

Abstract
Single-cell sequencing is a biotechnology to sequence one layer of genomic information for individual cells in a tissue sample. For example, single-cell DNA sequencing is to sequence the DNA from every single cell. Increasing in complexity, single-cell multi-omics sequencing, or single-cell multimodal omics sequencing, is to profile in parallel multiple layers of omics information from a single cell. In practice, single-cell multi-omics sequencing actually detects multiple traits such as DNA, RNA, methylation information and/or protein profiles from the same cell for many individuals in a tissue sample. Multi-omics sequencing has been widely applied to systematically unravel interplay mechanisms of key components and pathways in cell. This survey overviews recent developments in single-cell multi-omics sequencing, and their applications to understand complex diseases in particular the COVID-19 pandemic. We also summarize machine learning and bioinformatics techniques used in the analysis of the intercorrelated multilayer heterogeneous data. We observed that variational inference and graph-based learning are popular approaches, and Seurat V3 is a commonly used tool to transfer the missing variables and labels. We also discussed two intensively studied issues relating to data consistency and diversity and commented on currently cared issues surrounding the error correction of data pairs and data imputation methods. The survey is concluded with some open questions and opportunities for this extraordinary field.
Keywords: single-cell sequencing, single-cell multi-omics sequencing, COVID-19, integrative methods, variational inference, graph-based algorithms
Introduction
Single-cell sequencing was named the Method of the Year 2013 by Nature Methods to award its novel protocol to sequence a complete layer of genomic information such as the DNA or the RNA for individual cells from a tissue sample or a cell population [1]. The first single-cell RNA sequencing (scRNA-seq) was invented in 2009 [2]. Following that, the invention of microfluidics and droplet-based methods have markedly improved the efficiency and accuracy of scRNA-seq [3, 4]. The profiling of proteins at the single-cell level is performed by mass cytometry, mass spectrometry imaging or fluorescence-based imaging with high precision and throughput [5–7]. Developments in single-cell epigenomics include single-cell bisulfite sequencing [8], chromatin immunoprecipitation sequencing [9] and assay for transposase-accessible chromatin sequencing (ATAC-seq) [10]. In general, single-cell sequencing has five different purposes: single-cell genomics, single-cell transcriptomics, single-cell epigenomics, single-cell proteomics and single-cell metabolomics. These five essential components can be all coupled with their unique spatial information within the same cell to uncover more cellular information. In fact, DNA wraps around the histone octamer in the eukaryotic cell nucleus, whereas RNA and functional proteins exist in the cell cytoplasm; epigenetics is composed of DNA modification, histone modification, DNA accessibility and chromosome organization in the nucleus and cytoplasm and metabolism consists of immediate feedback by environmental stimulation.
Recent advances in single-cell sequencing have greatly deepened our understanding of complex biological systems. Previously, poorly characterized cell populations and rare cell types or tissues were challenging to comprehend because only the average molecular profiles were acquired by the conventional population-based sequencing strategy [11]. Phenotypically identical cells also posed challenges since they could exhibit discrepancies during their lifespan corresponding to variations in their molecular compositions [12]. Such challenges have now been largely resolved through recent developments in single-cell sequencing.
Why single-cell multi-omics sequencing?
Although single-cell sequencing methods have been well established to provide unprecedented resolution to uncover the heterogeneity of cells [12], scRNA-seq or single-cell DNA sequencing alone does not provide sufficient information to elucidate functional interplays of the cellular subpopulations [11]. At least, single-cell epigenomics is needed to add information about epigenetic transitions, including transcription factor binding, transcriptional response, active or repressive chromatin marks, DNA methylation or chromosome organization, which may occur on different time scales and indicate the past and future regulation state of the cell [13].
Furthermore, the overall state of a cell comprises its potential state and current functional state. The cell’s genotype, together with epigenomics, determines its potential state, whereas the cell’s proteins and metabolites are direct indicators of its current state. The potential and current exact state of a cell therefore rely on simultaneously extracting data from its genome, transcriptome, epigenome, proteome and/or metabolome [14]. In addition, DNA is transcribed to create RNAs, and then the coding RNAs are translated to create proteins. These different molecular items should have intensive communications with each other through signals within the same cell. Single-cell multi-omics techniques are therefore demanded to acquire these diverse molecular layers of omics information to build a much more comprehensive molecular view of the cell than through each layer individually [15]. This multi-omics approach can lead to truth-closer discoveries of cellular interplays and functions in specific cells. Attributed to this powerful full-coverage of the cellular elements and interactions, single-cell multi-omics sequencing was recently named the Method of the Year 2019 by Nature Methods [16].
Distinction between single-cell multi-omics sequencing and integrative multiple single-cell sequencing
Single-cell multi-omics sequencing is fundamentally different from those approaches that integrate data from multiple single-cell sequencing techniques. Consider that there are 100 cells from different patient samples. Through single-cell multi-omics sequencing, multiple types of omics data such as the transcriptome and proteome can be generated simultaneously from each of these 100 cells. In contrast, the integration of multiple single-cell sequencing methods is to apply different single-cell sequencing methods to different subsets of the cells to measure distinct layers of omics information. As for the above example of 100 cells, single-cell DNA sequencing may be applied to one half of the cells, and scRNA-seq to the other half to acquire integrated data for alternative splicing studies. However, these data integration approach may cause severe problems because the RNA profiles are not exactly mapped with the DNA data of the same cell.
The integration of multiple single-cell sequencing datasets is also prone to using different datasets across different single cells in the same or similar tissue sample. For example, to define the cell type in mouse frontal cortical neurons, the Linked Inference of Genomic Experimental Relationships (LIGER) method integrated two single-cell datasets: gene expression data of 55 803 cells and DNA methylation data of 33 78 cells from the same tissue [17]. Although the cells were from the same tissue, they were not necessarily exposed to the same spatial locations and might show different responses to the same stimuli. With such an integration approach, it is impossible to infer a cell’s multilayer responses to environmental stimuli or therapeutic treatment accurately. Hence, the genotype and phenotype correlations may only be accurately captured by single-cell multi-omics sequencing.
In this review, we present the latest developments in single-cell multi-omics sequencing and their application trends. We describe advanced data analysis methods used for processing single-cell multi-omics sequencing data, including unsupervised learning methods (e.g. matrix factorization, variational inference (VI), canonical correlation analysis and clustering) and graph-based learning models. We also discuss two intensively studied issues relating to data heterogeneity in the preprocessing of single-cell multi-omics sequencing data (consistency and diversity) and comment on two major currently cared issues (correction of data pairs and data point imputation). Moreover, we list open questions and suggest future perspectives about integrating diverse modalities at the single-cell level. This survey has a different focus comparing with three existing reviews on single-cell multi-omics sequencing [18–20] that all surveyed on critical technological details of the sequencing techniques. Instead, our focus is placed on the application trends particularly the up-to-date applications in the battle against COVID-19 and on pertinent issues relating to data analysis. A very recent survey [21] offered some interesting perspectives on bulk and single-cell multi-omics techniques and commented on their applications in precision medicine [21]. In contrast, our survey provides details on the medical applications of single-cell multi-omics sequencing as well as critical technical comments on data analysis methods and data preprocessing issues. We also provide fresh insights into open questions, challenges and future prospects of this exciting technology.
Developments in single-cell multi-omics sequencing
This section presents recent progresses of single-cell multi-omics sequencing and their applications over the last 5 years for genomic expression profiling, spatial information profiling, epigenomic profiling and protein profiling (Tables 1–4). We also describe COVID-19 studies that are based on single-cell multi-omics sequencing and comment on some limits of multi-omics sequencing.
Table 1.
Single-cell multi-omics techniques for genomic profiling together with their specific applications, conclusions and data sources
| Method | Data source | Molecular layers | Objective and outcome(s) | Platform(s) |
|---|---|---|---|---|
| G&T-seq [22] | Mouse data: Array Express (EERAD-381) Human data: EGA(EGAS000 01001204). | 192 Genomic DNA and 192 full-length mRNA sequencing in over 220 single cells from mice and humans. | To dissect genetic variation and its effects on gene expression. Cellular properties could not be inferred from DNA or RNA sequencing alone. | Illumina HiSeq X |
| DR-Seq [23] | GEO (GSE62952) | DR-Seq on E14 of mouse embryonic stem cell line and sequencing the mRNA from 13 single cells together with gDNA from 3 of these 13 cells. | To correlate DNA copy number variation to transcriptome variability among individual cells. Genes with high cell-to-cell variability in transcript numbers generally had lower genomic copy numbers, and vice versa. | Illumina HiSeq 2500 |
| DNTR-seq [24] | GEO (GSE144296). | DNTR-seq on 607 cells from two pediatric acute lymphoblastic leukemia (ALL) cases, human colon adenocarcinoma cell line HCT116, and melanoma cell line A375 using Whole-genome sequencing, transcriptomics at single-cell resolution. | To address how genetic alterations affect transcription and identify minor subclones within leukemia patients. Tumorigenic alterations had a large impact on gene expression, whereas natural X/Y chromosome differences were largely silent. | Illumina NextSeq 500 |
| Holo-Seq [25] | CRA001133, CRA001131 | Small RNAs and mRNAs of 32 human hepatocellular carcinoma single cells. | To overcome the hurdles that currently limit scRNA-seq methods. The RNA metabolism kinetics of core genes were different from housekeeping genes. | Illumina HiSeq 2500 |
| Wang et al. [26] | GSE 114071 | Cosequencing of microRNAs and mRNAs across 19 single cells that were phenotypically identical. | To study how miRNAs modulate nongenetic cell-to-cell variability posttranscriptionally. The predicted targets mRNAs were significantly anticorrelated with the variation of abundantly expressed microRNAs. | Illumina HiSeq 2000 |
Table 2.
Single-cell multi-omics sequencing for spatial information profiling together with their specific applications, results and data sources
| Method | Data source | Molecular layers | Objective and outcome(s) | Platform(s) |
|---|---|---|---|---|
| MERFISH [28] | GSE67685 | Copy numbers and spatial distributions of a large number of RNA species within single cells. | To overcome the obstacle of a limited number of RNA species that can be simultaneously imaged in individual cells. Thousands of RNA species could be imaged in single cells. | Illumina MiSeq |
| Moffitt et al. [29] | GSE113576 | Profiled about 31 000 cells using scRNA-seq and imaged about 1.1 million cells within intact tissues using MERFISH. | To identify molecularly distinct cell types and map their spatial and functional organization in the tissue. A combination MERFISH with scRNA-seq revealed the molecular, spatial and functional organization of neurons within the hypothalamic preoptic region. | Illumina NextSeq 500 |
| osmFISH [30] | http://linnarssonlab.org/osmFISH | Spatially resolved single-cell transcriptomics profiling. | To use spatial information and detect a large number of cell type–specific markers simultaneously in large tissue. The spatial information inherent to osmFISH could improve the interpretation of expression profiles. | N/A |
| seqFISH [31] | N/A | In situ profiling and visualization of transcription at the single-cell level. | To reveal spatial and temporal features of transcriptome. The sequential barcoding method enabled the transcriptome to be directly imaged at single-cell resolution in complex samples such as brain tissue. | N/A |
| seqFISH+ [32] | GSE98674 | Superresolution imaging and multiplexing of 10 000 genes in a single cell. | To overcome the optical crowding problem during implementation of spatial profiling experiments. With the genome coverage and spatial resolution of seqFISH+, it was possible to perform discovery-driven studies directly in situ. | Illumina HiSeq 2500 |
| FISSEQ [33] | http://arep.med.harv ard.edu/FISSEQ_Science:2014/ | Transcriptome-wide RNA sequencing with 8102 genes in situ in human primary fibroblasts with a simulated wound-healing assay. | To demonstrate imaging and analytic approaches across multiple specimen types and spatial scales. FISSEQ predominantly detected genes characterizing cell type and function. | Illumina humanRef–8 v2.0 expression beadchip,Harvard_Human _BeadChip_23K_Ref8v3, Illumina HiSeq 2000 |
| ST [34] | N/A | Quantitative gene expression data and visualization of the distribution of mRNAs within tissue sections. | To introduce positional molecular barcodes in the complementary DNA synthesis reaction in an intact tissue section before RNA-seq. ST revealed unexpected heterogeneity within a biopsy, which would not be possible to detect with regular transcriptome analysis and which may give more detailed prognostic information. | N/A |
| Visium Spatial Technology | https://www.10xgeno mics.com/products/ spatial–gene– expression | The whole transcriptome with morphological context within tissue sections. | To discover novel insights into normal development, disease pathology and clinical translational research. | Visium platform |
| Slide-seq [35] | https://portals.broad institute.org/single_cell/study/slide--seq –study | Spatially resolved gene expression data from individual cells. | To develop a high-throughput, genome-wide readout of gene expression within cellular resolution Slide-seq was easily integrated with large-scale scRNA-seq datasets and facilitated discovery of spatially defined gene expression patterns in normal and diseased tissues. | N/A |
| HDST [36] | GSE130682 | Transcript coupled spatial barcodes. | To develop high-resolution methods to capture both spatial and molecular characteristics for tissue function. Relating histopathology and transcriptional profiles could help improve understanding of disease biology, and patient diagnosis and treatment. | Illumina NextSeq 500 |
| STARmap [37] | www.starmapre sources.com | RNA quantity and 3D spatial information with more than 1000 genes over six imaging cycles at the single-cell level in intact tissue. | To uncover the integrated relationship between structure and function in complex biological tissues. | Fluorescent Nissl staining. |
| DBiT-seq [38] | GSE137986 | mRNAs and spatial information coupled with a panel of 22 proteins in mouse embryos tissue slide. | To dissect the initiation of early organogenesis at the whole embryo scale. Deterministic barcoding in tissue enabled NGS-based spatial multi-omics mapping. | Illumina HiSeq 4000 |
| Baccin et al. [39] | GSE122467 | Transcriptomics and spatial position information of distinct bone marrow sections at the single-cell level. | To define sources of prohematopoietic factors, infer bone marrow-resident cell types and localize their position. The cellular and spatial organization of bone marrow niches offered a systematic approach to dissect the complex organization of whole organs. | Illumina NextSeq 500 |
Technical progresses of single-cell multi-omics sequencing and its applications
Genomic profiling by single-cell multi-omics sequencing
Table 1 lists recent methods of single-cell multi-omics sequencing for genomic profiling together with their specific applications. By one of these studies, Genome and Transcriptome sequencing (G&T-seq) [22] and gDNA–mRNA sequencing (DR-Seq) [23] have been used to couple genomic DNA with mRNA information from the same cell to decipher the relationships between genetic variation and transcriptome variability. By another study, Direct Nuclear Tagmentation and RNA sequencing (DNTR-seq) [27] has been proposed to perform simultaneous whole-genome sequencing and transcriptomics to reveal the association of genetic alternation and transcription. Single-cell holo-transcriptome sequencing (Holo-Seq) [25] and the techniques introduced by [26] were shown to be useful for understanding miRNAs and mRNAs in a single cell simultaneously.
Spatial information profiling by single-cell multi-omics sequencing
Genomic spatial information represents another heritable dimension in single cells. Especially, tissue transcriptomes incorporating spatial information have opened up new opportunities for generating a comprehensive map of complex tissues such as the human brain. Such spatially resolved transcriptomics sequencing methods were recently selected by Nature Methods [40] as the Method of the Year 2020. Spatially resolved quantitative gene expression techniques and their applications were specifically reviewed elsewhere [41], whereas Table 2 summarizes important applications of spatial information of single-cell multi-omics sequencing. These imaging-based single-cell genomics and transcriptomics methods can be grouped into two broad categories: in situ sequencing and multiplexed fluorescence in situ hybridization (FISH) [42].
Table 3.
Single-cell multi-omics sequencing for epigenomic profiling together with their specific applications, results and data sources
| Method | Data source | Molecular layers | Objective and outcome(s) | Platform(s) |
|---|---|---|---|---|
| scCOOL-seq [45] | GSE78140 | Distinct layers of epigenomic information from 24 single mouse embryonic stem cells. | To simultaneously measure all the different layers of epigenomic information from the same individual cell. Factors other than DNA methylation are determinants of the heterogeneity of the open versus closed chromatin states of the promoter regions of genes. | Illumina HiSeq 2500. |
| scM&T-seq [46] | GSE76483 | DNA methylome and transcriptome from the same cell. | To understand the direct correlation of DNA methylation and gene expression within single cells. Methylation of non-CGI promoters was better anticorrelated with gene transcription, whereas gene body methylation of CGI promoter genes was better correlated with gene transcription. | Illumina Hiseq 2500 |
| scNMT-seq [47] | GSE109262 | Transcriptome, chromatin accessibility and DNA methylation within single cells. | To uncover complete information of connections and dependent relationships among epigenetic layers together with transcription. scNMT-seq was able to robustly profile gene expression, DNA methylation and chromatin accessibility within the same single cell. | Illumina NextSeq 500 |
| scTrio-seq [48] | GSE65364 | Genomic CNVs, DNA methylome and transcriptome of an individual mammalian cell. | To detect subpopulations of cancer cells and the relationships among the three different types of omics. Negative correlation between promoter methylation and RNA expression and positive correlation between gene body methylation and RNA expression were found in a single cell, and a strong positive correlation was found between the DNA copy number and gene expression within the affected genomic region. | Illumina HiSeq2000 or HiSeq 2500 Sequencer |
| SHARE-seq [49] | GSE140203 | Chromatin accessibility and gene expression within 84,426 cells across four different cell lines and three tissue types. | To recover fine but biologically important differences by measuring chromatin accessibility and gene expression within the same cell. The cell cycle was more associated with changes in gene expression and less prominent in chromatin accessibility profiles. | NextSeq 550 and Illumina NovaSeq 6000 |
| sci-ATAC-seq [50] | GSE118987 | Accessible chromatin landscape and transcriptomics information at 899 high-quality cells in cultured hippocampal neurons. | To comprehensively map the accessible chromatin landscape and transcriptomics in fresh and frozen hippocampal tissue samples. Cell type assignments with substantial concordance between the highest represented cell types across platforms. | Illumina NextSeq 500, Illumina HiSeq 2000, Illumina MiSeq and Illumina HiSeq 2500. |
| scCAT-seq [51] | SRA (SRP167062) and CNGB (CNP0000213). | Accessible chromatin and gene expression within the same single cell from a total of 192 samples. | To study how transcription factors and epigenomic features induce transcriptional outcomes that influence cell fate determinations. scCAT-seq data could recapitulate major features obtained by separately performed bulk ATAC-seq and RNA-seq. | BGISEQ-500 |
| SNARE-seq [52] | GSE126074 | Simultaneous profiling of gene expression and chromatin accessibility in each of thousands of single nuclei. | To enable highly parallel profiling of chromatin accessibility and mRNA from individual nuclei. SNARE-seq could effectively separate cell types on the basis of both their chromatin signatures and transcriptomes, with a high level of concordance. | Illumina HiSeq 2500, Illumina HiSeq 4000 |
| 10 Genomics scRNA +scATAC | https://www.10xge nomics.com/products/single-cell-multi ome–atac–plus–ge ne–expression | Simultaneous profiling of the transcriptome (using 3’ gene expression) and epigenome (using ATAC-seq) from single cells. | To deepen understanding of how genes are expressed and regulated across different cell types. | 10 Genomics |
In situ multiplexed FISH requires specialized knowledge and equipments as well as the upfront selection of gene sets for measurement [35]. Multiplexed error-robust FISH (MERFISH) [28] has been used for imaging thousands of RNA species in single cells by combinatorial FISH labeling coupled with encoding schemes used for detecting and/or correcting errors. For example, Moffitt et al. [29] has employed MERFISH and scRNA to detect gene expression of one million cells in situ. However, overlapping signal dots or the transcript length of Single-molecule fluorescence in situ hybridization (smFISH) pose challenges in targeting biologically relevant marker genes to map some cell types. To infer a cell type map in the mouse somatosensory cortex, a nonbarcoded and unamplified cyclic-ouroboros smFISH method (osmFISH) was proposed, so that gene expression and cluster localization can be visualized [30]. In addition, sequential fluorescence in situ hybridization (seqFISH) has been proposed, which imparts sequential barcoding for multiplexing different mRNAs. For each round of hybridization, each transcript with a set of FISH probes is labeled with a single type of fluorophore [31, 43, 44]. Based on this, an evolution of seqFISH+ has been invented [32] using sequential hybridizations and imaging with a standard confocal microscope. A sequencing technique was then developed by Baccin et al. [39], aiming to combine transcriptomics and spatial position information at the single-cell level to identify resident cell types in the mouse bone marrow and their localization.
On the other hand, for in situ sequencing, barcoded oligonucleotides have been used to capture spatially encoded RNA sequences. For example, fluorescent in situ RNA sequencing (FISSEQ), an untargeted in situ sequencing approach without any preselection, was utilized to capture all RNA species [33]. However, this approach might lead to lower detection efficiency compared with target expansion sequencing [42]. And a high-resolution in situ RNA sequencing (Slide-seq) was utilized for transferring RNA from tissue sections onto a surface covered in DNA-barcoded beads with known positions [35]. Then, high-definition spatial transcriptomics (HDST) was employed to capture RNA from tissue sections on a dense, smaller spatially barcoded bead array [36]. A spatial transcriptomics (ST) method has also been proposed to visualize and quantify the transcriptomic expression in individual tissue sections with spatial resolution [34]. Based on this, Visium Spatial Technology was developed by the 10 Genomics company to provide higher resolution for achieving more successful gene expression visualization platforms. Deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq) was used to couple the mRNAs and spatial information with proteins in a fixed tissue slide to uncover the spatial pattern of cells in the mouse embryo and define fine features such as brain microvascular networks [38]. Furthermore, spatially resolved Transcript Amplicon Readout mapping (STARmap) [37] has been used to combine mRNAs and 3D spatial information from intact tissue to identify the association between structure and function at the single-cell level.
Epigenomic profiling by single-cell multi-omics sequencing
The coupling of single-cell single-omics sequencing with epigenomic data can uncover the transition state of single cells as well as cell fate decision into distinct lineages [53]. Table 3 presents the current progress and applications of single-cell multi-omics sequencing for epigenomic profiling. For example, single-cell methylome and transcriptome sequencing (scM&T-seq) [46] can allow scBS-seq and RNA-seq to be performed simultaneously in the same single cell, which has the advantage of enabling intricate investigations of gene methylation and transcription relationship within a specific cell. Single-cell chromatin accessibility and transcriptome sequencing (scCAT) [51] has allowed simultaneous analysis on the accessible chromatin and gene expression of a single cell to reveal the association of these two molecular layers and their influence on cell fate. Single-cell combinatorial indexing assay for transposase accessible chromatin (sci-ATAC-seq) [50] has also been developed to combine the chromatin accessibility and transcriptomics information simultaneously to study cell clusters and regulatory networks in the mouse hippocampus at single-cell resolution. In addition, droplet-based single-nucleus chromatin accessibility and mRNA expression sequencing (SNARE-seq) has been utilized to find the association between a cell’s transcriptome and its accessible chromatin for sequencing at scale [52]. Recently, the standard operation research for single-cell Multiome ATAC + Gene Expression has been developed by the 10 Genomics company to associate gene expression with open chromatin from the same cell.
Table 4.
Single-cell multi-omics sequencing for protein profiling together with their specific applications, results and data sources.
| Method | Data source | Molecular layers | Objective and outcome(s) | Platform(s) |
|---|---|---|---|---|
| PEA [55] | N/A | Panel of up to 96 RNAs and proteins for individual cells from the same population. | To interrogate cell state and find the corresponding cell functions. For most gene products, only a small portion of the variation of protein levels could be explained by measuring mRNA levels in single cells. | the Fluidigm BioMark HD System |
| REAP-seq [56] | GSE100501 | mRNA expression level coupled with 82 antibodies among single cells. | To identify drug response and describe the unknown cell type. | Illumina HiSeq 2500 and 10X Genomics |
| CITE-seq [57] | GSE100866 | Cellular proteins and transcriptomes for thousands of single cells. | To combine highly multiplexed protein marker detection with unbiased transcriptome profiling for thousands of single cells. Multimodal data analysis could achieve a more detailed characterization of cellular phenotypes than transcriptome measurements alone. | Illumina HiSeq 2500 |
| Morita et al. [58] | GSE156934 | DNA mutation and cell-surface immunophenotype at the single-cell level in 26 AML patients. | To unravel clonal diversity and evolution patterns of AML. Systematic investigation of predictive and prognostic impact of clonal diversity in AML was possible. | Illumina Human Omni 2.5 BeadChip (hg19) and Mission Bio Tapestri Analysis Pipeline |
| DBiT-seq [38] | GSE137986 | mRNAs, proteins and spatial information in a formaldehyde-fixed tissue slide. | To dissect the initiation of early organogenesis at the whole embryo scale. Deterministic barcoding in tissue enabled NGS-based spatial multi-omics mapping. | Illumina HiSeq 4000 (Mus musculus) |
| ASAP-seq [59] | GSE156478 | Chromatin accessibility and protein levels in single cells. | To decipher the underlying regulatory mechanisms at their respective genomic loci. | NextSeq 550 |
| Miles et al.[60] | dbGAP (phs002049.v1.p1) | Protein expression and mutational information in 17 samples from patients with AML. | To find the correlation of somatic genotype and clonal architecture with immunophenotype. Multiple overlapping immunophenotypic states occurred across samples with divergent genotypes; no community was exclusive to an individual sample. | Illumina NovaSeq and Mission Bio Tapestri Insights |
To detect the subpopulations of cancer cells, single-cell triple omics sequencing (scTrio-seq) [48] has been developed to simultaneously profile genomic copy-number variations (CNVs), DNA methylome and transcriptome. To further gain a finer resolution about a single cell’s biological differences, simultaneous high-throughput ATAC and RNA expression (SHARE-seq) [49] was implemented for measuring in parallel chromatin accessibility and gene expression within the same cell. The integration of single-nucleus Droplet-based sequencing and single-cell transposome hypersensitive site sequencing [54] has also shown the possibilities to unravel regulatory elements and transcription factors related to cell-type distinctions, enabling the study of complex genetic programs in the brain as well as normal and pathogenic cellular processes.
Furthermore, epigenomic regulation was found to be useful for coupling with different components in the same cell to offer new possibilities for studying cellular heterogeneity. For example, single-cell chromatin overall omic-scale landscape sequencing (scCOOL-seq) [45] and single-cell nucleosome, methylation and transcription sequencing (scNMT-seq) [47] have offered such opportunities for understanding the epigenomic reprogramming and dependency relationships among these different types of omics.
Protein profiling by single-cell multi-omics sequencing
Proteins are critically important in the analysis of cell states [55]. Unbiased measurements of protein abundance levels play a key role in understanding cellular response to the environment or therapy and in modeling cellular dynamics [56]. The integration of protein and transcription information even has the potential to detect the dynamic change of RNA and protein abundance in the same cell. This has been illustrated by fluorescence-activated cell sorting and image-related approaches to measure RNAs and proteins in parallel [61, 62]. Table 4 presents recent studies on single-cell multi-omics sequencing of protein profiling. Examples of this technology also include proximity extension assay (PEA) [55], RNA expression and protein sequencing (REAP-seq) [56] and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) [57]; all of which have been applied to simultaneously explore the transcript and protein features of a cell, providing key information for identifying the cell state and response to treatments. ATAC with select antigen profiling by sequencing (ASAP-seq) [59] is another example capable of combining chromatin accessibility and protein profiling to interpret regulatory mechanisms in immune cells. Furthermore, the sequencing techniques developed by Miles et al. [60] can integrate mutational information and protein expression at single-cell resolution to reveal the clonal diversity and evolution patterns of specific diseases. Interestingly, reverse transcription and the proximity ligation assay are able to couple with quantitative polymerase chain reaction (PCR) to study cell dynamics and heterogeneity at single-cell resolution [63]. This method can be extended to quantify any combination of DNA, RNAs (such as mRNAs, microRNAs and noncoding RNAs) and proteins from the same single cell.
Metabolomics profiling by single-cell multi-omics sequencing: a brief highlight
Metabolomics constitutes another important molecular layer as well, which is particularly relevant to phenotypic diversity of single cells in response to environmental or chemical stimuli [14]. Mitochondria is a key component of cell metabolism, and mutations of mitochondrial genes have been reported to link to clinical phenotypes of the most common inherited metabolic disorders [64]. Mitochondrial single-cell Assay for transposase accessible chromatin with sequencing can be used to incorporate mitochondrial DNA (mtDNA) mutations and accessible chromatin information to deduce mtDNA heteroplasmy, which is an important factor for determining the severity of mitochondrial diseases.
Examples of integrative multiple single-cell sequencing and their distinction from single-cell multi-omics sequencing
Different to single-cell multi-omics approaches, the integration of multiple single-cell sequencing data sets generally involves merely merging multiple single-cell sequencing data sets from similar cells or tissues to establish correlations between distinct modalities [15, 65]. For instance, LIGER was developed such that single-cell RNA and epigenome datasets collected within the same tissue to reveal further information on cell types and the relationship between transcription and epigenomic regulation [17]. Stuart et al. [66] also conducted a study to integrate scATAC-seq and scRNA-seq datasets from similar tissues to identify subpopulations of cells. In a more complex scenario, a transfer learning method named scJoint was used to combine the CITE-seq and ASAP-seq datasets from different tissues. It was hoped that the joint profiling of gene expression and chromatin accessibility simultaneously with surface protein levels could generate a more comprehensive understanding of cellular phenotypes [67]. In addition, Clonealign [68] was used to infer gene expression profiles to its clone of origin. However, this tool has been used through single-cell RNA or DNA sequencing independently instead of simultaneous DNA and RNA profiling from the same cell.
As these different layers of genomic information are not from the same cell, we suggest that the integration of these single-cell sequencing datasets should be cautiously used for studies in systems biology.
Applications of single-cell multi-omics sequencing in the fight against COVID-19 and other diseases
Omics-based research into complex conditions including cancer [69], drug resistance [70] and neurobiology [71] has been intensively explored, ranging from the genome, transcriptome and proteome to metabolome [72]. However, these approaches can only reveal a modest level of the pathogenesis of complex disease due to the focus on a single omics layer at a time. As seen above, remarkable advances have been made in single-cell multi-omics techniques that can refine our understanding of complex diseases by gathering more than one modality at a time in the same cell [73]. For example, PEA has been used to perform parallel detection of RNA and protein traits for individual cells to reflect cancer cell function and feedback in response to BMP-4 treatment [55]. Morita et al. [58] integrated DNA mutation and protein profiling simultaneously at the single-cell level to study the clinical relevance and clonal diversity of acute myeloid leukemia (AML) disease, whereas Miles et al. [60] employed protein expression and mutational information at single-cell resolution to infer the clonal evolution of myeloid malignancies progression. Baccin et al. revealed the organization of the bone marrow niche by integrating transcriptomics and spatial information at single-cell resolution [39], whereas Lake et al. [54] inferred pathogenic cell types for brain-related diseases by integrating single-cell analysis of nuclear transcripts and DNA accessibility.
Recently, extraordinary effort taking single-cell multi-omics techniques has been dedicated to decipher the disease mechanisms of COVID-19. For example, single-cell multi-omics techniques have been used to investigate immune dysfunction in COVID-19 patients, particularly in peripheral blood mononuclear cells (PBMCs). An investigation by Stephenson et al. utilized full transcriptomes coupled with 188 cell surface proteins to co-profile over 800,000 PBMCs from 130 patients [74]. The researchers also utilized T and B lymphocyte antigen receptor repertoires with COVID-19 across disease severities ranging from asymptomatic to critical from three UK centers to distinguish the host immune response corresponding to SARS-CoV-2. Equipped with these multi-omics sequencing datasets, the authors found that plasmablasts and B cells were increased in severe and critical COVID-19 patients, whereas their mucosal associated invariant T cells were reduced, leading to the conclusion that the symptoms of critically ill COVID-19 patients fitted the main feature of prolonged infection course for critical disease. In another study, scRNA-seq was applied to PBMCs from 13 patients with COVID-19 ranging from moderate to severe symptoms as well as cells from five healthy donors [75]. The incorporation of single-cell T cell receptor (TCR) and B cell receptor (BCR) sequencing for each of the subjects in this study was shown to be useful for gaining an in-depth understanding of immune response and functional properties of immune cells during disease progression following SARS-CoV-2 infection. The authors also successfully assessed the immune responses during disease progression and found that COVID-19 patients had a strong interferon- response for most cell types and an overall acute inflammatory response. Furthermore, significant expansion of highly cytotoxic effector T cell subsets was found to be relevant in the recovery of moderate patients, whereas severe patients faced the challenge of a deranged interferon response, profound immune exhaustion with skewed TCR repertoire and broad T cell expansion. In another comprehensive study, data on immune response to SARS-CoV-2 were integrated to represent all levels of disease severity [76]. It involved single-cell multi-omics analysis of PBMCs, with coordinated profiling of the whole transcriptome, 192 surface markers, TCR and BCR at single-cell resolution from 16 healthy donors and 254 patients with COVID-19 as well as the incorporation of metabolomics and secretome from plasma, coupled with the patients’ clinical information from electronic health records. The association between the status and severity of COVID-19 infection revealed that the immune response was coupled with major plasma composition changes, and the clinical metrics of blood clotting were consistent with the sharp transition between mild and moderate disease.
response for most cell types and an overall acute inflammatory response. Furthermore, significant expansion of highly cytotoxic effector T cell subsets was found to be relevant in the recovery of moderate patients, whereas severe patients faced the challenge of a deranged interferon response, profound immune exhaustion with skewed TCR repertoire and broad T cell expansion. In another comprehensive study, data on immune response to SARS-CoV-2 were integrated to represent all levels of disease severity [76]. It involved single-cell multi-omics analysis of PBMCs, with coordinated profiling of the whole transcriptome, 192 surface markers, TCR and BCR at single-cell resolution from 16 healthy donors and 254 patients with COVID-19 as well as the incorporation of metabolomics and secretome from plasma, coupled with the patients’ clinical information from electronic health records. The association between the status and severity of COVID-19 infection revealed that the immune response was coupled with major plasma composition changes, and the clinical metrics of blood clotting were consistent with the sharp transition between mild and moderate disease.
Investigations on lung tissue and cells have been also an urgent priority for COVID-19 research because respiratory failure is the main reason for patient deaths [77]. From this respect, single-nucleus ATAC-seq and matched single-nucleus RNA-seq in nondiseased lungs from postnatal donors were used to analyze the SARS-CoV-2 host entry genes ACE2, TMPRSS2, CTSL, BSG and FURIN. This approach has also been used to infer age-associated dynamics in gene expression and chromatin accessibility as well as gene regulatory processes in human lungs [78]. Findings suggested that the gene expression of airway and alveolar epithelial cells played key roles in SARS-CoV-2 entry among the barrier cell types exposed to inhaled pathogens. Furthermore, simultaneous profiling of cell lineage protein markers and gene expression in single cells were useful for understanding asynchronization for innate and adaptive immune interaction in progressive COVID-19 patients, and effective for detecting type-1 interferon response across all immune cells [79]. Data analysis indicated that in progressive COVID-19 patients, the response of a dynamic type-1 interferon for all cell types would be dropping along with a decreasing viral load and that the clonal distribution of CD8 T cells and a primary B cell response might be skewed due to existing memory B cells.
On the side of single-cell sequencing (not multi-omics sequencing) for COVID-19 research, scRNA-seq has been applied to profile PBMCs from seven patients with COVID-19 and six healthy controls to reveal the peripheral immune response to severe COVID-19 infection and detect phenotype reconfiguration for peripheral immune cells [80], and scRNA-seq datasets of COVID-19 patients were used to establish a multilayer network integrating intercellular and intracellular signaling subnetworks [81]. Furthermore, the diverse changes in cellular responses and gene expression following SARS-CoV-2 infection were studied by analyzing the bulk transcriptome, bulk DNA methylome and single-cell transcriptome of peripheral blood samples [82]. To deduce the progression of SARS-CoV-2 infection within the body, the bulk-to-cell method focusing on ACE2 areas was employed to integrate the genome, transcriptome, and proteome levels in bulk tissues and single cells across species [83]. Although some conclusions were made regarding COVID-19 treatments in this study, using scRNA-seq alone might not generate as much information or offer as much precision as single-cell multi-omics sequencing data can.
Overall, the advances made by single-cell multi-omics sequencing have given researchers an unprecedented capacity to develop new therapeutic interventions, contributing to a global coordinated effort to win the fight against the COVID-19 pandemic.
Limitations of multi-omics sequencing: a brief discussion
Despite significant advances in the development of single-cell multi-omics sequencing methods, many challenges remain that are mainly associated with the limitations of individual omics methods.
The time at which omics information should be recorded shows discrepancies among different methods. While the genome is approximately static, the change time of the transcriptome and proteome is on the scale of minutes to hours, whereas the reaction time of the metabolome corresponding to environmental influences is in seconds or even milliseconds. In addition, single-cell multi-omics sequencing currently cannot offer significant detail in the identification of RNA isoforms and alternatively spliced variants [14, 84, 85].
Another challenge is that different sequencing methods have distinct throughput levels. Metabolomics and protein-based methods have lower throughput than scRNA-seq, limiting the throughput of multi-omics sequencing in the same cell [13]. To circumvent this problem, some methods have made full use of the existing single-cell datasets to conduct a relatively comprehensive analysis of cells. However, the poor quality of some layers of molecular data from single cells increases the burden of subsequent data analysis. Data problems such as missing data and mismatched data pairs from initial omics aggravate cumulatively when biological knowledge is combined among different layers at the single-cell resolution. This presents a primary obstacle for uncovering accurate and detailed information from heterogeneous data. For example, dropout means that a gene has been falsely identified as ‘unexpressed’ due to lacking detection of the corresponding transcript during the reverse-transcription step [86]. Dropout in gene expression issues is a common challenge encountered by distinct single-cell sequencing methods, particularly single-cell RNA droplet-based methods, leading to sparse expression [18, 87]. scRNA-seq often suffers from allelic dropout for library preparation, resulting in incorrect detection of monoallelic expression value [86]. In single-cell genomics, the traditional PCR and multiple displacement amplification methods utilized for amplification face the challenge of allelic and locus dropouts across the genome [18]. Furthermore, it is essential to note that single-cell chromatin accessibility expression is more sparse than single-cell RNA data. The expression of single-cell chromatin accessibility is nearly binary and has numerous dropouts [88]. For downstream analysis, abandon genes may narrow down the research field on highly expressed genes [18]. Thus, single-cell multi-omics sequencing and the integration of diverse modalities from single-cell datasets face the challenge of data fusion. Data analysis techniques are therefore an essential part of processing single-cell multi-omics sequencing data.
Advanced machine learning and bioinformatics approaches for preprocessing of single-cell multi-omics sequencing data
Parallel acquisition of several molecular layers of omics information from the same cell by single-cell multi-omics sequencing always results in a multitude of heterogeneous data with distinct data formats, which include but are not limited to substantially different numbers of variables, different distributions and scaling, diverse data modalities such as continuous and discrete data forms and ordered and unordered categorical data [94]. There is a critical unmet need in data science to develop methods that can excavate the shared and data-specific information in diverse single-cell measurements. Furthermore, since the missing data problem can escalate in single-cell multi-omics datasets, data imputation methods need to be used to prepare the data before analyzing. Another challenge arises from use of different molecular layers information at the same time. After projecting from one molecular layer to another, single-cell multi-omics sequencing techniques may face failures in data pair mapping, so that the original projection needs to be corrected and adapted to the heterogeneous data.
Figure 1 and Table 5 provide a summary of the advantages and disadvantages of nine popular tools for analyzing single-cell multi-omics data. Most of these methods can be adopted for data preprocessing including data imputation, batch effect removal and data integration. Some of them can be utilized for downstream analysis such as clustering, marker identification, cluster annotation, trajectory analysis and inferring pseudotime and regulators. These downstream analysis issues are presented in this section, whereas the data preprocessing issues are discussed in the next.
Figure 1 .
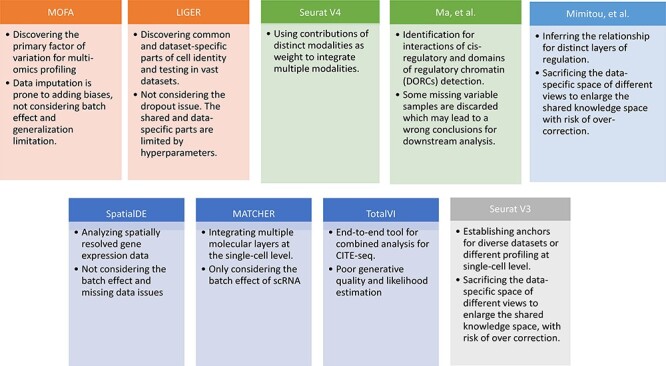
Advantages and disadvantages of nine single-cell multi-omics data analysis tools. The primary advantage and disadvantage of each method is represented by the 1st and 2nd points in each box, respectively. Orange denotes matrix decomposition, green denotes graph-based methods, light blue denotes clustering methods, navy blue denotes variational inference and gray denotes the CCA method. LIGER has employed both matrix decomposition and graph-based algorithms.
Table 5.
Tools for processing single-cell multi-omics sequencing data with their functions and programming language
| Tools | Methods | Functions | languages | References |
|---|---|---|---|---|
| MOFA | Matrix decomposition | Integration and data imputation, clustering. | R | [89] |
| LIGER | Matrix decomposition, SFN graph | Integration and batch effect, visualization, clustering, maker identification. | R | [17] |
| SpatialDE | VI, multivariate normal modeling | Clustering, identify spatially variable genes, spatial and/or temporal annotation, visualization. | Python | [90] |
| TotalVI | VAE | Integration, batch effect and data imputation,visualization,clustering. | Python | [91] |
| Seurat V3 | CCA, MNN | Integration, batch effect and data imputation, clustering, cluster annotation. | R | [66] |
| Seurat V4 | WNN graph | Integration, batch effect clustering, trajectory analysis, cluster annotation, response to vaccination. | R | [92] |
| Mimitou et al. | Seurat V3, harmony, LMM | Integration, data imputation, batch effect, clustering, trajectory analysis, multiplexed CRISPR perturbations in primary T cells. | R | [59] |
| MATCHER | Manifold learning | Integration, inferring pseudotime, trajectory analysis. | Python | [93] |
| Ma et al. | SNF, KNN | Integration and data imputation, clustering, visualization, pseudotime inference. | R and python | [49] |
Matrix factorization analysis
Matrix factorization-based approaches are unsupervised machine learning methods for simultaneous data integration and dimensional reduction. They are achieved by mapping the multidimensional data space of different dimensions and scales into a lower dimensional subspace of unified dimension and scale at the single-cell level.
Matrix decomposition has been adopted by Multi-Omics Factor Analysis (MOFA) [89] to aggregate transcriptome and epigenome profiling data among 61 mouse embryonic stem cells. An important step of this method is to decompose different data matrices 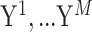 into the common factor matrix
into the common factor matrix 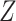 , the specific weight matrices
, the specific weight matrices 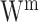 and the view-specific residual noise terms
and the view-specific residual noise terms 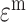 , as shown in Equation (1):
, as shown in Equation (1):
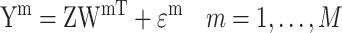 |
(1) |
where, 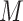 denotes the number of data matrices. Prior distributions are applied in all unobserved variables – the weights
denotes the number of data matrices. Prior distributions are applied in all unobserved variables – the weights 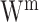 can be considered to be as a product of a normally distributed random variable and a Bernoulli distributed random variable; the residual noise terms
can be considered to be as a product of a normally distributed random variable and a Bernoulli distributed random variable; the residual noise terms 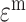 can be inferred following a Poisson model, a Bernoulli model, Gaussian noise model corresponding to count, binary, and continuous data types respectively and the factor matrix
can be inferred following a Poisson model, a Bernoulli model, Gaussian noise model corresponding to count, binary, and continuous data types respectively and the factor matrix  can be assumed using a standard normal prior.
can be assumed using a standard normal prior.
MOFA can be extended to resolve the missing data issue through observed variables. Actually the missing values 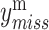 are not involved in updating parameters to compute the likelihood. Instead, the parameters
are not involved in updating parameters to compute the likelihood. Instead, the parameters 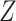 ,
, 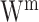 and
and 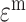 are updated by the observed variables
are updated by the observed variables 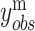 . Thus, the missing variable
. Thus, the missing variable 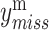 can be directly imputed by Equation (1).
can be directly imputed by Equation (1).
Another extension of matrix decomposition called LIGER has been developed for data integration [17]. In fact, the approach has been used to disentangle single-cell multi-omics datasets 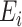 into the latent metagene factors matrix
into the latent metagene factors matrix 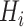 for each omics profiling
for each omics profiling 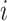 , shared modality metagenes
, shared modality metagenes 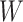 across distinct omics and dataset-specific metagenes
across distinct omics and dataset-specific metagenes 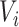 , which is shown in Equation (2) as follows.
, which is shown in Equation (2) as follows.
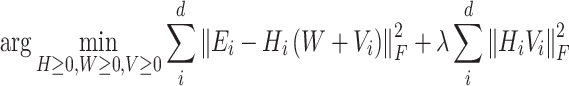 |
(2) |
Thus, these gene features can be classified into shared metagenes and data-specific metagenes of different modalities, thereby retaining data diversity of multi-omics profiling at the single-cell level.
VI-based analysis
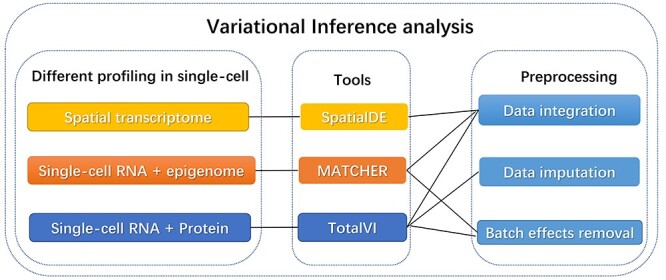
VI is a machine learning approach through approximating posterior probability densities for Bayesian models. Comparing with other methods, this algorithm is faster and easier to handle large data sets [95]. As shown in Figure 2, the three VI methods, SpatialDE, Manifold Alignment to CHaracterize Experimental Relationships (MATCHER) and TotalVI have been applied to combine data from distinct molecular layers at single-cell resolution. These tools can also be leveraged for data preprocessing.
Figure 2 .
Three different tools base on variational inference algorithm for single-cell multi-omics data. SpatialDE can be used for data integration of the spatial transcriptome; MATCHER can be used for data integration and batch effects removal of single-cell transcriptome coupled with epigenome and TotalVI can be used for data integration, batch effects removal and data imputation of single-cell transcriptome coupled with protein.
SpatialDE [90] has made use of VI and multivariate normal models to process the seqFISH data [31]. Genes 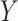 with spatial expression patterns
with spatial expression patterns 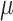 can be inferred. In addition,
can be inferred. In addition, 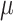 and
and 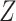 can be estimated using VI. The complete model covering all genes is formulated as:
can be estimated using VI. The complete model covering all genes is formulated as:
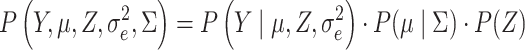 |
(3) |
where, the binary indicator matrix 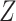 denotes the relationships between the genes and the patterns. The parameter
denotes the relationships between the genes and the patterns. The parameter 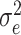 stands for the Gaussian distributed noise for the model and
stands for the Gaussian distributed noise for the model and 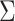 represents the spatial covariance matrix.
represents the spatial covariance matrix.
VI has been adopted by the incorporation of manifold and a shared Gaussian process for MATCHER [93]. The method can be used to map the high-dimensional gene data 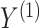 or methylation measurement data
or methylation measurement data 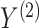 to a shared latent space
to a shared latent space  via different mapping functions
via different mapping functions  coupled the noise
coupled the noise 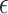 :
:
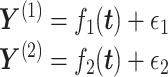 |
(4) |
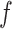 represents a Gaussian process function:
represents a Gaussian process function:
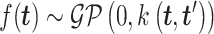 |
(5) |
MATCHER has used a radial basis function automatic relevance determination kernel, which has the advantage of enabling a different set of latent dimension weights for each data type. Then VI can be utilized to compute the posterior and optimize the value of different hyperparameters through evidence lower round (ELBO).
An important extension of VI is named variational autoencoders (VAE). VAE is a generative model. It has potential to integrate different molecular layers’ information at the single-cell level and can correct mismatched data pairs and impute the missing variables. For example, Gayoso et al. [91] generated the paired matrices for RNA and protein counts using a VAE model called TotalVI to integrate distinct omics knowledge in single cells and infer parameters for CITE-seq data. By this method, the batch index 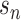 for the RNA expression
for the RNA expression 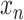 and the protein expression
and the protein expression 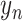 are taken to the encoder part to generate the approximate posterior parameters
are taken to the encoder part to generate the approximate posterior parameters 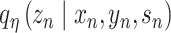 in shared latent space, the RNA size factor
in shared latent space, the RNA size factor 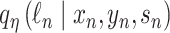 and the protein background factor
and the protein background factor 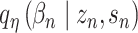 , which is shown in Equation (6) as follows:
, which is shown in Equation (6) as follows:
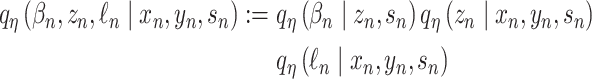 |
(6) |
The decoder part consist of three individual neural networks. The outputs of the encoder have been utilized as input terms to produce the likelihood parameters of the RNAs 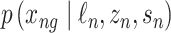 , and the proteins
, and the proteins 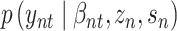 .
.
TotalVI [91] has been proposed to handle the batch effects for mismatched data pairs. TotalVI has a key step to generate a joint probabilistic representation for the RNA and protein data. In the encoder part, all the RNA expression, protein expression and batch index are used to produce the approximate posterior 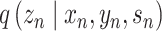 , which is robust to the batch effects. The parameters of the encoder and decoder part can be updated through the gradient of ELBO.
, which is robust to the batch effects. The parameters of the encoder and decoder part can be updated through the gradient of ELBO.
Furthermore, TotalVI has handled the merging of CITE-seq datasets with standard scRNA-seq datasets and has imputed the missing protein measurements simultaneously. The missing protein variables were all filled in with zeros, and the observed values of CITE-seq and scRNA-seq were all sent to the encoder part. Then, the gradient of ELBO was taken to update the parameters using the observed values 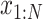 and
and 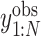 . Finally, the missing protein values
. Finally, the missing protein values 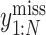 were inferred by the shared latent parameter
were inferred by the shared latent parameter 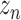 and the protein background value
and the protein background value 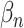 through Equation (6).
through Equation (6).
Canonical correlation analysis (CCA)
CCA is a general unsupervised approach to maximize a shared correlation space through the linear combination of features.
Seurat V3 [66] is a commonly used tool extended from the CCA data fusion method [96]. This tool has been applied to handle DBiT-seq datasets [38] for downstream data analysis. MAESTRO [97] has also employed this tool to integrate scRNA-seq and scATAC-seq datasets. Another application of Seurat V3 was to find the shared feature space and establish anchor pairs for the CITE-seq [57] and STARmap [37] datasets. The shared data part is defined as Equation (7):
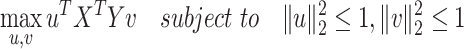 |
(7) |
where, 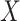 stands for one molecular layer matrix and
stands for one molecular layer matrix and 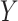 for another layer expression;
for another layer expression; 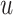 and
and 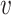 denote the projection vectors.
denote the projection vectors.
Stuart and colleagues found that incorrect anchor pairs could be detrimental for further analysis and could lead to a wrong conclusion. To overcome this problem, an idea of mutual nearest neighbors (MNN) [98] was implemented to adjust the mismatched anchor pairs through decreasing the scores and down-weighting in Seurat V3. The weight matrix was formulated as Equation (8):
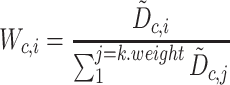 |
(8) |
where 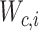 is normalized across all the
is normalized across all the 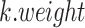 anchors using the weighted distance and the Gaussian kernel
anchors using the weighted distance and the Gaussian kernel 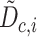 .
.
Seurat V3 is highly informative as the transfer learning framework which can propagate related information such as labels and variables from one omics layer to another. SHARE-seq [49] has utilized this idea to transfer cell-type labels from scRNA-seq to scATAC-seq. Similarly, Mimitou et al. [59] has employed this method to impute the RNA expression. Seurat V3 has also been applied by MAESTRO to transfer the label information from scRNA-seq to scATAC-seq [97]. After building the anchor pairs from the distinct omics layers, the prediction of feature expression 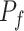 can be completed through the anchor feature-transferred matrix
can be completed through the anchor feature-transferred matrix 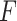 and the weight matrix
and the weight matrix 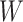 computed from Equation (8). Thus, this missing variable can be imputed, as shown in Equation (9):
computed from Equation (8). Thus, this missing variable can be imputed, as shown in Equation (9):
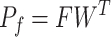 |
(9) |
Clustering analysis
Clustering is an unsupervised learning method that can find the internal structure among single-cell multi-omics data. By updating the input embedding and soft cluster, clustering methods can be employed to integrate different omics information at the single-cell level and correct errors in the data pairs.
This algorithm has been adopted by Mimitou et al. [59] coupled with the Harmony method [99] to merge ASAP-seq [59] and CITE-seq [57]. The principal component analysis result 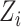 is utilized as the default input, then the Harmony approach maximizes a diversity clustering on the low-dimensional space. In addition, a k-means clustering with the restrictions of an entropy regularization term
is utilized as the default input, then the Harmony approach maximizes a diversity clustering on the low-dimensional space. In addition, a k-means clustering with the restrictions of an entropy regularization term 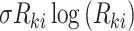 over the soft cluster’s assignment matrix
over the soft cluster’s assignment matrix 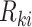 and a low batch-diversity penalization term
and a low batch-diversity penalization term 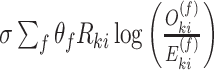 can be determined by
can be determined by
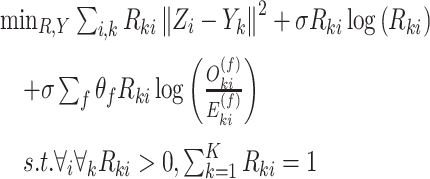 |
(10) |
It can be seen that this approach has added a penalty to maximize the variety of different single-cell multi-omics data for enlarging shared information, namely, maximizing the diversity of datasets within each cluster.
Mimitou and colleagues has also applied a Linear Mixture Model Correction idea to handle the batch effects on clusters in the low-dimensional space. They assumed that the low-dimensional expression 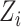 follows Gaussian Mixture Model via the cluster centroids
follows Gaussian Mixture Model via the cluster centroids 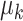 , the batch offset of cluster centroid
, the batch offset of cluster centroid 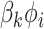 and the soft cluster assignment matrix
and the soft cluster assignment matrix 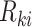 (Equation (11)). Under this assumption, the embedding expression
(Equation (11)). Under this assumption, the embedding expression 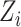 can be replaced (as by Equation (12)). Moreover, this Additive Batch Mixture Model is linked to every cluster, literately updating the soft cluster assignment matrix
can be replaced (as by Equation (12)). Moreover, this Additive Batch Mixture Model is linked to every cluster, literately updating the soft cluster assignment matrix 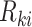 and the low-dimensional expression
and the low-dimensional expression 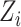 until it becomes convergent.
until it becomes convergent.
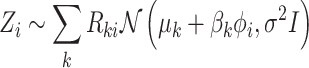 |
(11) |
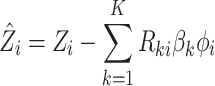 |
(12) |
where 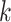 denotes the number of clusters.
denotes the number of clusters.
Graph-based learning analysis
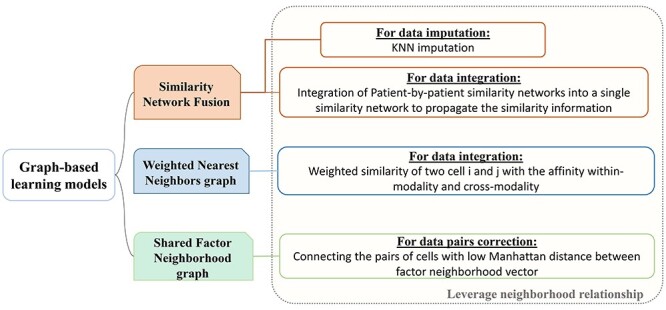
One common approach to tackling single-cell multi-omics sequencing data is to apply graph-based learning models. The graph’s neighborhood information can be utilized to correct the mismatched data pairs when considering the incorrect projection for data pairs. Furthermore, the graph can be employed to make a prediction according to the k-nearest neighbors (KNN) for within and cross-modality information. As shown in Figure 3, the established graph models can propagate the similarity information of different omics layers to preserve the diversity of different omics data at the single-cell level.
Figure 3 .
Three graph-based learning models for single-cell multi-omics sequencing. All of them have leveraged on the neighborhood relationship for data imputation, data integration and data pairs correction.
We present some graph-based models that have been applied to conduct data analysis for single-cell multi-omics sequencing datasets. Ma et al. [49] utilised similarity network fusion (SNF) [100] to aggregate ATAC and RNA signals of SHARE-seq into the whole network. They have established patient-by-patient similarity networks for each data type and adopted KNN relationship to represent local affinity. Then, these networks in different views have been integrated into a single weighted network. A normalized weight matrix 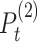 in
in 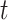 times and local affinity matrix
times and local affinity matrix  based on KNN relationships have been utilized to update the weight
based on KNN relationships have been utilized to update the weight 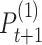 in
in 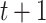 times for the joint network until it converges (formulated as Equation (13)).
times for the joint network until it converges (formulated as Equation (13)).
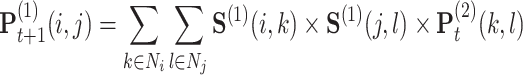 |
(13) |
That is using the local affinity matrix 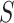 to find the common neighborhood in KNN (
to find the common neighborhood in KNN (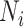 for vertices
for vertices 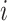 and
and 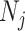 for
for 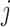 ). Hence, the similarity information can be propagated through the shared neighborhood, and the individual information can be preserved during a single SNF process.
). Hence, the similarity information can be propagated through the shared neighborhood, and the individual information can be preserved during a single SNF process.
To deal with the missing data problem, Ma et al. [49] adopted three strategies including case-wise deletion, feature-wise deletion and data imputation. The case-wise deletion can be utilized when missing parts account for more than 20% of features in a patient. Meanwhile, the feature-wise deletion can be adopted when missing values take up more than 20% of the patients’ specific biological features. For the rest of the missing data part, KNN imputation method [101] can be used to impute the value 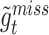 for missing gene
for missing gene 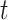 using the value
using the value 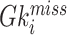 of KNN gene
of KNN gene 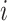 and the distance
and the distance 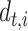 between gene
between gene 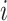 and
and 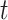 .
.
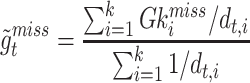 |
(14) |
Welch et al. [17] found that for highly divergent datasets, the maximum factor loading can generate spurious alignments using iNMF representation of cluster assignments. Then, they developed a shared factor neighborhood (SFN) graph for LIGER to address this problem, leveraging the KNN to increase the robustness of joint clustering results. Even though cells from different cell types may have factor loadings spuriously in distinct datasets, SFN can still reduce these false matches among pairs of distinct datasets since it is impossible for them to have the same factor neighborhoods. Hence, corrections has been made by connecting the pairs of cells with low Manhattan distance.
Furthermore, Seurat V4 [92] has been proposed taking a weighted nearest neighbors (WNN) graph to find the relationship between different molecular profiles for SHARE-seq [49], CITE-seq [57] and ASAP-seq [59] datasets. Firstly, the within-modality RNA prediction 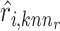 and protein prediction
and protein prediction 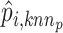 and cross-modality prediction
and cross-modality prediction 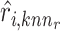 and
and 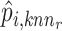 are calculated through the corresponding KNN. Then, with motivations from a Uniform Manifold Approximation and Projection (UMAP) weight function, this method also computes the weight of cell-specific modality for RNA
are calculated through the corresponding KNN. Then, with motivations from a Uniform Manifold Approximation and Projection (UMAP) weight function, this method also computes the weight of cell-specific modality for RNA 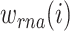 and protein
and protein 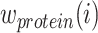 correspondingly. Meanwhile, the affinity within-modality and cross-modality for RNA
correspondingly. Meanwhile, the affinity within-modality and cross-modality for RNA 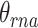 and protein
and protein 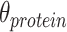 are calculated respectively. Finally, the weighted similarity of two cells
are calculated respectively. Finally, the weighted similarity of two cells 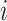 and
and 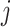 are used to build a WNN graph as shown in Equation (15).
are used to build a WNN graph as shown in Equation (15).
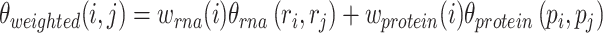 |
(15) |
This approach has the advantage of learning the varying information of each molecular perspective through each modality’s weight.
Issues and open questions for preprocessing of single-cell multi-omics data
Intensively studied issues: consistency and diversity
The integration of heterogeneous data of single-cell multi-omics sequencing usually faces a consistency issue (common or shared information) and a diversity issue (complement or data-specific information). While many single-cell multi-omics sequencing studies have been focused on data consistency research, diversity for different molecular layers’ information should not be overlooked. The main reason is that different biological layers convey their unique knowledge, so that the data-specific information for distinct omics layers at the single-cell level should be considered. Some methods attempt to maximize the shared space of different molecular profiles. However, these approaches are detrimental to the unique information of different omics sequencing at the single-cell level since they squeeze the data-specific space of different views to enlarge the shared knowledge space. For example, Seurat V3 [66] has been carried out through a variation of CCA to maximize a shared correlation space through the linear combinations of features across multi-sources datasets based on the same instances. Other unsolved problems include how much should be divided for data-specific and shared information to balance data consistency and diversity for distinct molecular layers in single cells. For example, LIGER [17] utilized iNMF to divide the single-cell multi-omics data into shared modality metagenes and dataset-specific metagenes, but balancing these two components was limited by the hyperparameter 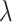 .
.
While these methods have achieved varying levels of success at dealing with consistency and diversity, they still encounter several issues. As an example, TotalVI [91] has been developed to take the advantage of flexible networks of VAE to map RNA and protein counts into shared latent embedding to preserve the consistency of RNA space and protein space. However, they faced challenges of poor generative quality and poor likelihood estimation. While MOFA [89] can be employed by matrix factorization to map the different omics datasets into a common factor matrix, the uninformative prior for different distribution has been restricted the solution space, thereby limiting the generalization.
Currently focused issues: mismatched and missing data
Ensuring data consistency and diversity in single-cell multi-omics sequencing is technically challenging. This is because that single-cell multi-omics sequencing often has difficulties in the management of mismatched and missing data. These two currently-focused issues are critical since multi-omics in the same cell need to integrate incomplete data from different biological layers [13]. This may lead to obstacles for the interpretation and therapeutic development of complex diseases. Thus, data imputation and correction are essential as preprocessing steps before downstream data analysis procedures.
Gene expression data often contain sequencing discrepancies after integrating distinct single-cell measurements. In earlier studies, batch correction methods has been employed for integration of scRNA-seq due to technical variability. To address incorrect data pairs, many researchers has learned from the experience of batch effects correction methods for single-cell RNA and applied this in single-cell multi-omics data since distinct molecular layers in single-cells also have technical variability among distinct sequencing measurements in the same cell. For example, Seurat V3 [66] has been inspired from MNN [98] for batch effects removal to solve the mismatched anchor pairs through decreasing scores and weights. Mimitou et al. [59] has utilized the Linear Mixture Model Correction approach [99] for batch effects method to correct mismatched pairs in low dimensional space. Furthermore, Seurat V4 [92] has been developed by MNN graph to find the corresponding weight for different modalities, which can be considered as batch effect correction.
Although many correction approaches to date overcoming the batch effects have achieved positive outcomes, there are always risk of overcorrection that may cover the true biological expression data and lead to wrong conclusions for downstream analysis.
Another issue in handling single-cell multi-omics data is about missing data. Data acquired from high-throughput single-cell sequencing platforms are known to have missing observations or variables due to various reasons, such as low coverage of next-generation sequencing (NGS), low sensitivity in protein detection and faltered metabolite measurement by tandem mass spectrometry. The problem of missing data in single-cell multi-omics sequencing can be aggravated since multi-omics in the same cell needs to integrate incomplete data from different biological layers [13], and missing values in single-cell multi-omics sequencing data can result in obstacles for downstream analysis. Thus, solving the missing values should be essential as a preprocessing step before the subsequent data analysis procedures are performed. Deletion is the simplest method for handling missing variables. However, it is difficult to represent the complete information using remaining cases, which may lead to a wrong conclusions. For example, Ma et al. [49] has employed deletion in multivariate and multimodel analyses, which involve many items and features. This can lead to a large number of samples and variate to be discarded and biased results. The remaining cases or features may not represent the complete multi-omics information in the same cell [102]. Moreover, TotalVI [91] and MOFA [89] have employed a similar strategy by using the observed sample and variable to compute the parameter. Then, inference parameters have been used to approximate the missing part. Although these single imputation methods appear to be more advantageous than case deletion and feature deletion, they are prone to adding biases and introduce difficulties in fitting well with the true distribution, since the imputation quality has a close relationship with the inference parameters and alleviates the variability of the missing data.
Open questions
Although single-cell multi-omics sequencing is showing a vital role in progressing our understanding of system biology and pathophysiology, the handling of heterogeneous data remains technically challenging. Compared with computer vision (CV) [103] and natural language processing (NLP) [104] applications, we believe that the sampling cost by single-cell multi-omics is higher than acquiring the image and language data, whereas that the technology of single-cell multi-omics sequencing is less mature than CV and NLP algorithms. The magnitude of data collection in single-cell multi-omics should be therefore much smaller than in CV and NLP applications. In addition, we know that many researchers collect distinct types of sequencing data as much as possible to make up for the lack of data samples in the attempt to convert the need for the number of samples to the demand for feature types of samples. Hence, distinct molecular layers at single-cell resolution can highlight the data heterogeneity. This brings up several open questions as follows.
i Do we need all of these data on different biological traits at the single-cell level for specific biological or medical applications? or more image data can be supplemented?
ii How to evaluate and benchmark the performance of single cell multi-omics tools when the ground-truth labels are not available? Whether NLP algorithms can help the understanding of the cross talks between the different layers of omics data from a large population of cells?
iii How to systematically interpret the downstream biological results from the single-cell multi-omics data?
Future perspectives
Every data processing method has its own advantages and disadvantages. To enable better decision-making or achieve greater performance, we may need to combine multiple machine learning methods into one model to amplify the advantages and compensate for the disadvantages of individual methods. Some relevant machine learning approaches that may lead to effective future developments in the single-cell multi-omics space are suggested below.
Multimodel is a specific example of multi-view algorithms. This method can utilize feature representations across different modalities from multiple sources [105]. The diverse molecular layers can be considered as the detailed multiple modalities including genetics, epigenetics, metabolism, protein and spatial information corresponding to different views. A joint embedding can be leveraged to map the distinct model data into shared latent space. Meanwhile, the neighborhood information of the local affinity can be exploited to establish a relationship with another modality corresponding to the other neighborhood information. Thus, sufficient biological information can be leveraged across and within molecular layers.
Deep learning is an exciting topic, where we have recently witnessed a massive expansion in Deep Neural Networks method development. These approaches have achieved ideal results in different fields while still greatly increasing the performance. The Convolutional Neural Network (CNN) [106], Recurrent Neural Network (RNN) [107] and transformer model [108] can be adopted for processing the heterogeneous data arising from single-cell multi-omics sequencing.
The input format of CNN is multidimensional, which is well suited for targeting the heterogeneous data simultaneously. Moreover, CNN can abstract the features through layer-by-layer filters to find the specific patterns. Some CNN models, such as DenseNet [109] and ResNet [110], also make it possible to alleviate the vanishing gradients. The loss landscapes of these models can become smoother, and the gradients can backpropagate the much further depth model, capturing a large amount of complex information.
The RNN is also a well-suited model for processing multi-omics sequencing data. The reason is that RNN can preserve great amounts of semantic context information about past states, such that it enables the learning of combined genetics effects corresponding to phenotypes. Moreover, the hidden state of the RNN model can be updated in intricate ways, thus making it possible to learn specific sequence properties.
The transformer model has recently attracted increasing attentions. Compared with the CNN and RNN, the transformer model can process sequencing data simultaneously and address long-term dependency problems. It can also utilize self-attention to update the embedding outcome and weight each word to represent the correlations of words. Thus, the transformer model is expected to achieve better performance in processing single-cell multi-omics data since it can find the gene-to-gene correlation without considering the long dependency of sequencing.
Transfer learning [111] can be used to address several challenges on missing variables, batch effects in single-cell multi-omics sequencing. Transfer learning can extract information from one molecular layer to correct or impute related information in another layer. By transferring the knowledge from the source domain to the target domain, we can make use of a vast amount of knowledge from biological layers to correct the error of some variables or impute missing values at different molecular layers.
Conclusion
Gathering information from different biological modalities at single-cell resolution offers the possibility of gaining deep insight into the cell state and function and refining our understanding of the relationship between cell genotypes and phenotypes. Recent studies have demonstrated the power of single-cell multi-omics sequencing in various biological and medical applications. Despite its extraordinary potential, challenges relating to the analysis of heterogeneous data arising from single-cell multi-omics sequencing remain to be addressed. The performance of fusing heterogeneous data in diverse molecular layers of single cells can vary depending on the trade-off between data consistency and diversity, correction of mismatched data pairs and imputation of the missing variables. Machine learning methods have great potential to improve the processing of single-cell multi-omics sequencing data as suggested in our understanding of their prospects.
Key Points
We presented technical developments and progresses of single-cell multi-omics sequencing made over the last 5 years and outlined the trends of their biological and medical applications.
We presented up-to-date applications of single-cell multi-omics sequencing for fighting against the COVID-19 pandemic.
We described advanced machine learning and bioinformatics methods used in data preprocessing of single-cell multi-omics sequencing data.
We discussed data analysis issues, and suggested open questions and prospects for the future research of single-cell multi-omics sequencing.
Lu Huo is a PhD student in the Faculty of Engineering and IT at the University of Technology Sydney (UTS). She has deep interest in single-cell multi-omics sequencing data preprocessing techniques and multimodal analysis.
Jiao Jiao Li is a lecturer in Biomedical Engineering at UTS, a National Health and Medical Research Council (NHMRC) Early Career Fellow, a chief investigator on the Australian Research Council Training Centre for Innovative BioEngineering, and a Science & Technology Australia 2021-22 Superstar of STEM. Her research interests are regenerative medicine and medical technologies.
Ling Chen is an associate professor with the UTS Priority Research Centre for Artificial Intelligence (CAI) and the Faculty of Engineering and Information Technology (FEIT) at the University of Technology Sydney. Her main research interests focus on data mining and machine learning.
Zuguo Yu is a professor in Hunan Key Laboratory for Computation and Simulation in Science and Engineering, Xiangtan University. His research is focused on fractals, bioinformatics and complex networks and geomagnetic data analysis.
Gyorgy Hutvagner is a professor in the School of Biomedical Engineering at the University of Technology Sydney. His research is focused on RNA mechanisms.
Jinyan Li is a professor at the Data Science Institute at University of Technology Sydney (UTS). He has a broad range of interests to invent data mining algorithms to solve challenging problems in genomics and computational biology.
Contributor Information
Lu Huo, Data Science Institute, University of Technology Sydney, Ultimo, NSW 2007, Australia; School of Computer Science, FEIT, University of Technology Sydney, Ultimo, NSW 2007, Australia.
Jiao Jiao Li, School of Biomedical Engineering, FEIT, University of Technology Sydney, Ultimo, NSW 2007, Australia.
Ling Chen, School of Computer Science, FEIT, University of Technology Sydney, Ultimo, NSW 2007, Australia.
Zuguo Yu, Key Laboratory of Intelligent Computing and Information Processing of Ministry of Education, Hunan Key Laboratory for Computation and Simulation in Science and Engineering, Xiangtan University, Hunan, 411105, P.R. China.
Gyorgy Hutvagner, School of Biomedical Engineering, FEIT, University of Technology Sydney, Ultimo, NSW 2007, Australia.
Jinyan Li, Data Science Institute, University of Technology Sydney, Ultimo, NSW 2007, Australia.
Funding
Australia Research Council Discovery Project (DP180100120); National Health and Medical Research Council (Australia; GNT1120249); National Natural Science Foundation of China (Grant No. 11871061).
References
- 1. Editorial. Method of the year 2013. Nat Methods 2014;11(1):1. [DOI] [PubMed] [Google Scholar]
- 2. Tang F, Barbacioru C, Wang Y, et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat Methods 2009;6(5):377–82. [DOI] [PubMed] [Google Scholar]
- 3. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015;161(5):1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klein AM, Mazutis L, Akartuna I, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015;161(5):1187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spitzer MH, Nolan GP. Mass cytometry: single cells, many features. Cell 2016;165(4):780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McDonnell LA, Heeren RMA. Imaging mass spectrometry. Mass Spectrom Rev 2007;26(4):606–43. [DOI] [PubMed] [Google Scholar]
- 7. Zrazhevskiy P, Gao X. Quantum dot imaging platform for single-cell molecular profiling. Nat Commun 2013;4(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smallwood SA, Lee HJ, Angermueller C, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 2014;11(8):817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rotem A, Ram O, Shoresh N, et al. Single-cell chip-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 2015;33(11):1165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buenrostro JD, Giresi PG, Zaba LC, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10(12):1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vitak SA, Torkenczy KA, Rosenkrantz JL, et al. Sequencing thousands of single-cell genomes with combinatorial indexing (report). Nat Methods 2017;14(3):302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saliba AE, Westermann AJ, Gorski SA, et al. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res 2014;42(14):8845–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelsey G, Stegle O, Reik W. Single-cell epigenomics: recording the past and predicting the future. Science 2017;358(6359):69–75. [DOI] [PubMed] [Google Scholar]
- 14. Zenobi R. Single-cell metabolomics: analytical and biological perspectives. Science 2013;342(6163):1243259. [DOI] [PubMed] [Google Scholar]
- 15. Stuart T, Satija R. Integrative single-cell analysis. Nat Rev Genet 2019;20(5):257–72. [DOI] [PubMed] [Google Scholar]
- 16. Editorial. Method of the year 2019: single-cell multimodal omics. Nat Methods 2020;17(1):1. [DOI] [PubMed] [Google Scholar]
- 17. Welch JD, Kozareva V, Ferreira A, et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 1873, 2019;177(7):1887.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu Y, An Q, Sheu K, et al. Single cell multi-omics technology: methodology and application. Front Cell Dev Biol 2018;6(28):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J, Hyeon DY, Hwang D. Single-cell multiomics: technologies and data analysis methods. Exp Mol Med 2020;52(9):1428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma A, McDermaid A, Xu J, et al. Integrative methods and practical challenges for single-cell multi-omics. Trends Biotechnol 2020;38(9):1007–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y, Ma L, Wu D, et al. Advances in bulk and single-cell multi-omics approaches for systems biology and precision medicine. Brief Bioinform 2021:bbab024. [DOI] [PubMed] [Google Scholar]
- 22. Macaulay IC, Haerty W, Parveen Kumar YI, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods 2015;12(6):519–522. [DOI] [PubMed] [Google Scholar]
- 23. Dey SS, Kester L, Spanjaard B, et al. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol 2015;33(3):285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zachariadis V, Cheng H, Andrews N, et al. Highly scalable method for joint whole-genome sequencing and gene-expression profiling of single cells. Mol Cell 2020;80(3):541–553.e5. [DOI] [PubMed] [Google Scholar]
- 25. Xiao Z, Cheng G, Jiao Y, et al. Holo-seq: single-cell sequencing of holo-transcriptome. Genome Biol 2018;19(1):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang N, Ji Z, Chen Z, et al. Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat Commun 2019;10(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zachariadis V, Cheng H, Andrews N, et al. A highly scalable method for joint whole-genome sequencing and gene-expression profiling of single cells. Mol Cell 2020;80(3):541–53. [DOI] [PubMed] [Google Scholar]
- 28. Chen KH, Boettiger AN, Moffitt JR, et al. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015;348(6233):aaa6090–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moffitt JR, Bambah-Mukku D, Eichhorn SW, et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 2018;362(6416). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Codeluppi S, Borm LE, Zeisel A, et al. Spatial organization of the somatosensory cortex revealed by osmfish. Nat Methods 2018;15(11):932–5. [DOI] [PubMed] [Google Scholar]
- 31. Lubeck E, Coskun AF, Zhiyentayev T, et al. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods 2014;11(4):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eng C-HL, Lawson M, Zhu Q, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqfish+. Nature 2019;568(7751):235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Je HL, Daugharthy ER, Scheiman J, et al. Highly multiplexed subcellular RNA sequencing in situ. Science 2014;343(6177):1360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ståhl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016;353(6294):78–82. [DOI] [PubMed] [Google Scholar]
- 35. Rodriques SG, Stickels RR, Goeva A, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019;363(6434):1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vickovic S, Eraslan G, Salmén F, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods 2019;16(10):987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Allen WE, Wright MA, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science Jul 2018;361(6400). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Y, Yang M, Deng Y, et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 2020;183(12):e18.1665–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baccin C, Al-Sabah J, Velten L, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol Jan 2020;22(1):38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Editorial. Method of the year 2020: spatially resolved transcriptomics. Nat Methods 2021;18(1):1–1. [DOI] [PubMed] [Google Scholar]
- 41. Waylen LN, Nim HT, Martelotto LG, et al. From whole-mount to single-cell spatial assessment of gene expression in 3D. Commun Biol 2020;3(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhuang X. Spatially resolved single-cell genomics and transcriptomics by imaging. Nature Methods 2021;18(1):18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shah S, Takei Y, Zhou W, et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqfish. Cell 2018;174(2):363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eng C-HL, Shah S, Thomassie J, et al. Profiling the transcriptome with RNA spots. Nat Methods 2017;14(12):1153–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo F, Li L, Li J, et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res 2017;27(8):967–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu Y, Huang K, An Q, et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 2016;17(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clark SJ, Argelaguet R, Kapourani CA, et al. Scnmt-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun 2018;9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hou Y, Guo H, Cao C, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 2016;26(3):304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma S, Zhang B, LaFave LM, et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 2020;183(4):1103–1116.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sinnamon JR, Torkenczy KA, Linhoff MW, et al. The accessible chromatin landscape of the murine hippocampus at single-cell resolution. Genome Res May 2019;29(5):857–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu L, Liu C, Quintero A, et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun 2019;1–10(12):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen S, Lake BB, Zhang K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol 2019;37(12):1452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moris N, Pina C, Arias AM. Transition states and cell fate decisions in epigenetic landscapes. Nat Rev Genet 2016;693–703(10):17. [DOI] [PubMed] [Google Scholar]
- 54. Lake BB, Chen S, Sos BC, et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 2018;36(1):70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Darmanis S, Gallant CJ, Marinescu VD, et al. Simultaneous multiplexed measurement of RNA and proteins in single cells. Cell Rep 2016;14(1):380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peterson VM, Zhang KX, Kumar N, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 2017;35(10):936–9. [DOI] [PubMed] [Google Scholar]
- 57. Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 2017;14(9):865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morita K, Wang F, Jahn K, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun 2020;11(1):5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mimitou L, Chen F, Takeshima L, et al. Scalable, multimodal profiling of chromatin accessibility and protein levels in single cells. bioRxiv. Cold Spring Harbor Laboratory, 2020;13.
- 60. Miles LA, Bowman RL, Merlinsky TR, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020;587(7834):477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Arrigucci R, Bushkin Y, Radford F, et al. Fish-flow, a protocol for the concurrent detection of mRNA and protein in single cells using fluorescence in situ hybridization and flow cytometry. Nat Protoc 2017;12(6):1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kochan J, Wawro M, Kasza A. Simultaneous detection of mRNA and protein in single cells using immunofluorescence-combined single-molecule RNA fish. Biotechniques 2015;59(4):209–21. [DOI] [PubMed] [Google Scholar]
- 63. Ståhlberg A, Thomsen C, Ruff D, et al. Quantitative PCR analysis of DNA, RNAs, and proteins in the same single cell. Clin Chem 2012;58(12):1682–91. [DOI] [PubMed] [Google Scholar]
- 64. Lareau CA, Ludwig LS, Muus C, et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol 2020;1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Adey AC. Integration of single-cell genomics datasets. Cell 2019;177(7):1677–9. [DOI] [PubMed] [Google Scholar]
- 66. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell (Cambridge) 2019;177(7):1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lin Y, Wu T-Y, Wan S, et al. scjoint: transfer learning for data integration of single-cell RNA-seq and atac-seq. bioRxiv. 2020.12.31.424916 2021.
- 68. Campbell KR, Steif A, Laks E, et al. clonealign: statistical integration of independent single-cell RNA and DNA sequencing data from human cancers. Genome Biol 2019;20(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weinstein John N, Collisson Eric A, Mills Gordon B, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet 2013;45(10):1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yagüe E, Raguz S. Drug resistance in cancer. Br J Cancer 2005;93(9):973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lodato MA, Woodworth MB, Lee S, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science (New York, NY) 2015;350(6256):94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gómez-López G, Dopazo J, Cigudosa JC, et al. Precision medicine needs pioneering clinical bioinformaticians. Brief Bioinform 2017;20(3):752–66. [DOI] [PubMed] [Google Scholar]
- 73. Yan J, Risacher SL, Shen L, et al. Network approaches to systems biology analysis of complex disease: integrative methods for multi-omics data. Brief Bioinform 2017;19(6):1370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stephenson E, Reynolds G, Botting RA, et al. The cellular immune response to COVID-19 deciphered by single cell multi-omics across three UK centres. medRxiv. Cold Spring Harbor Laboratory Press, 2021.
- 75. Zhang J-Y, Wang X-M, Xing X, et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat Immunol 2020;21(9):1107–18. [DOI] [PubMed] [Google Scholar]
- 76. Yapeng S, Chen D, Yuan D, et al. Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell 2020;183(6):1479–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. du Y, Tu L, Zhu P, et al. Clinical features of 85 fatal cases of COVID-19 from Wuhan. a retrospective observational study. Am J Respir Crit Care Med 2020;201(11):1372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang A, Chiou J, Poirion OB, et al. Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of sars-cov2 host genes. Elife 2020;9:e62522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Unterman A, Sumida TS, Nouri N, et al. Single-cell omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. medRxiv. Cold Spring Harbor Laboratory Press, 2020.
- 80. Wilk AJ, Rustagi A, Zhao NQ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 2020;26(7):1070–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cheng J, Zhang J, Wu Z, et al. Inferring microenvironmental regulation of gene expression from single-cell RNA sequencing data using scmlnet with an application to COVID-19. Brief Bioinform 2021;22(2):988–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bernardes JP, Mishra N, Tran F, et al. Longitudinal multi-omics analysis identifies responses of megakaryocytes, erythroid cells and plasmablasts as hallmarks of severe COVID-19 trajectories. medRxiv. Cold Spring Harbor Laboratory Press, 2020.
- 83. Du M, Cai G, Chen F, et al. Multiomics evaluation of gastrointestinal and other clinical characteristics of COVID-19. Gastroenterology 2020;158(8):2298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Murat A, Mettetal Jerome T, Van OA. Stochastic switching as a survival strategy in fluctuating environments. Nat Genet 2008;40(4):471. [DOI] [PubMed] [Google Scholar]
- 85. Weibel KE, Mor J-R, Fiechter A. Rapid sampling of yeast cells and automated assays of adenylate, citrate, pyruvate and glucose-6-phosphate pools. Anal Biochem 1974;58(1):208–16. [DOI] [PubMed] [Google Scholar]
- 86. Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet 2015;16(3):133–45. [DOI] [PubMed] [Google Scholar]
- 87. Lan T, Hutvagner G, Lan Q, et al. Sequencing dropout-and-batch effect normalization for single-cell mRNA profiles: a survey and comparative analysis. Brief Bioinform 2020;bbaa248. [DOI] [PubMed] [Google Scholar]
- 88. Fiers MWEJ, Minnoye L, Aibar S, et al. Mapping gene regulatory networks from single-cell omics data. Brief Funct Genomics 2018;17(4):246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Argelaguet R, Velten B, Arnol D, et al. Multi-omics factor analysis–a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol Jun 2018;14(6):e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Svensson V, Teichmann SA, Stegle O. Spatialde: identification of spatially variable genes. Nat Methods 2018;15(5):343–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Gayoso PA, Lopez R, Steier Z, et al. Joint probabilistic modeling of single-cell multi-omic data with totalVI. Nat Methods. 2021;18:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. bioRxiv. 2020.10.12.3353312020. [DOI] [PMC free article] [PubMed]
- 93. Welch JD, Hartemink AJ, Prins JF. Matcher: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol 2017;18(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mirza B, Wang W, Wang J, et al. Machine learning and integrative analysis of biomedical big data. Genes (Basel) 2019;10(2):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Blei DM, Kucukelbir A, McAuliffe JD. Variational inference: a review for statisticians. J Am Stat Assoc 2017;112(518):859–77. [Google Scholar]
- 96. Butler A, Hoffman P, Smibert P, et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36(5):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang C, Sun D, Huang X, et al. Integrative analyses of single-cell transcriptome and regulome using maestro. Genome Biol 2020;21(1):1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Haghverdi L, Lun ATL, Morgan MD, et al. Batch effects in single-cell RNA sequencing data are corrected by matching mutual nearest neighbours. Nat Biotechnol 2018;36(5):421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat Methods Dec 2019;16(12):1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang B, Mezlini AM, Demir F, et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods 2014;11(3):333. [DOI] [PubMed] [Google Scholar]
- 101. Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimation methods for DNA microarrays. Bioinformatics 2001;17(6):520–5. [DOI] [PubMed] [Google Scholar]
- 102. Schafer JL, Graham JW. Missing data: Our view of the state of the art. Psychol Methods 2002;7(2):147–77. [PubMed] [Google Scholar]
- 103. Forsyth DA, Ponce J. Computer Vision: A Modern Approach. UK: Pearson, 2012. [Google Scholar]
- 104. Cambria E, White B. Jumping nlp curves: a review of natural language processing research. IEEE Comput Intell Mag 2014;9(2):48–57. [Google Scholar]
- 105. Ngiam J, Khosla A, Kim M, et al. Multimodal deep learning. In: ICML, 2011;689–696. https://icml.cc/2011/papers/399_icmlpaper.pdf.
- 106. Krizhevsky A, Sutskever I, Hinton GE. Imagenet classification with deep convolutional neural networks. Adv Neural Inf Process Syst 2012;25:1097–105. [Google Scholar]
- 107. Rumelhart DE, Hinton GE, Williams RJ. Learning representations by back-propagating errors. Nature 1986;323(6088):533–6. [Google Scholar]
- 108. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. arXiv preprint arXiv:1706.03762. 2017.
- 109. Huang G, Liu Z, Van Der Maaten L, et al. Densely connected convolutional networks. Proc IEEE Conf Comput Vis Pattern Recognit 2017;4700–4708. [Google Scholar]
- 110. He K, Zhang X, Ren S, et al. Deep residual learning for image recognition. Proc IEEE Conf Comput Vis Pattern Recognit 2016;770–778. [Google Scholar]
- 111. Pan SJ, Yang Q. A survey on transfer learning. IEEE Trans Knowl Data Eng 2009;22(10):1345–59. [Google Scholar]