerance 5.0 ppm, product tolerance 5.0 ppm, intensity threshold: product ion 1.0%, m-score threshold 2.0). Results from positive and negative ion mode analyses were merged to generate a matched lipid dataset. The searched data were aligned with selected parameters (ExpType: LC-MS, Alignment Method: Median, Toprank Filter: on, Main node Filter: Main isomer peak, m-Score Threshold: 5.0, ID quality filter: A, B, C, D) and curated with species with high-confidence grade scores “A” and “B” for further analysis.
Electron Microscopy
Cells were seeded at 40% confluency onto thermanox coverslips (Fisher Scientific). After 48 hours, cells were treated with HBSS as described elsewhere in methods. Preparation of samples for transmission electron microscopy was done by the UVA Advanced Microscopy Facility. After treatment, cells were fixed in 2% paraformaldehyde/2.5% Glutaraldehyde in 1x cacodylate buffer. Fixed samples were washed in 1x cacodylate buffer and then incubated in 2% osmium tetroxide for 1 hour (postfixation). Samples were then dehydrated by incubating in increasing concentrations of EtOH (50%–100%) for 10 min each followed by incubation in 100% propylene oxide for 10 min. Samples were then incubated in 1:1 propylene oxide/epoxy resin overnight, followed by incubation in 1:2 and 1:4 propylene oxide/epoxy resin for approximately 3 hours each. Afterward, samples were incubated overnight in 100% epoxy resin. After dehydration, samples were embedded in fresh 100% epoxy resin and baked in a 65 degree oven. Following embedding, ultra-thin sections cut at 75 nm were made and placed on mesh copper grids. Samples were post stained for contrast with 0.25% lead citrate and 2% uranyl acetate. Images were taken on JEOL 1230 TEM (UVA Advanced Microscopy Facility.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism was used for graphical representation and statistical analysis of data. Two-way ANOVA with Tukey’s multiple comparison statistical analysis was applied to experiments unless stated otherwise in figure legends. Statistical significance is denoted in figures as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Biological replicates are denoted as n values and are listed in the figure legends for each experiment. n values represent the number of times an experiment was performed. Data are presented as mean ± SEM.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-RalA | BD Biosciences | Cat# 610221; RRID:AB_397618 |
| Rabbit monoclonal anti-β-Tubulin (9F3) | Cell Signaling Technology | Cat# 2128; RRID:AB_823664 |
| Rabbit monoclonal anti-β-Actin (D6AB) | Cell Signaling Technology | Cat# 8457; RRID:AB_10950489 |
| Rabbit monoclonal anti-GAPDH (D16H11) | Cell Signaling Technology | Cat# 5174; RRID:AB_10622025 |
| Rabbit monoclonal anti-4E-BP1 (53H11) | Cell Signaling Technology | Cat# 9644; RRID:AB_2097841 |
| Rabbit monoclonal anti-Phospho-4E-BP1 (Thr37/46) (236B4) | Cell Signaling Technology | Cat# 2855; RRID:AB_560835 |
| Rabbit polyclonal anti-LC3B | Cell Signaling Technology | Cat# 2775; RRID:AB_915950 |
| Mouse monoclonal anti-TIP47 (F-10) (Perilipin3) | Santa Cruz Biotechnology | Cat# sc-390968 |
| Mouse monoclonal anti-ADRP (B-6) (Perilipin2) | Santa Cruz Biotechnology | Cat# sc-377429 |
| Mouse monoclonal anti- RalBP-1 (H-10) | Santa Cruz Biotechnology | Cat# sc-48337; RRID:AB_628198 |
| Mouse monoclonal anti-Limp2 | Gift from John D. Castle | N/A |
| Rabbit polyclonal anti-Calnexin | Gift from John D. Castle | N/A |
| Rabbit polyclonal anti-Vinculin | Cell Signaling Technology | Cat# 4650; RRID:AB_10559207 |
| Chemicals, peptides, and recombinant proteins | ||
| pGEX-KG-RalBD | Pollock et al., 2019 | N/A |
| PLD Inhibitor, FIPI | Calbiochem | Cat# 528245 |
| Chloroquine | MP Biomedicals | Cat# ICN19391910 |
| Bafilomycin A1 | Sigma-Aldrich | Cat# B1793 |
| BODIPY 493/503 | Invitrogen | Cat# D3922 |
| BODIPY 558/568 C12 | Invitrogen | Cat# D3835 |
| HCS LipidTOX Deep Red | Invitrogen | Cat# H34477 |
| Monodansylpentane (MDH) | Abcepta | Cat# SM1000a |
| Fatty Acid-Free BSA | Fisher Scientific | Cat# BP9704100 |
| Oleic acid | Sigma-Aldrich | Cat# O1257 |
| Gluthathione Sepharose 4B | GE Healthcare | Cat# GE17-0756-01 |
| Critical commercial assays | ||
| Protein Assay Dye Reagent Concentrate | Bio-rad | Cat# 5000006 |
| Experimental models: Cell lines | ||
| HeLa Cells | ATCC | N/A |
| RalA flox/flox MEFs | Peschard et al., 2012. Gift from Christopher Marshall. | N/A |
| RalA KO MEFs | Peschard et al., 2012. Gift from Christopher Marshall. | N/A |
| Oligonucleotides | ||
| shRalA: 5′- AAGACAGGTTTCTG TAGAAGA-3′ | Lim et al., 2005 | N/A |
| shPLIN3: 5′- GAATGAGACATGT GTTTAA –3′ | Gu et al., 2019; Sztalryd et al., 2006 | N/A |
| shATG5: 5′- CCTTGGAACATCA CAGTACAT –3′ | Hollomon et al., 2013 | N/A |
| sgRalBP1–1: 5′-CACCGTCACTAGGGCTGCTGCTCGG –3′ | This study | N/A |
| sgRalBP1–2: 5′- CACCGCAGCTGATGTTGTTAAACAG –3′ | This study | N/A |
| sgRalBP1–1: 5′- CACCGAAAAGCAGCCTATGACCGAG –3′ | This study | N/A |
| Recombinant DNA | ||
| pSuperior-Retro-Puro shRalA | Lim et al., 2005 | N/A |
| pBabe-Puro myc-RalA | Gift from Chris Counter | N/A |
| pbabe puro myc-RalA S28N | This study | N/A |
| pbabe puro myc-RalA Q72L | This study | N/A |
| pbabe puro GFP-PLIN2 | This study | N/A |
| pbabe puro GFP-PLIN2-PLD1 | This study | N/A |
| pbabe neo mCherryTmem192 | This study | N/A |
| pbabe neo mCherryTMEM192-PLD1 | This study | N/A |
| pbabe neo mCherryTMEM192-LIPIN1 | This study | N/A |
| pEGFP C1 RalA | Pollock et al., 2019 | N/A |
| pmCherry-C1 RalA | This study | N/A |
| pEGFP-PLD1 | Gift from Michael Frohman | N/A |
| pEGFP-PLD1 (K898R) | Gift from Michael Frohman | N/A |
| pEGFP-Rab7 | Gift from James Casanova | N/A |
| pBabe puro mCherry-EGFP-LC3B | N’Diaye et al., 2009 | Addgene #22418 |
| pEX-YFP-hLC3WT | Tanida et al., 2008 | Addgene #24989 |
| pEYFP-N1-Lamp1 | Sherer et al., 2003 | Addgene #1816 |
| Software and algorithms | ||
| Fiji | Schindelin et al., 2012 | https://fiji.sc |
| GraphPad Prism | GraphPad Inc. | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Zen Microscopy Software | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
Highlights.
RalA and PLD1 are necessary for starvation-induced lipid droplet (LD) formation
RalA recruits PLD1 to lysosomes to promote LD accumulation during starvation
Inhibition of RalA or PLD1 prevents PLIN3 redistribution to growing LDs
PLD1 activity and PA are required for LD accumulation and PLIN3 localization on LDs
ACKNOWLEDGMENTS
We thank J. David Castle and Alex Kreutzberger for advice on lipid analysis and cell fractionation, Stacey Criswell and the UVA Advanced Microscopy Facility for assistance with imaging, and all Kashatus Lab members for their insightful comments and critical feedback. This work was supported by NCI grant number 1R01CA200755 to D.F.K.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109451.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alpy F, Rousseau A, Schwab Y, Legueux F, Stoll I, Wendling C, Spiegelhalter C, Kessler P, Mathelin C, Rio M-C, et al. (2013). STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell Sci 126, 5500–5512. [DOI] [PubMed] [Google Scholar]
- Andersson L, Boström P, Ericson J, Rutberg M, Magnusson B, Marchesan D, Ruiz M, Asp L, Huang P, Frohman MA, et al. (2006). PLD1 and ERK2 regulate cytosolic lipid droplet formation. J. Cell Sci 119, 2246–2257. [DOI] [PubMed] [Google Scholar]
- Athenstaedt K, and Daum G (1999). Phosphatidic acid, a key intermediate in lipid metabolism. Eur. J. Biochem 266, 1–16. [DOI] [PubMed] [Google Scholar]
- Bell M, Wang H, Chen H, McLenithan JC, Gong D-W, Yang R-Z, Yu D, Fried SK, Quon MJ, Londos C, and Sztalryd C (2008). Consequences of lipid droplet coat protein downregulation in liver cells: abnormal lipid droplet metabolism and induction of insulin resistance. Diabetes 57, 2037–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Peterson CWH, To M, Sahl SJ, Savikhin V, Grossman EA, Nomura DK, and Olzmann JA (2018). A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 44, 97–112.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou Y-H, Formstecher E, Maiti M, Hazelett CC, Wauson EM, Balakireva M, et al. (2011). RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasaemle DL, Barber T, Wolins NE, Serrero G, Blanchette-Mackie EJ, and Londos C (1997). Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J. Lipid Res 38, 2249–2263. [PubMed] [Google Scholar]
- Brown HA, Gutowski S, Moomaw CR, Slaughter C, and Sternweis PC (1993). ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 75, 1137–1144. [DOI] [PubMed] [Google Scholar]
- Brown FD, Thompson N, Saqib KM, Clark JM, Powner D, Thompson NT, Solari R, and Wakelam MJO (1998). Phospholipase D1 localises to secretory granules and lysosomes and is plasma-membrane translocated on cellular stimulation. Curr. Biol 8, 835–838. [DOI] [PubMed] [Google Scholar]
- Buers I, Robenek H, Lorkowski S, Nitschke Y, Severs NJ, and Hofnagel O (2009). TIP47, a lipid cargo protein involved in macrophage triglyceride metabolism. Arterioscler. Thromb. Vasc. Biol 29, 767–773. [DOI] [PubMed] [Google Scholar]
- Bulankina AV, Deggerich A, Wenzel D, Mutenda K, Wittmann JG, Rudolph MG, Burger KNJ, and Höning S (2009). TIP47 functions in the biogenesis of lipid droplets. J. Cell Biol 185, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor SB, Urano T, and Feig LA (1995). Identification and characterization of Ral-binding protein 1, a potential downstream target of Ral GTPases. Mol. Cell. Biol 15, 4578–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chardin P, and Tavitian A (1986). The ral gene: a new ras related gene isolated by the use of a synthetic probe. EMBO J 5, 2203–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X-W, Inoue M, Hsu SC, and Saltiel AR (2006). RalA-exocyst-dependent recycling endosome trafficking is required for the completion of cytokinesis. J. Biol. Chem 281, 38609–38616. [DOI] [PubMed] [Google Scholar]
- Chen X-W, Leto D, Chiang S-H, Wang Q, and Saltiel AR (2007). Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev. Cell 13, 391–404. [DOI] [PubMed] [Google Scholar]
- Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, and Langer T (2012). Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338, 815–818. [DOI] [PubMed] [Google Scholar]
- Corrotte M, Nyguyen APT, Harlay ML, Vitale N, Bader M-F, and Grant NJ (2010). Ral isoforms are implicated in Fc γ R-mediated phagocytosis: activation of phospholipase D by RalA. J. Immunol 185, 2942–2950. [DOI] [PubMed] [Google Scholar]
- Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, et al. (2010). The phospholipase D1 pathway modulates macroautophagy. Nat. Commun 1, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries JE, Vork MM, Roemen TH, de Jong YF, Cleutjens JP, van der Vusse GJ, and van Bilsen M (1997). Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J. Lipid Res 38, 1384–1394. [PubMed] [Google Scholar]
- Drizyte-Miller K, Schott MB, and McNiven MA (2020). Lipid Droplet Contacts With Autophagosomes, Lysosomes, and Other Degradative Vesicles. Contact (Thousand Oaks) 3, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Altshuller YM, Vitale N, Huang P, Chasserot-Golaz S, Morris AJ, Bader M-F, and Frohman MA (2003). Regulation of phospholipase D1 sub-cellular cycling through coordination of multiple membrane association motifs. J. Cell Biol 162, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei W, Shui G, Zhang Y, Krahmer N, Ferguson C, Kapterian TS, Lin RC, Dawes IW, Brown AJ, Li P, et al. (2011). A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet 7, e1002201–e1002211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg Z, Sweeney D, Siddhanta A, Bourgoin S, Frohman M, and Shields D (2001). Intracellular localization of phospholipase D1 in mammalian cells. Mol. Biol. Cell 12, 943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA (2015). The phospholipase D superfamily as therapeutic targets. Trends Pharmacol. Sci 36, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry LR, Martin TD, Reiner DJ, and Der CJ (2014). Ral small GTPase signaling and oncogenesis: More than just 15 minutes of fame. Biochim. Biophys. Acta 1843, 2976–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J-Q, Wang D-F, Yan X-G, Zhong W-L, Zhang J, Fan B, and Ikuyama S (2010). A Toll-like receptor 9-mediated pathway stimulates perilipin 3 (TIP47) expression and induces lipid accumulation in macrophages. Am. J. Physiol. Endocrinol. Metab 299, E593–E600. [DOI] [PubMed] [Google Scholar]
- Gu Y, Yang Y, Cao X, Zhao Y, Gao X, Sun C, Zhang F, Yuan Y, Xu Y, Zhang J, et al. (2019). Plin3 protects against alcoholic liver injury by facilitating lipid export from the endoplasmic reticulum. J. Cell. Biochem 120, 16075–16087. [DOI] [PubMed] [Google Scholar]
- Han JM, Kim Y, Lee JS, Lee CS, Lee BD, Ohba M, Kuroki T, Suh P-G, and Ryu SH (2002). Localization of phospholipase D1 to caveolin-enriched membrane via palmitoylation: implications for epidermal growth factor signaling. Mol. Biol. Cell 13, 3976–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri H, Rogers S, Ugrankar R, Liu YL, Feathers JR, and Henne WM (2018). Lipid droplet biogenesis is spatially coordinated at ER-vacuole contacts under nutritional stress. EMBO Rep 19, 57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri H, Speer N, Bowerman J, Rogers S, Fu G, Reetz E, Datta S, Feathers JR, Ugrankar R, Nicastro D, and Henne WM (2019). Mdm1 maintains endoplasmic reticulum homeostasis by spatially regulating lipid droplet biogenesis. J. Cell Biol 218, 1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Zhu L, Balogi Z, Stefan C, Pleiss JA, and Emr SD (2015). Mdm1/Snx13 is a novel ER-endolysosomal interorganelle tethering protein. J. Cell Biol 210, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger D, Burgoyne T, Sanchez-Heras E, Hartwig P, Colaco A, Newton J, Futter CE, Spiegel S, Platt FM, and Eden ER (2019). NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat. Commun 10, 4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollomon MG, Gordon N, Santiago-O’Farrill JM, and Kleinerman ES (2013). Knockdown of autophagy-related protein 5, ATG5, decreases oxidative stress and has an opposing effect on camptothecin-induced cytotoxicity in osteosarcoma cells. BMC Cancer 13, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itabe H, Yamaguchi T, Nimura S, and Sasabe N (2017). Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis 16, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier N, Choudhary V, Mari M, Toulmay A, Reggiori F, and Schneiter R (2011). Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Sci 124, 2424–2437. [DOI] [PubMed] [Google Scholar]
- Jiang H, Luo JQ, Urano T, Frankel P, Lu Z, Foster DA, and Feig LA (1995). Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature 378, 409–412. 10.1038/378409a0. [DOI] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro S-H, Kim Y-M, Otto NM, Cao J, Kundu M, and Kim D-H (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus DF, Lim K-H, Brady DC, Pershing NLK, Cox AD, and Counter CM (2011). RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol 13, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassan A, Herms A, Fernández-Vidal A, Bosch M, Schieber NL, Reddy BJN, Fajardo A, Gelabert-Baldrich M, Tebar F, Enrich C, et al. (2013). Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol 203, 985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, and Cuervo AM (2015). Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol 17, 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella BT, Erdman RA, and Maltese WA (1991). Carboxyl-terminal iso-prenylation of ras-related GTP-binding proteins encoded by rac1, rac2, and ralA. J. Biol. Chem 266, 9786–9794. [PubMed] [Google Scholar]
- Kroemer G, Mariño G, and Levine B (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribara H, Tago K, Yokozeki T, Sasaki T, Takai Y, Morii N, Narumiya S, Katada T, and Kanaho Y (1995). Synergistic activation of rat brain phospholipase D by ADP-ribosylation factor and rhoA p21, and its inhibition by Clostridium botulinum C3 exoenzyme. J. Biol. Chem 270, 25667–25671. [DOI] [PubMed] [Google Scholar]
- Kvam E, and Goldfarb DS (2004). Nvj1p is the outer-nuclear-membrane receptor for oxysterol-binding protein homolog Osh1p in Saccharomyces cerevisiae. J. Cell Sci 117, 4959–4968. [DOI] [PubMed] [Google Scholar]
- Lim K-H, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, and Counter CM (2005). Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 7, 533–545. [DOI] [PubMed] [Google Scholar]
- Lim C-Y, Davis OB, Shin HR, Zhang J, Berdan CA, Jiang X, Counihan JL, Ory DS, Nomura DK, and Zoncu R (2019). ER-lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann-Pick type C. Nat. Cell Biol 21, 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, and Schaffer JE (2003). Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 100, 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, and Anderson RGW (2004). Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J. Biol. Chem 279, 3787–3792. [DOI] [PubMed] [Google Scholar]
- Luo JQ, Liu X, Frankel P, Rotunda T, Ramos M, Flom J, Jiang H, Feig LA, Morris AJ, Kahn RA, and Foster DA (1998). Functional association between Arf and RalA in active phospholipase D complex. Proc. Natl. Acad. Sci. USA 95, 3632–3637. 10.1073/pnas.95.7.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Tanaka M, Nishina H, Murakami M, Kanaho Y, and Hanada K (2008). RalA functions as an indispensable signal mediator for the nutrient-sensing system. J. Biol. Chem 283, 35053–35059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesan D, Rutberg M, Andersson L, Asp L, Larsson T, Borén J, Johansson BR, and Olofsson S-O (2003). A phospholipase D-dependent process forms lipid droplets containing caveolin, adipocyte differentiation-related protein, and vimentin in a cell-free system. J. Biol. Chem 278, 27293–27300. [DOI] [PubMed] [Google Scholar]
- Martin TD, Mitin N, Cox AD, Yeh JJ, and Der CJ (2012). Phosphorylation by protein kinase Ca regulates RalB small GTPase protein activation, sub-cellular localization, and effector utilization. J. Biol. Chem 287, 14827–14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirheydari M, Rathnayake SS, Frederick H, Arhar T, Mann EK, Cocklin S, and Kooijman EE (2016). Insertion of perilipin 3 into a glycero(phospho) lipid monolayer depends on lipid headgroup and acyl chain species. J. Lipid Res 57, 1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, and Yoshimori T (2001). Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol 152, 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, Camonis J, and White MA (2003). Ral GTPases regulate exocyst assembly through dual subunit interactions. J. Biol. Chem 278, 51743–51748. [DOI] [PubMed] [Google Scholar]
- N’Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, and Brown EJ (2009). PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 10, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Banno Y, and Tamiya-Koizumi K (2005). Arf1-dependent PLD1 is localized to oleic acid-induced lipid droplets in NIH3T3 cells. Biochem. Biophys. Res. Commun 335, 117–123. [DOI] [PubMed] [Google Scholar]
- Neyraud V, Aushev VN, Hatzoglou A, Meunier B, Cascone I, and Camonis J (2012). RalA and RalB proteins are ubiquitinated GTPases, and ubiquitinated RalA increases lipid raft exposure at the plasma membrane. J. Biol. Chem 287, 29397–29405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, and Olzmann JA (2017). DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 42, 9–21.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, and Ohsumi Y (1998). Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem 273, 3963–3966. [DOI] [PubMed] [Google Scholar]
- Nose F, Yamaguchi T, Kato R, Aiuchi T, Obama T, Hara S, Yamamoto M, and Itabe H (2013). Crucial role of perilipin-3 (TIP47) in formation of lipid droplets and PGE2 production in HL-60-derived neutrophils. PLoS One 8, e71542–e71549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Suzuki N, Nakamura S, Hartwig JH, and Stossel TP (1999). The small GTPase RalA targets filamin to induce filopodia. Proc. Natl. Acad. Sci. USA 96, 2122–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Hennessy EJ, van Solingen C, Koelwyn GJ, Hussein MA, Ramkhelawon B, Rayner KJ, Temel RE, Perisic L, Hedin U, et al. (2016). miRNA Targeting of Oxysterol-Binding Protein-Like 6 Regulates Cholesterol Trafficking and Efflux. Arterioscler. Thromb. Vasc. Biol 36, 942–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagac M, Cooper DE, Qi Y, Lukmantara IE, Mak HY, Wu Z, Tian Y, Liu Z, Lei M, Du X, et al. (2016). SEIPIN Regulates Lipid Droplet Expansion and Adipocyte Development by Modulating the Activity of Glycerol-3-phosphate Acyltransferase. Cell Rep 17, 1546–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Roberts P, Chen Y, Kvam E, Shulga N, Huang K, Lemmon S, and Goldfarb DS (2000). Nucleus-vacuole junctions in Saccharomyces cerevisiae are formed through the direct interaction of Vac8p with Nvj1p. Mol. Biol. Cell 11, 2445–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Øvervatn A, Bjørkøy G, and Johansen T (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- Peschard P, McCarthy A, Leblanc-Dominguez V, Yeo M, Guichard S, Stamp G, and Marshall CJ (2012). Genetic deletion of RALA and RALB small GTPases reveals redundant functions in development and tumorigenesis. Curr. Biol 22, 2063–2068. [DOI] [PubMed] [Google Scholar]
- Pollock SR, Schinlever AR, Rohani A, Kashatus JA, and Kashatus DF (2019). RalA and RalB relocalization to depolarized mitochondria depends on clathrin-mediated endocytosis and facilitates TBK1 activation. PLoS One 14, e0214764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, and Lippincott-Schwartz J (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 108, 10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, and Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robenek H, Lorkowski S, Schnoor M, and Troyer D (2005). Spatial integration of TIP47 and adipophilin in macrophage lipid bodies. J. Biol. Chem 280, 5789–5794. [DOI] [PubMed] [Google Scholar]
- Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, and Neefjes J (2009). Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol 185, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudoni S, Colombo S, Coccetti P, and Martegani E (2001). Role of guanine nucleotides in the regulation of the Ras/cAMP pathway in Saccharomyces cerevisiae. Biochim. Biophys. Acta 1538, 181–189. [DOI] [PubMed] [Google Scholar]
- Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, De-Caprio JA, and Hahn WC (2007). The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell 129, 969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, and McNiven MA (2015). The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 61, 1896–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, and Bohnert M (2017). A different kind of love—lipid droplet contact sites. Mol. Cell Biol. Lipids 1862, 1188–1196. [DOI] [PubMed] [Google Scholar]
- Schulze RJ, Krueger EW, Weller SG, Johnson KM, Casey CA, Schott MB, and McNiven MA (2020). Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl. Acad. Sci. USA 117, 32443–32452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, and Mothes W (2003). Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4, 785–801. [DOI] [PubMed] [Google Scholar]
- Shipitsin M, and Feig LA (2004). RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol. Cell. Biol 24, 5746–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simicek M, Lievens S, Laga M, Guzenko D, Aushev VN, Kalev P, Baietti MF, Strelkov SV, Gevaert K, Tavernier J, and Sablina AA (2013). The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol 15, 1220–1230. [DOI] [PubMed] [Google Scholar]
- Singer WD, Brown HA, Jiang X, and Sternweis PC (1996). Regulation of phospholipase D by protein kinase C is synergistic with ADP-ribosylation factor and independent of protein kinase activity. J. Biol. Chem 271, 4504–4510. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh MK, Martin APJ, Joffre C, Zago G, Camonis J, Coppey M, and Parrini MC (2019). Localization of RalB signaling at endomembrane compartments and its modulation by autophagy. Sci. Rep 9, 8910–8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner JR, Shew TM, Schwartz DM, Tzekov A, Lepus CM, Abumrad NA, and Wolins NE (2009). Diacylglycerol enrichment of endoplasmic reticulum or lipid droplets recruits perilipin 3/TIP47 during lipid storage and mobilization. J. Biol. Chem 284, 30941–30948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorobogatko Y, Dragan M, Cordon C, Reilly SM, Hung C-W, Xia W, Zhao P, Wallace M, Lackey DE, Chen X-W, et al. (2018). RalA controls glucose homeostasis by regulating glucose uptake in brown fat. Proc. Natl. Acad. Sci. USA 115, 7819–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Yeku O, Olepu S, Genna A, Park J-S, Ren H, Du G, Gelb MH, Morris AJ, and Frohman MA (2009). 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol 75, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, and Frohman MA (1997). Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO J 16, 4519–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztalryd C, Bell M, Lu X, Mertz P, Hickenbottom S, Chang BH-J, Chan L, Kimmel AR, and Londos C (2006). Functional compensation for adipose differentiation-related protein (ADFP) by Tip47 in an ADFP null embryonic cell line. J. Biol. Chem 281, 34341–34348. [DOI] [PubMed] [Google Scholar]
- Szymanski KM, Binns D, Bartz R, Grishin NV, Li W-P, Agarwal AK, Garg A, Anderson RGW, and Goodman JM (2007). The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. USA 104, 20890–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanguy E, Wang Q, Moine H, and Vitale N (2019). Phosphatidic Acid: From Pleiotropic Functions to Neuronal Pathology. Front. Cell. Neurosci 13, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, and Hanada K (2008). Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 4, 131–134. [DOI] [PubMed] [Google Scholar]
- Toda K, Nogami M, Murakami K, Kanaho Y, and Nakayama K (1999). Colocalization of phospholipase D1 and GTP-binding-defective mutant of ADP-ribosylation factor 6 to endosomes and lysosomes. FEBS Lett 442, 221–225. [DOI] [PubMed] [Google Scholar]
- van Zutphen T, Todde V, de Boer R, Kreim M, Hofbauer HF, Wolinski H, Veenhuis M, van der Klei IJ, and Kohlwein SD (2014). Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 25, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N, Mawet J, Camonis J, Regazzi R, Bader M-F, and Chasserot-Golaz S (2005). The Small GTPase RalA controls exocytosis of large dense core secretory granules by interacting with ARF6-dependent phospholipase D1. J. Biol. Chem 280, 29921–29928. [DOI] [PubMed] [Google Scholar]
- Wang H, Wei E, Quiroga AD, Sun X, Touret N, and Lehner R (2010). Altered lipid droplet dynamics in hepatocytes lacking triacylglycerol hydrolase expression. Mol. Biol. Cell 21, 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-W, Miao Y-H, and Chang Y-S (2014). A sterol-enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast. J. Cell Biol 206, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng J-X, Graham M, Christiano R, Fröhlich F, et al. (2013). Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolins NE, Rubin B, and Brasaemle DL (2001). TIP47 associates with lipid droplets. J. Biol. Chem 276, 5101–5108. [DOI] [PubMed] [Google Scholar]
- Wolins NE, Quaynor BK, Skinner JR, Schoenfish MJ, Tzekov A, and Bickel PE (2005). S3–12, Adipophilin, and TIP47 package lipid in adipocytes. J. Biol. Chem 280, 19146–19155. [DOI] [PubMed] [Google Scholar]
- Wu JC, Chen TY, Yu CTR, Tsai SJ, Hsu JM, Tang MJ, Chou CK, Lin WJ, Yuan CJ, and Huang CYF (2005). Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J. Biol. Chem 280, 9013–9022. [DOI] [PubMed] [Google Scholar]
- Yan C, and Theodorescu D (2018). RAL GTPases: Biology and Potential as Therapeutic Targets in Cancer. Pharmacol. Rev 70, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M-S, Du G, Backer JM, Frohman MA, and Chen J (2011). Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol 195, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Madeo F, and Kratky D (2017). Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat. Rev. Mol. Cell Biol 18, 671–684. [DOI] [PubMed] [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, and Zechner R (2004). Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306, 1383–1386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared upon request by the lead contact. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.