

OBJECTIVES:
PFKFB3 regulates glycolysis in tumor cells, might function as an oncogene, and is associated with cancer metastasis. However, its role in gastric cancer (GC) remains largely unknown.
METHODS:
PFKFB3 expression was assessed by immunohistochemistry (IHC) in GC tissues and paired paracancerous histological normal tissues (PCHNTs). The associations of PFKFB3 expression with clinical features and HIF-1α, Ki-67, E-cadherin, Snail, and Vimentin expression levels were assessed. A series of in vivo and in vitro experiments were performed to investigate the effects of PFKFB3 on the growth, migration, and invasion of GC cells.
RESULTS:
We found that PFKFB3 expression was significantly higher in GC tissues compared with PCHNTs (P = 0.000). PFKFB3 expression was positively correlated with tumor size (P = 0.000), differentiation (P = 0.025), venous invasion (P = 0.084), nerve invasion (P = 0.014), lymphatic invasion (P = 0.000), local invasion (P = 0.000), invasive depth (P = 0.000), nodal metastasis (P = 0.000), tumor-node-metastasis stage (P = 0.000), and patient survival (P = 0.000). Notably, PFKFB3 upregulation was highly correlated with increased epithelial-mesenchymal transition (EMT) in GC samples. PFKFB3 overexpression positively modulated cell proliferation, migration, and EMT in GC cells in vitro, with concomitant activation of NF-κB signaling. Administration of an NF-κB inhibitor attenuated PFKFB3-induced EMT in GC cells. PFKFB3 overexpression promoted tumor development and EMT in nude mice, which were attenuated by PFK-15, a PFKFB3 inhibitor.
DISCUSSION:
PFKFB3 could potentiate malignancy in GC cells through NF-κB pathway–mediated EMT, suggesting PFKFB3 represents a potential target for GC therapy.
INTRODUCTION
The incidence and mortality of gastric cancer (GC) were ranked third and second among malignant tumors in China, representing a serious threat to human health (1). Invasion and metastasis are the main causes of poor prognosis of GC. Tumor cell infiltration and distant metastasis require adequate nutrition and energy. Therefore, tumor cells need to regulate energy metabolism through metabolic reprogramming, including glycolysis, glutamate-dependent biosynthesis, and lipid production to meet the need for rapid growth, invasion, and metastasis (2). Although previous studies have shown that metabolic disorders are an important feature of malignant tumors, it is unclear how metabolic changes occur in GC and play an important role in its tumorigenesis and progression. Therefore, exploring the metabolic changes and regulatory mechanisms of GC cells is helpful to clarify the molecular mechanism of invasion and metastasis of GC and to provide a new strategy for anti-invasion and metastasis of GC based on regulation of cellular metabolism. Tumor energy metabolism is a hot research field in recent years. In the 1920s, OttoWarburg et al. found that glucose metabolism of tumor cells is different from that of normal cells. Under hypoxia or even aerobic conditions, tumor cells use glycolysis as the main way to provide energy, known as aerobic glycolysis or Warburg effects (3). Among most tumor cells, glucose transporter receptors, mainly Glutl, are upregulated and glucose uptake is increased. Meanwhile, the expression level and activity of glycolytic-related enzymes such as HK2, PFK-1, PKM2, LDH-A, and PDK-1 are increased. The glycolysis ability of cancer cells is obviously enhanced (4). It has been demonstrated that glycolysis inhibition can significantly inhibit tumor proliferation, invasion, and metastasis, indicating that glycolysis plays an important role in the tumorigenesis and progression (5). Previous studies have confirmed that GC is one of the malignant tumors with abnormal glycolysis (6,7).
PFKFB3 is an enzyme that synthesizes fructose 2,6-diphosphate (F-2,6-DP), which participates in glycolysis and is widely found in many cells. F-2,6-DP is the most effective PFK-1 activator and the most potent inducer of glycolysis (8). Among all PFKFB3 isozymes, the activity of PFKFB3 is about 700 times higher than those of diphosphatase. Therefore, it is beneficial to cell survival, glycolysis, and F-2,6-DP synthesis (8). It has been shown that PFKFB3 can promote normal angiogenesis and tumor cell growth (9,10). Meanwhile, HIF-1α and p38/MK2 signaling pathways upregulate PFKFB3 through transcriptional regulation (11,12). Phosphorylation of serine at position 461 of PFKFB3 protein could upregulate its expression and promote tumor cell proliferation (13). Studies have shown that PFKFB3 expression in many tumor cells, such as colorectal cancer, is significantly increased (14,15), but the role of PFKFB3 in tumor invasion and metastasis is unclear, especially the possible mechanism is rarely reported.
In this study, we investigated PFKFB3 expression in GC and paired PCHNTs and constructed a GC cell model with PFKFB3 knockdown or overexpression to investigate the effects of PFKFB3 on glycolytic, proliferation, invasion, and metastasis in GC cells in vitro and tumorigenesis and metastasis in GC cells in vivo. The purpose of this study was to elucidate how enhanced PFKFB3 expression regulates glycolysis and promotes invasion and metastasis in GC cells, providing new insights into anti-invasive and antimetastatic therapy for GC.
MATERIAL AND METHODS
GC cell lines
Human GC cell lines (AGS, BGC-823, MGC-803, and SGC-7901 cells) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and grown in RPMI-1640, supplemented with 10% fetal bovine serum (FBS), 100-U/mL penicillin, and 100-U/mL streptomycin at 37°C and 5% CO2.
A complete description of the Materials and Methods is included in the Supplementary Methods (see Supplementary Digital Content 2, http://links.lww.com/CTG/A645), Supplementary Table 1 (see Supplementary Digital Content 3, http://links.lww.com/CTG/A646), and Supplementary Table 2 (see Supplementary Digital Content 4, http://links.lww.com/CTG/A647).
RESULTS
PFKFB3 is upregulated in GC tissues and cell lines
To evaluate PFKFB3 expression in patients with GC, we analyzed 319 samples by IHC, which showed PFKFB3 protein upregulation in GC tissues at a rate of 68.0% (217/319) (Figure 1a), in contrast to paired PCHNTs with a rate of 13.5% (43/319) (Figure 1b), indicating a statistically significant difference (χ2 = 23.361; P = 0.000). IHC results showed that PFKFB3 was expressed in the cytoplasm and nucleus of tumor cells and diffusely distributed. The expression levels of PFKFB3 in nuclei of tumor cells were significantly higher than those of paired PCHNTs (Figure 1c–e). However, there was no significant difference in PFKFB3 expression in PCHNTs as compared to noncancer controls (P < 0.05). The PFKFB3 protein was upregulated in all 4 GC cell lines detected by western blot (Figure 1f).
Figure 1.

Expression of PFKFB3 is significantly upregulated in primary gastric carcinomas. The protein level of PFKFB3 in 319 primary gastric cancer tissues and paired nonadjacent normal tissues as determined by IHC is shown in (a) tumor and (b) paired nonadjacent normal tissue samples (P = 0.000). An IHC score of 0–4 is considered to be no to low expression, 5–8 considered to be normal expression, and 9–12 considered to be high expression. Representative IHC images of PFKFB3 expression are shown for (c) high expression in cancer tissues, (d) moderate expression in cancer tissues, and (e) low expression in normal gastric mucosa. ×200 for all images. Western blot analysis of PFKFB3 expression in 4 GC cell lines is shown for (f). GC, gastric cancer; IHC, immunohistochemistry.
To investigate the significance of PFKFB3 overexpression, we analyzed the associations of PFKFB3 upregulation with clinicopathologic factors (Table 1). Upregulation of PFKFB3 protein was significantly associated with tumor size (≥5 cm), poor differentiation, venous invasion, etc. There was a weak association between PFKFB3 upregulation and Helicobacter pylori infection, which was close to statistical significance. However, PFKFB3 upregulation was not associated with gender, age at diagnosis, tumor site, and Lauren classification. Collectively, these data indicated that PFKFB3 upregulation at protein level was significantly associated with tumor progression and metastasis in GC.
Table 1.
Correlation between relative PFKEB3 expression level and clinicopathologic parameters of the patients
| Variables | N | PFKFB3 protein level | χ2 | P | |
| Low/normal, n (%) | High, n (%) | ||||
| Gender | |||||
| Male | 219 | 71 | 148 | 0.064 | 0.801 |
| Female | 100 | 31 | 69 | ||
| Age at diagnosis | |||||
| <60 | 165 | 59 | 106 | 2.248 | 0.134 |
| ≥60 | 154 | 43 | 111 | ||
| Tumor site | |||||
| Body/antrum | 231 | 77 | 154 | 0.71 | 0.399 |
| Cardia | 88 | 25 | 63 | ||
| Lauren classification | |||||
| Intestinal | 293 | 95 | 198 | 0.334 | 0.564 |
| Diffuse | 26 | 7 | 19 | ||
| Helicobacter pylori | |||||
| Negative | 62 | 26 | 36 | 3.51 | 0.061 |
| Positive | 257 | 76 | 181 | ||
| Tumor size | |||||
| <5 cm | 156 | 82 | 74 | 59.501 | 0.000 |
| ≥5 cm | 163 | 20 | 143 | ||
| Differentiation | |||||
| High/medium | 143 | 55 | 88 | 5.014 | 0.025 |
| Low | 176 | 47 | 129 | ||
| Venous invasion | |||||
| No | 146 | 63 | 83 | 15.459 | 0.000 |
| Yes | 173 | 39 | 134 | ||
| Nerve invasion | |||||
| No | 126 | 58 | 68 | 18.919 | 0.014 |
| Yes | 193 | 44 | 149 | ||
| Lymphatic invasion | |||||
| No | 73 | 52 | 21 | 67.704 | 0.000 |
| Yes | 246 | 50 | 196 | ||
| Local invasion | |||||
| No | 159 | 74 | 85 | 30.922 | 0.000 |
| Yes | 160 | 28 | 132 | ||
| Invasive depth | |||||
| T1/T2 | 49 | 41 | 8 | 71.138 | 0.000 |
| T3/T4 | 270 | 61 | 209 | ||
| Nodal metastasis | |||||
| No | 72 | 51 | 21 | 64.553 | 0.000 |
| Yes | 247 | 51 | 196 | ||
| Distant metastasis | |||||
| No | 303 | 102 | 201 | 7.918 | 0.005 |
| Yes | 16 | 0 | 16 | ||
| TNM stage | |||||
| I/II | 119 | 85 | 34 | 135.832 | 0.000 |
| III/IV | 200 | 17 | 183 | ||
The χ2 test was used for statistical analyses. P < 0.05 is considered statistically significant.
PFKFB3 protein high expression is defined as immunohistochemistry score ≥9.
TNM, tumor-node-metastasis.
Differential influence of PFKFB3 upregulation on survival in patients with GC
The median cumulative survival of patients with resected GC was 49.1 months. Patients with PFKFB3-overexpressing tumors had significantly shorter median survival compared with those showing normal or reduced PFKFB3 amounts (Figure 2a–c). Our data showed that PFKFB3 overexpression was associated with decreased survival time. In addition, we observed that all other factors, except patient gender, age at diagnosis, tumor site, etc, significantly influenced patient survival in univariate analysis (see Supplementary Table 3, Supplementary Digital Content 5, http://links.lww.com/CTG/A648), including tumor size, venous invasion, and PFKFB3 overexpression.
Figure 2.
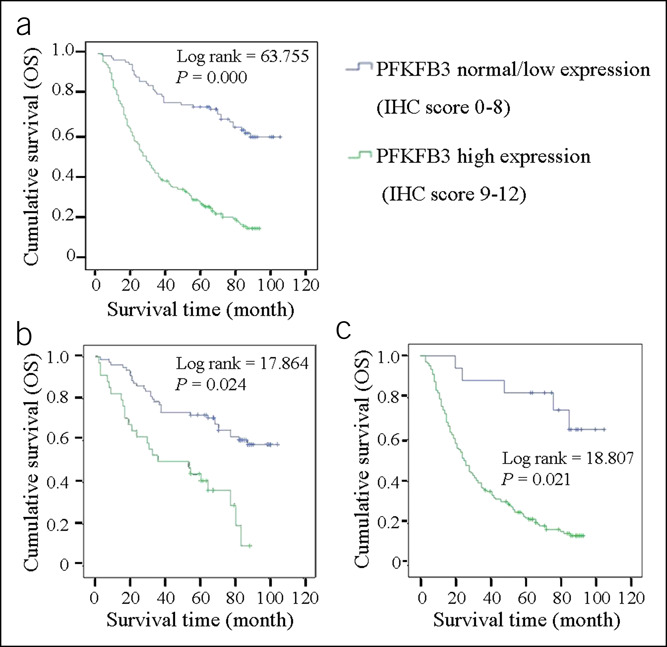
Correlation of the PFKFB3 expression level with survival of patients with gastric cancer. The Kaplan-Meier curves of overall survival (OS), in patients with gastric cancer, treated with primary gastrectomy according to the expression level of PFKFB3. OS is shown in (a) and its stratifications based on different tumor-node-metastasis stages are shown in (b) stage I/II and (c) stage III/IV. IHC, immunohistochemistry.
In contrast to univariate analysis, multivariate analysis using the Cox proportional hazards model showed that tumor site (Cardia) and PFKFB3 overexpression were independent prognostic factors for GC, in which their hazard ratio (HR) (95% confidence interval [CI]) was 1.478 (1.047–2.085) and 4.160 (2.593–6.673), respectively (see Supplementary Table 3, Supplementary Digital Content 5, http://links.lww.com/CTG/A648). Other factors, such as patient gender and age at diagnosis, were not prognostic factors.
Risk assessment of patients with GC based on PFKFB3 expression
We performed detailed analyses of risk assessment based on PFKFB3 expression in patients with GC (see Supplementary Table 4, Supplementary Digital Content 6, http://links.lww.com/CTG/A649). We used IHC scores of 0–8 and 9–12 for low to normal and high expression levels in these assays, respectively. Patients with high PFKFB3 expression levels had 17.6-fold elevated risk of increased invasive depth (HR 95% CI: 7.816–39.451), 9.3-fold higher risk of lymph node metastasis (HR 95% CI: 5.152–16.909], 11.8-fold elevated risk of either distant or lymph node metastasis (HR 95% CI: 6.271–22.071), and 29.9-fold increased risk of late stage of tumors (HR 95% CI: 14.240–50.860). Since no cases with distant metastasis in patients with downregulated PFKFB3 were found, it was impossible to perform statistical analysis. Therefore, it was not possible to assess the fold increase of risk of distant metastasis in patients with PFKFB3 overexpression. Collectively, these data further consolidated our findings that PFKFB3 overexpression was strongly associated with GC progression and metastasis.
PFKFB3 expression is associated with malignant phenotypic markers of GC
To investigate the potential function of PFKFB3 in GC progression, we analyzed HIF-1α, Ki-67, and epithelial-mesenchymal transition (EMT) markers, including E-cadherin, Snail, and Vimentin, in GC samples. IHC scores revealed that PFKFB3 overexpression was strongly associated with increased expression levels of HIF-1α (γ = 0.813, P = 0.000), Snail (γ = 0.712, P = 0.000), Ki-67 (γ = 0.748, P = 0.000), Vimentin (γ = 0.717, P = 0.000), and reduced expression of E-cadherin (γ = −0.512, P = 0.000) (see Supplementary Figure 1A–E, Supplementary Digital Content 1, http://links.lww.com/CTG/A644). These data, therefore, suggested that PFKFB3 overexpression was associated with increased malignancy, likely contributing to the observed aggravation of cell proliferation and metastasis in GC tissues with increased PFKFB3 expression.
PFKFB3 promotes migration and invasion in GC cells in vitro
To investigate whether PFKFB3 directly affects the growth and migration of GC cells, we analyzed PFKFB3 expression in SGC-7901, MGC-803, AGS, and BGC-823 cells by real-time reverse transcription polymerase chain reaction (RT-PCR) and western blot, respectively. Basal PFKFB3 mRNA and protein amounts were detected in the 4 GC cell lines (Figure 1f), which were increased or decreased significantly when cells were transfected with PFKFB3-overexpressing lentivirus or PFKFB3 shRNA (P < 0.001, Figure 3a; and P < 0.05, 3b). MTT assay showed that cell proliferation in SGC-7901 cells was significantly enhanced by PFKFB3 overexpression (P < 0.001, Figure 3c) and markedly inhibited by PFKFB3 knockdown in AGS cells (P < 0.001, Figure 3d). Consistently, colony formation in SGC-7901 cells was also significantly increased by PFKFB3 overexpression (P < 0.001, Figure 3e; and P < 0.01, 3g) and starkly reduced by PFKFB3 knockdown in AGS cells (P < 0.001, Figure 3f; and P < 0.01, 3h).
Figure 3.
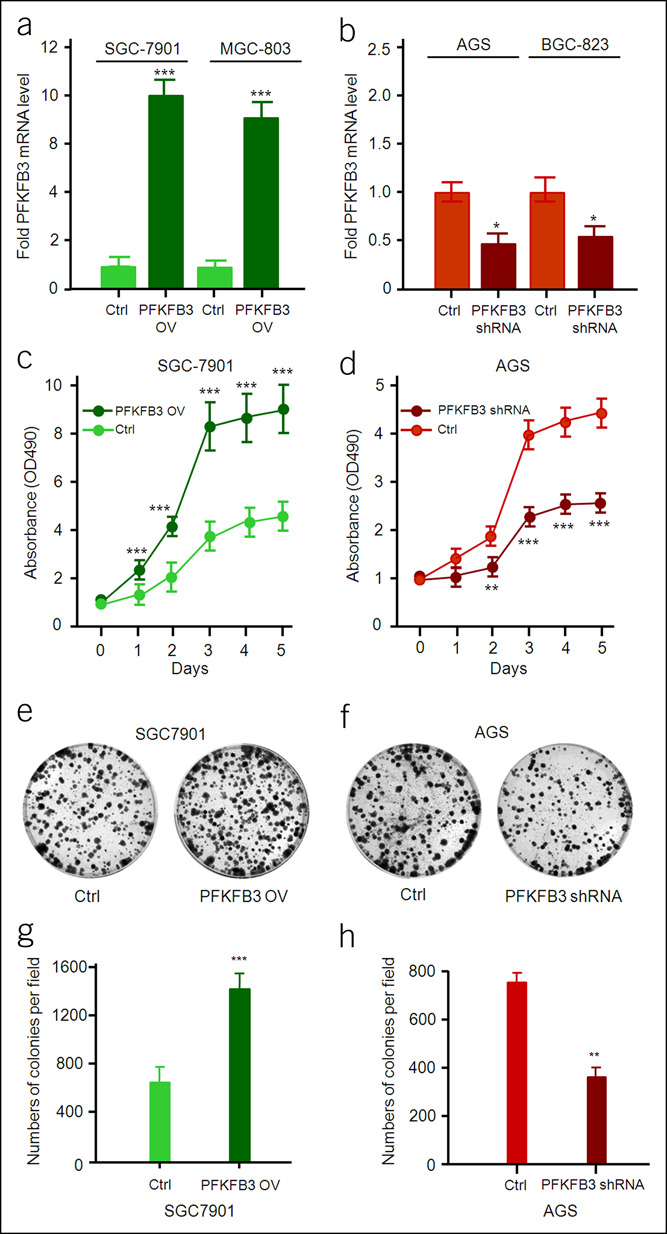
Effect of PFKFB3 on proliferation of gastric cancer cells. The mRNA level of PFKFB3 in PFKFB3-overexpressing or PFKFB3-specific shRNA gastric cancer cells was detected by quantitative RT-PCR. All data are shown as mean ± SD and *P < 0.05; ***P < 0.001 (a and b). The cell growth rate was measured by MTT assay with OD490 nm in gastric cancer cells (PFKFB3-overexpressing or PFKFB3-specific shRNA). All data are shown as mean ± SD and **P < 0.01; ***P < 0.001 (c and d). The colony formation assay in gastric cancer cells (PFKFB3-overexpressing or PFKFB3-specific shRNA) (e and f). All data are shown as mean ± SD and **P < 0.01; ***P < 0.001 (g and h). RT-PCR, reverse transcription polymerase chain reaction.
Then, wound-healing assay was performed to examine the role of PFKFB3 in regulating the migration abilities of SGC-7901 and AGS cells. PFKFB3-overexpressing SGC-7901 cells migrated much more to the wounded region at 24 hours after wound scratch compared with untreated cells (P < 0.001; Figure 4a,b). By contrast, opposite results were achieved by PFKFB3 knockdown in AGS cells (P < 0.001; Figure 4c,d). Furthermore, the impact of PFKFB3 on the invasive capacities of SGC-7901 and AGS cells was examined by Transwell invasion assay. On PFKFB3 overexpression, significantly more SGC-7901 cells passing through Transwell membrane were obtained compared with the control groups (P < 0.001; Figure 4e,g). On the contrary, PFKFB3 knockdown in AGS cells resulted in significantly reduced amounts of cells passing through Transwell membrane (P < 0.001; Figures 4f,g). Together, these results demonstrated that PFKFB3 promoted migration and invasion in PFKFB3-overexpressing GC cells.
Figure 4.

PFKFB3 regulates the migration of gastric cancer cells. The wound-healing assay in gastric cancer cells (PFKFB3-overexpressing or PFKFB3-specific shRNA) (a and c). All data are shown as mean ± SD and ***P < 0.001 (b and d). The Transwell assay in gastric cancer cells (PFKFB3-overexpressing or PFKFB3-specific shRNA) (e and f). All data are shown as mean ± SD, ***P < 0.001 (g).
PFKFB3 modulates EMT-related molecules in GC cells
To investigate whether EMT contributes to the enhanced GC cell migration and invasion induced by PFKFB3 overexpression, we examined the effect of PFKFB3 on the expression of EMT markers in SGC-7901 and AGS cells using real-time RT-PCR and western blot. In SGC-7901 cells, PFKFB3 overexpression resulted in downregulation of the epithelial marker E-cadherin, concomitant with upregulation of the mesenchymal markers N-cadherin and Vimentin at both mRNA and protein levels (P < 0.001, Figure 5a,b). Then, we explored the effect of PFKFB3 on the expression of EMT-related transcription factors in SGC-7901 cells. PFKFB3 overexpression significantly increased the mRNA and protein levels of Snail and Twist, but not Zeb1, in SGC-7901 cells (P < 0.01, Figures 5a,b). The opposite results were achieved by knockdown of PFKFB3 in AGS cells (P < 0.01, Figure 5c,d). Together, these data suggested that PFKFB3 overexpression could confer GC cells with EMT-like features, which might be responsible for PFKFB3-induced GC cell migration and invasion.
Figure 5.

PFKFB3 regulated the expression of EMT-related molecules in GC cells. Real-time RT-PCR and western blot analyses revealed PFKFB3 overexpression significantly promoted the expression of N-cadherin, Vimentin, Snail, and Twist, inhibited E-cadherin at both mRNA and protein levels in SGC-7901 cells, whereas the expression of Zeb1 did not show obvious changes (a and b). All data are shown as mean ± SD, ***P < 0.001 (a). SGC-7901 cells with stable PFKFB3 overexpression were pretreated with 1-mM helenalin for 60 minutes. Western blot analysis showed that helenalin pretreatment could suppressed PFKFB3-induced the expression of EMT-related molecules in SGC-7901 cells (b). RT-PCR and western blot analyses revealed PFKFB3 knockdown expression significantly inhibited the expression of N-cadherin, Vimentin, Snail, and Twist, promoted E-cadherin at both mRNA and protein levels in SGC-7901 cells, whereas the expression of Zeb1 did not show obvious changes (c and d). All data are shown as mean ± SD, **P < 0.01; ***P < 0.001 (c). AGS cells with PFKFB3 shRNA were incubated with 100-ng/mL NF-κB p65 for 24 hours, or AGS cells with stable PFKFB3 knockdown expression were transfected with NF-κB p65–overexpressing lentivirus. Western blot analysis showed that NF-κB p65 treatment could rescue PFKFB3 knockdown expression-mediated the expression of EMT-related molecules in AGS cells (d). EMT, epithelial-mesenchymal transition; RT-PCR, reverse transcription polymerase chain reaction.
NF-κB pathway is involved in PFKFB3-induced EMT process in GC cells
To assess whether NF-κB signaling pathway is involved in the effects of PFKFB3 on GC cells, an NF-κB inhibitor, helenalin, was used. Helenalin pretreatment abolished PFKFB3-mediated downregulation of E-cadherin and upregulation of N-cadherin, Snail, and Twist in SGC7901 and AGS cells (Figure 5b,d). Since Matrix Metallo Proteinase (MMP) family members are involved in tumor invasion and metastasis and can be regulated by NF-κB pathway, we further investigated the effect of PFKFB3 on MMPs. Western blot indicated that PFKFB3 overexpression would upregulate MMP2, MMP9, and MMP14 in SGC7901 cells (see Supplementary Figure 2A, Supplementary Digital Content 1, http://links.lww.com/CTG/A644). Moreover, pretreatment of SGC-7901 cells with helenalin significantly suppressed PFKFB3-induced alterations of MMPs. Conversely, downregulation of PFKFB3 expression by shRNA or helenalin could downregulate the expression of MMP-2, MMP-9, and MMP-14 in AGS cells (see Supplementary Figure 2B, Supplementary Digital Content 1, http://links.lww.com/CTG/A644). Together, these data indicated that NF-κB pathway contributed to PFKFB3-induced EMT in GC cells.
Inhibition of PFKFB3 enhances the sensitivity of DDP chemotherapy
PFK15 is a powerful inhibitor of PFKFB3. By inhibiting the enzyme activity of PFKFB3, the glycolysis rate and the synthesis of ATP are reduced (16,17). AGS cells were treated with PFKFB3 inhibitor PFK15 concentration gradient for 24 hours. It can be observed that PFKFB3 inhibitor PFK15 treatment at the concentration of 10 μm can significantly reduce the content of ATP in AGS cells (Figure 6a). The results of flow cytometry showed that PFK15, an inhibitor of PFKFB3 alone, could not effectively induce apoptosis of AGS cells. However, in the experimental group treated with DDP, the combined use of PFKFB3 inhibitor PFK15 could significantly enhance the apoptosis level of tumor cells induced by DDP. Consistent with the experimental results of knockout PFKFB3, inhibition of PFKFB3 can significantly enhance the sensitivity of tumor cells to chemotherapy with DDP and can improve the effect of DDP treatment on apoptosis of tumor cells (Figure 6b).
Figure 6.
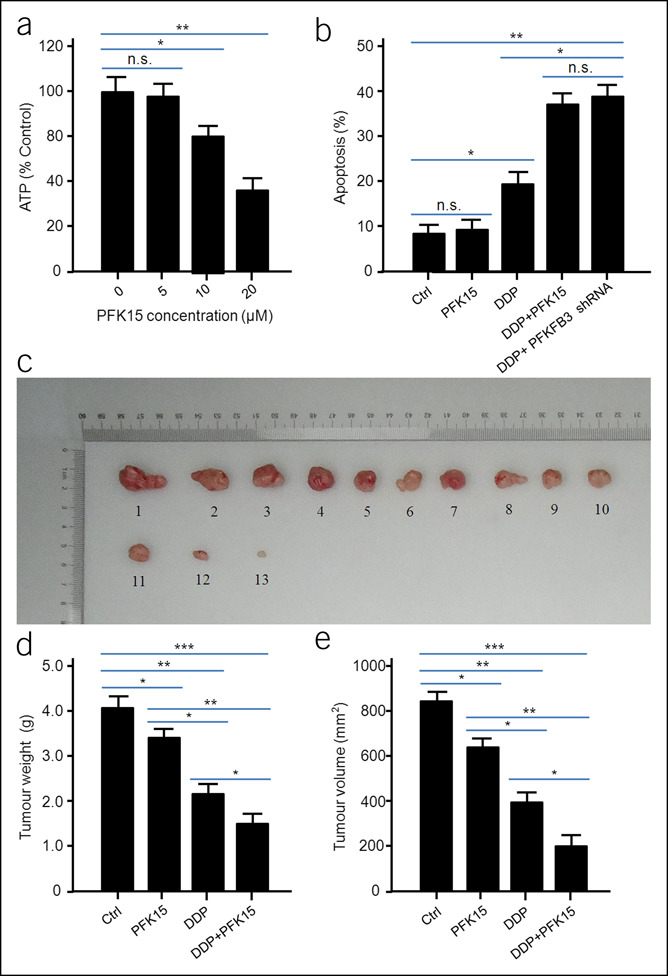
Combined use of cisplatin (DDP) and PFK15 is more efficient in inhibiting xenograft tumor growth than single treatment. PFK15 treatment could significantly reduce ATP content in cells. All data are shown as mean ± SD, *P < 0.05; **P < 0.01; (a). Combined use of PFK15 could significantly enhance the apoptosis of AGS cells induced by DDP. All data are shown as mean ± SD, *P < 0.05; **P < 0.01 (b). Representative images of the tumors isolated from the mice. 1#–4#, Ctrl; 5#–7#, PFK15 treatment group; 8#–10#, DDP treatment group; 11#–13#, DDP/PPK15 combined treatment group (c). The weight of the tumors from nude mice. All the data were shown as mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001 (d). The volume of the tumors from nude mice. All the data were shown as mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001 (e). The same experiment was performed 2 times with similar results. n.s., no significant.
The subcutaneous tumor model of AGS nude mice was established to observe the effect of combined use of PFKFB3 inhibitor PFK15 and DDP on the growth of nude mice. The nude mice were randomly divided into 4 groups with 4 mice in each group: control group, PFKFB3 inhibitor PFK15 treatment group, DDP treatment group, and PFKFB3 inhibitor PFK15/DDP combined treatment group. During the experiment, 1 mouse died in the PFKFB3 inhibitor PFK15 treatment group, DDP treatment group, and PFKFB3 inhibitor PFK15/DDP combined treatment group, respectively. After 30 days of treatment, the mice were killed, the subcutaneous tumors were collected, weighed, and the volume was measured and recorded, as shown in Figure 6c. The results of tumor weight and volume in nude mice showed that PFKFB3 inhibitor PFK15/DDP combined treatment could significantly inhibit the growth of tumor in nude mice compared with the DDP treatment group, and the tumor weight was lighter, and the tumor volume was smaller in the PFKFB3 inhibitor PFK15/DDP combined treatment group (Figure 6d,e). RT-PCR analysis of tumor in nude mice showed that PFKFB3 inhibitor PFK15 combined with DDP could significantly upregulate the expression of E-cadherin and downregulate the expression of N-cadherin, Twist, Snail, Vimentin, HIF-1α, and MMP9 (Figure 7a), suggesting that PFKFB3 inhibitor PFK15 combined with DDP could inhibit the EMT process mediated by PFKFB3 through NF-κB and inhibit the growth and metastasis of GC. The results also suggest that the application of PFKFB3 inhibitor PFK15 in mice could further enhance the therapeutic effect of DDP chemotherapy on nude mouse xenograft in vivo.
Figure 7.

Combined application of PFK15 could significantly promote the inhibition of tumor progress and metastasis by DDP in nude mice. Real-time RT-PCR analyses revealed DDP/PPK15 combined treatment significantly promoted the expression of E-cadherin and inhibited N-cadherin, Vimentin, Snail, Twist, NF-kB p65, MMP9, and HIF-1α in nude tumors. All the data were shown as mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001 (a). A schematic model of PFKFB3 promotes GC growth and metastasis through NF-κB–mediated EMT process, and the application of a PFKFB3 inhibitor could further enhance the therapeutic effect of DDP chemotherapy on GC (b). GC, gastric cancer; RT-PCR, reverse transcription polymerase chain reaction.
DISCUSSION
Glycolysis is the main energy source of tumor cells, and PFKFB is an important regulator of glycolysis (5,6,18,19). Recent studies have shown that PFKFB family members play an important role in tumorigenesis and progression through metabolic remodeling (20,21). PFKFB3 also plays an important role in the invasion and metastasis of some human cancers (9,10). However, little is known about the role of PFKFB3 in GC. Here, we showed that PFKFB3 was significantly overexpressed in GC tissues compared with PCHNTs. The expression of PFKFB3 in GC cells was overexpressed even under normoxic conditions. Furthermore, in vitro experiments showed that PFKFB3 overexpression significantly potentiated GC cell migration and invasion by promoting EMT. Moreover, PFKFB3-induced EMT in GC cells was completed through upregulation of Snail and Twist, which depend on NF-κB signaling activation. The results demonstrated a causal role of PFKFB3 in GC cell migration and invasion, which was further confirmed by the nude mouse xenograft model in vivo. However, the limitation of the study is that its exact molecular mechanism has not been fully elucidated and needs to be further explored in the future.
Previous studies have shown that PFKFB3 is overexpressed in colon cancer, which is associated with poor clinical outcomes (14,15,22,23). However, the level of PFKFB3 expression in GC and its significance remain elusive. Here, IHC analysis of a large cohort of 319 patients with GC demonstrated that PFKFB3 expression was significantly higher in tumor tissues compared with PCHNTs. The percentage of GC samples with grade 2 and the amounts of PFKFB3-overexpressing cells (IHC score ≥9) were significantly higher in GC tissues (68.0%, 217/319) compared with PCHNTs (13.5%, 43/319), whereas the percentage of samples with grade 1 (IHC score 0–8) was significantly lower in tumor tissues (32.0%, 102/319) compared with PCHNTs (86.5%, 276/319).
In vitro experiments found that PFKFB3 overexpresssion could markedly increase the migratory and invasive abilities of GC cells, with overt downregulation of the epithelial marker E-cadherin and upregulation of the mesenchymal marker N-cadherin, especially upregulation of transcription factors Snail and Twist, which are pivotal inducers of EMT (20,21,24,25). Snail and Twist have been reported to mediate EMT, resulting in tumor progression and poor survival in patients with GC (17,26,27). Our results showed that PFKFB3 overexpression led to significantly increased expression levels of Snail and Twist, but not Zeb1. These findings strongly suggested that PFKFB3 overexpression induced EMT through Snail and Twist upregulation, thereby favoring GC cell migration and invasion. Moreover, we found that PFKFB3 overexpression could upregulate MMP2, MMP9, and MMP14, indicating that increased expression of MMPs is involved in the mechanism by which PFKFB3 promotes tumor invasion and migration through the NF-κB pathway. Collectively, these findings suggest that PFKFB3 plays a key role in the process of GC metastasis.
Our data further revealed that PFKFB3 induced NF-κB p65 upregulation, indicating NF-κB signaling activation (28). Moreover, pharmacological inhibition of the NF-κB pathway attenuated PFKFB3-induced expression of EMT-related markers in GC cells in vitro. Therefore, it is reasonable to assume that NF-κB is involved in PFKFB3 overexpression-mediated invasion, migration, and EMT in GC cells. The nude mouse xenograft model in vivo further confirmed that PFKFB3 promotes GC growth and metastasis through NF-κB–mediated EMT process.
The effect of chemotherapy with DDP is limited by the drug resistance of tumor cells. How to improve the sensitivity of DDP chemotherapy to patients is an urgent problem to be solved. PFK15 is a powerful inhibitor of PFKFB3, known as the PKFKB3 inhibitor PFK15, reduces F2,6-DP, glucose uptake, and intracellular ATP levels in leukemia cells and lung adenocarcinoma cells, and also yields antitumor effects that are comparable with approved chemotherapeutic agents (16,17). Our results demonstrated that the combined use of PFKFB3 inhibitor PKF15 in GC cells can enhance the apoptosis induced by DDP in vitro. The results in vivo showed that the combined use of PFKFB3 inhibitor PFK15 could further enhance the inhibitory effect of DDP chemotherapy on the growth of subcutaneous tumor in nude mice, suggesting that targeted inhibition of PFKFB3 may become a new clinical treatment strategy.
In conclusion, we demonstrated that PFKFB3 is significantly overexpressed in GC, and PFKFB3 overexpression could lead directly to migration, invasion, and EMT in GC cells through NF-κB signaling pathway (Figure 7b). Therefore, PFKFB3-NF-κB may have a critical role in regulating the metastatic potential of GC cells, providing an attractive therapeutic target for GC.
CONFLICTS OF INTEREST
Guarantor of the article: Zhi-Qiang Ling, MD, PhD.
Specific author contributions: Lan Lei, MD, and Lian-Lian Hong, PhD, contributed equally to this work. Z.-Q.L.: conceived and designed the study. L.L. and L.-L.H.: performed the experiments and performed data analysis. L.L., Z.-N.L., Y.Z., X.-Y.H., and P.L.: performed statistical analysis. L.L. and L.-L.H.: wrote the manuscript. X.-Y.H.: participated in manuscript revision. Z.-Q.L.: performed manuscript editing and manuscript review. All authors read and approved the final manuscript.
Financial support: This work was funded by the National Natural Science Foundation of China (81372332, 81572822, and 81972908), Key Projects from the Zhejiang Provincial Natural Science Foundation (LZ13H160002 and LZ18H160002), General Project from the Zhejiang Provincial Natural Science Foundation (LQ19H160005 and LY17H160044), the Leading Talents in Scientific and Technological Innovation from Zhejiang Provincial Ten Thousand Talents Plan (Zhejiang Provincial CPC Committee Talents [2019]-3), Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents (Zjwjw2014-108), and the Major Training Personnel from Zhejiang Provincial Program for the Training and Development Project for 151 Talents (Zjhrss2014-150). Innovation Research Team (in Science and Technology) in University of Henan Province (20IRTSTHN026). Zhejiang Medical and Health Science and Technology Project (2019ZD025).
Potential competing interests: None to report.
Study Highlights.
WHAT IS KNOWN
✓ PFKFB3 regulates glycolysis in tumor cells, might function as an oncogene, and is associated with cancer metastasis. However, its role in gastric cancer (GC) remains largely unknown.
✓ The role of PFKFB3 in GC remains largely unknown, in particular, whether PFKFB3 is a useful therapeutic target in GC.
WHAT IS NEW HERE
✓ Our research first showed that PFKFB3 is significantly overexpressed in GC and PFKFB3-NF-κB may have a critical role in regulating the metastatic potential of GC, providing an attractive therapeutic target for patients with GC.
TRANSLATIONAL IMPACT
✓ Exploration the role of PFKFB3 in the laboratory setting may contribute to the understanding of underlying molecular mechanisms and may inspire the development of novel targeted therapeutic strategies for patients with GC.
Supplementary Material
ACKNOWLEDGMENT
We thank all individuals who participated in this study and donated samples.
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A644; http://links.lww.com/CTG/A645; http://links.lww.com/CTG/A646; http://links.lww.com/CTG/A647; http://links.lww.com/CTG/A648; and http://links.lww.com/CTG/A649.
Contributor Information
Lan Lei, Email: lan_lei@outlook.com.
Lian-Lian Hong, Email: hongll1988@hotmail.com.
Zhe-Nan Ling, Email: zhenanling@outlook.com.
Yi Zhong, Email: zhongyuyx@outlook.com.
Xuan-Yu Hu, Email: huxuanyu@gs.zzu.edu.cn.
Pei Li, Email: lipei19700415@outlook.com.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida GJ. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res 2015;34:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu XD, Shao SX, Jiang HP, et al. Warburg effect or reverse Warburg effect? A review of cancer metabolism. Oncol Res Treat 2015;38:117–22. [DOI] [PubMed] [Google Scholar]
- 4.Kalyanaraman B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol 2017;12:833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganapathy-Kanniappan S. Linking tumor glycolysis and immune evasion in cancer: Emerging concepts and therapeutic opportunities. Biochim Biophys Acta Rev Cancer 2017;1868:212–20. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Zhang Z, Wang J, et al. Metabolic reprogramming results in abnormal glycolysis in gastric cancer: A review. Onco Targets Ther 2019;12:1195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu DH, Li Q, Hu H, et al. Transmembrane protein GRINA modulates aerobic glycolysis and promotes tumor progression in gastric cancer. J Exp Clin Cancer Res 2018;37:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Goronzy JJ, Weyand CM. The glycolytic enzyme PFKFB3/phosphofructokinase regulates autophagy. Autophagy 2014;10:382–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi L, Pan H, Liu Z, et al. Roles of PFKFB3 in cancer. Signal Transduct Target Ther 2017;2:17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartrons R, Rodríguez-García A, Simon-Molas H, et al. The potential utility of PFKFB3 as a therapeutic target. Expert Opin Ther Targets 2018;22:659–74. [DOI] [PubMed] [Google Scholar]
- 11.Long Q, Zou X, Song Y, et al. PFKFB3/HIF-1α feedback loop modulates sorafenib resistance in hepatocellular carcinoma cells. Biochem Biophys Res Commun 2019;513:642–50. [DOI] [PubMed] [Google Scholar]
- 12.Novellasdemunt L, Bultot L, Manzano A, et al. PFKFB3 activation in cancer cells by the p38/MK2 pathway in response to stress stimuli. Biochem J 2013;452:531–43. [DOI] [PubMed] [Google Scholar]
- 13.Bando H, Atsumi T, Nishio T, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res 2005;11:5784–92. [DOI] [PubMed] [Google Scholar]
- 14.Yu H, Zhang H, Dong M, et al. Metabolic reprogramming and AMPKα1 pathway activation by caulerpin in colorectal cancer cells. Int J Oncol 2017;50:161–72. [DOI] [PubMed] [Google Scholar]
- 15.Han J, Meng Q, Xi Q, et al. Interleukin-6 stimulates aerobic glycolysis by regulating PFKFB3 at early stage of colorectal cancer. Int J Oncol 2016;48:215–24. [DOI] [PubMed] [Google Scholar]
- 16.Clem BF, O'Neal J, Tapolsky G, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 2013;12:1461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li FL, Liu JP, Bao RX, et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat Commun 2018;9:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li HM, Yang JG, Liu ZJ, et al. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res 2017;36:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantelmo AR, Conradi LC, Brajic A, et al. Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 2016;30:968–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kocemba KA, Dulińska-Litewka J, Wojdyła KL, et al. The role of 6-phosphofructo-2-kinase (PFK-2)/fructose 2,6-bisphosphatase (FBPase-2) in metabolic reprogramming of cancer cells. Postepy Hig Med Dosw (Online) 2016;70:938–50. [DOI] [PubMed] [Google Scholar]
- 21.Ros S, Schulze A. Balancing glycolytic flux: The role of 6-phosphofructo-2- kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab 2013;1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atsumi T, Chesney J, Metz C, et al. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6- bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res 2002;62:5881–7. [PubMed] [Google Scholar]
- 23.Klarer AC, O'Neal J, Imbert-Fernandez Y, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab 2014;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goossens S, Vandamme N, Van Vlierberghe P, et al. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim Biophys Acta Rev Cancer 2017;1868:584–91. [DOI] [PubMed] [Google Scholar]
- 25.Zhang P, Hu P, Shen H, et al. Prognostic role of twist or snail in various carcinomas: A systematic review and meta-analysis. Eur J Clin Invest 2014;44:1072–94. [DOI] [PubMed] [Google Scholar]
- 26.Krzysiek-Maczka G, Targosz A, Szczyrk U, et al. Role of Helicobacter pylori infection in cancer-associated fibroblast- induced epithelial-mesenchymal transition in vitro. Helicobacter 2018;23:e12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosivatz E, Becker I, Specht K, et al. Differential expression of the epithelial-mesenchymal transition regulators snail, SIP1, and twist in gastric cancer. Am J Pathol 2002;161:1881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanigur Sultuybek G, Soydas T, Yenmis G. NF-κB as the mediator of metformin's effect on ageing and ageing-related diseases. Clin Exp Pharmacol Physiol 2019;46:413–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.