

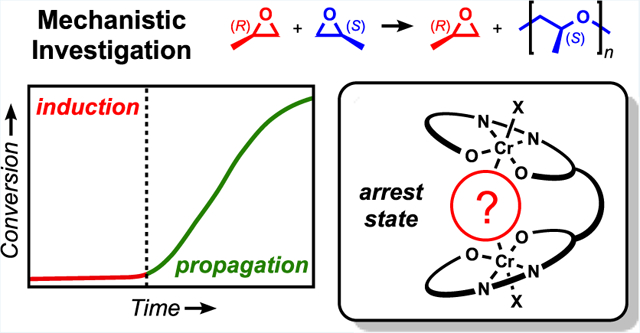
Abstract
Initial catalyst dormancy has been mitigated for the enantioselective polymerization of propylene oxide using a tethered bimetallic chromium(III) salen complex. A detailed mechanistic study provided insight into the species responsible for this induction period and guided efforts to remove them. High-resolution electrospray ionization–mass spectrometry and density functional theory computations revealed that a μ-hydroxide and a bridged 1,2-hydroxypropanolate complex are present during the induction period. Kinetic studies and additional computation indicated that the μ-hydroxide complex is a short-lived catalyst arrest state, where hydroxide dissociation from one metal allows for epoxide enchainment to form the 1,2-hydroxypropanolate arrest state. While investigating anion dependence on the induction period, it became apparent that catalyst activation was the main contributor for dormancy. Using a 1,2-diol or water as chain transfer agents (CTAs) led to longer induction periods as a result of increased 1,2-hydroxyalkanolate complex formation. With a minor catalyst modification, rigorous drying conditions, and avoiding 1,2-diols as CTAs, the induction period was essentially removed.
Keywords: isotactic polyethers, arrest state, mechanism, bimetallic catalysis, enantioselective, polyols
Graphical Abstract
INTRODUCTION
Atactic poly(propylene oxide) (aPPO) is an industrially ubiquitous polymer used in polyurethane foam production.1,2 Although aPPO is an amorphous polymer with a Tg of −70 °C, its semicrystalline analogue, isotactic PPO (iPPO), has a Tm of 67 °C. A recent report from Coates highlights the robust mechanical strength of iPPO and its susceptibility toward photodegradation.3 This material is suggested for high strength, environmentally susceptible applications as its ultimate tensile strength is comparable to that of nylon-6,6. With photodegradable3–6 and potentially biodegradable pathways,7–10 it is anticipated that this material could help alleviate the environmental burden of persistent plastics.
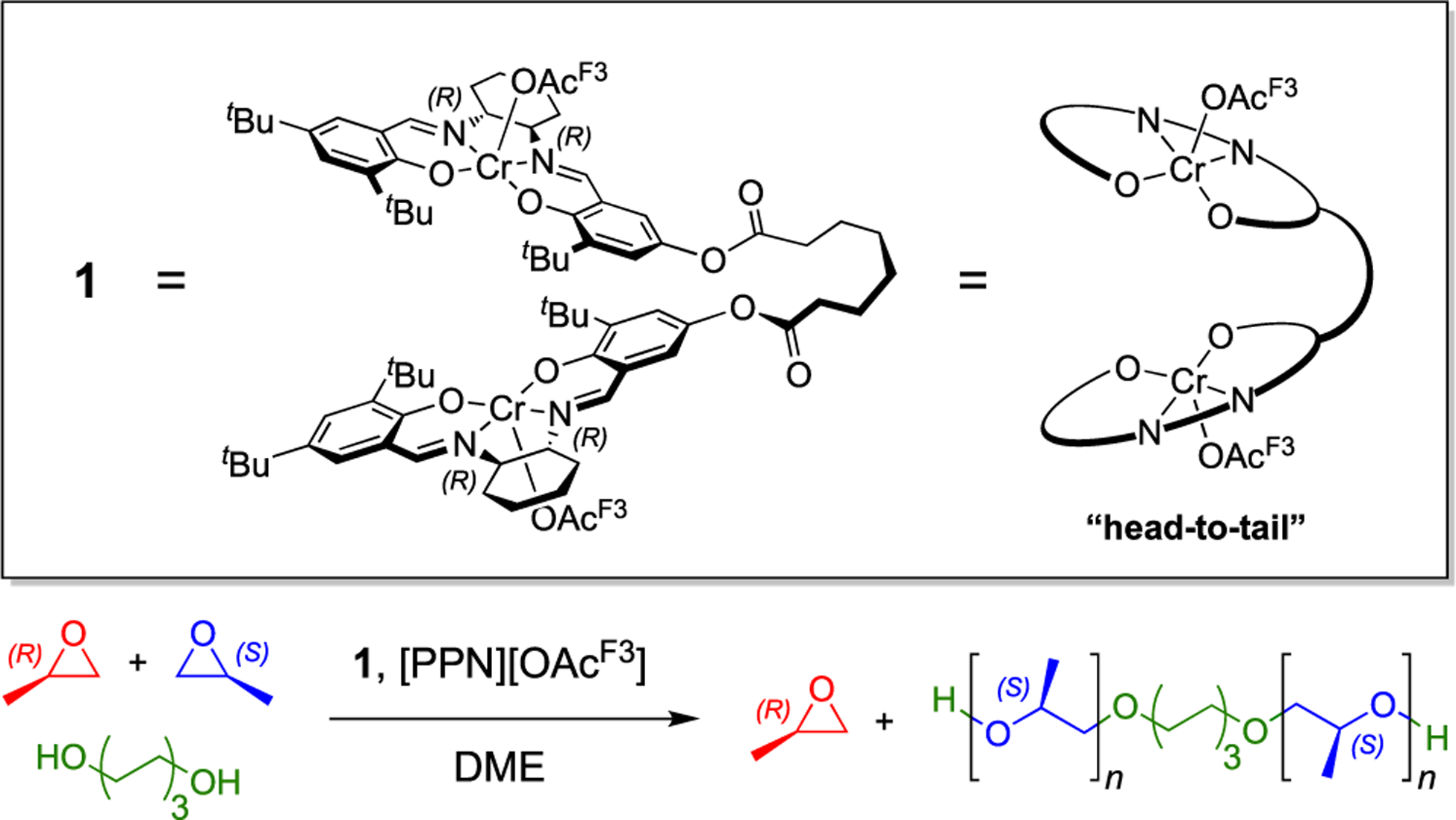
Several decades of catalyst development have led to improved isotacticity,11–13 dispersity,14,15 and molecular weight control14,15 for iPPO. The chiral bimetallic chromium(III) salen complex (1, Scheme 1) efficiently produces iPPO with high selectivity.14 Complex 1 was inspired by Jacobsen’s azido analogue developed for the enantioselective ring opening of epoxides.16–19 His work highlights that the two salen moieties of the ligand framework must adopt a “head-to-tail” arrangement for the metal centers to act cooperatively. Computational studies of similar chromium- and cobalt-bimetallic systems for epoxide polymerization also propose a cooperative mechanism in which one metal center provides an alkoxide chain end to ring-open an activated monomer, datively coordinated by the second metal.15,20 Previous work demonstrates that alcohol chain transfer agents (CTAs) in conjunction with 1 afford an immortal polymerization in which molecular weight is readily controlled and dispersities remain low through rapid alcoholysis.14 Although the 1-catalyzed system demonstrates exquisite control over polymerization, it is subject to an induction period (~20% of the total reaction time) during which there is little to no conversion.
Scheme 1.
Chromium Salen-Catalyzed iPPO Synthesis
Induction periods have been previously observed in similar salen-catalyzed systems.17,21,22 Lee observed induction periods during PO/CO2 copolymerization with salen–cobaltate complexes where the relative humidity and small catalyst modifications led to varied induction times.23 Wu and Darensbourg observed a similar induction period in PO/CO2 copolymerization catalyzed by a monometallic cobalt salen complex when using water as a CTA.24 During the induction period, they observed the formation of 1,2-propanediol (1,2-PD), the suspected species responsible for the bimodality of molecular weight distributions in this and similar systems.14,23,25–28 When 1,2-PD was substituted as the CTA, polymerization began immediately, demonstrating 1,2-PD’s innocence on catalyst dormancy. In light of Lee’s and Darensbourg’s work, every effort was made to exclude water from the 1-catalyzed system. Despite rigorous drying conditions, low concentrations of adventitious water remained, potentially contributing to the observed induction period. Herein, we report an investigation of the induction period for the enantioselective polymerization of PO facilitated by 1.
RESULTS AND DISCUSSION
Because of paramagnetic broadening by chromium, NMR spectroscopy was not used to characterize inorganic species for this mechanistic study. Instead, high-resolution electrospray ionization–mass spectrometry (ESI–MS) was used to help identify species present during the polymerization. This analytical method provided the first insight into the catalyst arrest states and oligomers formed during the induction period.29–32 Kinetic studies were used to corroborate MS findings and answer persistent mechanistic questions. Computation reinforces the experimental findings and provides additional structural insight.
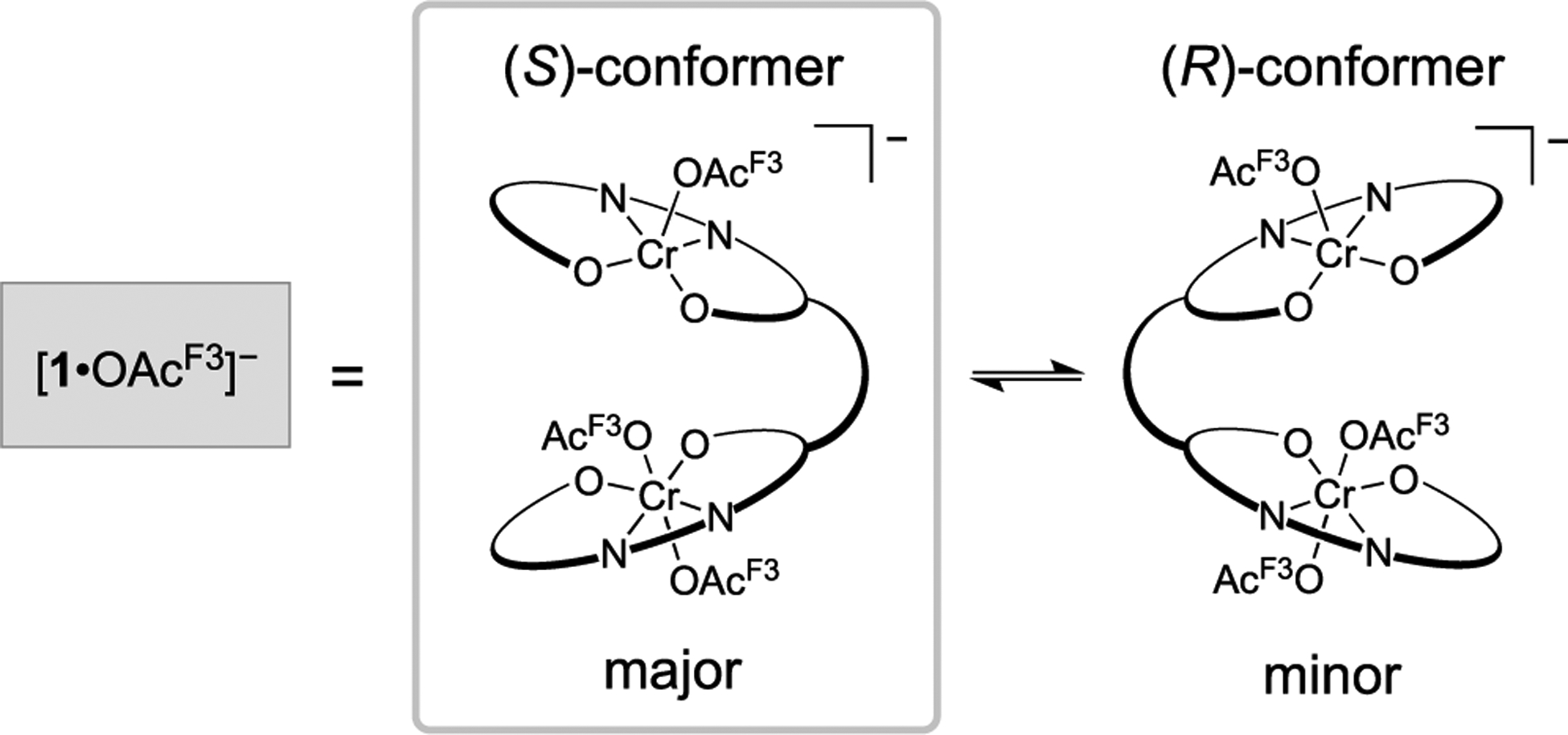
To elucidate species formed early during the polymerization, several reactions were analyzed by ESI–MS. In the absence of PO, [1·OAcF3]− (1589.5775 m/z) was the primary ion resulting from the reaction mixture containing 1, catalyst activator, and 1,6-hexanediol (1,6-HD) (Figure 1A). This signal is recognized as the activated complex, where the counter ion [PPN]+ ([Ph3P–N=PPh3]+) was not observed while operating in the negative detection mode. Although 1 was originally portrayed as the (S)-conformer based on literature precedent,19,20 we did not ignore the possibility of 1 or [1·OAcF3]− resting as the (R)-conformer (Scheme 2). Quantum chemical calculations conducted using density functional theory (DFT) support that polymer propagation as the (S)-conformer is preferred (vide infra, Figure S19). The favorability of a single conformation is attributed to the 1,2-cyclohexanediamine chirality. With the additional support from computation, all remaining calculations and analogues of 1 are presented as the (S)-conformer.
Figure 1.
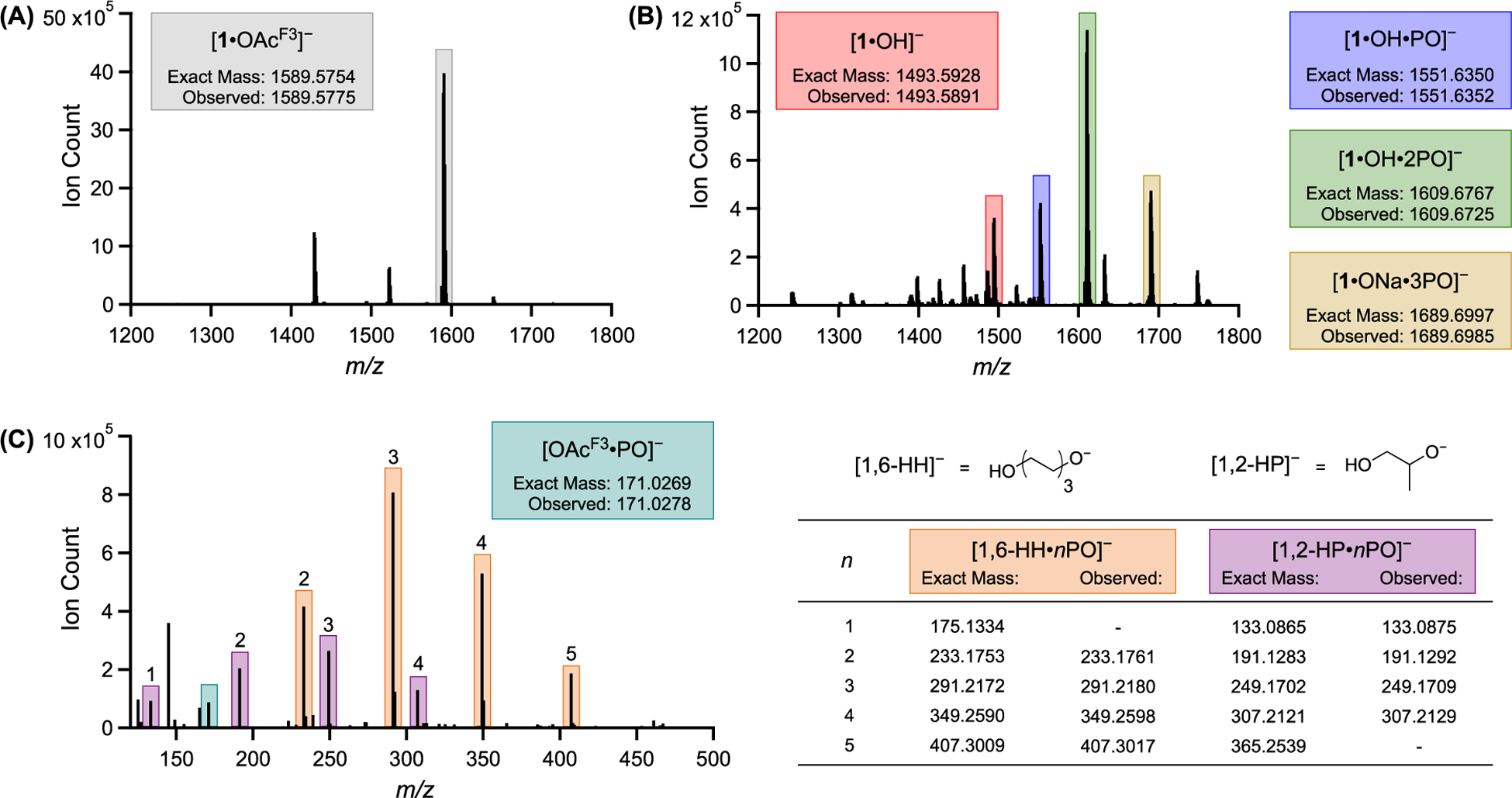
Mass spectra: (A) Reaction mixture prior to addition of PO, consisting of 1.3 μmol 1, 1.3 μmol [PPN][OAcF3], and 13 μmol 1,6-HD in 2 mL of DME. (B) Reaction mixture 0.5 h after 10.7 mmol PO was added. (C) Oligomers present (labeled with a degree of polymerization, n) 1.0 h after PO addition. Because of the off-scale signal of [OAcF3]− at 112.9859 m/z, 100–120 m/z was omitted.
Scheme 2.
Proposed Activated Diastereomeric Complex Conformation
Within 30 min of adding PO to the reaction containing [1·OAcF3]−, ESI–MS analysis revealed an ion at 1493.5891 m/z, corresponding to the hydroxide complex ([1·OH]−) (Figure 1B, red). The ion’s dependence on PO addition is consistent with adventitious water protonating an alkoxide chain end to form [1·OH]− (Scheme 3). This process results in [OH]− coordinated within the catalyst active site, proximate to the second metal center. A μ-hydroxide complex of an analogous nontethered chromium salen (Figure 2, crystallization and structural parameter details are in the Supporting Information) suggests a probable arrest state for the tethered bimetallic system. In an effort to determine [OH]− arrangement, [1·OH]− was subjected to collision-induced dissociation (CID) MS. The resulting fragmentation pattern showed cleavage of [OAcF3]− rather than [OH]− (Figure S12). The stronger interaction with [OH]− reinforces its possible coordination with both metal centers, consistent with a μ-hydroxide species. Calculations further support the assignment as a μ-hydroxide (vide infra).
Scheme 3.
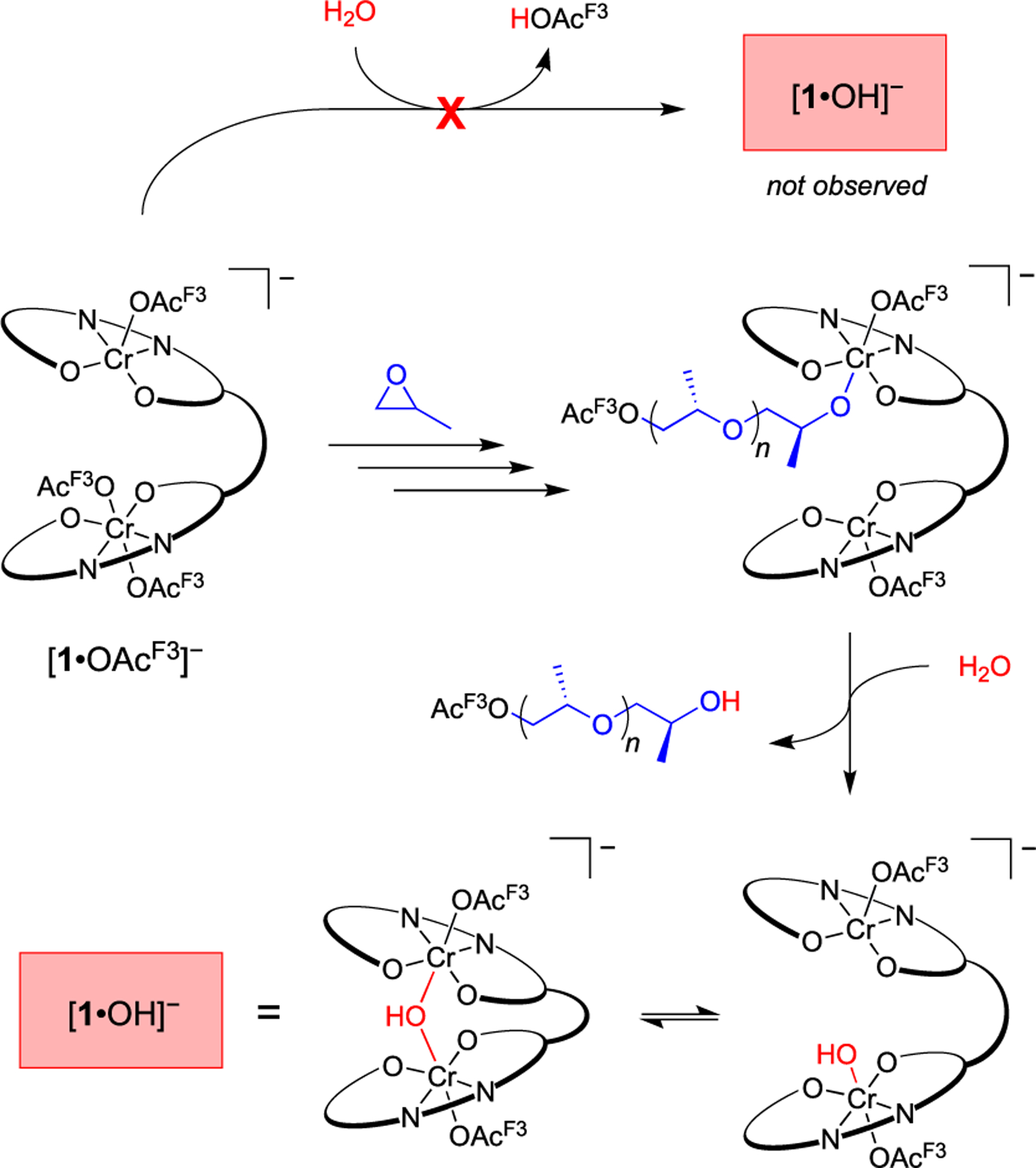
Proposed Pathway for [1·OH]− Formation from Adventitious Water
Figure 2.
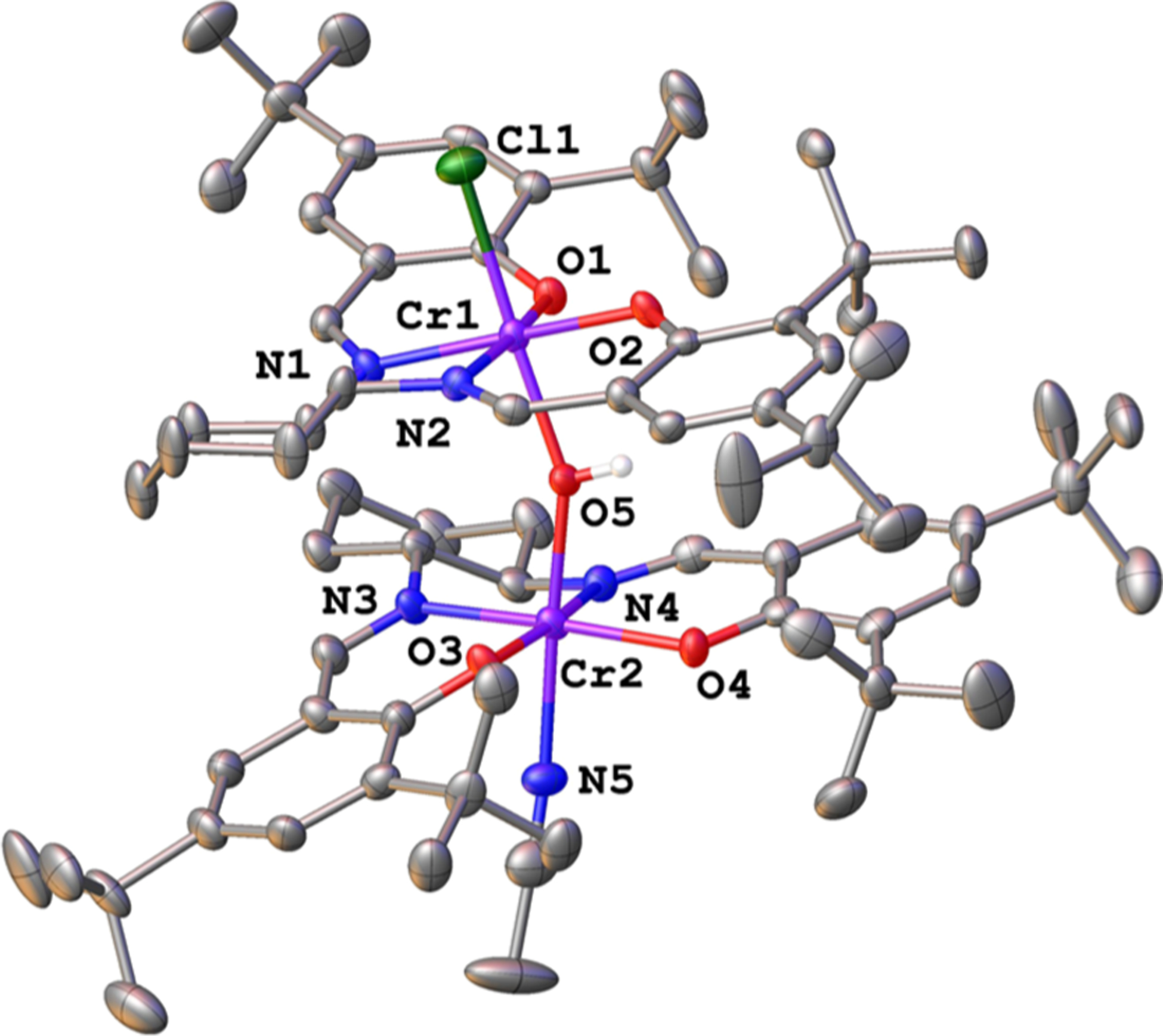
Solid-state structure of the nontethered chromium salen, crystallized as the μ-hydroxide complex. Hydrogen atoms except that on the hydroxide fragment are omitted for clarity. Ellipsoids shown at 50% probability.
In addition to [1·OH]−, three signals (1551.6352, 1609.6725, and 1689.6985 m/z) corresponding to the incorporation of 1–3 monomers from [1·OH]− or [1·ONa]− were observed (Figure 1B). In an effort to distinguish between PO enchainment and dative coordination, these ions were subjected to CID MS analysis.33,34 The ion corresponding to [1·OH·PO]− primarily resulted in a loss of 58.04 m/z, the mass of a single PO, suggesting dative coordination (Scheme 4A, Figure S13). In a control experiment, the activated complex ([1·OAcF3][PPN]) and 1,2-PD produced the isoelectronic ring-opened complex in situ in the absence of PO (Scheme 4B). When the ion was subjected to CID, the same fragmentation pattern and loss of 58.04 m/z were observed (Figure S14). Although these results support ring-closing and loss of PO by CID, it was not possible to discern if the PO was ring-opened or -closed for [1·OH·PO]−.
Scheme 4.
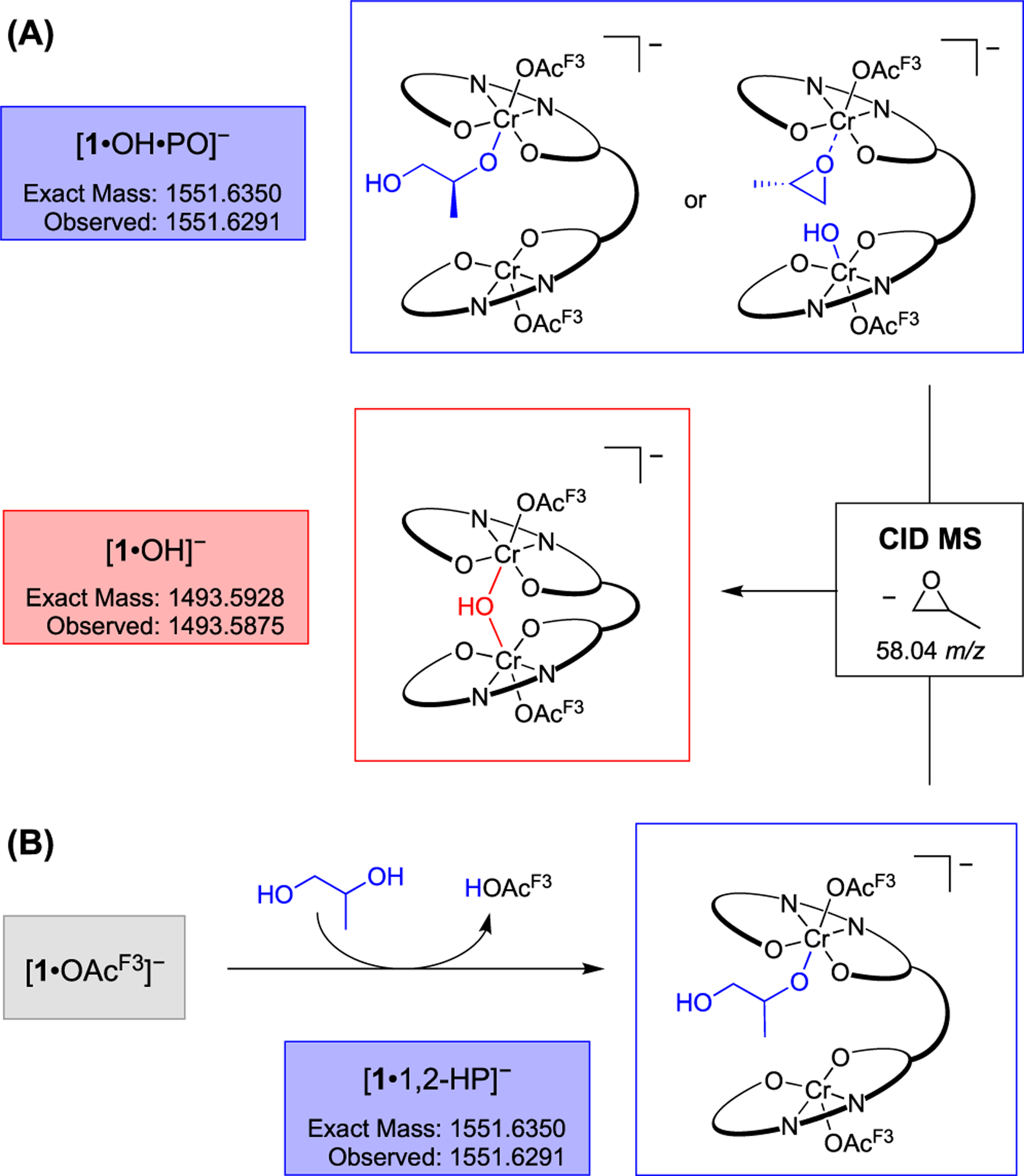
Failure to Determine the PO Enchainment of [1·PO·OH]− by CID MS
To augment CID MS findings about the complex, we examined the small molecules present during the induction period by ESI–MS.35 Dormant chains initiated by 1,6-hydroxyhexanolate ([1,6-HH]−), 1,2-hydroxypropanolate ([1,2-HP]−), and [OAcF3]− were formed within the first hour of polymerization (Figure 1C, nomenclature assignment described in the Supporting Information). The presence of [1,2-HP]−-derived chains in the absence of exogenous 1,2-PD, suggests that [1·OH]− ring-opens PO to generate [1·1,2-HP]−. Similar to the equilibrium of the μ-hydroxide and single coordination for [1·OH]−, we suspected that [1·1,2-HP]− could exist in a bridged coordination (Scheme 5). The bridged [1,2-HP]− complex is coordinatively saturated, yielding a catalyst arrest state in addition to the μ-hydroxide complex.
Scheme 5.
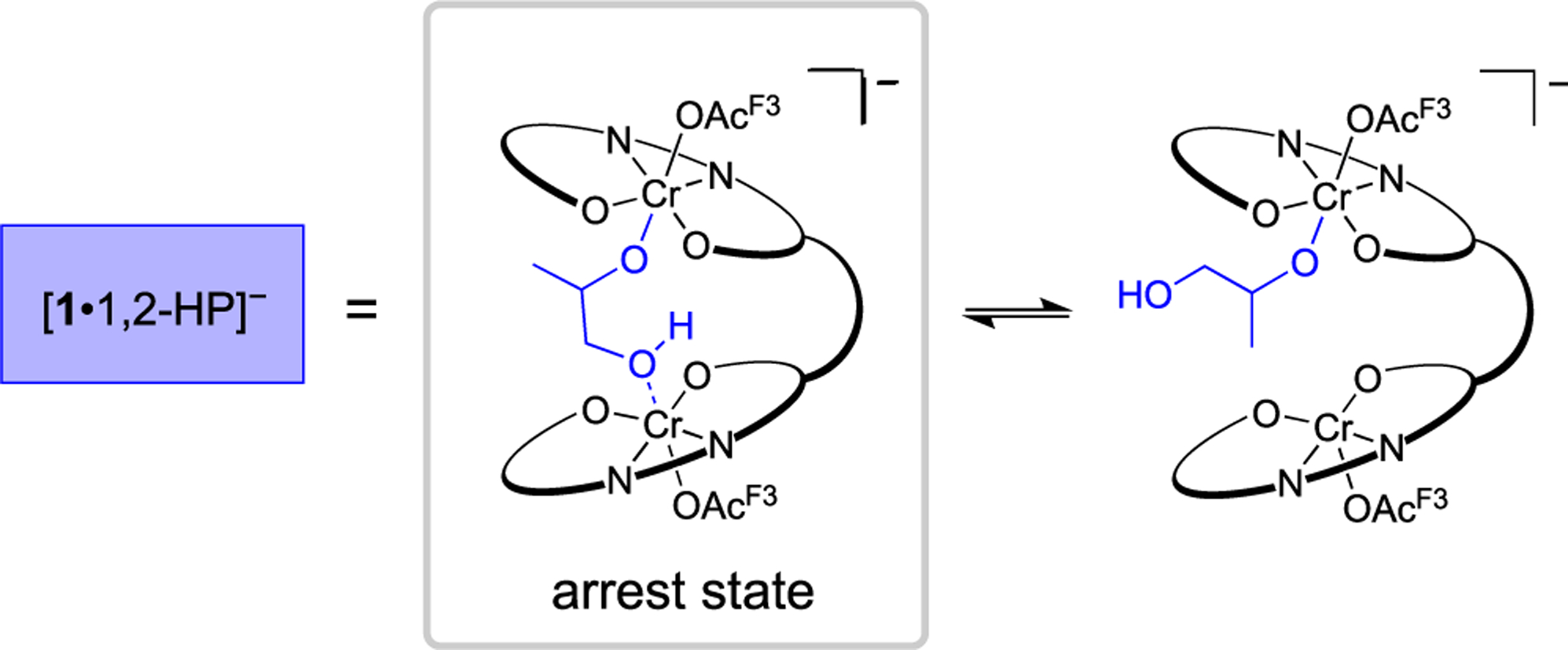
Proposed [1·1,2-HP]− Arrest State
To determine qualitative coordination strengths of potential inhibitors, solutions of 1,2-, 1,4-, and 1,6-diols were combined with the activated complex ([1·OAcF3][PPN]) and the resulting mixtures were analyzed by ESI–MS. Upon introducing 10 equiv of 1,2-PD or 1,2-butanediol (1,2-BD), the respective 1,2-hydroxyalkanolate complexes were observed (Figures S9 and S10). When the CTA was not a vicinal diol (e.g., 1,4-BD and 1,6-HD), no hydroxyalkanolate complex was detected (Figures S8 and 1A). This observation that only 1,2-diols form hydroxyalkanolate complexes suggests a chromium–hydroxy interaction between the vicinal alcohol and second metal.
Kinetic analysis featuring various protic CTAs corroborates the coordination effects identified by ESI–MS. Polymerizations with 1,6-HD exhibited the same induction period (~1.0 h) as those performed in the absence of a CTA (Figure 3, red and blue, respectively). These results are consistent with the ESI–MS data, which suggests that the bridged 1,6-hydroxyalkanolate complex is not formed. By contrast, polymerizations performed in the presence of 1,2-BD or water did not reach appreciable conversion before 3 h (Figure 3, green and orange, respectively). The additional induction times resulted only from arrest states intrinsically derived by each CTA. When introducing water as a CTA, the catalyst forms both the μ-hydroxide and 1,2-hydroxyalkanolate arrest states, whereas 1,2-BD’s implementation only forms the latter (Scheme 6). With each CTA promoting the same induction period, we recognize that the μ-hydroxide complex must be short-lived and the 1,2-hydroxyalkanolate species is the rate-limiting arrest state.
Figure 3.
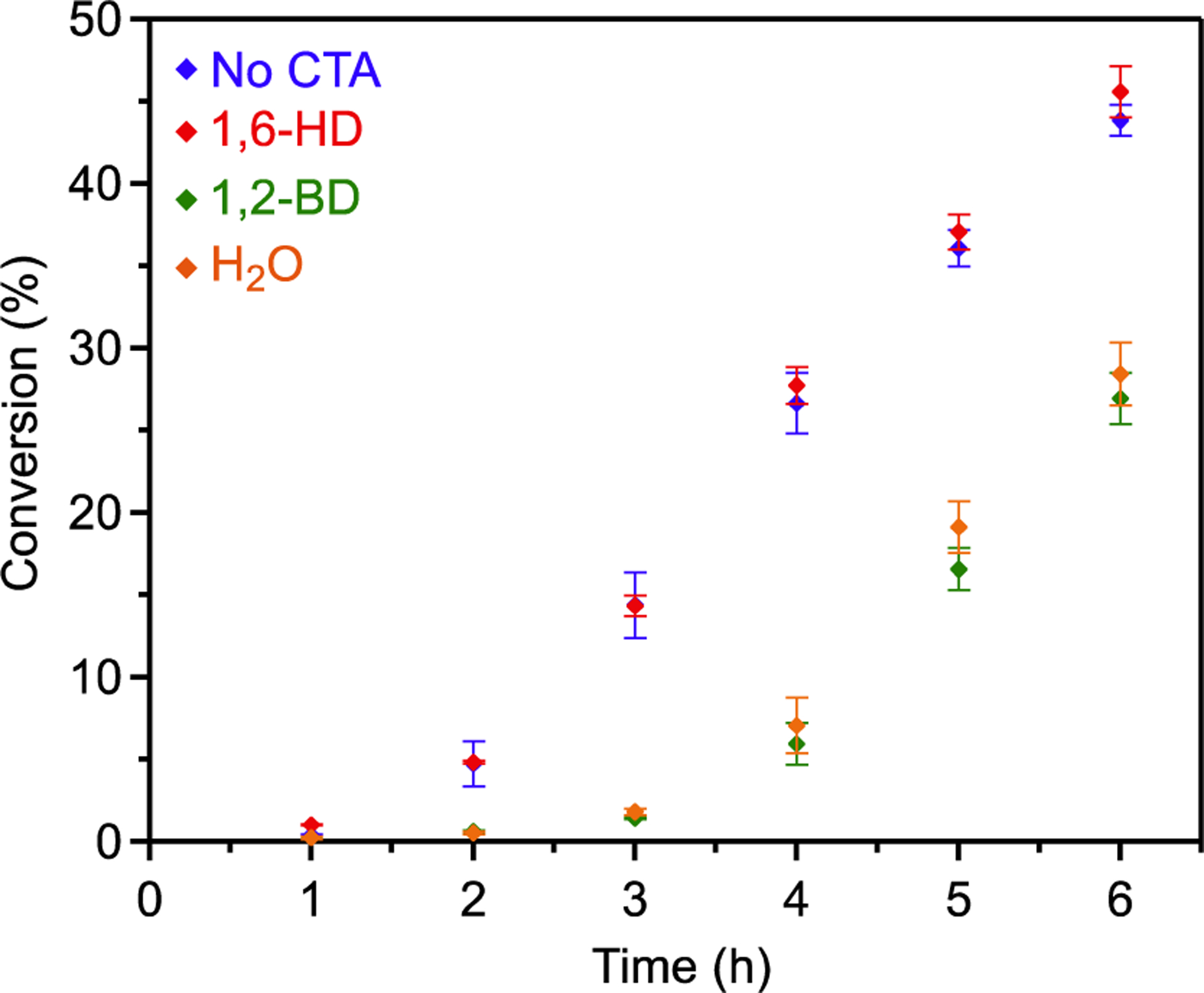
Conversion of PO vs time for the enantioselective polymerization using no CTA, 1,6-HD, 1,2-BD, and H2O as CTAs (1.8 μmol 1, 1.8 μmol [PPN][OAcF3], 18 μmol CTA, and 14.3 mmol PO in 2 mL of DME) detected by 1H NMR. Error represents a standard deviation from the average conversion of three polymerizations.
Scheme 6.
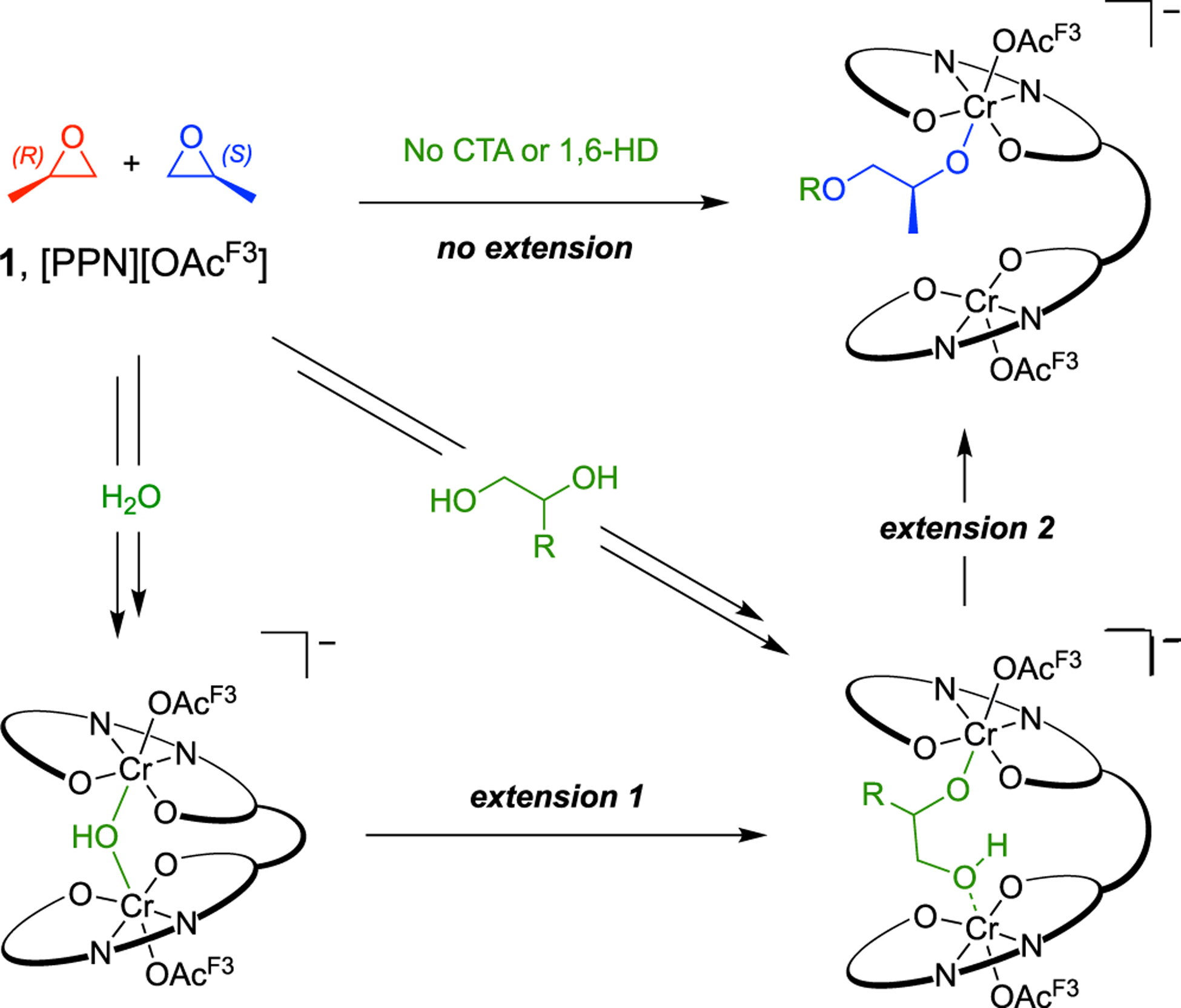
Pathways for Extended Induction Time by CTA
Quantum chemical calculations were conducted to further investigate the catalyst dormancy. Several intermediates present during the induction period were optimized with the B3LYP density functional36 and a mixed 6–31G/6–31G* basis set.37 A truncation of the para-tBu groups on the catalyst was applied to reduce computation time only after quantitative agreement was achieved between select full and truncated models (Figure S17). Additionally, [OAcF3]− anions were exchanged for [Cl]− to further reduce computation time. Minimum energy reaction pathways and transition states were elucidated using the growing string method.38–40 The energies of the [OH]−-bound catalyst intermediates (Figure 4, Series 1) and [1,2-HP]− complexes (Figure 4, Series 2) were compared. The activation barrier to form the PO-bound complex is 40.2 kcal/mol higher for Series 2, supporting 5 as the rate-limiting arrest state. Although the activation barrier from 5 to 7 is quite high, we suspect that intramolecular hydrogen bonding from the terminal alcohol to the Cr alkoxide could reduce the energies of 6 and 7. Both series are well below the energies of propagation, which exhibit moderate activation barriers for catalyst turnover (Figure S19).
Figure 4.
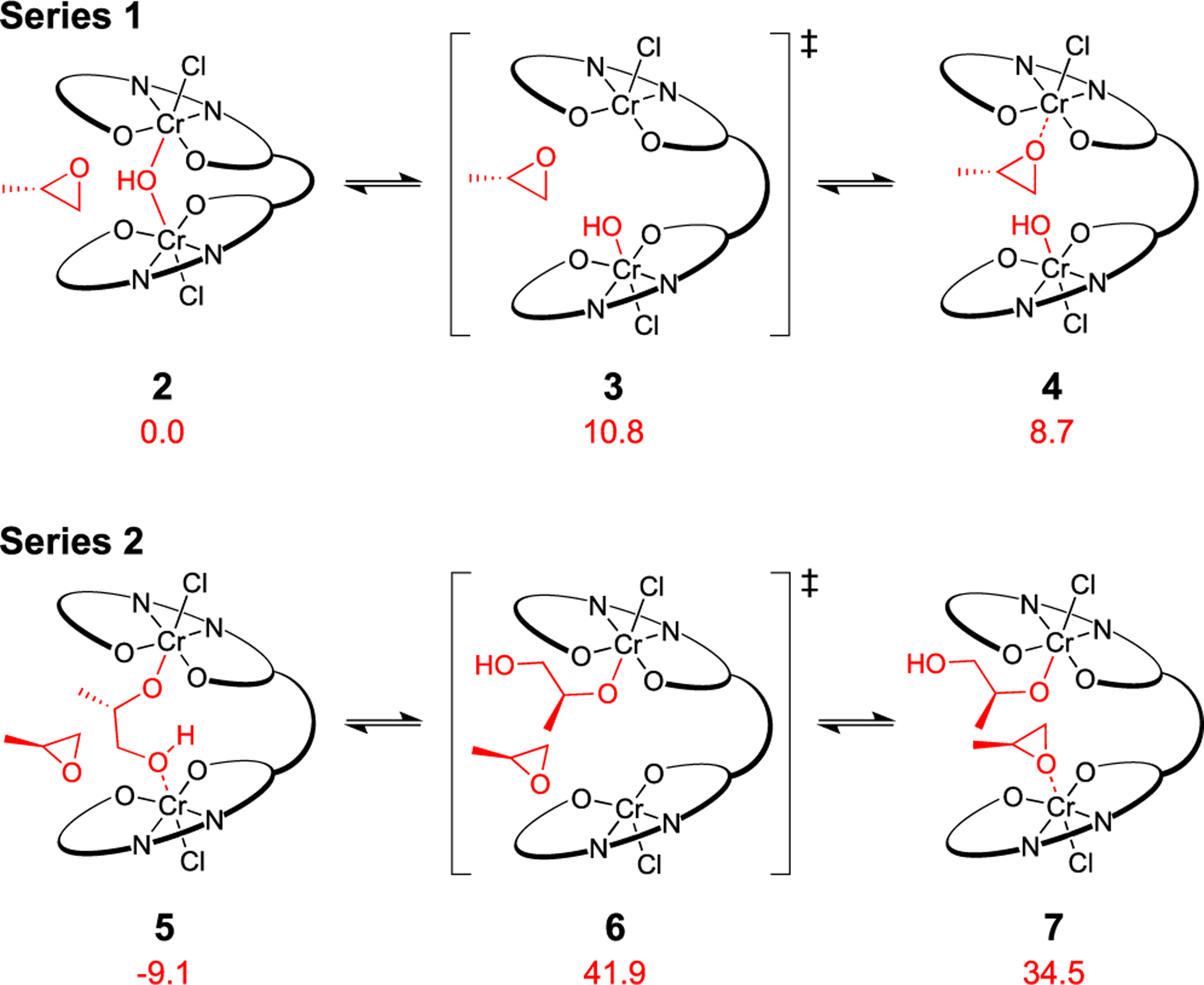
DFT-calculated Gibbs free energies (kcal/mol) of the competing proposed catalyst arrest states referenced to 2 (see the Supporting Information for full diagrams). Series 1 and 2 show epoxide binding from the [OH]− and [1,2-HP]− complexes, respectively.
Although confident that the extended induction period (an additional ~2.0 h) in the presence of vicinal diols or water is due to the 1,2-hydroxyalkanolate complex, we were surprised that the total induction period approximately tripled. Based on our CTA loading of 10 equiv, this suggests that these polymerizations could have contained about 5 equiv of adventitious water relative to 1. The gel permeation chromatogram for a polymerization without a CTA shows that the water content under these standard drying conditions was <1:1 (H2O:1, Figure S1). This led us to investigate catalyst activation as another potential contributor to the induction period. Although 1 was originally portrayed with both anions exterior to the ligand framework (exo–exo), this species could also exist with either one (endo–exo) or both (endo–endo) anions within the active site (Scheme 7). Previous mechanistic work20 suggests that this catalyst requires exo/endo–exo coordination to be active. We hypothesized that a bridged [OAcF3]− complex (bridged exo/endo–exo) could also inhibit propagation acting as another arrest state. Although we anticipate this bridging to occur through Cr–O interactions, the possibility of a Cr–F interaction has not been ruled out.41,42 We suspected that interconversion of [1·OAcF3]− to the active diastereomer and the first PO enchainment could also contribute to the induction period.
Scheme 7.
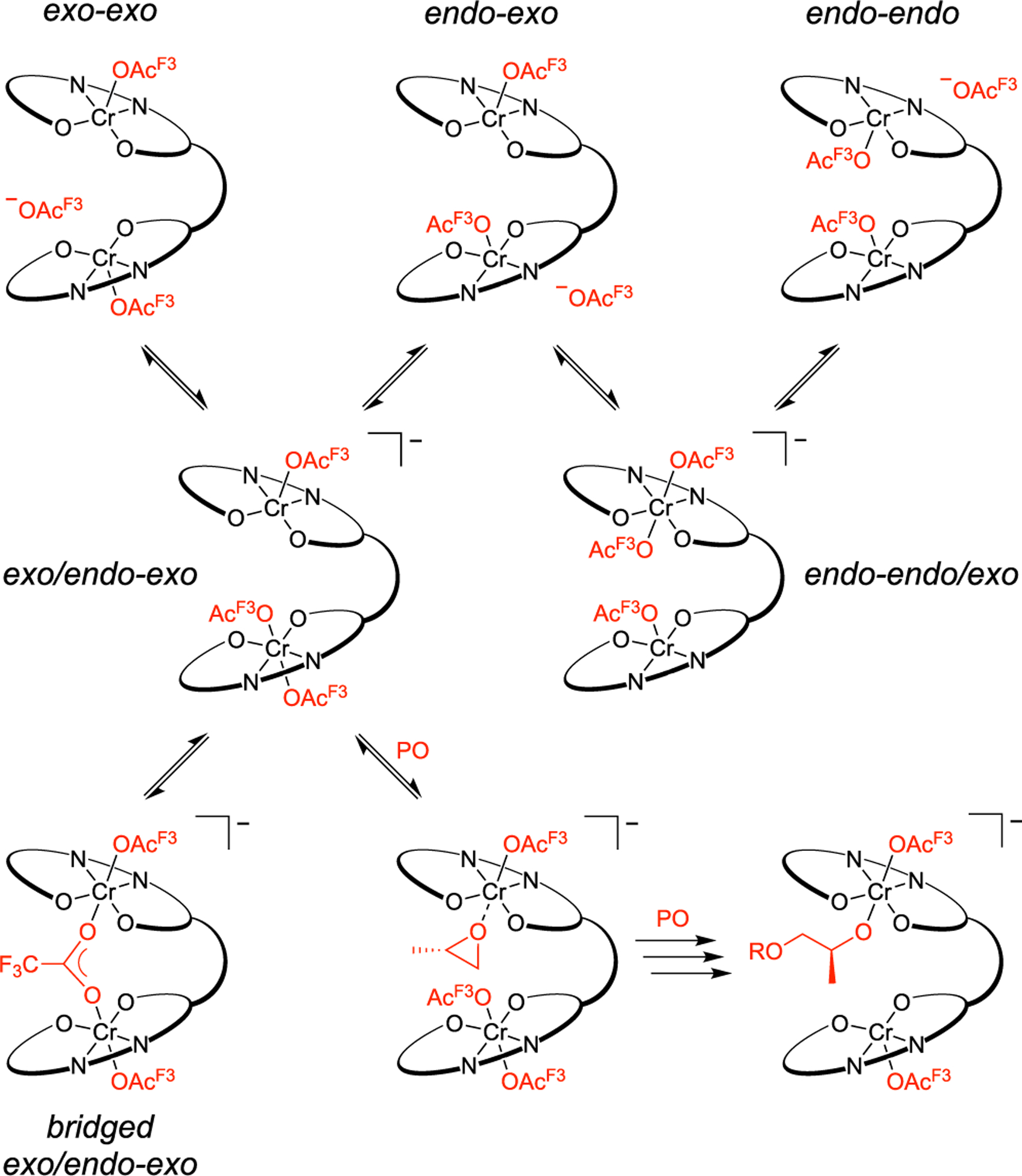
Anion Coordination Interconversion of [1·OAcF3]− to an Active Catalyst
To investigate this hypothesis, complex 8, the dichloride analogue of 1, and [PPN]Cl were subjected to the same kinetic analysis as 1 in the absence of a CTA (Figure 5, blue). The polymerization began almost immediately, providing 8% conversion after the first hour. A previous report featuring a similar bimetallic Co catalyst shows that the ground states of coordination interconversion are closer in energy when using [Cl]− anions compared to [OAc]−.20 Based on the almost nonexistent induction period when using 8, we conclude that the [Cl]− system experiences an overall faster interconversion to the active diastereomer and first PO enchainment. Introducing 1,2-PD as a CTA in the [Cl]− system caused an induction period attributed to the [1,2-HP]− arrest state (Figure 5, purple). Notably, the duration of this induction period matches the extended portion of induction time observed with 1 and 1,2-BD or H2O. The [Cl]− system suffers from the same [1,2-HP]− arrest state as the [OAcF3]− system, but it substantially reduced the induction time when no CTA was used.
Figure 5.
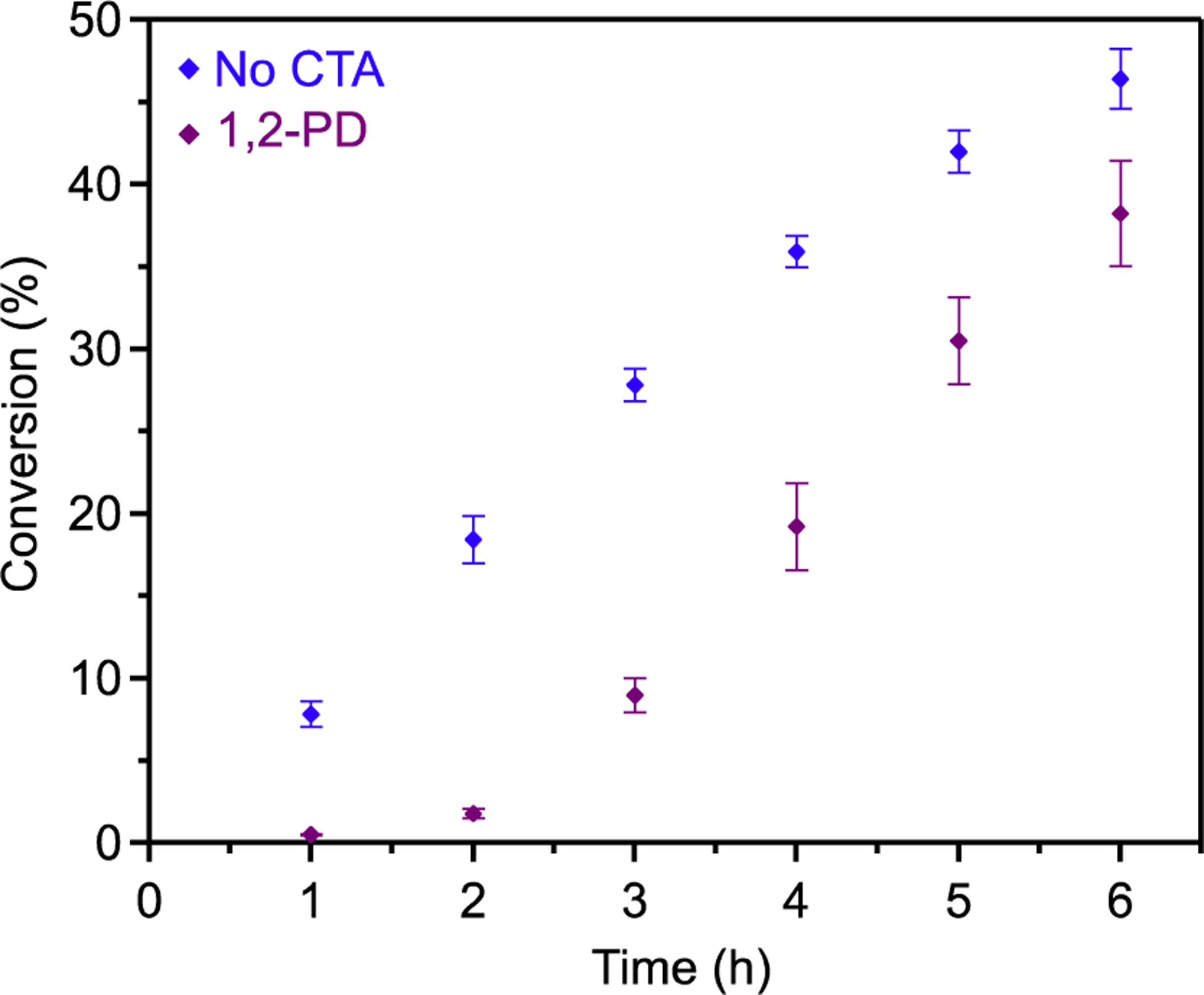
Conversion of PO vs time for the enantioselective polymerization using no CTA and 1,2-PD as a CTA (1.8 μmol 8, 1.8 μmol [PPN]Cl, 18 μmol CTA, and 14.3 mmol PO in 2 mL of DME) detected by 1H NMR. Error represents a standard deviation from the average conversion of three polymerizations.
With ESI–MS, computational, and kinetic data in hand, we proposed a general mechanism explaining this induction period (Scheme 8). First, 1 or 8 reacts with [PPN]X to generate a mixture of diastereomers. Following the addition of PO, monomer coordination to the exo/endo-exo diastereomer yields 9. Subsequent ring-opening yields a small-molecule alkoxide bound to a chromium center (10). The lack of induction period when using 8 with [PPN]Cl supports the fast interconversion of [Cl]− coordination and PO enchainment. Conversely, the standard induction period (~1.0 h) observed with 1 and [PPN][OAcF3] is primarily attributed to the conversion of 1 to 10. In the presence of either adventitious water or water introduced as a CTA, protonation and dissociation of the resulting alcohol produce the μ-hydroxide complex (11). Overcoming a minor activation barrier to cleave a chromium–hydroxide bond, PO becomes datively coordinated to the unsaturated Lewis acid. Facile attack by the hydroxide ring-opens the monomer to form the bridged [1,2-HP]− arrest state (12). Although this arrest state is short-lived relative to the conversion of 1 to 10, using 1,2-diols or H2O as a CTA results in a substantial addition to the induction period. Following chromium–hydroxy bond cleavage of 12 to form 10, PO coordination can remove the catalyst from the chain transfer arrest cycle to form 13. Enchainment of the PO consumes the vicinal alcohol, allowing propagation to occur (14). Although not emphasized in the proposed mechanism, the alcohols generated by chain transfer participate as CTAs during the polymerization.
Scheme 8.
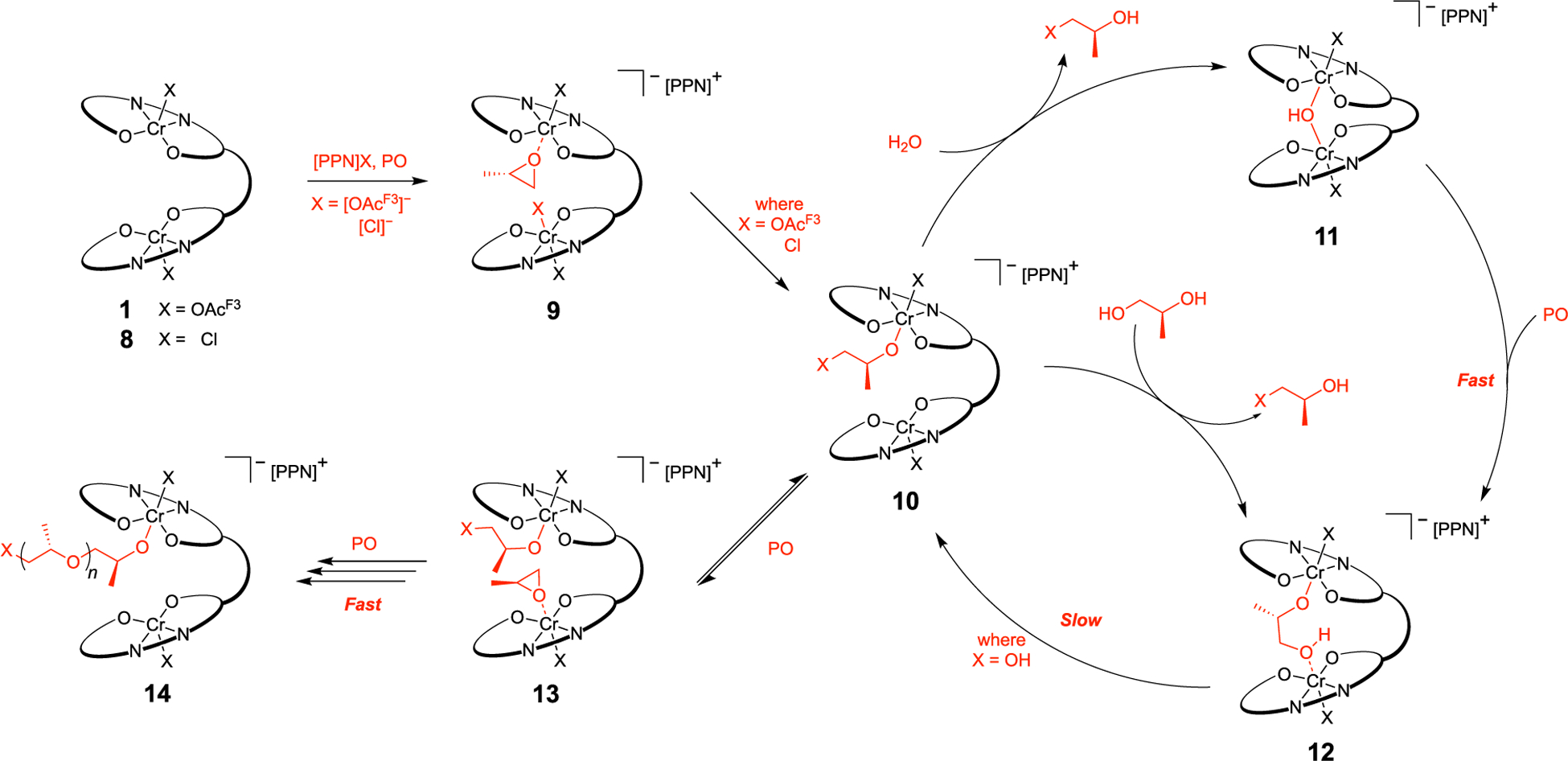
Proposed General Mechanism of iPPO Formation in the Presence of Water and 1,2-PD
CONCLUSIONS
In conclusion, we have identified induction period arrest states of a tethered bimetallic chromium catalyst used in the synthesis of iPPO. Through complementary ESI–MS and kinetic studies, we identified the conversion of adventitious water to [1,2-HP]− and its inhibiting effects on this system. Experimental and computational results indicate that the bridged [1,2-HP]− complex (12), not the μ-hydroxide species (11), is the predominant arrest state responsible for the extended induction period when using water as a CTA. The original induction period was essentially removed when substituting [OAcF3]− with [Cl]−. This effect was attributed to the faster interconversion to the active diastereomer and its initial PO enchainment. Both 1 and 8 were susceptible to forming the bridged [1,2-HP]− arrest state (12), but 8 only generated a noticeable induction period in the presence of an inhibiting CTA. By avoiding water and vicinal diols when using 8, narrowly disperse iPPO can be synthesized without an induction period. This mechanistic study allows for a greater understanding of these enantioselective catalysts and fuels ongoing investigations to further develop these systems.
Supplementary Material
ACKNOWLEDGMENTS
Synthesis and kinetic studies were supported by the Center for Sustainable Polymers, a National Science Foundation (NSF) Center for Chemical Innovation (CHE-1901635; B.M.L., L.S.M., and G.W.C.). Mass spectrometry studies were supported by the Office of Naval Research (ONR N000141410551; N.E.C. and R.M.W.), the NSF-GFRP (K.L.W.), and the Air Force Office of Scientific Research through a Basic Research Initiative grant (AFOSRFA9550-16-1-0113; R.N.Z.). Computation was supported by the National Institute of Health through an Outstanding Investigator Award (R35-GM-128830; T.M.E.J., A.G.R., and P.M.Z.). Solid-state structure analysis was supported by Cornell University (Y.D.Y.L.G. and S.N.M.). This work made use of the NMR Facility at Cornell University, which is supported, in part, by the NSF under the award CHE-1531632.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.0c02135
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c02135.
Crystallization and structural parameter details of a μ-hydroxide complex (CIF)
General experimental considerations, detailed procedures, and characterization spectra (PDF)
The authors declare no competing financial interest.
Contributor Information
Bryce M. Lipinski, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853-1301, United States;.
Lilliana S. Morris, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853-1301, United States;.
Richard N. Zare, Department of Chemistry, Stanford University, Stanford, California 94305-5080, United States;.
Paul M. Zimmerman, Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109-1382, United States;.
Robert M. Waymouth, Department of Chemistry, Stanford University, Stanford, California 94305-5080, United States;.
Geoffrey W. Coates, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853-1301, United States;.
REFERENCES
- (1).Engels H-W; Pirkl H-G; Albers R; Albach RW; Krause J; Hoffmann A; Casselmann H; Dormish J Polyurethanes: Versatile Materials and Sustainable Problem Solvers for Today’s Challenges. Angew. Chem., Int. Ed 2013, 52, 9422–9441. [DOI] [PubMed] [Google Scholar]
- (2).Wegener G; Brandt M; Duda L; Hofmann J; Klesczewski B; Koch D; Kumpf R-J; Orzesek H; Pirkl H-G; Six C; Steinlein C; Weisbeck M Trends in industrial catalysis in the polyurethane industry. Appl. Catal., A 2001, 221, 303–335. [Google Scholar]
- (3).Lipinski BM; Morris LS; Silberstein MN; Coates GW Isotactic Poly(Propylene Oxide): A Photodegradable Polymer with Strain Hardening Properties. J. Am. Chem. Soc 2020, 142, 6800–6806. [DOI] [PubMed] [Google Scholar]
- (4).Gardette J-L; Mailhot B; Posada F; Rivaton A; Wilhelm C Photooxidative Degradation of Polyether-Based Polymers. Macromol. Symp 1999, 143, 95–109. [Google Scholar]
- (5).Gauvin P; Lemaire J; Sallet D Photo-Oxydation de Polyether-Bloc-Polyamides. Makromol. Chem 1987, 188, 1815–1824. [Google Scholar]
- (6).Morlat S; Gardette J-L Phototransformation of Water-Soluble Polymers. I: Photo- and Thermooxidation of Poly(Ethylene Oxide) in Solid State. Polymer 2001, 42, 6071–6079. [Google Scholar]
- (7).Kawai F Microbial Degradation of Polyethers. Appl. Microbiol. Biotechnol 2002, 58, 30–38. [DOI] [PubMed] [Google Scholar]
- (8).Beran E; Hull S; Steininger M The Relationship Between the Chemical Structure of Poly(Alkylene Glycol)s and Their Aerobic Biodegradability in an Aqueous Environment. J. Polym. Environ 2013, 21, 172–180. [Google Scholar]
- (9).West RJ; Davis JW; Pottenger LH; Banton MI; Graham C Biodegradability Relationships among Propylene Glycol Substances in the Organization for Economic Cooperation and Development Ready- and Seawater Biodegradability Tests. Environ. Toxicol. Chem 2007, 26, 862–871. [DOI] [PubMed] [Google Scholar]
- (10).Zgola-Grzeskowiak A; Grzeskowiak T; Zembrzuska J; Franska M; Franski R; Kozik T; Lukaszewski Z Biodegradation of Poly(Propylene Glycol)s under the Conditions of the OECD Screening Test. Chemosphere 2007, 67, 928–933. [DOI] [PubMed] [Google Scholar]
- (11).Childers MI; Longo JM; Van Zee NJ; LaPointe AM; Coates GW Stereoselective Epoxide Polymerization and Copolymerization. Chem. Rev 2014, 114, 8129–8152. [DOI] [PubMed] [Google Scholar]
- (12).Ghosh S; Lund H; Jiao H; Mejía E Rediscovering the Isospecific Ring-Opening Polymerization of Racemic Propylene Oxide with Dibutylmagnesium. Macromolecules 2017, 50, 1245–1250. [Google Scholar]
- (13).Ferrier RC; Pakhira S; Palmon SE; Rodriguez CG; Goldfeld DJ; Iyiola OO; Chwatko M; Mendoza-Cortes JL; Lynd NA Demystifying the Mechanism of Regio- and Isoselective Epoxide Polymerization Using the Vandenberg Catalyst. Macromolecules 2018, 51, 1777–1786. [Google Scholar]
- (14).Morris LS; Childers MI; Coates GW Bimetallic Chromium Catalysts with Chain Transfer Agents: A Route to Isotactic Poly(propylene oxide)s with Narrow Dispersities. Angew. Chem., Int. Ed 2018, 57, 5731–5734. [DOI] [PubMed] [Google Scholar]
- (15).Childers MI; Vitek AK; Morris LS; Widger PCB; Ahmed SM; Zimmerman PM; Coates GW Isospecific, Chain Shuttling Polymerization of Propylene Oxide Using a Bimetallic Chromium Catalyst: A New Route to Semicrystalline Polyols. J. Am. Chem. Soc 2017, 139, 11048–11054. [DOI] [PubMed] [Google Scholar]
- (16).Konsler RG; Karl J; Jacobsen EN Cooperative Asymmetric Catalysis with Dimeric Salen Complexes. J. Am. Chem. Soc 1998, 120, 10780–10781. [Google Scholar]
- (17).Nielsen LPC; Zuend SJ; Ford DD; Jacobsen EN Mechanistic Basis for High Reactivity of (salen)Co–OTs in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Org. Chem 2012, 77, 2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nielsen LPC; Stevenson CP; Blackmond DG; Jacobsen EN Mechanistic Investigation Leads to a Synthetic Improvement in the Hydrolytic Kinetic Resolution of Terminal Epoxides. J. Am. Chem. Soc 2004, 126, 1360–1362. [DOI] [PubMed] [Google Scholar]
- (19).Ford DD; Nielsen LPC; Zuend SJ; Musgrave CB; Jacobsen EN Mechanistic Basis for High Stereoselectivity and Broad Substrate Scope in the (Salen)Co(III)-Catalyzed Hydrolytic Kinetic Resolution. J. Am. Chem. Soc 2013, 135, 15595–15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ahmed SM; Poater A; Childers MI; Widger PCB; LaPointe AM; Lobkovsky EB; Coates GW; Cavallo L Enantioselective Polymerization of Epoxides Using Biaryl-Linked Bimetallic Cobalt Catalysts: A Mechanistic Study. J. Am. Chem. Soc 2013, 135, 18901–18911. [DOI] [PubMed] [Google Scholar]
- (21).Lu X-B; Shi L; Wang Y-M; Zhang R; Zhang Y-J; Peng X-J; Zhang Z-C; Li B Design of Highly Active Binary Catalyst Systems for CO2/Epoxide Copolymerization: Polymer Selectivity, Enantioselectivity, and Stereochemistry Control. J. Am. Chem. Soc 2006, 128, 1664–1674. [DOI] [PubMed] [Google Scholar]
- (22).Chukanova OM; Perepelitsina EO; Belov GP The influence of reagent concentration on the kinetics of carbon dioxide-propylene oxide copolymerization in the presence of a cobalt complex. Polym. Sci. B 2014, 56, 547–552. [Google Scholar]
- (23).Na SJ; S S; Cyriac A; Kim BE; Yoo J; Kang YK; Han SJ; Lee C; Lee BY Elucidation of the Structure of a Highly Active Catalytic System for CO2/Epoxide Copolymerization: A salen-Cobaltate Complex of an Unusual Binding Mode. Inorg. Chem 2009, 48, 10455–10465. [DOI] [PubMed] [Google Scholar]
- (24).Wu G-P; Darensbourg DJ Mechanistic Insights into Water-Mediated Tandem Catalysis of Metal-Coordination CO2/Epoxide Copolymerization and Organocatalytic Ring-Opening Polymerization: One-Pot, Two Steps, and Three Catalysis Cycles for Triblock Copolymers Synthesis. Macromolecules 2016, 49, 807–814. [Google Scholar]
- (25).Sugimoto H; Inoue S Copolymerization of Carbon Dioxide and Epoxide. J. Polym. Sci., Part A: Polym. Chem 2004, 42, 5561–5573. [Google Scholar]
- (26).Nakano K; Kamada T; Nozaki K Selective Formation of Polycarbonate over Cyclic Carbonate: Copolymerization of Epoxides with Carbon Dioxide Catalyzed by a Cobalt(III) Complex with a Piperidinium End-Capping Arm. Angew. Chem., Int. Ed 2006, 45, 7274–7277. [DOI] [PubMed] [Google Scholar]
- (27).Kember MR; White AJP; Williams CK Highly Active Di- and Trimetallic Cobalt Catalysts for the Copolymerization of CHO and CO2 at Atmospheric Pressure. Macromolecules 2010, 43, 2291–2298. [Google Scholar]
- (28).Liu B; Gao Y; Zhao X; Yan W; Wang X Alternating copolymerization of carbon dioxide and propylene oxide under bifunctional cobalt salen complexes: Role of Lewis base substituent covalent bonded on salen ligand. J. Polym. Sci., Part A: Polym. Chem 2010, 48, 359–365. [Google Scholar]
- (29).Ingram AJ; Boeser CL; Zare RN Going beyond electrospray: mass spectrometric studies of chemical reactions in and on liquids. Chem. Sci 2016, 7, 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yunker LPE; Stoddard RL; McIndoe JS Practical approaches to the ESI-MS analysis of catalytic reactions. J. Mass Spectrom 2014, 49, 1–8. [DOI] [PubMed] [Google Scholar]
- (31).Vikse KL; Ahmadi Z; Scott McIndoe J The application of electrospray ionization mass spectrometry to homogeneous catalysis. Coord. Chem. Rev 2014, 279, 96–114. [Google Scholar]
- (32).Rao D-Y; Li B; Zhang R; Wang H; Lu X-B Binding of 4-(N,N-dimethylamino)pyridine to Salen- and Salan-Cr(III) Cations: A Mechanistic Understanding on the Difference in Their Catalytic Activity for CO2/Epoxide Copolymerization. Inorg. Chem 2009, 48, 2830–2836. [DOI] [PubMed] [Google Scholar]
- (33).Davis DC; Walker KL; Hu C; Zare RN; Waymouth RM; Dai M Catalytic Carbonylative Spirolactonization of Hydroxycyclopropanols. J. Am. Chem. Soc 2016, 138, 10693–10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rudenko AE; Clayman NE; Walker KL; Maclaren JK; Zimmerman PM; Waymouth RM Ligand-Induced Reductive Elimination of Ethane from Azopyridine Palladium Dimethyl Complexes. J. Am. Chem. Soc 2018, 140, 11408–11415. [DOI] [PubMed] [Google Scholar]
- (35).Gruendling T; Weidner S; Falkenhagen J; Barner-Kowollik C Mass spectrometry in polymer chemistry: a state-of-the-art up-date. Polym. Chem 2010, 1, 599–617. [Google Scholar]
- (36).Becke AD Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (37).Rassolov VA; Pople JA; Ratner MA; Windus TL 6–31G* basis set for atoms K through Zn. J. Chem. Phys 1998, 109, 1223–1229. [Google Scholar]
- (38).Zimmerman PM Growing string method with interpolation and optimization in internal coordinates: Method and examples. J. Chem. Phys 2013, 138, 184102. [DOI] [PubMed] [Google Scholar]
- (39).Zimmerman P Reliable Transition State Searches Integrated with the Growing String Method. J. Chem. Theory Comput 2013, 9, 3043–3050. [DOI] [PubMed] [Google Scholar]
- (40).Zimmerman PM Single-ended transition state finding with the growing string method. J. Comput. Chem 2015, 36, 601–611. [DOI] [PubMed] [Google Scholar]
- (41).Batsanov AS; Dillon KB; Gibson VC; Howard JAK; Sequeira LJ; Yao JW Synthesis and Crystal Structures of Chromium and Molybdenum Complexes Containing the 2,4,6-Tris(Trifluoromethyl)Phenyl Ligand. J. Organomet. Chem 2001, 631, 181–187. [Google Scholar]
- (42).Plenio H The Coordination Chemistry of the CF Unit in Fluorocarbons. Chem. Rev 1997, 97, 3363–3384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.