


Abstract
The transcriptomic diversity of cell types in the human body can be analysed in unprecedented detail using single cell (SC) technologies. Unsupervised clustering of SC transcriptomes, which is the default technique for defining cell types, is prone to group cells by technical, rather than biological, variation. Compared to de-novo (unsupervised) clustering, we demonstrate using multiple benchmarks that supervised clustering, which uses reference transcriptomes as a guide, is robust to batch effects and data quality artifacts. Here, we present RCA2, the first algorithm to combine reference projection (batch effect robustness) with graph-based clustering (scalability). In addition, RCA2 provides a user-friendly framework incorporating multiple commonly used downstream analysis modules. RCA2 also provides new reference panels for human and mouse and supports generation of custom panels. Furthermore, RCA2 facilitates cell type-specific QC, which is essential for accurate clustering of data from heterogeneous tissues. We demonstrate the advantages of RCA2 on SC data from human bone marrow, healthy PBMCs and PBMCs from COVID-19 patients. Scalable supervised clustering methods such as RCA2 will facilitate unified analysis of cohort-scale SC datasets.
INTRODUCTION
Since its first usage in 2009 (1), single cell (SC) RNA sequencing (scRNA-seq) has quickly become the method of choice for profiling gene expression in complex samples (2). Due to the unprecedented resolution of scRNA-seq data, cell type-specific analysis of gene expression can now be performed easily and at low cost. SC transcriptomes are well-suited to characterizing heterogeneous biological specimens, e.g. tumors (3).
Clustering is an essential step in SC data analysis, since each cell cluster in transcriptome space represents a distinct cell type or state. There are two established paradigms to address the SC clustering problem: (i) unsupervised (de-novo) clustering (4), which is the most prevalent, and (ii) supervised clustering, which exploits a panel of reference transcriptomes (5). In addition, reference transcriptomes are used to classify single cells, which is by definition a supervised approach (6–10). Among the unsupervised methods, the Louvain graph-based clustering algorithm is the most prevalent, since it offers better scalability than hierarchical clustering (11–13).
Despite the existence of multiple algorithms, SC clustering is still challenging: (i) cells may cluster by technical variation and batch effects rather than biological properties (4), (ii) scRNA-seq data tend to be noisy, primarily due to sampling noise and (iii) the gene expression matrix can be very large, since modern datasets commonly include > 100,000 cells. Consequently, different algorithms can return highly divergent clusterings, i.e. partitions of cells into clusters, of the same input dataset (14). Moreover, de-novo clustering requires an error-prone, time-consuming manual step of assigning cell clusters to cell types (annotation) based on subjective evaluation of the expression of marker genes. Supervised clustering and supervised cell type annotation algorithms have been developed to address these limitations.
Previously, we proposed Reference Component Analysis (RCA) for supervised clustering of scRNA-seq data guided by a panel of reference transcriptomes (5).
Unlike the above-mentioned methods, RCA was not primarily designed for cell type annotation. Rather, the objective of RCA is to cluster single cells in the space of reference transcriptome projections. This is fundamentally different from unsupervised clustering approaches, which cluster cells in the space defined by over a thousand feature genes (15,16).
To the best of our knowledge, RCA is the only supervised clustering algorithm for scRNA-seq data. However, the original version of RCA could not scale to datasets larger than 20,000 cells on a high-end laptop, used only a single reference panel, did not implement methods to identify differential gene expression, did not offer KEGG and Gene Ontology (GO) enrichment analysis, was benchmarked on only a single Smart-seq dataset and could not be easily integrated into existing data analysis workflows.
To fully leverage the merits of supervised clustering, we present RCA2, the first algorithm that combines reference projection with graph-based clustering. The former provides accuracy and robustness, whereas the latter provides scalability and speed. We show the unique advantages of RCA2 by analyzing diverse scRNA-seq data sets: a publicly available 10× PBMC data set, a novel human bone marrow data set and a recently published data set of PBMCs from COVID-19 positive patients (17). In addition, by providing multiple new human and mouse reference panels, as well as the option to generate custom panels from user-supplied bulk transcriptomes, we significantly broadened the applicability of RCA2 compared to the previous version.
Furthermore, we demonstrate using diverse sample types that supervised clustering can be accurate even when the dataset contains cell types not contained in the reference panel. In summary, RCA2 is well suited to the task of robustly clustering large scRNA-seq datasets, which are typically generated in multiple batches or across multiple sites.
MATERIALS AND METHODS
Projection to a reference
Given a reference data set 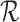 containing n cell types and
containing n cell types and 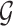 marker genes as well as a query data set
marker genes as well as a query data set 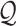 containing k single cells and
containing k single cells and 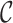 genes, we determine a marker gene set
genes, we determine a marker gene set 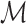 by intersecting
by intersecting 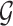 and
and 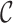 :
:
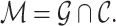 |
(1) |
The reference matrix 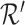 and set
and set 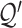 are generated by extracting the gene set
are generated by extracting the gene set 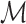 from
from 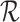 and
and 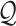 , respectively. Next, RCA2 computes the reference projection matrix
, respectively. Next, RCA2 computes the reference projection matrix 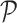 with an entry
with an entry 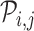 denoting the correlation (default: Pearson (r)) between
denoting the correlation (default: Pearson (r)) between 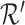 and
and 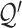 for a single cell i (column
for a single cell i (column 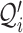 ) and cell type j (column
) and cell type j (column 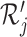 ):
):
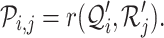 |
(2) |
The projection matrix 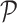 is modified according to
is modified according to
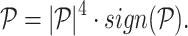 |
(3) |
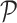 is scaled to zero mean and unit variance. All matrices are represented as sparse matrix R objects. The projection is computed using the fastcor package (18).
is scaled to zero mean and unit variance. All matrices are represented as sparse matrix R objects. The projection is computed using the fastcor package (18). 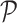 can be visualized as a 2D and 3D UMAP.
can be visualized as a 2D and 3D UMAP.
Clustering and interpreting the projection
RCA2 offers three clustering algorithms: (i) hierarchical clustering using the memory efficient fastcluster (19) package, (ii) shared-nearest neighbour (SNN) clustering using dbscan (20) and (iii) graph-based clustering using the Louvain algorithm (11). The depth to cut the dendrogram in hierarchical clustering is a parameter (default 1). The SNN algorithm used in dbscan has three parameters: k (neighborhood size of the SNN graph), eps (two cells are only reachable from each other if they share at least eps nearest cells) and min − pts (minimum number of nearest neighbours for a cell to be considered a core cell). To guide the users choice on parameters for graph-based clustering, a 3D figure illustrating how the final number of clusters depends to the used parameters can be generated. The Louvain algorithm requires only the resolution parameter. A line-plot illustrating how the resolution influences the number of identified clusters can be generated. As input, all clustering methods use either a distance matrix 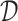 computed from
computed from 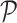 according to
according to
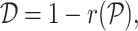 |
(4) |
where 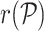 is the cell-to-cell similarity using correlation (Pearson (default in this manuscript), Spearman or Kendal) as a metric in the cell type space or an embedding of cells in PC space computed on the reference projection (not available with hierarchical clustering). The clustering result is visualized in a heatmap, including quality control (QC) metrics: number of detected genes (NODG), the percentage of mitochondrial genes (pMito) and the number of unique molecular identifiers (NUMI). Reference cell types with a low variance across all query cells are not shown. Figures are returned as ggplot2 (21) objects, allowing further modifications by the user.
is the cell-to-cell similarity using correlation (Pearson (default in this manuscript), Spearman or Kendal) as a metric in the cell type space or an embedding of cells in PC space computed on the reference projection (not available with hierarchical clustering). The clustering result is visualized in a heatmap, including quality control (QC) metrics: number of detected genes (NODG), the percentage of mitochondrial genes (pMito) and the number of unique molecular identifiers (NUMI). Reference cell types with a low variance across all query cells are not shown. Figures are returned as ggplot2 (21) objects, allowing further modifications by the user.
Reference panels
RCA2 includes ten human reference panels as well as two mouse reference panels (Supplementary Section 1). Multiple panels can be used for reference projections simultaneously. Furthermore, RCA2 provides users with the option to generate their own reference panel: the buildReferencePanel function considers a bulk gene expression matrix (genes as rows and replicates as columns) of raw counts and returns a reference panel that can be used with RCA2. Details are provided in Supplementary Section 2.1.
Annotation of cell types
RCA2 implements cell type assignment at the SC level following a strategy inspired by SingleR (6). From the projection matrix 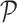 , we identify, for each cell i, the cell type t associated to the highest score Pi according to
, we identify, for each cell i, the cell type t associated to the highest score Pi according to
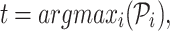 |
(5) |
where argmaxi returns the column index corresponding to the cell type of the projection matrix 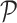 holding the highest correlation for cell i. Cluster composition plots elucidating the cell identity per cluster both in terms of absolute numbers and relative proportions can be generated as well.
holding the highest correlation for cell i. Cluster composition plots elucidating the cell identity per cluster both in terms of absolute numbers and relative proportions can be generated as well.
To annotate cell types on the cluster level, we consider the cell type composition for each cluster based on the SC cell type assignment described above. If the cell type distribution within a cluster is heterogeneous and the proportion of the major cell type is below a user defined threshold (default 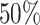 ), the cluster is labelled as Unknown. Further details are provided in Supplementary Section 2.2.
), the cluster is labelled as Unknown. Further details are provided in Supplementary Section 2.2.
Cluster specific quality control
Quality of scRNA-seq data is usually assessed using NODG, nUMI and pMito metrics. In applying uniform QC cutoffs across all cells, scRNA-seq datasets can suffer from cell type depletion. To alleviate this issue, RCA2 provides cluster-specific QC, allowing to impose upper/lower bounds on QC metrics for each cluster independently.
Differentially expressed gene computation and enrichment analysis
Differentially expressed genes (DEGs) are calculated between clusters, either in a 1 versus all (default) or a pairwise fashion using a modified version of Seurat’s DEG calling module. We incorporated a mean expression threshold, which is either a user defined value or automatically determined as a trimmed mean excluding the top n (default: 5) genes with the highest expression. Gene’s with a cluster specific expression below the threshold are not considered for the DE test. In RCA2 the user can use one of the following tests for DEG calling: Wilcoxon rank sum test (default), likelihood-ratio test, ROC analysis and t-test. DEGs are used to perform enrichment analysis of GeneOntology (GO) terms or KEGG Pathways, for which RCA2 incorporates the ClusterProfiler R-package (22). We retrieve the version number of the latest GO annotation from the org.Hs.eg.db R package. For KEGG, we send a GET request to the KEGG API at the time of analysis to retrieve the version number of the KEGG database used if the user decides to not use the version available in ClusterProfiler. By changing the version of org.Hs.eg.db or ClusterProfiler, the user can change the GO and KEGG database versions, respectively. By default, we consider cluster-specific background sets composed of genes expressed within each cluster. Alternatively, all genes expressed across all clusters or simply all genes available in the used annotation can be considered. Several options are available to distinguish expressed from not expressed genes: a numeric threshold, the 1st quartile, the mean, the median or the 3rd quartile of the distribution of mean gene-expression values within one or across all clusters. For each tested cluster, RCA2 generates barplots, dotplots and GoPlots (if applicable). Further details are provided in Supplementary Section 2.3.
Considered scRNA-seq data sets and data processing
10X PBMC data sets
We downloaded scRNA-seq data of 5025 PBMCs generated using Chromium SC 3′ Reagent Kits v3 from 10× (single-cell-gene-expression/datasets/ 3.0.2/5k_pbmc_protein_v3). We applied the following QC thresholds (min, max): NODG (300, 4500), nUMI (100, ∞), pMito (0.025, 0.1). In total, 4,249 cells passed QC (Supplementary Table S1). The data set was projected against the Novershtern reference panel comprised of 15 hematopoietic cell types (23) (Supplementary Section 1).
The resolution parameter used for Louvain clustering was determined using a grid-search with a step-size of 0.05. DEGs between clusters are computed in a pairwise-manner using the parameters:min.pct = 0.5, logfc.threshold = 0.5 and p_val_adj ≤ 0.05. GO terms were computed using ClusterProfiler utilising the org.Hs.eg.db database, and a q-value threshold of 0.05.
CITE-seq PBMC data sets
A Drop-Seq data set with 29,929 genes profiled in 7,985 cells and 10 antibodies was obtained from Stoeckius et al. (25). A CITE-seq data set with 7,865 cells profiled on 33,538 genes and 17 antibodies was obtained from 10× (single-cell-gene-expression/datasets/3.0.0/pbmc_10k_protein_v3). Both data sets were processed using Seurat. To obtain a ground truth, we clustered cells in antibody-derived tags (ADT) space. ADT data was normalized using the centered log ratio transformation (satijalab.org/seurat/v3.2/multimodal_vignette.html). All PCs were selected for clustering using Louvain clustering. Since the Drop-Seq data did not include a control for antibody detection, clusters exhibiting noisy antibody detection or those clusters not representing known immune cell type signature (23,24) were removed. In the 10x data, clusters showing IgG1, IgG2a or IgG2b and clusters showing promiscuous antibody expression were discarded. After QC (Supplementary Table S1), the Drop-Seq and 10× datasets contained 5,925 and 6,744 cells, respectively. The Drop-Seq and 10× data sets were next merged with respect to their common genes (13,267). The merged data set was provided as input to all clustering methods for benchmarking. Using Seurat’s FindMarker function, we computed batch specific marker genes for each ADT cluster in the merged data set using the parameters: min.pct = 0.5, logfc.threshold = 1.5 and p_val_adj ≤ 0.05. GO terms for batch specific clusters are computed using ClusterProfiler utilising the org.Hs.eg.db database, and a q-value threshold of 0.05.
Rheumatoid arthritis scRNA-seq data set
The scRNA-seq data set of 10,001 cells from Rheumatoid Arthritis (RA) samples, obtained from Zhang et al. (26), was processed using Seurat. Cells were filtered based on the QC criteria provided in Supplementary Table S1. This data was generated using CEL-Seq2 (27) after sorting for B cells (CD45+CD3-CD19+), T cells (CD45+CD3+), monocytes (CD45+CD14+), and stromal fibroblasts (CD45–CD31–PDPN+) from synovial tissues of ultrasound-guided biopsies or joint replacements of RA patients. Cell type annotation based on the authors cell sorting strategy is used as a ground truth. The resulting data set was used as input to all clustering methods for benchmarking.
Bone-marrow scRNA-seq data set
We obtained eight human bone marrow (BM) specimens from STEMCELL technologies and generated ten scRNA-seq data sets using the 10× 5’scRNA-seq protocol (see Supplementary Section 2.4) by separating the cells into CD34+ and CD34– cell fractions followed by sequencing on a HiSeq4000. Preprocessing was done using the 10× CellRanger pipeline (3.0.1) using the hg38 reference genome resulting in a data set comprised of 45,363 cells, capturing 24,206 genes. Considering only an initial requirement of at least 1000 nUMI, and a pMito rate between 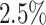 and
and 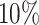 , we projected the data against RCA’s global panel obtaining a classification into major groups (resolution 0.1) to perform cluster specific QC (Supplementary Table S2). Cluster specific QC values are chosen based on the outer most layer of the computed densities in the scatter plots. Final cell types were identified upon QC using a resolution of 0.5.
, we projected the data against RCA’s global panel obtaining a classification into major groups (resolution 0.1) to perform cluster specific QC (Supplementary Table S2). Cluster specific QC values are chosen based on the outer most layer of the computed densities in the scatter plots. Final cell types were identified upon QC using a resolution of 0.5.
Doublets were removed using DoubletFinder (28). DoubletFinder was run separately on the CD34+ and CD34- populations using 20 PCs and a pNN value of 0.25 as well as pk values of 0.005 and 0.01, respectively. The obtained pANN values were merged to rank cells based on their doublet neighbourhood. A pANN threshold was derived considering both the expected number of doublets (∼1,560) and by examining the proportion of possible doublets in each cluster.
COVID-19 PBMCs
A Seurat object with scRNA-seq data studied by Wilk et al. (17) was obtained from the COVID-19 Cell Atlas (covid19cellatlas.org). All 44,721 cells were projected against the global, Monaco, Novershtern and CITE-seq panel and clustered using the Louvain algorithm (resolution 1.3). Clusters were annotated according to reference projection profiles and marker gene expression. Marker genes for each subset of developing neutrophils are computed using the following DEG parameters: min.pct = 0.25, logfc.threshold = 1, and p_val_adj ≤ 0.05. Sub-clustering and cell embedding of CD14 monocytes, intermediate monocytes, CD16 monocytes, myeloid dendritic cells (mDC), myelocytes, neutrophils and plasmablasts was conducted using Seurat. The union set of pair-wise DEGs between these cell types was used as feature genes for PCA. DEGs were determined using the parameters: min.pct = 0.25, logfc.threshold = 1.5, and p_val_adj ≤ 0.05. For downstream clustering we used 18 PCs and annotated clusters using marker genes.
AML dataset
The AML data scRNA-seq data set 809653 was obtained from the zenodo archive of Petti et al. (29) at 10.5281/zenodo.3345981. No additional QC was performed. The data was projected using RCA2s Multi Panel Projection function with default parameters. The data was clustered using hierarchical clustering at deep split 1.
Methods used for batch effect benchmarking
To benchmark the batch effect robustness of RCA2, we considered Seurat, Seurat Integration, SCTransform, SCTransform Integration, Scran (30), Scanpy, MNNCorrect (31) and Scanorama (32). Below, we briefly describe how we utilized each method in the benchmarking experiments. Code is available in our Zenodo archive (10.5281/zenodo.4686335). We refer the reader to the respective publications for further method details.
Seurat
Seurat (version 3.2) was used as recommended in its documentation (satijalab.org/seurat/vignettes.html). We used the CreateSeuratObject function to create the Seurat object at default parameter settings. Then, we log-normalized the raw counts using NormalizeData, identified highly variable genes using FindVariableFeatures, scaled all genes using ScaleData, and ran principal component analysis using RunPCA, all at default settings. We then plotted an ElbowPlot of the variance explained versus number of principal components (PCs) to select the number of PCs to cluster cells and to be used in UMAP reductions.
Seurat Integrated
We split the Seurat object created above into its various batches using the SplitObject at default parameter settings. Then, we log-normalized the raw counts using NormalizeData and identified highly variable genes using FindVariableFeatures for each batch at default settings. We then ran FindIntegrationAnchors using dims = 1:30. The resulting anchors were used for IntegrateData with the aforementioned 30 dimensions. Then, we scaled all genes using ScaleData, and ran principal component analysis using RunPCA, all at default settings.
SCTransform
We used SCTransform (33) as part of our benchmarking approach. After creating the Seurat object, we used the SCTransform function at default settings, and ran PCA on the result. We used the ElbowPlot function to determine the optimal number of PCs, which we deemed to be 20.
SCTransform Integrated
We followed a similar approach as mentioned in the ‘Seurat Integrated’ subsection to split the object, and ran the SCTransform function on each batch separately. We then selected the top 3000 features for integration using the SelectIntegrationFeatures function, and ran the PrepSCTIntegration function to ensure that all necessary Pearson residuals have been calculated. We then ran FindIntegrationAnchors, specifying the normalization method used as ‘SCT’. The resulting anchors were used for IntegrateData.Then, we scaled all genes using ScaleData, and ran principal component analysis using RunPCA, all at default settings. We selected the top 30 PCs, as recommended in SCT Integration Workflow Vignette (https://satijalab.org/seurat/archive/v3.0/integration.html).
Scran
For Scran (version 1.18.1), we followed the tutorial from bioconductor (bioconductor.org/packages/ release/bioc/html/scran.html). We used the SingleCellExperiment function to create a SingleCellExperiment object. We computed sum factors using computeSumFactors, log-normalized the counts using logNormCounts and modeled the variance of each gene using modelGeneVar. We then identified highly variable genes using getTopHVGs (FDR ≤ 0.05) and ran PCA using these features. PCs corresponding to technical noise were removed using the denoisePCA function at default settings.
MNNCorrect
We used MNNCorrect (batchelor R package (version 1.6)) according to bioconductor.org/packages/release/bioc/html/batchelor.html. We create a separate SingleCellExperiment object for each batch, and for each object, computed sum factors, normalized, modeled gene variance using trendVar and decomposeVar - all at default settings. We then used combineVar to merge the decomposed variance objects, and identified chosen HVGs as those with a biological component >0. Using these chosen HVGs, we ran fastMNN to obtain a reduced dimensional representation of the integrated dataset.
Scanpy
Furthermore, we used Scanpy (version 1.5.1) (scanpy.readthedocs.io/en/stable/). To transfer the raw counts from R to Python, we saved them into a CSV file, and loaded the CSV file into an AnnData object using the read_csv() function. We normalized the counts using normalize_total, with 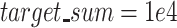 , and log-transformed these normalized counts using the log1p function. We then identified HVGs using the highly_variable_genes function, and scaled these genes with
, and log-transformed these normalized counts using the log1p function. We then identified HVGs using the highly_variable_genes function, and scaled these genes with 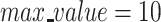 . Then, PCA was performed using Scanpy’s pca function at default settings, and used the pca_variance_ratio function to determine the elbow point of 20 PCs for both the CITE-Seq and RA datasets.
. Then, PCA was performed using Scanpy’s pca function at default settings, and used the pca_variance_ratio function to determine the elbow point of 20 PCs for both the CITE-Seq and RA datasets.
Scanorama
Scanorama (version 1.6) was used for this analysis, using the tutorial provided at nbisweden.github.io/workshop-scRNAseq. We split the AnnData object generated above into batches, and used the list of batches as input to the integrate_scanpy function. We used the resulting integrated dataset as the reduced dimensional representation for both the CITE-Seq and RA datasets.
Silhouette Index for quantifying batch effect
The Silhouette Index (SI) of a cell measures how similar a cell is to other cells within its own cluster, relative to cells in other clusters (34). We compute SI S(x) for each cell x in the DE gene-space defined by CITE-Seq antibody tags:
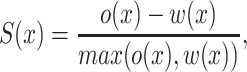 |
(6) |
where o(x) is the smallest mean between-cluster distance and 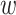 (x) is the mean within-cluster distance for cell x defined as
(x) is the mean within-cluster distance for cell x defined as
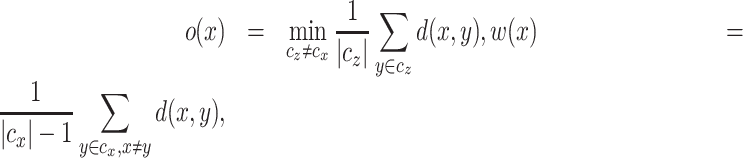 |
(7) |
where we use Euclidean distance to compute the distance d(x, y) between cell x and cell y, 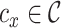 is the cluster assigned to cell x and |cx| is the size of that cluster. We obtain the average SI for each cluster by averaging the SI values over all cells in that cluster. Thereby, each cell type is given equal weight in the final SI score.
is the cluster assigned to cell x and |cx| is the size of that cluster. We obtain the average SI for each cluster by averaging the SI values over all cells in that cluster. Thereby, each cell type is given equal weight in the final SI score.
For Seurat, Seurat Integration, scran, MNNCorrect and Scanpy, cell-cell distances are calculated in principal component (PC) space considering the top 20 PCs. For Scanorama, the dimensionality was fixed to 100, as recommended by the authors. For Rca, cell-cell distances were calculated in the reference projection space.
Implementation
RCA2 is freely available at www.github.com/prabhakarlab/RCAv2. The github contains detailed tutorials in the README as well as in the vignettes. RCA2 is extensively tested with R versions ≥3.6 on Windows, Linux and Mac devices. To ensure robustness and correctness, we have incorporated unit tests and have also ensured that all CRAN and devtools checks are passed.
RESULTS
Novel and improved features of RCA2
The RCA2 workflow is shown in Figure 1. As input, RCA2 takes either raw or pre-processed scRNA-seq data and facilitates QC either as a single operation on all cells or in a cluster-specific manner (Supplementary Figures S1 and S2). Unlike the original Rca, RCA2 provides a function to directly load the raw output of the 10× CellRanger software, which is a commonly used preprocessing pipeline for prevalent single cell 10× data.
Figure 1.

RCA2 takes two types of scRNA-seq data as input: (i) CellRanger output files and (ii) data preprocessed elsewhere, which can be loaded as a gene × cell count matrix. Reference datasets in RCA2 for human and mouse are based on bulk RNA-seq, microarray and scRNA-seq assays. RCA2 can also generate custom reference panels from user-supplied raw count matrices. RCA2 computes a correlation matrix representing the similarity of each SC transcriptome to each reference transcriptome. Correlations are calculated using marker (DE) genes from the reference panel. Cells are clustered and visualized in the space of reference projections. After DE gene analysis, enriched GO terms and KEGG pathways can be identified.
While the first release of Rca provided one human reference panel only, RCA2 includes eleven panels, for instance a microarray-based human cord blood cell panel with 15 immune cell types (23), one related RNA-seq panel with 28 human immune cell types (24), a panel based on CITE-seq data containing 34 primary human cell types (35), one human primary cell type panel based on ENCODE (36) RNA-seq data containing 97 cell types and a mouse ENCODE panel with 15 cell types. A list of all panels as well as guidelines on how to choose panels are provided in Supplementary Section 1. In contrast to the previous version, RCA2 offers means for de-novo panel generation from user-provided transcriptomes (Supplementary Section 2.4). Another novel feature of RCA2 is that it allows SC data to be projected against several reference panels at the same time. Furthermore, RCA2 offers a significant speed up of several folds in computing the reference projection compared to the previous release. (Figure 2A, Supplementary Section 2.5).
Figure 2.

(A) Speedup of the reference projection step. (B) Memory consumption of graph-based clustering compared to hierarchical clustering. Benchmarking was performed with a notebook using an Intel i9-9980 CPU(2.40 GHz) and 64GB RAM. Projecting using RCAv1 and hierarchical clustering ran out of memory using 100k cells and 50k cells, respectively.
RCA is based on hierarchical clustering, which is challenging on large datasets since the memory complexity scales as the square of the number of cells. One fundamental change is that RCA2 uses Louvain graph-based clustering as the default, instead of hierarchical clustering. The graph-based clustering approach requires orders of magnitude less memory (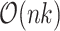 versus
versus 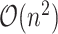 , where n is the number of cells and k is the number of nearest neighbours (Figure 2 B; Supplementary Section 2.5)). As an additional option, RCA2 provides a memory-efficient implementation of hierarchical clustering (37). To aid in parameter selection for graph-based clustering RCA2 provides visualizations on how parameter settings influence the number of clusters (Supplementary Figures S3 and S4). Further improvements for scalability include parallelization and use of sparse data structures. Together, these modifications allow RCA2 to scale in principle to datasets comprising millions of cells.
, where n is the number of cells and k is the number of nearest neighbours (Figure 2 B; Supplementary Section 2.5)). As an additional option, RCA2 provides a memory-efficient implementation of hierarchical clustering (37). To aid in parameter selection for graph-based clustering RCA2 provides visualizations on how parameter settings influence the number of clusters (Supplementary Figures S3 and S4). Further improvements for scalability include parallelization and use of sparse data structures. Together, these modifications allow RCA2 to scale in principle to datasets comprising millions of cells.
Note that, unlike other SC clustering frameworks, Rca utilizes the reference projection to cluster cells in a cell type space instead of a high-dimensional feature gene-space. Clustering results can be visualised using newly implemented 2D and 3D UMAP (Supplementary Figures S5 and S6) representations of the reference projection (cell type) space. Compared to the previous heatmap visualization of the reference projection matrix, RCA2 shows additional QC information (NODG, nUMI, pMito) and removes cell types not showing significant variation in the correlation score to facilitate data interpretation and outlier detection.
To further enrich the functionality of RCA2 compared to its predecessor, we incorporated singleR/scMatch-like assignment of cell types to individual cells (6,7). Exploiting RCA2’s clustering algorithms, we also allow cell type annotation on the cluster level (see Materials and Methods).
For further biological interpretation, which was not in the scope of the previous version of RCA, RCA2 offers various statistical tests to identify DEGs in either a 1 versus all or a pairwise scheme. DEGs are visualized in heatmaps following the established Seurat color scheme (Supplementary Figure S7) and can be used as input for a GO-term (38) enrichment and KEGG pathway (39) analysis, providing biological insights on clusters beyond lists of marker genes (Supplementary Figure S8).
A summary of new features and methodological advances in RCA2 compared to the initial release is provided in Table 1.
Table 1.
Methodological improvements and new features in RCA2 compared to the prototype release
| Rca | RCA2 | |
|---|---|---|
| Reference panels | 1 | 11 |
| Species supported | Human | Human, mouse |
| Data import | Count matrix | 10x file import, Count matrix |
| Custom panel usage | Yes | Yes |
| Custom panel generation | Not supported | Automated |
| Clustering | Hierarchical (basic) | Hierarchical (divide-and-conquer), graph-based |
| Data structures | Standard | Sparse data structures |
| Clustering in | full projection space | full or PCA reduced projection space |
| Cell type/cluster specific QC | No | Yes |
| Parallelized correlation computation | No | Yes, if applicable |
| UMAP visualizations | 2D | 2D,3D |
| Cell type annotation | Not supported | Yes |
| Easy integration in existing workflows | No | Yes |
| DEG calling | No | 1 vs all and pairwise |
| Enrichment analysis | None | KEGG and GO |
| Number of UNIT tests | 0 unit tests | 90 unit tests |
| Tutorials and documentation | Limited documentation | Extensive manual and R-vignette |
| Applicable to datasets >20 000 cells | No | Yes |
Supervised clustering is robust to batch effects
One of the major advantages of supervised clustering is its ability to reduce the contribution of unwanted variation, which manifests in the form of noise or technical variation. This is crucial in the prevention of batch effects. By projecting SC data onto a reference panel of purified transcriptomes, supervised clustering is able to preserve the cell type-specific variation and ignore variation from other sources. This is based on the concept shown in Supplementary Figure S9, i.e. the batch effect expression signature is likely to be orthogonal to the signature of cell type marker genes, because two randomly selected vectors are likely to be orthogonal in a high dimensional space.
We benchmarked RCA2’s robustness to batch effects by comparing its performance against several other commonly used scRNA-seq clustering algorithms (see Methods): Seurat, Seurat with SCTransform normalization, Seurat Integration, SCTransform Integration, Scran, Scanpy, MNN Correct and Scanorama, significantly exceeding the benchmarking of the initial RCA version.
We benchmarked RCA2 on two data sets that provide a ground truth for cell type identity that is independent from scRNA-seq data: (i) a dataset of joint synovial tissues from Rheumatoid Arthritis (RA) patients with plate- and donor-specific batch effects (26) and a (ii) PBMC CITE-seq data sequenced using (a) Drop-Seq and (b) 10× Chromium platforms (25). Further details are provided below. We used the Silhouette Index (SI) to quantify grouping of cells by batch or by cell type (34). Note that a robust method should have low batch SI and high cell type SI, indicating that cells are separated by cell type rather than by batch.
Rheumatoid arthritis (RA) data set benchmarking
We use a data set by Zhang et al. (26) comprised of 5,829 cells from 51 RA samples sequenced on 24 384-well plates. Cells have been FACS sorted into T cells, B cells, monocytes, and fibroblasts, which we use as ground-truth cell type labels. Zhang et al. detected plate-specific batch effects while clustering cells using the Seurat package. These plate-specific batch effects were more pronounced in some plates as compared to others. Here, RCA2 considerably outperforms the other tested algorithms in terms of cell type separation (Figure 3A) but also in terms of batch robustness. While data from different plates is readily merged together (Figure 3B) cells cluster well according to their FACS determined cell type (Figure 3C). We observed that using SCTransform in the Seurat Integration workflow worsened both separation by batch and separation by cell type.
Figure 3.

(A) Silhouette Index (SI) measuring separation of cells in RA data by plate and cell type. (B, C) UMAP visualization of RCA2 clustering of RA data colored by (B) plate and (C) cell type. (D) SI measuring separation of cells in CITE-Seq data by protocol and cell type. (E, F) UMAP visualization of RCA2 clustering of CITE-Seq data colored by (E) protocol and (F) cell type.
Cite-seq data set benchmarking
Next, we considered PBMC CITE-seq data sequenced using (a) Drop-Seq and (b) 10× Chromium that were analysed together as a single dataset. CITE-Seq data contains both the protein abundance of cells and their transcriptomic profile. The protein quantification can be used to define ground-truth cell type labels independently of the transcriptome data. Specifically, we used Seurat to cluster SCs in protein abundance space and defined each resulting cluster as a ground-truth cell type (Supplementary Figure S10) (Materials and Methods).
Benchmarking on the CITE-Seq data showed that Seurat Integrated, MNN Correct and Scanorama successfully reduced batch effects, similar to RCA2. However, that was achieved at the cost of worsening cell type separation (Figure 3D). Seurat (Supplementary Figure S11), Scran and Scanpy produced clustering results significantly affected by protocol, with Scanpy performing as the best among all unsupervised approaches without explicit batch correction in terms of cell type separation. We note that the usage of SCTransform worsened the batch susceptibility in this use-case if the data integration feature was not used. RCA2 was among the top methods in terms of batch robustness (Figure 3E) and provided the best separation of cell types (Figure 3F).
Using ADT-tags of the CITE-seq data, we are able to characterize the batch not only from a technical perspective using the SI, but also from a biological point of view: we computed the set of DEGs (see Methods) that characterizes the SC capture protocol batch within each ADT cluster (Figure 4A, Supplementary Table S3). While the majority of DEGs are ribosomal genes, we also find several genes that are both cell type and batch specific markers, such as H3F3A, IGKC, IFI30 or IGLC2. The latter three are related to immune reaction and gamma-interferon signaling. Another interesting gene that is associated to the batch is FOS, which has been associated to several molecular processes and has been linked to cancer progression (40). As it is known that the expression of FOS can be easily changed by external stimuli (41), it might be more likely that, in our data set, the observed differences in FOS expression are of technical instead of biological nature. Indeed, the RCA2 projection, shown in Figure 4 B, is not affected by any of the DEGs linked to the batch effects and is supporting the antibody based clustering well. The latter is also backed up by the expression of the genes targeted by the antibodies (Supplementary Figure S12).
Figure 4.

(A) Expression of DEG computed for sequencing protocol batch within ADT clusters. (B) Reference projection of the CITE-seq data against RCA2’s global panel.
To characterize the genes defining the observed batch further, we investigated their GO term enrichment separately for genes expressed in the 10X batch (Supplementary Figure S13) and the Drop-seq batch (Supplementary Figure S14). All of the observed GO terms can be exclusively explained by the difference in sequencing protocol and therefore potentially misguide down-stream analysis, if less robust clustering methods are used.
We investigated this hypothesis by computing cluster specific marker genes for clusters identified with various scRNA-seq pipelines, using the same parameters and settings for the DEG tests in all methods (Wilcox test, 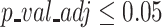 ). We compared those method specific DEGs to the set of batch specific DEGs, shown in Figure 4A, using an Upset plot (Supplementary Figure S15). Indeed, RCA2 shows the lowest overlap between cluster specific marker genes and the set of batch DEGs compared to the other methods, underlining the robustness of supervised clustering as implemented in RCA2 towards batch effects even further.
). We compared those method specific DEGs to the set of batch specific DEGs, shown in Figure 4A, using an Upset plot (Supplementary Figure S15). Indeed, RCA2 shows the lowest overlap between cluster specific marker genes and the set of batch DEGs compared to the other methods, underlining the robustness of supervised clustering as implemented in RCA2 towards batch effects even further.
Use-case on 10X PBMC data set comprising 5,000 cells
We obtained a 10X Genomics dataset containing 5,025 peripheral blood mono nuclear cells (PBMCs) from a healthy donor. We imported the CellRanger output directly into RCA, considering cells with a UMI count ≥ 100.
Upon QC using RCA2’s QC functionality (Supplementary Figure S15, Table S1), the scRNA-seq data is projected against a new, manually curated reference panel of immune cell types, based on purified populations of human hematopoietic cells (23). We utilized the novel integration of the Louvain graph-based clustering algorithm into RCA2 to cluster the data using a resolution of 0.1, which leads to a sensible separation of cells in terms of the projection heatmap (Supplementary Figure S16) as well as in the resulting number of clusters (Supplementary Figure S4). We obtained nine clusters forming four disconnected islands in a UMAP based on the cell to cell type correlation space obtained by the reference projection (Supplementary Figure S17a).
RCA2’s new, SingleR-inspired automated cluster annotation function determines cell types, shown in Supplementary Figure S17b. While the B-cell cluster (red) is very distinct from all remaining clusters, T and natural killer cells form a continuum (blue, brown, yellow, green). Monocytes (turquiose) and non-classical monocytes (black) appear to be well separated within a major myeloid cluster. In close proximity in UMAP space, small populations of myeloid (pink) and plasmacytoid (magenta) dendritic cells were identified. While the automatically determined labels agree with the projection heatmap shown in Supplementary Figure S17a, this can be expected by the design of RCA2 and the projection step. Therefore, we use canonical marker genes, which are considered for instance also in the Seurat tutorial, to verify cell types. As shown in Supplementary Figure S17c–j, the abundance of the various marker genes corresponds well to the identified clusters. Note that for some cell types more than one marker gene should be considered, e.g. CD14 and CD16 for non-classical monocytes or CD8A and NKG7 for natural killer cells, respectively.
To further characterize the clusters, we compute DEGs using RCA2’s default settings in a pair-wise manner (Supplementary Figure S7b). Supplementary Table S4 lists all identified DEGs. As shown in Supplementary Figure S18, we obtain several significant terms in GO term analysis using these DE genes for the natural killer cell cluster including cytolysis, cell killing and cellular defense response. These are well matching to the expected biological function of natural killer cells. Also, for the naive CD4+ T-cell cluster we obtain sensible terms such as adaptive immune response, immune response-activating cell surface receptor signaling pathways, and activation of immune response (Supplementary Figure S19).
This example illustrates that RCA2 allows a hassle-free analysis to characterize clusters with minimal manual efforts. The example can be reproduced by following the tutorial provided both in the github READMe as well as in the packages vignette.
Cluster specific quality control is essential to retain high-quality cells in complex data sets
Here, we consider four novel human bone marrow specimens separated into CD34+ and CD34– fractions (Materials and Methods). By clustering the RCA2 reference projection using Louvain clustering with a resolution of 0.1, we find ten clusters representing major cell types (Supplementary Figure S20). With RCA2’s new cluster specific QC function, we observed that the various cell types included in the dataset do require different QC thresholds (Supplementary Figure S21). For instance, the average NODG for the lymphoid population, e.g. B or T cells, is around 1,000, while the NODG of progenitor cells can be up to three fold higher. Similarly, the percentage of mitochondrial reads shows different distributions. While it has low standard deviation for Pro-B cells, its values spread out widely e.g. for Classical Monocytes. As indicated by the color code in Figure 5A, cluster agnostic thresholds (Supplementary Figure S22) would result in a substantial loss of cells, which is quantified for the final clusters in Figure 5B and clearly illustrates the importance of a (major) cell type specific QC. Final cell type specific QC thresholds are listed in Supplementary Table S2. They were chosen based on the outer most layer of the computed densities in the cluster specific scatter plots (Supplementary Figure S21).
Figure 5.

(A) Cluster-specific QC based on NODG and pMito. Colors indicates whether cells are discarded (red, blue) or retained (black) if general, cluster-unspecific QC would be used. (B) Proportions of cells discarded per cell type using cluster unspecific QC. (C) UMAP reduction of a multi panel RCA2 projection coloured by cell type using a resolution of 0.5.
Upon QC on the level of major cell types, we used RCA2 to define cell types on a more detailed level. Using Louvain clustering, we found the most convincing clustering in terms of the projection heatmap using a resolution of 0.5. Doublets have been removed at this stage with DoubletFinder (28) using the 0.97 quantile of all pANN values as a threshold, resulting in the identification and removal of 906 doublets (Supplementary Figure S23) retaining 31,081 cells. Final cell type annotations, based on projection scores (Supplementary Figure S24) and backed up with DEGs (Supplementary Figure S25, Table S5) as well as canonical markers (Supplementary Figure S26), are indicated in the UMAP representation of the RCA2 projection shown in Figure 5C.
We separated the bone marrow data into two populations using magnetic bead selection as cells that are either positive or negative for the progenitor marker CD34 (42). As shown in Supplementary Figure S27, the RCA2 reference projection based UMAP of the scRNA-seq data shows distinct levels of CD34 MACS labels. These match well to the identified cell types shown in Figure 5 C.
For example, hematopoietic stem/progenitor clusters (HSPC), i.e. HSC/MPP, LyP-1, ERP, MEP, MyP-1 and MyP-2, representing progenitor populations are almost completely composed of cells with a CD34+ MACS label, while clusters such as B cells or Classical Monocytes, that are composed of differentiated cells are enriched for cells with a CD34- label (Supplementary Figure S28a). However, we note that some clusters like naive T cells and non-classical monocytes also had a small contribution of 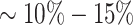 from cells labelled as CD34+ cells by our MACS sorting strategy. This is not unexpected because our workflow for purifying HSPCs lacks a prior conventional lineage-depletion (lin-) step in which cells are immuno-depleted for differentiated cells such as T, B, NK and monocytes by incubating them with antibody-cocktails recognising these cell types. CD34+ populations within the bone marrow are known to be heterogeneous and lin- CD34+ populations were shown to mainly harbour stem cell activity (43). Hence, our MACS sorting strategy is expected to deliver false positives in the form of cells that are retained in the CD34+ magnetic columns but are in reality differentiated cells. However, RCA2 is able to identify such lineage+CD34+ cells and clusters them correctly based on their transcriptome. Thus, RCA2 offers a more precise in-silico alternative to the conventional lineage-depletion step for HSPC studies. Compared to an analysis with Seurat using default parameters (Supplementary Figure S28b), RCA2 achieves a better purity: In only
from cells labelled as CD34+ cells by our MACS sorting strategy. This is not unexpected because our workflow for purifying HSPCs lacks a prior conventional lineage-depletion (lin-) step in which cells are immuno-depleted for differentiated cells such as T, B, NK and monocytes by incubating them with antibody-cocktails recognising these cell types. CD34+ populations within the bone marrow are known to be heterogeneous and lin- CD34+ populations were shown to mainly harbour stem cell activity (43). Hence, our MACS sorting strategy is expected to deliver false positives in the form of cells that are retained in the CD34+ magnetic columns but are in reality differentiated cells. However, RCA2 is able to identify such lineage+CD34+ cells and clusters them correctly based on their transcriptome. Thus, RCA2 offers a more precise in-silico alternative to the conventional lineage-depletion step for HSPC studies. Compared to an analysis with Seurat using default parameters (Supplementary Figure S28b), RCA2 achieves a better purity: In only 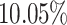 of RCA2 clusters, the impurity is
of RCA2 clusters, the impurity is 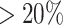 , compared to
, compared to 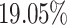 using Seurat. The validity of our approach is also supported by the fact that cell type proportions are in agreement with earlier studies (Supplementary Figure S29).
using Seurat. The validity of our approach is also supported by the fact that cell type proportions are in agreement with earlier studies (Supplementary Figure S29).
This example illustrates the ability of RCA2 to seamlessly derive meaningful annotations and dimensionality reductions even in large, highly complex datasets where cells are placed in a continuum and the reference set might not contain exactly matching cell types.
RCA2 clusters PBMCs from COVID-19 patients more robustly than de-novo clustering
Using PBMC scRNA-seq data obtained from seven COVID-19 patients and six healthy donors, Wilk et al. reported a (myeloid) developing neutrophil (DN) population apparently derived from (lymphoid) plasmablasts (17). This interpretation, which was based on a UMAP plot, deviated from the prior expectation that terminally differentiated lymphoid cells would not trans-differentiate to a myeloid lineage (44). We therefore re-clustered the 44,721 cells analyzed by Wilk et al. using RCA2 (Supplementary Figures S30 and S31a). Notably, the UMAP plot based on RCA2’s reference projection did not support trans-differentiation of plasmablasts into DN cells (Figure 6A). Rather, the cells annotated as DN cells were grouped with other myeloid cells. Specifically, RCA2 placed some DNs within the CD14+ monocyte cluster (Supplementary Figure S32b), a majority in a cluster resembling myelocytes (immature granulocytes) and some within a debris-like cluster that bridged the two (Figure 6; Supplementary Figures S31b–d and S32). Thus, supervised clustering using RCA2 yielded cluster assignments for DN cells that were more consistent with known markers and also more consistent with the known developmental separation between myeloid and lymphoid lineages.
Figure 6.
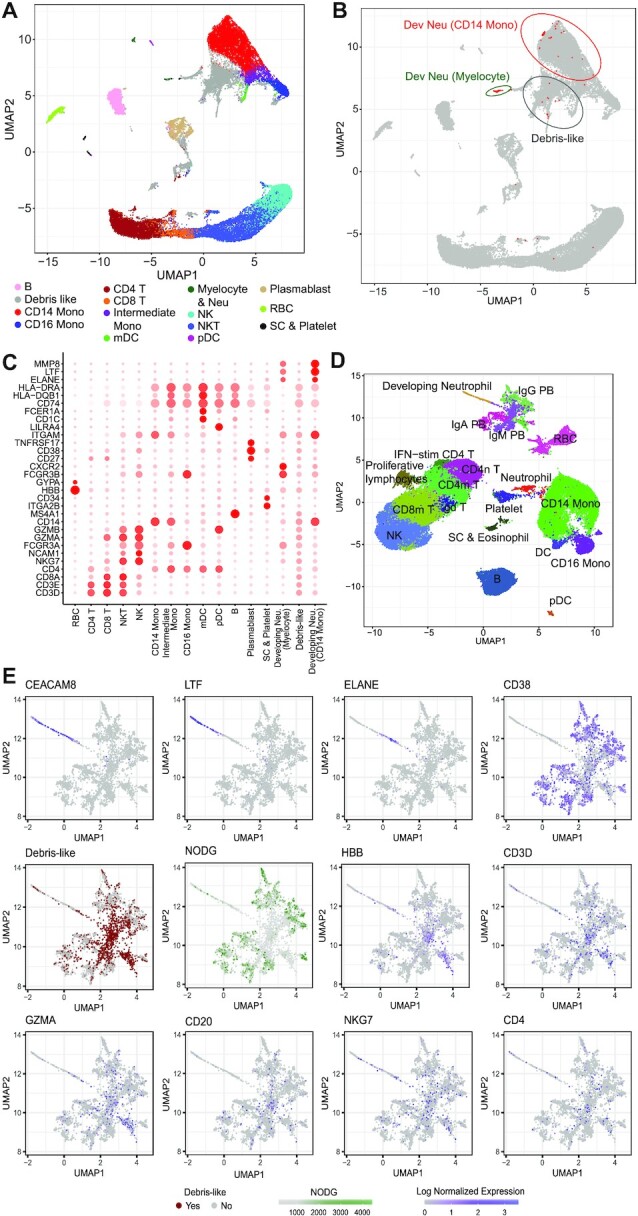
(A) UMAP shows the RCA2 clustering of cells from the COVID-19 study by Wilk et al. (B) The location of developing neutrophils annotated by Wilk et al. are marked as red dots in the RCA2 UMAP. (C) Bubble plot shows the marker gene expression levels across the cell types shown in a. Bubble size indicates expression percentage within each cell type, while color intensity represents scaled expression levels. (D) UMAP plot showing the cell clustering using the de-novo analysis pipeline and cell-type annotation by Wilk et al. (E) UMAPs showing marker gene expression and data quality. Markers are shown for developing neutrophils (CEACAM8, LTF, ELANE), plasmablasts (CD38), red blood cells (HBB), T cells (CD4, CD3D), B cells (CD20), NK and cytotoxic T cells (GZMA, NKG7). Number of detected genes (NODG, orange) is shown as a measure of cell quality and debris-like cells that co-express markers of diverse cell types are indicated in red.
We then attempted to ascertain the cause of the unexpected clustering result reported by Wilk et al., which had connected myeloid DN cells to lymphoid plasmablasts. To this end, we used de-novo clustering and reproduced their UMAP plot, which showed a population of bridging cells between DNs and IgM plasmablasts (Figure 6D, Supplementary Figure S33). As shown in Figure 6E, DN cells expressed the general neutrophil marker CEACAM8 (45) as well as neutrophil subtype markers LTF (46) and ELANE (47), indicating that they did indeed belong to the neutrophil lineage. However, they did not express the key plasmablast marker CD38 (48). CD38 was also absent in the bridging cells. Most surprisingly, CD38 was missing even in the adjacent IgM plasmablast population. Since the bridging cells and IgM plasmablasts expressed neither neutrophil nor plasmablast markers, we hypothesized that these two populations may in fact have been mis-annotated. Indeed, we found that the debris-like cells described above accounted for most of the bridging cells and IgM plasmablasts (Figure 6C, Supplementary Figure S31b-d). Consistently with their classification as debris-like, these cells had lower data quality (NODG) than other plasmablasts and expressed markers of multiple distinct cell types, such as T, B, NK and red blood cells (HBB, CD3D, CD20 and NKG7; Figure 6E).
Thus, our results suggest that the previously reported association of myeloid DN cells with lymphoid plasmablasts could potentially have represented a clustering artifact arising from the presence of mixed-lineage debris in the dataset. Overall, the above results suggest that, in addition to reducing batch effects, supervised clustering using RCA2 is also robust to data artifacts resulting from debris.
Reference based clustering is able to capture disease states of cells
To address a prevalent misconception that supervised clustering algorithms are unable to identify novel cell types and cell states, we used RCA2 to project and to cluster two publicly available data sets: one Acute Myeloid Leukemia (AML) data set (29) as well as the already introduced COVID-19 dataset (17). Note that no additional QC was performed and data is used as provided by the authors. We refer to the Methods section for further processing details.
As shown in Figure 7A, we observe that PBMCs from the COVID-19 data occur both in condition specific and in shared neighbourhoods. This is an expected behaviour and corresponds well to the original findings of Wilk et al. (17). Upon clustering the data in RCA2, we obtained 28 clusters. As shown in Supplementary Figure S34a, several clusters are depleted for cells from COVID-19 patients, whereas five clusters are composed of more than 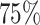 of cells from COVID-19 patients, despite no disease specific reference cell types are included in our panels.
of cells from COVID-19 patients, despite no disease specific reference cell types are included in our panels.
Figure 7.

(A) UMAP embedding of a reference projection for the COVID-19 PBMC data set from (17). (B) UMAP embedding of a reference projection for the AML dataset 809653 from (29). AML and control cells are well separated in reference space.
For the AML data set we obtain a clearer picture. According to the authors classification of cells, AML and healthy cells separate almost perfectly in the RCA2 projection although no AML samples are included in the reference panels (Figure 7B.). This separation is also reflected in the cluster composition plot (Supplementary Figure S34b).
DISCUSSION AND CONCLUSIONS
Although de-novo clustering is currently the predominant strategy to cluster scRNA-seq data, it does have some disadvantages, the most important of which is vulnerability to batch effects and other data quality artifacts. Consequently, de-novo clustering necessitates use of supporting algorithms for explicit batch-effect correction (4). However, one fundamental problem with batch correction algorithms is that they cannot easily distinguish between technical variation and genuine biological differences. Hence, when batch and biology are confounded, there is a risk of erroneously suppressing biological variation (49). Since reference-based methods can mitigate this problem, mapping of SCs to a reference atlas has recently been identified as one of the grand challenges in the SC field (50). RCA2 directly addresses this challenge.
Indeed, our benchmarking of batch effect robustness supports the above expectation. In two independent benchmarks that rely on a robust, independent ground truth, RCA2 was the best performer in clustering cells by cell type rather than batch, even without explicit batch correction (Figures 3 and 4). Consistently with this finding, DEGs between clusters reflected cell type identity in the case of RCA2, but batch effects in the case of de-novo clustering. Importantly, in addition to being robust to batch effects, RCA2 is able to detect cell types and states not present in the reference panel (Figures 5 and 7). This capability of RCA2 implies that novel cell types can potentially be discriminated even when data are projected onto the transcriptomes of related known cell types.
One inter-operability advantage of RCA2 is that count matrices from Seurat can be imported. In return, RCA2 results can be incorporated into a Seurat object. In terms of scalability, one key improvement is that RCA2 memory usage grows linearly with the number of cells, unlike the quadratic scaling of the original RCA version (Figure 2). Also, reference projection is now over ten-fold faster on large datasets. Consequently, RCA2 scales easily to > 100,000 cells on a conventional laptop.
In addition, RCA2 incorporates multiple new reference panels for human and mouse and also supports generation of new panels from user-supplied transcriptome data. RCA2 also provides multiple features for data visualization and interpretation, such as generation of editable (ggplot2) figures, KEGG and GO enrichment analysis (Figure 1). Lastly, RCA2 simplifies cluster-specific data QC, which is essential for discarding low-quality cells and doublets in SC data from heterogeneous samples (Figure 5).
Technical variation can have a severe effect on de-novo clustering. For example, a recent single cell study of PBMCs used de-novo clustering to conclude that plasmablasts could trans-differentiate into developing neutrophils in COVID patients. However, when we reproduced their clustering pipeline, we noticed that the putative trans-differentiating cells showed multiple hallmarks of mixed-lineage cell debris. Thus, it is likely that the surprising finding of lymphoid-to-myeloid trans-differentiation in COVID PBMCs arose from the vulnerability of de-novo clustering to data quality artifacts. In contrast, reference-based clustering using RCA2 was robust to the presence of debris-like artifacts in the data and yielded a result more consistent with the prevailing view that circulating myeloid cells remain myeloid (Figure 6a, Supplementary Figures S30 and S31).
In summary, RCA2 is the first algorithm to combine the batch effect robustness of reference projection with the scalability of graph-based clustering. Our detailed benchmarking of RCA2 demonstrates that reference-based clustering of scRNA-seq data has unique advantages and provides a complementary strategy to widely-used unsupervised approaches. With RCA2, which is freely available on github (https://github.com/prabhakarlab/RCAv2), we provide the single-cell community with the first robust, scalable and easy-to-use R-package that can be easily integrated into existing workflows to leverage the advantages of supervised clustering. We will continue to maintain and enhance RCA2, for example by expanding the set of reference panels and by adding more clustering strategies downstream of reference projection. Given the potential of reference-based methods for SC data analysis, we believe that such methods may in future also prove useful in analyzing multi-modal SC data.
DATA AVAILABILITY
Data analysis scripts, code to create the main figures and RDS files with R objects for the batch effect benchmarking, the 10X PBMC data, the novel BM data and the COVID-19 data are available at Zenodo (10.5281/zenodo.4686335). Fastq files for the BM data are part of a large scale single cell project which requires controlled access. Access requests should be directed to Shyam Prabhakar (prabhakars@gis.a-star.edu.sg) and Sin Tiong Ong (sintiong.ong@duke-nus.edu.sg). We note that processed BM data is available on Zenodo.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank all members of the Prabhakar lab for testing and providing feedback on the functionality and user-friendliness of RCA2. This publication is part of the Human Cell Atlas – www.humancellatlas.org/publications.
Contributor Information
Florian Schmidt, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Bobby Ranjan, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Quy Xiao Xuan Lin, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Vaidehi Krishnan, DUKE-NUS Medical School, 8 College Rd, 169857, Singapore.
Ignasius Joanito, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Mohammad Amin Honardoost, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore; Department of Medicine, School of Medicine, National University of Singapore, 1 Kent Ridge Road, level 10, NUHS Tower Block, 119228, Singapore.
Zahid Nawaz, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Prasanna Nori Venkatesh, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Joanna Tan, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Nirmala Arul Rayan, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
Sin Tiong Ong, DUKE-NUS Medical School, 8 College Rd, 169857, Singapore; Department of Medicine, Duke University Medical Center, Durham, NC 27710, USA.
Shyam Prabhakar, Laboratory of Systems Biology and Data Analytics, Genome Institute of Singapore, A*STAR, 60 Biopolis St, 138672, Singapore.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agency for Science, Technology and Research, Singapore [CDAP201703-172-76-00056, IAF-PP-H18/01/a0/020]; National Medical Research Council, Singapore [MOH-CSASI18may-0002, NMRC/CIRG/1468/2017]. Funding for open access charge: Core funding of GIS.
Conflict of interest statement. None declared.
REFERENCES
- 1.Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A.et al.. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009; 6:377–382. [DOI] [PubMed] [Google Scholar]
- 2.Editorial Method of the year 2013. Nat. Methods. 2014; 11:1. [DOI] [PubMed] [Google Scholar]
- 3.Lawson D.A., Kessenbrock K., Davis R.T., Pervolarakis N., Werb Z.. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018; 20:1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiselev V.Y., Andrews T.S., Hemberg M.. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019; 20:273–282. [DOI] [PubMed] [Google Scholar]
- 5.Li H., Courtois E.T., Sengupta D., Tan Y., Chen K.H., Goh J. J.L., Kong S.L., Chua C., Hon L.K., Tan W.S.et al.. Nat GenetReference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017; 49:708–718. [DOI] [PubMed] [Google Scholar]
- 6.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R.et al.. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019; 20:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou R., Denisenko E., Forrest A. R.R.. scMatch: a single-cell gene expression profile annotation tool using reference datasets. Bioinformatics. 2019; 35:4688–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alquicira-Hernandez J., Sathe A., Ji H.P., Nguyen Q., Powell J.E.. scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019; 20:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiselev V.Y., Yiu A., Hemberg M.. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods. 2018; 15:359–362. [DOI] [PubMed] [Google Scholar]
- 10.Zhang A.W., O’Flanagan C., Chavez E.A., Lim J. L.P., Ceglia N., McPherson A., Wiens M., Walters P., Chan T., Hewitson B.et al.. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods. 2019; 16:1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E.. Fast unfolding of communities in large networks. J. Stat. Mech.: Theory Exp. 2008; 2008:P10008. [Google Scholar]
- 12.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R.. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018; 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf F.A., Angerer P., Theis F.J.. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018; 19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freytag S., Tian L., Lönnstedt I., Ng M., Bahlo M.. Comparison of clustering tools in R for medium-sized 10x Genomics single-cell RNA-sequencing data. F1000Res. 2018; 7:1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A.. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015; 33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranjan B., Sun W., Park J., Mishra K., Xie R., Fatemeh A., Singhal V., Schmidt F., Joanito I., Arul Rayan N.et al.. DUBStepR: correlation-based feature selection for clustering single-cell RNA sequencing data. 2021; bioRxiv doi:18 November 2020, preprint: not peer reviewed 10.1101/2020.10.07.330563. [DOI] [PMC free article] [PubMed]
- 17.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T.et al.. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020; 26:1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badr H.S., Zaitchik B.F., Dezfuli A.K.. A tool for hierarchical climate regionalization. EARTH Sci. Inform. 2015; 8:949–958. [Google Scholar]
- 19.Müllner D.fastcluster: fast hierarchical, agglomerative clustering routines for R and Python. J. Stat. Softw. 2013; 53:1–18. [Google Scholar]
- 20.Hahsler M., Piekenbrock M., Doran D.. dbscan: fast density-based clustering with R. J. Stat. Softw. 2019; 91:1–30. [Google Scholar]
- 21.Wickham H.ggplot2: Elegant Graphics for Data Analysis. 2016; Springer-Verlag. [Google Scholar]
- 22.Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novershtern N., Subramanian A., Lawton L.N., Mak R.H., Haining W.N., McConkey M.E., Habib N., Yosef N., Chang C.Y., Shay T.et al.. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011; 144:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monaco G., Lee B., Xu W., Mustafah S., Hwang Y.Y., Carré C., Burdin N., Visan L., Ceccarelli M., Poidinger M.et al.. RNA-Seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019; 26:1627–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoeckius M.et al.. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017; 14:865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang F.et al.. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. immunol. 2019; 20:928–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimshony T., Senderovich N., Avital G., Klochendler A., de Leeuw Y., Anavy L., Gennert D., Li S., Livak K.J., Rozenblatt-Rosen O.et al.. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016; 17:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGinnis C.S., Murrow L.M., Gartner Z.J.. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019; 8:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petti A.A., Williams S.R., Miller C.A., Fiddes I.T., Srivatsan S.N., Chen D.Y., Fronick C.C., Fulton R.S., Church D.M., Ley T.J.. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat. Commun. 2019; 10:3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lun A.T.et al.. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research. 2016; 5:2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haghverdi L., Lun A. T.L., Morgan M.D., Marioni J.C.. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018; 36:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hie B., Bryson B., Berger B.. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 2019; 37:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hafemeister C., Satija R.. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019; 20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rousseeuw P.J.Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987; 20:53–65. [Google Scholar]
- 35.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M. 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M.et al.. Integrated analysis of multimodal single-cell data. Cell. 2021; 184:3753–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murtagh F., Legendre P.. Ward’s hierarchical agglomerative clustering method: which algorithms implement Ward’s criterion?. J. Classif. 2014; 31:274–295. [Google Scholar]
- 38.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.et al.. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tulchinsky E.Fos family members: regulation, structure and role in oncogenic transformation. Histol. Histopathol. 2000; 15:921–928. [DOI] [PubMed] [Google Scholar]
- 41.Patronas P., Horowitz M., Simon E., Gerstberger R.. Brain ResDifferential stimulation of c-fos expression in hypothalamic nuclei of the rat brain during short-term heat acclimation and mild dehydration. Brain Res. 1998; 798:127–139. [DOI] [PubMed] [Google Scholar]
- 42.Civin C.I., Strauss L.C., Brovall C., Fackler M.J., Schwartz J.F., Shaper J.H.. Antigenic analysis of hematopoiesis. III. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG-1a cells. J. Immunol. 1984; 133:157–165. [PubMed] [Google Scholar]
- 43.Pellin D., Loperfido M., Baricordi C., Wolock S.L., Montepeloso A., Weinberg O.K., Biffi A., Klein A.M., Biasco L.. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 2019; 10:2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alquicira-Hernandez J., Powell J.E., Phan T.G.J.. No evidence that plasmablasts transdifferentiate into developing neutrophils in severe COVID-19 disease. Clin. Transl. Immunology. 2021; 10:e1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Opasawatchai A., Amornsupawat P., Jiravejchakul N., Chan-In W., Spoerk N.J., Manopwisedjaroen K., Singhasivanon P., Yingtaweesak T., Suraamornkul S., Mongkolsapaya J.et al.. Neutrophil Activation and Early Features of NET Formation Are Associated With Dengue Virus Infection in Human. Front. Immunol. 2018; 9:3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao X., Ting S.M., Sun G., Roy-O’Reilly M., Mobley A.S., Bautista Garrido J., Zheng X., Obertas L., Jung J.E., Kruzel M.et al.. Beneficial role of neutrophils Through function of lactoferrin after intracerebral hemorrhage. Stroke. 2018; 49:1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.James M.N.Barrett A.J., Rawlings N.D., Woessner J.F.. Handbook of proteolytic enzymes. Protein Science. 1999; 8:London: Academic Press; 693–694. [Google Scholar]
- 48.Fink K.Origin and function of circulating plasmablasts during acute viral infections. Front. Immunol. 2012; 3:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran H. T.N., Ang K.S., Chevrier M., Zhang X., Lee N. Y.S., Goh M., Chen J.. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020; 21:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lähnemann D., Köster J., Szczurek E., McCarthy D.J., Hicks S.C., Robinson M.D., Vallejos C.A., Campbell K.R., Beerenwinkel N., Mahfouz A.et al.. Eleven grand challenges in single-cell data science. Genome Biol. 2020; 21:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data analysis scripts, code to create the main figures and RDS files with R objects for the batch effect benchmarking, the 10X PBMC data, the novel BM data and the COVID-19 data are available at Zenodo (10.5281/zenodo.4686335). Fastq files for the BM data are part of a large scale single cell project which requires controlled access. Access requests should be directed to Shyam Prabhakar (prabhakars@gis.a-star.edu.sg) and Sin Tiong Ong (sintiong.ong@duke-nus.edu.sg). We note that processed BM data is available on Zenodo.