

Abstract
Epstein-Barr Virus (EBV) was the first discovered human tumor virus and is the etiological agent of B cell lymphomas and also epithelial cancers. Indeed, nearly 10% of gastric cancers worldwide are EBV-positive and display unique molecular, epigenetic, and clinicopathological features. EBV-positive gastric cancers display the highest rate of host genome methylation of all tumor types studied and harbor recurrent mutations activating PI3Kα, silencing ARID1A, and amplifying PD-L1. While EBV infection of B cells can be studied efficiently, de novo epithelial cell infection is much more difficult. We propose that new culture models including 3D-based gastric organoids and xenografts can bring new insight into EBV-induced gastric carcinogenesis and will lead to improved precision medicine-based therapies for patients with EBV-positive gastric cancer.
Keywords: Epstein-Barr virus, EBV, gastric cancer, PI3K, methylation, immunotherapy
Epstein-Barr Virus and cancer
Epstein-Barr Virus (EBV) is a ubiquitous human herpesvirus present in approximately 95% of the population worldwide [1]. The virus infects through the oral epithelium where it primarily replicates and produces viral progeny. Primary infection often occurs during early childhood and is asymptomatic, though delayed exposure can result in acute infectious mononucleosis in young adults [2]. Viral particles infect naïve B cells and establish latency mimicking normal B-cell maturation through a germinal center-like reaction [3,4]. EBV persists as a lifelong infection in the memory B cell pool and while latent infection is usually asymptomatic, immunosuppression or concurrent HIV infection can lead to the development of B-cell malignancies [1]. EBV was first discovered in 1964 in the biopsy of a Burkitt lymphoma tumor, making it the first virus shown to cause cancer in humans [5]. Since then, EBV has been linked to Hodgkin’s Lymphoma, post-transplant lymphoproliferative disease, diffuse large B-cell lymphoma and T-cell and NK-cell lymphomas [3]. Additionally, EBV is present in several epithelial cell cancers including approximately 90% of nasopharyngeal carcinoma (NPC) cases and 8% of gastric carcinoma (GC) cases [1,4,6]. In total, EBV-related cancers account for approximately 1.8% of the global cancer death burden [7].
EBV de novo infection has been extensively studied in B cells where upon initial infection, viral latency proteins promote uncontrolled B-cell proliferation leading to transformation into lymphoblastoid cell lines (LCLs). Infected B cells express virally-encoded latent proteins in a state known as ‘latency III’. The EBV-encoded latency proteins include six EBV nuclear antigens (EBNAs 1, 2, 3A, 3B, 3C, and -LP) and three latent membrane proteins (LMPs 1, 2A, and 2B). Upon initial infection of B cells, EBNA-LP and EBNA2 are expressed and these transcriptional activators turn on the other EBNA genes, and ultimately the LMPs, most of which are crucial for immortalization of B cells in vitro [8]. Additionally, non-coding RNAs are expressed including two non-coding RNAs, EBER1 and EBER2, as well as 44 short miRNAs transcribed from the BamHI A rightward transcripts and the BHRF1 locus, called the BART and BHRF1 miRNAs, respectively [1]. During latency III, and initial B cell infection, all latency proteins and non-coding RNAs are expressed. In contrast, other tumor types as well as latently-infected cells in asymptomatic carriers display more restricted gene expression patterns. For example, EBV-positive Hodgkin’s Lymphomas express a ‘latency IIa’ phenotype, which is EBNA1, LMP1, LMP2A, and the non-coding RNAs. Infected memory B cells switch between ‘latency 0’ where no latent proteins are expressed and ‘latency I’ where EBNA1 is the only protein expressed in order to evade immune recognition [1]. Most EBV-positive endemic Burkitt Lymphoma B cells and also EBV-positive gastric cancer cells also display latency I, where EBNA1 (from a distinct viral promoter called Qp), the EBERs, and the BART miRNAs are the only latency genes expressed [4]. As detailed below, some gastric cancers also express LMP2A.
EBV-associated Gastric Cancer (EBVaGC)
Gastric cancer (GC) is the fourth most common cancer and the second leading cause of cancer deaths worldwide with a median overall survival of less than one year [9]. Poor prognosis is often due to diagnosis at an advanced stage of disease. In many parts of the world where gastric cancer is common and the population is at a high risk, there is no regular screening process due to the invasive nature of diagnostic methods [10]. Treatment is usually surgical resection of the tumor combined with chemotherapy [9]. The majority of gastric cancer cases are associated with Helicobacter pylori, a class I carcinogen that infects approximately half of the world’s population [11]. However, H. pylori is not the only infectious agent that has been shown to be involved in the development of GC. EBV was first detected in a lymphoepithelioma gastric tumor in 1990 via polymerase chain reaction and has since been detected in approximately 8% of GC cases worldwide [12,13]. EBV-positive tumors are identified using EBER in situ hybridization, though this screening is not regularly performed during diagnosis of the tumor [14]. Although only ~8% of GC cases are EBV positive, this cancer represents the largest global death burden of all EBV-positive cancers accounting for nearly 70,000 deaths from 1990–2010 [7]. While gastric cancer is generally more common in Asian countries, EBV-positive tumors are not specifically associated with any geographic region. Nevertheless, Asian countries represent the largest global death burden of EBV-associated malignancies [7,9].
EBVaGC displays a restricted latency I gene expression program where only a few crucial genes are expressed including EBNA1, the BART miRNAs, EBERs and occasionally LMP2A. EBNA1 is responsible for tethering EBV episomes to host chromosomes and is expressed in all latency programs and all infected cells. EBNA1 can also act as a transcriptional regulator and bind to viral promoters including its own promoter resulting in self-regulation [1]. The BART miRNAs are highly expressed in latency I neoplasms and epithelial tumor xenografts and can promote tumor growth in vivo [15]. Additionally, it has been shown that the BART miRNAs can prevent apoptosis through interactions with pro-apoptotic proteins of the BH3 only protein family and promote epithelial cell survival [16–18]. LMP2A, which is expressed in approximately 50% of epithelial tumors, can upregulate DNA methyltransferase 1 (DNMT1) and increase CpG island methylation of tumor suppressor promoters [19]. Monoclonal EBV episomes in the tumors indicate that EBV infection occurs early in tumor development, and the restricted latency program suggests that there are likely additional oncogenic factors that play a role in tumor development. Sub-populations of epithelial cells may be pre-malignant, or more susceptible to EBV infection and transformation due to prior genetic perturbations [4,20].
A recent study by The Cancer Genome Atlas Research Network (TCGA) identified four distinct subtypes of gastric cancer using genotypic, phenotypic, and clinicopathological features. This study described EBV-positive tumors as a distinct subtype of gastric cancer in comparison to EBV-negative cancers which were divided into the additional three subtypes (Fig. 1 and [12]). When genomic profiles of the gastric cancer subtypes were compared, EBV-positive tumors displayed a distinct mutational profile including high mutation rates of PIK3CA and ARID1A, findings that have been validated by multiple other groups [12,21–23]. Additionally, EBV-positive tumors displayed extreme DNA hypermethylation of tumor suppressor promoter regions within a broad CpG island methylator phenotype (CIMP) of the host genome as well as within the viral genome, which is commonly observed [12].
Figure 1: Key features of EBV-associated gastric cancer.

EBV-positive gastric cancers are molecularly, pathologically and epigenetically distinct from EBV-negative gastric cancers. EBV-associated gastric tumors display an extreme CpG island hypermethylator phenotype (CIMP), and are the most hypermethylated of all tumor types. EBV-positive tumors display an 80% rate of PIK3CA activating mutation and 55% rate of ARID1A silencing mutation which frequently co-occurs with PTEN inactivation. Tumors are made up of EBV-positive gastric epithelial cells that up-regulate PD-L1 and densely infiltrating lymphocytes contributing to an inflammatory microenvironment. EBV infected gastric cells display an extremely restricted latency profile where only a few crucial proteins are expressed.
Along with a unique genomic profile, EBV-positive tumors have a characteristic histologic appearance. EBV-positive tumors are most commonly found in the fundus or body of the stomach, though they can arise in any anatomical compartment of the stomach [12]. EBV-positive tumors are either lymphoepithelioma-like carcinomas (LELC), similar to NPC, or poorly to moderately differentiated diffuse type with intense lymphoid infiltration of CD8- and CD4-positive T lymphocytes and CD68-positive macrophages [10]. Given that both gastric and NPC tumors are made up of poorly differentiated epithelial cells, it has been proposed that EBV infects either stem cells or poorly differentiated epithelial cells and prevents further differentiation leading to monoclonal EBV-positive tumors with stem-like properties [24]. These infected cells in patients with atrophic gastritis or regional inflammation confer a survival advantage to pre-malignant cells, which then acquire additional mutations and CIMP leading to tumor development [24]. While this is the most widely accepted hypothesis in the field, it has been difficult to demonstrate the ordering of oncogenic hits in gastric cancer due to the lack of an efficient model system in which to study EBV infection of gastric epithelial cells and tumorigenesis.
Mutation profile and distinct molecular features
EBV-positive gastric tumors display an 80% rate of activating PIK3CA mutations compared to approximately 20% of all gastric cancers independent of EBV status [12]. The PI3K/mTOR/AKT intracellular signaling pathway plays a crucial role in cell proliferation, survival, differentiation, and metabolism (Fig. 2). The upstream component of this pathway, PI3K, is comprised of an 85kDa regulatory subunit which stabilizes the 110kDa catalytic subunit, called p110α for PI3Kα, encoded by the PIK3CA gene [25]. PIK3CA is the second most commonly mutated gene in cancer following TP53, and mutations often occur at one of three hotspots, E542K, E545K, or H1047R [26]. These hotspots are located in exons 9 and 20 of PIK3CA corresponding with the helical and kinase domains of the PI3Kα protein, respectively. EBVaGC tumors display recurrent somatic mutations dispersed throughout the PI3Kα protein, including at hotspot residues [12]. Although PIK3CA mutant tumors do not show a different prognosis than PIK3CA wild type tumors, cellular dependence on the PI3K/mTOR/AKT pathway represents a potential target for therapeutics. PI3Kα is a well-characterized therapeutic target in multiple cancers including breast cancer, non-small cell lung carcinoma, lymphomas, and head and neck squamous cell carcinoma [27]. Small molecules that target components of the PI3K/mTOR/AKT pathway have been approved for a number of these other cancers, and several of these have entered clinical trials for gastric cancer as well [28].
Figure 2: PI3K pathway signaling and drug treatments.
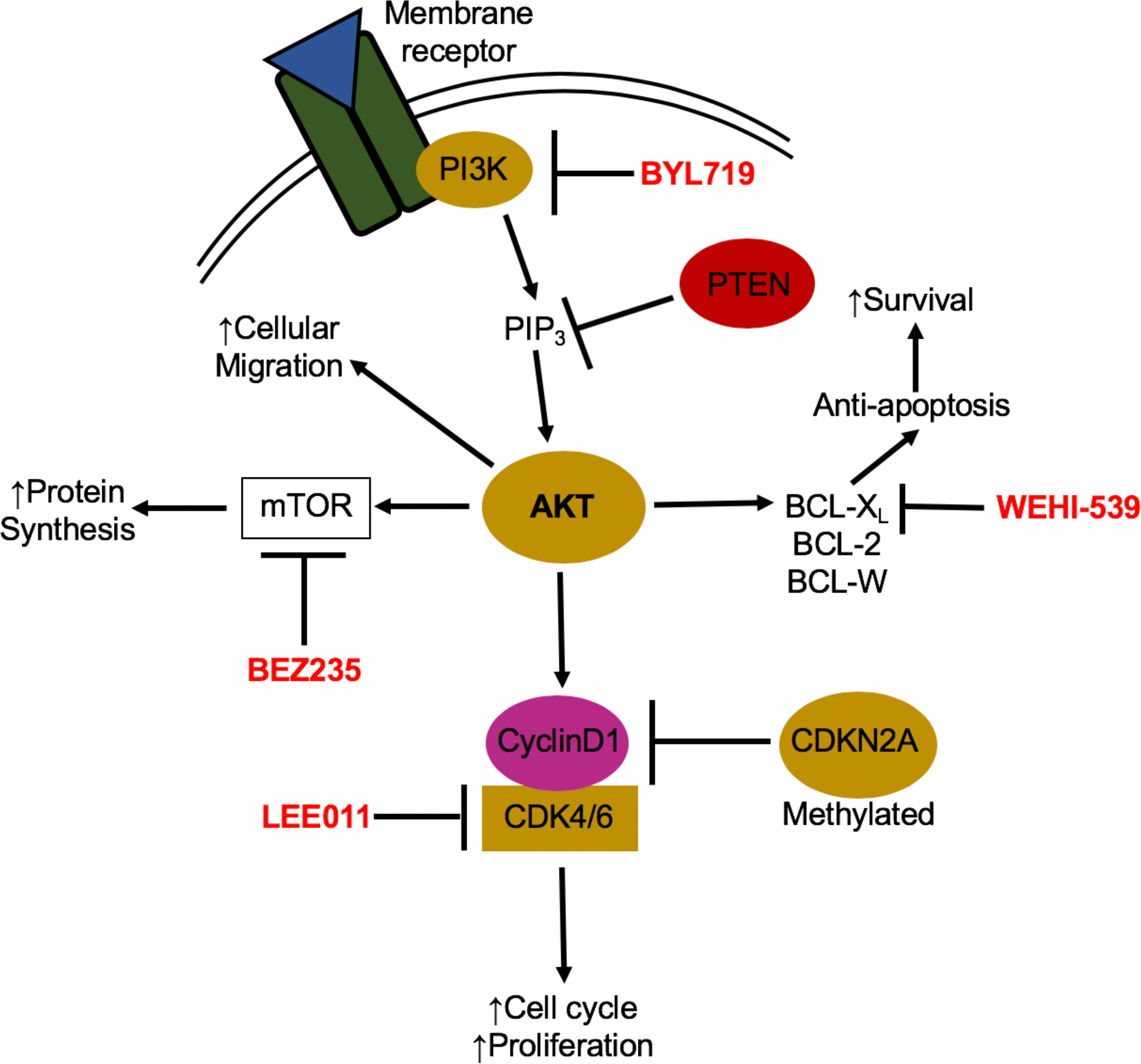
Activation of the PI3K/AKT signaling pathway occurs through binding of an external stimuli to a cellular receptor such as a receptor tyrosine kinase, cytokine receptor or G-coupled protein receptor. Mutations in the catalytic domain of PI3K, PIK3CA, can lead to constitutive activation of the pathway resulting in increased proliferation, protein synthesis, and cell survival and migration. When an external stimulus binds to its appropriate cellular receptor, PI3K is recruited to the intracellular domain of the receptor and phosphorylates PIP2 to PIP3. PIP3 then activates AKT leading to a number of downstream protein functions including activation of cyclin D1, mTOR and BCL-xL. There are a number of developed drugs that target different members of the PI3K/AKT pathway and have been shown to be effective in PIK3CA mutant breast cancers and can potentially be used in PIK3CA mutant EBV-positive gastric cancers as well.
Activation of the PI3K/mTOR/AKT pathway also occurs in other EBV-associated malignancies including NPC and Burkitt Lymphoma suggesting that EBV may play a role in pathway activation to promote survival of infected cells. LMP2A activates PI3K signaling and provides pro-survival signals in B cells and human foreskin keratinocytes [29,30]. In the case of NPC, activation of the PI3K/mTOR/AKT pathway occurs due to chromosomal amplification and increased transcription of PIK3CA rather than activating mutations. PIK3CA amplification in NPC is associated with increased metastasis and advanced tumor stage leading to poor prognosis [31].
EBV-positive gastric tumors additionally harbor a 55% rate of silencing ARID1A mutations [12]. The ARID1A (AT-rich interactive domain-containing protein 1A) gene encodes a non-catalytic subunit of the Switch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex and has been shown to be silenced in a number of cancers including ovarian cancer, Burkitt lymphoma, endometroid cancer and EBV-associated gastric cancer [32–35]. Thus, ARID1A is emerging as a crucial tumor suppressor due to the broad range of cancers in which it is silenced. ARID1A silencing has been linked to poor prognosis in gastric tumors as well as increased cell migration and invasion and enhanced proliferation [36]. Importantly, restoration of ARID1A expression significantly decreased gastric cancer cell proliferation and tumor growth, while silencing ARID1A in wild-type cells increased proliferation [33,37]. Loss of ARID1A expression is common in early stage gastric cancers, and to date EBV infection has not been found to promote silencing of ARID1A expression in vitro [22]. It is possible that mutational silencing of ARID1A occurs prior to EBV infection of epithelial cells or comes about via an alternate mechanism. ARID1A silencing mutations is also present in nearly 25% of Burkitt Lymphoma tumors [34]. Loss of ARID1A protein expression is common in precursor lesions of ovarian carcinoma as well as esophageal adenocarcinomas leading to questions regarding how early stage ARID1A loss contributes to tumorigenesis [35].
There is an increasing body of evidence that concurrent mutation of ARID1A and PIK3CA can contribute to the development of ovarian cancer and gastric cancer through the complementary interactions between ARID1A and the PI3K/mTOR/AKT pathway [35,36]. Concurrent loss of ARID1A and PTEN, a tumor suppressor downstream of PI3K, induced tumor formation and epithelial hyperplasia in a mouse model of ovarian cancer [32]. Furthermore, ARID1A silencing is associated with PI3K activation in gastric adenocarcinoma [37]. While there is evidence for pathway crosstalk in tumor cells, it remains to be determined when ARID1A loss, PI3K activation, and EBV infection occur in the development of EBVaGC, strongly providing rationale for the generation of robust experimental models of this disease.
Epithelial cells are typically the site for lytic replication of EBV rather than long-term latent infection as is the case for B cell infection. Normal nasopharyngeal epithelial cells are unable to maintain the EBV episome and viral latency and it is has been proposed that a premalignant cellular genotype is necessary for the development of EBV-associated epithelial tumors [38]. Nasopharyngeal epithelial cells that display specific genetic alterations including CDKN2A (p16) deletion and cyclin D1 overexpression occur prior to EBV infection leading to a premalignant cell capable of sustaining latent infection [39]. Gastric epithelial cells with prior oncogenic mutations such as activating PIK3CA mutation or ARID1A loss may be more likely to survive or inhibit lytic replication and maintain latent EBV infection. Additionally, it is possible that either abortive lyitc or primarily latent EBV infection is the primary oncogenic hit conferring a slight survival advantage to those cells infected. The infected cells may subsequently acquire mutations in PIK3CA, ARID1A or other genes that ultimately result in EBVaGC tumor outgrowth [24].
Aberrant hypermethylation and epigenetic abnormalities
EBV-positive gastric tumors display the highest rate of genome-wide DNA methylation compared to all other tumor types and tissues resulting in what is called an extreme CpG island hyper-methylator phenotype (CIMP) [12]. CpG dinucleotides are densely present in promoter regions of approximately half of all genes. These regions are called CpG islands and are generally unmethylated to allow for downstream gene transcription. In contrast, EBVaGC tumors display CIMP with aberrant hypermethylation causing gene silencing of tumor suppressor genes (Fig. 3b and c) [40]. Multiple groups have completed genome-wide sequencing and identified methylation sensitive genes, where hypermethylation is present throughout the promoter region and within +2kb of the transcriptional start site (TSS) [41]. Notably, CDKN2A, which encodes p14 and p16, is hypermethylated and silenced in all EBV-positive gastric tumors [12]. Silencing of CDKN2A leads to cyclin D/CDK4 activation and increased proliferation, thus contributing to epithelial carcinogenesis (Fig. 2). Hypermethylation is also present throughout the EBV genome and methylation of EBV lytic promoters Z and R, encoding BZLF1 and BRLF1 immediate early lytic proteins respectively, is important in establishing latency in nasopharyngeal epithelial cells [42]. Induction of methylation programs to ensure proper control of viral gene expression may result in the inhibition of tumor suppressors and contribute as an early event in gastric carcinogenesis [24].
Figure 3: Mechanism of hypermethylation in EBV-associated gastric cancer.

CpG islands located upstream of the coding region are normally non-methylated to allow for transcription, but are methylated by DNA-methyltransferases (DNMTs) to control gene transcription and expression. EBV infection of gastric epithelial cells induces hypermethylation by taking advantage of existing cellular methylation mechanisms. A) Cytosine bases are methylated on the 5-position carbon by DNMTs, changing 5-C to 5mC. Methylated cytosines can be oxidized by ten-eleven translocation (TET) family enzymes changing 5mC to 5hmC. B) CpG islands are located throughout promoter regions and surrounding the transcription start sites (TSS) of genes. CpG islands are normally un-methylated to allow for transcription of genes including tumor suppressors such as PTEN and cell cycle genes including CDKN2A. C) Virally encoded LMP2A can phosphorylate transcription factor STAT3, which moves to the nucleus and activates transcription of DNMT1 leading to induction of hypermethylation. Additionally, LMP2A inhibits TET2 oxidation of methylated cytosines to maintain hypermethylation phenotype.
EBV infection of the low methylation gastric epithelial cell lines GES1 and MKN7 induced de novo hypermethylation comparable to the CIMP observed in gastric cancer tissue [43]. Moreover, a time course following infection indicates that both the host and viral genome methylation occurs within a month post-infection, providing some evidence that hypermethylation is in fact an early event in tumorigenesis [43,44]. Other work supports the model where the EBV-induced hypermethylator phenotype is sustained long-term and also that cells maintain epigenetic alterations induced by EBV even after losing the viral episome [45]. EBV-induced epigenetic alterations may therefore be permanent and potentially heritable between cell generations. This suggests that EBV may establish latent infection in an epithelial cell, creating a so-called ‘founder clone’, which will proliferate and pass on the hypermethylator phenotype to daughter cells creating a monoclonal population of heavily methylated EBV-positive tumor cells. [24,46].
EBV-infected tumor cells display an extremely restricted latency gene expression pattern leading to questions regarding the mechanism by which viral infection induces hypermethylation and contributes to tumorigenesis. The Kaneda group attempted to answer this question by introducing each latent gene expressed during latency I (EBNA1, EBER1, EBER2, LMP2A and BARF0) individually, in order to assay whether a single gene product may be responsible for the induction of methylation. Expression of no single latency gene replicated the phenotype; however, additional studies support a role for the viral protein LMP2A (Fig. 3c) [43]. The Fukayama group found that LMP2A expression in stomach epithelial cells induced phosphorylation and activation of the transcription factor STAT3 leading to upregulation of DNA methyltransferase 1 (DNMT1) [19]. DNMT1 is a member of the DNMT family of enzymes responsible for DNA methylation and gene silencing during mammalian development. DNMTs function by adding a methyl group to the 5-position of cytosine leading to 5-methylcytosine (5mC) [47]. The up-regulation of DNMT1 during EBV infection of gastric epithelial cells may thus lead to increased CpG island methylation resulting in CIMP seen in EBV-positive tumors (Fig. 3) [19]. EBV has also been shown to repress ten-eleven translocation (TET) family enzymes during infection of gastric epithelial cells [48]. TET family enzymes are demethylases that oxidize 5mC to 5-hydroxymethylcytosine (5hmC), which is a key part of the demethylation pathway (Fig. 3a) [49]. RNA-seq of the low methylation cell line MKN7 following infection with EBV showed a significant decrease of TET family enzyme expression levels, especially TET2. Repression of TET2 was shown to be caused by seven host miRNAs as well as viral latent proteins LMP2A and BARF0, giving LMP2A another role in induction of hypermethylation during infection (Fig. 3c) [48]. TET enzymes also demethylate the lytic Z and R promoters in nasopharyngeal epithelial cells resulting in lytic reactivation. Through this mechanism, EBV-mediated TET repression may also play an important role in promoting maintenance of latency in epithelial cells [42].
While LMP2A may play a role in the induction of hypermethylation through DNMT1 up-regulation and TET2 repression, expression of LMP2A in EBV-positive gastric tumors is heterogenous and only occurs in approximately 40% of EBVaGC cases [50]. It is possible that early LMP2A expression may establish the CIMP program, but LMP2A expression is lost in more advanced tumors. Alternatively, additional viral mechanisms may be responsible for the hypermethylation phenotype. It has been shown that the BART miRNAs play an important role in the maintenance of latency and survival of epithelial cells through inhibition of pro-apoptotic protein expression [51,52]. To date, BART miRNA expression and induction of the hypermethylation phenotype has not been extensively studied. However, BART knockout viral strains can be used to infect gastric epithelial cells to study induction of hypermethylation in the absence of the miRNAs. Expression of the miRNAs is highly variable in gastric epithelial cells; however, miRNA expression remains higher in EBVaGC than that observed in EBV-infected B cells in vitro [53,54]. This suggests that EBV miRNAs may play an important role in both epithelial cell survival as well as de novo methylation in EBVaGC.
Histopathology and the Immune Response
EBV-associated gastric tumors contain monoclonal EBV-positive populations of gastric epithelial cells and dense infiltration of CD4- and CD8-positive T cells and CD68-positive macrophages [55]. T lymphocytes are not normally present in healthy gastric tissue, however local or regional inflammation or atrophic gastritis can trigger the development of mucosal-associated lymphoid tissue (MALT) in the submucosa of the stomach. Gastric inflammation can be caused by many sources, although the most common is infection with H. pylori. MALT is secondary lymphoid tissue that contains memory B cells, CD4+ T helper cells, and CD8+ cytotoxic T cells [56]. It is thought that the memory B cells present in MALT may harbor latent EBV that could be reactivated leading to infection of epithelial cells in the gastric mucosa [24]. MALT-associated CD8+ T cells and macrophages contribute to the inflammatory microenvironment through the release of cytokines including interferon-γ (IFNγ), interleukin(IL)-23, IL-12, and IL-27 [12]. IL-23 and IL-12 are structurally similar and drive proliferation of effector T cells that release IFNγ in response to viral infection. The IFNγ/IL-12/IL-23 pathway remains active through autocrine signaling, contributing to maintenance of the inflammatory microenvironment, which is important early in tumor development driven by EBV infection [24,57].
EBV-positive gastric epithelial cells display amplification of chromosome 9p24.1, leading to overexpression of programmed death ligand 1 and 2 (PD-L1/2) and janus kinase 2 (JAK2) [12,58]. Up-regulation of PD-L1 is also common in other viral malignancies including diffuse large B-cell lymphoma (DLBCL), NPC and adult T-cell leukemia/lymphomas [59–61]. PD-L1 over-expression has been shown to be involved in tumor immune cell evasion through interaction with its cellular receptor PD-1, expressed on tumor infiltrating T cells. It is thought that overexpression of PD-L1 on the EBV-positive tumor cells contributes to anti-tumor immunity and attenuation of the cytotoxic response of infiltrating CD8+ T cells abundant in EBV-positive gastric tumors [18]. For that reason, PD-L1 overexpression is thought to be involved in the progression of EBVaGC as an early tumorigenic event [58]. EBV-positive gastric tumors with a microenvironment dominated by IFNγ signaling and over expression of PD-L1 may make strong candidates for checkpoint inhibitor therapies including PD-1 blockage [58,62,63].
Culture Models: 2D and 3D models of infection
Epithelial cells lack the EBV B-cell receptor, CD21, and there are several proposed modes by which epithelial infection can occur in vivo independent of CD21 binding [50]. Due to the absence of CD21, in vitro infection of epithelial cells is inefficient, whereas EBV easily infects and establishes latency in B cells. EBV infection of B cells in vitro is highly successful despite the fact that only 10–15% of viral genomes reach the nucleus while a large percentage of genomes remain bound to the surface of the B cell [64]. It has been hypothesized that those surface bound genomes allow for a B cell-mediated method of infecting epithelial cells. An in vitro method of infection has thus been developed using B cells as a transfer vehicle with which to infect epithelial cells showing an 800-fold increase in infection efficiency compared to using a cell free method of infection (Fig. 4b) [65]. Multiple groups have taken advantage of this technique to study the biology of EBV infection of diverse epithelial cells including nasopharyngeal carcinoma cells, keratinocytes, and gastric carcinoma cells [66–69]. The inefficiency of in vitro infection of epithelial cells with EBV has made it difficult to find cell lines that can sustain latent infection, and only a few of gastric epithelial cell lines have been studied extensively. To date, the most commonly studied EBV-negative gastric cell lines include GES1, a SV40 transformed fetal gastric epithelial cell line, and AGS and MKN7, both derived from gastric adenocarcinomas [40,43,70]. These cell lines have been used to study de novo methylation and epigenetic alterations, miRNA and latent gene expression and cellular outgrowth following infection [41,43,44,46,71].
Figure 4: Culture model systems in which to study tumorigenesis and potential therapies for EBVaGC.

EBV does not readily infect and establish latency in epithelial cells in vitro and multiple culture model systems are in development to allow for study of EBVaGC tumorigenesis both in vitro and in vivo. A) Naturally occurring EBV-positive GaCa-derived cell lines can be treated with PI3K pathway drugs shown to be effective in other solid tumors and cellular growth is assayed via growth inhibition assays and xenograft tumor volume and growth. B) Using the co-culture method, EBV negative GaCa cell lines can be infected with rEBV and selected for using drug resistance and GFP expression. In vitro infected cells can also be treated with PI3K pathway drugs to assay growth and cell death. Additionally, EBV infected cell lines can be sequenced to understand induction of hypermethylation and genomic and transcriptomic changes that occur during infection. C) Stem cells can be differentiated into gastric organoids or directly infected to study proliferation and survival of differentiated cell types of the stomach.
Recently, ephrin receptor A2 (EphA2) was identified as a potential epithelial cell receptor for EBV infection. EphA2 is important for fusion and entry of EBV into epithelial cells and it was shown that over-expression of EphA2 increased epithelial cell infection while a shEphA2 or CRISPR/Cas9 knockout decreases infection rates [72,73]. The discovery of this receptor means that additional gastric cell lines can be infected and used to further research EBVaGC tumor development and outgrowth. While 2D cell line infection models can be useful, 3D culture systems can potentially provide a more physiologically accurate model of infection and cellular response to therapy in the stomach. This is primarily due to the presence of diverse cell types and crypt architecture, which allows for cell-to-cell interactions not present in monolayer culture systems.
Traditionally, mouse xenograft models have been the gold standard of 3D culture. The EBV-positive gastric cancer cell lines KT and SNU719 established from primary tumors have been successfully transplanted into SCID mice and nude mice, respectively. The resulting xenograft tumors were EBV-positive as determined by EBER in situ hybridization and displayed the latency I viral gene expression profile as seen in the primary tumors [74,75]. The successful establishment of mouse xenograft models provides an in vivo system in which to study EBVaGC. However, mouse models can be costly due to specific breeding requirements and the need for isolated facilities and full-time technicians. In the past decade, in vitro stem cell-derived organoids have gained popularity as an emerging 3D culture model in which to study human tissue development, stem cell behavior and disease progression [76–78].
Recently, the Clevers group at the Hubrecht Institute discovered populations of multipotent Lgr5+ve stem-cells present in the base of gastric crypts, responsible for the long-term renewal of stomach tissue in vivo. These adult gastric stem cells can be grown in matrigel in vitro with specific growth factors and develop into long-lived, self-renewing pyloric gastric organoids (Fig. 4c) [79]. Additionally, the Wells group at the Cincinnati Children’s Hospital has developed a protocol to differentiate human induced pluripotent stem cells (iPSCs) into fully developed antral gastric organoids [76,80,81]. Gastric organoids are currently being used to study host-pathogen interactions following infection with Helicobacter pylori and rotavirus, though very little research is being done with EBV [78,80,82]. A majority of EBVaGC and NPC cases are poorly differentiated cancers and it has been hypothesized that less differentiated cells, such as stem cells or those in the process of differentiation, may be more likely to become latently infected with EBV and develop into EBV-positive tumors. Adult gastric stem cells are located throughout the isthmus and base regions of the gastric gland and consistently replicate, differentiate, and replace other cell types as needed [78]. The organoid model is an effective system in which to identify undifferentiated cell populations that are most susceptible to latent EBV infection and track monoclonal outgrowth of infected cells. Organoid models can be used to better understand how EBV infection and proliferation is occurring during tumor development, and how EBV infected cells are interacting with other gastric cell types.
Therapeutics and translational research
Gastric cancers usually have a poor prognosis due to diagnosis at an advanced stage leading to a 5-year overall survival rate of less than 30% [9]. Most commonly, treatment is surgical resection combined with chemotherapy. Currently, EBV-positive tumors are not treated differently than EBV-negative tumors. However, given the distinct characteristics of EBV-positive tumors, it is possible that patient specific treatments can be developed to target affected oncogenic pathways. The field of cancer therapeutics has changed dramatically with the improvement of precision medicine and development of small molecule inhibitors [83]. PI3K inhibitors are being developed due to the overwhelming number of cancers with common PI3K activating mutations [27]. Pan-PI3K as well as PI3KCA specific inhibitors show promise in treatment of solid tumors with PIK3CA mutation and a newly developed PI3KCA inhibitor, BYL719, is currently in clinical trials for gastric cancer, breast cancer and neuroendocrine cancers (Fig. 2) [84–86]. However, recent work from the Wood lab at Duke has shown that solid breast cancer tumors displaying PIK3CA activating mutations cannot be effectively targeted with single agents and must be treated with a combination of PI3K pathway drugs. PIK3CA mutant tumors often depend on several different pro-survival mechanisms and thus single agent inhibition is ineffective as tumor cells shift survival dependence to a new pathway, conferring drug resistance. By targeting additional members of the PI3K/mTOR/AKT pathway such as BCL2 and BCL-XL, or proteins involved in cell cycle regulation such as CDK4/6, solid tumors can be sensitized to apoptosis through combination drug therapies [87–89]. Small molecule inhibitors such as WEHI-539, BEZ235 and LEE011, which target BCL-XL, PI3K and mTOR, and CDK4/6 respectively, have shown promise treating PIK3CA mutant solid breast tumors [87,88]. Given that 80% of EBV-positive gastric tumors display activating PIK3CA mutations, these PI3K pathway drug combinations should be assessed in this setting (Fig. 4a).
EBV-positive gastric cancers may also be good candidate tumors for PD-1 blockade due to an inflammatory microenvironment dominated by IFNγ signaling [58]. In normal tissue, PD-L1 expression is important during inflammation, and functions as a control mechanism to prevent damage to normal body tissue. In tumor tissue, T cells express PD-1 and bind to PD-L1 on cancer cells, inhibiting tumor cell killing through cytotoxic T-cell effector functions (Fig. 5a) [63]. Neutralizing antibodies that bind the PD-1 receptor or PD-L1 allow tumor infiltrating T cells to recognize and kill tumor cells via tumor antigen and major histocompatibility complex (MHC) interactions (Fig. 5b) [90]. Several anti–PD-1 and anti–PD-L1 antibodies are currently being tested in clinical trials for cancers including DLBCL, NPC, ovarian cancer and advanced gastric adenocarcinoma [63]. EBV-positive tumors are strong candidates for PD-1 targeted therapies due to the dense and homogenous infiltration of CD8+ T cells and inflammatory microenvironment of these tumors [18,58,62,63].
Figure 5: PD-1 blockade therapy for EBV-associated gastric tumors.

Disruption of PD-1/PD-L1 interactions between gastric tumor cells and infiltrating CD8+ T cells can be an effective therapy for the treatment of EBV-positive gastric cancers due to overexpression of PD-L1 and a IFNγ dominated inflammatory microenvironment. A) PD-L1 on tumor cells is recognized by PD-1 expressed by cytotoxic T cells preventing normal T cell effector functions even if the TCR is presented with a tumor specific antigen. Through this interaction, the tumor cells can evade the immune response. B) Anti-PD-1 antibodies can be used to prevent binding of PD-L1 to PD-1 allowing the CD8 T cells to recognize tumor cells through antigen presentation and perform effector functions to kill tumor cells.
Adoptive T-cell immunotherapies may also be viable for EBVaGC. Autologous cytotoxic T cells (CTL) expanded on EBV transformed B cells expressing all latency proteins can be curative in PTLD, a latency III neoplasm [91,92]. However, EBV-positive gastric tumors only express EBNA1, which is not strongly displayed to CD8 T cells, albeit somewhat to CD4 T cells, and occasionally LMP2A. Those tumors with LMP2A expression may be candidates for EBV-CTL therapy.
In addition to the development of targeted drug therapies specific to the molecular landscape of a patient’s tumor, new screening methods are being developed that can provide accurate early diagnosis of a cancer. Over 80% of gastric cancers are diagnosed during the middle to late stage of disease highlighting a need to improved screening techniques [93]. Circulating tumor DNA (ctDNA) and miRNAs can provide information about the molecular landscape of a tumor to begin treatment with specific drugs without needing an invasive tumor biopsy [93,94]. Both ctDNA and miRNAs can be detected in the peripheral blood due to normal necrosis and apoptosis of tumor cells using a minimally invasive procedure called a liquid biopsy. Liquid biopsies can be taken throughout treatment allowing for consistent monitoring of tumor progression and adjustments to therapy methods as needed. ctDNA screening depends on specifically methylated genes to determine presence of a certain tumor. Due to the extreme methylation phenotype in EBV-positive tumors, ctDNA may provide an efficient method for earlier diagnosis and precision treatment options as discussed above [94].
Executive Summary:
EBV and cancer:
EBV was the first human tumor virus discovered in the biopsy of a Burkitt Lymphoma patient in 1964.
Since its discovery, EBV has been linked to Hodgkin’s Lymphoma, post-transplant lymphoproliferative disorder (PTLD), NK and T cell lymphomas, gastric cancer, and nasopharyngeal carcinoma.
EBV-associated cancers account for approximately 1.8% of cancer deaths worldwide.
EBV is a human herpesvirus that latently infects over 95% of the adult population, and is typically asymptomatic, though it can reactivate and cause cancer in immunosuppressed patients.
EBV-positive gastric cancer:
5–10% of gastric cancers worldwide show monoclonally infected EBV-positive gastric epithelial cells indicating that EBV infection is an early event that contributes to tumor formation.
EBV-positive tumors display an extremely restricted latency pattern entitled latency I, where only a key few viral proteins are expressed.
A TCGA study on gastric adenocarcinomas showed that EBV-positive cancers are molecularly, epigenetically, and pathologically distinct from EBV-negative tumors.
Mutational profile and distinct molecular features:
EBV-positive tumors display an 80% activating mutation rate of PIK3CA and a 55% silencing mutation rate of ARID1A.
PIK3CA is a known oncogene that is the second most mutated gene in cancer following p53.
ARID1A is a member of the SWI/SNF chromatin-remodeling complex that is silenced in a number of additional cancers leading to its emergence as a potent tumor suppressor.
PIK3CA and ARID1A mutations have been shown to occur simultaneously in a number of cancers and there is increasing evidence showing pathway crosstalk between these proteins.
It is unknown whether PIK3CA and ARID1A mutation occurs prior to or following EBV infection and how the order of oncogenic events contributes to tumorigenesis.
Epigenetic abnormalities and aberrant methylation:
EBV-positive tumors display an extreme CpG island methylator phenotype and are the most heavily methylated tumor genomes of all cancers.
EBV has been shown to induce de novo methylation that is long-lived even after cells have lost the viral episome.
The viral latency protein LMP2A may play a role in the induction of methylation through interactions with DNMT1 and TET family enzymes though LMP2A is not expressed in an EBV-positive gastric tumors.
It is possible that other viral mechanisms are playing a role in hypermethylation.
Histopathology and the immune response:
EBV-positive tumors are made up of monoclonal EBV-positive gastric epithelial cells, and infiltrating CD8+ and CD4+ T cells and macrophages.
Infiltrating lymphocytes contribute to an inflammatory tumor microenvironment through release of interferonγ and are considered tissue resident memory T cells due to expression of αEβ7 integrin that binds to E-cadherin on epithelial cells.
Gastric tumor cells display over-expression of PD-L1, which is thought to attenuate CD8 effector activity and contribute to immune evasion.
Culture models: 2D and 3D models of infection:
EBV infection of epithelial cells is inefficient and to date, only a couple cell lines have been successfully infected and characterized.
Organoid culture models are emerging as a way to study host-pathogen interactions in vitro that are more accurate than typical cell culture models, and more accessible than traditional mouse models.
Organoid cultures can be grown from adult gastric stem cells or induced pluripotent stem cells, and both approaches have advantages and disadvantages in the study of EBV infection of the gastric epithelium.
Translation:
The advancement of precision medicine in cancer has allowed for patient specific therapies that can be potentially more effective than traditional surgery and chemotherapy.
Using the specific features of EBVaGC, combination drug therapies can be developed to specifically target affected oncogenic pathways including the PI3K/mTOR/AKT pathway, and interactions between PD-L1 and PD-1.
Better screening methods such as sequencing and analysis of circulating tumor DNA and microRNAs can provide faster diagnosis and increase patient survival through more accurate therapies.
Future Directions:
Since the discovery of EBV’s association with gastric cancer, many studies have been published that characterize distinct features of EBV-positive tumors in comparison to EBV-negative tumors [12,23,95]. The development of methods for infection of epithelial cells in culture has increased the extent of research and in vitro experiments to study the role of EBV in tumorigenesis [67,71]. To date, most work has been done with a few gastric cancer cell lines and a single Burkitt Lymphoma-derived strain. As culture models continue to develop and improve, many additional gastric epithelial cell lines can be infected in vitro in order to query effects of the virus on cell proliferation, survival, gene expression and hypermethylation (Fig. 4b). There is an emerging body of work examining diversity of viral strains focused primarily on B-cell infections and whole genome sequencing [96]. While whole genome sequencing analysis of EBVaGC strains is well underway, in vitro epithelial cell infection with diverse viral strains is still in progress [95,97]. An NPC-derived strain of EBV, M81, is considered to be epitheliotropic and is therefore more likely to establish latent infection in epithelial cells. There are several gastric cancer-derived strains of the virus that may be more likely to display phenotypes upon latent infection of epithelial cells relative to analogous experiments with Burkitt lymphoma-derived strains [98], but that remains to be seen. Querying the role of the BART lncRNA, miRNAs, and other gene products within the context of gastric cancer-associated strains will provide clear evidence of the roles of these molecules in tumorigenesis and other salient phenotypes found in EBVaGC.
Advances in sequencing technology have allowed for extensive profiling of the induction of hypermethylation following EBV infection. Whole genome and whole methylome sequencing can provide insight into the mechanisms of EBV-induced hypermethylation including which genes may be responsible, and when epigenetic changes are occurring during the infection process [41,43,48]. The EBV genome is also methylated during infection and it is thought that hypermethylation of the EBV genome can control the expression of latency proteins and regulate lytic reactivation of the virus [14,42]. It is likely that hypermethylation of both the host and viral genomes plays a key role during epithelial cell infection, though precisely what mechanistic role is still unclear and needs to be further studied.
EBV-positive gastric cancers account for approximately 50% of all deaths associated with EBV malignancies and improved therapeutic methods are necessary for affected patients [7]. Currently, gastric tumors are not routinely screened for EBV status prior to starting treatment. As we understand more about the distinct features of EBV-positive gastric cancer, it is crucial to screen tumors for EBV to identify patients who are most likely to respond to specific therapies designed for those with EBV-associated cancers. Additionally, early diagnostic methods can improve patient prognosis and provide information about the distinct features of a patient’s tumors. New methods using circulating tumor DNA (ctDNA) and miRNAs have been developed for a number of cancers and can be beneficial for gastric cancers as well. [93]. For example, a minimally invasive liquid biopsy can be used for detection of CpG methylation markers in ctDNA that differ from normal methylation patterns [94]. This method can be especially useful for EBVaGC given the hypermethylator phenotype seen in these tumors. The continuing advancements of precision medicine can bring new treatment options for patients with a specific molecular tumor profile related to EBV-tumor status. Combination drug therapies can be developed using information known about treating other solid tumors. New therapies need to be tested in mouse models and clinical trials before being brought into practice. Patient-derived xenograft models and traditional cell line-derived xenografts can provide optimal models in which to study EBVaGC therapies and interactions with the immune system. Gastric organoid systems can provide an additional system in which to assay proliferation of EBV infected gastric epithelial cells and effect of EBV infection on cellular differentiation.
Acknowledgements:
We thank our colleagues Drs. Katie Garman and David Hsu for reading and editing the manuscript and Patrick Tan and Nisha Padmanabhan for discussions and collaboration on EBVaGC. We acknowledge support from a Duke Cancer Institute/Nicholas School for the Environment Pilot Award and a Duke/Duke-NUS Pilot Award for studies of EBVaGC.
References:
- 1.Young LS, Yap LF & Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat. Rev. Cancer 16, 789–802, (2016). [DOI] [PubMed] [Google Scholar]
- 2.Niedobitek G, Agathanggelou A, Steven N & Young LS. Epstein-Barr virus (EBV) in infectious mononucleosis: detection of the virus in tonsillar B lymphocytes but not in desquamated oropharyngeal epithelial cells. Mol. Pathol 53, 37–42, (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price AM et al. Epstein-Barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young LS & Dawson CW. Epstein-Barr virus and nasopharyngeal carcinoma. Chin. J. Cancer 33, 581–590, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epstein M, Achong B & Barr Y Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1, (1964). [DOI] [PubMed] [Google Scholar]
- 6.Ko YH. EBV and human cancer. Exp. Mol. Med 47, e130, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan G & Hashim MJ. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agent Cancer 9, 38, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price AM & Luftig MA. Dynamic Epstein-Barr virus gene expression on the path to B-cell transformation. Adv. Virus Res 88, 279–313, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carcas LP. Gastric cancer review. J. Carcinog 13, 14, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukayama M & Ushiku T Epstein-Barr virus-associated gastric carcinoma. Pathol. Res. Pract 207, 529–537, (2011). [DOI] [PubMed] [Google Scholar]
- 11.Wroblewski LE, Peek RM Jr. & Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin. Microbiol. Rev 23, 713–739, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Research, N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209, (2014).** Identification and characterization of four subtypes of gastric cancer including EBV-positive gastric cancer. EBV-positive gastric cancers display CIMP, amplification of PD-L1, activating mutations of PIK3CA and silencing mutations of ARID1A.
- 13.Burke AP, Yen TS, Shekitka KM & Sobin LH. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod. Pathol. 3, 377–380, (1990). [PubMed] [Google Scholar]
- 14.Nishikawa J et al. Epstein-barr virus in gastric carcinoma. Cancers (Basel) 6, 2259–2274, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu J, Smith P, Leahy L & Thorley-Lawson DA. The Epstein-Barr virus encoded BART miRNAs potentiate tumor growth in vivo. PLoS Pathog. 11, e1004561, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang D, Skalsky RL & Cullen BR. EBV BART MicroRNAs Target Multiple Pro-apoptotic Cellular Genes to Promote Epithelial Cell Survival. PLoS Pathog. 11, e1004979, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marquitz AR, Mathur A, Nam CS & Raab-Traub N The Epstein-Barr Virus BART microRNAs target the pro-apoptotic protein Bim. Virology 412, 392–400, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giudice A et al. Role of Viral miRNAs and Epigenetic Modifications in Epstein-Barr Virus-Associated Gastric Carcinogenesis. Oxid. Med. Cell Longev 2016, 6021934, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hino R et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 69, 2766–2774, (2009).*Expression of LMP2A in EBVaGC tumors plays a role in induction of methylation through activation of DNMTs leading to silencing of tumor supressors.
- 20.Imai S et al. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc. Natl. Acad. Sci. U S A 91, 9131–9135, (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boger C et al. Epstein-Barr virus-associated gastric cancer reveals intratumoral heterogeneity of PIK3CA mutations. Ann. Oncol 28, 1005–1014, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abe H et al. ARID1A expression loss in gastric cancer: pathway-dependent roles with and without Epstein-Barr virus infection and microsatellite instability. Virchows Arch. 461, 367–377, (2012). [DOI] [PubMed] [Google Scholar]
- 23.Wang K et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet 46, 573–582, (2014). [DOI] [PubMed] [Google Scholar]
- 24.Fukayama M, Kunita A & Kaneda A Gastritis-Infection-Cancer Sequence of Epstein-Barr Virus-Associated Gastric Cancer. Adv. Exp. Med. Biol 1045, 437–457, (2018). [DOI] [PubMed] [Google Scholar]
- 25.Martini M, De Santis MC, Braccini L, Gulluni F & Hirsch E PI3K/AKT signaling pathway and cancer: an updated review. Ann. Med 46, 372–383, (2014). [DOI] [PubMed] [Google Scholar]
- 26.Karakas B, Bachman KE & Park BH. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 94, 455–459, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massacesi C et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco. Targets Ther 9, 203–210, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chivu-Economescu M et al. New therapeutic options opened by the molecular classification of gastric cancer. World J. Gastroenterol 24, 1942–1961, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Portis T & Longnecker R Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 23, 8619–8628, (2004). [DOI] [PubMed] [Google Scholar]
- 30.Morrison JA, Klingelhutz AJ & Raab-Traub N Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J. Virol 77, 12276–12284, (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fendri A et al. PIK3CA amplification is predictive of poor prognosis in Tunisian patients with nasopharyngeal carcinoma. Cancer Sci. 100, 2034–2039, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan B et al. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J. Natl. Cancer Inst 106, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang DD et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLoS One 7, e40364, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love C et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet 44, 1321–1325, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu RC, Wang TL & Shih Ie M The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther 15, 655–664, (2014).*ARID1A mutation occurs frequently in cancer and has a role in tumor suppression. Additionally, mutations of ARID1A and PIK3CA co-occur in many cancers.
- 36.Yan HB et al. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis 35, 867–876, (2014). [DOI] [PubMed] [Google Scholar]
- 37.Zang ZJ et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet 44, 570–574, (2012). [DOI] [PubMed] [Google Scholar]
- 38.Tsang CM et al. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 33, 549–555, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsang CM et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. U S A 109, E3473–3482, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsusaka K, Funata S, Fukayama M & Kaneda A DNA methylation in gastric cancer, related to Helicobacter pylori and Epstein-Barr virus. World J. Gastroenterol 20, 3916–3926, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Funata S et al. Histone modification alteration coordinated with acquisition of promoter DNA methylation during Epstein-Barr virus infection. Oncotarget 8, 55265–55279, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wille CK et al. 5-hydroxymethylation of the EBV genome regulates the latent to lytic switch. Proc. Natl. Acad. Sci. U S A 112, E7257–7265, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsusaka K et al. Epstein-Barr virus infection induces genome-wide de novo DNA methylation in non-neoplastic gastric epithelial cells. J. Pathol 242, 391–399, (2017).**The authors show that EBV induces methylation of the host genome following de novo infection of gastric epithelial cells. Methylation of the host genome is completed within a month following infection.
- 44.Matsusaka K et al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 71, 7187–7197, (2011). [DOI] [PubMed] [Google Scholar]
- 45.Birdwell CE et al. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J. Virol 88, 11442–11458, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Queen KJ, Shi M, Zhang F, Cvek U & Scott RS. Epstein-Barr virus-induced epigenetic alterations following transient infection. Int. J. Cancer 132, 2076–2086, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin B & Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp. Med. Biol 754, 3–29, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Namba-Fukuyo H et al. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget 7, 81512–81526, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohli RM & Zhang Y TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iizasa H, Nanbo A, Nishikawa J, Jinushi M & Yoshiyama H Epstein-Barr Virus (EBV)-associated gastric carcinoma. Viruses 4, 3420–3439, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giudice A et al. Role of Viral miRNAs and Epigenetic Modifications in Epstein-Barr Virus-Associated Gastric Carcinogenesis. Oxidative Medicine and Cellular Longevity, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lo AK, Dawson CW, Jin DY & Lo KW. The pathological roles of BART miRNAs in nasopharyngeal carcinoma. J. Pathol 227, 392–403, (2012). [DOI] [PubMed] [Google Scholar]
- 53.Marquitz AR, Mathur A, Edwards RH & Raab-Traub N Host Gene Expression Is Regulated by Two Types of Noncoding RNAs Transcribed from the Epstein-Barr Virus BamHI A Rightward Transcript Region. J. Virol 89, 11256–11268, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shinozaki-Ushiku A et al. Profiling of Virus-Encoded MicroRNAs in Epstein-Barr Virus-Associated Gastric Carcinoma and Their Roles in Gastric Carcinogenesis. J. Virol 89, 5581–5591, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cho J, Kang MS & Kim KM. Epstein-Barr Virus-Associated Gastric Carcinoma and Specific Features of the Accompanying Immune Response. J. Gastric Cancer 16, 1–7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cesta MF. Normal structure, function, and histology of mucosa-associated lymphoid tissue. Toxicol. Pathol 34, 599–608, (2006). [DOI] [PubMed] [Google Scholar]
- 57.Pirhonen J, Matikainen S & Julkunen I Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J. Immunol 169, 5673–5678, (2002). [DOI] [PubMed] [Google Scholar]
- 58.Saito R et al. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1(+) immune cells in Epstein-Barr virus-associated gastric cancer: the prognostic implications. Mod. Pathol 30, 427–439, (2017). [DOI] [PubMed] [Google Scholar]
- 59.Zhou Y et al. PD-L1 predicts poor prognosis for nasopharyngeal carcinoma irrespective of PD-1 and EBV-DNA load. Sci. Rep 7, 43627, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gravelle P et al. Mechanisms of PD-1/PD-L1 expression and prognostic relevance in non-Hodgkin lymphoma: a summary of immunohistochemical studies. Oncotarget 8, 44960–44975, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miyoshi H et al. PD-L1 expression on neoplastic or stromal cells is respectively a poor or good prognostic factor for adult T-cell leukemia/lymphoma. Blood 128, 1374–1381, (2016). [DOI] [PubMed] [Google Scholar]
- 62.Derks S et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget 7, 32925–32932, (2016).*EBV-positive gastric epithelial cells display amplification of PD-L1 while CD8 T cells high in surface PD-1 infiltrate the tumor.
- 63.Alsaab HO et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 8, 561, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shannon-Lowe C et al. Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J. Gen. Virol 86, 3009–3019, (2005). [DOI] [PubMed] [Google Scholar]
- 65.Imai S, Nishikawa J & Takada K Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human epithelial cells. J. Virol 72, 4371–4378, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shannon-Lowe C & Rowe M Epstein-Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell-mediated transfer infection. PLoS Pathog. 7, e1001338, (2011).**Resting B cells can be used as a transfer vehicle to increase infection of epithelial cells 800 fold. Diverse epithelial cells can be infected with EBV and develop latent infection.
- 67.Shannon-Lowe CD, Neuhierl B, Baldwin G, Rickinson AB & Delecluse HJ. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. U S A 103, 7065–7070, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsao SW et al. The biology of EBV infection in human epithelial cells. Semin. Cancer Biol 22, 137–143, (2012). [DOI] [PubMed] [Google Scholar]
- 69.Tsang CM et al. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: regulation of infection and phenotypic characterization. Int. J. Cancer 127, 1570–1583, (2010). [DOI] [PubMed] [Google Scholar]
- 70.Ke Y, Ning T & Wang B [Establishment and characterization of a SV40 transformed human fetal gastric epithelial cell line-GES-1]. Zhonghua Zhong Liu Za Zhi 16, 7–10, (1994). [PubMed] [Google Scholar]
- 71.Shannon-Lowe C et al. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J. Virol 83, 7749–7760, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen J et al. Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol 3, 172–180, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang H et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol 3, 1–8, (2018). [DOI] [PubMed] [Google Scholar]
- 74.Iwasaki Y et al. Establishment and characterization of a human Epstein-Barr virus-associated gastric carcinoma in SCID mice. J. Virol 72, 8321–8326, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oh ST, Cha JH, Shin DJ, Yoon SK & Lee SK. Establishment and characterization of an in vivo model for Epstein-Barr virus positive gastric carcinoma. J. Med. Virol 79, 1343–1348, (2007). [DOI] [PubMed] [Google Scholar]
- 76.McCracken KW et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mahe MM et al. Establishment of Gastrointestinal Epithelial Organoids. Curr. Protoc. Mouse Biol 3, 217–240, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pompaiah M & Bartfeld S Gastric Organoids: An Emerging Model System to Study Helicobacter pylori Pathogenesis. Curr. Top. Microbiol. Immunol 400, 149–168, (2017). [DOI] [PubMed] [Google Scholar]
- 79.Barker N et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 6, 25–36, (2010).**The discovery of Lgr5 as a marker of gastric stem cells led to isolation of gastric glands and differentiation of gastric organoids from adult gastric stem cells.
- 80.Finkbeiner SR et al. Stem cell-derived human intestinal organoids as an infection model for rotaviruses. MBio 3, e00159–00112, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Munera JO & Wells JM. Generation of Gastrointestinal Organoids from Human Pluripotent Stem Cells. Methods Mol. Biol 1597, 167–177, (2017).**Protocol that outlines the pathway to differentiate iPSCs into gastric organoids.
- 82.Schlaermann P et al. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut 65, 202–213, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoelder S, Clarke PA & Workman P Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol. Oncol 6, 155–176, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mayer IA et al. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-Specific Inhibitor, with Letrozole in ER+/HER2− Metastatic Breast Cancer. Clin. Cancer Res 23, 26–34, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nolting S et al. The selective PI3Kalpha inhibitor BYL719 as a novel therapeutic option for neuroendocrine tumors: Results from multiple cell line models. PLoS One 12, e0182852, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh SS et al. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol 21, 12261–12273, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anderson GR et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci. Transl. Med 8, 369ra175, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vora SR et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26, 136–149, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fritsch C et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther 13, 1117–1129, (2014).*The PIK3CA inhibitor BYL719 can be used as a therapeutic for mutant tumors.
- 90.Tran PN, Sarkissian S, Chao J & Klempner SJ. PD-1 and PD-L1 as emerging therapeutic targets in gastric cancer: current evidence. Gastrointest. Cancer 7, 1–11, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Craddock J & Heslop HE. Adoptive cellular therapy with T cells specific for EBV-derived tumor antigens. Update Cancer Ther 3, 33–41, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kanakry JA & Ambinder RF. EBV-related lymphomas: new approaches to treatment. Curr. Treat. Options Oncol 14, 224–236, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu HS & Xiao HS. MicroRNAs as potential biomarkers for gastric cancer. World J. Gastroenterol 20, 12007–12017, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu RH et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Mater 16, 1155–1161, (2017). [DOI] [PubMed] [Google Scholar]
- 95.Borozan I, Zapatka M, Frappier L & Ferretti V Analysis of Epstein-Barr Virus Genomes and Expression Profiles in Gastric Adenocarcinoma. J. Virol 92, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsai MH et al. The biological properties of different Epstein-Barr virus strains explain their association with various types of cancers. Oncotarget 8, 10238–10254, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tan P & Yeoh KG. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 149, 1153–1162 e1153, (2015). [DOI] [PubMed] [Google Scholar]
- 98.Kanda T, Furuse Y, Oshitani H & Kiyono T Highly Efficient CRISPR/Cas9-Mediated Cloning and Functional Characterization of Gastric Cancer-Derived Epstein-Barr Virus Strains. J. Virol 90, 4383–4393, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]