

Abstract
Osteoblast-specific gene transcription requires interaction between bone cell-specific transcription factors and more widely expressed transcriptional regulators. This is particularly evident for the basic domain-leucine zipper factor Activating Transcription Factor 4 (ATF4), whose activity can be enhanced or inhibited through interaction with other leucine zipper proteins, intermediate filament proteins, components of the basic transcriptional machinery, nuclear matrix attachment molecules, or ubiquitously expressed transcription factors. We discuss the results supporting the relevance of these interactions and present the first evidence of a functional interaction between ATF4, FIAT (Factor Inhibiting ATF4-mediated Transcription) and αNAC (Nascent polypeptide associated complex And Coactivator alpha), three proteins that have been previously shown to associate using various protein-protein interaction assays.
Keywords: Activating Transcription Factor 4 (ATF4), Factor Inhibiting ATF4-mediated Transcription (FIAT), Nascent polypeptide associated complex And Coactivator alpha (αNAC), osteoblast, transcription
Our understanding of tissue-specific gene transcription has increased considerably since the purification and cloning of the first sequence-specific DNA binding transcription factor.1, 2 From the naïve notion that gene expression in each tissue would be controlled by factors uniquely expressed in that tissue, the knowledge has evolved to include a plethora of coregulatory proteins acting as scaffolds to recruit multiunit complexes bridging the DNA-binding factors to the general transcriptional machinery. Many of these cofactors exhibit enzymatic activity that impacts chromatin structure.3
In addition to the assembly of these macro-molecular complexes that are crucial to differential gene transcription, nature has developed other means to restrict gene transcription in specific cell types. The interaction of distinct transcription factor families to regulate gene expression is an established concept, and this interaction may require promoter DNA binding by each partner or not. These combinatorial interactions have been shown to involve not only tissue-restricted proteins but also ubiquitously expressed transcription factors.
Initially, the interaction between distinct families of DNA-binding transcription factors was observed both in the context of transcriptional activation or repression, and this notion remains valid nowadays. Early examples include the interaction between AP-1 family members and nuclear hormone receptors leading to inhibition of nuclear receptor-mediated transcription by cFOS/cJUN.4-7 Cooperative activation of gene transcription through protein-protein combination of heterologous classes of DNA-binding factors was first clearly demonstrated by the interaction between MEF2 and myogenic bHLH proteins in muscle gene expression. In this context, the cooperativity required direct protein-protein interaction, but only one of the factors needed to be bound to DNA.8 Many combinatorial interactions regulating bone-cell specific transcription have been described involving the osteoblast-specific factor RUNX2. Discussing every characterized interaction is beyond the scope of this perspective and the reader is referred to other reviews.9-11 Here, we will restrict the topic to interactions involving the recent arrival on the scene of osteoblast biology, the basic domain-leucine zipper (bZIP) transcription factor ATF4.
ATF4 is a member of the activating transcription factor (ATF)/cyclic adenosine monophosphate responsive element-binding (CREB) family. This gene family consists of transcription factors that bind the CRE (cAMP response element) through highly related bZIP dimerization domains.12 There is extensive sequence similarity between the different family members within the basic DNA binding domain and the leucine zipper dimerization interface, but members of the family that are not part of the same subgroup do not share much similarity other than the bZIP motif. ATF4 plays a pivotal role in the regulation of osteoblast function. While expression of the Atf4 mRNA is ubiquitous, the ATF4 protein is unstable and degraded in most cell types through ubiquitination by the SCFbTrCP ubiquitin ligase,13 except in osteoblasts where it accumulates.14 In the bone-forming cell, ATF4 affects several key functions such as the synthesis of type I collagen15 and the transcriptional control of several major osteoblastic genes: Osteocalcin, Rankl (Receptor activator of NF-kB ligand), and Esp (Embryonic stem cell phosphatase).15-17 In addition to Osteocalcin, ATF4 targets include genes involved in amino acid import, metabolism, and assimilation.18 This role of ATF4 in regulating amino acid import appears responsible for the decrease in type I collagen synthesis measured in ATF4-deficient osteoblasts.15
Combinatorial interactions activating ATF4
a). Dimerization partners
The ATF4 leucine zipper dimerization interface consists of heptad repeats of leucine residues which align along one face of an alpha helix. When aligned in parallel, the hydrophobic faces of two complementary helices form a coiled coil.19 Leucine zipper dimerization serves to juxtapose adjacent regions of each of the dimer's partners that are rich in basic amino acid residues and that serve as the DNA binding domain of the dimer.20, 21 ATF4 can form homodimers22, 23 but can also heterodimerize with a variety of partners. Indeed, the first ATF4 cDNAs (identified under several different names such as CREB2, TAXREB67, C/ATF, or mTR67) were cloned through the interaction of the ATF4 bZIP domain with the leucine zipper of other transcription factors.22, 24 The dimerization partner appears to influence specificity of DNA binding, with a preference for the CRE binding site: cJUN/ATF4 and cFOS/ATF4 dimers bind the CRE site, while the FRA-1/ATF4 heterodimer can interact with both CRE and AP-1 binding sites.23 Interaction of ATF4 with members of the CCAAT/Enhancer-binding protein (C/EBP) family diverts C/EBP factors from CCAAT-box binding sites to CRE motifs.22
The dimerization of ATF4 with C/EBP family members is significant in bone biology. A C/EBPβ/ATF4 dimer binds a response element within the proximal promoter of the collagen receptor, discoidin domain receptor tyrosine kinase (DDR2).25 This dimer enhances Ddr2 transcription in cultured cells.25 More importantly, mice deficient for C/EBPβ exhibit delayed bone formation and one of the mechanisms characterized also involves heterodimerization of C/EBPβ with ATF4.26 A number of sophisticated assays confirmed the interaction of the two proteins and their association with the proximal Osteocalcin gene promoter at the ATF4 binding site (osteocalcin-specific element 1 or OSE1) to activate Osteocalcin transcription.26 Interestingly, C/EBPβ allowed ATF4 to form a complex and synergize with RUNX2 to increase Osteocalcin expression (Fig 1A, panel i).26 The osteoblast appears to use several accessory molecules such as C/EBPβ to promote the formation of complexes containing both ATF4 and RUNX2 to activate Osteocalcin gene transcription, as will be further detailed below.
Figure 1:

Combinatorial control of ATF4 activity. Schematized, partial depiction of the proximal promoter fragment of the Osteocalcin gene: the RUNX2 binding site, OSE2 (Osteoblast Specific Element 2) and the ATF4 binding site, OSE1 (Osteoblast Specific Element 1) are shown. A. activating combinations: i) dimerization of ATF4 with C/EBPβ allows formation of a complex with RUNX2 to increase Osteocalcin expression; ii) TFIIAγ acts as a protein bridge between ATF4, RUNX2, and general transcription factors (GTFs); iii) SATB2 stabilizes the interaction of RUNX2 and ATF4 with DNA, leading to enhanced transcription; iv) transcriptional cooperativity between FoxO1 and ATF4. B. inhibitory combinations: i, ii) interaction of ATF4 with ICER or FIAT forms inactive dimers that cannot bind the OSE1 site; iii) steric hindrance at the proximal promoter caused by the binding of αNAC prevents ATF4 dimers from binding to the OSE1 element; iv) αNAC or FIAT could recruit repressor molecules to the promoter to block ATF4 activity.
b). Cooperative interactions
Soon after the initial description of the role of ATF4 in osteoblast-specific gene expression15, it was observed that ATF4 activates transcription from the proximal Osteocalcin gene promoter via cooperative interaction with RUNX2.27 The co-stimulation required an intact RUNX2 DNA binding site, and while it was maximal when both the ATF4 and RUNX2 elements were present, cooperativity was still observed in the absence of the ATF4 binding site (OSE1), suggesting that some of the effects of ATF4 occur in the absence of DNA binding.27 RUNX2 and ATF4 could be co-immunoprecipitated from osteoblastic nuclear extracts, but assays using purified recombinant proteins failed to show a direct interaction between the two factors. This data was interpreted to mean that accessory factors are involved in stabilizing the interaction between the two molecules.27 A follow-up study by the same investigators identified the smallest subunit of the general transcription factor IIA, TFIIAγ, as a protein bridge between the two factors.28 Protein-protein interactions were demonstrated between RUNX2 and TFIIAγ, as well as between ATF4 and TFIIAγ, and the same region of RUNX2 was necessary for interaction with TFIIAγ or for association with ATF4. All three proteins associated with the same region of the Osteocalcin gene promoter, and co-transfection of all three factors synergistically enhanced transcription of the endogenous Osteocalcin gene or of reporter constructs under the control of the proximal Osteocalcin promoter.28 Interestingly, one of the mechanisms through which TFIIAγ increases Osteocalcin expression involves stabilization of the ATF4 protein.28 Thus the general transcription factor TFIIAγ, just like C/EBPβ, maximizes Osteocalcin gene transcription by promoting the formation of multimeric complexes containing ATF4 and RUNX2 (Fig 1A, panel ii). The association of components of the general transcriptional machinery with osteoblast-relevant sequence-specific DNA binding transcription factors has been described before, such as the interaction of the vitamin D receptor or OSTERIX with TFIIB.29, 30 However, the mechanism through which TFIIAγ increases Osteocalcin expression, i.e. stabilization of the half-life of the ATF4 protein,28 is a first.
The regulation of gene transcription occurs in a specific subcellular compartment, the nucleus, and the role of the nuclear matrix in controlling transcription is now clearly recognized.31 RUNX2 associates with the nuclear matrix, which impacts on its subnuclear localization and function.32 While its interaction with the nuclear matrix may be direct, RUNX2 could be targeted to subnuclear regions through its association with SATB2, a member of the special AT-rich binding protein family that binds to nuclear matrix-attachment regions.33, 34 Satb2-deficient mice have compromised osteoblast differentiation and function, with decreased expression of osteoblast-specific genes, including, amongst others, the RUNX2 and ATF4 targets Osteocalcin and Bone Sialoprotein (Bsp).35 Chromatin immunoprecipitation assays confirmed that SATB2 is recruited to the Osteocalcin and Bsp promoters. While SATB2 directly binds the Bsp gene 5’-flanking region, its association with the Osteocalcin promoter was shown to be indirect. Direct protein-protein interactions between SATB2 and RUNX2, as well as between SATB2 and ATF4, were demonstrated using pull-downs with recombinant molecules, and their association in cells was confirmed by co-immunoprecipitation. SATB2 appears to stabilize the interaction of RUNX2 and ATF4 with their cognate DNA binding sites within the Osteocalcin promoter, leading to enhanced transcription. The model proposed, once again, involves the formation of complexes associating SATB2, RUNX2, and ATF4 (Fig 1A, panel iii).35
As previously mentioned, ATF4 controls the transcription of genes involved in amino acid import, metabolism, and assimilation18 in addition to Osteocalcin and Bsp. A link between oxidative stress, amino acid import, and osteoblast biology was uncovered by the study of the phenotype of mice with osteoblast-specific inactivation of the transcription factor FoxO1.36 The ubiquitously expressed FoxO family of transcription factors is involved in the response of cells to reactive oxygen species and oxidative stress.37, 38 Inactivating the FoxO1 gene specifically in osteoblasts reduces their number and leads to a decrease in bone volume and bone formation rates.36 These effects can be rescued by treating the mice with the antioxidant N-acetyl L-cysteine, demonstrating that the phenotype results from oxidative stress. Antioxidant administration had no effect on the reduced levels of glutathione measured in FoxO1-deficient bones, however. Since ATF4 is part of the pathway controlling amino acid import, leading to glutathione synthesis, these observations suggested a link between FoxO1 and ATF4. Such a link was further supported by the findings that in FoxO1-deficient osteoblasts, just like in ATF4-deficient bone cells,15 type I collagen protein synthesis was reduced while the level of α1(I) collagen mRNA was not affected.36 Similarly, a high protein diet corrected the phenotype of FoxO1 osteoblast-deficient mice,36 as has been described in mutants that affect ATF4 function.39, 40 In studying the mechanisms involved, it was found that ATF4 and FoxO1 co-localize in osteoblasts and that both proteins co-immunoprecipitated from nuclear extracts of primary osteoblasts or bone tissue. Transcriptional cooperativity was observed between FoxO1 and ATF4 in the expression of reporter constructs that were either FoxO targets or controlled by the Osteocalcin proximal promoter region (Fig 1A, panel iv).36 These results show that FoxO1 affects osteoblast proliferation and function by controlling redox balance, and by regulating amino acid import through its interaction with ATF4. Moreover, they implicate a ubiquitously expressed transcription factor in the regulation of osteoblast function, in addition to the better-studied osteoblast-specific transcriptional regulators.
Combinatorial interactions inhibiting ATF4
Partnering can also inhibit ATF4 function. In a search for functional partners of ATF4 that utilized His-ATF4 affinity chromatography with ROS17/2.8 osteosarcoma cells41 nuclear extracts followed by mass spectrometry of the purified partners, the intermediate filament protein, vimentin, was identified.42 It was confirmed that vimentin can localize to the nucleus and interacts with ATF4 through a putative leucine zipper comprised of amino acid residues 124 to 138. The interaction of vimentin with ATF4 prevents the binding of ATF4 to its cognate response element and inhibits osteoblast differentiation and the transcription of the ATF4 targets, Bsp and Osteocalcin.42 Inhibition of vimentin expression using siRNA-mediated knockdown induces endogenous Osteocalcin expression in preosteoblasts. These results suggest a novel mechanism through which a cytoskeletal protein re-localizes to the nucleus to interact with ATF4 and act as an inhibitor of osteoblastogenesis.42
Dimerization with nuclear transcriptional regulators have also been shown to inhibit ATF4 activity. Co-transfection of ATF4 with ICER (inducible cAMP early repressor) specifically represses ATF4-dependent transcription.43 The ICER proteins are differentially-spliced products of the cAMP responsive element modulator (Crem) gene that act as transcriptional repressors.44 All four ICER isoforms are induced in osteoblasts following PTH treatment45, 46 and the measured inhibition of ATF4 transcriptional activity by ICER was invoked as a mechanism to explain the reduced bone mass and impaired osteoblast differentiation observed in ICER transgenic mice (Fig. 1B, panel i).43
Our laboratory has cloned and characterized FIAT (Factor Inhibiting ATF4-mediated Transcription, also named γ-taxilin),47, 48 a leucine-zipper protein devoid of DNA-binding activity but capable of heterodimerizing with ATF4 to form inactive dimers and inhibit ATF4 transcriptional activity (Fig. 1B, panel ii).48 Stable overexpression of a Fiat transgene was shown to inhibit transcription from the Osteocalcin gene promoter and to reduce mineralization, both in cultures of primary osteoblasts or in established osteoblastic cells.48, 49 Conversely, siRNA-mediated inhibition of FIAT expression enhanced all ATF4 functions tested: Osteocalcin transcription and promoter occupancy, Bsp gene transcription, as well as type I collagen synthesis.50 FIAT-depleted osteoblasts also displayed increased mineralization and an increased number of nodules.50 Bones from FIAT transgenic animals were osteopenic with decreased bone mineral density, bone volume, mineralized volume, mineral apposition rates, and reduced trabecular thickness, trabecular number, and rigidity of long bones.48 The exhaustive phenotype analysis of the FIAT transgenic mice48 combined with a number of in vitro experiments48-50 support the interpretation that FIAT interacts with ATF4 to repress its transcriptional activity, thus regulating bone mass.
We cloned FIAT in a yeast two-hybrid screen for proteins interacting with the αNAC (Nascent polypeptide associated complex And Coactivator alpha) transcriptional coregulator.48, 51 This interaction was independently confirmed52 but its biological relevance has remained elusive. In the course of our exhaustive structure-function analysis of the αNAC protein,53-60 we have identified a mutation affecting a putative phosphoacceptor site that define a functional interaction between ATF4, FIAT, and αNAC. The αNAC protein is extensively post-translationally modified by phosphorylation events that modulate its half-life, subcellular localization, and activity.56-58 Our recent mutational analysis involved the replacement of a putative phosphoacceptor site, residue serine132, by a charged aspartic acid (D) moiety, thus mimicking a permanent phosphorylated state for the mutant S132D protein. When co-transfected with ATF4 and FIAT, the S132D αNAC mutant potentiated the FIAT-mediated repression of ATF4 activity, leading to complete suppression of transcription from a synthetic promoter containing six copies of the OSE1 binding site for ATF4 (Fig. 2). Transfected alone, FIAT and the αNAC mutant had no effect on reporter gene expression (Fig. 2), and the wild-type αNAC protein did not affect FIAT activity in this assay (not shown). These results are the first demonstration of a functional interaction between ATF4, FIAT, and αNAC, proteins that had been previously shown to associate using various protein-protein interaction assays.48, 51, 52 We have previously shown that αNAC binds the Osteocalcin proximal promoter.53 Thus the interaction of FIAT with αNAC could lead to steric hindrance at the proximal promoter and contribute to the FIAT-mediated inhibition of ATF4-dependent Osteocalcin gene transcription (Fig. 1B, panel iii).
Figure 2:
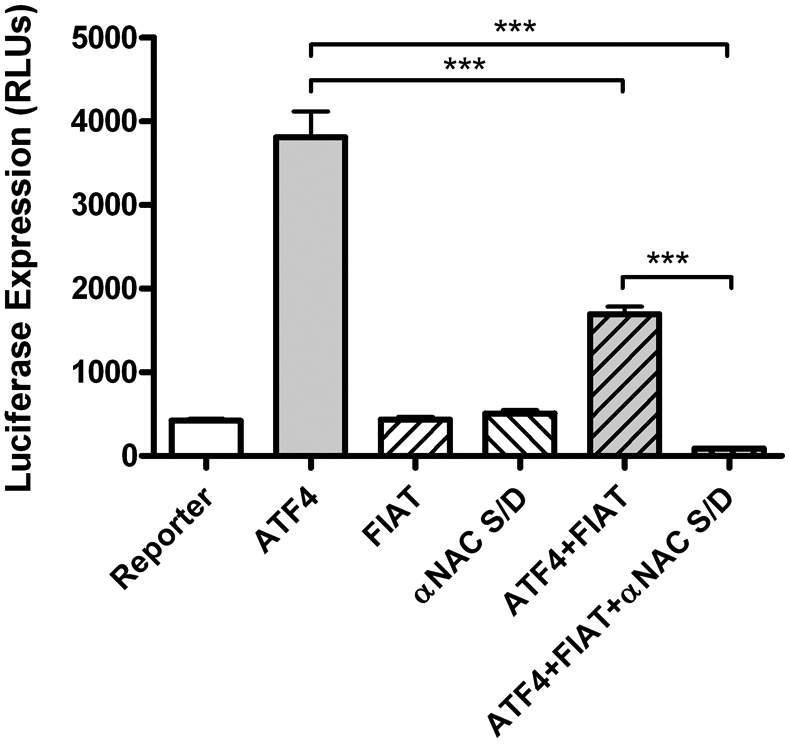
Functional interaction between FIAT and αNAC completely inhibits ATF4-dependent transcription. Osteoblastic cells were transfected with a reporter construct under the control of multiple copies of the ATF4 binding site, OSE1 (Osteoblast Specific Element 1), together with expression vectors for ATF4, FIAT, and a site-directed mutant (S/D) of αNAC, alone or in combination. FIAT inhibited ATF4 transcriptional activity, and this effect was further enhanced by addition of the αNAC S/D mutant. RLUs, relative light units; ***, p<0.001.
Summary and perspectives
Considering its critical roles in osteoblast biology, the activity of ATF4 needs to be tightly regulated. Cells have evolved several mechanisms to this effect, including ubiquitinylation,13, 14 post-translational modifications,15, 16 and interaction with specific partners.25, 26, 28, 35, 42, 48 It is likely that the list of accessory factors that modulate ATF4 function is not complete yet.
While the studies discussed herein all involved sophisticated experiments that support the relevance of the observed interactions with ATF4, incontrovertible genetic evidence of the physiological relevance of the interaction was only provided for SATB2. The observation that compound Satb2+/−;Atf4+/− as well as Satb2+/−;Runx2+/− heterozygous animals exhibit a reduced bone formation phenotype provided genetic proof of the synergy between SATB2, ATF4, and RUNX2.35 Such breeding experiments remain to be performed to confirm that all the identified ATF4-interacting molecules form part of a common genetic pathway, and these might require tissue-specific inactivation in the case of ubiquitously expressed proteins such as vimentin, TFIIAγ or FoxO1.
Other unanswered questions concern the intricacies of the reported protein-protein interactions. What mechanisms control the nuclear re-localization of vimentin? Are the interactions direct or indirect? Some of the assays used to demonstrate ATF4-partner interactions do not allow to unequivocally conclude that the two proteins directly interact. Moreover, it is never evident to determine if a given partner interacts with a monomeric ATF4 protein or an ATF4 dimer. This question could be addressed using single-chain ATF4 dimers in which monomers are joined via a flexible polypeptide tether to force pairing.61 Finally, considering the relevance of post-translational modifications for ATF4 activity, it will prove interesting to determine whether the observed associations are modulated through phosphorylation or other means. This is particularly relevant for the functional interaction between ATF4, FIAT, and αNAC, which was only unraveled through a mutational mimic of phosphorylation. The characterization of the kinase regulating this interaction could identify novel signal transduction pathways involved in the control of osteoblastic gene transcription. Attractive, testable mechanisms include the recruitment of corepressors to the chromatin by differentially phosphorylated αNAC (Fig. 1B, panel iv).
At any rate, it has become clear that combinations of transcriptional regulators are the norm rather than the exception and that they contribute significantly to achieve the exquisite fine control of gene expression observed during osteoblastic differentiation and in the mature bone-forming cell.
Acknowledgements
Work in the laboratory of the authors is supported by the Shriners of North America, the Network for Oral and Bone Health Research, and by a grant from the U.S. National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR53287) to R.St-A.
References
- 1.Briggs MR, Kadonaga JT, Bell SP, et al. 1986. Purification and biochemical characterization of the promoter-specific transcription factor, Sp1. Science. 234: 47–52. [DOI] [PubMed] [Google Scholar]
- 2.Kadonaga JT, Carner KR, Masiarz FR, et al. 1987. Isolation of cDNA encoding transcription factor Sp1 and functional analysis of the DNA binding domain. Cell. 51: 1079–1090. [DOI] [PubMed] [Google Scholar]
- 3.Kumar R, Wang RA & Barnes CJ. 2004. Coregulators and chromatin remodeling in transcriptional control. Mol. Carcinog 41: 221–230. [DOI] [PubMed] [Google Scholar]
- 4.Pfahl M 1993. Nuclear receptor/AP-1 interaction. Endocr. Rev 14: 651–658. [DOI] [PubMed] [Google Scholar]
- 5.Jonat C, Rahmsdorf HJ, Park KK, et al. 1990. Antitumor promotion and antiinflammation: down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell. 62: 1189–1204. [DOI] [PubMed] [Google Scholar]
- 6.Yang-Yen HF, Chambard JC, Sun YL, et al. 1990. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 62: 1205–1215. [DOI] [PubMed] [Google Scholar]
- 7.Schule R, Rangarajan P, Kliewer S, et al. 1990. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 62: 1217–1226. [DOI] [PubMed] [Google Scholar]
- 8.Molkentin JD, Black BL, Martin JF, et al. 1995. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell. 83: 1125–1136. [DOI] [PubMed] [Google Scholar]
- 9.Hassan MQ, Saini S, Gordon JA, et al. 2009. Molecular switches involving homeodomain proteins, HOXA10 and RUNX2 regulate osteoblastogenesis. Cells Tissues Organs. 189: 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Javed A, Afzal F, Bae JS, et al. 2009. Specific residues of RUNX2 are obligatory for formation of BMP2-induced RUNX2-SMAD complex to promote osteoblast differentiation. Cells Tissues Organs. 189: 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franceschi RT, Ge C, Xiao G, et al. 2009. Transcriptional regulation of osteoblasts. Cells Tissues Organs. 189: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hai T & Hartman MG. 2001. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 273: 1–11. [DOI] [PubMed] [Google Scholar]
- 13.Lassot I, Segeral E, Berlioz-Torrent C, et al. 2001. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol Cell Biol. 21: 2192–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X & Karsenty G. 2004. ATF4, the osteoblast accumulation of which is determined post-translationally, can induce osteoblast-specific gene expression in non-osteoblastic cells. J Biol Chem. 279: 47109–47114. [DOI] [PubMed] [Google Scholar]
- 15.Yang X, Matsuda K, Bialek P, et al. 2004. ATF4 Is a Substrate of RSK2 and an Essential Regulator of Osteoblast Biology; Implication for Coffin-Lowry Syndrome. Cell. 117: 387–398. [DOI] [PubMed] [Google Scholar]
- 16.Elefteriou F, Ahn JD, Takeda S, et al. 2005. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 434: 514–520. [DOI] [PubMed] [Google Scholar]
- 17.Yoshizawa T, Hinoi E, Jung DY, et al. 2009. The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J. Clin. Invest 119: 2807–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harding HP, Zhang Y, Zeng H, et al. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 11: 619–633. [DOI] [PubMed] [Google Scholar]
- 19.Landschulz WH, Johnson PF & McKnight SL. 1988. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 240: 1759–1764. [DOI] [PubMed] [Google Scholar]
- 20.Gentz R, Rauscher FJ 3rd, Abate C, et al. 1989. Parallel association of Fos and Jun leucine zippers juxtaposes DNA binding domains. Science. 243: 1695–1699. [DOI] [PubMed] [Google Scholar]
- 21.Glover JN & Harrison SC. 1995. Crystal structure of the heterodimeric bZIP transcription factor c-Fos- c-Jun bound to DNA. Nature. 373: 257–261. [DOI] [PubMed] [Google Scholar]
- 22.Vallejo M, Ron D, Miller CP, et al. 1993. C/ATF, a member of the activating transcription factor family of DNA- binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to cAMP response elements. Proc Natl Acad Sci U S A. 90: 4679–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hai T & Curran T. 1991. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 88: 3720–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chevray PM & Nathans D. 1992. Protein interaction cloning in yeast: identification of mammalian proteins that react with the leucine zipper of Jun. Proc Natl Acad Sci U S A. 89: 5789–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin KL, Chou CH, Hsieh SC, et al. 2010. Transcriptional upregulation of DDR2 by ATF4 facilitates osteoblastic differentiation through p38 MAPK-mediated Runx2 activation. J. Bone Miner. Res 25: 2489–2503. [DOI] [PubMed] [Google Scholar]
- 26.Tominaga H, Maeda S, Hayashi M, et al. 2008. CCAAT/enhancer-binding protein beta promotes osteoblast differentiation by enhancing Runx2 activity with ATF4. Mol. Biol. Cell 19: 5373–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao G, Jiang D, Ge C, et al. 2005. Cooperative interactions between activating transcription factor 4 and Runx2/Cbfa1 stimulate osteoblast-specific osteocalcin gene expression. J Biol Chem. 280: 30689–30696. [DOI] [PubMed] [Google Scholar]
- 28.Yu S, Jiang Y, Galson DL, et al. 2008. General transcription factor IIA-gamma increases osteoblast-specific osteocalcin gene expression via activating transcription factor 4 and runt-related transcription factor 2. J Biol Chem. 283: 5542–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatta M, Yoshimura Y, Deyama Y, et al. 2006. Molecular characterization of the zinc finger transcription factor, Osterix. Int. J. Mol. Med 17: 425–430. [PubMed] [Google Scholar]
- 30.MacDonald PN, Sherman DR, Dowd DR, et al. 1995. The vitamin D receptor interacts with general transcription factor IIB. J Biol Chem. 270: 4748–4752. [DOI] [PubMed] [Google Scholar]
- 31.Zaidi SK, Young DW, Javed A, et al. 2007. Nuclear microenvironments in biological control and cancer. Nat Rev Cancer. 7: 454–463. [DOI] [PubMed] [Google Scholar]
- 32.Zaidi SK, Javed A, Pratap J, et al. 2006. Alterations in intranuclear localization of Runx2 affect biological activity. J. Cell. Physiol 209: 935–942. [DOI] [PubMed] [Google Scholar]
- 33.Bode J, Benham C, Knopp A, et al. 2000. Transcriptional augmentation: modulation of gene expression by scaffold/matrix-attached regions (S/MAR elements). Crit. Rev. Eukaryot. Gene Expr 10: 73–90. [PubMed] [Google Scholar]
- 34.Dobreva G, Dambacher J & Grosschedl R. 2003. SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin mu gene expression. Genes Dev. 17: 3048–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobreva G, Chahrour M, Dautzenberg M, et al. 2006. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 125: 971–986. [DOI] [PubMed] [Google Scholar]
- 36.Rached MT, Kode A, Xu L, et al. 2010. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 11: 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salih DA & Brunet A. 2008. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol 20: 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sedding DG 2008. FoxO transcription factors in oxidative stress response and ageing--a new fork on the way to longevity? Biol. Chem 389: 279–283. [DOI] [PubMed] [Google Scholar]
- 39.Elefteriou F, Benson MD, Sowa H, et al. 2006. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 4: 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sowa H & Karsenty G. 2007. ATF4 is a key molecule linking food intake and skeletal development. J Musculoskelet Neuronal Interact. 7: 326–327. [PubMed] [Google Scholar]
- 41.Majeska RJ, Rodan SB & Rodan GA. 1980. Parathyroid hormone-responsive clonal cell lines from rat osteosarcoma. Endocrinology. 107: 1494–1503. [DOI] [PubMed] [Google Scholar]
- 42.Lian N, Wang W, Li L, et al. 2009. Vimentin inhibits ATF4-mediated osteocalcin transcription and osteoblast differentiation. J Biol Chem. 284: 30518–30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandhoke TK, Huang YF, Liu F, et al. 2008. Osteopenia in transgenic mice with osteoblast-targeted expression of the inducible cAMP early repressor. Bone. 43: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Molina CA, Foulkes NS, Lalli E, et al. 1993. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell. 75: 875–886. [DOI] [PubMed] [Google Scholar]
- 45.Tetradis S, Nervina JM, Nemoto K, et al. 1998. Parathyroid hormone induces expression of the inducible cAMP early repressor in osteoblastic MC3T3-E1 cells and mouse calvariae. J. Bone Miner. Res 13: 1846–1851. [DOI] [PubMed] [Google Scholar]
- 46.Nervina JM, Tetradis S, Huang YF, et al. 2003. Expression of inducible cAMP early repressor is coupled to the cAMP-protein kinase A signaling pathway in osteoblasts. Bone. 32: 483–490. [DOI] [PubMed] [Google Scholar]
- 47.Nogami S, Satoh S, Tanaka-Nakadate S, et al. 2004. Identification and characterization of taxilin isoforms. Biochem. Biophys. Res. Commun 319: 936–943. [DOI] [PubMed] [Google Scholar]
- 48.Yu VW, Ambartsoumian G, Verlinden L, et al. 2005. FIAT represses ATF4-mediated transcription to regulate bone mass in transgenic mice. J Cell Biol. 169: 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu VW, Gauthier C & St-Arnaud R. 2008. FIAT represses bone matrix mineralization by interacting with ATF4 through its second leucine zipper. J. Cell. Biochem 105: 859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu VW, El-Hoss J & St-Arnaud R. 2009. FIAT inhibition increases osteoblast activity by modulating Atf4-dependent functions. J. Cell. Biochem 106: 186–192. [DOI] [PubMed] [Google Scholar]
- 51.Yu VW, Gauthier C & St-Arnaud R. 2006. Inhibition of ATF4 transcriptional activity by FIAT/gamma-taxilin modulates bone mass accrual. Ann. N. Y. Acad. Sci 1068: 131–142. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida K, Nogami S, Satoh S, et al. 2005. Interaction of the taxilin family with the nascent polypeptide-associated complex that is involved in the transcriptional and translational processes. Genes Cells. 10: 465–476. [DOI] [PubMed] [Google Scholar]
- 53.Akhouayri O, Quelo I & St-Arnaud R. 2005. Sequence-Specific DNA Binding by the {alpha}NAC Coactivator Is Required for Potentiation of c-Jun-Dependent Transcription of the Osteocalcin Gene. Mol Cell Biol. 25: 3452–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meury T, Akhouayri O, Jafarov T, et al. 2010. Nuclear alpha NAC influences bone matrix mineralization and osteoblast maturation in vivo. Mol Cell Biol. 30: 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moreau A, Yotov WV, Glorieux FH, et al. 1998. Bone-specific expression of the alpha chain of the nascent polypeptide- associated complex, a coactivator potentiating c-Jun-mediated transcription. Mol Cell Biol. 18: 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quelo I, Akhouayri O, Prud'homme J, et al. 2004. GSK3beta-dependent phosphorylation of the alphaNAC coactivator regulates its nuclear translocation and proteasome-mediated degradation. Biochemistry. 43: 2906–2914. [DOI] [PubMed] [Google Scholar]
- 57.Quelo I, Gauthier C, Hannigan GE, et al. 2004. Integrin-linked kinase regulates the nuclear entry of the c-Jun coactivator alpha-NAC and its coactivation potency. J Biol Chem. 279: 43893–43899. [DOI] [PubMed] [Google Scholar]
- 58.Quelo I, Gauthier C & St-Arnaud R. 2005. Casein kinase II phosphorylation regulates alphaNAC subcellular localization and transcriptional coactivating activity. Gene Expr. 12: 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quelo I, Hurtubise M & St-Arnaud R. 2002. alphaNAC requires an interaction with c-Jun to exert its transcriptional coactivation. Gene Expr. 10: 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yotov WV & St-Arnaud R. 1996. Differential splicing-in of a proline-rich exon converts alphaNAC into a muscle-specific transcription factor. Genes Dev. 10: 1763–1772. [DOI] [PubMed] [Google Scholar]
- 61.Bakiri L, Matsuo K, Wisniewska M, et al. 2002. Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol. 22: 4952–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]