

Abstract
Introduction:
Pediatric patients, especially neonates and infants, are more susceptible to adverse drug events as compared to adults. In particular, immature small molecule drug metabolism and excretion can result in higher incidences of pediatric toxicity than adults if the pediatric dose is not adjusted.
Area covered:
We reviewed the top 29 small molecule drugs prescribed in neonatal and pediatric intensive care units and compiled the mechanisms of their metabolism and excretion. The ontogeny of Phase I and II drug metabolizing enzymes and transporters (DMETs), particularly relevant to these drugs, are summarized. The potential effects of DMET ontogeny on the metabolism and excretion of the top pediatric drugs were predicted. The current regulatory requirements and recommendations regarding safe and effective use of drugs in children are discussed. A few representative examples of the use of ontogeny-informed physiologically-based pharmacokinetic (PBPK) models are highlighted.
Expert opinion:
Empirical prediction of pediatric drug dosing based on body weight or body-surface area from the adult parameters can be inaccurate because DMETs are not mature in children and the age-dependent maturation of these proteins is different. Ontogeny-informed-PBPK modeling provides a better alternative to predict the pharmacokinetics of drugs in children.
Keywords: Pediatric precision medicine, pediatric pharmacology, developmental toxicity, drug metabolism, drug transport, ontogeny
1. Introduction
Children, especially neonates and infants, are more susceptible to adverse drug events (ADEs) as compared to adults [1-4]. Particularly, immature drug detoxification, i.e., metabolism and excretion, are associated with higher incidences of ADEs in children (Figure 1). For example, the low expression of uridine 5'-diphospho-glucuronosyltransferase (UGT) enzymes in neonates resulted in greater exposure of chloramphenicol when dosed based on empirical scaling of adult data, i.e., body-weight or body-surface area (BSA) normalized dosing. This was linked to toxicity, termed grey baby syndrome, which resulted in several deaths [5,6]. Subsequent studies demonstrated that chloramphenicol is metabolized by UGT2B7, which is primarily expressed in the liver, and its expression increases by 20-fold from neonates to adults [7,8]. Individual drug metabolizing enzymes and transporters (DMETs) show unique developmental patterns or ontogeny. Such non-monotonic changes in DMET abundance is another challenge for drugs that are metabolized by multiple enzymes, e.g., acetaminophen which is metabolized by UGTs and sulfotransferases (SULTs). Although hepatic UGT levels change dramatically with age during the first two years of life, acetaminophen metabolizing SULT (SULT2A1) exhibits a biphasic age-dependent expression in liver with higher levels in children (1-12 years) as compared to adults [9]. This results in a higher contribution of SULTs over UGTs in acetaminophen detoxification in children as compared to adults and vice versa [10]. Therefore, it is important to characterize the ontogeny and differential abundance of DMETs during development for the safe use of drugs in children.
Figure 1.

Summary of age-associated changes in absorption, distribution, metabolism, and excretion (ADME) processes from neonates to adults (the artwork was supported by Servier Medical Art, licensed under CC BY 3.0).
Empirical scaling of drug dosing from adult data is not optimal, since developmental differences in DMET abundance may result in toxicities that would not be observed in adults. In a recent analysis of 290 pediatric studies ranging from 1997 to 2010, 50% of the total drugs prescribed to the pediatric population are available without clinical testing and more than 90% of the drugs prescribed to neonates and infants are without safety and efficacy data [3]. The incidence of off-label medicines prescribed in neonatal intensive care units (NICU) reaches as high as 95.6% [11], resulting in increased incidences of ADE or reduced drug efficacy [12,13]. In the absence of clinical studies on pediatric drug disposition, pediatricians must select the first-in-child dose based on the empirical scaling of adult data, which is often associated with ADEs or failed clinical trials [14]. Considering this challenge, the Food and Drug Administration (FDA) and other regulatory agencies now require that drug developers provide a written plan of conducting pediatric studies before the completion of phase II clinical trials. Furthermore, the regulatory agencies have initiated strong incentives for drug developers to conduct pediatric clinical trials that include 180 days additional market exclusivity [15-18]. However, it is still logistically and ethically difficult to conduct clinical trials in children as it requires consent from parents and the risks associated with ADEs.
An ideal approach would be to prospectively predict drug disposition and potential ADEs in children early in drug development by careful consideration of ontogeny of drug disposition processes, including DMET abundance. The research into pediatric DMET ontogeny in the past two decades allows us to better explain why therapeutic drug response can be substantially different in children versus adults [7,9,13,19-24]. Moreover, the recent emergence of physiologically-based pharmacokinetic (PBPK) modeling has resulted in promising applications of ontogeny information to predict first-in-child dose and design better pediatric clinical trials to avoid ADEs and save on the costs for failed clinical trials.
In this review, we first compiled the commonly used small molecule drugs in the neonatal and pediatric intensive care units (NICU and PICU, respectively) and then reviewed the mechanisms of metabolism and excretion of these drugs in adults. Finally, we investigated the potential implications of DMET ontogeny on the disposition of commonly prescribed drugs in the NICU and PICU. Some examples of the application of DMET ontogeny in prospectively predicting drug disposition by PBPK modeling are provided. The regulatory requirements and recommendations regarding the pediatric pharmacotherapy are also summarized.
2. Mechanisms of drug metabolism and excretion
Typically, metabolism inactivates drugs, but sometimes the metabolites can be pharmacologically active (i.e., in case of prodrugs) or reactive (leading to toxicity). Drugs undergo metabolism through various pathways such as oxidation, reduction, hydrolysis, and conjugation to facilitate excretion from the body. Enzymes involved in metabolism are present throughout the body but are concentrated in the liver. The extent of drug metabolism varies depending on many factors such as age, sex, genetics, or disease states. In children, the development of some liver enzymes are not present until the age of 9 years [25,26]. Typically, drug metabolism reactions are classified into two categories, Phase I and II. In the Phase I metabolism, a drug is converted into an inactive, active, or reactive moiety by adding or modifying a functional group through oxidation, reduction, or hydrolysis. The Phase II reactions involve conjugating a polar compound (e.g., glucuronic acid or sulfate) to a reactive group, either already present or created by a Phase I reaction. This makes the compound more easily excreted via kidneys or liver into urine or bile, respectively. Not all drugs undergo both Phase I and II reactions, some may involve just one path and not a linear sequence. The common drug metabolizing enzymes are cytochrome P450s (CYP), UGTs and SULTs [27].
Drug transporters are proteins that regulate entry and exit of drugs to or from a cell and thus can influence metabolism, intercellular concentration, toxicity and efficacy of drugs [28-31]. Often, drug transporters from the ATP-binding cassette (ABC) and solute carrier (SLC) transporter families are rate-limiting in the uptake and efflux of drugs in the liver, intestine, and kidney [32]. The common transporters are organic anion transporting polypeptides (OATPs) in the liver, organic anion and organic cation transporters (OATs and OCTs) in the kidney, and P-glycoprotein (P-gp) and breast cancer resistance protein (BRCP) expressed ubiquitously [33,34]. In the kidney, drugs are eliminated by glomerular filtration, active secretion, or reabsorption. Active secretion and reabsorption are typically mediated by transporters such as OATs, OCTs and ABC efflux transporters [28].
3. Commonly prescribed drugs in PICU/NICU
We analyzed a list of commonly prescribed drugs from the NICU and PICU of Providence Sacred Heart Medical Center and Children’s Hospital in Spokane, WA, USA. The data were collected for the top 30 drugs each prescribed in NICU and PICU between January to December 2019. Out of these drugs, eight were common in both categories. The pharmacokinetics (PK) of a total of 29 drugs was critically investigated for the mechanisms of drug metabolism and excretion (Table 1 and Supplemental Table S1). The following drugs or compounds were excluded from the top 30 drug lists: i) large molecules, e.g., heparin, ii) drugs used for topical or local applications, and iii) vitamins and supplements (Supplemental Table S2). When free drug as well as its salt-forms were used, the data were combined for both the forms. The top 5 prescribed drugs in the NICU were morphine, levalbuterol, caffeine, ampicillin, and lorazepam (Figure 2a) and the top 5 dosed drugs in the PICU were albuterol (racemic), acetaminophen, famotidine, furosemide, and levalbuterol (Figure 2b).
Table 1.
Adult pharmacokinetic parameters of the most prescribed drugs used in NICU/PICU
| Drug | F (%) |
t1/2 (hr) |
Vd (L) |
CLH (ml/min ) |
CLR (ml/min ) |
Mechanism of elimination |
Primary hepatic enzymes# |
Renal mechanism |
Critical mechanism(s) leading to age- dependent PK changes |
References |
|---|---|---|---|---|---|---|---|---|---|---|
| Acetaminophen | 88 | 2 | 67 | 340* | 11* | Hepatic | UGT1s | Reabs. | UGTs are more important in adults, whereas SULTs are more important in children | [74,149,150] |
| Albuterol (Racemic, R+S) | R: 30 | R: 2 | R: 140 | R: 389* | R: 332* | Equal | SULT1A3 | Sec. | Ontogeny of hepatic SULT1A3 and renal OATs are the likely determinants of the age-effect | [151,152] |
| S: 71 | S: 2.8 | S: 124 | S: 205* | S: 250* | ||||||
| Levalbuterol | 30 | 2.5 | 124 | 383* | 400 | [153,154] | ||||
| Ampicillin | 89 | 1.8 | 25 | 47 | 234* | Renal | NA | Sec. | Ontogeny of renal OATs is the likely rate-determining factor | [155-157] |
| Budesonide | 10 | 3 | 245 | 1186* | 147* | Hepatic | CYP3A4 | Sec. | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [158] |
| Caffeine | 108 | 5.2 | 30 | 53* | 1 | Hepatic | CYP1A2 | Reabs. | Hepatic CYP1A2 ontogeny is the likely rate determining factor | [4,41,159-162] |
| Cefepime | 0 | 2 | 18 | 21* | 105 | Renal | NA | GF | Age-dependent CL likely correlate with the GFR | [157,163] |
| Clonidine | 95 | 12 | 147 | 87* | 141 | Equal | CYP2D6 | Sec. | Ontogeny of hepatic CYP2D6 and renal transporters are both important | [164-166] |
| Dexamethasone | 65 | 4 | 66 | 95* | 11* | Hepatic | CYP3A4 | Reabs. | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [167] |
| Dexmedetomidine | 16 | 2.1 | 1.6 | 736* | 7* | Hepatic | UGT1A4, UGT2B10 | Reabs. | Ontogeny of UGT1A4 and UGT2B10 is the likely rate determining factor | [168,169] |
| Diazepam | 100 | 43 | 77 | 26* | 0.2* | Hepatic | CYP3A4 | Reabs. | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [170-172] |
| Famotidine | 37 | 3 | 87 | 292* | 768 | Renal | NA | Sec. | Ontogeny of renal transporters is the likely determinant of the age-effect | [173-177] |
| Fentanyl | 50 | 3.7 | 280 | 837* | 73 | Hepatic | CYP3A4 | Sec. | Hepatic CYP3A4 ontogeny is the rate determining factor | [77,178,179] |
| Furosemide | 71 | 1.3 | 9.1 | 34* | 83 | Renal | UGT1A9 | Sec. | Ontogeny of renal OATs is the likely determinant of the age-effect | [114,180-183] |
| Gentamicin | 100 | 2.5 | 22 | 8* | 76 | Renal | NA | Reabs. | Ontogeny of apical renal transporters is the likely determinant of the age-effect | [157,184,185] |
| Hydrocortisone | 96 | 1.8 | 34 | 77 | 223 | Renal | CYP3A4 | Sec. | Ontogeny of renal transporters is the likely determinant of the age-effect | [186-189] |
| Ibuprofen | >80 | 2 | 11 | 48* | 0.4* | Hepatic | CYP2C9 | Reabs. | Hepatic CYP2C9 ontogeny is the likely rate determining factor | [190-192] |
| Levetiracetam | 95 | 7 | 42 | 2* | 30 | Renal | Hydrolysis | Reabs. | Ontogeny of apical renal transporters is the likely determinant of the age-effect | [193,194] |
| Lorazepam | 93 | 14 | 91 | 71* | 0.7* | Hepatic | UGT2B7 | Reabs. | Hepatic UGT2B7 ontogeny is the likely rate determining factor | [83,195,196] |
| Methadone | 92 | 27 | 252 | 83* | 26 | Hepatic | CYP3A4 | Sec. | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [197-199] |
| Methylprednisolone | 82 | 2.3 | 84 | 1425* | 75 | Hepatic | CYP3A4 | Sec. | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [187,189,200] |
| Midazolam | 30 | 2 | 77 | 366* | 4 | Hepatic | CYP3A4 | Sec. | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [4,201,202] |
| Morphine | 24 | 2 | 231 | 1613* | 67 | Hepatic | UGT2B7 | GF | Hepatic UGT2B7 ontogeny is the likely rate determining factor | [77,79,203] |
| Ondansetron | 62 | 3.5 | 133 | 549* | 29 | Hepatic | CYP1A2 | GF | Hepatic CYP1A2 ontogeny is the likely rate determining factor | [204-206] |
| Oxycodone | 42 | 2.6 | 140 | 868 | NA | Hepatic | CYP3A4 | NA | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [77,207-209] |
| Prednisolone | 82 | 2.2 | 30 | 616* | 217 | Hepatic | CYP3A4 | Sec. | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [187,200,210] |
| Propranolol | 26 | 4 | 301 | 1811* | 9 | Hepatic | CYP2D6 | Reabs. | Hepatic CYP2D6 ontogeny is the likely rate determining factor | [211-214] |
| Sildenafil | 38 | 2.4 | 84 | 420* | NA | Hepatic | CYP3A4 | NA | Hepatic CYP3A4 ontogeny is the likely rate determining factor | [215,216] |
| Vancomycin | NA | 5 | 27 | 28* | 103 | Renal | NA | GF | Age-dependent CL likely correlate with the GFR | [157,217-219] |
Rac: racemic; S: sinister stereochemistry; R: rectus stereochemistry; Sec: secretion; Reabs: reabsorption; F: oral bioavailability; t1/2: plasma half-life; Vd: volume of distribution; CLH: hepatic clearance; CLR: renal clearance; GF: glomerular filtration; NA: not applicable; OAT: organic anion transporter
denotes derived values
denotes primary enzymes in adults.
All values normalized to 70 kg adult weight
Figure 2.
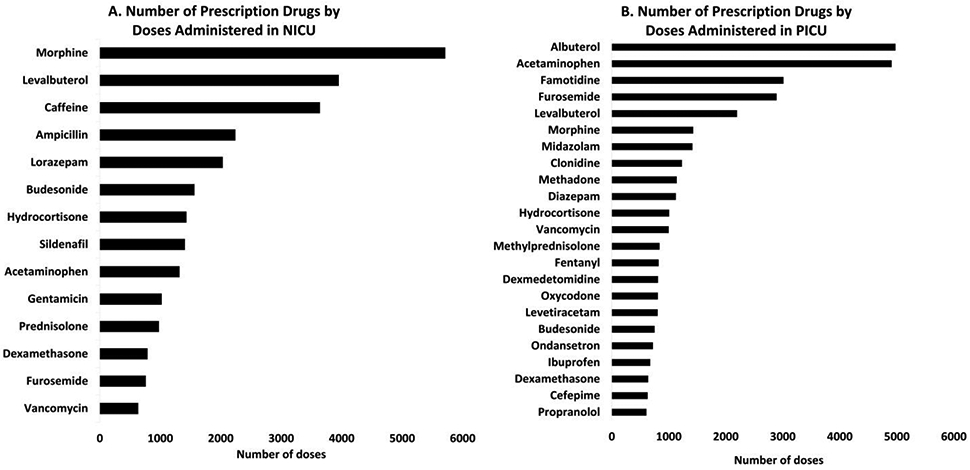
Top drugs in NICU (A) and PICU (B) by number of doses administered from January 2019 to December 2019 at Providence Sacred Heart Medical Center and Children’s Hospital, Spokane, WA, USA.
Key PK parameters normalized to 70 kg adult weight for the shortlisted drugs (n=29) are presented in Table 1 and additional parameters for these drugs are listed in Supplemental Table S1. Specifically, we compiled the data for bioavailability (F), half-life (t1/2), volume of distribution (Vd), protein binding (fb), renal filtration (RF), renal mechanism of drug elimination, liver extraction (EH), fraction of drug excreted unchanged in urine (fe), hepatic clearance (CLH), renal clearance (CLR), total clearance (CLT), prominent route of elimination and primary enzyme responsible for biotransformation. If the data was not available in the literature, it was derived using equations presented below (Equation 1-5). These properties include EH, fe, CLH, CLR, CLT, and renal filtration. The ratio between RF and CLRenal was calculated to determine the likely mechanism of renal excretion. For example, if the fold difference between renal filtration clearance (CL) versus total renal CL was less than 0.8 or greater than 1.25, active secretion or reabsorption was considered as the likely rate-determining mechanism of drug excretion, respectively. The ratio of the total hepatic CL and total renal CL indicates the likely site of drug elimination in adults. For example, if the fold difference (CLH/CLR) is less than 0.5 or greater than 2, the renal or hepatic routes was considered as likely site of elimination, respectively (Equation 7). When CL was reported from intravenous (IV) PK data, it was considered as the total systemic CL. However, when IV data were not available, the oral CL (CL/F) was multiplied by bioavailability (F) data to derive CLT.
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
Of the 29 shortlisted drugs, 18 and 8 drugs are primarily cleared by liver and kidney, respectively. Whereas, both liver and kidney contribute equally to the elimination of three drugs (Figure 3a). Of the renally or hepatically plus renally cleared drugs, secretion and reabsorption are important for the elimination of 7 and 2 drugs, respectively (Figure 3b). Of the hepatically cleared drugs, 15 are primarily metabolized by the CYP isoform family, whereas UGTs and SULTs contributed to the metabolism of 4 and 2 drugs, respectively (Figure 3c). CYP3A4, CYP1As, CYP2D6, UGT2B7 and SULT1A3 are amongst the top enzymes metabolizing NICU and PICU drugs (Figure 3d).
Figure 3.

Relative importance of drug elimination mechanisms of top dosed drugs in the NICU/PICU, i.e., (A) prominent route of elimination, (B) prominent excretory pathways of renally eliminated drugs, (C) prominent enzyme families of hepatically eliminated drugs, and (D) prominent enzymes of hepatically eliminated drugs.
Finally, the adult PK data were interpreted for the top 29 drugs in conjunction with the ontogeny of individual drug metabolism or excretion pathways. The critical mechanisms leading to age-dependent PK changes are summarized in Table 1.
4. Ontogeny of DMEs and transporters
The Phase I and II DMEs and transporters are generally immature and show unique ontogeny patterns, thus leading to an inconsistent impact on drug PK. Metabolite profile of some drugs can be different between children and adults. In young children, acetaminophen is predominantly metabolized via sulfation, but in adults, glucuronidation is the primary elimination metabolism [9]. Additionally, ontogeny of the renal system is critical in drug disposition. Nephrogenesis may be complete by 36 weeks gestation, but post-birth maturation of renal transporters and variability in blood flow and vascular resistance require a detailed mechanistic understanding of the ontogeny of renal functions such as glomerular filtration rate (GFR), secretion and reabsorption [27,35].
The ontogeny patterns of DMEs can be classified into three categories based on their trajectories. Class I enzymes are expressed during the first trimester and remain constant or gradually decrease as gestation progresses. They are reduced to low levels post-partum within days or up to 2 years of age. Class 2 enzymes remain constant throughout development and into adulthood. Class 3 enzymes are expressed at low levels during development and increase after birth to adult levels within weeks to several years [2]. Although Phase I reactions are frequently observed to be CYP-dependent, other enzyme families such as flavin mono-oxygenases and alcohol dehydrogenases contribute significantly to drug metabolism in children [36].
4.1. Phase I DMEs
4.1.1. Cytochrome P450s (CYPs)
The CYP450 superfamily consists of over 50 proteins and play an important role in the metabolism of >50% of the 200 most prescribed drugs [28,37]. The total CYP450 content in fetal liver tissue (per gram) is as low as 30% as compared to adult liver tissue and increases with age from birth to 10 years old [13,38]. There are significant variability in the ontogeny profiles of individual cytochrome P450 enzymes [2,13,39]. CYP1A1 expression is detectable in fetal liver between 6 and 12 weeks, however, the expression is not detectable in adult tissues [13]. CYP1A1 is responsible for metabolic activation of polycyclic aromatic hydrocarbons which are environmental pollutants that can cause neurodevelopmental toxicity in children [40]. CYP1A2 demonstrates a slow postnatal developmental trajectory reaching 50% of the adult value by 1 year of age. In fetal and neonatal livers, CYP1A2 expression is limited. [27,41]. Caffeine, often prescribed as a respiratory stimulant to treat apnea in neonates, is a substrate of CYP1As, which shows ~10-fold lower CL in neonates as compared to adults [42]. CYPs contribute to metabolism of ondansetron and caffeine, which are prevalently used in NICU and PICU and thus their ontogeny should be considered in the pediatric pharmacotherapy (Table 1). CYP2A6 is not detected in fetal liver tissue and displays a moderate increase in abundance and activity with age [38,43]. CYP2C9 is the likely rate determining mechanism involved in ibuprofen metabolism (Table 1), which increases from 1% to 30% relative adult concentration from the first to third trimesters and continues to increase postnatally to achieve adult levels by 2 years of age [2,44]. Genetic variants of CYP2C9 have shown an impact on the response of preterm infants to ibuprofen and have been associated with sudden infant death syndrome (SIDS) [45,46]. CYP2D6 is highly polymorphic and involved in metabolism of a multitude of drugs including propranolol and clonidine used in NICU and PICU (Table 1) [27,47]. In fetal livers, CYP2D6 is present at low levels and increases to adult values by the first year of life [48,49]. Since CYP2D6 is highly polymorphic, both ontogeny and genetic polymorphisms must be considered when dosing drugs metabolized by CYP2D6 [50]. CYP3A4 is the most abundant CYP in the adult intestine and liver and is the rate determining mechanism in case of several NICU and PICU drugs, i.e., diazepam, fentanyl, and oxycodone (Table 1) [2,51]. The ontogeny profile of CYP3A4 shows a concentration of 1% of adult values prenatally, delayed development for the first month postpartum, and then a steady increase to adult values by 2 to 3 years of age [49,52]. Similarly, increases in the intestinal abundance of CYP3As are also age-dependent [53]. CYP3A4-mediated fentanyl CL is a function of gestational age at birth with higher trajectories and higher CL for values at a postnatal age of 28 and 31 weeks compared to 25 weeks gestational age [54]. CYP3A5 expression is limited in fetal liver and is independent of age. CYP3A7 is highly expressed in the fetal liver and accounts for 10 to 40% of total CYP3A levels [27]. CYP3A4 and CYP3A7 display developmental switch, where after birth the levels of these enzymes increase and decrease, respectively [55].
4.1.2. Flavin-monooxygenases (FMOs)
The flavin-monooxygenases (FMO) are critical for nicotinamide adenine dinucleotide phosphate (NADPH) dependent oxidative metabolism and are encoded by a six-member gene family (FMO1–6) [13]. In adult kidneys, FMO1 is the most abundant isoform but it is not present in other adult tissues [56-59]. FMO1 protein levels decrease ~20-fold from fetal to adulthood, with peak levels during the first and second trimesters [56,60]. The ontogeny of FMO1 and FMO3 also exhibit metabolic switch similar to that of CYP3A4 and CYP3A7 [61]. For instance, FMO1 is suppressed within 3 days after birth and the levels of FMO3 begin to increase 2.2-fold to adulthood [56,62].
4.1.3. Alcohol dehydrogenases (ADH)
The ADH family of enzymes catalyzes the interconversion between alcohols to aldehydes or ketones [63]. A steep increase in expression of ADH1A, ADH1B, and ADH1C has been observed during development of the fetus. ADH1A is detected in the fetal liver as soon as 11 weeks of gestation [64]. By the age of 17 gestational weeks, ADH1B is detected in fetal livers and by 36 gestational weeks, it progresses to become the most dominant isoform [64]. ADH1B and ADH1C levels continue to increase with age whereas ADH1A levels decrease by 40% in adults as compared to childhood [65]. The levels of ADH1A, ADH1B, ADH1C in neonates are lower than the adults by 3-, 8-, and 146-fold levels, respectively [65].
4.2. Phase II DMEs
4.2.1. UDP glucuronosyltransferases (UGTs)
The UGT enzyme family is responsible for the glucuronidation of many hydrophobic xenobiotics and endogenous compounds by conjugating uridine 5'-diphosphoglucuronic acid (UDPGA) to functional groups. This is created as a result of oxidative metabolism, forming β-D-glucopyranosiduronic acids [7]. The glucuronide product is typically the inactive form of the parent compound and is removed through renal or biliary elimination. However, acyl glucuronides are sometimes toxic [66]. UGTs are categorized into two gene families, UGT1 with nine and UGT2 with seven functional genes, accounting for almost 15% of drug CL in adults [67].
UGT proteins have been found in the liver and spleen at gestational age but at very low levels [68]. UGT1A1 plays an important role in bilirubin metabolism and increases by ~8-fold from neonates to adults in the human liver [8]. Activity assays assessed by bilirubin conjugation indicate that UGT1A1 is undetectable in fetal liver but it is detectable at birth [68]. Allelic variations and mutated promoter regions in UGT1A1 (*28) lead to reduced enzymatic activity causing hyperbilirubinemia characterized as Criglere-Najjar syndrome or Gilbert’s syndrome, and can lead to serious liver injury and death [69-72]. UGT1A6 increases by 35-fold from neonates to adults [8,73,74]. Both UGT1A6 and UGT1A1 are critical in the metabolism of acetaminophen in adults, whereas SULTs are more important in children. Glucuronidation of acetaminophen is minimal in the fetus, then low at birth and slowly increases with age, which corroborates with the UGT1A6 abundance [8].
Morphine is the most commonly prescribed drug in NICU (Figure 2a). Age-dependent morphine metabolism to morphine-6-glucuronide and morphine-3-glucuronide by UGT2B7 is well characterized, which should be taken into account in NICU and PICU [75-80]. As discussed earlier, failure in understanding the impact of ontogeny on the metabolism of chloramphenicol, also a substrate for UGT2B7 led to deaths in neonates [5,6]. The CL of morphine in premature infants is 5-fold lower and the neonatal expression of UGT2B7 is 10-20% of the adult value, both of which increase to reach the adult values between 2 to 6 months of age [7,80-82]. UGT2B15, involved in lorazepam metabolism, increases 3-fold from neonates to adults [8,83]. UGT2B17 is expressed at almost undetectable levels in children under 9 years but displays a steep developmental trajectory during pubertal development increasing ~10 fold to adult levels [26]. Thus, the use of a drug primarily metabolized by UGT2B17 should be prohibited in children below 9 years of age.
4.2.2. Sulfotransferases (SULTs)
SULTs catalyze the conjugation of 3′-phosphoadenosine-5′-phosphosulfate to various exogenous and endogenous substances [37]. The sulfate conjugates are typically inactive and are removed through renal or biliary elimination. SULTs are categorized into three families, phenol SULTs (SULT1) encoded by seven genes, hydroxysteroid SULTs (SULT2) encoded by two genes, and SULT4A1 encoded by a single brain-specific gene [37,84,85]. The neonatal abundance of SULT1A1, SULT1B1, and SULT2A1 are 24%, 19%, and 38% of the adult values, respectively [9]. SULT1A family is involved in the metabolism of acetaminophen, albuterol, and levalbuterol. The ontogeny of SULT1A family are implicated as the rate determining mechanisms in acetaminophen metabolism in children whereas both hepatic SULT1A and renal OAT ontogeny are the rate determining factors for albuterol and levalbuterol elimination (Table 1). The expression of SULT1A1 and SULT2A1 are at higher levels in childhood but then decrease by 40% from adolescence to adulthood [9]. However, SULT1A3 activity is generally independent of age with high activity observed in both fetuses and adults [9,86-89]. SULT2A1 protein abundance is 4-fold higher in early childhood as compared to neonatal and 2-fold higher than in adolescence [9].
4.2.3. Other DMEs
Glutathione S-transferases (GSTs) catalyze the conjugation of glutathione resulting in detoxification and elimination of xenobiotics. GSTs belong to five different classes (GST1-5) most of which have been identified in fetal livers at 16-18 weeks of gestation [90]. GST1 and GST2 have been identified as early as 10 weeks of gestational age [25]. GST1 was found at around 20% of adult levels and rapidly increased to adult levels by 42 weeks of age [91]. GST2 is also expressed at 10 weeks of gestational age and increases to adult levels after birth [92]. GST3 is expressed early on at 10 weeks of gestational age, but the levels are nearly non-detectable in adults [91,92]. The total distribution of GSTs is uniform throughout various organs with average fetal liver activity at about 60% compared to adults [93,94].
Many drugs and toxic agents are metabolized by N-Acetyltransferase (NATs) expressed in cell cytosols, via conjugation to an acetyl moiety. NATs are ubiquitously expressed in several tissues and play an important role in the metabolism of caffeine. Para-aminobenzoic acid confirmed as a probe substrate for NAT activity, indicates NAT activity in neonates, increasing for infants then decreasing for children [95]. The maturation of NAT activity is not completed before 1 year of age and NAT activity was found to be 3-fold lower in fetal vs adult livers [95,96].
4.3. Hepatic transporters
Transporters are membrane-bound proteins involved in absorption, distribution, or uptake into tissues, and excretion of several drugs and physiological substrates throughout the body. Some data exists on the ontogeny of hepatic and renal transporters. For instance, we and others quantified the ontogeny of hepatic transporters and observed that the abundance of OCT1, P-gp, and OATP1B3 are age-dependent [33,97]. Particularly, hepatic OCT1 show a 5-fold increase in protein expression with age. OATP1B3 and MRP3 are also expressed significantly lower in infants when compared to adults. P-gp is found to reach 50% of the adult expression by ~3 years of age [33]. The most important NICU drug, morphine, is primarily transported to the liver on OCT1. Age- and genotype-dependent expression of OCT1 is associated with morphine disposition in children [98-100].
4.4. Ontogeny of kidney anatomy and function
The impact of DME ontogeny is critical in children as it relates directly to the development and function of their renal system. The maturation of the kidney plays a profound role especially in drugs that rely on enzymes not fully developed for metabolism. The renal system has three mechanisms by which drugs are cleared: GFR, tubular secretion, and tubular reabsorption. The development of the renal system begins around 9 weeks gestational age and is continues through 36 weeks of gestation [101,102]. The mechanisms involved in renal CL is not fully developed at birth, reducing the renal elimination capacity of newborns [103]. Renal elimination is much improved in the first few months after birth due to major changes in anatomy and function of the kidney reaching adult levels by around 1 year of age [101,104-106]. Renal CL is heavily influenced by the unbound protein fraction (fu) of a drug and is a critical PK variable to evaluate its relation to DME ontogeny [107]. The development of the kidney postpartum allows for improved renal blood flow, more efficacious GFR, and continued maturation of various tubular processes [108].
4.4.1. Ontogeny of Glomerular Filtration (GFR)
Renal CL is often quantitively assessed using GFR. GFR remains relatively low during gestation and increases to adult values by 1-2 years of age. GFR continues to increase to above adult values until around 6 years of age, then returns to adult values by 12 years of age [104]. Nephrogenesis leads to an increase in glomeruli which cause a rapid increase of GFR during gestation. Postpartum, the increase of GFR is thought to be due to increased cardiac output and higher renal blood flow [109]. GFR, estimated using the Cockcroft-Gault equation or the Schwartz equation for children, is normalized based on adult values of 70 kg and a body surface area of 1.73 m2 [110]. In case of NICU/PICU drugs such as vancomycin and cefepime, which have high fe, age-dependent CL is likely correlated with GFR (Table 1).
4.4.2. Ontogeny of renal tubular secretion
Renal tubular secretion is important in the excretion of many pediatric drugs including penicillins and cephalosporins; however, tubular secretion is not fully developed in neonates and infants and reaches adult capacity only by around 7 months of age [111,112]. Furosemide, a renal tubular secretion pathway substrate, has a half-life of 0.5 hours in adults compared to 7.7 hours in infants [113,114], clearly indicating that renal CL of organic anions is almost 15-fold lower in infants. Furosemide and other organic anionic drugs are substrates of OATs expressed at the basolateral membrane of proximal tubules. Organic cation transporters are also important at the basolateral side, whereas P-glycoprotein, multidrug and toxin extrusion transporters (MATEs), and multidrug resistance-associated proteins (MRPs) at the apical side are important determinants of renal secretions of several pediatric drugs [115,116]. Although the human data on tissue abundance of transporters is limited due to the lack of pediatric kidney specimens, animal studies indicate age-dependent transport in kidneys [32]. The disproportional maturation of GFR versus renal tubular mechanism must be considered in scaling down the adult data of renally eliminated drugs to predict pediatric doses. Moreover, renal transporters may lead to fluctuating CL of drugs dependent on renal secretion for excretion depending on co-administration with inducing or inhibitory substances [117-119].
Although it is well known that kidney transporters play an important role in drug CL and dosing requirement, limited knowledge exist regarding transporter ontogeny in kidneys except a limited understanding of ontogeny of OATs in rodents [34,120-122]. The effects of OAT ontogeny in the kidney have been demonstrated by para-aminohippurate (PAH) as a model to describe OAT1-mediated tubular secretion [123]. The maximum tubular secretory capacity of PAH is low postnatally until around 2 years of age at which it begins to increase to 50% of adult value by 8 years of age [35]. Per our review, the ontogeny of renal transporters is the likely determinant of the age-effect in the PK of hydrocortisone, famotidine, ampicillin, and furosemide (Table 1).
4.4.3. Tubular reabsorption
More liposoluble drugs that are not metabolized generally undergo passive renal tubular reabsorption [111]. Transporters are expressed on the basolateral and apical membranes of the renal tubule and are involved in tubular reabsorption as well [124]. Concentrations of the retinol-binding protein as a marker for glomerular development and microalbumin as a marker for maturation have been analyzed in the urine of healthy subjects aged from birth to 60 [125]. Glomerular permeability and renal tubular reabsorption functions continue to develop and mature gradually from birth to adolescence with critical stages at years 1 and 3 of age. Moreover, urinary microalbumin and retinol-binding protein assays indicate the rate of glomerular development may be faster than tubular development [125]. The capacity of a young kidney for reabsorption is lower, due to lower tubular mass, leading to increased dehydration risks [110]. The ontogeny of apical renal transporters involved in tubular reabsorption could be the determinant of the age-effect in gentamicin and levetiracetam (Table 1).
4.5. Further considerations of the impact of DME and transporter ontogeny on drug dosing in NICU and PICU
Drug metabolism and transport are affected by both genetic and non-genetic factors. While non-invasive genotyping allows for the prospective characterization of genetic variability, the effect of non-genetic factors such as age, disease condition, sex and environment is understudied and should be considered for optimum drug dosing [126]. Particularly, the ontogeny of hepatic and renal drug elimination processes has been shown to influence toxicity and efficacy of drugs such as zidovudine, oseltamivir, lorazepam, and chloramphenicol [7,19,127]. Furthermore, perturbations in these processes by drug-drug interactions or genetic polymorphism have significantly different impacts in children than in adults. For example, a drug equally metabolized by CYP3A7 and CYP3A5 will be affected by a genetic polymorphism in CYP3A5 (*3) in adults when CYP3A7 is absent, but the same polymorphism will not show significant impact on the drug metabolism in neonates when CYP3A7 is highly expressed [27]. Thus, not only age-dependent expression in DMETs is important, the difference in the fractional contribution of individual enzymes should be considered for ensuring safe and effective drug dosing.
Retrospective clinical studies on 40 pediatric drugs between 1998 and 2015 resulted in a change in the labeling for 90% of the drugs. Moreover, 20 of these drugs were associated with severe adverse effects, whereas deaths were reported during testing of 9 drugs [128]. Another study reviewing data from 2010 to 2014 found that the common pediatric drugs such as acetaminophen, ampicillin, gentamicin, ibuprofen, metronidazole, and morphine were amongst the top 10 most associated with medication errors [129]. The results of these studies further strengthen the need for integrating ontogeny data into PBPK modeling prior to first-in-child dosing. Furthermore, understanding of ontogeny of DMET proteins is also relevant for those drugs which are not prescribed to children but are consumed by pregnant women or nursing mothers. For example, CYP2D6 and CYP3A4 are responsible for metabolism of selective serotonin reuptake inhibitors (SSRIs) such as fluoxetine or sertraline. Pregnant women who took SSRIs had newborns that exhibited a hyper-serotonergic state with similar ADEs as seen in serotonin syndrome which can be attributed to immature development of CYP enzyme pathways [126]. Similarly, the prodrug codeine, which was frequently used by nursing mothers, is metabolized to morphine by polymorphic CYP2D6. The latter is pharmacologically active and eliminated by UGT2B7 to morphine-glucuronide. In CYP2D6 extensive metabolizer mothers, a higher concentration of morphine is produced which is readily transferred to infants through breast milk and has been associated with severe ADEs as well as deaths [130]. Since UGT2B7 abundance in infants is significantly lower (around 5% of adult levels), infants are highly vulnerable to morphine toxicity as compared to mothers [131].
5. Application of PBPK modeling in predicting drug disposition and dosing in pediatric population
The number of new drug approvals with the application of PBPK modeling has increased more than 2-fold in 2019 as compared to 2013 [132]. Pediatric PBPK modeling accounts for 20% of the total FDA submissions. PBPK modeling is routinely applied in predicting drug disposition in the pediatric population, including neonates and infants (Figure 4). Successful applications of DMET ontogeny data in predicting neonatal and infant PK have been shown for a number of drugs such as acetaminophen, morphine, zidovudine, bupropion, epirubicin, linezolid, midazolam, oseltamivir and propofol [9,19,129,133-138]. For example, Ladumor et al. developed a proteomics-informed PBPK model of acetaminophen (substrate of UGTs, SULTs, and CYPs) for different age groups, including neonatal, infancy, early-childhood, middle-childhood, adolescence, and adulthood [9]. In this model, the protein abundance information on UGT, SULT, and CYP enzymes was incorporated in the PBPK model for predicting age-dependent change in the fraction of acetaminophen metabolized by individual enzymes (fm). The model confirmed age-dependent metabolic switching in acetaminophen elimination. Since fm is directly proportional to the relative DME abundance, the non-monotonic pattern of enzyme maturation across different age groups results in different metabolites profiling with age. For example, the model integrated with drug-specific physiochemical, biochemical, and physiological changes along with ontogeny-based information predicted acetaminophen fm,ratio values (i.e., fm,UGT/fm,SULT) of 0.46, 0.56, and 1.71 in neonates, children, and adults, respectively. This suggests an increased role of sulfation over glucuronidation in the metabolism of acetaminophen in children. Consistent with these findings, Mian et al. demonstrated the phenomenon of metabolic switching in acetaminophen where the sulfation is 13-fold higher as compared to CYP2E1-mediated NAPQI in the fetus [139].
Figure 4.

Schematic workflow of DMET ontogeny linked-pediatric PBPK model development for extrapolation of adult data to predict drug disposition in children. The model structure is based on a middle-out approach, where reported clinical pharmacokinetic (PK) data and in-house experimental data can be used as input parameters (i.e., system- and drug-specific parameters). DMET proteins ontogeny data are crucial for pediatric model development. SAD: single ascending dose, MAD: multiple ascending doses.
Bhatt et al. and others presented another example of the successful use of proteomics-informed PBPK modeling to assess age-dependent changes in morphine exposure to neonates and infants [8,99]. Morphine is widely used for pain management in the pediatric population; however, significant variability is observed in its PK in children. The morphine PBPK model was developed with age-dependent changes in organic cation transporter 1 (OCT1) and UGT2B7 along with changes associated with physiology such as organ size, blood flow to organs, microsomal protein concentration per gram of tissue, plasma protein concentration (i.e., albumin and alpha-1-acid glycoprotein), hematocrit level, and GFR. Similarly, Emoto et al. predicted the effect of OCT1 genotype and liver blood flow on morphine CL after intravenous administration using a ontogeny-informed PBPK modeling [140]. Particularly, PBPK modeling helps to identify the key determinants explaining the variability in drug PK. Strougo et al. also show that the accurate prediction of drug PK and the estimation of ‘first dose in children’ can be achieved by integrating ontogeny and drug- and physiology-specific properties [141].
The application of quantitative proteomics to extrapolate drug CL from adult to pediatric population is illustrated in Equation 6, the obtained value is then integrated in the PBPK model.
| (Equation 6) |
There are a few limitations of the ontogeny-informed PBPK modeling. In particular, demonstration of model validation using pediatric clinical data for multiple drugs is critical to achieve confidence in prospective PK predictions in children. Nevertheless, the level of validation required for pediatric PBPK modeling depends on the application of these models. For example, to apply for a clinical study waiver of a new drug, a rigorously validated model using similar drugs is required. Whereas potential for a drug-drug interaction in case of a wide therapeutic index drug can be assessed based on fm prediction. The uncertainty associated with differences in the physiology-related factors is important to be evaluated through sensitivity analysis. However, the confidence on the individual optimized values is low when more than one uncertain parameters are fitted based on the in vivo data. Despite these limitations, better understanding of DMET ontogeny has a potential to more accurately predict age-related changes in drug metabolism, transport and PK. Although this method is not fully developed to allow clinical trial waivers, such models can be used in making informed decisions during clinical trials for safer and effective use of drugs in children. The ongoing efforts to develop best practices to integrate complex drug disposition pathways in children will be crucial for wider applications of pediatric PBPK modeling in the future.
6. Regulatory requirements and recommendations in pediatric pharmacotherapy
The potential for ADEs in children is increased nearly 3-fold with off-label drug administration versus approved labeling use [11]. Patients in NICU and PICU are often administered multiple drugs for various disease states, suffer comorbidities, and have dynamic physiology making it difficult to recognize an ADE. The FDA is encouraging pharmaceutical companies to consider pediatric studies early in drug development and a plan to conduct these studies during phase II of adult clinical studies [142]. In 2012 the FDA enacted the Food and Drug Administration Safety and Innovation Act (FDASIA) which renewed and made permanent three critical laws to improve safety and efficacy of pediatric medical care: Best Pharmaceuticals for Children Act (BPCA), the Pediatric Research Equity Act (PREA) and the Pediatric Medical Device Safety and Improvement Act, which had been in place since 1997, 2003 and 2007, respectively [15-18]. With these regulatory changes, particularly the BPCA, the FDA is offering 180 days of additional market exclusivity if pharmaceutical companies perform pediatric clinical trials for their investigation drug. Typically, FDA grants 5 years of market exclusivity with no generic competition from the date of marketing [143]. PREA requires that applications submitted for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration must contain a pediatric assessment unless the applicant has obtained either a waiver or deferral, or the requirement is inapplicable. The waivers are granted if the study is not feasible, the drug would be unsafe or ineffective in pediatrics, no meaningful therapeutic benefit exists, or if an age-appropriate formulation cannot be made [144]. Drug developers who are applying for a new drug or biologic must submit an initial pediatric study plan (PSP) that outlines the pediatric studies the applicant plans to conduct. The PSP must be submitting before the beginning of phase 3 clinical trials and should convey to the FDA a pediatric clinical trial plan [145].
7. Conclusion
Children continue to be at risk for ADE due to the prevalent off-label dosing of drugs based on empirical scaling from adult data. Particularly, neonates, infants and critically ill children are highly vulnerable to drug toxicity due to immature or compromised drug detoxification and the use of polypharmacy. We recommend that the ontogeny of drug metabolism and excretion should be taken into consideration during pharmacotherapy of NICU and PICU patients. The FDA recognizes the gap in pediatric clinical testing and is encouraging drug companies conduct such trials. Pediatric clinical trials come with inherent risk to both study participants and the drug developer. To address this limitation, prospective prediction of drug disposition and potential ADEs using PBPK modeling in children is emerging as a pivotal approach in drug development that carefully considers the ontogeny of drug disposition processes, including DMET abundance. However, PBPK modeling is data hungry and needs tissue DMET abundance and clinical PK data of the model compounds in children for the model development and validation. Newer approaches such as non-invasive endogenous biomarkers and the use of exosomes isolated from biofluids (liquid biopsy) for DMET characterization are the complementary approaches to PBPK modeling to support pediatric precision medicine [146,147].
8. Expert opinion
Children, especially neonates and infants, are more vulnerable to drug toxicity due to immature drug metabolism and excretion. By understanding ontogeny in drug metabolism and excretion, the fate of a drug in neonates and infants can be predicted based on the adult data. The ontogeny data in conjunction with physiologically-based PK modeling provides a tool to simulate drug plasma profiles, which thereby help to predict the potential of altered PK or drug toxicity in children. Since clinical trials are rarely conducted in children and most of the pediatric drugs are used off-label, the ontogeny-based prediction of drug dosing in children could offer an important public health benefit. Such approaches are currently recommended by the regulatory bodies to design pediatric dosing prior to clinical trials; however, the routine clinical implementation of this approach for the prescription drugs needs further validation. Nevertheless, there is a consensus among regulators and clinical pharmacologists that for children older than 2 years old, allometry would be a reasonable approach for predicting PK, given that enzyme/transporter expression and activity would approximate their counterparts in adults [148].
The scarcity of pediatric clinical data for the validation of the proposed PBPK approach is one of the key limitations. Recognizing this limitation, the regulatory agencies now incentivize pharmaceutical industry to generate pediatric clinical data for new drugs. The lack of tissue samples from pediatric population for the characterization of the ontogeny data is another limitation. These challenges can be overcome by working in partnership with pediatric hospitals and clinicians to i) obtain opportunistic PK studies in children, and ii) establishing pediatric tissue repositories.
Non-invasive methods of ontogeny characterization using biofluids and endogenous metabolites as biomarkers are encouraging approaches. Advances in mass spectrometry are encouraging as the quantification of drugs and metabolites is possible at low concentrations and volumes of pediatric urine and blood samples, e.g., using dry blood spot technique. Further research on robust characterization of the ontogeny of drug metabolism and transport is needed for some proteins, particularly drug transporters in extrahepatic tissues. PBPK models can be developed and validated for all the pediatric drugs using the ontogeny data. When validated, such models may be used more routinely in clinical decision making and be incorporated into more general pediatric clinical systems. There is a large potential to increase positive pediatric health outcomes using real-time patient sampling with continuous feedback from PBPK simulations.
For pediatrics drug labeling, the future lies in robust clinical studies in children, which requires PBPK modeling to ensure safety and efficacy of drugs. Children are a vulnerable population to drug toxicity and planning or conducting in vivo pediatric clinical trials are challenging. Drug developers will rely heavily on the guidance provided through PBPK modeling in selecting first-in-human dose. Identifying non-invasive endogenous biomarkers of drug metabolizing enzymes and transporters in biofluids (blood or urine) that are specific and stable, is a promising complementary approach to PBPK modeling. Similarly, the characterization of interindividual variability in enzyme and transporter proteins using exosome approach is another promising complementary approach.
Ontogeny-informed modeling has evolved tremendously in the past decade and with current developmental trends in technology the field will continue to grow for the foreseeable future. Continued enrichment of tissue enzyme and transporter abundance data and in vivo pediatric PK data for model development and validation will allow more accurate prediction of drug disposition in children.
The field of pediatric PBPK modeling is rapidly evolving. The future has great promise to not only support making decision on the first-in-child dosing and pediatric clinical trial design, but the modeling has a potential to enable precision medicine in clinic by predicting real-time interindividual and intraindividual (e.g., within days in neonates) variability in drug disposition. Given the rate of technological advancement in computers and medical sciences, the field of PBPK modeling may soon move out of research laboratories and into hospitals. Particularly, the non-invasive estimation of the capacity of an individual patient to eliminate drugs can be integrated real-time into mathematical models. Such data can be fed in the medical devices to support real-time recommendations for drug dosing. Basically, the PBPK platform can be brought to the user like genetic testing available for several drugs now. While the scope of this review article was limited to pediatric population, PBPK modeling will be critical in all areas of precision medicine revolution.
Supplementary Material
Article highlights.
Pediatric patients, especially neonates and infants, are more susceptible to adverse drug events as compared to adults, in part due to immature drug detoxification mechanisms.
Such developmental differences may result in toxicities that would not be observed in adults. This, along with prevalent off-label use of drugs in children without clinical pharmacokinetic data is a critical challenge in pediatric pharmacotherapy. This requires a critical understanding and application of the ontogeny of drug metabolism and excretion in predicting drug dosing for children.
Ontogeny-informed physiologically-based pharmacokinetic modeling is an emerging alternative to empirical scaling of drug dosing, which can be considered not only in the first-in-child dosing of new drugs but also to predict the safety and efficacy of drugs in neonatal and pediatric intensive care units.
Acknowledgments
Funding
The work was partially supported by the Department of Pharmaceutical Sciences, WSU, Spokane, WA and NIH/NICHD grant (R01-HD081299).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Articles of special interest have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- [1].Implementation of the Food Quality Protection Act. Food Drug Law J [Internet]. 1997;52:525. Available from: https://heinonline.org/HOL/Page?handle=hein.journals/foodlj52&id=563&div=&collection=. [PubMed] [Google Scholar]
- [2]*.Hines RN. Developmental expression of drug metabolizing enzymes: Impact on disposition in neonates and young children. Int J Pharm [Internet]. 2013;452:3–7. Available from: 10.1016/j.ijpharm.2012.05.079. [DOI] [PubMed] [Google Scholar]
- [3]**.Laughon MM, Avant D, Tripathi N, et al. Drug labeling and exposure in neonates. JAMA Pediatr. 2014;168:130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45:931–956. [DOI] [PubMed] [Google Scholar]
- [5].Sutherland JM. Fatal Cardiovascular Collapse of Infants Receiving Large Amounts of Chloramphenicol. AMA J Dis Child. 1959;97:761–767. [DOI] [PubMed] [Google Scholar]
- [6].Weiss CF, Glazko AJ, Weston JK. Chloramphenicol in the newborn infant. N Engl J Med [Internet]. 1960;262:787–794. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7993409. [DOI] [PubMed] [Google Scholar]
- [7].De Wildt SN, Kearns GL, Leeder JS, et al. Glucuronidation in humans. Pharmacogenetic and developmental aspects. Clin Pharmacokinet. 1999;36:439–452. [DOI] [PubMed] [Google Scholar]
- [8]**.Bhatt DK, Mehrotra A, Gaedigk A, et al. Age-and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin Pharmacol Ther. 2019;105:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]**.Ladumor MK, Bhatt DK, Gaedigk A, et al. Ontogeny of hepatic sulfotransferases and prediction of age-dependent fractional contribution of sulfation in acetaminophen metabolism. Drug Metab Dispos. 2019;47:818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Age-related differences in salicylamide and acetaminophen conjugation in man. J Pediatr [Internet]. 1977. [cited 2020 May 6];90:130–135. Available from: http://www.sciencedirect.com/science/article/pii/S0022347677807877. [DOI] [PubMed] [Google Scholar]
- [11].De Lima Costa HTM, Costa TX, Martins RR, et al. Use of off-label and unlicensed medicines in neonatal intensive care. PLoS One. 2018;13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McCarver DG, Hines RN. The Ontogeny of Human Drug-Metabolizing Enzymes : Phase II Conjugation Enzymes and Regulatory Mechanisms. 2002;300:361–366. [DOI] [PubMed] [Google Scholar]
- [13].Hines RN, Mccarver DG. The Ontogeny of Human Drug-Metabolizing Enzymes : Phase I Oxidative Enzymes. 2002;300:355–360. [DOI] [PubMed] [Google Scholar]
- [14].Hay M, Thomas DW, Craighead JL, et al. Clinical development success rates for investigational drugs. Nat Biotechnol [Internet]. 2014;32:40–51. Available from: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- [15].Harkin TS 3187 - 112th Congress (2011-2012): Food and Drug Administration Safety and Innovation Act. 2012. [cited 2020 Jul 29]; Available from: https://www.congress.gov/bill/112th-congress/senate-bill/3187.
- [16].Dodd CJ. S.1789 - 107th Congress (2001-2002): Best Pharmaceuticals for Children Act. 2002. [cited 2020 Jul 29]; Available from: https://www.congress.gov/bill/107th-congress/senate-bill/1789.
- [17].DeWine MS 650 - 108th Congress (2003-2004): Pediatric Research Equity Act of 2003. 2003. [cited 2020 Jul 29]; Available from: https://www.congress.gov/bill/108th-congress/senate-bill/650.
- [18].Dodd CJ. S.830 - 110th Congress (2007-2008): Pediatric Medical Device Safety and Improvement Act of 2007. 2007. [cited 2020 Jul 29]; Available from: https://www.congress.gov/bill/110th-congress/senate-bill/830.
- [19].Bhatt DK, Mehrotra A, Gaedigk A, et al. Age- and Genotype-Dependent Variability in the Protein Abundance and Activity of Six Major Uridine Diphosphate-Glucuronosyltransferases in Human Liver. Clin Pharmacol Ther. 2019;105:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ladumor MK, Thakur A, Sharma S, et al. A repository of protein abundance data of drug metabolizing enzymes and transporters for applications in physiologically based pharmacokinetic (PBPK) modelling and simulation. Sci Rep. 2019;9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Salem F, Johnson TN, Barter ZE, et al. Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug-drug interactions. J Clin Pharmacol. 2013;53:857–865. [DOI] [PubMed] [Google Scholar]
- [22].Blake MJ, Gaedigk A, Pearce RE, et al. Ontogeny of dextromethorphan O-and N-demethylation in the first year of life. Clin Pharmacol Ther. 2007;81:510–516. [DOI] [PubMed] [Google Scholar]
- [23].de Wildt SN, Kearns GL, Leeder JS, et al. Cytochrome P450 3A: ontogeny and drug disposition. Clin Pharmacokinet. 1999;37:485–505. [DOI] [PubMed] [Google Scholar]
- [24].Gail McCarver D, Hines RN. The ontogeny of human drug-metabolizing enzymes: Phase II conjugation enzymes and regulatory mechanisms. J Pharmacol Exp Ther. 2002;300:361–366. [DOI] [PubMed] [Google Scholar]
- [25].Saghir SA, Khan SA, Mccoy AT. Ontogeny of mammalian metabolizing enzymes in humans and animals used in toxicological studies. 2012;42:323–357. [DOI] [PubMed] [Google Scholar]
- [26].Bhatt DK, Basit A, Zhang H, et al. Hepatic abundance and activity of androgen- and drug-metabolizing enzyme UGT2B17 are associated with genotype, age, and sex. Drug Metab Dispos. 2018;46:888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther. 2008;118:250–267. [DOI] [PubMed] [Google Scholar]
- [28].Saravanakumar A, Sadighi A, Ryu R, et al. Physicochemical Properties, Biotransformation, and Transport Pathways of Established and Newly Approved Medications: A Systematic Review of the Top 200 Most Prescribed Drugs vs. the FDA-Approved Drugs Between 2005 and 2016. Clin Pharmacokinet [Internet]. 2019;58:1281–1294. Available from: 10.1007/s40262-019-00750-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hu S, Leblanc AF, Gibson AA, et al. Identification of OAT1/OAT3 as Contributors to Cisplatin Toxicity. Clin Transl Sci. 2017;10:412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Guo Y, Chu X, Parrott NJ, et al. Advancing Predictions of Tissue and Intracellular Drug Concentrations Using In Vitro, Imaging and Physiologically Based Pharmacokinetic Modeling Approaches. Clin Pharmacol Ther. 2018;104:865–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Endres CJ, Moss AM, Ke B, et al. The role of the equilibrative nucleoside transporter 1 (ENT1) in transport and metabolism of ribavirin by human and wild-type or Ent1−/− mouse erythrocytes. J Pharmacol Exp Ther. 2009;329:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sweeney DE, Vallon V, Rieg T, et al. Functional maturation of drug transporters in the developing, neonatal, and postnatal kidney. Mol Pharmacol. 2011;80:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]*.Prasad B, Gaedigk A, Vrana M, et al. Ontogeny of Hepatic Drug Transporters as Quantified by LC-MS / MS Proteomics . Clin Pharmacol Ther. 2016;100:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34]**.Brouwer KLR, Aleksunes LM, Brandys B, et al. Human Ontogeny of Drug Transporters: Review and Recommendations of the Pediatric Transporter Working Group. Clin Pharmacol Ther. 2015;98:266–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Momper JD, Yang J, Gockenbach M, et al. Dynamics of organic anion transporter-mediated tubular secretion during postnatal human kidney development and maturation. Clin J Am Soc Nephrol. 2019;14:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Strolin Benedetti M, Baltes EL. Drug metabolism and disposition in children. Fundam Clin Pharmacol. 2003;17:281–299. [DOI] [PubMed] [Google Scholar]
- [37].Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. 2008;118:250–267. [DOI] [PubMed] [Google Scholar]
- [38].Shimada T, Yamazaki H, Mimura M, et al. Characterization of microsomal cytochrome P450 enzymes involved in the oxidation of xenobiotic chemicals in human fetal liver and adult lungs. Drug Metab Dispos. 1996;24:515–522. [PubMed] [Google Scholar]
- [39].Shimada T, Yamazaki H, Mimura M, et al. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- [40].Jedrychowski WA, Perera FP, Camann D, et al. Prenatal exposure to polycyclic aromatic hydrocarbons and cognitive dysfunction in children. Environ Sci Pollut Res. 2015;22:3631–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Alsabri SG, Mari WO, Younes S, et al. Kinetic and Dynamic Description of Caffeine. J Caffeine Adenosine Res. 2018;8:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aranda JV, Cook CE, Gorman W, et al. Pharmacokinetic profile of caffeine in the premature newborn infant with apnea. J Pediatr. 1979;94:663–668. [DOI] [PubMed] [Google Scholar]
- [43].Tanner J-A, Prasad B, Claw KG, et al. Predictors of variation in CYP2A6 mRNA, protein, and enzyme activity in a human liver bank: influence of genetic and nongenetic factors. J Pharmacol Exp Ther. 2017;360:129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Koukouritaki SB, Manro JR, Marsh SA, et al. Developmental expression of human hepatic CYP2C9 and CYP2C19. J Pharmacol Exp Ther. 2004;308:965–974. [DOI] [PubMed] [Google Scholar]
- [45].Treluyer JM, Cheron G, Sonnier M, et al. Cytochrome P-450 expression in sudden infant death syndrome. Biochem Pharmacol. 1996;52:497–504. [DOI] [PubMed] [Google Scholar]
- [46].Figueiras A, Estany-Gestal A, Aguirre C, et al. CYP2C9 variants as a risk modifier of NSAID-related gastrointestinal bleeding: A case-control study. Pharmacogenet Genomics. 2016;26:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ingelman-Sundberg M Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5:6–13. [DOI] [PubMed] [Google Scholar]
- [48].Stevens JC, Marsh SA, Zaya MJ, et al. Developmental changes in human liver CYP2D6 expression. Drug Metab Dispos. 2008;36:1587–1593. [DOI] [PubMed] [Google Scholar]
- [49].Johnson TN, Tucker GT, Rostami-Hodjegan A. Development of CYP2D6 and CYP3A4 in the first year of life. Clin Pharmacol Ther. 2008;83:670–671. [DOI] [PubMed] [Google Scholar]
- [50].Gaedigk A, Dinh JC, Jeong H, et al. Ten years’ experience with the CYP2D6 activity score: a perspective on future investigations to improve clinical predictions for precision therapeutics. J Pers Med. 2018;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Drozdzik M, Busch D, Lapczuk J, et al. Protein abundance of clinically relevant drug-metabolizing enzymes in the human liver and intestine: a comparative analysis in paired tissue specimens. Clin Pharmacol Ther. 2018;104:515–524. [DOI] [PubMed] [Google Scholar]
- [52].Hines RN. Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol. 2007;21:169–175. [DOI] [PubMed] [Google Scholar]
- [53].Johnson TN, Tanner MS, Taylor CJ, et al. Enterocytic CYP3A4 in a paediatric population: developmental changes and the effect of coeliac disease and cystic fibrosis. Br J Clin Pharmacol. 2001;51:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Völler S, Flint RB, Andriessen P, et al. Rapidly maturing fentanyl clearance in preterm neonates. Arch Dis Child - Fetal Neonatal Ed. 2019;104:F598–F603. [DOI] [PubMed] [Google Scholar]
- [55].Vyhlidal CA, Bi C, Ye SQ, et al. Dynamics of cytosine methylation in the proximal promoters of CYP3A4 and CYP3A7 in pediatric and prenatal livers. Drug Metab Dispos. 2016;44:1020–1026. [DOI] [PubMed] [Google Scholar]
- [56].Koukouritaki SB, Simpson P, Yeung CK, et al. Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr Res. 2002;51:236–243. [DOI] [PubMed] [Google Scholar]
- [57].Dolphin CT, Cullingford TE, Shcphard EA, et al. Differential developmental and tissue-specific regulation of expression of the genes encoding three members of the flavin-containing monooxygenase family of man, FMO1, FMO3 and FMO4. Eur J Biochem. 1996;235:683–689. [DOI] [PubMed] [Google Scholar]
- [58].Yeung CK, Lang DH, Thummel KE, et al. Immunoquantitation of FMO1 in human liver, kidney, and intestine. Drug Metab Dispos. 2000;28:1107–1111. [PubMed] [Google Scholar]
- [59].Zhang J, Cashman JR. Quantitative analysis of FMO gene mRNA levels in human tissues. Drug Metab Dispos. 2006;34:19–26. [DOI] [PubMed] [Google Scholar]
- [60].Chen Y, Zane NR, Thakker DR, et al. Quantification of flavin-containing monooxygenases 1, 3, and 5 in human liver microsomes by UPLC-MRM-based targeted quantitative proteomics and its application to the study of ontogeny. Drug Metab Dispos. 2016;44:975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zane NR, Chen Y, Wang MZ, et al. Cytochrome P450 and flavin-containing monooxygenase families: age-dependent differences in expression and functional activity. Pediatr Res. 2018;83:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xu M, Bhatt DK, Yeung CK, et al. Genetic and nongenetic factors associated with protein abundance of flavin-containing monooxygenase 3 in human liver. J Pharmacol Exp Ther. 2017;363:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Edenberg HJ, Bosron WF. Comprehensive Toxicology: biotransformation. In: Guengerich FP, editor. 3rd ed. New York: Pergamon; 1997. p. 119–131. [Google Scholar]
- [64].Smith M, Hopkinson DA, Harris H. Developmental changes and polymorphism in human alcohol dehydrogenase. Ann Hum Genet. 1971;34:251–271. [DOI] [PubMed] [Google Scholar]
- [65].Bhatt DK, Gaedigk A, Pearce RE, et al. Age-dependent protein abundance of cytosolic alcohol and aldehyde dehydrogenases in human liver. Drug Metab Dispos. 2017;45:1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Smith DA, Hammond T, Baillie TA. Safety assessment of acyl glucuronides—a simplified paradigm. Drug Metab Dispos. 2018;46:908–912. [DOI] [PubMed] [Google Scholar]
- [67].Williams JA, Hyland R, Jones BC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUC 1/AUC) ratios. Drug Metab Dispos. 2004;32:1201–1208. [DOI] [PubMed] [Google Scholar]
- [68].Kawade N, Onishi S. The prenatal and postnatal development of UDP-glucuronyltransferase activity. Biochem J. 1981;196:257–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ciotti M, Obaray R, Martín MG, et al. Genetic defects at the UGT1 locus associated with Crigler-Najjar type I disease, including a prenatal diagnosis. Am J Med Genet. 1997;68:173–178. [PubMed] [Google Scholar]
- [70].Clarke DJ, Moghrabi N, Monaghan G, et al. Genetic defects of the UDP-glucuronosyltransferase-1 (UGT1) gene that cause familial non-haemolytic unconjugated hyperbilirubinaemias. Clin Chim Acta. 1997;266:63–74. [DOI] [PubMed] [Google Scholar]
- [71].Bosma PJ, Chowdhury JR, Bakker C, et al. The genetic basis of the reduced expression of bilirubin udp-glucuronosyltransferase 1 in gilbert’s syndrome. N Engl J Med. 1995;333:1171–1175. [DOI] [PubMed] [Google Scholar]
- [72].Blake MJ, Castro L, Leeder JS, et al. Ontogeny of drug metabolizing enzymes in the neonate. Semin Fetal Neonatal Med. 2005;10:123–138. [DOI] [PubMed] [Google Scholar]
- [73].King CD, Rios GR, Assouline JA, et al. Expression of UDP-glucuronosyltransferases (UGTs) 2B7 and 1A6 in the human brain and identification of 5-hydroxytryptamine as a substrate. Arch Biochem Biophys. 1999;365:156–162. [DOI] [PubMed] [Google Scholar]
- [74].Bock KW, Forster A, Gschaidmeier H, et al. Paracetamol glucuronidation by recombinant rat and human phenol UDP-glucuronosyltransferases. Biochem Pharmacol. 1993;45:1809–1814. [DOI] [PubMed] [Google Scholar]
- [75].Blanchet M, Bru G, Guerret M, et al. Routine determination of morphine, morphine 3-β-D-glucuronide and morphine 6-β-D-glucuronide in human serum by liquid chromatography coupled to electrospray mass spectrometry. J Chromatogr A. 1999;854:93–108. [DOI] [PubMed] [Google Scholar]
- [76].Coffman BL, Rios GR, King CD, et al. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab Dispos. 1997;25:1–4. [PubMed] [Google Scholar]
- [77].Smith HS. Opioid Metabolism. Mayo Clin Proc. 2009;84:613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Venkatasubramanian R, Fukuda T, Niu J, et al. ABCC3 and OCT1 genotypes influence pharmacokinetics of morphine in children. Pharmacogenomics. 2014;15:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Glare PA, Walsh TD. Clinical pharmacokinetics of morphine. Ther Drug Monit. 1991;13:1–23. [DOI] [PubMed] [Google Scholar]
- [80].Pacifici GM, Säwe J, Kager L, et al. Morphine glucuronidation in human fetal and adult liver. Eur J Clin Pharmacol. 1982;22:553–558. [DOI] [PubMed] [Google Scholar]
- [81].Pacifici GM, Franchi M, Giuliani L, et al. Development of the glucuronyltransferase and sulphotransferase towards 2-naphthol in human fetus. Dev Pharmacol Ther. 1990;14:108–114. [PubMed] [Google Scholar]
- [82].The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther [Internet]. 2008. [cited 2020 May 6];118:250–267. Available from: http://www.sciencedirect.com/science/article/pii/S0163725808000375. [DOI] [PubMed] [Google Scholar]
- [83].Uchaipichat V, Suthisisang C, Miners JO. The glucuronidation of R- and S-lorazepam: Human liver microsomal kinetics, UDP-glucuronosyltransferase enzyme selectivity, and inhibition by drugs. Drug Metab Dispos. 2013;41:1273–1284. [DOI] [PubMed] [Google Scholar]
- [84].Blanchard RL, Freimuth RR, Buck J, et al. A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenet Genomics. 2004;14:199–211. [DOI] [PubMed] [Google Scholar]
- [85].Hume R, Barker EV, Coughtrie MWH. Differential expression and immunohistochemical localisation of the phenol and hydroxysteroid sulphotransferase enzyme families in the developing lung. Histochem Cell Biol. 1996;105:147–152. [DOI] [PubMed] [Google Scholar]
- [86].Richard K, Hume R, Kaptein E, et al. Sulfation of thyroid hormone and dopamine during human development: ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J Clin Endocrinol Metab. 2001;86:2734–2742. [DOI] [PubMed] [Google Scholar]
- [87].Adjei AA, Gaedigk A, Simon SD, et al. Interindividual variability in acetaminophen sulfation by human fetal liver: implications for pharmacogenetic investigations of drug-induced birth defects. Birth Defects Res A Clin Mol Teratol. 2008;82:155–165. [DOI] [PubMed] [Google Scholar]
- [88].Duanmu Z, Weckle A, Koukouritaki SB, et al. Developmental expression of aryl, estrogen, and hydroxysteroid sulfotransferases in pre-and postnatal human liver. J Pharmacol Exp Ther. 2006;316:1310–1317. [DOI] [PubMed] [Google Scholar]
- [89].Saghir SA, Khan SA, McCoy AT. Ontogeny of mammalian metabolizing enzymes in humans and animals used in toxicological studies. Crit Rev Toxicol. 2012;42:323–357. [DOI] [PubMed] [Google Scholar]
- [90].Mitra A, Hilbelink DR, Dwornik JJ, et al. A novel model to assess developmental toxicity of dihaloalkanes in humans: Bioactivation of 1, 2-dibromoethane by the isozymes of human fetal liver glutathione S-transferase. Teratog Carcinog Mutagen. 1992;12:113–127. [DOI] [PubMed] [Google Scholar]
- [91].Strange RC, Davis BA, Faulder CG, et al. The human glutathione S-transferases: developmental aspects of the GST1, GST2, and GST3 loci. Biochem Genet. 1985;23:1011–1028. [DOI] [PubMed] [Google Scholar]
- [92].Strange RC, Howie AF, Hume R, et al. The developmental expression of alpha-, mu-and pi-class glutathione S-transferases in human liver. Biochim Biophys Acta. 1989;993:186–190. [DOI] [PubMed] [Google Scholar]
- [93].Pacifici GM, Rane A. Metabolism of styrene oxide in different human fetal tissues. Drug Metab Dispos. 1982;10:302–305. [PubMed] [Google Scholar]
- [94].Pacifici GM, Franchi M, Colizzi C, et al. Glutathione S-transferase in humans: development and tissue distribution. Arch Toxicol. 1988;61:265–269. [DOI] [PubMed] [Google Scholar]
- [95].Pacifici GM, Bencini C, Rane A. Acetyltransferase in humans: development and tissue distribution. Pharmacology. 1986;32:283–291. [DOI] [PubMed] [Google Scholar]
- [96].Pons G, Rey E, Carrier O, et al. Maturation of AFMU excretion in infants. Fundam Clin Pharmacol. 1989;3:589–595. [DOI] [PubMed] [Google Scholar]
- [97].Elmorsi Y, Barber J, Rostami-Hodjegan A. Ontogeny of Hepatic Drug Transporters and Relevance to Drugs Used in Pediatrics. Drug Metab Dispos. 2016;44:992–998. [DOI] [PubMed] [Google Scholar]
- [98].Balyan R, Zhang X, Chidambaran V, et al. OCT1 genetic variants are associated with postoperative morphine-related adverse effects in children. Pharmacogenomics. 2017;18:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99]*.Emoto C, Johnson TN, Neuhoff S, et al. PBPK Model of Morphine Incorporating Developmental Changes in Hepatic OCT1 and UGT2B7 Proteins to Explain the Variability in Clearances in Neonates and Small Infants. CPT pharmacometrics Syst Pharmacol. 2018;7:464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Fukuda T, Chidambaran V, Mizuno T, et al. OCT1 genetic variants influence the pharmacokinetics of morphine in children. Pharmacogenomics. 2013;14:1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Haycock GB. Development of glomerular filtration and tubular sodium reabsorption in the human fetus and newborn. Br J Urol. 1998;81:33–38. [DOI] [PubMed] [Google Scholar]
- [102].Bueva A, Guignard JP. Renal function in preterm neonates. Pediatr Res. 1994;36:572–577. [DOI] [PubMed] [Google Scholar]
- [103].Alcorn J, McNamara PJ. Pharmacokinetics in the newborn. Adv Drug Deliv Rev. 2003;55:667–686. [DOI] [PubMed] [Google Scholar]
- [104]**.Kearns GL, Abdel-Rahman SM, Alander SW, et al. Developmental pharmacology - Drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–1167. [DOI] [PubMed] [Google Scholar]
- [105].Hook JB, Hewitt WR. Development of mechanisms for drug excretion. Am J Med. 1977;62:497–506. [DOI] [PubMed] [Google Scholar]
- [106].Fetterman GH, Shuplock NA, Philipp FJ, et al. The growth and maturation of human glomeruli and proximal convolutions from term to adulthood: studies by microdissection. Pediatrics. 1965;35:601–619. [PubMed] [Google Scholar]
- [107].Rowland M Protein binding and drug clearance. Clin Pharmacokinet. 1984;9:10–17. [DOI] [PubMed] [Google Scholar]
- [108].Hayton WL. Maturation and growth of renal function: dosing renally cleared drugs in children. AAPS PharmSci. 2000;2:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Gruskin AB, Edelmann CM, Yuan S. Maturational changes in renal blood flow in piglets. Pediatr Res. 1970;4:7–13. [DOI] [PubMed] [Google Scholar]
- [110].Bueters R, Bael A, Gasthuys E, et al. Ontogeny and Cross-species Comparison of Pathways Involved in Drug Absorption, Distribution, Metabolism, and Excretion in Neonates (Review): Kidney. Drug Metab Dispos. 2020;48:353–367. [DOI] [PubMed] [Google Scholar]
- [111].Strolin M, Whomsley R, Baltes EL. Differences in absorption, distribution, metabolism and excretion of xenobiotics between the paediatric and adult populations. Expert Opin Drug Metab Toxicol. 2005;1:447–471. [DOI] [PubMed] [Google Scholar]
- [112].Brown RD, Campoli-Richards DM. Antimicrobial therapy in neonates, infants and children. Clin Pharmacokinet. 1989;17:105–115. [DOI] [PubMed] [Google Scholar]
- [113].Peterson RG, Simmons MA, Rumack BH, et al. Pharmacology of furosemide in the premature newborn infant. J Pediatr. 1980;97:139–143. [DOI] [PubMed] [Google Scholar]
- [114].Kelly MR, Cutler RE, Forrey AW, et al. Pharmacokinetics of orally administered furosemide. Clin Pharmacol Ther. 1974;15:178–186. [PubMed] [Google Scholar]
- [115].Li CY, Hosey-Cojocari C, Basit A, et al. Optimized Renal Transporter Quantification by Using Aquaporin 1 and Aquaporin 2 as Anatomical Markers: Application in Characterizing the Ontogeny of Renal Transporters and Its Correlation with Hepatic Transporters in Paired Human Samples. AAPS J [Internet]. 2019;21:88. Available from: https://pubmed.ncbi.nlm.nih.gov/31297641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Cheung KWK, van Groen BD, Spaans E, et al. A Comprehensive Analysis of Ontogeny of Renal Drug Transporters: mRNA Analyses, Quantitative Proteomics, and Localization. Clin Pharmacol Ther. 2019;106:1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Jacobs RF, Kearns GL, Brown AL, et al. Renal clearance of imipenem in children. Eur J Clin Microbiol. 1984;3:471–474. [DOI] [PubMed] [Google Scholar]
- [118].Linday LA, Engle MA, Reidenberg MM. Maturation and renal digoxin clearance. Clin Pharmacol Ther. 1981;30:735–738. [DOI] [PubMed] [Google Scholar]
- [119].Koren G, Hesslein PS, MacLeod SM. Digoxin toxicity associated with amiodarone therapy in children. J Pediatr. 1984;104:467–470. [DOI] [PubMed] [Google Scholar]
- [120].Nigam SK, Bhatnagar V. How much do we know about drug handling by SLC and ABC drug transporters in children? Clin Pharmacol Ther. 2013;94:27–29. [DOI] [PubMed] [Google Scholar]
- [121].Pavlova A, Sakurai H, Leclercq B, et al. Developmentally regulated expression of organic ion transporters NKT(OAT1), OCT1, NLT(OAT2), and Roct. Am J Physiol - Ren Physiol. 2000;278:635–643. [DOI] [PubMed] [Google Scholar]
- [122].Eraly SA, Vallon V, Vaughn DA, et al. Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. J Biol Chem. 2006;281:5072–5083. [DOI] [PubMed] [Google Scholar]
- [123].Nigam SK, Bush KT, Martovetsky G, et al. The organic anion transporter (OAT) family: A systems biology perspective. Physiol Rev. 2015;95:83–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Yin J, Wang J. Renal drug transporters and their significance in drug-drug interactions. Acta Pharm Sin B [Internet]. 2016;6:363–373. Available from: 10.1016/j.apsb.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Hua MJ, Kun HY, Jie CS, et al. Urinary microalbumin and retinol-binding protein assay for verifying children’s nephron development and maturation. Clin Chim Acta. 1997;264:127–132. [DOI] [PubMed] [Google Scholar]
- [126].Leeder JS. Ontogeny of drug-metabolizing enzymes and its influence on the pathogenesis of adverse drug reactions in children. Curr Ther Res - Clin Exp. 2001;62:900–912. [Google Scholar]
- [127].Shi D, Yang D, Prinssen EP, et al. Surge in expression of carboxylesterase 1 during the post-neonatal stage enables a rapid gain of the capacity to activate the anti-influenza prodrug oseltamivir. J Infect. 2011;203:937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Avant D, Baer G, Moore J, et al. Neonatal safety information reported to the FDA during drug development studies. Ther Innov Regul Sci. 2018;52:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Rishoej RM, Almarsdóttir AB, Christesen HT, et al. Medication errors in pediatric inpatients: a study based on a national mandatory reporting system. Eur J Pediatr. 2017;176:1697–1705. [DOI] [PubMed] [Google Scholar]
- [130].Dean L Codeine therapy and CYP2D6 genotype. Med Genet Summ [Internet]. National Center for Biotechnology Information (US); 2017. [PubMed] [Google Scholar]
- [131].Food and Drug Administration (FDA). Use of Codeine and Tramadol Products in Breastfeeding Women [Internet]. 2019. [cited 2020 Aug 1]. Available from: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/use-codeine-and-tramadol-products-breastfeeding-women-questions-and-answers.
- [132].Perry C, Davis G, Conner TM, et al. Utilization of Physiologically Based Pharmacokinetic Modeling in Clinical Pharmacology and Therapeutics: an Overview. Curr Pharmacol Reports. 2020;6:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Pearce RE, Gaedigk R, Twist GP, et al. Developmental expression of CYP2B6: a comprehensive analysis of mRNA expression, protein content and bupropion hydroxylase activity and the impact of genetic variation. Drug Metab Dispos. 2016;44:948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Zaya MJ, Hines RN, Stevens JC. Epirubicin glucuronidation and UGT2B7 developmental expression. Drug Metab Dispos. 2006;34:2097–2101. [DOI] [PubMed] [Google Scholar]
- [135].Kearns GL, Jungbluth GL, Abdel-Rahman SM, et al. Impact of ontogeny on linezolid disposition in neonates and infants. Clin Pharmacol Ther. 2003;74:413–422. [DOI] [PubMed] [Google Scholar]
- [136].Payne K, Mattheyse FJ, Liebenberg D, et al. The pharmacokinetics of midazolam in paediatric patients. Eur J Clin Pharmacol. 1989;37:267–272. [DOI] [PubMed] [Google Scholar]
- [137].Boberg M, Vrana M, Mehrotra A, et al. Age-dependent absolute abundance of hepatic carboxylesterases (CES1 and CES2) by LC-MS/MS proteomics: application to PBPK modeling of oseltamivir in vivo pharmacokinetics in infants. Drug Metab Dispos. 2017;45:216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Smits A, Annaert P, Allegaert K. Biomarkers of propofol metabolism in neonates: the quest beyond ontogeny. Biomark. Med England; 2017. p. 933–936. [DOI] [PubMed] [Google Scholar]
- [139].Mian P, Allegaert K, Conings S, et al. Integration of Placental Transfer in a Fetal-Maternal Physiologically Based Pharmacokinetic Model to Characterize Acetaminophen Exposure and Metabolic Clearance in the Fetus. Clin Pharmacokinet. 2020;59:911–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Emoto C, Fukuda T, Johnson TN, et al. Characterization of Contributing Factors to Variability in Morphine Clearance Through PBPK Modeling Implemented With OCT1 Transporter. CPT pharmacometrics Syst Pharmacol. 2017;6:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Strougo A, Eissing T, Yassen A, et al. First dose in children: physiological insights into pharmacokinetic scaling approaches and their implications in paediatric drug development. J Pharmacokinet Pharmacodyn. 2012;39:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Food and Drug Administration. General clinical pharmacology considerations for pediatric studies for drugs and biological products (Draft). 2014;1–25. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm425885.pdf. [Google Scholar]
- [143].Food and Drug Administration (FDA). 21 CFR 314.108 - New drug product exclusivity. [Internet]. [cited 2020 Jul 29]. Available from: https://www.govinfo.gov/app/details/CFR-2012-title21-vol5/CFR-2012-title21-vol5-sec314-108.
- [144].Food and Drug Administration. Good Review Practices [Internet]. [cited 2020 Jul 29]. Available from: https://www.fda.gov/drugs/guidance-compliance-regulatory-information/good-review-practices-grps.
- [145].FDA. Draft Guidance for Industry - Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Pediatric Study Plans. FDA Guid [Internet]. 2016;115:13. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM360507.pdf. [Google Scholar]
- [146].Rodrigues D, Rowland A. From Endogenous Compounds as Biomarkers to Plasma-Derived Nanovesicles as Liquid Biopsy; Has the Golden Age of Translational Pharmacokinetics-Absorption, Distribution, Metabolism, Excretion-Drug-Drug Interaction Science Finally Arrived? Clin Pharmacol Ther. 2019;105:1407–1420. [DOI] [PubMed] [Google Scholar]
- [147].Rowland A, Ruanglertboon W, Dyk M, et al. Plasma extracellular nanovesicle (exosome)-derived biomarkers for drug metabolism pathways: a novel approach to characterize variability in drug exposure. Br J Clin Pharmacol. 2019;85:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Wagner C, Zhao P, Pan Y, et al. Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: Report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst Pharmacol. 2015;4:226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Forrest JAH, Clements JA, Prescott LF. Clinical Pharmacokinetics of Paracetamol. Clin Pharmacokinet. 1982;7:93–107. [DOI] [PubMed] [Google Scholar]
- [150].Wang C, Allegaert K, Tibboel D, et al. Population pharmacokinetics of paracetamol across the human age-range from (pre)term neonates, infants, children to adults. J Clin Pharmacol. 2014;54:619–629. [DOI] [PubMed] [Google Scholar]
- [151].Mohamed MHN, Lima JJ, Eberle LV., et al. Effects of gender and race on albuterol pharmacokinetics. Pharmacotherapy. 1999;19:157–161. [DOI] [PubMed] [Google Scholar]
- [152].Boulton DW, Fawcett JP. Enantioselective Disposition of Albuterol in Humans. Clin Rev Allergy Immunol. 1996;14:115–138. [DOI] [PubMed] [Google Scholar]
- [153].Boulton DW, Fawcett JP. Enantioselective disposition of salbutamol in man following oral and intravenous administration. Br J Clin Pharmacol. 1996;41:35–40. [DOI] [PubMed] [Google Scholar]
- [154].Boulton DW, Fawcett JP. The pharmacokinetics of levosalbutamol: What are the clinical implications? Clin Pharmacokinet. 2001;40:23–40. [DOI] [PubMed] [Google Scholar]
- [155].Rafailidis PI, Ioannidou EN, Falagas ME. Ampicillin/sulbactam: Current status in severe bacterial infections. Drugs. 2007;67:1829–1849. [DOI] [PubMed] [Google Scholar]
- [156].Yokoyama Y, Matsumoto K, Yamamoto H, et al. Pharmacokinetics of ampicillin-sulbactam and the renal function-based optimization of dosing regimens for prophylaxis in patients undergoing cardiovascular surgery. J Infect Chemother. 2012;18:878–882. [DOI] [PubMed] [Google Scholar]
- [157].Kucers A Kucers’ the Use of Antibiotics, a Clinical Review of Antibacterial, Antifungal, Antiparasitic and Antiviral Drugs. 2012. [Google Scholar]
- [158].Edsbäcker S, Andersson T. Pharmacokinetics of budesonide (Entocort™ EC) capsules for Crohn’s disease. Clin Pharmacokinet. 2004;43:803–821. [DOI] [PubMed] [Google Scholar]
- [159].Blanchard J, Sawers SJA. The absolute bioavailability of caffeine in man. Eur J Clin Pharmacol. 1983;24:93–98. [DOI] [PubMed] [Google Scholar]
- [160].Ha HR, Chen J, Krähenbühl S, et al. Biotransformation of caffeine by cDNA-expressed human cytochromes P-450. Eur J Clin Pharmacol. 1996;49:309–315. [DOI] [PubMed] [Google Scholar]
- [161].Kaplan GB, Greenblatt DJ, Ehrenberg BL, et al. Dose-dependent pharmacokinetics and psychomotor effects of caffeine in humans. J Clin Pharmacol. 1997;37:693–703. [DOI] [PubMed] [Google Scholar]
- [162].Brachtel D, Richter E. Absolute bioavailability of caffeine from a tablet formulation. J Hepatol. 1992;16:385. [DOI] [PubMed] [Google Scholar]
- [163].Rybak M The pharmacokinetic profile of a new generation of parenteral cephalosporin. Am J Med. 1996;100:39S–44S. [DOI] [PubMed] [Google Scholar]
- [164].Claessens AJ, Risler LJ, Eyal S, et al. CYP2D6 mediates 4-hydroxylation of clonidine in vitro: Implication for pregnancy-induced changes in clonidine clearance. Drug Metab Dispos. 2010;38:1393–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Lowenthal DT, Matzek KM, MacGregor TR. Clinical pharmacokinetics of clonidine. Clin Pharmacokinet. 1988;14:287–310. [DOI] [PubMed] [Google Scholar]
- [166].Davies DS, Wing LMH, Reid JL, et al. Pharmacokinetics and concentration-effect relationships of intravenous and oral clonidine. Clin Pharmacol Ther. 1977;21:593–601. [DOI] [PubMed] [Google Scholar]
- [167].Spoorenberg SMC, Deneer VHM, Grutters JC, et al. Pharmacokinetics of oral vs. intravenous dexamethasone in patients hospitalized with community-acquired pneumonia. Br J Clin Pharmacol. 2014;78:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Anttila M, Penttilä J, Helminen A, et al. Bioavailability of dexmedetomidine after extravascular doses in healthy subjects. Br J Clin Pharmacol. 2003;56:691–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Weerink MAS, Struys MMRF, Hannivoort LN, et al. Clinical Pharmacokinetics and Pharmacodynamics of Dexmedetomidine. Clin Pharmacokinet. 2017;56:893–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Mandelli M, Tognoni G, Garattini S. Clinical Pharmacokinetics of Diazepam. Clin. Pharmacokinet 1978. p. 72–91. [DOI] [PubMed] [Google Scholar]
- [171].Friedman H, Greenblatt DJ, Peters GR, et al. Pharmacokinetics and pharmacodynamics of oral diazepam: Effect of dose, plasma concentration, and time. Clin Pharmacol Ther. 1992;52:139–150. [DOI] [PubMed] [Google Scholar]
- [172].Greenblatt DJ, Divoll Allen M, Harmatz JS, et al. Diazepam disposition determinants. Clin Pharmacol Ther. 1980;27:301–312. [DOI] [PubMed] [Google Scholar]
- [173].Krishna DR, Klotz U. Newer H 2-receptor antagonists. Clinical pharmacokinetics and drug interaction potential. Clin Pharmacokinet. 1988;15:205–215. [DOI] [PubMed] [Google Scholar]
- [174].Maples HD, James LP, Stowe CD, et al. Famotidine disposition in children and adolescents with chronic renal insufficiency. J Clin Pharmacol. 2003;43:7–14. [DOI] [PubMed] [Google Scholar]
- [175].Howden CW, Tytgat GNJ. The tolerability and safety profile of famotidine. Clin Ther. 1996;18:36–54. [DOI] [PubMed] [Google Scholar]
- [176].Lin JH. Pharmacokinetic and Pharmacodynamic Properties of Histamine H2-Receptor Antagonists: Relationship Between Intrinsic Potency and Effective Plasma Concentrations. Clin Pharmacokinet. 1991;20:218–236. [DOI] [PubMed] [Google Scholar]
- [177].Ohbuchi M, Noguchi K, Kawamura A, et al. Different effects of proton pump inhibitors and famotidine on the clopidogrel metabolic activation by recombinant CYP2B6, CYP2C19 and CYP3A4. Xenobiotica. 2012;42:633–640. [DOI] [PubMed] [Google Scholar]
- [178].Olkkola KT, Hamunen K, Maunuksela E-L. Clinical pharmacokinetics and pharmacodynamics of opioid analgesics in infants and children. Clin Pharmacokinet. 1995;28:385–404. [DOI] [PubMed] [Google Scholar]
- [179].Scholz J, Steinfath M, Schulz M. Clinical pharmacokinetics of alfentanil, fentanyl and sufentanil. An update. Clin Pharmacokinet. 1996;31:275–292. [DOI] [PubMed] [Google Scholar]
- [180].Andreasen F, Hansen U, Husted SE, et al. The pharmacokinetics of frusemide are influenced by age. Br J Clin Pharmacol. 1983;16:391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Ponto LL, Schoenwald RD. Furosemide (frusemide). A pharmacokinetic/pharmacodynamic review (Part I). Clin Pharmacokinet. 1990;18:381–408. [DOI] [PubMed] [Google Scholar]
- [182].Waller ES, Hamilton SF, Massarella JW, et al. Disposition and absolute bioavailability of furosemide in healthy males. J Pharm Sci. 1982;71:1105–1108. [DOI] [PubMed] [Google Scholar]
- [183].Kerdpin O, Knights KM, Elliot DJ, et al. In vitro characterisation of human renal and hepatic frusemide glucuronidation and identification of the UDP-glucuronosyltransferase enzymes involved in this pathway. Biochem Pharmacol. 2008;76:249–257. [DOI] [PubMed] [Google Scholar]
- [184].Zaske DE, Cipolle RJ, Rotschafer JC, et al. Gentamicin pharmacokinetics in 1,640 patients: Method for control of serum concentrations. Antimicrob Agents Chemother. 1982;21:407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Regamey C, Gordon RC, Kirby WMM. Comparative pharmacokinetics of tobramycin and gentamicin. Clin Pharmacol Ther. 1973;14:396–403. [DOI] [PubMed] [Google Scholar]
- [186].Derendorf H, Mollmann H, Barth J, et al. Pharmacokinetics and oral bioavailability of hydrocortisone. J Clin Pharmacol. 1991;31:473–476. [DOI] [PubMed] [Google Scholar]
- [187].Czock D, Keller F, Rasche FM, et al. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44:61–98. [DOI] [PubMed] [Google Scholar]
- [188].Sarkar U, Rivera-Burgos D, Large EM, et al. Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab Dispos. 2015;43:1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Furuta T, Mori C, Suzuki A, et al. Simultaneous determination of 6β-hydroxycortisol and cortisol in human urine by liquid chromatography with ultraviolet absorbance detection for phenotyping the CYP3A activity determined by the cortisol 6β-hydroxylation clearance. J Chromatogr B Anal Technol Biomed Life Sci. 2004;801:165–171. [DOI] [PubMed] [Google Scholar]
- [190].Mazaleuskayaa LL, Thekena KN, Gong L, et al. PharmGKB summary: ibuprofen pathways. Pharmacogenet Genomics. 2015;25:96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Lee EJ, Williams KM, Day R, et al. Stereoselective disposition of ibuprofen enantiomers in man. Br J Clin Pharmacol. 1985;19:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Lockwood GF, Albert KS, Gillespie WR, et al. Pharmacokinetics of ibuprofen in man. I. Free and total area/dose relationships. Clin Pharmacol Ther. 1983;34:97–103. [DOI] [PubMed] [Google Scholar]
- [193].Strolin Benedetti M, Whomsley R, Nicolas JM, et al. Pharmacokinetics and metabolism of 14C-levetiracetam, a new antiepileptic agent, in healthy volunteers. Eur J Clin Pharmacol. 2003;59:621–630. [DOI] [PubMed] [Google Scholar]
- [194].Patsalos PN. Clinical pharmacokinetics of levetiracetam. Clin Pharmacokinet. 2004;43:707–724. [DOI] [PubMed] [Google Scholar]
- [195].Greenblatt DJ. Clinical pharmacokinetics of oxazepam and lorazepam. Clin Pharmacokinet. 1981;6:89–105. [DOI] [PubMed] [Google Scholar]
- [196].Anderson GD, Lynn AM. Optimizing pediatric dosing: A developmental pharmacologic approach. Pharmacotherapy. 2009;29:680–690. [DOI] [PubMed] [Google Scholar]
- [197].Dyer KR, Foster DJR, White JM, et al. Steady-state pharmacokinetics and pharmacodynamics in methadone maintenance patients: Comparison of those who do and do not experience withdrawal and concentration-effect relationships. Clin Pharmacol Ther. 1999;65:685–694. [DOI] [PubMed] [Google Scholar]
- [198].Inturrisi CE, Colburn WA, Kaiko RF, et al. Pharmacokinetics and pharmacodynamics of methadone in patients with chronic pain. Clin Pharmacol Ther. 1987;41:392–401. [DOI] [PubMed] [Google Scholar]
- [199].Kharasch ED, Hoffer C, Whittington D, et al. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76:250–269. [DOI] [PubMed] [Google Scholar]
- [200].Rohatagi S, Barth J, Möllmann H, et al. Pharmacokinetics of methylprednisolone and prednisolone after single and multiple oral administration. J Clin Pharmacol. 1997;37:916–925. [DOI] [PubMed] [Google Scholar]
- [201].Thummel KE, O’Shea D, Paine MF, et al. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther. 1996;59:491–502. [DOI] [PubMed] [Google Scholar]
- [202].Miller A, McKee A, Mazer CD. Sedation, Analgesia, and Related Topics [Internet]. Second Edi. Cardiothorac. Crit. Care. Elsevier Inc.; 2007. Available from: 10.1016/B978-0-7506-7572-7.50007-2. [DOI] [Google Scholar]
- [203].Berkowitz BA. The relationship of pharmacokinetics to pharmacological activity: morphine, methadone and naloxone. Clin Pharmacokinet. 1976;1:188–219. [DOI] [PubMed] [Google Scholar]
- [204].Roila F, Del Favero A. Ondansetron clinical pharmacokinetics. Clin Pharmacokinet. 1995;29:95–109. [DOI] [PubMed] [Google Scholar]
- [205].Yang SH, Lee MG. Effects of cytochrome P450 (CYP) inducers and inhibitors on ondansetron pharmacokinetics in rats: involvement of hepatic CYP2D subfamily and 3A1/2 in ondansetron metabolism. J Pharm Pharmacol. 2008;60:853–861. [DOI] [PubMed] [Google Scholar]
- [206].Culy CR, Bhana N, Plosker GL. Ondansetron A Review of its Use as an Antiemetic in Children. Paediatr Drugs. 2001;3:441–479. [DOI] [PubMed] [Google Scholar]
- [207].Ordóñez Gallego A, González Barón M, Espinosa Arranz E. Oxycodone: A pharmacological and clinical review. Clin Transl Oncol. 2007;9:298–307. [DOI] [PubMed] [Google Scholar]
- [208].Benziger DP, Kaiko RF, Miotto JB, et al. Differential effects of food on the bioavailability of controlled-release oxycodone tablets and immediate-release oxycodone solution. J Pharm Sci. 1996;85:407–410. [DOI] [PubMed] [Google Scholar]
- [209].Takala A, Kaasalainen V, Seppälä T, et al. Pharmacokinetic comparison of intravenous and intranasal administration of oxycodone. Acta Anaesthesiol Scand. 1997;41:309–312. [DOI] [PubMed] [Google Scholar]
- [210].Frey BM, Frey FJ. Clinical pharmacokinetics of prednisone and prednisolone. Clin Pharmacokinet. 1990;19:126–146. [DOI] [PubMed] [Google Scholar]
- [211].Colangelo PM, Blouin RA, Steinmetz JE, et al. Age and propranolol stereoselective disposition in humans. Clin Pharmacol Ther. 1992;51:489–494. [DOI] [PubMed] [Google Scholar]
- [212].Walle T, Conradi EC, Walle UK, et al. 4-Hydroxypropranolol and its glucuronide after single and long-term doses of propranolol. Clin Pharmacol Ther. 1980;27:22–31. [DOI] [PubMed] [Google Scholar]
- [213].Masubuchi Y, Hosokawa S, Horie T, et al. Cytochrome P450 isozymes involved in propranolol metabolism in human liver microsomes. The role of CYP2D6 as ring-hydroxylase and CYP1A2 as N-desisopropylase. Drug Metab Dispos. 1994;22:909–915. [PubMed] [Google Scholar]
- [214].Mansur AP, Avakian SD, Paula RS, et al. Pharmacokinetics and pharmacodynamics of propranolol in hypertensive patients after sublingual administration: Systemic availability. Brazilian J Med Biol Res. 1998;31:691–696. [DOI] [PubMed] [Google Scholar]
- [215].Walker DK. Pharmacokinetics and metabolism of sildenafil in mouse, rat, rabbit, dog and man. Xenobiotica. 1999;29:297–310. [DOI] [PubMed] [Google Scholar]
- [216].Zhao P, Vieira M de LT, Grillo JA, et al. Evaluation of Exposure Change of Nonrenally Eliminated Drugs in Patients With Chronic Kidney Disease Using Physiologically Based Pharmacokinetic Modeling and Simulation. J Clin Pharmacol. 2012;52:91S–108S. [DOI] [PubMed] [Google Scholar]
- [217].Marsot A, Boulamery A, Bruguerolle B, et al. Vancomycin: A review of population pharmacokinetic analyses. Clin Pharmacokinet. 2012;51:1–13. [DOI] [PubMed] [Google Scholar]
- [218].Leader WG, Chandler MHH, Castiglia M. Pharmacokinetic optimisation of vancomycin therapy. Clin Pharmacokinet. 1995;28:327–342. [DOI] [PubMed] [Google Scholar]
- [219].Golper TA, Noonan HM, Elzinga L, et al. Vancomycin pharmacokinetics, renal handling, and nonrenal clearances in normal human subjects. Clin Pharmacol Ther. 1988;43:565–570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.