

 This work is licensed under a
This work is licensed under a Abstract
Objective
Hypoglycemia is associated with an increased risk of cardiovascular disease including cardiac arrhythmias. We investigated the effect of hypoglycemia in the setting of acute glycemic fluctuations on cardiac rhythm and cardiac repolarization in insulin-treated patients with type 2 diabetes compared with matched controls without diabetes.
Design
A non-randomized, mechanistic intervention study.
Methods
Insulin-treated patients with type 2 diabetes (n = 21, age (mean ± s.d.): 62.8 ± 6.5 years, BMI: 29.0 ± 4.2 kg/m2, HbA1c: 6.8 ± 0.5% (51.0 ± 5.4 mmol/mol)) and matched controls (n = 21, age: 62.2 ± 8.3 years, BMI 29.2 ± 3.5 kg/m2, HbA1c: 5.3 ± 0.3% (34.3 ± 3.3 mmol/mol)) underwent a sequential hyperglycemic and hypoglycemic clamp with three steady-states of plasma glucose: (i) fasting plasma glucose, (ii) hyperglycemia (fasting plasma glucose +10 mmol/L) and (iii) hyperinsulinemic hypoglycemia (plasma glucose < 3.0 mmol/L). Participants underwent continuous ECG monitoring and blood samples for counterregulatory hormones and plasma potassium were obtained.
Results
Both groups experienced progressively increasing heart rate corrected QT (Fridericia’s formula) interval prolongations during hypoglycemia ((∆mean (95% CI): 31 ms (16, 45) and 39 ms (24, 53) in the group of patients with type 2 diabetes and controls, respectively) with similar increases from baseline at the end of the hypoglycemic phase (P = 0.43). The incidence of ventricular premature beats increased significantly in both groups during hypoglycemia (P = 0.033 and P < 0.0001, respectively). One patient with type 2 diabetes developed atrial fibrillation during recovery from hypoglycemia.
Conclusions
In insulin-treated patients with type 2 diabetes and controls without diabetes, hypoglycemia causes clinically significant and similar increases in cardiac repolarization that might increase vulnerability for serious cardiac arrhythmias and sudden cardiac death.
Introduction
Patients with type 2 diabetes have an increased risk of cardiovascular disease and death (1). The cornerstone in the treatment of type 2 diabetes is lifestyle intervention, glucose-lowering therapy along with the treatment of traditional cardiovascular risk factors including hypertension and dyslipidemia. Several large-scale clinical trials that investigated the effect of strict glycemic control failed to demonstrate any cardiovascular risk reduction (2), and in the ACCORD trial, strict glycemic control resulted in a three-fold increase in severe hypoglycemia and excess mortality (3). The notion of an association between hypoglycemia and cardiovascular disease is supported by multiple studies; however, a causal relationship has not been proven (4).
In individuals with type 2 diabetes and high cardiovascular risk, current consensus recommends treatment with a sodium-glucose cotransporter 2 (SGLT2) inhibitor or a glucagon-like peptide 1 (GLP-1) receptor agonist with proven cardiovascular disease benefit and low risk of hypoglycemia (5). Nevertheless, insulin therapy is often necessary to achieve satisfactory glycemic control thereby increasing the risk of hypoglycemia.
Potential mechanisms by which hypoglycemia may promote cardiovascular disease include blood coagulation abnormalities, inflammation, endothelial dysfunction and hemodynamic changes (4). Additionally, the counterregulatory response during hypoglycemia with pronounced sympathoadrenal activation is suspected to cause abnormal cardiac repolarization and induce cardiac arrhythmias (6). It is well established that heart rate-corrected QT interval (QTc) is prolonged during hypoglycemia in patients with type 1 diabetes and type 2 diabetes (7). QTc prolongation is associated with an increased risk of ventricular arrhythmias and is a strong predictor of cardiovascular mortality in type 2 diabetes (8, 9), which provides a potential explanation for the increased mortality observed in the ACCORD trial (3). In insulin-treated patients with type 2 diabetes, spontaneous episodes of hypoglycemia are associated with an increased incidence of ventricular premature beats and nocturnal bradycardia (10).
In previous experimental studies of insulin-induced hypoglycemia, the effect of hypoglycemia on cardiac repolarization has been compared to cardiac repolarization during hyperinsulinemic euglycemia (11, 12, 13, 14). Hence, hypoglycemia was preceded by a relatively small decline in plasma glucose (PG). It has also been suggested that diabetes may aggravate the risk of cardiac rhythm abnormalities (11, 15); however, in the only study comparing the electrophysiological response to hypoglycemia in patients with type 2 diabetes with the response in controls without diabetes, only a fraction of the patients with type 2 diabetes were insulin-treated (11). Consequently, it is uncertain whether the results from this study translate into a risk population of insulin-treated patients.
In the present study, we aimed to investigate the effect of hypoglycemia in the setting of marked (but clinically relevant), acute glycemic fluctuations on cardiac rhythm, QTc interval and hormonal counterregulatory response in patients with insulin-treated type 2 diabetes compared to matched controls with normal glucose tolerance.
Subjects and methods
Approvals and registrations
The present study was carried out at Steno Diabetes Center Copenhagen and Center for Clinical Metabolic Research, Gentofte Hospital, Hellerup, Denmark from May 2017 to July 2019. The study was conducted in accordance with the Declaration of Helsinki as revised in 2008 and was approved by the Scientific Ethical Committee of the Capital Region of Denmark (ID No. H-16046212) and the Danish Data Protection Agency (ID No. HGH-2017-030) and registered at ClinicalTrials.gov (NCT03150030). Oral and written consent was obtained from all participants prior to inclusion in the study.
Design and study population
The present study consisted of a combined hyperglycemic and hypoglycemic clamp. Twenty-one adults with insulin-treated type 2 diabetes with good glycemic control (HbA1c ≤ 58 mmol/mol (7.5%)) and at least one microvascular complication were included. Complications to diabetes were defined as: peripheral neuropathy with vibration perception threshold of >25 mV determined by biothesiometry, moderate to severe retinopathy, and/or nephropathy (creatinine > 130 μmol/L and/or albuminuria). A group of 21 individuals with HbA1c ≤ 42 mmol/mol (6.0%) and fasting PG (FPG) ≤ 6.1 mmol/L matched for sex, age and BMI served as controls. Individuals in the control group were excluded if they had any first-degree relatives with diabetes. Individuals with a medical history of episodes of arrhythmia, implantable cardiac defibrillator or pacemaker, severe heart failure (defined by the protocol as left ventricular ejection fraction < 25%) or structural heart disease (e.g. Wolf–Parkinson–White syndrome, congenital heart disease, severe valve disease) were excluded in both groups to ensure safety and avoid bias from any underlying arrhythmic substrate. Individuals with anemia or thyroid dysfunction (except well-regulated myxedema) were also excluded. Patients with type 2 diabetes were screened for hypoglycemia unawareness by asking the patients to which extent they were able to recognize episodes of hypoglycemia with answers categorized as (i) always (ii) sometimes or (iii) never. The latter two were categorized as impaired awareness.
The clamp procedure
All participants were instructed to fast from 22:00 h and to avoid strenuous physical activity the day before the experimental day and not to take any medication (including insulin) on the experimental day. None of the participants measured plasma glucose levels < 3.0 mmol/L 24 h prior to the start of the experiment. A FPG of ≥ 4.0 and ≤ 12.0 mmol/L was required by protocol to initiate the experimental day. A peripheral intravascular catheter was placed in an anterior cubital vein of each forearm. One arm was heated throughout the clamp to facilitate repeated blood sampling and to achieve measures closer to arterial blood concentrations, and the intravascular catheter on the contralateral side was used for infusions. The clamp experiment was started between 8:30 h and 11:00 h. To avoid volume depletion due to fasting and blood drawing, an isotonic saline infusion was initiated at time 0 (infusion rate 200 mL/h) and kept at a constant infusion rate. Blood was drawn and analyzed for PG every 5 min. After 30 min of monitoring at FPG level (FPG phase), an individually adjusted infusion of 20% (weightt/volume) glucose was initiated and PG was increased to FPG + 10 mmol/L over a period of 50 min and kept constant for 30 min (hyperglycemic phase). At time 110 min, rapid acting insulin (Actrapid®, Novo Nordisk) was administrated as an i.v. bolus and an i.v. infusion was initiated. Insulin bolus dose was calculated as 9.1 IU times body surface area (m2) (BSA) estimated by the Mosteller formula (16) for patients with diabetes and 2.5 IU times BSA for controls. The corresponding insulin infusion rates were calculated as 20 IU/h times BSA for patients with diabetes and 5 IU/h times BSA for controls. If judged necessary by the principal investigator (per protocol), the insulin dose was halved in participants with diabetes and low daily insulin dose. A steady-state PG < 3.0 mmol/L was targeted to occur from 160 min to 190 min (hypoglycemic phase) followed by a spontaneous recovery period from 190 min to 220 min. The insulin infusion was discontinued after 10 min of PG < 3.0 mmol/L. If PG was < 2.2 mmol/L or the trend indicated that the next measurement would be below this level, PG was immediately corrected by glucose infusion.
ECG monitoring and analysis
Participants underwent ECG monitoring with a three-lead (XYZ) Holter monitor (Spiderview version 3.03A, Microport CRM, Clamart, France). All three leads were bipolar with a positive (+) and a negative (−) component. The electrodes generating the X-, Y-, and Z-lead were placed in the right (−) and left mid-axillary line (+), the superior part of manubrium (−) and below xiphoid process (+), and at the fourth intercostal space left to manubrium(+) and posterior to fourth intercostal space (−), respectively. Signals were sampled at 1000 Hz and band filtered with a low pass 50 Hz filter. Pre-processing and data analysis were performed with SyneScope version 3.10, Microport CRM, Clamart, France. Holter recordings were manually reviewed for clinically relevant cardiac arhrythmias including atrial fibrillation, bradyarrhythmias (sinus arrest for more than 3 s, frequency below 30 b.p.m, or high grade atrioventricular (AV) block) and tachyarrhythmias (ventricular tachycardia). QTc interval was calculated as means within 10 min intervals from all three leads by an automated algorithm determining a mean complex waveform based on 30 s of ECG. The peak of the T-wave was determined by the parabola method and the end of the T-wave by the intersection between the maximum decreasing tangent and the isoelectric line (17). Heart rate correction was performed by both Fridericia’s correction (QTcF) and Bazett’s correction (QTcB). Mean heart rate was calculated for each phase in 1-min intervals. The software automatically detected supraventricular premature beats (prematurity threshold < 75%) and ventricular premature beats (prematurity threshold < 85%), and all events were subsequently manually validated.
Blood samples and analyses
Blood samples for PG measurement were drawn into fluoride tubes and centrifuged immediately at 8200 g for 30 s before analyzed bedside using YSI model 2300 or 2900 biochemistry analyzer (Xylem Analytics, OH, USA). All other samples were drawn in lithium-heparin tubes (potassium), EDTA tubes (glucagon), tubes containing serum clot activator (cortisol and somatotropin), and iced serum tubes containing EGTA/glutathione preservation (noradrenaline). Plasma potassium was analyzed by the indirect ion-selective electrodes method (Atellica CH 930 Analyzer, Siemens Healthineers). Glucagon was analyzed by RIA, and cortisol and somatotropin were analyzed using a quantitative electrochemiluminescence assay (Cobas 8000, Roche Diagnostics). Noradrenalin was analyzed with an ELISA (ELISA) (Labor Diagnostika Nord, Nordhorn, Germany).
Statistical analysis
Statistical analyses were performed with SAS studio version 3.71 (SAS Institute Inc., Cary, NC, USA). When describing baseline characteristics of the clamp procedure, data that followed an approximate normal distribution were presented as mean ± s.d., and skewed data were summarized as median (interquartile range). When describing endpoints, data were presented as mean and 95% CI or depicted as mean and s.e. in figures. To evaluate changes in QTc intervals, a general linear mixed model with phase as a repeated factor, ID as a random factor and an unrestricted covariance structure was applied. A similar general linear mixed model was applied to compare counts of ventricular and supraventricular premature beats during hyperglycemia and hypoglycemia with the FPG phase, and data were fitted to a negative binomial model to allow modeling of over-dispersed data and presented as incident rate ratios (IRR). Furthermore, a sensitivity analysis with the elimination of single participants with high count numbers demonstrated was performed. Hormonal counterregulatory response was measured as a baseline-subtracted area under the curve, with baseline defined as the end of the hyperglycemic phase. Differences in hormonal counterregulatory response were evaluated by an unpaired t-test. A P-value < 0.05 was considered statistically significant.
Results
Participants
The groups were well matched for age, sex and BMI (Table 1). Mean blood pressure was similar between groups, but antihypertensive therapy was more prevalent in the group of patients with type 2 diabetes. None of the participants had pre-existing ischemic heart disease or heart failure. Four participants in the group of patients with type 2 diabetes were unable to complete the full experimental day due to vasovagal symptoms (e.g. nausea) during the decline in PG before reaching hypoglycemia. One participant in the control group experienced a transient (2–3 min) seizure immediately after reaching hypoglycemia at a PG level of 2.4 mmol/L, but the seizure was immediately resolved when normal PG was restored. The participant was subsequently admitted for observation for 24 h before discharged without sequela. No explanation for the apparently low seizure threshold was found. All five participants, who were unable to complete the experimental day, were excluded from the analysis and replaced, resulting in 21 participants completing the experimental procedure in each group (Table 1).
Table 1.
Baseline characteristics of the group of insulin-treated patients with type 2 diabetes and the group of controls matched according to age, sex and BMI. Binary data are presented as n (%) and continuous variables are presented as mean (s.d.) or median (interquartile range).
| Type 2 diabetes (n = 21) | Controls (n = 21) | P | |
|---|---|---|---|
| Age (years) | 62.8 (6.5) | 62.2 (8.3) | 0.805 |
| Female | 5 (24%) | 5 (24%) | 1.000 |
| BMI (kg/m2) | 29.0 (4.2) | 29.2 (3.5) | 0.873 |
| HbA1c (mmol/mol) | 51.0 (5.4) | 34.3 (3.3) | <0.001 |
| HbA1c (%) | 6.8 (0.5) | 5.3 (0.3) | <0.001 |
| FPG (mmol/L) | 7.2 (1.7) | 5.7 (0.3) | <0.001 |
| C-peptide (pmol/L) | 259 (174–370)a | 440 (344–573) | 0.006 |
| Insulin treatment | 21 (100%) | NA | |
| Basal | 12 (57%) | ||
| Basal/bolus | 4 (19%) | ||
| Insulin mix | 5 (24%) | ||
| Daily insulin dose (IU) | 38 (25–60) | NA | |
| Oral glucose-lowering drugs | 16 (76%) | NA | |
| Metformin | 15 (71%) | ||
| SGLT2i | 7 (33%) | ||
| DDP-4i | 4 (19%) | ||
| Diabetes duration (years) | 15.3 (6.6) | NA | |
| Impaired awareness | 6 (29%) | NA | |
| Neuropathy | 18 (86%) | NA | |
| Retinopathy | 7 (33%) | NA | |
| Nephropathy | 3 (14%) | NA | |
| Hypertension | 18 (86%) | 4 (19%) | <0.001 |
| Beta-blockers | 4 (19%) | 1 (5%) | |
| Non-dihydropyridine calcium antagonist | 1 (5%) | 0 (%) | |
| Systolic blood pressure (mmHg) | 138.0 (12.4) | 137.0 (11.0) | 0.794 |
| Diastolic blood pressure (mmHg) | 80.3 (7.5) | 82.6 (7.3) | 0.324 |
| Heart rate (b.p.m) | 67.1 (10.7) | 60.0 (9.6) | 0.027 |
| Creatinine (µmol/L) | 87.6 (25.2) | 83.0 (10.3) | 0.452 |
| Potassium (mmol/L) | 4.1 (0.4) | 4.1 (0.3) | 0.778 |
aFasting levels while on active insulin treatment.
DDP-4i, dipeptidyl peptidase-4 inhibitor; FPG, fasting plasma glucose; SGLT2i, sodium-glucose transport protein 2 inhibitor; T2D, type 2 diabetes.
The clamp procedure
During the FPG phase and the hyperglycemic phase, PG levels were higher in the group of patients with type 2 diabetes compared with controls (6.9 ± 1.5 8 mmol/L vs 5.4 ± 0.5 8 mmol/L, P < 0.0001 and 16.4 ± 1.8 mmol/L vs 15.6 ± 1.3 mmol/L, P < 0.0001, respectively) (Fig. 1A ). Mean PG was similar between groups during hypoglycemia (2.5 ± 0.4 mmol/L in both groups, P = 0.53). At the end of the hyperglycemic phase, the cumulative amount of glucose infused was 53.0 (41.4–59.0) g and 62.8 (55.2–80.2) g in patients with type 2 diabetes and controls, respectively. Corresponding results at the end of the hypoglycemic phase were 53.6 (47.8–59.8) g and 69.9 (60.8–83.4) g, respectively. Time from ending the hyperglycemic phase until reaching PG ≤ 3.0 mmol/L exceeded the planned 50 min and was 72 ± 17 min and 59 ± 13 min in the group of patients with type 2 diabetes and controls, respectively. The PG decline rate within this period was 0.20 ± 0.04 mmol/L/min in the group of patients with type 2 diabetes and 0.22 ± 0.04 mmol/L/min in controls. Due to the delay in time to hypoglycemia, data on the recovery phase were not systematically obtained and are, therefore, not provided.
Figure 1.
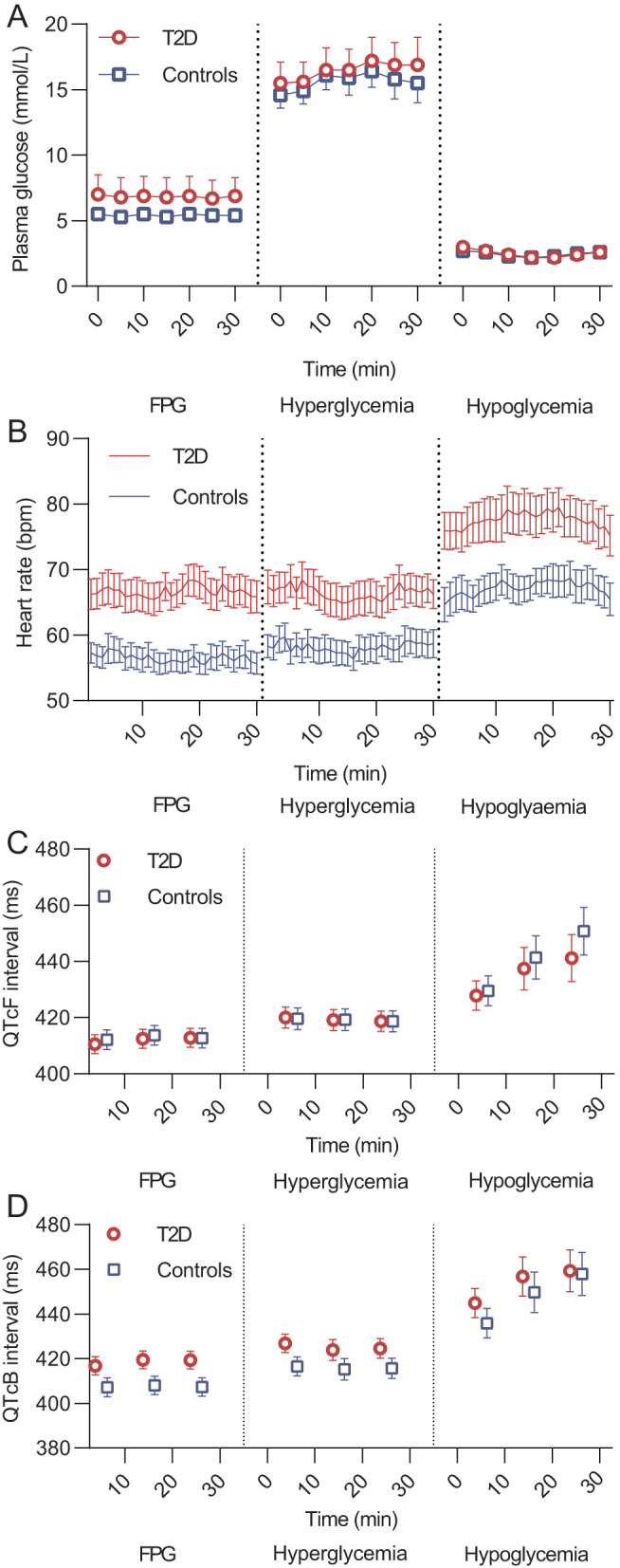
Plasma glucose, heart rate and QTc. (A) Plasma glucose (mean ± s.d., n = 21 in both groups), (B) heart rate (mean ± s.e., n = 21 (both groups)), (C) QTcF interval (mean ± s.e., n = 21 (type 2 diabetes) and n = 20 (controls)), (D) QTcB interval (mean ± s.e., n = 21 (type 2 diabetes) and n = 20 (controls)) during each steady-state of the experimental day in patients with type 2 diabetes (red circles) and controls (blue squares).FPG, fasting plasma glucose; T2D, type 2 diabetes; QTcB, heart rate-corrected QT interval by Bazett’s formula; QTcF, heart rate-corrected QT interval by Fridericia’s formula.
Blood pressure and heart rate
Mean heart rate was higher in the group of patients with type 2 diabetes compared with controls during the FPG phase (Fig. 1B and Table 2). A marked increase in heart rate was observed in both groups during hypoglycemia. Systolic blood pressure remained unchanged in both groups when compared with the FPG phase, whereas diastolic blood pressure declined significantly during the hypoglycemic phase in both groups (Table 2).
Table 2.
Changes in mean heart rate, blood pressure, plasma potassium and QTc interval during acute hyperglycemia and acute hypoglycemia compared with fasting plasma glucose in the group of insulin-treated patients with type 2 diabetes and the group of controls.
| Parameter/group | FPG | Hyperglycemia | Hypoglycemia | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | 95% CI | ∆Mean | ∆95% CI | P | ∆Mean | ∆95% CI | P | |
| Heart rate (b.p.m) | ||||||||
| T2D | 67 | 63; 71 | −1 | −2; 0 | 0.157 | 11 | 8; 14 | <0.0001 |
| Controls | 56 | 52; 61 | 2 | 1; 3 | 0.002 | 11 | 8; 13 | <0.0001 |
| ∆Groups | −2 | −1, −4 | 0.002 | 0 | −3, 4 | 0.835 | ||
| Systolic BP (mmHg) | ||||||||
| T2D | 133 | 125; 141 | 2 | −4; 8 | 0.504 | 3 | −4, 8 | 0.514 |
| Controls | 131 | 123; 139 | 6 | 0; 12 | 0.055 | −6 | −15; 3 | 0.199 |
| ∆Groups | −4 | −13, 5 | 0.363 | 9 | −4, 22 | 0.173 | ||
| Diastolic BP (mmHg) | ||||||||
| T2D | 78 | 73; 83 | −1 | −5; 2 | 0.456 | −10 | −16; −4 | 0.002 |
| Controls | 80 | 75; 85 | 1 | −3; 5 | 0.613 | −10 | −16; −4 | 0.002 |
| ∆Groups | −2 | −8, 3 | 0.377 | 0 | −8, 9 | 0.946 | ||
| Plasma potassium (mmol/L) | ||||||||
| T2D | 4.1 | 4.0; 4.2 | 0.0 | −0.1; 0.1 | 0.545 | −1.0 | −1.1; −0.9 | <0.0001 |
| Controls | 4.1 | 4.0; 4.3 | −0.2 | −0.3; −0.1 | 0.002 | −0.9 | −1.0; −0.7 | <0.0001 |
| ∆Groups | 0.1 | 0.0, 0.3 | 0.074 | −0.2 | −0.3, 0.0 | 0.053 | ||
| QTcF30 min (ms) | ||||||||
| T2D | 410 | 404, 418 | 8 | 5, 11 | <0.0001 | 31 | 16, 45 | <0.0001 |
| Controls | 412 | 405, 419 | 7 | 3, 10 | <0.001 | 39 | 24, 53 | <0.0001 |
| ∆Groups | 1 | −3, 6 | 0.529 | −8 | −28, 12 | 0.434 | ||
| QTcFmax (ms) | ||||||||
| T2D | 447 | 432, 462 | 15 | −6, 36 | 0.162 | 49 | 18, 79 | <0.001 |
| Controls | 440 | 425, 455 | 3 | −19, 25 | 0.790 | 55 | 24, 86 | <0.001 |
| ∆Groups | 12 | −18, 43 | 0.426 | −6 | −50, 37 | 0.765 | ||
| QTcB30 min (ms) | ||||||||
| T2D | 417 | 408, 425 | 8 | 3, 12 | <0.001 | 42 | 27, 58 | <0.0001 |
| Controls | 407 | 398, 416 | 8 | 4, 13 | <0.001 | 51 | 35, 66 | <0.0001 |
| ∆Groups | −1 | −7, 5 | 0.799 | −8 | −30, 14 | 0.448 | ||
| ∆QTcBmax (ms) | ||||||||
| T2D | 453 | 437, 470 | 16 | −5, 37 | 0.133 | 60 | 30, 90 | <0.001 |
| Controls | 449 | 432, 465 | 7 | −14, 29 | 0.486 | 67 | 37, 97 | <0.0001 |
| ∆Groups | 8 | −28, 32 | 0.574 | −7 | −50, 36 | 0.747 | ||
FPG, fasting plasma glucose; QTcB, heart rate-corrected QT by Bazett’s correction; QTcF, heart rate-corrected QT by Fridericia’s correction, ref, reference; T2D, type 2 diabetes.
Cardiac rhythm and QTc
None of the participants evolved any clinically significant arrhythmias during hypoglycemia. One participant with type 2 diabetes evolved asymptomatic atrial fibrillation immediately following the hypoglycemic phase before normal PG had been restored. The episode lasted for 5 h before spontaneously converting to sinus rhythm without any medical intervention. In one participant in the control group, QTc analysis was not possible due to technical issues. There was no difference in QTcF between the two groups during the FPG phase (Fig. 1C). QTcF increased progressively during hypoglycemia in both groups without any between group difference (Table 2). The increase in QTc was higher when applying Bazett’s correction. The maximal registered QTcF and QTcB increased significantly in both groups during hypoglycemia compared to the FPG phase without any between group difference (Table 2). When applying Bazett’s correction, maximal QTcB exceeded 500 ms in 48% of patients with type 2 diabetes and 55% of controls. During the hypoglycemic phase, the incidence of ventricular premature beats increased significantly in both groups when compared with the FPG phase (Fig. 2 and Table 3).
Figure 2.
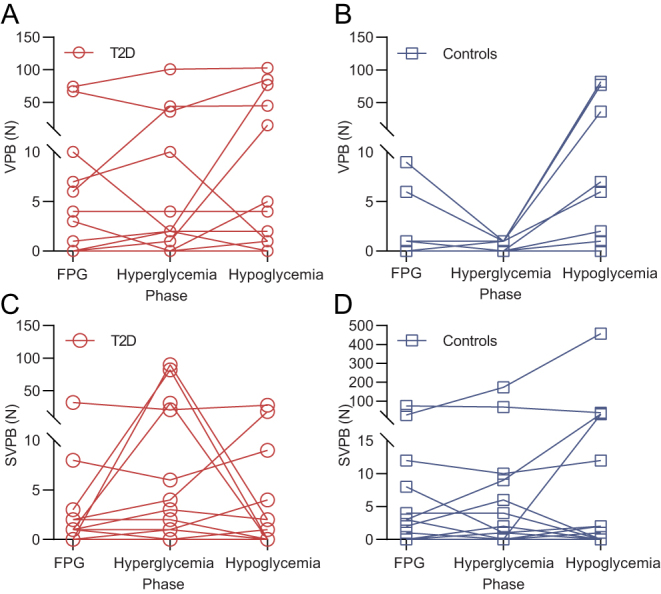
Ventricular and supraventricular premature beats. Individual counts of ventricular and supraventricular premature beats in patients with type 2 diabetes (A and C (red circles)) and controls (B and D (blue squares)). FPG, fasting plasma glucose; SVPB, supraventricular premature beats; T2D, type 2 diabetes; VPB, ventricular premature beats.
Table 3.
Incident rate ratio of ventricular and supraventricular premature beats during hyperglycemia and hypoglycemia compared with fasting plasma glucose in the group of insulin-treated patients with type 2 diabetes and the group of controls.
| Parameter/group | Hyperglycemia | Hypoglycemia | ||||
|---|---|---|---|---|---|---|
| IRR | ∆95% CI | P | IRR | ∆95% CI | P | |
| VPB | ||||||
| T2D | 1.2 | (0.7; 2.1) | 0.583 | 2.0 | (1.1; 3.7) | 0.033 |
| Controls | 0.2 | (0.1; 0.4) | <0.0001 | 10.6 | (3.4; 33.0) | <0.0001 |
| SVPB | ||||||
| T2D | 4.6 | (1.1; 19.3) | 0.035 | 1.2 | (0.7, 2.3) | 0.511 |
| Controls | 2.0 | (0.7; 5.7) | 0.194 | 4.2 | (0.9; 19.4) | 0.065 |
FPG, fasting plasma glucose; IRR, incident rate ratio; SVPB, supraventricular premature beats; T2D, type 2 diabetes; VPB, ventricular premature beats.
Counterregulatory hormones – glucagon, noradrenaline, cortisol and somatotropin
Fasting plasma glucagon, noradrenaline, somatotropin and cortisol were similar between groups (Fig. 3). During hyperglycemia, plasma glucagon was equally suppressed, although significantly higher in the group of patients with type 2 diabetes compared to controls (Fig. 3A). During hypoglycemia, plasma glucagon, noradrenaline and somatotropin increased in both groups, without any difference in the baseline-subtracted area under the curve. When the hypoglycemic phase was initiated, serum cortisol was already markedly elevated in the group of patients with type 2 diabetes but not in controls (Fig. 3C). Accordingly, the baseline-subtracted area under the curve for serum cortisol was significantly higher in the group of patients with type 2 diabetes.
Figure 3.
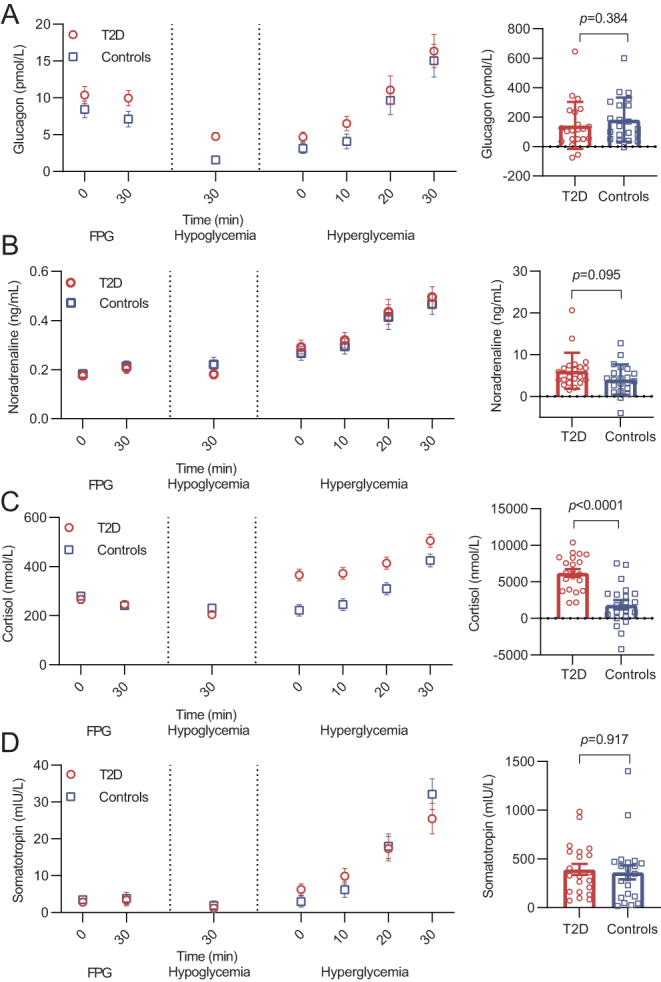
Hormonal counterregulatory response. Levels of (A) glucagon, (B) noradrenaline, (C) cortisol and (D) somatotropin during each steady-state phase of the experimental day in patients with type 2 diabetes (red circles) and controls (blue squares). On the left side, levels of the respective counterregulatory hormones during the clamp procedure are depicted (mean ± s.e., n = 21 (both groups)). On the right side, hormonal counterregulatory response during the hypoglycemic phase is depicted as baseline subtracted area under the curve with the hyperglycemic phase applied as baseline and differences between groups evaluated by an unpaired t-test. FPG, fasting plasma glucose; T2D, type 2 diabetes.
Plasma potassium
The two groups had similar levels of plasma potassium during the FPG phase (Fig. 4 and Table 2). During hyperglycemia, a significant decrease in plasma potassium was found in controls, whereas it remained unchanged in the group of patients with type 2 diabetes. During the hypoglycemic phase, plasma potassium decreased in both groups compared with the FPG phase with no difference between the groups at the end of the hypoglycemic phase. However, plasma potassium reached a significantly lower nadir during hypoglycemia in the group of patients with type 2 diabetes compared with controls (3.1 mmol/L (2.9, 3.2) and 3.2 mmol/L (3.1, 3.3), respectively, P = 0.023).
Figure 4.
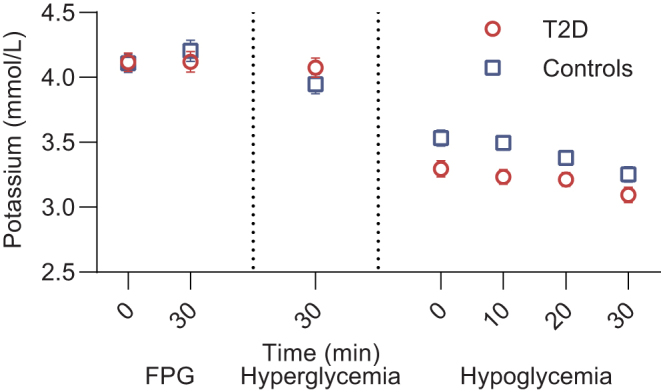
Plasma potassium. Levels of plasma potassium (mean ± s.e., n = 21 in both groups) during each steady-state phase of the experimental day in patients with type 2 diabetes (red circles) and controls (blue squares). FPG, fasting plasma glucose; T2D, type 2 diabetes.
Discussion
In the present study, QTc interval increased significantly during insulin-induced acute hypoglycemia in both insulin-treated patients with type 2 diabetes and matched controls with no difference between groups. In a previous study comparing streptozotocin-treated diabetic rats to non-diabetic rats, diabetes was found to aggravate QTc prolongation during hypoglycemia, indicating increased susceptibility to QTc abnormalities in patients with diabetes (15). Similarly, Chow et al. found that QTc prolongation during hypoglycemia was greater in patients with type 2 diabetes compared to controls, although the difference was not significant (11). This difference could be explained by longer diabetes duration, the criteria of insulin treatment and a potentially higher incidence of cardiac autonomic neuropathy in the present study (18). The observed increase in QTc interval during hypoglycemia in patients with type 2 diabetes was smaller than what has previously been reported (11, 12, 13) even when applying Bazett’s correction which is known to overcorrect for high heart rates (19). This is likely to reflect the older age and long diabetes duration in the present study, but also a shorter period of hypoglycemia since QTc has been demonstrated to increase progressively for up to 80 min of hypoglycemia (18). Nevertheless, both groups experienced significant maximal QTcB and QTcF interval prolongations above normal range during hypoglycemia and, in half of the individuals in each group, the maximal QTcB interval exceeded 500 ms, which has been identified as an important threshold for risk of syncope, cardiac arrest and sudden cardiac death in patients with the long-QT syndrome (20). Thus, the observed changes should be considered clinically important.
Spontaneous hypoglycemia has previously been demonstrated to increase the incidence of premature ventricular beats in patients with type 2 diabetes (10). In the present study, the incidence of ventricular premature beats increased in both groups during hypoglycemia. Although premature ventricular beats are common in healthy individuals (21), frequent premature ventricular beats have been associated with a substantial increase in the risk of sudden cardiac death (22). The development of atrial fibrillation immediately following hypoglycemia in a single patient in the group of patients with type 2 diabetes adds to a small number of reported cases of hypoglycemia-related atrial fibrillation (23, 24). Although no conclusions can be drawn from these anecdotal cases, frequent hypoglycemia may add to the atrial fibrillation burden in insulin-treated patients with paroxysmal atrial fibrillation. Our findings support recommendations to avoid hypoglycemia in patients with established cardiovascular disease (25).
Both groups had marked counterregulatory responses during hypoglycemia. Interestingly, the cortisol response was significantly higher in patients with type 2 diabetes. High levels of plasma insulin might potentiate cortisol, adrenaline and noradrenaline secretion during hypoglycemia (26, 27) and, in the present study, higher doses of insulin were required to induce hypoglycemia in patients with type 2 diabetes due to pronounced insulin resistance. Hence, it seems likely that the difference in cortisol secretion between the groups, which was evident already when initiating the hypoglycemic phase, may be explained by differences in plasma insulin. Another potential contributing factor could be a higher prevalence of obstructive sleep apnea in the group of patients with type 2 diabetes (sleep apnea is associated with increased cortisol levels (28)). Thus, a limitation of the current study is that we did not include screening for sleep apnea. Nevertheless, there was no difference in cortisol levels during the FPG phase nor the hyperglycemic phase, and the effect of sleep apnea on the hormonal counterregulatory response has not previously been investigated.
The sympathoadrenal response to hypoglycemia is the main mediator of QTc prolongation during hypoglycemia (29). In accordance with previous observations, the response in plasma noradrenaline was similar between patients with type 2 diabetes and controls (11). The threshold for the sympathetic and hormonal counterregulatory response is generally elevated in patients with type 2 diabetes (30, 31), whereas the counterregulatory threshold is normalized when insulin therapy is initiated (32). Furthermore, previous episodes of hypoglycemia attenuate the counterregulatory response during subsequent episodes of hypoglycemia (33). In contrast to previous studies, the present study included only insulin-treated patients with type 2 diabetes with a low HbA1c and, accordingly, a high risk of hypoglycemia. Nevertheless, noradrenaline response to hypoglycemia was unaffected indicating a preserved sympathoadrenal response.
In a previous study in patients with type 2 diabetes and controls, a significant decrease in plasma potassium during hypoglycemia was found only in patients with type 2 diabetes (11). The decline in plasma potassium during insulin-induced hypoglycemia is almost equally mediated by insulin-induced and catecholamine-induced cellular potassium uptake (34). In the present study, there was no significant difference in the decrease in plasma potassium at the end of the hypoglycemic phase between the two groups, whereas plasma potassium nadir was significantly lower in the group of patients with type 2 diabetes. Since no between-group difference in noradrenaline was observed, the difference in plasma potassium nadir is most likely to be explained by the difference in insulin dose between the groups. Since the effect of insulin on PG and plasma potassium is independent (35), insulin-induced cellular potassium uptake is not affected by insulin resistance in type 2 diabetes, which could explain the difference in plasma potassium in spite of almost similar changes in PG. Since hypokalemia is an underlying mechanism behind hypoglycemia-induced abnormal cardiac repolarization (29), large therapeutic doses of insulin to overcome insulin resistance may amplify the degree of hypokalemia during iatrogenic insulin-induced hypoglycemia in type 2 diabetes and, thereby, increase the risk of ventricular arrhythmias. Importantly, even mild hypokalemia is associated with an increased risk of cardiac arrhythmias and death (36, 37).
The present study has some limitations. A rather high insulin dose was applied to induce hypoglycemia within a short period. Although this may not fully resemble most hypoglycemic episodes in real life, cardiac arrhythmia induced by hypoglycemia is likely to be a relatively rare event. In this context, exploring the response to a potent stimulus is relevant. Insulin concentrations were not measured, since the available assay would not detect concentrations of some long-acting insulin analogs in the patients with type 2 diabetes, which would complicate interpretation of data. The difference in time to hypoglycemia and insulin dose between groups complicates the interpretation of between-group differences, although the rate of decline in plasma glucose was almost similar between the two groups. The difference in the dose of insulin required to induce hypoglycemia reflects a relatively high daily insulin dose in these high insulin-resistant patients. The period of hypoglycemia was relatively short mimicking a daytime episode of hypoglycemia, potentially ending before the full effect on cardiac rhythm and repolarization could be observed (18). Lastly, due to over-dispersed data, estimates of the relative incidence of premature beats should be interpreted with caution.
In conclusion, hypoglycemia causes significant and clinically relevant QTc prolongations in patients with type 2 diabetes. The findings add to the evidence supporting a role for hypoglycemia in the induction of cardiac arrhythmias and sudden cardiac death. Noteworthy, any relevant difference in QTc interval prolongation during hypoglycemia between high-risk, insulin-treated patients with type 2 diabetes and controls without diabetes does not appear to exist. Hence, type 2 diabetes itself does not appear to aggravate the repolarization abnormalities caused by hypoglycemia.
Declaration of interest
A A, M P A B, M B C, K U A, J F, T B L, G G declare that they have no competing interests. J I B has received lecture fee from Novo Nordisk. U P B has served on advisory boards for AstraZeneca/Bristol Myers Squibb, Sanofi Aventis, Novo Nordisk and Zealand Pharma and has received lecture fees and research grants from Novo Nordisk. J J H has served on scientific advisory panels for Novo Nordisk. F K K has served on scientific advisory panels and/or been part of speaker’s bureaus for, served as a consultant to and/or received research support from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Carmot Therapeutics, Eli Lilly, Gubra, MedImmune, MSD/Merck, Mundipharma, Norgine, Novo Nordisk, Sanofi and Zealand Pharma. T V has served on scientific advisory panels, been part of speaker’s bureaus for, served as a consultant to and/or received research support from Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Gilead, Mundipharma, MSD/Merck, Novo Nordisk, Sanofi and Sun Pharmaceuticals.
Funding
This work was supported by unrestricted grants from the Novo Nordisk Foundation (grant number NNF 16230) and the Capital Region of Denmark (grant number E-19280-51-07).
Availability of data and materials
The datasets generated and analyzed during the current study are not publicly available due to Danish data protection laws but are available from the corresponding author on reasonable request.
Author contribution statement
A A, J I B, M B C, T B L, G G, F K K and T V designed the study and wrote the protocol. U P B contributed with participant recruitment. A A, M P A B and K U A performed the study. J J H and J F measured glucagon and noradrenaline, respectively. A A performed the data analysis and wrote the first draft. All authors critically revised the manuscript and approved the final version. A A and T V are the guarantors of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Acknowledgements
The authors gratefully thank the study participants for their contribution. The authors also thank co-workers in our department for valuable support and, especially, J V Lauritsen and M Seghieri from Center for Clinical Metabolic Research for invaluable assistance in conducting the study.
References
- 1.Raghavan S, Vassy JL, Ho YL, Song RJ, Gagnon DR, Cho K, Wilson PWF, Phillips LS. Diabetes mellitus-related all-cause and cardiovascular mortality in a national cohort of adults. Journal of the American Heart Association 2019. 8 e011295. ( 10.1161/JAHA.118.011295) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen A, Jørgensen PG, Knop FK, Vilsbøll T. Hypoglycaemia and cardiac arrhythmias in diabetes. Therapeutic Advances in Endocrinology and Metabolism 2020. 11 2042018820911803. ( 10.1177/2042018820911803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, Goff DC, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F. et al. Effects of intensive glucose lowering in type 2 diabetes. New England Journal of Medicine 2008. 358 2545–25. ( 10.1056/NEJMoa0802743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Hypoglycaemia Study Group, Aschner P, Childs B, Cryer PE, de Galan BE, Frier BM, Gonder-Frederick L, Heller SR, Jones T, Khunti K. Hypoglycaemia, cardiovascular disease, and mortality in diabetes: epidemiology, pathogenesis, and management. Lancet: Diabetes and Endocrinology 2019. 7 385–3. ( 10.1016/S2213-8587(1830315-2) [DOI] [PubMed] [Google Scholar]
- 5.Buse JB, Wexler DJ, Tsapas A, Rossing P, Mingrone G, Mathieu C, D’Alessio DA, Davies MJ. 2019 update to: management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2020. 43 487–4. ( 10.2337/dci19-0066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonaventura A, Montecucco F, Dallegri F. Update on strategies limiting iatrogenic hypoglycemia. Endocrine Connections 2015. 4 R37–R. ( 10.1530/EC-15-0044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzpatrick C, Chatterjee S, Seidu S, Bodicoat DH, Ng GA, Davies MJ, Khunti K. Association of hypoglycaemia and risk of cardiac arrhythmia in patients with diabetes mellitus: a systematic review and meta-analysis. Diabetes, Obesity and Metabolism 2018. 20 2169–21. ( 10.1111/dom.13348) [DOI] [PubMed] [Google Scholar]
- 8.Al-Khatib SM, LaPointe NMA, Kramer JM, Califf RM. What clinicians should know about the QT interval. JAMA 2003. 289 2120–212. ( 10.1001/jama.289.16.2120) [DOI] [PubMed] [Google Scholar]
- 9.Cox AJ, Azeem A, Yeboah J, Soliman EZ, Aggarwal SR, Bertoni AG, Carr JJ, Freedman BI, Herrington DM, Bowden DW. Heart rate-corrected QT interval is an independent predictor of all-cause and cardiovascular mortality in individuals with type 2 diabetes: the diabetes heart study. Diabetes Care 2014. 37 1454–14. ( 10.2337/dc13-1257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chow E, Bernjak A, Williams S, Fawdry RA, Hibbert S, Freeman J, Sheridan PJ, Heller SR. Risk of cardiac arrhythmias during hypoglycemia in patients with type 2 diabetes and cardiovascular risk. Diabetes 2014. 63 1738–17. ( 10.2337/db13-0468) [DOI] [PubMed] [Google Scholar]
- 11.Chow E, Bernjak A, Walkinshaw E, Lubina-Solomon A, Freeman J, Macdonald IA, Sheridan PJ, Heller SR. Cardiac autonomic regulation and repolarization during acute experimental hypoglycemia in type 2 diabetes. Diabetes 2017. 66 1322–13. ( 10.2337/db16-1310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landstedt-Hallin L, Englund A, Adamson U, Lins PE. Increased QT dispersion during hypoglycaemia in patients with type 2 diabetes mellitus. Journal of Internal Medicine 1999. 246 299–307. ( 10.1046/j.1365-2796.1999.00528.x) [DOI] [PubMed] [Google Scholar]
- 13.Marques JL, George E, Peacey SR, Harris ND, Macdonald IA, Cochrane T, Heller SR. Altered ventricular repolarization during hypoglycaemia in patients with diabetes. Diabetic Medicine 1997. 14 648–6. ( 10.1002/(SICI)1096-9136(199708)14:8<648::AID-DIA418>3.0.CO;2-1) [DOI] [PubMed] [Google Scholar]
- 14.Lindström T, Jorfeldt L, Tegler L, Arnqvist HJ. Hypoglycaemia and cardiac arrhythmias in patients with type 2 diabetes mellitus. Diabetic Medicine 1992. 9 536–5. ( 10.1111/j.1464-5491.1992.tb01834.x) [DOI] [PubMed] [Google Scholar]
- 15.Reno CM, VanderWeele J, Bayles J, Litvin M, Skinner A, Jordan A, Daphna-Iken D, Fisher SJ. Severe hypoglycemia–induced fatal cardiac arrhythmias are augmented by diabetes and attenuated by recurrent hypoglycemia. Diabetes 2017. 66 3091–309. ( 10.2337/db17-0306) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verbraecken J, Van de Heyning P, De Backer W, Van Gaal L. Body surface area in normal-weight, overweight, and obese adults. A comparison study. Metabolism: Clinical and Experimental 2006. 55 515–5. ( 10.1016/j.metabol.2005.11.004) [DOI] [PubMed] [Google Scholar]
- 17.Postema PG, Wilde AAM. The measurement of the QT interval. Current Cardiology Reviews 2014. 10 287–2. ( 10.2174/1573403x10666140514103612) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipponen JA, Kemppainen J, Karjalainen PA, Laitinen T, Mikola H, Kärki T, Tarvainen MP. Dynamic estimation of cardiac repolarization characteristics during hypoglycemia in healthy and diabetic subjects. Physiological Measurement 2011. 32 649–660. ( 10.1088/0967-3334/32/6/003) [DOI] [PubMed] [Google Scholar]
- 19.Vandenberk B, Vandael E, Robyns T, Vandenberghe J, Garweg C, Foulon V, Ector J, Willems R. Which QT correction formulae to use for QT monitoring? Journal of the American Heart Association 2016. 5 e003264. ( 10.1161/JAHA.116.003264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G. et al. Risk stratification in the long-QT syndrome. New England Journal of Medicine 2003. 348 1866–18. ( 10.1056/NEJMoa022147) [DOI] [PubMed] [Google Scholar]
- 21.Brodsky M, Wu D, Denes P, Kanakis C, Rosen KM. Arrhythmias documented by 24 hour continuous electrocardiographic monitoring in 50 male medical students without apparent heart disease. American Journal of Cardiology 1977. 39 390–39. ( 10.1016/s0002-9149(7780094-5) [DOI] [PubMed] [Google Scholar]
- 22.Ataklte F, Erqou S, Laukkanen J, Kaptoge S. Meta-analysis of ventricular premature complexes and their relation to cardiac mortality in general populations. American Journal of Cardiology 2013. 112 1263–12. ( 10.1016/j.amjcard.2013.05.065) [DOI] [PubMed] [Google Scholar]
- 23.Collier A, Matthews DM, Young RJ, Clarke BF. Transient atrial fibrillation precipitated by hypoglycaemia: two case reports. Postgraduate Medical Journal 1987. 63 895–89. ( 10.1136/pgmj.63.744.895) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Odeh M, Oliven A, Bassan H. Transient atrial fibrillation precipitated by hypoglycemia. Annals of Emergency Medicine 1990. 19 565–56. ( 10.1016/s0196-0644(0582191-2) [DOI] [PubMed] [Google Scholar]
- 25.American Diabetes Association. 6. Glycemic targets: standards of medical care in diabetes – 2020. Diabetes Care 2020. 43 (Supplement 1) S66–S. ( 10.2337/dc20-S006) [DOI] [PubMed] [Google Scholar]
- 26.Davis SN, Goldstein RE, Jacobs J, Price L, Wolfe R, Cherrington AD. The effects of differing insulin levels on the hormonal and metabolic response to equivalent hypoglycemia in normal humans. Diabetes 1993. 42 263–2. ( 10.2337/diab.42.2.263) [DOI] [PubMed] [Google Scholar]
- 27.Davis SN, Goldstein RE, Price L, Jacobs J, Cherrington AD. The effects of insulin on the counterregulatory response to equivalent hypoglycemia in patients with insulin-dependent diabetes mellitus. Journal of Clinical Endocrinology and Metabolism 1993. 77 1300–130. ( 10.1210/jcem.77.5.8077324) [DOI] [PubMed] [Google Scholar]
- 28.Vgontzas AN, Pejovic S, Zoumakis E, Lin HM, Bentley CM, Bixler EO, Sarrigiannidis A, Basta M, Chrousos GP. Hypothalamic-pituitary-adrenal axis activity in obese men with and without sleep apnea: effects of continuous positive airway pressure therapy. Journal of Clinical Endocrinology and Metabolism 2007. 92 4199–4. ( 10.1210/jc.2007-0774) [DOI] [PubMed] [Google Scholar]
- 29.Robinson RTCE, Harris ND, Ireland RH, Lee S, Newman C, Heller SR. Mechanisms of abnormal cardiac repolarization during insulin-induced hypoglycemia. Diabetes 2003. 52 1469–14. ( 10.2337/diabetes.52.6.1469) [DOI] [PubMed] [Google Scholar]
- 30.Spyer G, Hattersley AT, MacDonald IA, Amiel S, MacLeod KM. Hypoglycaemic counter-regulation at normal blood glucose concentrations in patients with well controlled type-2 diabetes. Lancet 2000. 356 1970–197. ( 10.1016/s0140-6736(0003322-5) [DOI] [PubMed] [Google Scholar]
- 31.Levy CJ, Kinsley BT, Bajaj M, Simonson DC. Effect of glycemic control on glucose counterregulation during hypoglycemia in NIDDM. Diabetes Care 1998. 21 1330–133. ( 10.2337/diacare.21.8.1330) [DOI] [PubMed] [Google Scholar]
- 32.Korzon-Burakowska A, Hopkins D, Matyka K, Lomas J, Pernet A, Macdonald IA, Amiel S. Effects of glycemic control on protective responses against hypoglycemia in type 2 diabetes. Diabetes Care 1998. 21 283–2. ( 10.2337/diacare.21.2.283) [DOI] [PubMed] [Google Scholar]
- 33.Davis SN, Mann S, Briscoe VJ, Ertl AC, Tate DB. Effects of intensive therapy and antecedent hypoglycemia on counterregulatory responses to hypoglycemia in Type 2 diabetes. Diabetes 2009. 58 701–70. ( 10.2337/db08-1230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen KG, Schlüter KJ, Kerp L. Regulation of serum potassium during insulin-induced hypoglycemia. Diabetes 1982. 31 615–61. ( 10.2337/diab.31.7.615) [DOI] [PubMed] [Google Scholar]
- 35.Nguyen TQ, Maalouf NM, Sakhaee K, Moe OW. Comparison of insulin action on glucose versus potassium uptake in humans. Clinical Journal of the American Society of Nephrology 2011. 6 1533–153. ( 10.2215/CJN.00750111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bird JG, McCully RB, Pellikka PA, Kane GC. Dobutamine stress echocardiography: impact of abnormal blood potassium levels on cardiac arrhythmias. Journal of the American Society of Echocardiography 2017. 30 595–601. ( 10.1016/j.echo.2017.01.017) [DOI] [PubMed] [Google Scholar]
- 37.Collins AJ, Pitt B, Reaven N, Funk S, McGaughey K, Wilson D, Bushinsky DA. Association of serum potassium with all-cause mortality in patients with and without heart failure, chronic kidney disease, and/or diabetes. American Journal of Nephrology 2017. 46 213–2. ( 10.1159/000479802) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analyzed during the current study are not publicly available due to Danish data protection laws but are available from the corresponding author on reasonable request.