

Abstract
Uterine artery endothelium undergoes a form of functional adaptation during pregnancy because of an increase in Cx43 communication, resulting in increased Ca2+/IP3 exchange and more synchronous and sustained vasodilator production. We have shown previously that acute exposure to growth factors and TNF can block this adaptation through ERK and/or Src-mediated Cx43 phosphorylation. In preeclampsia such adapted function is already missing, but while elevated TNF is associated with this condition, particularly after 28 weeks (late PE), elevated circulating VEGF165 is not. Given PE is a long term condition emerging in the second half of pregnancy, and is often associated with added edema, we now compare the chronic effects of these two factors on the cell monolayer in order to establish if the breakdown of junctional adherens and tight junctional assemblies in which Cx43 resides could also explain loss of vasodilatory function. We report that while TNF can degrade monolayer integrity even in the 0.1 to 1 ng/ml physiologic range, VEGF up to 10 ng/ml does not. In addition, the progressive action of TNF is mediated through Src and ERK signaling to promote internalization and destruction of VE-Cadherin (VE-Cad) and ZO-1, as well as the expression and secretion of a variety of proteases. At least one protein degraded from the extracellular space is VE-Cad, resulting in release of a shed VE-Cad protein product, and consistent with monolayer breakdown being sensitive to both Src and MEK/ERK kinase inhibitors and the general protease inhibitor GM6001. We conclude that the greater association of TNF with ‘late’ PE is as much due to its longer term destabilizing effects on junctional assemblies as it is to acute closure of Cx43 channels themselves. New therapies aimed at stabilizing these junctional assemblies may help treat this hypertensive condition.
Keywords: TNF-alpha, pregnancy, endothelial dysfunction, junctional proteins, edema, hypertension
Introduction
Within the vasculature, the endothelial cell’s ability to form a monolayer provides a physical barrier between circulating blood and the underlying vessel wall, but it also favors high levels of gap junction communication between cells necessary for maximal vasodilator production. We have previously shown in uterine artery endothelial cells (UAEC) that the increased maternal vasodilatory capacity in normal pregnancy is achieved through increased Cx43 gap junction communication between endothelial cells, resulting in more cells participating in a synchronous Ca2+ response, and each cell maintaining Ca2+ signaling necessary for NO output for longer (Yi et al., 2010; Yi et al., 2011). We have also shown that short term (<1 hr), those functional Cx43 gap junctions are closed in direct response to hormones such as growth factors and cytokines commonly associated with non-healing wounding, and locally elevated in the vessel wall with the onset of hypertension, and/or elevated in the serum of women with preeclampsia (PE) (Bird et al., 2013). In a number of recent studies we have compared the immediate actions of VEGF and TNF to inhibit Cx43 gap junction function in UAEC from pregnancy (P-UAEC). We have shown that this closure of functional gap junctions between endothelial cells is achieved through Src- and ERK-mediated signaling kinases (Boeldtet al., 2015; Boeldt et al., 2017; Ampey et al., 2019). These same reports found that VEGF-mediated inhibition is primarily though ERK signaling, while TNF is mediated primarily through Src and secondary downstream ERK signaling. Our Cx43 studies are consistent with data from a number of cell types that have shown Cx43 is subject to complex post-translational regulation through a number of kinases (Solan & Lampe, 2014), and it would be tempting to think this is the end of the story. Nonetheless, Cx43 gap junctions continued existence in the plasma membrane is also dependent on stabilizing interactions with other junctional proteins. The junctional proteins which may bind Cx43 directly include ZO-1, while those that stabilize cell-cell contact in general include VE-Cadherin (VE-Cad). Together, direct binding to ZO-1 and overall integration with tight and adherens junctional assemblies allows Cx43 plaques to couple as a functional channel between cells. The expression and continued placement of these junctional proteins are also under the control of a variety of signaling kinases, including Src and ERK, which may act on them directly and/or may further promote additional expression and release of metalloproteinases that can cause junctional proterin degradation from outside of the cell (Wettschureck et al., 2019). Pregnancy and the onset of PE are both long term events, and PE is not only characterized by a loss of vasodilatory response, but also by edema , a state where endothelial monolayer cohesion is clearly lost. We have already made the case that PE can be considerd an abnormal wounding reponse associated with elevation of cytokines and growth factors and a loss of Cx43 function (Bird et al., 2013). In this study we examine the relative importance of growth factors and cytokines in destabilizing the endotheial monolayer itself longer term at the level of junctional protein assemblies within which Cx43 plaques reside. Specifically, in this study we ask if VEGF and/or TNF have the ability to destablize the P-UAEC monolayer longer term, and particularly in the physiologic dose range. If that is so, then what cell mechanisms mediate this adverse response, and are they also related to ERK and Src activation? Such knowledge specific to uterine artery endothelium may help us understand the damage being done to endothelium and identify possible common drug targets to prevent such adverse events. Such mechanistic knowledge would take us one step closer to identifying drug targets that could prevent and/or reverse this common pregnancy condition. Herein we report the outcomes of that study, and discuss how such new knowledge informs potential future therapy.
Materials and Methods
General Reagents:
Minimal essential medium (MEM) and all other cell culture reagents unless otherwise stated were obtained from Life Technologies (Carlsbad, CA). Ultrapure water with 0.1% gelatin and GM6001 were from EMD Millipore (Billerica, MA). ATP (disodium salt), TPA, PP2 (a selective inhibitor of Src family kinases), and VE-Cad antibody (#V1514) along with all other basic chemicals, unless noted otherwise, were from Sigma (St. Louis, MO). Cyclohexamide was from Tocris Bioscience (Pittsburgh, PA). TNF-alpha and vascular endothelial growth factor (VEGF165) were from R&D Systems, Inc. (Minneapolis, MN). U0126 (a selective inhibitor of MEK) was from Promega Corp (Fitchburg, WI). For ECIS experiments, 96-well plates were provided by Applied Biophysics (Troy, NY). Affymetrix Ovine ST1.1 chips were from Thermo Fisher Scientific (Waltham, MA). Human MMP Antibody Array was from Abeam (Cambridge, MA).
Isolation of uterine artery endothelial cells (UAEC):
Procedures and protocols for animal handling were approved by the University of Wisconsin-Madison Research Animal Care Committees of both the School of Medicine and Public Health and the College of Agriculture and Life Sciences and followed the recommended American Veterinary Medicine Association guidelines for humane treatment and euthanasia of laboratory farm animals. Uterine arteries were obtained from mixed Western breed pregnant ewes at 120–130 days of gestation during non-survival surgery, and UAEC were prepared by collagenase dispersion as previously described (Yi et al., 2010). Cells were recovered by trypsin digest at the end of passage 3 and stored in MEM media containing 10% DMSO and 20% serum in liquid nitrogen. For general protein quantification studies and ICC, a single vial of cells was grown to near confuence on a T75 flask and then passaged to 4-6 x 60 mm dishes as required each time. Experiments were replicated using cells from a 4-6 different animals as needed. For ECIS, corresponding shed VE-Cad/lysate VE-Cad and MMP Western blots, as well as the Affymetrix microarray experiments, pools of cells were created by growing 1 x T75 of cells each from 4 different NP- or P- sheep and then combining into a P- Pool before freezing down at a 1:4 split ratio.
Endothelial cell monolayer permeability assays:
Monolayer permability was assessed electrically using the ECIS system (Applied Biophysics, Troy, NY). Pooled ovine P-UAEC were plated on a gelatin-coated, stabilized 96-well ECIS plate, and cells were grown up to ~90% confluence, typically around 75-85 hr since plating. Cells were then serum starved by replacing growth media with serum-free media, and incubation continued for at least 3 hr. Cells were then treated with or without 0.1, 1, and 10 ng/mL of VEGF165 or TNF-alpha, or with 0.1, 1, and 10 nM TPA, and resistance was continuously recorded for a further 20 hr. For U0126 (10 uM), PP2 (10 uM) and GM6001 (10, 50, or 100 uM) experiments, cells were pretreated first for 30 min with inhibitor after which vehicle or 10 ng/mL VEGF165 or TNF-alpha or 1 or 10 nM TPA (control) was added. Resistance values (measured in ohms) for each single well were first normalized to resistance values 25 min before initial treatments were added, and then grouped treatment data were averaged together and normalized so control/untreated wells were expressed as 1, plotted as a function of time. Of note, our raw data is expressed as fold of control from electrical resistance at 4000 Hz, and not TEER. The reason is simply that TEER calculations require cells to begin at 100% confluence, and unlike HUVEC, P-UAEC at 100% will both lay over each other and form a monolayer that peels from the well. As such, we begin expriments at 90% confluence, so the conditions are not fully met for TEER. For those wishing to know the equivalent TEER values, we include Sub Table 1 of representative TEER values for comparison to the figures in this paper.
Table 1: Effects of TNF in the presence or absence of PP2 or U016 on ZO-1, VE-Cad, and Cx43 protein concentration.
ICC Staining for ZO-1, VE-Cad, and Cx43 show in Fig 3 was quantified per cell with respect to total cell, periphery, and cytosol. Data is combined from n=3–5 independent experiments and shown as mean +− SEM. Significance by comparison to control or ‘same drug’ control is as indicated.
| Control | Control + PP2 | Control + U0126 | TNF-alpha | TNF-alpha + PP2 | TNF-alpha + U0126 | ||
|---|---|---|---|---|---|---|---|
| Z0-1 | |||||||
| Total cell Mean | 1.00 | 1.19 | 1.13 | 0.82 | 1.48 | 1.14 | |
| SE | 0.00 | 0.05 | 0.29 | 0.07 | 0.03 | 0.23 | |
| P<0.05 | 0.039 | 0.640 | 0.012 | 0.00004 | 0.527 | ||
| P<0.05 vs same control | 0.00004 | 0.192 | |||||
| Periphery Mean | 1.00 | 1.20 | 1.16 | 0.76 | 1.47 | 1.14 | |
| SE | 0.00 | 0.08 | 0.34 | 0.07 | 0.06 | 0.23 | |
| P<0.05 | 0.052 | 0.601 | 0.003 | 0.0002 | 0.516 | ||
| P<0.05 vs same control | 0.0001 | 0.129 | |||||
| Cytosol Mean | 1.00 | 1.32 | 1.21 | 1.26 | 1.82 | 1.50 | |
| SE | 0.00 | 0.20 | 0.26 | 0.16 | 0.47 | 0.42 | |
| P<0.05 | 0.078 | 0.39 | 0.0475 | 0.100 | 0.212 | ||
| P<0.05 vs same control | 0.615 | 0.099 | |||||
| VE-Cadherin | |||||||
| Total cell Mean | 1.00 | 1.84 | 1.65 | 0.98 | 1.63 | 1.11 | |
| SE | 0.00 | 0.24 | 0.07 | 0.11 | 0.27 | 0.02 | |
| P<0.05 | 0.008 | 0.00002 | 0.832 | 0.045 | 0.0002 | ||
| P<0.05 vs same control | 0.053 | 0.408 | |||||
| Periphery Mean | 1.00 | 1.81 | 1.63 | 0.93 | 1.56 | 1.15 | |
| SE | 0.00 | 0.22 | 0.16 | 0.12 | 0.21 | 0.03 | |
| P<0.05 | 0.006 | 0.002 | 0.591 | 0.030 | 0.0002 | ||
| P<0.05 vs same control | 0.033 | 0.237 | |||||
| Cytosol Mean | 1.00 | 2.18 | 2.09 | 1.36 | 2.22 | 1.02 | |
| SE | 0.00 | 0.46 | 0.28 | 0.13 | 0.72 | 0.16 | |
| P<0.05 | 0.033 | 0.002 | 0.023 | 0.129 | 0.883 | ||
| P<0.05 vs same control | 0.272 | 0.148 | |||||
| Cx43 | |||||||
| Total cell Mean | 1.00 | 0.42 | 1.30 | 0.55 | 0.36 | 0.53 | |
| SE | 0.00 | 0.04 | 0.14 | 0.05 | 0.06 | 0.22 | |
| P<0.05 | 0.0000003 | 0.025 | 0.00001 | 0.00001 | 0.026 | ||
| P<0.05 vs same control | 0.038 | 0.888 | |||||
| Periphery Mean | 1.00 | 0.38 | 1.44 | 0.55 | 0.29 | 0.55 | |
| SE | 0.00 | 0.09 | 0.11 | 0.08 | 0.06 | 0.26 | |
| P<0.05 | 0.0001 | 0.002 | 0.0003 | 0.000002 | 0.055 | ||
| P<0.05 vs same control | 0.026 | 0.998 | |||||
| Cytosol Mean | 1.00 | 0.51 | 1.35 | 0.62 | 0.50 | 0.55 | |
| SE | 0.00 | 0.10 | 0.55 | 0.07 | 0.07 | 0.18 | |
| P<0.05 | 0.001 | 0.416 | 0.0009 | 0.00007 | 0.014 | ||
| P<0.05 vs same control | 0.276 | 0.708 | |||||
Western blot analysis:
Frozen P-UAEC at passage 3 were grown in a T75 flask to 90% confluence and passaged to 60-mm dishes. Cells were maintained in growth medium until >80% confluent (24-48 hr). Prior to treatment with either agonist or vehicle, cells were rinsed twice and then incubated without serum for 1 hr with 0.01% BSA. Antagonists or inhibitors U0126, PP2, or GM6001 (10 uM or as stated) or vehicle were added to cells 30 min prior to agonist treatment (10 ng/mL VEGF165, 10 ng/mL TNF-alpha, or 10 nM TPA). At the end of a further 6 or 18 hr, media was collected and cells were washed with ice cold PBS, and then cells on aspirated dishes were snap frozen in liquid nitrogen. Media was centrifuged at 2,000 x g for 10 min at 4 C, and supernatant stored at −20 C. Upon thawing, cells were scraped and solubilized in 100ul lysis buffer [50mM HEPES (pH 7.5), 4mM Na4P2O7-10H2O, 100mM NaCl, 10mM EDTA, 10mM NaF, 2mM Na3(VO4)2, 1mM phenylmethylsulfonulfluoride, 1%Triton X-100, 5ug/mL leupeptin, and 5ug/mL aprotinin], briefly sonicated, and centrifuged at 12,000 x g for 5 min. Solubilized protein concentration was determined using the bicinchoninic acid assay (BCA).
Collected media from experiments to be used for shed VE-Cad and MMP assays was concentrated using a Fischer Amicon Ultra-2 Centrifugal Filter Devices with 10,000 nominal molecular weight limit. Concentrated media was used to detect shed VE-Cad (~90 kDa) by loading 20ug per lane (based on BCA assay detecting the amount of 0.01% BSA in media) on the same type of gels used for cell lysates (below). Western blotting of cell lysates was completed using 15-25 ug protein per lane as appropriate for the protein of interest on 7.5% polyacrylamide gels (110 V, 2 hr; Mini-Protein II, Bio-Rad Laboratories Inc, Hercules, CA) and transfer to Immobilon P membrane (100V, 2 hr; EMD Millipore For blocking, Starting Block TBS blocking buffer (Thermo Fisher Scientific) was used for all antibodies: VE- Cad (#V1514; Sigma-Aldrich), Cx43 (#c6219, Sigma-Aldrich), and ZO-1 (#33-9100, Life Technologies). Primary antibodies were in turn detected using specific goat anti-rabbit HRP-conjugated secondary antibody (#7074; Cell Signaling Technology, Inc., Danvers, MA) at 1:3250 for all primary antibodies unless otherwise noted. ZO-1 antibody was detected using a specific rabbit anti-mouse IgG HRP-conjugated secondary antibody (#AQ160P, EMD Millipore). To ensure consistent protein loading, data were normalized to corresponding levels of Hsp90 (#4874, Cell Signaling Technology, 1:2600). For media, the amount of nonspecific antibody binding to BSA was quantified and used as the loading control. In all cases, specific binding was detected by enhanced chemiluminescence (ECL) reagent detection system (GE Healthcare BioSciences, Piscataway, NJ). Bands were captured on Hyperfilm and quantified using the HP Deskscan system in transmission scanning mode (Hewlett-Packard, Palo Alto, CA). Densitometry was performed using Molecular Analyst software (version 1.4 Bio-Rad Laboratories, Inc).
MMP and TIMP microarray analysis:
Concentrated media from shed VE-Cadherin experiments (after treatments with 10 ng/mL TNF-alpha, VEGF165, or 10 nM TPA for 18 hr) was also used for MMP protein detection. Human MMP Antibody Array Membranes (Abcam; Cambridge, United Kingdom #ab134004) were used to measure the MMP and TIMP proteins found in media. These assays consist of arrays of immobilized antibody spots with each specific to the desired target protein subtype. Detection uses a complimentary antibody cocktail that is biotinylated (in the same manner as a sandwich assay), and streptavidin-HRP detection was achieved using ECL reagents. All incubations were performed overnight and all supplies and dilution buffers were as included in the kit. Film was developed as for western blots with multiple exposure times. Intensity of each MMP/TIMP spot was typically observed at 1 min exposure, and quantified as density values using Image J.
Immunocytochemistry analysis:
Passage 4 P-UAEC cells were plated at 7.2 x 103 cells/well onto eight-chamber slides (Ibidi, Fitchburg, WI; #80826) after treating wells with 0.1% gelatin solution (EMD Millipore; #SF008) for 45 min, followed by air-drying. After 3 days, media was removed and replaced with 0.01% BSA media (MEM containing 1% penicillin-streptomycin and 1% gentamycin). After 1 hr, cells were treated with antagonists; 40 min later agonists were added. After an 18-hr treatment, media was removed and cells were washed twice with PBS. All subsequent steps were at room temperature unless noted. Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA; #15710-S) in 0.2 M sodium cacodylate buffer (Sigma; #C0250) for 8 min. Cells were then washed 3 times with PBS and then blocked for 1 hr in blocking buffer consisting of 2% goat serum (Sigma; #G9023), 1% BSA (Sigma; #A7906) and 0.5% Triton X-100 in 50 mM glycine PBS (pH 7.5). Primary antibody solutions were made in blocking buffer diluted 1:5 in 50 mM glycine PBS (BB2), in which cells were incubated overnight at 4C in the dark with rocking. Primary antibody was then removed and cells were washed 5 times for 5 min each with BB2. Secondary antibody solutions in BB2 containing DAPI (100 ug/ml; 1:250; Sigma; #D8417) were added to wells and cells were incubated at room temperature for 1 hr in the dark with rocking. After 1 hr, cells were washed twice for 5 min each with PBS. PBS was removed and mounting medium (Ibidi; #50001) applied for visualization. For each antibody, control wells of cells were treated with no primary antibody or an antibody isotype control. Sources and dilutions of primary and corresponding secondary antibodies and isotype controls used were as follows: Cx43 IF1 (Ms IgG2a), Fred Hutchison Cancer Research Center, Seattle, WA, 1:500; Alexa Fluor 568 Goat Anti-Mouse IgG2a, Life Technologies, Grand Island NY, #A21134, 1:500; Ms IgG2a, R&D Systems, 1:500. ZO-1 (MsIgG1), Life Technologies, #33-9100, 1:500; Alexa Fluor 488 Goat Anti-Mouse IgG1, Life Technologies, # A21121, 1:750. VE-Cadherin (Rb), Life Technologies, #36-1900, 1:250; Alexa Fluor 647 Goat Anti-Rabbit, Life Technologies; # A21245; 1:750; Rb IgG, Sigma, 1:250. Confocal images of the three protein signals and DAPI together were acquired sequentially to eliminate bleed-through of signal on a Nikon A1R-SI+ confocal microscope (Nikon Instruments, Melville, NY), equipped with a 60 ×/1.4 numerical aperture oil objective (Leica, Buffalo Grove, IL) with a 1.0 airy unit pinhole. Preliminary analyses for each fluorophore confirmed no bleed-through of signal. Images were then analyzed by the surface rendering tools within Imaris software (Bitplane, Concord, MA). For each image, cells were selected as regions of interest and then thresholded for ZO-1, Cx43 and VE-Cad to generate the respective surfaces. The same threshold settings were used within a given experiment. Total intensity within a thresholded surface was determined for each protein. Membrane intensity was determined using the distance transformation function within Imaris that allows for selection of a region at the perimeter of the cell of determined thickness (2.5 um used for all experiments). The intensity for each protein within that periphery surface was measured, and the cytosol intensity was then calculated by subtraction of the periphery intensity from the total cell intensity. Within a given experiment, the intensity values for each protein, averaged per number of cells within a region analyzed, from treatment wells were expressed as a ratio with the similar average intensity from control cells. The ratios were then used to combine data across experiments. Object-based overlap, hereafter called colocalization, of proteins was determined from the same thresholded surfaces used for intensity data. A colocalized surface was generated equaling the intersection of the surfaces of two proteins via the Imaris surface-surface colocalization MATLAB extension. Percent colocalization was determined by calculating the number of voxels in the intersection surface versus the total number of voxels in the surface for a given protein.
Affymetrix microarray analysis of RNA:
Pooled P-UAEC were grown to confluence in a T75 flask before passage to 6 x 60 mm dishes. Cells were then grown to 80-90 % confluence and then washed twice and treated with serum-free medium containing 0.01% BSA for 1.5 hr. Cells were then treated in a final 2.5 ml volume with vehicle or 10 ng/ml VEGF165, 10ng/ml TNF-alpha, 10 nM TPA, 10 uM PP2 or 10 uM U0126 for 12 hr. Media was then removed, cells were very gently rinsed in ice cold PBS, and dishes were snap frozen in liquid nitrogen. RNA was subsequently recovered using a Direct-zol RNA MiniPrep Isolation Kit (Zymo Research, Irvine CA). Briefly, cells were directly solubilized in 1 ml TRizol and left for 5 min at room temperature before recovery with scraping. The recovered TRizol/ethanol mix was then processed as recommended by the manufacturer including in-column DNAse treatment. All recovered RNA samples showed consitent yields and a 260/280 absorbance ratio in excess of 2.0. Further quality control using an Agilent 2100 bioanalyzer (Santa Clara, CA) showed the expected RNA size profile with prominent 18s and 28s peaks. RNA was then stored at −80C before analysis. Succesful probe generation using the Ambion WT Expression kit (Thermo Fisher) in conjunction with the Affymetrix GeneChip WT Terminal Labeling Kit (Thermo Fisher), exactly as recommended by Affymetrix, was uniformly achieved for all samples. Probes were succesfully hybridized to Affymetrix ST 1.0 ovine chips, and final data was combined and analyzed using TAC 3.0 (Affymetrix). This experiment was undertaken as part of a parallel combined transcriptomics/proteomics study; only the data related to metaloproteinases is shown here.
Statistics:
For ECIS data, treatments were replicated on multiple wells (minimum 4) in each plate and averaged per experiment. Each experiment was replicated in turn (n= 6-11, depending on the magnitude of the response). Resistance values for each well were normalized to control (as the ratio of measured resistance to baseline resistance) and then data was normalized such that the control resistance value at the same time point was defined as 1. Normalized final data was plotted as a function of time, and mean +/− standard error (SE) calculated. Students t-test was initially applied. A 1-way ANOVA test was subsequently applied to the full data set using Sigma Plot software at single time points (usually at 3,5, 10, 15 and 20 hr) to determine significance between treatments. For all other methods, data was combined from 4-7 different experiments and presented as means +/− SE. Data were analyzed by Student’s t-test, paired t-test, or analysis of variance, as appropriate. A value of P<0.05 was considered statistically significant.
Results
Impact of agonist and antagonists on monolayer resistance:
As shown in Fig 1, maximal doses of TPA and TNF have a clear impact to decrease monolayer resistance which is immediate for TPA or delayed (by 3 hr) for TNF. In contrast, VEGF did not significantly alter the monolayer at any time or dose (note the expanded scale in panel B) even though VEGF can acutely inhibit gap junction function through Src and ERK (Boeldt et al., 2015; Ampey et al., 2019). We performed statistical comparison of responses at 3, 5,10, 15 and 20 hr on the basis that changes up to 3 hr would mostly reflect movement/changes in location of any existing protein. By 10-15 hr, movement and removal/destruction of old protein would be more significant, and any new expression of mRNA would be largely ongoing, even if new protein translation may still be trailing behind. By 15-20 hr, however, the cell may be in a very different state as there has been ample time for protein destruction, but also changes in new message and synthesis and delivery of new/replacement proteins. Using this approach, the effects of TPA to reduce monolayer resistance show the expected dose and time dependency (Fig 1E). While TNF was also effective at damaging the monolayer in the 1-10 ng/ml range (Fig 1G), the lowest dose (0.1 ng/ml) was actually a mild stimulant to monolayer resistance, achieving significance above control by 20 hr.
Figure 1: Dose dependent effects of TPA, VEGF165, and TNF-alpha on monolayer resistance.
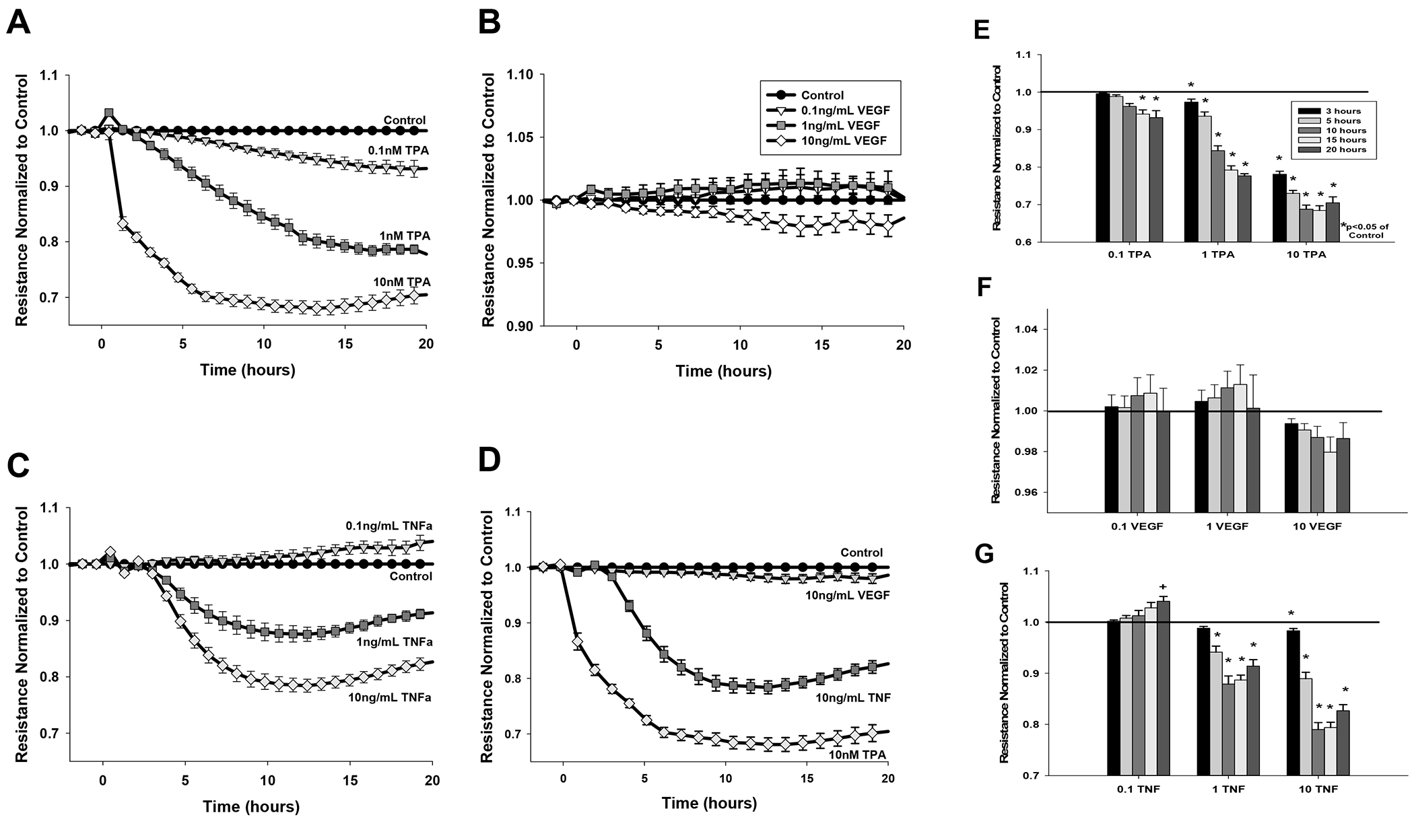
Cells grown to 90% confluence were serum-starved for 3 hr and monolayer resistance monitored by ECIS for a further 20 hr. Data shows resistance (ohms) normalized to control and adjusted for falling baseline for (A) 0.1, 1, 10 nM TPA (B) 0.1, 1, and 10 ng/mL VEGF165 (C) 0.1, 1, 10 ng/mL TNF-alpha; (D) the highest doses of VEGF165, TNF-alpha, and TPA placed on the same graph for comparison. Data is means of n= 9–14 plates with multiple wells per treatment per plate. Panels E, F, and G display specific time points for specific treatments TPA, VEGF165, and TNF-alpha respectively, and displays the significant difference at hours 3, 5, 10, 15, or 20 (key in E) compared to control. Data are expressed as mean ± SEM. *P<0.05 denotes significant difference below untreated control (1 fold). +P<0.05 denotes significant difference above untreated control.
Consistent with the known involvement of Src and ERK regulating junctional protein turnover in the plasma membrane of other cells (Wettschureck et al., 2019), inhibitors of Src and MEK/ERK applied alone to P-UAEC had positive effects on monolayer resistance (Fig 2A, 2B) and their actions were both immediate and continued to 20 hr. Both PP2 and U0126 afforded some degree of rescue from the 1nM TPA response (Fig 2C, Fig 2D). At the 10nM TPA dose, U0126 co-addition provided partial rescue of monolayer resistance. Regarding the physiologic agonists, given that VEGF alone had little effect on the monolayer (Fig 1, 2E, 2F), it was perhaps no surprise the co-addition of PP2 or U0126 with VEGF (Fig 2E, 2F) had largely the same positive effect as the use of these antagonists alone (Fig 2A, 2B). In contrast, when combined with TNF (Fig 2G, 2H), both PP2 and U0126 each provided partial ‘rescue’ of the monolayer destruction long term when compared to TNF alone, but there were also subtle differences. PP2 had no acute rescue effect with TNF, but thereafter promoted a more rapid recovery from the TNF induced minimum. In contrast, U0126 combined with TNF resulted in an immediate improvement in resistance, much like the action of U0126 alone, and a subsequent lesser reduction in resistance was seen thereafter relative to TNF alone.
Figure 2: Rescue ability of U0126 or PP2 on the effects of VEGF165, TNF-alpha, and TPA on monolayer resistance.
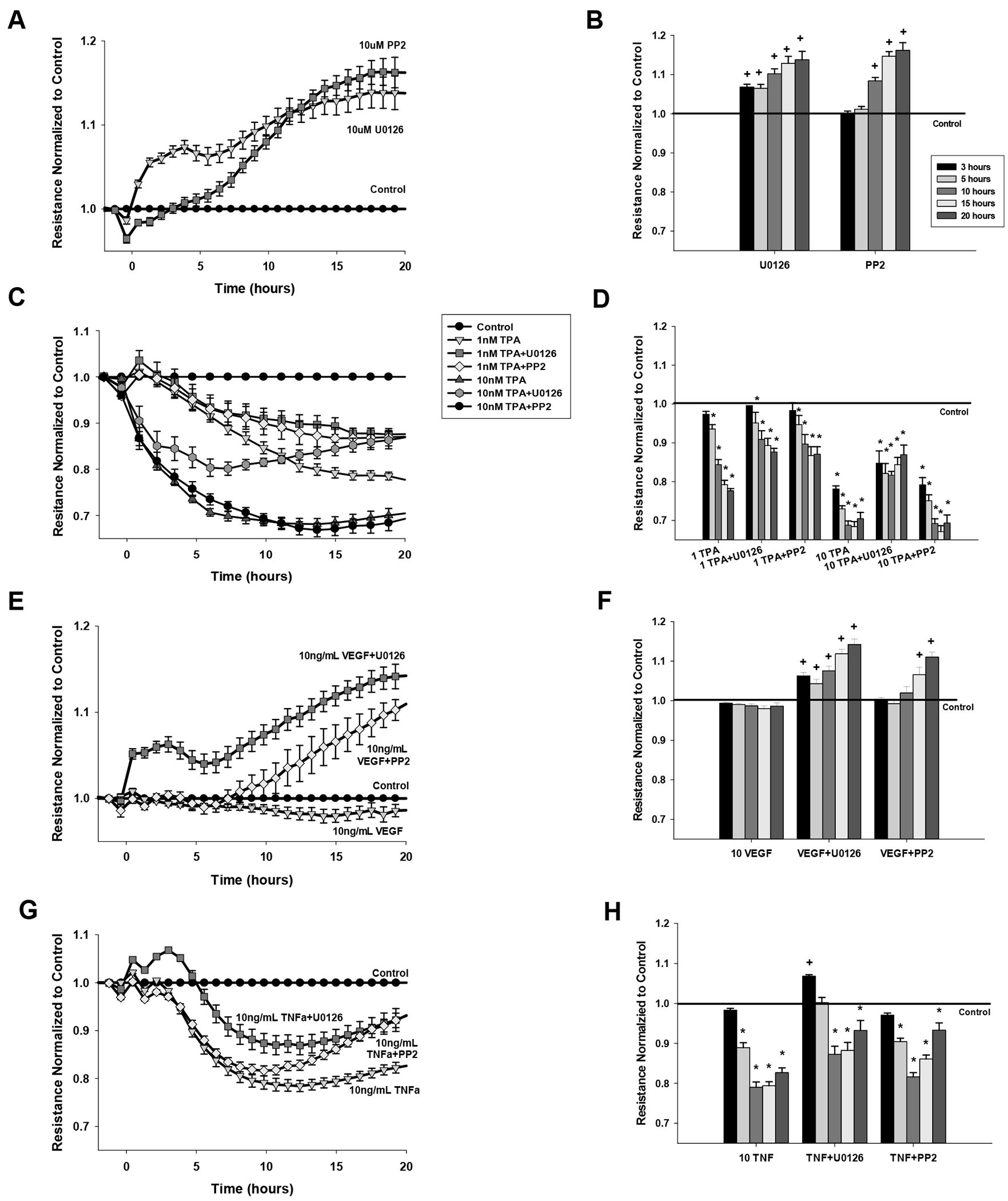
Cells grown to 90% confluence were serum-starved for 3 hr and each experiment recorded for a further 20 hr. Data is normalized to control and adjusted for falling baseline setting untreated, control wells at 1. Data are expressed as mean ± SEM of n= 8 plates with multiple wells per treatment per plate for (A,B) 10 uM PP2 or U0126 alone (C,D) 1 or 10 nM TPA with or without PP2 or U0126, (E,F) 10 ng/mL VEGF165 with or without PP2 or U0126, and (G,H) 10 ng/mL TNF-alpha with or without PP2 or U0126. Panels B,D,F and H show the average normalized resistance at a specific time point of that treatment at 3, 5, 10, 15, or 20 hr (legend in A). *P<0.05 denotes significant decrease from untreated control at 1 fold. +P<0.05 denotes significant increase above untreated control.
Impact of agonist and antagonists on protein level and cell localization:
We next studied more directly the impact of these same agonists and antagonists on junctional protein expression and turnover by immunocytochemistry (ICC) and Western blot. Both ZO-1 and VE-Cad showed distinct membrane-associated staining under florescence microscopy, as would be expected in endothelial cells in the basal state (Fig 3). Both PP2 and U0126 alone had no effect on ZO-1 staining, but did significantly increase the intensity of VE-Cad staining. TPA alone had the expected negative impact on both ZO-1 and VE-Cad staining, while PP2 but not U0126 was effective at rescuing both ZO-1 and VE-Cad to above control levels. Consistent with its lack of impact on monolayer resistance (above), VEGF also had little effect on ZO-1 or VE-Cad ICC staining, but surprisingly the positive effects of PP2 and U0126 were not seen in combination with VEGF. In contrast, TNF reduced ZO-1 staining but not VE-Cad, and PP2 co-addition drove ZO-1 and VE-Cad to control or above control, while U0126 restored ZO-1 and VE-Cad to control levels.
Figure 3: Effects of TPA, VEGF165, or TNF-alpha alone or with added PP2 or U0126 on ZO-1, VE-Cad and Cx43 protein levels in P-UAEC, as detected by Immunocytochemistry.
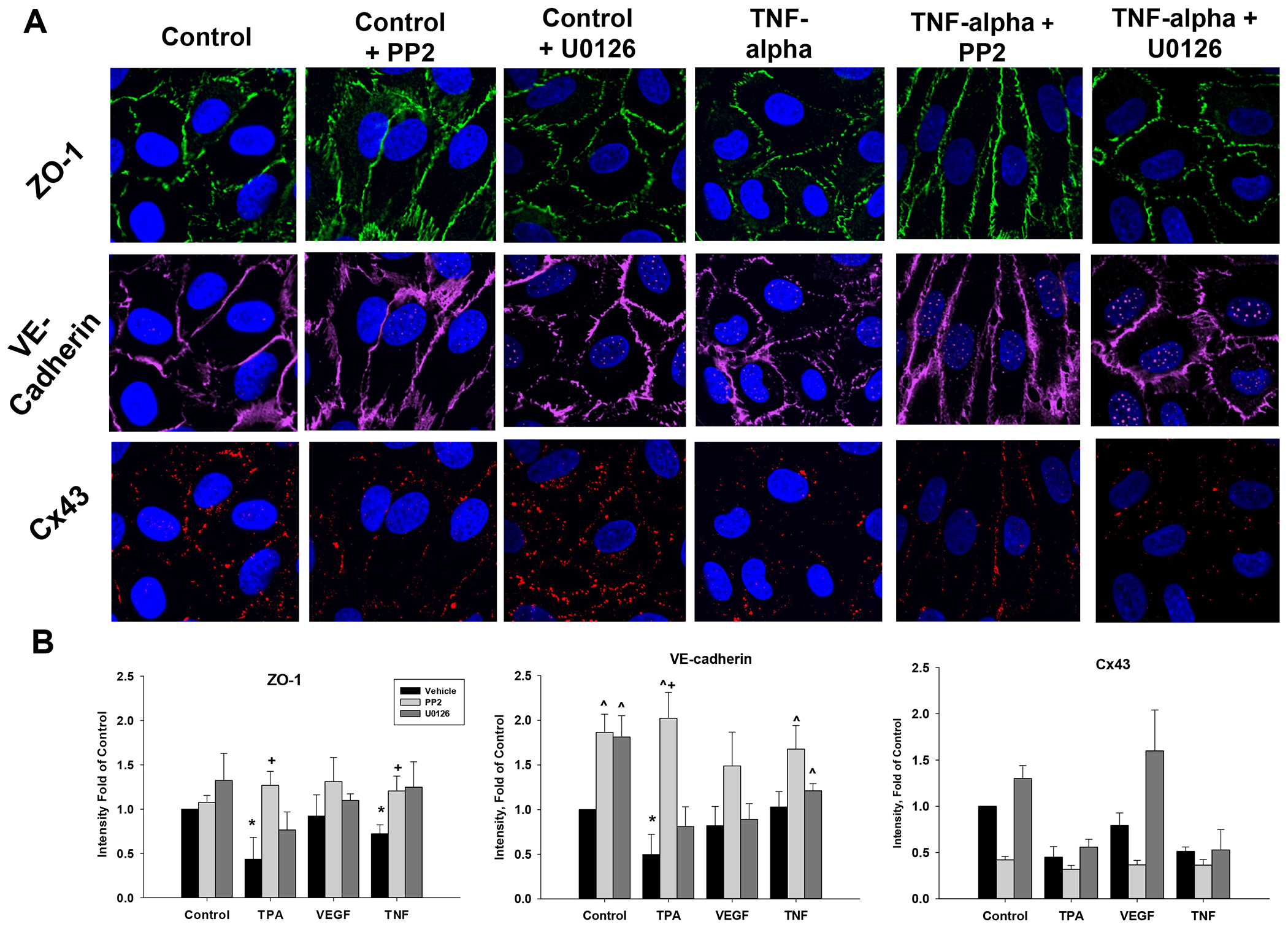
P-UAEC grown to 90% confluence on multi chamber glass slides were pretreated with vehicle, PP2 or U0126 (10 uM) for 40 min prior to addition of vehicle (control), TPA (10 nM), VEGF165 (10 ng/mL) or TNF-alpha (10 ng/mL) as described. After 18 hr, media was removed, and cells fixed prior to ICC staining as described in Methods. All data is normalized as fold of control in the same experiment, and is shown as mean ± SEM of n=3–5 independent experiments. Panel A is a representative ICC image of ZO-1, VE-Cad and Cx43 after control or TNF-alpha treatments with or without PP2 or U0126. Negative controls with no primary antibody or with an isotype control were also performed, but consistently showed no fluorescence and so images are not included (to maximize space for images where staining was seen). Panel B shows quantification (Imaris) of the ICC agonist experiments presented in bar graph form along with statistical differences (bar legend at top). * P<0.05 indicates decrease from control; ^P<0.05 increase from control; +P<0.05 increase from same agonist treatment; #P<0.05 decrease from same agonist treatment.
As expected, Cx43 staining in P-UAEC in the basal state did not show the same intensity or continuity as ZO-1 and VE-Cad. Staining for Cx43 also declined with the addition of PP2, but was increased by U0126. TPA or TNF alone significantly decreased fluorescence of Cx43, and co-treatment of PP2 with TPA or TNF had no rescue effect. Surprisingly, while VEGF alone had little effect on Cx43 as expected, co-treatment with PP2 further reduced fluorescence compared to VEGF alone, while U0126 had no significant effect.
We next quantified relative fluorescence at the periphery (plasma membrane focused staining) vs the whole cell or intracellular space (without plasma membrane) as a crude measure of changes in the balance of new protein synthesis and delivery vs internalization with recycling or internalization with possible destruction. Overall, total ZO-1 cell staining (Table 1) was consistent with that shown in Fig 3, and peripheral ZO-1 changed in parallel with total ZO-1 for each treatment - consistent with most ZO-1 being in the plasma membrane. TNF reduced peripheral staining, and PP2 was effective at rescuing ZO-1 from these TNF effects. In contrast U0126 was ineffective as a rescue agent. In the ‘cytosolic’ region, PP2 and U0126 treatments alone had no significant effect, but TNF did promote an increase in ZO-1, suggesting possible internalization had occurred. PP2 and U0126 were ineffective at altering this response further in the cytosolic region.
For VE-Cad, treatment with PP2 and U0126 alone resulted in an increased total cell staining and increases in both ‘plasma membrane’ and ‘cytosolic’ staining. While TNF alone had little effect on total or peripheral VE-Cad staining, it did increase staining in the cytosolic region. Co-addition of PP2 or U0126 with TNF were able to protect against the negative impact of TNF alone, but rescue effects did not exceed the positive effects of PP2 or U0126 alone.
The findings for Cx43 were inconsistent with those for ZO-1 or VE-Cad. PP2 alone reduced staining in total or at the periphery while U0126 had the opposite effect. TNF reduced Cx43 in total or at the periphery and while PP2 did not rescue Cx43 from the impact of TNF (and arguably made things worse), U0126 had little to no rescue effect on the response to TNF. This is consistent again with the far more dynamic regulation of Cx43 by a wide variety of signaling kinases acting to control both its cell location and junctional opening state (Solan & Lampe 2014).
The Importance of colocalization of ZO-1 and VE-Cad for monolayer resistance.
So far we have considered the two major junctional proteins location separately, but there is also the possibility that tight junctions and adherens junctions can colocalize. To that end, we re-analyzed the ICC data for % colocalization in the plasma membrane. Panel A of Fig 4 further shows in control cells that staining of ZO-1, VE-Cad, and Cx43 can distribute individually or colocalize. ZO-1 and VE-Cad were often found colocalized, as indicated by yellow or white color. Given the more continuous staining of VE-Cad and ZO-1 vs the punctate staining of Cx43, it is no surprise that ZO-1 and VE-Cad are not always found in the same location as Cx43, but Cx43 was often found colocalized with either ZO-1 or VE-Cad. In panel B we also show the quantification of % colocalization in each case. PP2 and U0126 alone both increased % ZO-1 with VE-Cad, but not VE-Cad with ZO-1. TPA was the only treatment that reduced % ZO-1 with VE-Cad, while co-addition of either PP2 or U0126 promoted significant increases in % ZO-1 with VE-Cad to levels above control. Although VEGF alone and indeed TNF alone had no significant effect on % colocalization, co-addition of PP2 further increased % ZO-1 with VE-Cad in each case, but U0126 no longer had a similar effect.
Figure 4: Effects TPA, VEGF165, and TNF-alpha with or without PP2 or U0126 on colocalization of ZO-1, VE-Cad and Cx43 in P-UAEC.
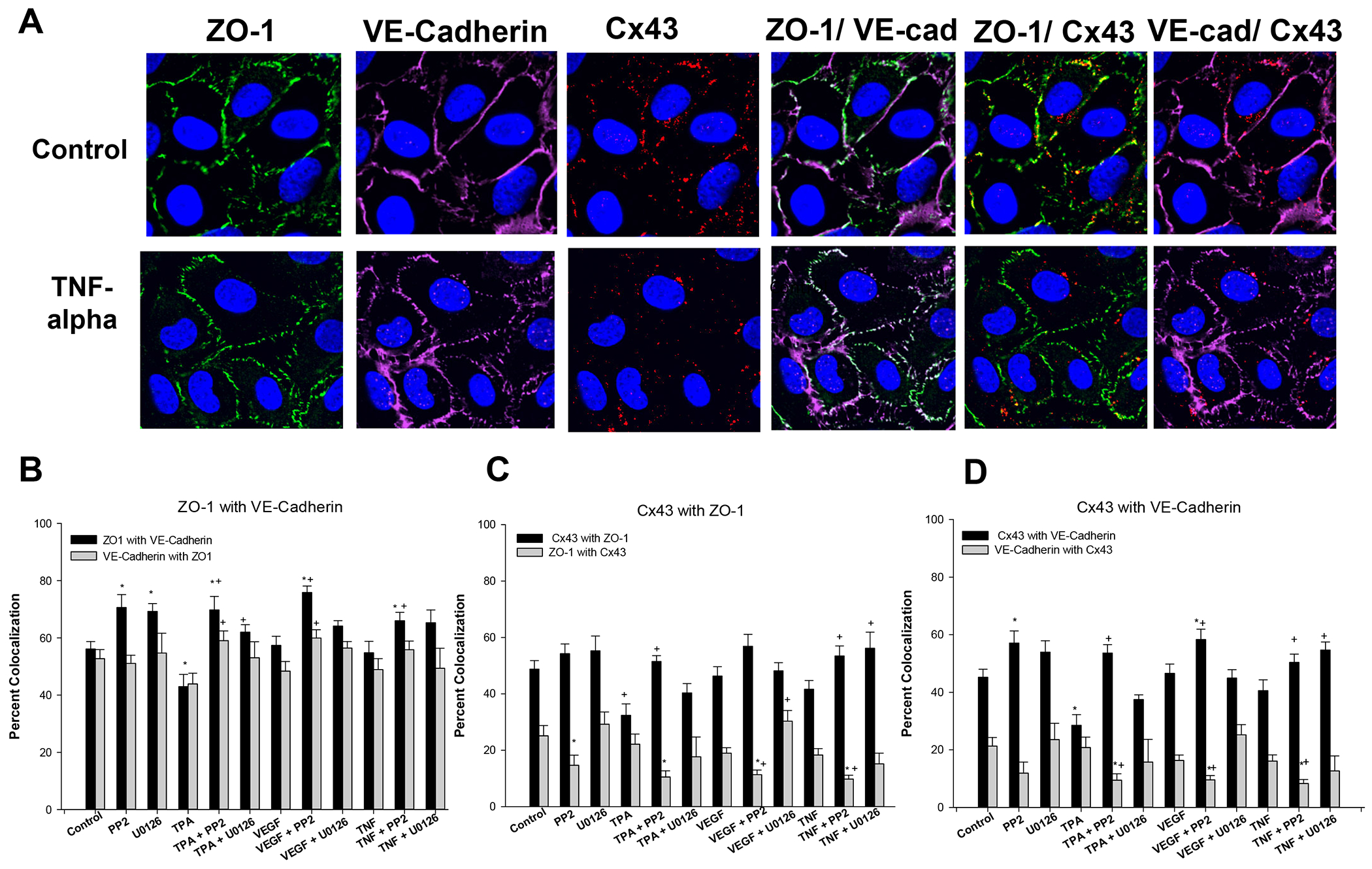
The same images from Fig 3 were reprocessed to consider % of detected protein that was colocalized with another protein. For ease of illustration, Panel A includes the representative ICC image of ZO-1, VE-Cad and Cx43 after control or TNF-alpha treatments as shown in Fig 3, and also the colocalization of the proteins. Panels B-D represent data from all the ICC agonist experiments with or without TPA, VEGF, TNF, and cotreated with or without PP2, or U0126, and is presented in bar graph form for ease of comparison. Data is expressed as means ± SEM of values from n=3–5 independent experiments * P<0.05 indicates significant from control; + P<0.05 significant from same agonist treatment.
As a comparison, we also studied Cx43 colocalization with ZO-1. Once again, TPA alone was the only treatment that decreased % colocalization of Cx43 and ZO-1. Of relevance, PP2 alone, U0126 alone and VEGF or TNF alone all had no significant effect on % colocalization of Cx43 with ZO-1. When co-treating with antagonists, PP2 but not U0126 improved the % colocalization in cells treated with TPA, but both PP2 and U0126 improved the % colocalization of Cx43 with ZO-1 in response to TNF. Finally, a similar analysis of colocalization of Cx43 with VE-Cad showed that TPA was the only treatment to significantly reduce % colocalization, and PP2 alone was the only antagonist that could significantly increase % colocalization above control. As above, while VEGF had no effect alone, PP2 with VEGF increased % colocalization to a degree similar to PP2 alone. TNF alone also had no effect on % colocalization of Cx43 with VE-Cad but PP2 or U0126 co-treatment increased % colocalization.
To further confirm what was suggested by the colocalization data, the mean lowest ECIS resistances for each treatment group were plotted against mean % colocalization by ICC. This analysis (Fig 5) shows most clearly there was no linear relationship between the mean lowest ECIS resistance and the mean % colocalization data for the ZO-1 with Cx-43 and VE-Cad with Cx-43 combinations. Nonetheless, there was a clear linear relationship (P<0.024) between mean lowest ECIS resistance and mean % colocalization of ZO-1 with VE-Cad. This relationship suggests that colocalization of ZO-1 and VE-Cad is a significant overall determinant of monolayer integrity, and while TNF does not reduce monolayer integrity primarily through changes in ZO-1 and VE-Cad % colocalization alone, addition of PP2 or U0126 can provide some rescue effect in a manner associated with an increase in % colocalization.
Figure 5: Relationship between monolayer integrity as determined by ECIS resistance vs colocalization of ZO-1, VE-Cad, or Cx43.
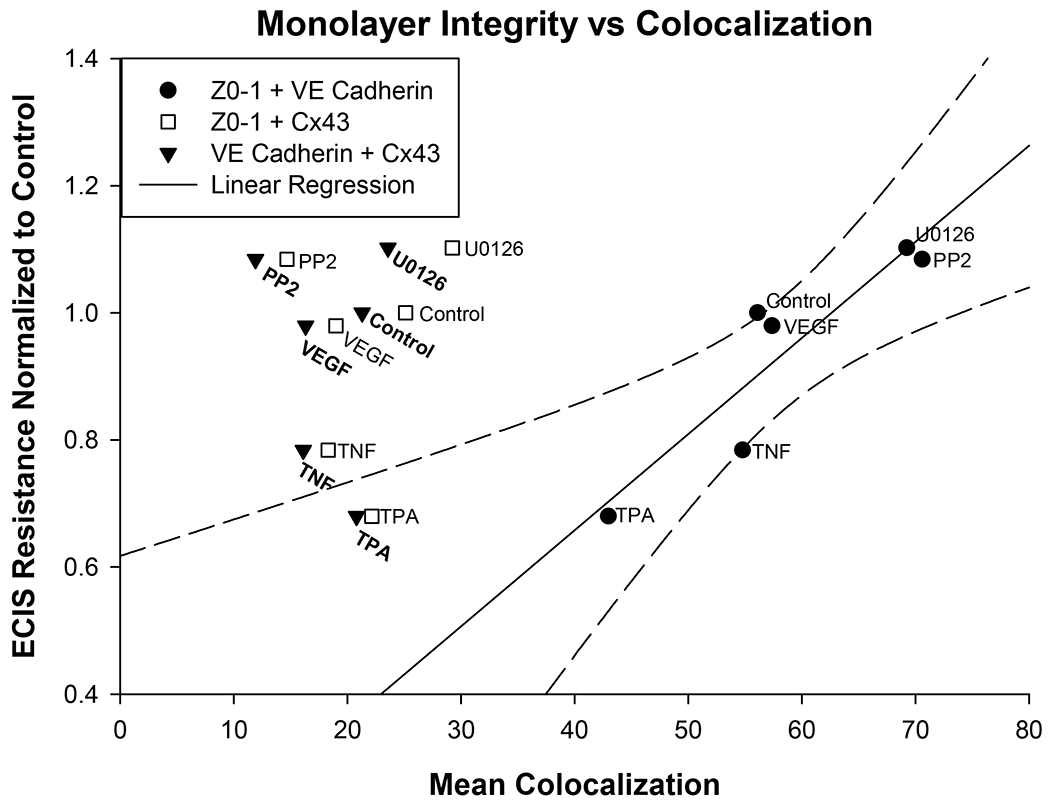
The data for mean lowest ECIS resistance normalized to the control from Fig 2 was compared to the mean % colocalization of the three protein pairings as shown in Fig 4. While ZO-1 with Cx43 and VE-Cad with Cx43 did not show a significant regression relative to monolayer resistance, Z0–1 with VE-Cad colocalization showed a clear and significant linear regression (P< 0.024) against the lowest ECIS resistance for each group.
Evidence for a potential role for protein turnover vs proteolytic degradation of junctional assemblies:
We also evaluated junctional protein expression by Western blot, and found (Fig 6) that ZO-1 expression at 18 hr (Panel A) is significantly lower than control for TPA and VEGF, but not TNF treatment groups. The decrease in response to TPA was around 25% while that for VEGF was 10-15%. While ZO-1 is only subject to turnover inside the cell, VE-Cad spans the plasma membrane and is accessible to extracellular degradation. As such, we also tested the possibility that shed VE-Cad is released to the media through the stimulated release of metalloproteases. Panels B-D show the release of the extracellular domain (shed VE-Cad) to media vs the remaining VE-Cad in cell lysate, and the ratio of shed VE-Cad to intact VE-Cad in lysate. Of note, while TPA had little to no effect increasing shed VE-Cad in the media, it did promote VE-Cad loss from cell lysate. VEGF had no detectable effect on shed VE-Cad or on VE-Cad in cell lysate. However, TNF both increased shed VE-Cad and reduced the level of intact VE-Cad in cell lysate. As such, a ratio of the effects of both TPA and TNF in media vs lysate were significantly elevated, while VEGF had no effect.
Figure 6: Effect of TPA, VEGF165, and TNF-alpha on ZO-1 and VE-Cad protein in P-UAEC and shed VE-Cad in cell media.
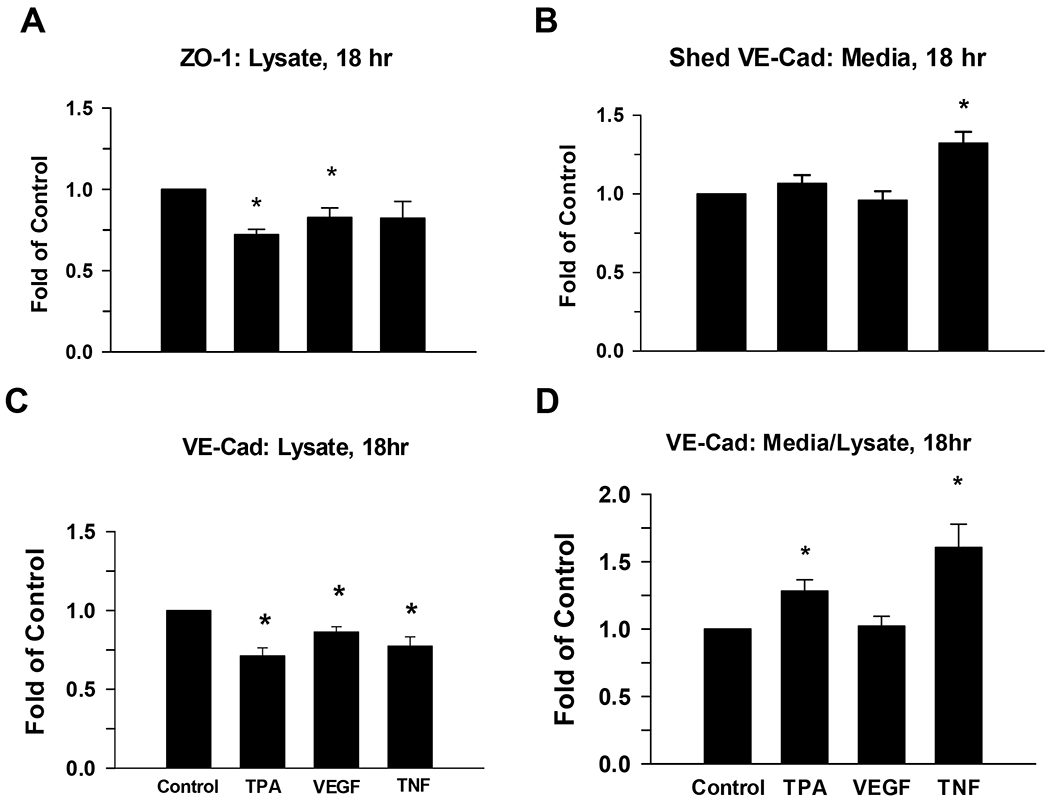
P-UAEC grown to near confluence were pretreated for 20 min with PP2 (10 uM) or vehicle and then treated for 18 hr with TPA (10 nM), VEGF165 (10 ng/mL), or TNF-alpha (10 ng/ml). The ZO-1 and VE-Cad remaining in the cells and shed VE-Cad release to the media were then measured. Results for cell lysate were normalized to Hsp90 protein, and shed VE-Cad in media was normalized to BSA in media. All data are expressed as fold change over unstimulated control. Values shown are means ±SEM of n=6–9 independent experiments. Changes were highly significant (*P<0.01) compared to control.
The next question was how much are these events a balance of synthesis and destruction in the basal state, and further what are the relative rates at which ZO-1 and VE-Cad turn over? To address this, we incubated the cells for 18h with an inhibitor of protein synthesis, cycloheximide (CHX). Our results (Fig 7) show that the % decline in VE-Cad far exceeds that of ZO-1 or indeed a housekeeping protein such as Hsp90. While a simple approach, this does suggest % VE-Cad turnover is both substantial and ongoing even in the basal state, and far exceeds that of ZO-1.
Figure 7: Effect of cycloheximide on VE-Cad (media and lysate) and ZO-1 (lysate) at 18 hr.
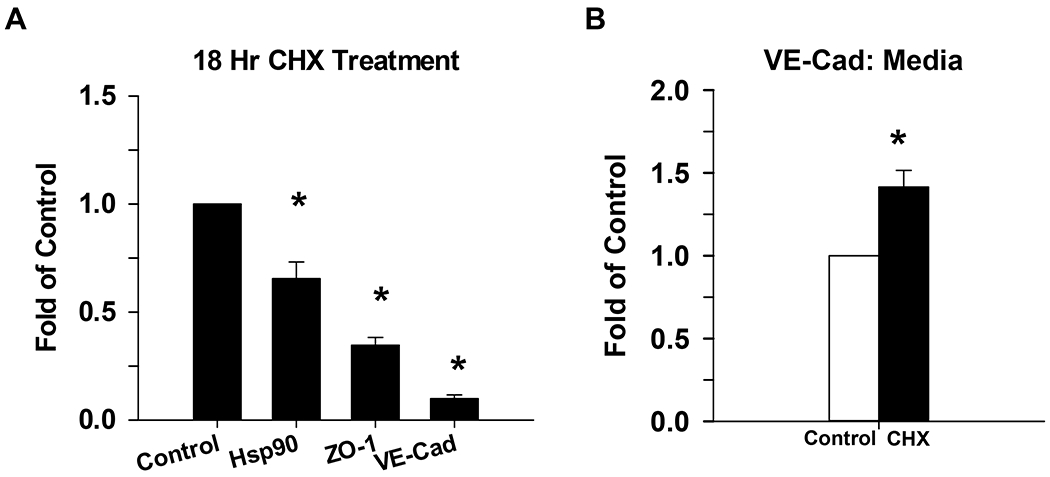
P-UAEC grown to near confluence were treated with cycloheximide (CHX), a protein synthesis inhibitor, for 18 hr. The expression of VE-Cad and ZO-1 were then measured in cell lysates by western blot, and normalized to Hsp90 (Panel A). Shed VE-Cad expression was also measured in the media (Panel B) and was normalized to nonspecific staining of BSA. Data is consistent with findings for changes in both VE-Cad vs ZO-1 in Figs 3 and 4. Values shown are mean ±SEM of 4–5 independent experiments and expressed as fold of control. *P<0.05 compared with control; +P<0.05 compared with same agonist treatment.
Evidence that agonists regulate monolayer resistance through the action of MMPs:
Given our finding that TNF drives shed VE-Cad release to media, we further examined this response using a broad spectrum MMP inhibitor, GM6001 (Fig 8). When GM6001 was present alone, shed VE-Cad in the media decreased. With agonist treatment, the effects on shed VE-Cad were uniformly blocked.
Figure 8: Effect of MMP Inhibitor GM6001 on reversal of the effects of TPA, VEGF165, or TNF-alpha on Shed VE-Cad in media and VE-Cad in P-UAEC cell lysate.
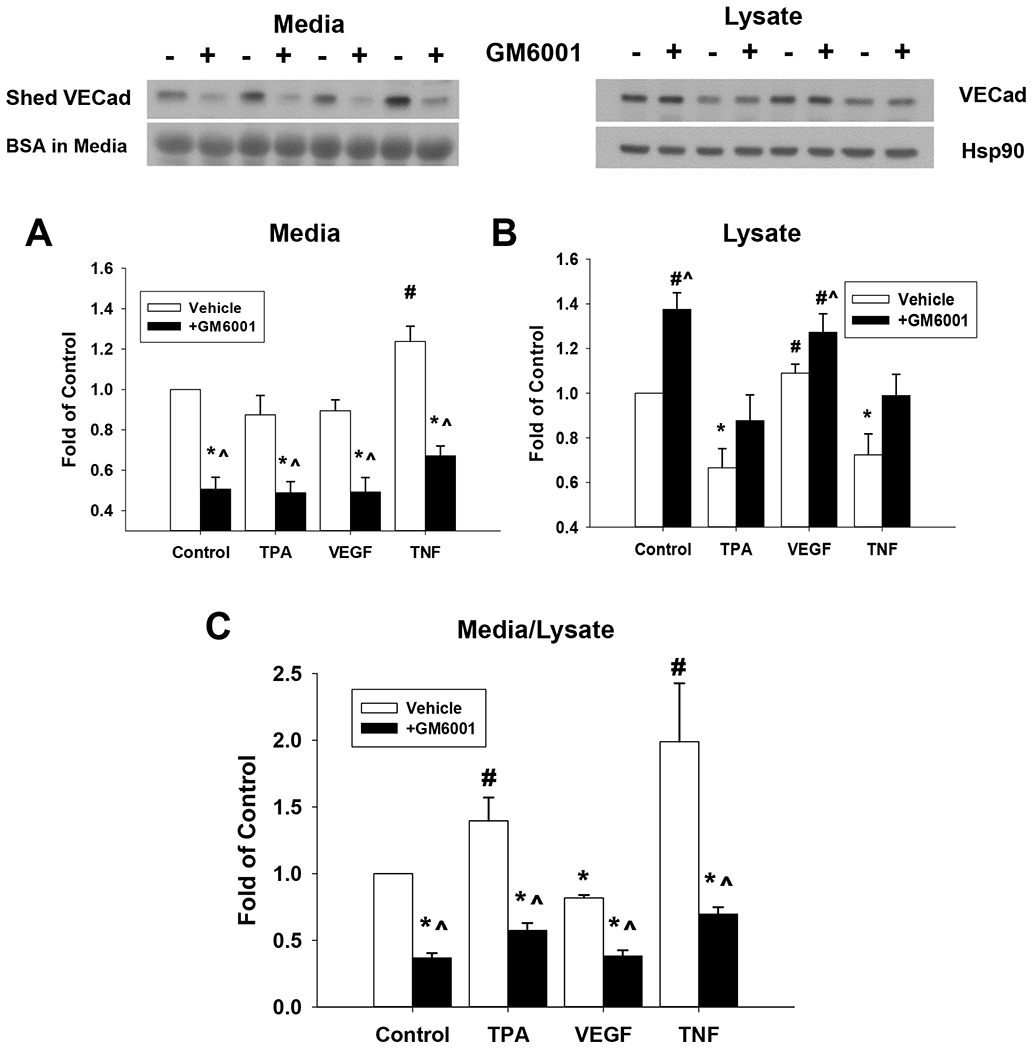
Cells grown to near confluence were pretreated for 20 min with MMP inhibitor GM6001 (10 uM) or vehicle before being treated with TPA (10 nM), VEGF165 (10 ng/mL) or TNF-alpha (10 ng/mL) for 18 hr. Shed VE-Cad was assayed in the media (A) and intact VE-Cad was measured in the cell lysate (B) by western blot as described. Shed VE-Cad expressed as a ratio as VE-Cad in media/VE-Cad in lysate is also shown (C). Results were normalized to Hsp90 protein (lysates) or BSA (media), and data expressed as fold change over unstimulated control. Values shown are means ±SE of n=7 independent experiments. *P<0.05 decrease compared to control; #P<0.05 increase compared to control; ^P<0.05 compared with same agonist treatment.
Consistent with the implications from CHX studies that VE-Cad degradation is ongoing, GM6001 also increased intact VE-Cad in cell lysate for all treatment groups, most likely by blocking ongoing MMP synthesis necessary for VE-Cad degradation to shed VE-Cad. Similar conclusions are indicated by the shed VE-Cad media/lysate ratio. The positive effects of TPA and TNF are reversed by GM6001, consistent with both TPA and TNF acting through MMPs, albeit to different degrees. Certainly, the implication of MMP action could explain the loss of monolayer integrity.
Our next step was to investigate the expression of MMP by P-UAEC in basal and stimulated states. After treating cells for 12 hours, we extracted the cell RNA and analyzed it using the Affymetrix Ovine ST1.1 chip. We clearly detected the transcripts of multiple MMP isoforms and TIMPS, and saw major and significant fold changes in MMP1, MMP3, and MMP12 on TNF stimulation, and substantial decreases of the same targets in response to PP2 or U0126 alone (Table 2). While TPA stimulated a similar and more robust response and also increased the transcript for MMP9, VEGF had no such effect. The medium from the cells treated for 18 hr and analyzed for shed VE-Cad (above) was also further analyzed for MMP and TIMP proteins. Our data (Fig 9) confirmed that many of the same MMP isoforms identified at the level of dominant RNA transcripts were detectable in the media, including MMP1, MMP3, MMP9 and MMP13. Of note, MMP 12 was not on this protein array. Multiple TIMP isoforms were detected with TIMP 4 being the most abundant, but note TIMP3 is also missing from this array. Agonist or antagonist treatment did not significantly alter protein levels, but these proteins are not regulated by expression alone. To that end we further examined the ability of the MMP inhibitor GM6001 to reverse the negative effects of TNF on monolayer integrity using the ECIS method (Fig 10). When submaximal doses of TNF were combined with MMP inhibitor GM6001, there was an improvement in resistance, and the optimal dose to increase resistance was 50uM. When maximally effective (10 ng/ml) doses of TNF were added, the 10uM GM6001 provided partial recovery, while the 50uM and 100uM GM6001 provided maximal rescue.
Table 2: Affymetrix chip analysis of MMP transcripts expressed in P-UAEC treated with VEGF165, TNF-alpha, TPA, U0126, or PP2.
Pooled P-UAEC were grown to 80–90% confluence and treated with serum free medium before adding vehicle or 10 ng/ml VEGF165, 10 ng/ml TNF-alpha, 10 nM TPA, 10 uM PP2 or 10 uM U0126 for 12 hr. RNA was recovered and analyzed using Ovine ST1.0 Affymetrix chips as described in Methods. Data shown is that for MMP transcripts and is the mean fold response relative to control. Significance is as determined using TAC console. Data is from n=6 independent experimental replicates. Bold indicates values significant at P<0.05 and grey highlight indicates changes of 2-fold or more.
| Cluster ID | Gene | TPA Fold | ANOVA | VEGF Fold | ANOVA | TNF Fold | ANOVA | PP2 Fold | ANOVA | U0126 Fold | ANOVA | Gene Symbol | Description |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14831868 | MMP1 | 9.26 | 0.000380 | 1.27 | 0.577285 | 3.79 | 0.010092 | −1.32 | 0.410276 | −2.01 | 0.022409 | MMP1 | matrix metallopeptidase 1 (interstitial collagenase) |
| 14747122 | MMP2 | −1.04 | 0.289801 | 1.05 | 0.192794 | −1.10 | 0.02258 | −1.00 | 0.749364 | 1.04 | 0.103107 | MMP2 | matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase) |
| 14831857 | MMP3 | 1.89 | 0.036519 | 1.25 | 0.250130 | 2.74 | 0.001004 | −7.23 | 0.000003 | −4.87 | 0.000025 | MMP3 | matrix metallopeptidase 3 (stromelysin 1, progelatinase) |
| 14831911 | MMP7 | 1.11 | 0.489874 | 1.08 | 0.609593 | 1.05 | 0.457661 | 1.03 | 0.935785 | 1.10 | 0.779365 | MMP7 | matrix metallopeptidase 7 (matrilysin, uterine); matrilysin |
| 14831879 | MMP8 | 1.08 | 0.951225 | −1.11 | 0.252445 | −1.03 | 0.896687 | −1.06 | 0.723733 | −1.24 | 0.405728 | MMP8 | matrix metallopeptidase 8 (neutrophil collagenase) |
| 14740499 | MMP9 | 4.26 | 0.000182 | 1.18 | 0.54346 | 1.62 | 0.133407 | −1.86 | 0.013284 | −1.45 | 0.041844 | MMP9 | matrix metallopeptidase 9 (gelatinase B, 92kDa gelatinase, 92kDa type IV collagenase) |
| 14719461 | MMP11 | 1.32 | 0.018937 | 1.03 | 0.683522 | −1.01 | 0.820525 | 1.08 | 0.488398 | 1.12 | 0.183513 | MMP11 | matrix metallopeptidase 11 (stromelysin 3) |
| 14831846 | MMP12 | 24.72 | 0.000319 | 1.26 | 0.752276 | 7.12 | 0.009823 | −2.23 | 0.042057 | −2.21 | 0.043927 | MMP12 | matrix metallopeptidase 12 (macrophage elastase) |
| 14831835 | MMP13 | 33.89 | 0.000682 | 1.23 | 0.958025 | 4.34 | 0.065957 | −2.00 | 0.119886 | −2.31 | 0.040849 | MMP13 | matrix metallopeptidase 13 (collagenase 3) |
| 14770048 | MMP14 | 1.86 | 0.000432 | 1.05 | 0.836455 | −1.12 | 0.375081 | −1.04 | 0.534162 | −1.09 | 0.210136 | MMP14 | matrix metallopeptidase 14 (membrane-inserted) |
| 14747730 | MMP15 | 1.06 | 0.235088 | −1.01 | 0.995443 | −1.02 | 0.831112 | 1.08 | 0.456359 | −1.24 | 0.106583 | MMP15 | matrix metallopeptidase 15 (membrane-inserted) |
| 14783462 | MMP16 | −1.06 | 0.549559 | 1.00 | 0.857197 | 1.01 | 0.310671 | −1.32 | 0.803501 | 1.26 | 0.109459 | MMP16 | matrix metallopeptidase 16 (membrane-inserted) |
| 14791388 | MMP17 | −1.02 | 0.492428 | 1.03 | 0.88133 | −1.02 | 0.272677 | −1.08 | 0.080985 | −1.04 | 0.543826 | MMP17 | matrix metallopeptidase 17 (membrane-inserted) |
| 14806051 | MMP19 | 1.38 | 0.135573 | 1.04 | 0.802994 | 1.12 | 0.669149 | −1.44 | 0.039653 | 1.13 | 0.838978 | MMP19 | matrix metallopeptidase 19 |
| 14831900 | MMP20 | 1.09 | 0.261885 | −1.14 | 0.019253 | −1.02 | 0.904140 | −1.05 | 0.197874 | −1.05 | 0.751797 | MMP20 | matrix metallopeptidase 20 |
| 14833393 | MMP24 | 1.12 | 0.061920 | −1.06 | 0.314654 | 1.04 | 0.369383 | −1.09 | 0.882969 | −1.01 | 0.408488 | MMP24 | matrix metallopeptidase 24 (membrane-inserted) |
| 14771712 | MMP25 | 1.09 | 0.375893 | 1.05 | 0.276963 | 1.13 | 0.055192 | 1.02 | 0.583339 | −1.10 | 0.536965 | MMP25 | matrix metallopeptidase 25 |
| 14831889 | MMP27 | 1.03 | 0.744258 | −1.24 | 0.005127 | 1.05 | 0.524123 | −1.03 | 0.544841 | −1.17 | 0.135851 | MMP27 | matrix metallopeptidase 27 |
| 14883630 | MMP28 | −1.16 | 0.184595 | −1.03 | 0.957346 | −1.33 | 0.036173 | 1.02 | 0.550302 | 1.31 | 0.004269 | MMP28 | matrix metallopeptidase 28 |
| 14793298 | TIMP1 | 1.24 | 0.028553 | 1.03 | 0.84644 | 1.14 | 0.21381 | −1.39 | 0.097836 | −1.11 | 0.223265 | TIMP1 | TIMP metallopeptidase inhibitor 1 |
| 14748370 | TIMP2 | 1.35 | 0.123444 | 1.05 | 0.800133 | 1.21 | 0.324644 | −1.25 | 0.258587 | 1.35 | 0.120889 | TIMP2 | TIMP metallopeptidase inhibitor 2 |
| 14744606 | TIMP3 | −1.28 | 0.001912 | −1.05 | 0.525343 | −1.03 | 0.896624 | −1.42 | 0.000707 | −1.26 | 0.014062 | TIMP3 | TIMP metallopeptidase inhibitor 3 |
| 14770758 | TIMP4 | −1.03 | 0.988126 | 1.02 | 0.904207 | −1.07 | 0.561512 | 1.03 | 0.815771 | 1.54 | 0.008201 | TIMP4 | TIMP metallopeptidase inhibitor 4 |
Figure 9: Identification of MMP and TIMP isoforms secreted into media.
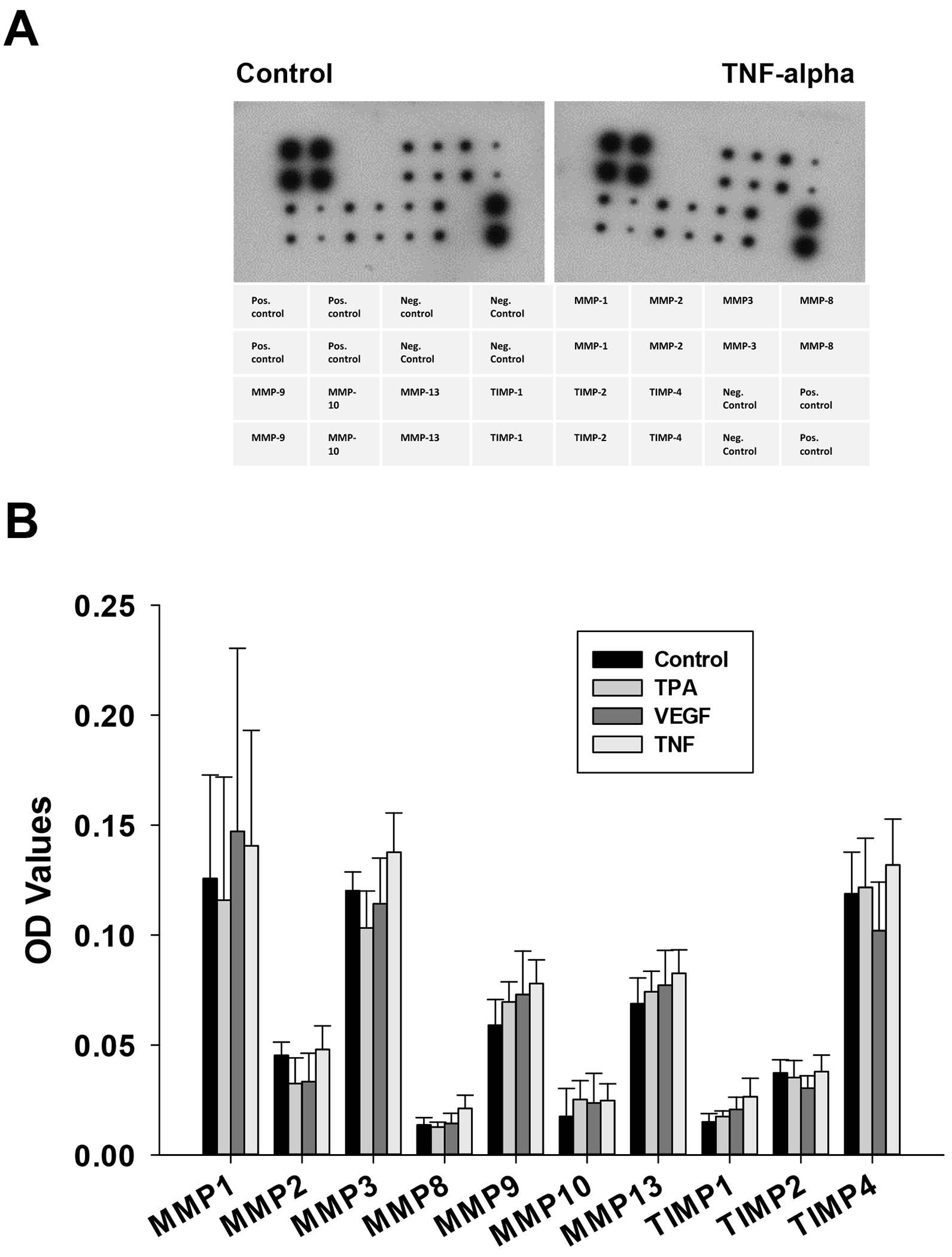
Human MMP antibody array membranes were also used to measure the MMPs and TIMPs found in media of experiments from Fig 6. Quantifiable data was obtained from n= 4 experiments. Film was developed with multiple exposure times, and intensity of each MMP/TIMP spot was typically observed and quantified at 1 min exposure. Optical density (OD) values determined in Image J are shown as mean ± SEM. While abundance of different MMP isoforms were clearly different and in close agreement with mRNA transcripts (Table 2), no further changes in protein level were seen in response to the different endocrine treatments.
Figure 10: Recovery effect of GM6001 on TNF-alpha-treated P-UAEC monolayer integrity.
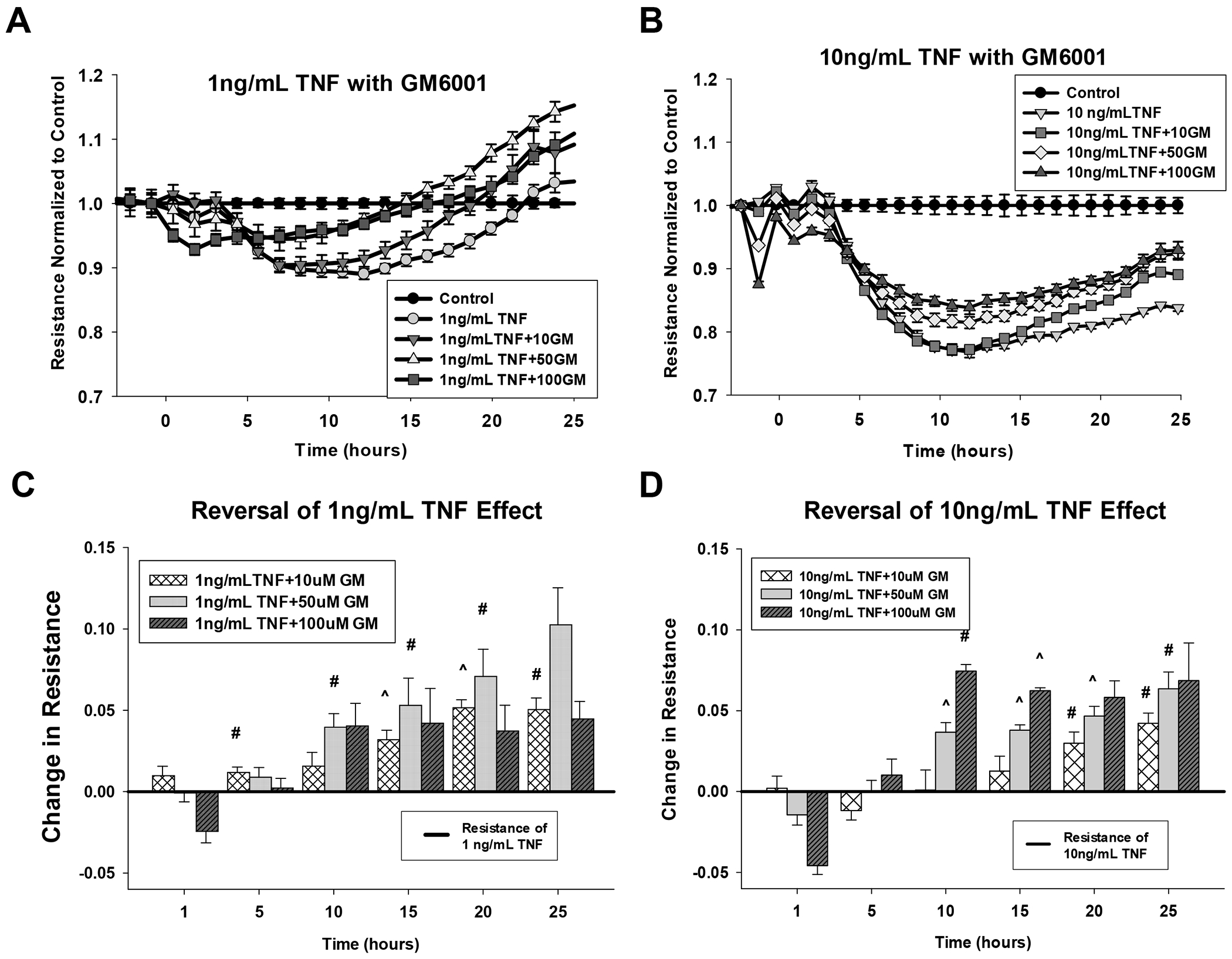
P-UAEC grown to near confluence on specialized 96 well ECIS plates were treated with doses of GM6001 (10, 50, and 100 uM) for 30 min as indicated. The addition of 10 ng/mL TNF-alpha is marked as time 0. Panels A and B show the impact on resistance relative to untreated control wells as above. Panels C and D show the data as incremental recovery with GM6001 relative to the effect of TNF alone. The black line at 0 represents the resistance of cells treated with 1 ng/mL (Panel C) or 10 ng/ml (Panel D) TNF-alpha alone. The bars represent the recovery GM6001 gives at specific times (1, 5, 10, 15, 20, or 25 hr - see methods). Data are means ± SEM from 9 independent experiments with multiple wells per treatment per plate. Significant differences are as indicated (# P<0.05 and ^ P<0.01) compared to TNF-alpha alone.
Discussion
On initial examination, our data confirm our proposal that agents which regulate Src and ERK to acutely close Cx43 within gap junctions on the cell surface are also capable of longer term regulation of junctional proteins that hold cells together and provide the microenvironment in which Cx43 operates at the cell surface. Clearly both TPA (a widely used control in studies of wounding responses in which Cx43 is intimately involved) and the inflammatory cytokine TNF (also involved in wounding responses in vivo) promote a rapid and sustained breakdown of P-UAEC monolayers. This breakdown can also be partially blocked/rescued by inhibitors of Src and MEK/ERK. What is most interesting however is that (i) VEGF is not equally as effective on monolayer breakdown long term, even though it can close Cx43 via Src and ERK within 1 hr, and (ii) while PP2 and U0126 can help protect UAEC against the action of these agents, they cannot fully protect cells against such actions. There are other physiologically interesting observations that stand out, including (i) 0.1 ng/ml TNF is stimulatory to monolayer cohesion, (ii) the collective finding that many processes are involved in the control of junction protein levels and distribution in these cells, and (iii) that control of ZO-1 and VE-Cad junctional proteins are clearly distinct from control of Cx43.
Beginning with the relative lack of effect of VEGF compared to TNF and TPA on the monolayer, some clues why this is the case were described in our recent paper on Cx43 phosphorylation and closure (Ampey et al., 2019). We previously found that activation of Src and ERK in P-UAEC by VEGF is not sustained, and this seems to be true of growth factors in other endothelial cell models. In contrast, TNF activation of Src is sustained, and activation of Src can cross-activate the MEK/ERK pathway (Ampey et al,. 2019). So while transient activation of Src or ERK can close Cx43 communication, the monolayer itself is under the control of other inflammatory mediators including TNF that mount more sustained responses. Such dependency on TNF action long term is interesting because it also relates to the physiologic concentration range. In most pregnant individuals, the circulating concentration of TNF is reported in the low pg/ml range, and is slightly elevated relative to healthy nonpregnant subjects (Redman & Sargent 2003). As such our data suggests the mild elevation of circulating TNF in normal pregnancy would actually support a healthy, intact, and tightly coupled monolayer. It is important to note that absolute quantification is difficult given circulating TNF is relatively unstable, so it is not clear what the true concentration is at the surface of endothelial cells. Furthermore, in hypertensive states including PE, further local elevation through the 0.1-1 ng/ml range may well occur in the local vessel wall environment due to immune cell attachment/local immune cell invasion, or in the decidual/uteroplacental space where TNF may be secreted locally. Such local secretion in PE subjects would cause local TNF to easily rise beyond 0.1 ng.ml where our data suggests loss of monolayer cohesion will begin to occur and present in PE subjects as edema. Thus our dose dependency data does support a role for local TNF above the normal range as a mediator of the negative effects of PE in vivo.
Given that TNF treatment can degrade the monolayer, the next question is how? The answer is clearly ‘at multiple levels’. We know from studies in other endothelial and non-endothelial cells that junctional proteins ZO-1 and VE-Cad can be tagged by phosphorylation through Src or ERK, and such tagged proteins can then be internalized from the plasma membrane. What happens next depends on the cell and associated signaling processes. In simple terms, proteins can be reprocessed for recycling through the outer Golgi complex to return to the plasma membrane, or they can be further tagged for destruction in the lysozome. It should be noted that Src in particular can also stimulate the expression of new junctional proteins by stimulating new or further gene expression. The balance of recycling or destruction of existing plasma membrane proteins vs new protein synthesis provides a powerful and sophisticated regulatory system that can cause the level of junctional proteins in the plasma membrane to dramatically increase or decrease in a short period of time (Wettschureck et al., 2019). In addition, there is also the possibility of protein destruction from the outside of the cell. Membrane-spanning proteins such as VE-Cad are known targets for MMPs (Antalis et al., 2016), and we can clearly see that shed VE-Cad is a product of TNF action, but not VEGF, in P-UAEC. It is also clear from studies in other cells (Sondergaardy et al., 2009) that Src can mediate the increased expression and secretion of MMPs, so the one agonist, TNF, could achieve all these responses in a way VEGF cannot.
Regarding the exact mechanism at play in P-UAEC, we needed both ICC and protein quantification in media and lysate to dissect the likely general mechanisms. Starting with ZO-1 as the simpler example, the action of TNF but not VEGF to reduce the ZO-1 level in cells, and the ability of PP2 more than U0126 to block this effect is entirely consistent with TNF activation of Src being necessary to tag ZO-1 for internalization (Wettschureck et al., 2019). The small but significant elevation of staining inside the plasma membrane (‘cytosolic’) may reflect the recycling of such protein through the Golgi complex, or may reflect protein destruction and compensatory resynthesis of new protein - our experimental design cannot distinguish these two events.
The story for VE-Cad is more complicated, and not only due to the added destruction as shed VE-Cad. Unlike ZO-1, the level of VE-Cad does not drop significantly in response to TNF at 18 hr, but the co-addition of PP2 in particular does increase the level determined by ICC by almost two fold to achieve full rescue. It is highly unlikely given the clear increase in shed VE-Cad that TNF is having no effect alone and so we must ask how the level can be maintained in the face of ongoing destruction. Our data that PP2 and U0126 cause a dramatic increase in staining of the plasma membrane and inside the cell is consistent with substantial ongoing basal protein synthesis and delivery, which is supported by the fact VE-Cad is reduced 90% in the presence of CHX protein synthesis inhibitor, even in the absence of any stimulus. So TNF alone appearing to have no effect on levels of VE-Cad simply means its rapid removal from the plasma membrane is balanced by rapid delivery of new and possibly also recycled protein to counterbalance VE-Cad destruction by MMP action. Indeed this cell is always geared to maintain VE-Cad in the face of even basal extracellular MMP degradation, and TNF makes those compensatory processes work even harder.
With regard to Cx43 placement in the plasma membrane, while TPA and TNF (but not VEGF) were able to redistribute Cx43 back into the cell, the relatively poor rescue effect of PP2 and U0126 on plasma membrane staining suggest other kinases may play a more prominent role. This again is consistent with the very different life cycle of Cx43 (Solan & Lampe, 2014) in its membrane placement and removal compared to other junctional proteins such as VE-Cad and ZO-1 (Wettschureck et al., 2019).
Looking at the data as a whole, we can also ask what the causal relationship is between the levels of individual junctional proteins and the decline in monolayer resistance in response to TNF. Superficially it would be tempting to just say that the resistance is mostly a consequence of changes in VE-Cad, but this is not entirely the case. Healthy endothelium requires both tight cell-cell coupling and Cx43 insertion to make functional gap junctions. The consensus in the Cx43 field is that ZO-1 has particular significance because its PDZ domains can bind directly to Cx43. Alterations of interaction of ZO-1 with Cx43 in this way can impact Cx43 plaque size in the plasma membrane, and this can be influenced by multiple kinases (Solan & Lampe, 2014). This would infer that Cx43 can only exist in tight junctions, but experimental observation says that is not always true. First, tight junctions and adherens junctions have been reported to overlap and functionally influence each other (Wettschureck et al., 2019). Secondly, Cx43 is also observed in adherens junctions (Dianati et al., 2016). So while there is a great deal of data regarding the relationship of Cx43 with tight junctions, that does not mean they cannot exist in adherens junctions, or that these two junctional assemblies are exclusive of each other in any given region of the plasma membrane. A mechanistic explanation of this has recently been proposed by Campbell et al. (2017) who point out that ZO-1 can bind to proteins in both complexes. The existence of colocalized adherens and tight junctions may thus be a sign of the cell being in a state of longer term stability and the stimuli to promote internalization and degradation of ZO-1 is largely minor or absent. As such, more extensive junctional macro assemblies can be formed. In the event of adverse cytokine exposure, they are destroyed by ERK or Src mediated ZO-1 tagging and internalization, and so colocalization is lost. It should be noted that VE-Cad does also undergo additional dramatic and dynamic changes in turnover in response to TNF relative to ZO-1, but this also does not have a dramatic impact on % colocalization of VE-Cad with ZO-1. The additional substantial role for extracellular proteases in mediating the fall in monolayer resistance via extracellular degradation of VE-Cad does explain this, and this is verified by the clear evidence that P-UAEC are capable of making a variety of MMP transcripts and corresponding proteins (MMP1, 3, 9, 12 and 13 in particular), and the rescue ability of the general MMP inhibitor GM6001 on both monolayer resistance and release of shed VE-Cad in response to TNF. Of clinical relevance, these same MMP isoforms are also associated with hypertension in general (Wang & Khalil, 2017) and PE in particular (Chen & Khalil, 2017).
So where does this leave us in considering treatments for endothelial failure in PE? In cases that are mild and present as a transient episode of hypertension that is reasonably responsive to anti-hypertensives, then the likely endothelial failure is a transient loss of gap junction function and strategies directed at re-opening Cx43 channels themselves may be effective in cases where smooth muscle relaxants (L-type voltage gated Ca2+ channel blockers) are not enough. But in more severe cases, and particularly with added edema, we may be seeing evidence that monolayer cohesion is being lost in a way that no longer offers tight and adherens junctional protein complexes sufficient to hold cells close enough together to allow stable Cx43 gap junctions to achieve optimal functional coupling. If the loss of abundant and particularly colocalized VE-Cad and ZO-1 is counter to Cx43 plasma membrane insertion and optimal functional coupling between cells, then recovery of pregnancy-enhanced vasodilation will not be possible until junctional protein placement, and ideally colocalization in the plasma membrane is restored. Future therapy may only be possible if we address the chronic inflammatory ‘wounding’ processes that otherwise destabilize the monolayer in this way.
In summary, and in response to our initial question, while in the short term (1 hr) VEGF and TNF are both effective inhibitors of Cx43 gap junction function through the actions of Src and ERK (Boeldt et al., 2015; Ampey et al., 2019), the longer-term action of TNF reported here is distinct from that of VEGF on monolayer integrity because of its more sustained ability to activate Src and ERK. TNF directly impacts both tight and adherens junctions in our cells. While both ZO-1 and VE-Cad may both be regulated in their internalization and trafficking as well as possible destruction inside the cell, VE-Cad is also subject to significant destruction by the action of extracellular proteinases, most likely metalloproteinases. Our findings are consistent with known junction regulatory mechanism in other endothelial and non endothelial cells. Of particular note, Cx43 may not be a major regulator of cell monolayer integrity, but it is intimately associated with both tight and adherens junctions. As such our proposal TNF may not only act to regulate Cx43 channel activity by direct and reversible phosphorylation through Src and ERK, but may also act longer term to actively disrupt the local environment in which Cx43 is found in the plasma membrane is supported by this study. As such, the actions of TNF can explain the loss of vasodilatory capacity and loss of monolayer integrity seen in subjects with PE, and Src and ERK are clearly involved. To that end, further study of the action of TNF on maternal endothelial function and dysfunction is warranted as well as the impact of other ‘wounding’ hormones that may synergize with TNF through the same and complementary signaling pathways. Such studies are now underway in this laboratory.
Supplementary Material
Highlights:
In this manuscript we investigate how TNF but not VEGF may impact on junctional assemblies in uterine artery endothelial cells. Our data supports the argument that cytokines are more damaging than growth factors in loss of vasodilation in the hypertensive condition, preeclampsia. We demonstrate the multiple mechanisms through which TNF causes such damage, and discuss how this may guide much needed new therapy in future.
Funding:
This work was supported by NIH P01 award HD038843. This paper reports partial fulfillment of the requirements for ACA and RLD towards a PhD in the University of Wisconsin Endocrinology and Reproductive Physiology Training Program. ACA and RLD were also supported by a T32 predoctoral training grant (T32-HD41921) from NICHD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Ampey AC, Boeldt DS, Clemente L, Grummer MA, Yi FX, Magness RR, Bird IM 2019. TNF-alpha inhibits pregnancy-adapted Ca2+ signaling in uterine artery endothelial cells. Mol Cell Endocrinol. 488, 4–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antalis TM, Conway GD, Peroutka RJ, and Buzza MS 2016. Membrane-anchored proteases in endothelial cell biology. Curr Opin Hematol. 23(3), 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird IM, Boeldt DS, Krupp J, Grummer MA, Yi FX, Magness RR 2013. Pregnancy, programming and preeclampsia: gap junctions at the nexus of pregnancy-induced adaptation of endothelial function and endothelial adaptive failure in PE. Curr Vase Pharmacol. 11(5), 712–29. [DOI] [PubMed] [Google Scholar]
- Boeldt DS, Grummer MA, Yi FX, Magness RR, Bird IM 2015. Phosphorylation of Ser-279/282 and Tyr-265 positions on Cx43 as possible mediators of VEGF-165 inhibition of pregnancy-adapted Ca2+ burst function in ovine uterine artery endothelial cells. Mol Cell Endocrinol. 412, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeldt DS, Krupp J, Yi FX, Khurshid N, Shah DM, Bird IM 2017. Positive versus negative effects of VEGF165 on Ca2+ signaling and NO production in human endothelial cells. Am J Physiol Heart Circ Physiol. 312(1), H173–H81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell HK, Maiers JL, DeMali KA 2017. Interplay between tight junctions & adherens junctions. Experimental Cell Research. 358, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Khalil RA 2017. Matrix Metalloproteinases in Normal Pregnancy and Preeclampsia. Progress in Molecular Biology and Translational Science 48, 87–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianati E, Poiraud J, Weber-Ouellette A, Plante I 2016. Connexins, E-cadherin, Claudin-7 and β-catenin transiently form junctional nexuses during the postnatal mammary gland development. Developmental Biology 416, 52–68. [DOI] [PubMed] [Google Scholar]
- Redmana CWG, Sargent IL. 2003. Pre-eclampsia, the Placenta and the Maternal Systemic Inflammatory Response—A Review. Placenta (2003), 24, Supplement A, Trophoblast Research, Vol. 17, S21–S27. [DOI] [PubMed] [Google Scholar]
- Solan JL, Lampe PD 2014. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 588(8), 1423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondergaardy BC, Schultzy N, Madseny SH, Bay-Jenseny AC, Kassemz M and Karsdaly MA 2010. MAPKs are essential upstream signaling pathways in proteolytic cartilage degradation e divergence in pathways leading to aggrecanase and MMP-mediated articular cartilage degradation. Osteoarthritis and Cartilage 18, 279–288. [DOI] [PubMed] [Google Scholar]
- Wang X, Khalil RA 2017. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Advances in Pharmacology 81, Ch8, 241–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettschureck N, Strilic B, Offermanns S 2019. Passing the vascular barrier: endothelial signaling processes controlling extravasation. Physiol Rev 99, 1467–1525. [DOI] [PubMed] [Google Scholar]
- Yi FX, Boeldt DS, Gifford SM, Sullivan JA, Grummer MA, Magness RR, and Bird IM 2010. Pregnancy Enhances Sustained Ca2+ Bursts and eNOS Activation in Ovine Uterine Artery Endothelial Cells Through Increased Connexin 43 Function. Biol Reprod. 82(1), 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi FX, Boeldt DS, Magness RR, Bird IM 2011. [Ca2+]i signaling vs. eNOS expression as determinants of NO output in uterine artery endothelium: relative roles in pregnancy adaptation and reversal by VEGF165. AJP Heart Circ Physiol. 300(4), H1182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.