

Abstract
Organic electrosynthesis is an increasingly popular tool for driving and probing redox reactions. Recent advances in this field often employ an electrocatalyst to enhance the selectivity and efficiency of electrochemical reactions. A laboratory experiment was developed to introduce students to relevant mechanistic techniques in electrochemistry for analysis of electrocatalytic reactions using aminoxyl-catalyzed alcohol oxidation as a case study. This lab activity employs cyclic voltammetry for qualitative assessment of catalytic turnover prior to introducing students to chronoamperometry, an underutilized technique that facilitates quantitative determination of the rate of catalysis. Students identify and rationalize the important features of reversible electron transfer and a catalytic reaction in a cyclic voltammogram, probe the origin of scan rate effects on these traces, and calculate turnover frequency using a series of chronoamperograms. The method employs safe and readily available reagents: basic aqueous buffer solution, alcohol substrate, and an inexpensive organic aminoxyl catalyst. Student data presented herein were obtained from a course attended by undergraduate students, graduate students, and pharmaceutical chemists.
Keywords: Upper-Division Undergraduate, Graduate Education, Continuing Education, Interdisciplinary, Hands-On Learning, Laboratory Equipment, Electrochemistry, Catalysis
GRAPHICAL ABSTRACT
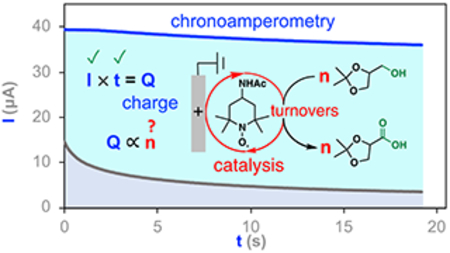
INTRODUCTION
Electrochemistry is one of the oldest and most fundamental methods to promote and analyze redox reactions.1 Recent advances in the electrosynthesis of organic compounds demonstrate that electrochemistry is a powerful tool to access unique reactivity and investigate complex mechanisms.2-8 Investigation of these redox reactions and their coupled chemical reactions can play a valuable role in improving electrochemical reactions and chemical reactions that involve electron transfer and/or redox steps.8 Cyclic voltammetry (CV) is the most widely used electrochemical technique for studying electrode processes.9-12 The flexible time window and forward and reverse scans of CV make it a powerful technique to study the mechanism of reactions that occur at an electrode surface; however, extracting quantitative information from CV data can be complicated.13,14 Chronoamperometry (CA) is another standard electrochemical technique for understanding electrode reaction processes.15 Although CA data contain less mechanistic information than CV data, the former can be more straightforward to analyze than CV data to obtain quantitative information from electrochemical processes.16 CA is comparatively underexplored in instructional electrochemistry curricula and limited to theoretical discussions.17 The present article outlines laboratory experiments to demonstrate how CA represents a versatile complement to CV for analysis of electrochemical reactions.
In a CA experiment, the electrode potential steps from a potential where no electron transfer occurs (Einit) to one at which a faradaic process occurs (Eapp). The resulting current is monitored as a function of time (Figure 1b).18 In the case of an oxidative electrochemical reaction (Figure 1), Eapp is sufficiently more positive than E° (large ΔE is applied) where the rate of electron transfer no longer influences the current.19 In the case of a reductive reaction, Eapp would be more negative than E°.
Figure 1.

The applied potential waveform (a) and the resulting current–time trace (b) for a chronoamperometric experiment of a general oxidation reaction.
The current is controlled (limited) by the flux of the redox-active species to the electrode (Figure 1b) or the kinetics of the reaction that (re)generates the redox-active species.15,20 Under these limiting conditions, extracting quantitative information from the CA for electrode processes is straightforward. When the current is controlled by diffusion, the diffusion coefficient of the electroactive species can be derived based on the Cottrell equation (eqn. 1).
| (Eqn. 1) |
where I is current (amperes, A), n is the number of electrons transferred in the half reaction, F is the Faraday constant (96,485 C mol−1), A is the area of the electrode (in cm2), DA and CA are the diffusion coefficient (cm2 s−1), and the initial (bulk) concentration of electroactive species (mol cm−3), and t is time (s).15,21 When the current is reaction-controlled, quantitative information about the reaction kinetics can be derived by analysis of the amperometric responses. The goal of this experiment is to introduce students to electroorganic chemistry and teach students how to employ CV for mechanistic investigation of catalysis and CA for quantitative analysis of catalytic turnover rates.
2,2,6,6-tetramethylpiperidin-N-oxyl (TEMPO) and its derivatives represent a class of organic radicals (Scheme 1a) known as aminoxyl radicals that find widespread applications as catalysts for the oxidation of organic molecules.22-24 Oxidation of aminoxyl compounds results in formation of the corresponding oxoammonium species (Scheme 1a), which is the reactive form of the catalysts. Oxoammonium oxidizes the substrate, herein alcohol, and undergoes reduction to hydroxylamine (Scheme 1b). The turnover of these catalysts may be accomplished by using stoichiometric oxidants such as oxygen or bleach (Scheme 1b).25,26 Aminoxyl radicals undergo facile redox reactions at electrode surfaces (Scheme 1a).27 Aminoxyl-catalyzed electrochemical oxidation reactions generate hydrogen gas as the sole byproduct of the oxidation reactions and avoid stoichiometric chemical oxidants altogether (Scheme 1c).27-29 Moreover, the electrochemical activity of aminoxyl radicals provide insights into the origin of their reactivity and structure-reactivity correlations.30-33
Scheme 1.

Structures and oxidation reaction of ACT and TEMPO (a). Anelli–Montonari oxidation (b) and TEMPO-catalyzed electrochemical alcohol oxidation (c) with blue arrows showing the catalytic turnover.
In this report we describe a study of electrochemical alcohol oxidation catalyzed by 4-AcNH-TEMPO (ACT). This laboratory exercise introduces students to the interdisciplinary concepts of electroorganic chemistry, electrocatalysis, organocatalysis, and kinetic analysis. In contrast to many undergraduate education resources that focus only on examining direct electron transfer to and from inorganic molecules using CV,12,34 this experiment teaches students to conduct a mechanistic investigation of an electrocatalytic organic reaction. The experiment is well suited for upper level undergraduate laboratory courses, including physical chemistry, organic chemistry, instrumental analysis, and electrochemistry. The procedure has been successfully completed by 16 graduate students and scientists from pharmaceutical companies as part of the “Organic Electrochemistry Short Course” 35 lab section held during summer 2019 and by 12 students in the University of Missouri - Kansas City physical chemistry lab during spring 2020.
EXPERIMENTAL OVERVIEW
A variety of aminoxyl radicals, including TEMPO, are capable of catalyzing alcohol oxidation under both chemical and electrochemical conditions. For this laboratory experiment, the catalyst is the low-cost aminoxyl derivative ACT that has been shown to be one of the most effective aminoxyls for electrochemical oxidation of alcohols.30 Sufficiently basic conditions (pH 8 – 11) are required for fast oxidation of alcohols by oxoammonium. Aqueous carbonate buffer is a suitable reaction medium and the pH can easily be adjusted by changing the ratio of carbonate/bicarbonate. For this experiment, identical quantities of carbonate and bicarbonate are used, resulting in a pH 10 solution29,30 This electrolyte provides simple and safe reaction conditions that avoid the need for more costly organic electrolytes and bases, making this experiment ideal for educational purposes. The substrate 1,2-isopropylideneglycerol (solketal) is a protected form of glycerol and is soluble in the buffered aqueous solution conditions.36,37 The reversible electrode reaction for ACT and its corresponding oxoammonium (ACT+) under these experimental conditions results in a well-defined voltammetric signal (Figure 2, trace a) that may be analyzed by students.38 Electrochemical oxidation of ACT also exhibits an ideal diffusion controlled chronoamperometric response (Figure 2, trace c). The substantial changes in voltammetric (Figure 2, trace b) and chronoamperometric responses (Figure 2, trace d) of ACT in the presence of solketal match diagnostic criteria for electrocatalytic behavior.15 We will show that the enhancement of the oxidative CA current and the additional charge during the CA experiment can be used to derive the turnover frequency (TOF)39 of ACT-catalyzed electrochemical oxidation of solketal. The experimental procedure outlined below was readily completed by students within a three-hour laboratory period. All experiments were performed at ambient temperature (ca. 25 °C).
Figure 2.

Cyclic voltammograms (left) and chronoamperograms (right) of 1.0 mM ACT in the absence (red traces, a and c) and presence of 4 mM solketal (blue traces, b and d). Solution conditions: aqueous solution with NaHCO3/Na2CO3 electrolyte (0.1/0.1 M, pH 10), scan rate for cyclic voltammetry 10 mV s−1, and applied potential for chronoamperometry 0.8 V vs Ag/AgCl.
A typical experiment was carried out in the following manner. After making the buffer solutions, the students conducted an initial voltammetric experiment with a solution of ACT. After observing the oxidation and reduction peaks, the potential range was adjusted for further voltammetric experiments. For the CA experiment, the initial voltage (Einit, cf. Figure 1) should be a value at which no oxidation was observed for the CV experiment, or approximately 200 mV less than oxidation peak potential of ACT. The applied potential to measure the oxidation current (Eapp) should be 100 – 150 mV more positive than the oxidation peak potential, which was determined by the students from the initial CV experiment. CV data at various scan rates and CA data were collected for solutions containing ACT in the absence and presence of various concentrations of solketal. The CA experiment and one CV experiment were then conducted with a buffered solution containing only solketal as a “blank” experiment. The same CA and CV experiments were also performed for ACT in the presence of 4 mM of solketal in a NaCl solution, instead of carbonate/bicarbonate buffer to examine the effect of basic conditions on this catalytic reaction.
EXPERIMENTAL PROCEDURE
The students were given a 0.2 M stock solution of solketal and were asked to prepare the following additional stock solutions: 10 mL of 20 mM ACT solution, 100 mL of 0.2 M NaHCO3 solution, 100 mL of 0.2 M Na2CO3 solution, and 50 mL of 0.2 M NaCl solution. The required solutions for electrochemical studies were prepared by mixing the required volumes of solketal stock solution, 0.5 mL ACT, 4 mL stock solution of NaHCO3, and 4 mL stock solution of Na2CO3 followed by dilution with deionized water in a 10 mL volumetric flask. To study the effect of base, a separate solution was prepared with unbuffered electrolyte as follows: 0.2 mL stock solution of solketal, 0.5 mL ACT, 8 mL stock solution of NaCl, followed by dilution with deionized water in a 10 mL volumetric flask. The blank solution where no ACT was added was prepared as follows: 0.2 mL stock solution of solketal, 8 mL stock solution of NaCl, followed by dilution with deionized water in a 10 mL volumetric flask. The entire contents of each solution were transferred and analyzed independently in the electrochemical cell. A Pine Wavenow XV 100 Potentio/Galvano-Stat and a Pine Low Volume Three Electrode Cell were used to perform the electrochemical experiments.40-43 The cell was equipped with a Ag/AgCl (internal solution 3M KC1) reference electrode, a Pt wire counter electrode, and a glassy carbon disk (2 mm) as the working electrode.44-47 The reference and working electrodes should be placed close together to minimize solution resistance. The working electrode was polished using alumina powder on a polishing pad before each chronoamperometric experiment.48
HAZARDS
Proper laboratory attire, gloves, and approved safety goggles should be worn in the laboratory at all times. Sodium carbonate, bicarbonate, and solketal can cause eye irritation. Solketal is flammable and should be kept away from heat or open flame. 4-Acetamido-TEMPO (ACT) has oral toxicity. All chemicals should be handled carefully. Flush eyes or skin with plenty of water for at least 15 minutes in the case of contact.
RESULTS AND DISCUSSION
We initiated the CV and CA study of ACT in the absence of solketal to examine an electrochemically reversible redox reaction. The CV of ACT shows one anodic peak corresponding to oxidation of ACT to the corresponding oxoammonium (ACT+) and a cathodic peak for the reduction of electrogenerated ACT+ to ACT (Figure 3a). The potential difference between the cathodic peeak and anodic peaks, also known as the peak to peak separation, is 59 mV, and the anodic to cathodic peak current ratio is near unity. The peak height of both the anodic and cathodic peaks, Ip (A), is best described by the Randles-Ševčík equation (at 25 °C),
| (Eqn. 2) |
where n is the number of electrons, A is the electrode surface area (cm2), DACT is the diffusion coefficient of ACT (cm2 s−1), CACT is the concentration of ACT in the bulk solution (mol cm−3), and v is the scan rate of voltammetric experiment (V s−1).49 Plotting the peak currents versus the square root of scan rate (Figure 3b) shows a linear correlation – evidence of an electron transfer process involving freely diffusing redox species.50 These CV features agree with the characteristics of a reversible, one-electron reaction at the electrode. The CA of ACT in the absence of solketal shows that the current-time profile follows the diffusion-controlled correlation described by the Cottrell equation (Figure 2c).51
Figure 3.

Cyclic voltammograms (a) of 1.0 mM ACT at different scan rates; scan rates (mV s−1) are depicted for each CV. The blank CV (black dashed trace) is the CV of 4 mM solketal substrate in the absence of ACT at a scan rate of 80 mV s−1. The plot of anodic and cathodic peak currents versus square root of scan rate (b). Solution conditions: aqueous solution with NaHCO3/Na2CO3 electrolyte 0.1/0.1 M, and pH 10.
CVs of ACT in the presence of solketal were then considered to show the characteristics of an electrocatalytic mechanism (Figure 4). Compared to the CV of ACT alone, two significant differences were observed for the CV of ACT in the presence of solketal. First, the cathodic peak disappears, indicating that ACT+ reacts with solketal and doesn’t persist as the oxidized form. Therefore, it is not present and able to be reduced at the electrode in the reverse scan. Second, the anodic peak current exhibits an increase in magnitude. This increase indicates that the reduced form of ACT (i.e., the hydroxylamine form, ACTH), generated upon reaction of oxoammonium with substrate, can be oxidized again at the electrode surface on the time scale of the CV scan. It should be noted that solketal does not undergo direct electron transfer within this potential range (Figure 3a black dashed trace). Thus, the enhancement in oxidation current does not arise from direct oxidation of solketal at the electrode. If the reaction between excess solketal and ACT+ continuously generates ACTH, the CV traces will exhibit a plateau current instead of peak. Under these conditions, the current is controlled by the kinetics of the reaction between solketal and ACT+ and is called reaction-controlled current. The scan rate shows minimal influence on the current (Figure 4a). Instead, these reaction-controlled currents now depend on solketal concentration as demonstrated in Figure 4b.52
Figure 4.

Cyclic voltammograms of 1.0 mM ACT in the absence (dotted line) and presence of 9 mM solketal at different scan rates (25–120 mV s−1) (a), and cyclic voltammograms of 1.0 mM ACT in the presence of various concentrations of solketal (b). Scan rate for dotted trace is 80 mV s−1 and for traces in (b) is 25 mV s−1. Solution conditions: aqueous solution with NaHCO3/Na2CO3 electrolyte 0.1/0.1 M, and pH 10.
After collecting information about the electrochemical reaction mechanism using CV, CA analysis was performed for the quantitative analysis of the reaction kinetics. The CAs of ACT in the presence of various concentrations of solketal are shown in Figure 5a. The total consumed charge (Q) for each CA experiment was derived by integrating the current-time trace.53 For the “ACT-only” solution, the consumed charge (QA) is related to the number of ACT molecules in the electrode diffusion layer that are oxidized to ACT+. In the presence of solketal, ACT+ reacts with solketal to generate the corresponding carbonyl product and ACTH (see Figure 5b), and the latter is re-oxidized at the electrode surface via a two-electron, one-proton oxidation process.54
Figure 5.

Chronoamperograms for a blank solution and ACT in the absence and presence of various concentrations of solketal (a) with applied chronoamperometry potential of 0.8 V vs Ag/AgCl, the consumed charges are shown on each chronoamperogram, the consumed charge for blank solution is 12 μC (not depicted on its plot). Proposed mechanism for ACT catalyzed electrochemical solketal oxidation (b).
The total consumed charge (QT) contains the initial charge required for ACT oxidation (QA) plus the charge consumed as a result of catalytic turnover of ACTH at the electrode surface, QC, such that QT = QC + QA. The ratio of QC to QA is proportional to the turnover numbers (TONs) of ACT under these conditions. To precisely determine TOFs, the consumed charged for a blank solution (QB) needs to be subtracted from QA to account for non-faradaic charging of the electrode.55 Considering the number of electrons corresponding to one turnover (n = 2) and conversion factor of the time unit for turnover frequency (one hour or 3600 s) over the CA experiment time (t), the TOF can be calculated as shown in equation 3:
| Eq. 3 |
The unit of the resulting TOF value is per hour (h−1). The TOFs derived from the CA experiments in Fig. 5a are shown in Table 1. The voltammetric and chronoamperometric studies of the reaction under neutral conditions were performed to demonstrate the effect of base on the reaction kinetics. As demonstrated in entry 5 of Table 1, the catalytic activity of ACT in the absence of base is negligible.56 The presence of base favors formation of the alkoxide adduct between ACT+ and solketal and favors the electrochemical oxidation of ACTH.30,37
Table 1.
TOF (h−1) of ACT for different concentrations on solketal in the absence and presence of base
| Entry | Solketal concentration | TOF (h−1)c |
|---|---|---|
| 1 | 2 mMa | 346 |
| 2 | 4 mMa | 548 |
| 3 | 9 mMa | 802 |
| 4 | 16 mMa | 1024 |
| 5 | 4 mMb | 36 |
Reaction Conditions: aqueous solution with NaHCO3/Na2CO3 electrolyte 0.1/0.1 M, pH 10, 1.0 mM ACT, applied potential 0.8 V, experiment duration 20 sec.
Reaction Conditions: aqueous solution with NaCl electrolyte 0.2 M, pH 6.7, 1.0 mM ACT.
"Turnover Frequency", defined as the molecules of solketal converted per molecule of catalyst per hour. The standard deviation of reported TOFs (h−1) for four groups of students were (8-14%).
The CA and CV experiments demonstrated here form the basis for several inquiry labs or an undergraduate research project. For example, CV analysis of ACT under a range of basic buffers pH 8-11 visually demonstrates the basicity-reactivity relationship. CA analysis of the reaction between ACT and diverse substrates, such as secondary benzylic alcohols and primary and secondary aliphatic alcohols, is a simple and quantitative way to explore substrate structure-reactivity relationships. Experimenting with electronically/structurally distinct aminoxyl catalysts, such as commercially available TEMPO or 9-azabicyclo-[3.3.1]nonane N-oxyl (ABNO), allows a research student to begin making connections between catalyst structure and activity.30
CONCLUSION
This lab allows students to explore the concepts of chronoamperometry, cyclic voltammetry, and electrocatalysis. The operationally simple experiments give students experience in voltammetric and chronoamperometric analysis of both direct and mediated (catalytic) electron transfer reactions. The emphasis on chronoamperometry provides students with an example of how a simple electrochemical technique can be used for quantitative analysis of a catalytic reaction and for deriving the turnover frequency (TOF). The pedagogically rich protocol reported herein is an example of an interdisciplinary experiment that is well suited for incorporation into various upper level undergraduate laboratory courses, including instrumental analysis, physical chemistry, organic chemistry, and electrochemistry.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the attendees and teaching assistants of our first Organic Electrochemistry Short Course, held during Aug. 2019 at the University of Wisconsin-Madison and to the students and teaching assistant of Chem 437WI (Physical Chemistry Laboratory) laboratory course at University of Missouri – Kansas City (UMKC) for sharing their observations and feedback on this experiment. The authors would like to thank the NIH (R35 GM134929; SSS) for financial support to develop this laboratory experiment at the University of Wisconsin-Madison, the NSF for a predoctoral fellowship to JEN (DGE-1747503), and the Chemistry Department of the (UMKC) for its support in purchasing equipment and chemicals to run this experiment in the Chem 437WI. We also thank Pine Research for providing equipment for the Rafiee-Stahl Organic Electrochemistry Short Course lab section (at University of Wisconsin-Madison) and Timothy Paschkewitz and Alex Peroff (Pine Research Instrument) for demonstrating the Aftermath software’s functions that facilitate post analysis of voltammetric and amperometric results.
REFERENCES
- 1.Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Remick AE Electrochemical Methods Available for Reaction Mechanism Studies. J. Chem. Educ 1956, 33, 564–574. [Google Scholar]
- 3.Ibanez JG; Singh MM; Pike RM; Szafran Z Laboratory Experiments on Electrochemical Remediation of the Environment. Part 2: Microscale Indirect Electrolytic Destruction of Organic Wastes. J. Chem. Educ 1997, 74, 1149–1450. [Google Scholar]
- 4.Manchanayakage R Designing and Incorporating Green Chemistry Courses at a Liberal Arts College to Increase Students’ Awareness and Interdisciplinary Collaborative Work. J. Chem. Educ 2013, 90, 1167–1171. [Google Scholar]
- 5.An editorial note for an open access virtual issue focused on electrochemical concepts and their spread into different areas of research and development: Gollas, B. Advances in Electrochemistry, ChemistryOpen 2017, 6, 299–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubner I; Berry AJ; Grofe T; Oetken M Educational Modules on the Power-to-Gas Concept Demonstrate a Path to Renewable Energy Futures. J. Chem. Educ 2019, 96, 248–255. [Google Scholar]
- 7.Sandford C; Edwards MA; Klunder KJ; Hickey DP; Li M; Barman K; Sigman MS; White HS and Minteer SD A Synthetic Chemist’s Guide to Electroanalytical Tools for Studying Reaction Mechanisms. Chem. Sci 2019, 10, 6404–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam CH; Jackson JE Teaching Electrochemistry with Common Objects: Electrocatalytic Hydrogenation of Acetol with U.S. Coins. J. Chem. Educ 2020, 97, 172–177. [Google Scholar]
- 9.For leading reference see: Nicholson RS; Shain I Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem 1964, 36, 706–723. [Google Scholar]
- 10.Bard AJ The Rise of Voltammetry: From Polarography to the Scanning Electrochemical Microscope. J. Chem. Educ 2007, 84, 644–650. [Google Scholar]
- 11.Khalafi L; Rafiee M Cyclic Voltammetry. In Encyclopedia of Physical Organic Chemistry; Wang Z, Ed.; Wiley: Hoboken, NJ, 2017; Vol. 5, pp 3437–3478. [Google Scholar]
- 12.Elgrishi N; Rountree KJ; McCarthy BD; Rountree ES; Eisenhart TT; Dempsey JL A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ 2018, 95, 197–206. [Google Scholar]
- 13.To see the example of voltammetric working curves for deriving the quantitative information see Ref. 9 and the following ref: Andrieux CP; Blocman C; Dumas-Bouchiat JM; M'Halla F; Savéant JM Homogeneous Redox Catalysis of Electrochemical Reactions: Part V. Cyclic voltammetry. J. Electroanal. Chem. Interf. Electrochem 1980, 11, 19–40. [Google Scholar]
- 14.Messersmith SJ Cyclic Voltammetry Simulations with DigiSim Software: An Upper-Level Undergraduate Experiment. J. Chem. Educ 2014, 91, 1498–1500. [Google Scholar]
- 15.Bard AJ; Faulkner LR Electrochemical Methods. Fundamentals and Applications; John Wiley & Sons, Inc.: New York, 2001. [Google Scholar]
- 16.Savéant JM; Vianello E Potential-Sweep Chronoamperometry. Kinetic Currents for First-Order Chemical Reaction Parallel to Electron-Transfer Process (Catalytic Currents). Electrochim. Acta 1965, 10, 905–920. [Google Scholar]
- 17.Chronoamperometry is typically only discussed in basic electroanalytical chemistry literature and is less well known by students/scientists from other disciplines. For example, there is only one paper published in J. Chem. Educ. on this subject, and it focuses on its mathematical foundations, rather than application to understanding chemical reactions: Ryswyk HV Simulation of Single-Step Chronoamperometry via Spreadsheet. J. Chem. Educ 1991, 68, A283–A285. [Google Scholar]
- 18.This is the description of a normal or single-step CA experiment, but application of more than one potential step in a CA experiment is also possible. Depending on the number of steps, these experiments are called double step or multi-step CA.
- 19.The region of interest at which the potential is held is typically one where the electrochemical reaction is complete and the concentration of one of the electroactive species is nearly zero at the electrode surface. For example, the applied potential (Eapp) should be 120 mV more positive (ΔE) than the redox potential (E°) for the oxidation reaction of interest, and 120 mV more negative than the redox potential for reduction reactions, to meet the requirement of simplified boundary conditions.
- 20.Stephens LI; Mauzeroll J Demystifying Mathematical Modeling of Electrochemical Systems. J. Chem. Educ 2019, 96, 2217–2224. [Google Scholar]
- 21.See Section 6 and Figure S3 of the Supporting Information.
- 22.Sheldon RA; Arends IWCE Organocatalytic Oxidations Mediated by Nitroxyl Radicals. Adv. Synth. Catal 2004, 346, 1051–1071. [Google Scholar]
- 23.Vogler T; Studer A Applications of TEMPO in Synthesis. Synthesis 2008, 2008, 1979–1993. [Google Scholar]
- 24.Hill NJ; Hoover JM; Stahl SS Aerobic Alcohol Oxidation Using a Copper(I)/TEMPO Catalyst System: A Green, Catalytic Oxidation Reaction for the Undergraduate Organic Chemistry Laboratory. J. Chem. Educ 2013, 90, 102–105. [Google Scholar]
- 25.Straub TS A Mild and Convenient Oxidation of Alcohols; Benzoin to Benzil and Borneol to Camphor. J. Chem. Educ 1991, 68, 1048–1049. [Google Scholar]
- 26.Bobbitt JM; Brückner C; Merbouh N Oxoammonium- and Nitroxide-Catalyzed Oxidations of Alcohols. In Organic Reactions; Wiley: Hoboken, NJ, 2009; pp 103–424. [Google Scholar]
- 27.For leading reference see: Semmelhack MF; Chou CS; Cortes DA Nitroxyl-Mediated Electrooxidation of Alcohols to Aldehydes and Ketones. J. Am. Chem. Soc 1983, 105, 4492–4494. [Google Scholar]
- 28.For review see: Nutting JE; Rafiee M; Stahl SS Tetramethylpiperidine N- Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rafiee M; Konz ZM; Graaf MD; Koolman HF; Stahl SS Electrochemical Oxidation of Alcohols and Aldehydes to Carboxylic Acids by 4-Acetamido-TEMPO: An Alternative to “Anelli” and “Pinnick” Oxidations. ACS Catal. 2018, 8, 6738–6744. [Google Scholar]
- 30.Rafiee M; Miles KC; Stahl SS Electrocatalytic Alcohol Oxidation with TEMPO and Bicyclic Nitroxyl Derivatives: Driving Force Trumps Steric Effects. J. Am. Chem. Soc 2015, 137, 14751–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickey DP; Schiedler DA; Matanovic I; Doan PV; Atanassov P; Minteer SD; Sigman MS Predicting Electrocatalytic Properties: Modelling Structure-Activity Relationships of Nitroxyl Radicals. J. Am. Chem. Soc 2015, 137, 16179–16186. [DOI] [PubMed] [Google Scholar]
- 32.Siu JC; Sauer GS; Saha A; Macey RL; Fu N; Chauvire T; Lancaster KM; Lin S Electrochemical Azidooxygenation of Alkenes Mediated by a TEMPO–N3 Charge-Transfer Complex. J. Am. Chem. Soc 2018, 140, 12511–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rafiee M; Alherech M; Karlen SD; Stahl SS Electrochemical Aminoxyl-Mediated Oxidation of Primary Alcohols in Lignin to Carboxylic Acids: Polymer Modification and Depolymerization. J. Am. Chem. Soc 2019, 141, 15266–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toma HE; Araki K; Dovidauskas S A Cyclic Voltammetry Experiment Illustrating Redox Potentials, Equilibrium Constants and Substitution Reactions in Coordination Chemistry. J. Chem. Educ 2000, 77, 1351–1353. [Google Scholar]
- 35.http://128.104.71.100/content/organic-electrochemistrv-short-course, accessed Jun 30, 2020.
- 36.Solketal has a chiral center. Our recent study shows that ACT-catalyzed electrochemical oxidation of S-solketal to the corresponding carboxylic acid retains the enantiomeric excess of the starting alcohol. Solketal also contains a heterocycle that makes it a challenging substrate for metal catalyzed alcohol oxidation. This aminoxyl-mediated electrochemical method produces the carboxylic acid efficiently. These features make solketal an appealing substrate for students and scientists oriented in organic chemistry. However, a variety of other primary and secondary alcohols can be used as the substrate. For water-insoluble substrates, the mixture of acetonitrile and carbonate buffer results in sufficient solubility for the substrate. For other possible substrates and the reactions conditions, see the substrate scopes in Refs 27, 29 and 37.
- 37.Rafiee M; Karimi B; Alizadeh S Mechanistic Study of the Electrocatalytic Oxidation of Alcohols by TEMPO and NHPI. ChemElectroChem 2014, 1, 455–462. [Google Scholar]
- 38.In this paper we use IUPAC convention for representation of cyclic voltammograms. Some of the potentiostats use a different notation called Texas or classical notation. See Refs 11 and 12 which describe both conventions.
- 39.The definition of Turnover Frequency (TOF) is “the number of reactant molecules converted per a time unit (hour, minute or second) per catalyst molecule (for given reaction conditions)”. For more details about the definition of TOF and its related topics please see: Kozuch S; Martin JML “Turning Over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2, 2787–2794. [Google Scholar]
- 40.Pine Potentiostat is an example of standard equipment for electrochemical studies. For the Physical Chemistry course at UMKC we purchased only one potentiostat and one group of students worked with the potentiostat each week/section. However, if the cost of the potentiostat represents a barrier, a homemade potentiostat provides a cost-effective option. Examples of the latter are included in Refs 41-43. Moreover, lower cost potentiostats are available for educational purposes. One example of an open-source potentiostat is Rodeostat: https://iorodeo.com/collections/cheapstat-open-source-potentiostat, accessed September 4, 2020.
- 41.Meloni GN Building a Microcontroller Based Potentiostat: An Inexpensive and Versatile Platform for Teaching Electrochemistry and Instrumentation. J. Chem. Educ 2016, 93, 1320–1322. [Google Scholar]
- 42.Li YC; Melenbrink EL; Cordonier GJ; Boggs C; Khan A; Isaac MK; Nkhonjera LK; Bahati D; Billinge SJ; Haile SM; Kreuter RA; Crable RM; Mallouk TE An Easily Fabricated Low-Cost Potentiostat Coupled with User-Friendly Software for Introducing Students to Electrochemical Reactions and Electroanalytical Techniques. J. Chem. Educ 2018, 95, 1658–1661. [Google Scholar]
- 43.Glasscott MW; Verber MD; Hall JR; Pendergast AD; McKinney CJ; Dick JE SweepStat: A Build-It-Yourself, Two-Electrode Potentiostat for Macroelectrode and Ultramicroelectrode Studies. J. Chem. Educ 2020, 97, 265–270. [Google Scholar]
- 44.The three required electrodes are also commercially available from different vendors. The more cost-effective options for educational purpose are homemade electrodes. For examples, see Refs 45-47.
- 45.King D; Friend J; Kariuki J, Measuring Vitamin C Content of Commercial Orange Juice Using a Pencil Lead Electrode. J. Chem. Educ 2010, 87, 507–509. [Google Scholar]
- 46.Masse RC; Gerken JB Assembly of a Robust and Economical MnO2-Based Reference Electrode. J. Chem. Educ 2015, 92, 110–115. [Google Scholar]
- 47.Schmidt B; King D; Kariuki J Designing and Using 3D-Printed Components that Allow Students to Fabricate Low-Cost, Adaptable, Disposable, and Reliable Ag/AgCl Reference Electrodes. J. Chem. Educ 2018, 95, 2076–2080. [Google Scholar]
- 48.For details of polishing and other experimental conditions, see the Supporting Information. Based on the observed results, polishing the electrode once is enough to perform all the CV and CA experiments for each solution. Discarding the data for the first experiment conducted after polishing and repeating it without polishing results in more reproducible responses.
- 49.The voltammetric currents normalized by square root of the scan rate should be identical to a reversible electron transfer at various scan rates. The students were asked to normalize the CV currents and compare them: see Student’s Handout in Supporting Information.
- 50.The plot of peak current (IP) as a function of the square root of scan rate should be linear. The slope of the line is proportional to the diffusion coefficient of electroactive species. See Supporting Information, Section 6, and Figure S4.
- 51.When the current is diffusion-controlled it should be inversely proportional to t1/2. An example of a linear correlation was plotted by students and is shown in Figure S2 and S3 (Supporting Information). The diffusion coefficient derived from amperometric results, based on the Cottrell equation, is in good agreement with the value derived from voltammetric analysis, See Supporting Information, Section 6 and Figure S3.
- 52.Based on the proposed mechanism, the height of the anodic peak is proportional to the concentration of solketal and the number of catalytic cycles. The slope of the linear plot for the anodic plateau current versus the square root of the solketal concentration is proportional to the rate constant of the chemical reaction between ACT+ and solketal. See Ref. 36 and Figure S5 and Figure S6 (Supporting Information).
- 53.All the chronoamperometric measurements must be taken in exactly the same timeframe to enable subtraction and division of the consumed charge for different chronoamperograms. To derive the consumed charge for each plot, see Section 8 of Supporting Information and the Supporting Information Excel file. The optimal time for a chronoamperometry experiment is 15-20 sec. See Ref. 15.
- 54.Taitt BJ; Bender MT; Choi K-S Impacts of the Regeneration Pathways of the Oxoammonium Cation on Electrochemical Nitroxyl Radical-Mediated Alcohol Oxidation. ACS Catal. 2020, 10, 265–275. [Google Scholar]
- 55.Since solketal is not electroactive at this potential range (e.g. Figure 3a), the blank experiment can be a solution consisting only of supporting electrolyte/buffer or may include solketal.
- 56.Examples of more results by students are presented in Tables S2 and Table S3 of the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.