

A time-saving and deletion-prone method facilitates functional study of maternal factors in zebrafish.
Abstract
Maternal products are exclusive factors to drive oogenesis and early embryonic development. As disrupting maternal gene functions is either time-consuming or technically challenging, early developmental programs regulated by maternal factors remain mostly elusive. We provide a transgenic approach to inactivate maternal genes in zebrafish primary oocytes. By introducing three tandem single guide RNA (sgRNA) expression cassettes and a green fluorescent protein (GFP) reporter into Tg(zpc:zcas9) embryos, we efficiently obtained maternal nanog and ctnnb2 mutants among GFP-positive F1 offspring. Notably, most of these maternal mutants displayed either sgRNA site–spanning genomic deletions or unintended large deletions extending distantly from the sgRNA targets, suggesting a prominent deletion-prone tendency of genome editing in the oocyte. Thus, our method allows maternal gene knockout in the absence of viable and fertile homozygous mutant adults. This approach is particularly time-saving and can be applied for functional screening of maternal factors and generating genomic deletions in zebrafish.
INTRODUCTION
Maternally supplied mRNAs and proteins, produced by over half of the coding genes in zebrafish (1), control oogenesis and the earliest processes of embryonic development, especially those before zygotic genome activation (2–6). Some maternal factors can also perform zygotic functions during early development (7–9). Thus, eliminating both maternal and zygotic products is essential to elucidate functional roles of genes with maternal expression. However, most maternal gene products are cell autonomously deposited in primary oocytes that have the same genotype as somatic cells. As a result, to generate maternal (M) or maternal-zygotic (MZ) mutant offspring by traditional genetic approach, viable and fertile homozygous mutant females are indispensable. These homozygous mutants are classically produced in a time-consuming manner by repeated crossing and screening through typically three generations (almost 9 months in zebrafish). Nevertheless, if loss of zygotic gene functions causes death or sterility, then it becomes particularly difficult to produce maternal mutants. One way to circumvent these obstacles is to rescue phenotype defects by injecting the wild-type mRNA, such as the case for the generation of Moep mutants (8), but this approach was proved to be possible only for a small proportion of zygotic lethal genes. Several alternatives have also been developed, such as germline replacement (9–12), oocyte microinjection in situ (13), generation of genetically mosaic females (7), or the Bacterial Artificial Chromosome-rescue-based Knockout (BACK approach) using a BAC plasmid containing the wild-type gene that is sent “BACK” to the genome for rescue (14). These methods remain technically challenging, time-consuming, or less efficient. Therefore, a rapid and straightforward system to inactivate genes in the oocyte may break the barrier to study their maternal functions.
The CRISPR-Cas9 system has become a powerful tool in genome editing, promoting extensive functional analyses (15–17). CRISPR-Cas9–based conditional knockout has been tested in several species, including zebrafish, mouse, and mosquito (18–21). The strategy to achieve tissue-specific or temporally controlled gene inactivation relies on the transgenesis and the use of promoters to drive the spatiotemporal expression of Cas9 protein (18–28). However, the application of Cas9-mediated conditional knockout is still limited, probably due to the low efficiency and mosaicism or failure to ensure complete biallelic mutations (18, 19, 25, 29–31). These often result in incomplete removal of tissue-specific gene products or functional compensation from wild-type or heterozygous cells, thus generating animals without detectable mutant phenotypes. Distinct from tissues or organs in which component cells function as a whole, every oocyte with biallelic maternal gene disruption in the ovary can lead to the generation of a homogeneous maternal mutant animal. Therefore, we reasoned that CRISPR-Cas9–mediated oocyte-specific conditional knockout should still be highly applicable to create maternal mutants, despite the moderate efficiency and mosaic feature of the CRISPR-Cas9 system in transgenesis.
Zona pellucida gene zp3b (also known as zpc) is specifically expressed at early stages of oocytes, and its promoter can drive a high level of maternal expression of green fluorescent protein (GFP) (32). We have previously created a transgenic line Tg(zpc:zcas9) that specifically expresses zebrafish codon-optimized cas9 in the oocytes and tested its application in routine gene inactivation by injection of single guide RNAs (sgRNAs) into the embryo (33, 34). In this study, we developed a rapid conditional knockout strategy to generate maternal mutants by just one generation (less than 3 months). By introducing a single or multiple sgRNA expression modules into Tg(zpc:zcas9) embryos via I-SceI–mediated transgenesis, we successfully generated maternal mutants for nanog and ctnnb2 (β-catenin2) genes among F1 embryos. We also discovered a marked tendency for genome editing in zebrafish oocytes to frequently produce heritable large deletions when expressing multiple sgRNAs. Therefore, our study demonstrates the promising potential of this conditional knockout strategy to circumvent current technical restrictions in functional studies of maternal factors.
RESULTS
Experimental design of a rapid oocyte-specific conditional knockout system
As Tg(zpc:zcas9) zebrafish has adequate maternal Cas9 protein for genome editing after injection of synthetic sgRNAs into fertilized eggs (34), we were wondering whether it was possible to directly inactivate genes of interest in the oocytes by introducing a transgenic sgRNA expression cassette to produce maternal mutants by just one generation. We thus designed a transgenic vector pGGDestISceIEG-1sgRNA containing the U6a promoter to drive the expression of a single sgRNA and an ef1α promoter to drive the ubiquitous expression of GFP that will serve as a selection marker. These two modules are flanked by Tol2 inverse repeat sequences (TIR) and I-SceI restriction sites (Fig. 1A). By Golden Gate ligation, we can also connect three sgRNA expression modules driven by U6a or U6b promoter to create a pGGDestISceIEG-3sgRNA vector (Fig. 1B). This endows the vector with the ability to express three sgRNAs simultaneously targeting a single gene of interest, potentially improving the efficiency of biallelic gene disruption (35).
Fig. 1. Experimental design of a CRISPR-Cas9 system to generate maternal mutants in F1 embryos.
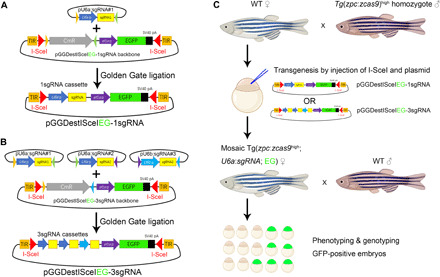
(A) Construction of pGGDestISceIEG-1sgRNA plasmid that contains a single sgRNA expression cassette and a GFP expression module driven by the ef1α promoter. I-SceI restriction sites and TIR are designed to flank sgRNA and GFP sequences for transgenesis. EGFP, enhanced GFP. (B) Construction of the transgenic pGGDestISceIEG-3sgRNA vector expressing three tandem sgRNAs via Golden Gate ligation. (C) Pipeline to generate maternal mutants in F1 embryos. The sgRNA expression cassette is introduced into Tg(zpc:zcas9) embryos by I-SceI–mediated transgenesis, and the phenotype of the resulting GFP-positive F1 embryos is examined to identify developmental defects. CmR represents the chloramphenicol resistant gene, while EG highlighted in green designates the GFP expression cassette driven by the ef1α promoter. WT, wild type.
Because Tg(zpc:zcas9) zebrafish line was created by Tol2 transposition, we decided to use meganuclease-mediated transgenesis to integrate the sgRNA expression cassette into the genome (36), such that an accidental removal of the zpc:zcas9 transgene could be avoided. Embryos from a wild-type female and a homozygous Tg(zpc:zcas9) male are used for further I-SceI–mediated transgenesis. After coinjection of I-SceI and sgRNA transgenic vector, mosaic embryos with widespread GFP expression were selected and raised to adulthood. The mosaic F0 female fish will be subsequently outcrossed with wild-type males to check sgRNA expression in the embryos by monitoring the expression of maternal GFP. Last, maternal mutants will be identified by phenotyping and genotyping GFP-positive offspring (Fig. 1C).
A proof of principle to produce maternal nanog mutants in F1 embryos
As a proof of principle, we selected nanog gene as a target because its maternal products are required for zygotic transcription of many essential genes involved in pattern formation and cell movements during gastrulation (37–39). As a result, maternal nanog mutant (Mnanog) embryos can be readily identified from 5 hours postfertilization (hpf) onward by the extremely delayed epiboly phenotype, whereas zygotic homozygous mutant embryos develop normally. An sgRNA target was selected in the first exon of nanog gene by CRISPRscan (40) and was found to be highly efficient (66.3% of mutation rate) following its coinjection with cas9 mRNA into wild-type embryos (fig. S1). We then constructed a pGGDestISceIEG-1sgRNA vector containing this sgRNA. By injecting this plasmid and I-SceI enzyme into the blastodisc of 1-cell stage embryos, we created mosaic transgenic sgRNA lines in the Tg(zpc:zcas9) or wild-type background. In a representative experiment, we obtained 3 female adults with germline GFP expression in a total of 25 F0 fish under the Tg(zpc:zcas9) background (12% germline transmission rate). After outcross, we observed an average of 16.2% GFP-positive embryos displaying characteristic phenotypes of Mnanog mutants (Fig. 2, A to K), with severely delayed epiboly already prominent at the shield stage (Fig. 2, A to D and F to I), and the absence of visible dorsoventral and anteroposterior axes at 24 hpf (Fig. 2, E and J), reminiscent of previously reported developmental defects (37–39). In contrast, embryos with only sgRNA expression did not show any developmental defects (Fig. 2K and fig. S2).
Fig. 2. Rapid generation of nanog maternal mutants by transgenic expression of one or three sgRNAs.
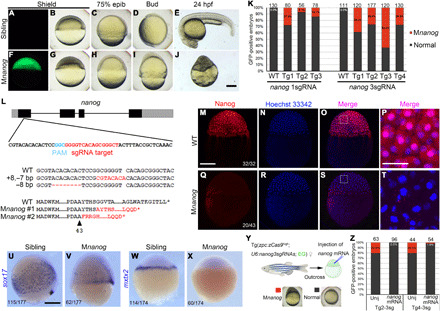
(A to E) GFP-negative F1 sibling embryos developed normally. (F to J) Typical Mnanog defective phenotypes observed among GFP-positive embryos. All embryos are lateral view with the dorsal region on the right (A to D and F to I) or upward (E and J). (K) Stacked columns show the ratio of Mnanog-like defective embryos among GFP-positive offspring in Tg(zpc:zcas9) background. Higher efficiency in generating maternal mutants was observed by expressing three sgRNAs. Numbers on the top of each column represent the total GFP-positive embryos scored. WT, wild-type fish; Tg1 to Tg4, different mutation-carrying F0 founders. (L) Single sgRNA expression in Tg(zpc:zcas9) leads to indel in nanog maternal transcripts, resulting in frameshift and premature termination of translation. The four exons of nanog are represented by black boxes, with gray indicating untranslated regions (UTRs). Mutant transcripts were identified in two independent embryos at 3 hpf. The dashed line corresponds to deleted nucleotides; red bases indicate insertions. Red letters in protein sequences indicates amino acid residues translated after frameshift mutation. The arrowhead represents the position of amino acid in wild-type protein. (M to P) Nuclear accumulation of Nanog protein in 3-hpf wild-type embryos revealed by immunofluorescence. (Q to T) Absence of Nanog protein expression in 3-hpf Mnanog embryos. (P and T) Magnified images of framed regions in (O) and (S). Hoechst 33342 (blue) was used to stain nuclei. (U to X) In situ hybridization (ISH) analysis shows the expression of sox17 at 7.5 hpf and mxtx2 at the sphere stage. (Y) Procedure to rescue Mnanog phenotype by mRNA injection. (Z) Stacked columns show a nearly complete rescue of Mnanog mutant defects after injection of the wild-type nanog-myc mRNA. Unij, Uninjected. Numbers represent total embryos scored from two experiments. Scale bars, 50 μm (P and T) and 250 μm (others).
We then randomly selected 10 GFP-positive cleavage stage embryos from two mutant-producing female fish and individually extracted total RNAs to analyze their genotypes. After reverse transcription polymerase chain reaction (RT-PCR), the products were cloned into the pZeroback vector, and individual clones were subjected to Sanger sequencing. We found that 2 embryos among the 10 GFP-positive offspring carried mutations in nanog transcripts (Fig. 2L). For embryo #1, all 33 sequenced clones had an indel of 8–base pair (bp) insertion and 7-bp deletion; while for embryo #2, all 29 sequenced clones showed an 8-bp deletion (Fig. 2L). Theoretically, as four copies of the haploid genome emerged in the primary oocyte and were edited independently, four different mutant nanog RNAs should exist in each maternal mutant embryo. The bias in the emergence of the mutant transcripts may represent a differential RNA stability between these mutant variants. Nevertheless, both types of indels lead to frameshift and premature stop that should produce nonfunctional proteins (Fig. 2L). These results illustrate the feasibility of this transgenic method to generate maternal mutants by only one generation, with less than 3 months.
Multiple sgRNAs increase the efficiency to produce nanog maternal mutants
We next designed two additional sgRNAs in the first and second exons of the nanog gene. Following verification of their efficiency (fig. S1), the three sgRNAs were cloned tandemly into the pGGDestISceIEG-3sgRNA vector and integrated into the genome of Tg(zpc:zcas9) fish. We found that 40% of F0 females (n = 10) produced GFP-positive offspring. On average, 38.8% of these GFP expressing Tg(zpc:zcas9) embryos displayed typical Mnanog phenotypes, while transgenic embryos expressing the three sgRNAs and GFP but without Cas9 did not show any developmental defect (Fig. 2K and fig. S2). The efficiency was hence about 2.5-fold higher than that obtained by just one sgRNA.
To further test whether the embryos with aberrant phenotypes are bona fide nanog maternal mutants, we performed whole-mount immunofluorescence staining on wild-type and GFP-positive embryos at 3 hpf with a commercially available Nanog antibody. As zygotic transcription is not activated at this stage, only maternal Nanog protein exists in these embryos. All wild-type embryos showed a strong Nanog protein signal, with an apparent enrichment in the nuclei (Fig. 2, M to P). However, 46.5% (n = 43) of GFP-positive embryos were negative for Nanog antibody staining, indicating loss of maternally deposited Nanog protein in these mutants (Fig. 2, Q to T). To determine the expression of Nanog target genes, we examined sox17 and mxtx2 transcripts by whole-mount in situ hybridization (ISH). Sox17 is a transcription factor specifically expressed in the endoderm and the dorsal forerunner cells. Compared to wild-type embryos, sox17 expression in the endoderm of defective embryos was substantially reduced, and its expression in the dorsal forerunner cells expanded laterally (Fig. 2, U and V). Mxtx2 is another transcription factor expressed exclusively in the yolk syncytial layer and is essential for epiboly movement (41). Its expression completely disappeared in presumed Mnanog mutant embryos at the sphere stage (Fig. 2, W and X). These disrupted gene expression patterns were identical to those observed in the genuine Mnanog mutants (37–39), suggesting that these defects were caused by loss of maternal Nanog function. To further demonstrate the specific loss of maternal nanog gene products, we tried to rescue the defective epiboly phenotype by supplementing wild-type nanog-myc mRNA to GFP-positive embryos (Fig. 2Y). The result showed that there was almost a complete rescue of epiboly defects (Fig. 2Z). Thus, these data demonstrate a remarkable increase in the efficiency of producing maternal mutants by targeting a single gene with multiple transgenic sgRNAs.
Expression of multiple sgRNAs in the oocyte tends to generate large deletions
We next tried to genotype nanog maternal mutants created by oocyte expression of multiple sgRNAs. As each oocyte is surrounded by a large number of follicle cells and contains a minimal amount of genomic DNA, it is almost impossible to perform DNA extraction from a single oocyte without contamination by genomic DNA from somatic follicle cells. Hence, we designed a pipeline based on the regulative development of zebrafish early embryos and used RT-PCR analysis to genotype the maternal mutants. GFP-positive embryos were raised to 3 hpf when dozens of cells were aspired from the blastodisc and subjected to RNA extraction. These cells only express maternal mRNAs because they were collected before zygotic genome activation occurring at 3.5 hpf. Thus, we can genotype the donor embryo by performing RT-PCR analysis of the transcripts of interest. All donor embryos were allowed to develop to appropriate stages for phenotype analysis. If an embryo presents the maternal mutant phenotype, then the total RNAs from collected cells were extracted, and RT-PCR was performed to amplify the coding sequence (CDS) of concerned transcripts. The resulting PCR products were directly analyzed by agarose gel electrophoresis and further amplified by a pair of nested primers for constructing a mini expression library. The reverse primer sequence is located immediately before the stop codon and ensures in-frame cloning of the open reading frame (ORF) from the wild-type mRNA with the six myc epitopes in the pCS2-MT vector. So if mutant sequences with frameshifts or large deletions are cloned, then they will not produce any protein with correct molecular weight (see also Materials and Methods and table S1). This library can be examined by sequencing individual clones or can be injected into wild-type embryos to verify whether the mutant transcripts produce protein products by Western blot analysis (Fig. 3A).
Fig. 3. The deletion-biased tendency in Mnanog mutant embryos induced by multiple Cas9 RNPs.
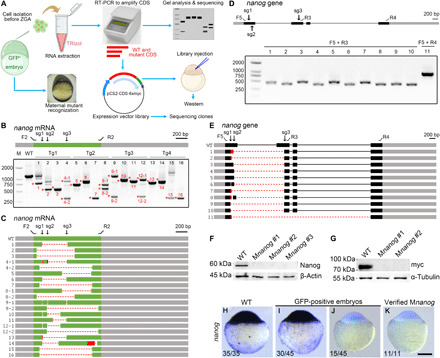
(A) Strategy combining phenotyping and genotyping. Cells are isolated from GFP-positive embryos at 3 hpf and stored in TRIzol. When donors developed maternal mutant phenotype, total RNAs from isolated cells are extracted and analyzed by RT-PCR. Primers are designed to amplify the entire wild-type CDS. PCR products are either analyzed by gel electrophoresis or subcloned into pCS2-MT vector, generating a mini expression library. A control library is made from a wild-type embryo using the same set of primers. These primers ensure the in-frame ligation of the wild-type CDS and the myc epitopes. The libraries are either injected to express protein or subjected to sequencing. (B) RT-PCR analysis of nanog CDS region from 16 Mnanog embryos shows the presence of truncated transcripts. Red asterisks and numbers indicate PCR products subjected to Sanger sequencing. F2 and R2 represent primers to amplify the CDS region. The positions of sgRNA targeting sites are marked as sg1 to sg3. Gray boxes indicate UTRs, and the green represents the CDS region. (C) Diagram summarizes the sequencing results. Dashed lines and red boxes represent deletions and insertions, respectively. (D) Analysis of genomic deletions in Mnanog mutants using different primer sets as displayed on the nanog gene. Grey and black boxes designate UTR and CDS regions of exons, respectively; thin lines indicate introns. (E) Sequencing of PCR products reveals various patterns of genomic deletions. (F) Absence of Nanog protein in verified Mnanog embryos at 3 hpf. (G) Injection of expression libraries constructed from two Mnanog mutants produced no myc-tagged protein product. (H to K) ISH was used to examine the absence of nanog transcripts in Mnanog mutants among GFP-positive embryos at the sphere stage. Scale bar, 250 μm.
Using this strategy, we examined 16 embryos with apparent Mnanog phenotypes from four independent F0 founders. By PCR amplification of nanog CDS from collected cells, we found that all these embryos had truncated nanog maternal transcripts (Fig. 3B). Some embryos displayed multiple patterns of deletions, as revealed by the presence of PCR products with different sizes. Note that although maternal nanog mRNAs are independently produced by four genomic copies of nanog gene during the prophase of meiosis I, not all four mutant maternal transcripts in each embryo could be detected by PCR amplification of cDNA pools. This may be due to the reduced stability of some mutant mRNAs and/or the absence of binding sites for primers to amplify the ORF following unintended deletions. The deletions in nanog maternal transcripts were further confirmed by directly sequencing their corresponding PCR products (Fig. 3C). Except for deletions spanning the sgRNA targeting sites, we also frequently detected truncated transcripts with large deletions that extended several hundred base pairs downstream of the sgRNA3 site (Fig. 3C; bands 4-2, 5, 7, 8-1, 9-2, 12-2, 14, 15, and 16). This deletion-prone phenomenon was lastly confirmed by directly examining genomic DNA in other Mnanog mutant embryos, where both sgRNA site–spanning and unusual large deletions were detected on the genome (Fig. 3, D and E). In an extreme situation, we observed a deletion of 3.8 kb extending from the 3′ region of exon 1 to the beginning of exon 4, whereas there is only 1.49 kb between the two extreme sgRNA targeting sites (Fig. 3E; embryo #11). We next examined Nanog protein expression in 3-hpf embryos by Western blot analysis. A clear band with molecular weight lower than 60 kDa was detected in a wild-type sample, while it was absent in samples from verified Mnanog embryos (Fig. 3F). Consistently, Western blot analysis following injection of myc-tagged expression libraries constructed from two of these Mnanog mutant embryos into wild-type embryos failed to detect the presence of any protein product (Fig. 3G).
This deletion bias of genome editing with multiple Cas9 ribonucloproteins (RNPs) in the oocyte was further confirmed by sequencing individual clones (n = 57) from the two mutant libraries. We detected four different large deletions in a large proportion of clones, as well as other indels, but not wild-type nanog transcripts (fig. S3). The deletion of a large portion of nanog transcripts could also be confirmed by ISH using a probe encompassing the two extreme sgRNA sites. We found that one-third of GFP-positive embryos (n = 45) were devoid of hybridization signal in a representative experiment, whereas all wild-type embryos were strongly positive (Fig. 3, H to J). After genotyping using collected cells at 3 hpf, ISH analysis of all verified Mnanog mutant donor embryos (n = 11) clearly showed an absence of staining (Fig. 3K). Thus, ISH experiments could help identify maternal mutant embryos or even mutant oocytes induced by multiple sgRNAs.
Because possible off-target sequences of the three sgRNAs are not present elsewhere in the genomic region flanked by F2 and R2 PCR primers (Fig. 3C), it is unlikely that these unintended large deletions identified in Mnanog mutant embryos are produced because of off-target effects. This was confirmed by injecting the three Cas9 RNPs into fertilized wild-type eggs. At 24 hpf, we sequenced different genomic regions covered by the large deletion (fig. S4A, numbers 1 to 11). Genome editing activity was only detected in the vicinity of sgRNA sites but not at those loci distantly located from different sgRNA target sites (fig. S4B). Hence, these data revealed an apparent feature of the genome editing in zebrafish primary oocytes characterized by the frequent occurrence of on-target site-spanning and unintended large deletions.
Maternal Cas9 RNPs fail to edit zygotic wild-type nanog allele
As maternally inherited Cas9 RNPs are still possible to mutate the paternal wild-type allele, new mutations may continue to be generated, which will complicate the analysis of maternal phenotypes. We thus tried to test the editing activity of maternal Cas9 RNPs on the wild-type allele from the sperm. To discriminate paternally originated DNA from the maternally inherited allele, we set to analyze Mnanog embryos with maternally inherited deletions that prevent the amplification of regions flanking each sgRNA target due to the absence of primer binding sites, so that only the paternal genome can be examined. To identify deletion-bearing Mnanog embryos, we crossed mutation-carrying F0 female with wild-type male and extracted genomic DNAs from individual Mnanog embryos at 10 hpf, a critical time just before massive embryonic death. By PCR amplification using primers F5 and R3 that should not allow the detection of PCR products in wild-type embryos or mutants carrying small indels due to their large distance on the genome (fig. S5A), we found that 88.9% of Mnanog embryos (n = 9) had on-target large deletions between the two extreme sgRNA sites (fig. S5B).
The site-spanning deletion may result from genome editing activity after fertilization, and this may mislead our judgment for the presence of maternally inherited deletion alleles. To exclude this possibility, we analyzed the sequencing chromatogram data of these PCR products and found that they all showed a stringent single-peaked pattern at the joining points after site-spanning deletions (fig. S5, C and D), indicating that the deletion was homogeneous in different embryos. This is not supportive to the postfertilization origin for the large deletions, because genome editing in different embryonic cells should lead to a heterogeneous deletion pattern. In addition, quantitative PCR (qPCR) analysis using primers flanking different sgRNA sites indicated that they were likely present as haploid alleles in the eight Mnanog embryos (fig. S5E), suggesting that only maternal nanog allele should have the large deletion.
We next analyzed the sgRNA targeting region from the paternal allele. Sequence analysis of genomic regions around the three sgRNA sites showed that they were indeed wild-type, without any hint of mutation, as shown by the chromatogram data (fig. S5F). Because the wild-type allele could not be descended from the mother as those embryos all displayed maternal mutant phenotype, it must originate from the wild-type sperm. Thus, these data suggest that the expression level of maternally deposited Cas9 RNPs is not sufficient to function on paternal wild-type nanog alleles in the early embryo.
We then supplemented exogenous Cas9 protein, sgRNAs, or both to Mnanog embryos with maternally inherited large deletions and compared the mutation rates occurring on the paternal nanog allele at 10 hpf. Moderate mutation rates ranging from 14 to 29% were observed at all three sgRNA targets following injection of Cas9 protein alone or synthetic sgRNAs alone (fig. S6). These mutation rates were not saturated because they were significantly lower than the editing efficiency observed after coinjecting Cas9 protein and synthetic sgRNAs at comparable doses (fig. S6). These observations suggest that maternal nanog allele should have been mutated in the primary oocytes within a confined period when Cas9 protein and sgRNA expression is high enough to reach a threshold.
Generation of nanog MZ mutants
As defective embryos obtained by outcrossing mosaic F0 transgenic fish with wild-type males were indeed maternal mutants of nanog, we next tried to obtain MZ mutant embryos (MZnanog) through two approaches. First, we crossed a mutation-producing female F0 fish with a nanog zygotic homozygous mutant male identified during the study to generate MZnanog in their offspring (fig. S7A). As expected, the resulting genuine MZnanog embryos showed epiboly retardation, widespread apoptosis, and disrupted pattern formation (fig. S7A). Alternatively, MZnanog mutants could also be obtained by injecting the same set of three Cas9 RNPs into GFP-positive embryos derived from outcross between mosaic F0 transgenic fish and wild-type male, which eliminates the remaining wild-type allele. This strategy is particularly time-saving when adult heterozygous or homozygous mutant male fish are not available. We first expressed three nanog Cas9 RNPs in wild-type embryos as a control and did not observe any toxic effect as shown by the normal development of injected embryos (fig. S7, B and C). After injection of these Cas9 RNPs in Mnanog embryos, we examined nanog transcripts at 10 hpf by sequencing individual clones prepared from RT-PCR products. As expected, we detected five wild-type sequences among 34 clones in uninjected Mnanog embryos (fig. S7D). However, in injected Mnanog embryos, all clones contained frameshift mutations or large deletions, suggesting a near-complete removal of wild-type nanog allele in these injected embryos (fig. S7E). This was further supported by Western blot analysis because we failed to observe any zygotic wild-type Nanog protein at the shield stage after injecting three Cas9 RNPs into Mnanog embryos (fig. S7F). Conversely, we did observe wild-type zygotic Nanog protein in uninjected Mnanog embryos at the shield stage (fig. S7F). Phenotypic analyses indicated that MZnanog mutants generated by these two approaches exhibited similar developmental defects as Mnanog embryos (fig. S8).
Rapid generation of ctnnb2 maternal mutants with large deletions
To further test the performance of this oocyte-specific conditional knockout strategy, we designed three highly efficient sgRNAs to simultaneously target ctnnb2 (β-catenin2) gene in exons 3, 4, and 9 (Fig. 4S and fig. S9). Because ctnnb2 is required for activation of maternal Wnt signaling and is essential for the formation of the dorsal organizer, the disruption of its maternal expression leads to a ventralized phenotype (42, 43). After plasmid construction and transgenesis as described above, two female fish were found to produce GFP-positive offspring (20% germline transmission rate, n = 10). Examination of their phenotypes indicated that an average of 25.6% GFP-positive embryos displayed various degrees of ventralization at 24 hpf (Fig. 4, A to K). There were no such axis defective phenotypes in embryos only expressing ctnnb2 sgRNAs and GFP (Fig. 4K and fig. S2). When these ventralized embryos were grouped into four categories (V1 to V4) as described (44, 45), we found that 59.8% of them displayed the most severely affected phenotype (V4) with the complete absence of dorsal axis (Fig. 4K). This proportion is much higher than the 34% of V4 phenotype reported for ichabod mutants that almost completely lack ctnnb2 transcription (42).
Fig. 4. Generation of ctnnb2 maternal mutants and large deletions by transgenic expression of multiple Cas9 RNPs.
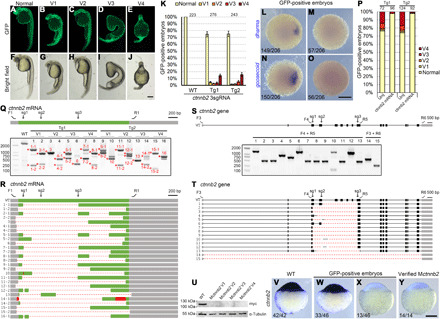
(A to J) Phenotypes of GFP-positive embryos simultaneously expressing three ctnnb2 sgRNAs. (K) Statistical analysis of ventralized phenotypes in GFP-positive offspring from two mutation-carrying F0 fish. Numbers designate total embryos analyzed. Error bars indicate SDs from three independent spawnings. (L to O) Absence of dharma and goosecoid expression in GFP-positive Mctnnb2 embryos. (P) The ventralized phenotype in GFP-positive embryos can be rescued by overexpressing the wild-type ctnnb2-myc mRNA. Numbers on top represent total embryos scored. Tg1 and Tg2 are two independent mutation-carrying F0 fish. (Q) RT-PCR analysis of the CDS from 16 Mctnnb2 mutant embryos. Wild-type embryos served as a control. Deletion alleles were present in all mutant embryos. V1 to V4 indicate degrees of ventralized phenotypes, as shown in (A) to (J). Asterisks and numbers designate PCR products subjected to Sanger sequencing. F1 and R1 are primers to amplify the CDS of ctnnb2 transcripts. The positions of the three sgRNA sites are indicated as sg1 to sg3. Grey boxes indicate UTRs, the green box represents the CDS region, dashed lines represent deletions, and red boxes indicate insertions. (R) Sequencing results of PCR products show extensive deletion events. (S) Analysis of deletion events in the genome of Mctnnb2 mutant embryos using indicated primers. Gray boxes indicate UTRs, and black boxes represent coding regions. Primer positions and sgRNA targeting sites are indicated on the wild-type allele. (T) Sequencing results show different deletion patterns at the ctnnb2 locus. (U) No Ctnnb2 protein product was produced after injecting expression libraries from Mctnnb2 embryos with V1-V4 phenotypes. (V to Y) ISH was used to examine the absence of ctnnb2 transcripts in Mctnnb2 mutants among GFP-positive embryos at the sphere stage. Scale bars, 250 μm.
We further verified the ventralized phenotype by analyzing the expression of dharma and goosecoid, two organizer genes directly controlled by maternal Wnt signaling. The result showed a complete loss of dharma and goosecoid expression in 27.7 and 27.2% of GFP-positive embryos at the dome stage, respectively (Fig. 4, L to O). The ventralized phenotype can also be efficiently rescued by injection of wild-type ctnnb2-myc mRNA (200 pg per embryo) into GFP-positive embryos at 1-cell stage (Fig. 4P). All rescued embryos displayed wild-type phenotype but were not dorsalized. This is probably because overexpression of wild-type β-catenin at such a dose was not sufficient to cause excess activation of maternal Wnt signaling. Extensive dorsalization only occurred after injecting a constitutively active form of Ctnnb1 (fig. S10). Together, these results demonstrate that ctnnb2 maternal mutant embryos can be efficiently created at an acceptable efficiency through transgenic oocyte expression of CRISPR-Cas9 and multiple sgRNAs.
We next performed RT-PCR analysis of ctnnb2 coding region in 16 maternal mutant embryos with different degrees of ventralization by using isolated cells. Unintended large deletions were detected in all Mctnnb2 mutants (Fig. 4, Q and R). Up to this point, 100% F0 founders carrying mutations either in nanog or ctnnb2 gene gave rise to embryos with large genomic deletions. In half of these Mctnnb2 embryos, there was even a near complete removal of ctnnb2 coding region, as revealed in PCR products 1-2, 2-2, 4-2, 6, 8-2, 11-2, 14-2, and 15-2 (Fig. 4R). This extremely high frequency of inducing deletion alleles was also directly confirmed by analyzing genomic sequences in ventralized mutant embryos (Fig. 4, S and T). In addition to sgRNA site–spanning deletions, we also detected deletions of huge DNA fragment extending distantly from the sgRNA targeting sites. Notably, the deleted regions covered more than 20 kb, whereas there was only 4.2 kb between the two extreme sgRNA sites (Fig. 4T; embryos #14 and #15). Similar to the specific editing of nanog locus, large deletions resulted from oocyte-specific conditional knockout of ctnnb2 were not a consequence of off-target effects, because injection of ctnnb2 Cas9 RNPs into fertilized wild-type eggs did not lead to editing of genomic regions distantly located from the sgRNA target sites (fig. S11).
We then constructed ctnnb2 mini expression libraries from four ventralized embryos with V1 to V4 phenotypes by using a small group of cells. Sequencing of individual clones did not detect the presence of wild-type transcripts (fig. S12). Consistently, injecting these libraries into wild-type embryos produced no protein product (Fig. 4U), suggesting a complete loss of maternal ctnnb2 products in either mildly or severely ventralized embryos. Like Mnanog mutants generated by multiple sgRNAs, Mctnnb2 mutants could also be identified among GFP-positive embryos at the sphere stage by ISH using a probe hybridizing the ctnnb2 coding region between the two extreme sgRNA sites (Fig. 4, V to Y).
Efficient inheritance of large deletions generated by oocyte-specific genome editing
To further demonstrate the high efficiency in generating heritable large-deletion alleles by oocyte transgenic expression of CRISPR-Cas9 and multiple sgRNAs, we compared it with the conventional method by injecting Cas9 protein and sgRNA mixture targeting nanog into fertilized wild-type eggs. Injected embryos were raised to adulthood, which were outcrossed for further analysis. Although all founders (n = 17) produced mutant offspring, only 17.6% of them gave rise to descendants with large deletions (fig. S13A). We also tested the ctnnb2 locus and found that only 18.8% of mutant carriers (n = 16) produced embryos with large deletion alleles (fig. S13A). Therefore, the germline transmission rate of large deletions at nanog and ctnnb2 loci by the conventional method is far lower than the 100% transmission ratio (fig. S13A) observed in oocyte-specific expression of multiple Cas9 RNPs.
Moreover, founders generated by the conventional method only produced a tiny proportion (9.7 to 12.4%) of offspring bearing large deletions among mutant embryos, much less than the percentage (92.3 to 94.1%) following this oocyte-specific conditional knockout (fig. S13B). Sequence analysis indicated that all deletions induced by injection of Cas9 RNPs were sgRNA site spanning, and we never detected unusual larger truncations as observed in the oocyte-specific genome editing via multiple Cas9 RNPs (fig. S13, C to F). These apparent differences clearly illustrate the high efficiency in generating genomic deletions in developing oocytes using transgenic expression of CRISPR-Cas9 and multiple sgRNAs, at least for the two loci tested here.
We next examined the transmission of large deletions in the following generations. The lethal phenotype of Mnanog mutants was first rescued by injecting wild-type nanog-myc mRNA (150 pg per embryo) into GFP-positive offspring (Fig. 5, A and B). Injected embryos were genotyped by RT-PCR analysis performed on a group of cells isolated from the blastodisc at 3 hpf, using primers that specifically bind to the 5′ and 3′ untranslated regions (UTRs) of endogenous nanog transcripts but do not recognize synthetic nanog mRNAs (Fig. 5A and table S1). In a representative experiment, we found that 46.7% (14 of 30) of GFP-positive embryos were rescued Mnanog mutants that displayed different patterns of deletions in nanog transcripts (Fig. 5, C and D). They were raised to sexual maturation and genotyped by PCR amplification of tail fin genomic DNA using different primer pairs to detect wild-type and small indel alleles (F5 and R7) or large deletions (other primer combinations), respectively (Fig. 5E). The results showed that 92.9% of Mnanog fish (13 of 14) had large deletions on the nanog locus (Fig. 5F). These deletions were transmitted to the offspring when crossed with wild-type fish (Fig. 5G). In particular, unintended large deletions that extended distantly from the extreme sgRNA site were detected in 21.4% (3 of 14) of rescued Mnanog adults (#2, #11, and #14) and were faithfully recovered in descendant embryos (Fig. 5H). These data demonstrate that the genomic deletions generated by oocyte-specific genome editing can be inherited efficiently. Thus, this important deletion-biased trend can be potentially applied to many research purposes such as maternal gene knockouts and functional studies of noncoding regions on the genome.
Fig. 5. Highly efficient inheritance of large deletions.
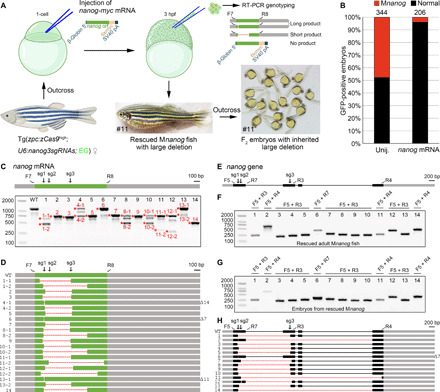
(A) Diagram demonstrates the rescue of Mnanog lethal phenotype by injection of wild-type nanog-myc mRNA, identification of rescued maternal mutants, and the resulted deletion-containing F1 fish and F2 embryos. A pair of primers was designed to specifically amplify the CDS of endogenous nanog mRNA. The rescued F1 adult female fish shown in the diagram carries an inherited unintended large deletion in one allele. Thus, F2 offspring derived from outcross with a wild-type fish developed normally. (B) Stacked bars show the efficient rescue of Mnanog lethal phenotype by injecting 150 pg per embryo nanog-myc mRNA. (C) RT-PCR analysis of nanog coding region in 14 rescued Mnanog embryos. Asterisks and numbers designate bands subjected to Sanger sequencing. In the schema of nanog mRNA, gray boxes indicate UTRs, and the green box represents the CDS region. F7 and R8 represent the primer pair used for amplification of nanog ORF. The three sgRNA target sites are marked by sg1, sg2, and sg3. (D) Sequencing results of PCR products as indicated in (C). Dashed lines represent deletions. Numbers of deleted nucleotides in small indels are indicated on the right. (E) Schema of nanog gene with sgRNA target sites and genotyping primers indicated. Gray boxes indicate UTRs, and black boxes represent coding regions. (F) PCR analysis of deletions in 14 rescued Mnanog mutant adults. Primer pairs are indicated on the top of each lane and their positions can be found in (E). (G) Similar patterns of genomic deletions in offspring derived from outcrosses of 14 rescued F1 Mnanog mutant adult fish with wild-type fish. (H) Sequence analyses of PCR products from 14 rescued F1 Mnanog adults. Their outcrossed offspring displayed identical deletions in the genome. Photo credit: Chong Zhang, Shandong University.
DISCUSSION
We have developed a rapid and feasible approach to inactivating maternal gene functions in the developing oocytes by transgenic expression of multiple Cas9 RNPs. As a proof of principle, we successfully created Mnanog and Mctnnb2 mutants with high efficiency. Moreover, we observed an apparent bias to generate large deletion alleles in the primary oocytes by this approach. Thus, these data highlight the effectiveness and rapidness of oocyte-specific multiple Cas9 RNPs for the functional study of maternal factors and illustrate the potential of this method for generating deletions in the genome.
Two aspects critically hamper the generation of maternal mutants in zebrafish by the traditional approach. First, obtaining maternal mutants is time-consuming, which generally requires crossing and screening for three generations. Second, the occurrence of zygotic lethal or infertile phenotype represents an obstacle in obtaining the next generation. Much effort has been made to circumvent zygotic mutant lethality, such as germline replacement, oocyte microinjection in situ, mosaic, and BACK approaches (7, 9–14). To some extent, these methods have contributed to the understanding of maternal gene functions. However, they are either technically demanding, time-consuming, or less efficient.
The method presented here has several advantages. First, it is much simple and particularly labor-saving. To target a gene of interest, one only has to construct an sgRNA expression vector via a well-established Golden Gate method followed by standard microinjection. In particular, only one generation of fish rearing is required to generate maternal mutant embryos, reducing the time by at least half compared with conventional methods and germline replacement. Second, the efficiency in obtaining maternal mutants is satisfactory. By introducing three sgRNAs, we could expect an average maternal mutation rate of more than 25% in GFP-positive F1 offspring. For some F0 females, the percentage of maternal mutant embryos can even exceed 50%. Third, F0 female fish can be used repeatedly in their life span, although they may produce maternal mutants with a fluctuation of 10% between different spawnings. Fourth, the present method ensures a more efficient removal of maternal products from targeted genes. This efficient removal probably results from the precocious expression of cas9 driven by the zpc promoter, which starts in stage I oocytes. Thus, targeted genes could be inactivated early enough to prevent an eventual production of trace amounts of corresponding proteins. In support of this possibility, we found that Mctnnb2 mutants generated by this method presented a more severely ventralized phenotype than the ichabod mutants, which may not be utterly devoid of ctnnb2 maternal products (43). Last, but not least, maternal functions of certain genes, such as piwil1, vasa, and dazl, can only be studied by the present oocyte-specific conditional knockout strategy, because zygotic mutation of these genes leads to germ cell death before the diplotene stage (46–48). As a result, even using germline replacement, transplanted germ cells carrying homozygous mutation for these genes are still unable to get rid of premature death. In this situation, the oocyte-specific conditional knockout strategy may be the only possible way to circumvent this difficulty. Therefore, it will be interesting to obtain maternal mutants of these well-known germ plasm components by the method presented here in a future study.
We found a prominent trend in generating large deletions by oocyte expression of multiple Cas9 RNPs. For nanog and ctnnb2 genes examined here, 100% of F0 fish with germline mutations presented large deletions (over 1 kb in the genome). By comparison, large deletions generated by conventional methods, for example, injection of Cas9 RNPs to fertilized eggs or early embryos, are notably less efficient, with germline transmission rates varied from 7.1 to 31.3%, according to published data (49–53). Consistently, only 17.6 to 18.8% of deletion transmission rates were observed by the routine method using the same Cas9 RNPs targeting nanog and ctnnb2 in this study. Moreover, deletion-harboring embryos among mutant siblings are scarce by Cas9 RNPs injection into wild-type embryos. Data from this work and a recent publication (54) showed that only 3.2 to 12.4% F1 offspring contained large deletions, whereas more than 90% ratio was observed in Mnanog embryos after the oocyte-specific conditional knockout method presented in this study. We also rescued the lethal phenotype of Mnanog embryos and successfully recovered the large deletions in F1 adults and F2 embryos following the oocyte-specific genome editing strategy. Hence, these results highlight the strong potential of multiple Cas9 RNPs for genome editing in primary oocytes to efficiently generate heritable deletion alleles. This transgenic method should have a number of important applications. It could create deletions in the promoters, which should prevent potential genetic compensations that hamper the study of gene functions (55, 56). It may also be helpful to remove transcriptional or posttranscriptional elements from the genome. Therefore, it will be interesting to further evaluate the performance of oocyte-specific multiple Cas9 RNPs in generating even larger deletions covering one or multiple gene loci. An improvement of this system should substantially enhance the tool kit of functional genomics.
The mechanisms underlying this deletion-prone tendency are not clear and need further investigation. The prolonged action of Cas9, the disparate chromatin status, and a distinct DNA repair mechanism for double-strand breaks (DSBs) in early-stage oocytes might collectively contribute to the occurrence of site-spanning and those unusual large deletions. In particular, homologous recombination (HR) DNA repair was reported to dominate in meiosis I (57), while nonhomologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ) are more active in primordial germ cells (PGCs) and somatic cells. As a result, DSBs generated in the oocytes might need substantially longer time to be repaired by NHEJ or MMEJ, which may help increase the occurrence of sgRNA site–spanning deletions. However, the unintended large deletions extending well beyond the sgRNA targeting sites observed in this study probably rely on a vigorous exonuclease activity, which does not present in NHEJ and MMEJ mechanisms but does play critical roles in the resection process of HR (58). It is thus conceivable that multiple Cas9 RNPs might disrupt the normal HR process. They might introduce additional DSBs on the repair template or newly synthesized DNA strands during strand invasion and extension, leading to an abortive HR. As a result, the single DNA strand generated during the resection process of HR may be trimmed, and DNA repair might switch to other mechanisms such as MMEJ to generate unintended and complicated patterns of deletion alleles (59). These unintended genomic alterations, termed on-target effects or OnTEs, were demonstrated to happen frequently in embryonic stem cells, induced pluripotent stem cells, and early embryos of mouse and human origins (59–65). In zebrafish, however, unintended on-target effects were only extensively observed when performing oocyte-specific genome editing but seldom happened in early embryos injected with Cas9 RNPs. Thus, it is very likely that DNA repair mechanisms used to heal CRISPR-Cas9–induced DSBs by zebrafish oocytes may be similar to that in mammalian early embryos but are somewhat different from somatic cells and PGCs in zebrafish embryos.
It should be mentioned that in our oocyte-specific conditional knockout method, transgenic zcas9 expression may vary among female fish. We found that the expression level of zcas9 in some Tg(zpc:zcas9) fish was too low to generate mutations in the oocytes or offspring. However, the use of homozygous Tg(zpc:zcas9) females could ensure the efficiency and the reproducibility of genome editing in oocytes. In general, Tg(zpc:zcas9) adult female fish with sufficient genome editing efficiency can be identified relatively easily. For example, in our previous study, we have injected bmp2b sgRNA in the embryos and examined the proportion of dorsalized phenotype (34). Another critical quality control step should be set up before phenotyping GFP-positive embryos. The presence of Cas9 activity could be confirmed by testing the efficiency of bmp2b sgRNA in GFP-negative siblings. These measures should guarantee the success of maternal knockout experiments. For versatile applications of this method, further efforts should be made to improve the efficiency of oocyte-specific multiple Cas9 RNPs in genome editing. It has been shown that injection of high amounts of Cas9 protein and multiple in vitro synthesized sgRNAs targeting a single gene in the embryos was efficient to disrupt most of the wild-type alleles and produced phenotypes as observed in homozygous zygotic mutants (35, 53). Thus, there is a possibility that suboptimal expression of Cas9 protein or sgRNAs in transgenic lines may limit the efficiency of oocyte conditional knockout. Our data clearly showed that the expression level of maternally inherited Cas9 RNPs was below a threshold for efficient mutagenesis on the wild-type allele from the sperm, demonstrating that the method in its present form is highly specific for knockout of maternal factors without affecting zygotic gene products. In another aspect, however, it also highlights a promising potential to increase the efficiency of this and other conditional knockout systems using Cas9 RNPs by improving the expression level of Cas9 protein and sgRNAs. It is possible to increase cas9 transcription via either gal4-UAS or Suntag system (31, 66). At the same time, modification of the cas9 sequence may help enhance its translation efficiency. Alternatively, sgRNA expression could be improved by strong conventional promoters followed by maturation via Csy4, ribozyme, or transfer RNA processing system (67–69). These procedures could likely increase the efficiency in generating maternal mutants for single or even multiple genes.
In summary, our work provides evidence that maternal gene inactivation can be achieved by oocyte transgenic expression of Cas9 and sgRNAs in an important time-saving and deletion-prone manner. The accessibility of this approach should open a new avenue for large-scale functional screening of maternal factors in zebrafish.
MATERIALS AND METHODS
Zebrafish husbandry and ethic statement
Wild-type and Tg(zpc:zcas9) fish were raised at 28.5°C in standard housing systems (Haisheng). Offspring from Tg(zpc:zcas9) fish were screened for genome editing efficiency by injecting bmp2b sgRNA followed by analyzing the dorsalized phenotype at 11 hpf (34). Only female fish with more than 50% of dorsalized rate in their offspring were kept for breeding and maternal knockout experiments. All experiments were designed and performed following the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines issued by the Ethics Committee for Animal Research of Life Science of Shandong University (permit number SYDWLL-2018-05).
Design and efficiency of sgRNAs
The sgRNAs for nanog and ctnnb2 were designed via online software CRISPRscan (40) and listed in table S1. Their efficiency was tested before the construction of sgRNA expression vectors. DNA templates for sgRNAs were synthesized by fill-in PCR. After in vitro transcription, the purified sgRNA (150 pg) was injected along with zebrafish codon–optimized cas9 mRNA (200 pg) into 1-cell stage embryos. At 10 hpf, injected embryos were lysed in 50 mM NaOH for 20 min at 95°C, followed by neutralization using 1/10 volume of 1 M tris-HCl (pH 7.5). The DNA fragment containing the sgRNA targeting site was amplified by PCR from the lysate and subjected to Sanger sequencing. Sequencing chromatogram files were uploaded to https://ice.synthego.com/#/ to quantify the efficiency of the sgRNA.
Construction of plasmids for transgenic expression of sgRNAs
pU6x:sgRNA plasmids were constructed as previously described (18). Briefly, primers containing sense and antisense sequences of sgRNA protospacer were synthesized and annealed. The sequence of sense primer is TTC plus G(N)19, where G(N)19 represents the protospacer, while the sequence of antisense primer is AAAC plus reverse-complement sequence of (N)19. Sense and antisense primers were diluted to 100 μM and mixed as follows: 1 μl of sense primer, 1 μl of antisense primer, 2 μl of 10x buffer 2.1 from New England Biolabs (NEB), and 16 μl of H2O. The mixture was incubated in a PCR thermocycler with the following program: 95°C for 15 min, slow rampling to 50°C at 0.1°C/s, 50°C for 10 min, and cooling quickly to 4°C. The annealed primers (1 μl) were then mixed in a 10-μl system with 100 ng of pU6x:sgRNA empty plasmid (pU6a:sgRNA#1, pU6a:sgRNA#2, or pU6b:sgRNA#3), 1 μl of 10× CutSmart buffer, 1 μl of 10× T4 ligase buffer, 0.3 μl of T4 ligase, 0.3 μl of BsmBI, 0.2 μl of PstI, and 0.2 μl of SalI. They were incubated in a PCR thermocycler using the following program: 37°C for 20 min and 16°C for 15 min for 6 cycles, 37°C for 10 min, 55°C for 15 min, and 80°C for 15 min. The ligation products were then used to transform DH5α Escherichia coli. After spreading on Spec+ plates, positive clones were identified by PCR using the M13 forward and the protospacer antisense primers. The corresponding plasmids were extracted for further construction of the final transgenic vector.
Golden Gate assembly was used to ligate single or three sgRNA expression cassettes into the pGGDestISceIEG-1sgRNA or pGGDestISceIEG-3sgRNA backbone plasmid. Here, we take the cloning of three sgRNAs as an example. The pU6a:sgRNA#1, pU6a:sgRNA#2, and pU6b:sgRNA#3 plasmids (100 ng each) were mixed with 2 μl of 10× CutSmart buffer, 2 μl of 10× T4 ligase buffer, 50 ng of empty pGGDestISceIEG-3sgRNA, 1 μl of T4 DNA ligase, and 1 μl of BsaI in a 20-μl reaction mixture. It is incubated using the following PCR program: 37°C for 20 min and 16°C for 15 min for 3 cycles and then 80°C for 15 min. The ligation products were transformed to DH5α E. coli and plated on Amp+ LB medium. Positive clones were identified by colony PCR using primers: 5′-TTCTTGTTTAAGCTTTTAATCTCAAAAAAC-3′ and 5′-GGCTGTTTACATCTGATAGTGG-3′. The annealing primers for nanog and ctnnb2 sgRNAs are listed in table S1. pGGDestISceIEG-1sgRNA plasmid was constructed similarly. The only difference lies in the ligation mixture components, where only 100 ng of pU6a:sgRNA#1 was added, and the pGGDestISceIEG-3sgRNA should be replaced with pGGDestISceIEG-1sgRNA.
I-SceI–mediated transgenesis
A mixture (1 to 2 nl) containing pGGDestISceIEG-1sgRNA or pGGDestISceIEG-3sgRNA plasmid (10 ng/μl), I-SceI (1 U/μl; NEB), and 0.5× CutSmart buffer (NEB) was injected into the blastodisc of 1-cell stage embryos (36). At 4 dpf, the larva showing strong and extensive green fluorescence, reflecting early integration event, were picked up and raised to adulthood. The overall germline transmission rate of I-SceI–mediated transgenesis may depend on whether an early integration event occurs in the embryos. As shown in the present study, the germline transmission rates varied from 12 to 40%. If only embryos with widespread GFP expression at 4 dpf are selected, then the germline transmission rate will be normally higher than 50%. Typically, injection of 200 embryos will produce about 10 to 20 embryos with early integration and will be sufficient to screen for maternal mutant–carrying founders.
Quality control before screening of maternal mutants
GFP-negative sibling embryos from transgenic F0 female founders were injected with 100 pg of bmp2b sgRNA. The phenotype of GFP-positive embryos was further analyzed only when their GFP-negative counterparts were efficiently dorsalized.
Rescue of mutant phenotypes by wild-type mRNAs
The nanog CDS was amplified and inserted into the BamHI and ClaI sites of the pCS2-MT vector using T4 DNA ligase. The ctnnb2 CDS was inserted into the BamHI site of the pCS2-MT vector by Gibson assembly. We also constructed mouse ΔN-ctnnb1-pCS2 for comparison of dorsalizing effects. PCR primers are listed in table S1. Synthetic mRNAs were in vitro transcribed using the mMESSAGE mMACHINE SP6 Kit (Invitrogen, AM1340). For rescue, 200 pg of nanog-myc or ctnnb2-myc mRNA was injected into 1-cell stage GFP-positive embryos. Their phenotypes were analyzed at the bud stage (Mnanog) or 24 hpf (Mctnnb2).
Cell isolation, RNA extraction, and cDNA synthesis
To check the genotype of maternal mRNAs, we aspirated about 30 to 40 cells from 1 k-cell stage embryos using a pneumatic microinjector (12). They were lysed in 200 μl of TRIzol, and total RNAs were extracted by adding 40 μl of chloroform and precipitated by isopropanol. To facilitate RNA precipitation, 1 μl of glycogen solution (20 mg/ml) was added to each sample. Then, cDNAs were synthesized using the First-Strand cDNA Synthesis kit (Transgene, AT301) and subjected to PCR amplification using primers listed in table S1.
Gel analysis and sequencing
The CDS in genes of interest were amplified by PCR from embryos with defective phenotypes and subjected to agarose gel electrophoresis. DNA fragments of different sizes were sequenced and aligned with the wild-type sequence using SnapGene software.
Library construction and single-clone sequencing
CDS amplicons from intact embryos or isolated blastomeres were ligated into pCS2-MT vector in-frame with the six myc epitopes. The reverse primer used for library construction was designed immediately before the stop codon. A 15-bp 5′-homology arm was designed for Gibson ligation and ensures the in-frame connection between the ORF of the wild-type mRNA and the 6x myc epitope sequence in the pCS2-MT vector. Following transformation in E. coli, individual clones were subjected to Sanger sequencing. The primers used for constructing the library were listed in table S1.
Western blot
The plasmid library (30 to 40 pg) was injected into 1-cell stage embryos. At 12 hpf, at least 30 injected embryos were dechorionated, and the yolk was removed. Cell pellets were then lysed in the lysis buffer [100 mM NaCl, 0.5% NP-40, 5 mM EDTA, and 10 mM tris-HCl (pH 7.5)] containing a cocktail of protease inhibitors and boiled in Laemmli sample buffer. Protein sample were subjected to polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and probed with anti-Myc (1:1000; Abcam, ab32) or anti-α-Tubulin (1:1000; GeneTex, GTX124303) antibodies. Following a similar procedure for yolk removal and protein extraction, we also used anti-Nanog (1:500; ZDB-ATB-191003-3, GTX132491, GeneTex) to detect endogenous Nanog protein, where anti–β-actin (1:5000; 66009-1-Ig, Proteintech) served as a loading control.
Whole-mount immunofluorescence staining
Embryos were fixed at 3 hpf in 4% paraformaldehyde. Antigen retrieval was performed by treating embryos in 0.1 M tris-HCl (pH 9.5) for 15 min at 70°C. The embryos were then subjected to immunofluorescence staining with anti-Nanog (1:500) and counterstained with Hoechst 33342 (Cell Signaling Technology, 4082S) as described (70). Images were acquired using a spinning disc confocal microscope (Olympus SpinSR10) with its Z-stack function.
RNA probe design and ISH
ISH was performed as previously described (45). Probes for dharma and goosecoid were reported previously (45). The coding regions for sox17 and mxtx2 and cDNA sequences within the two extreme sgRNA targets for nanog and ctnnb2 were amplified by PCR and cloned in pZeroback vector. Digoxigenin-labeled probes were in vitro–transcribed using T7 or SP6 RNA polymerase in the presence of Dig-labeling mix (Roche).
Quantitative PCR
Wild-type embryos and Mnanog mutants were lysed at 10 hpf, and their genomic DNAs were extracted. qPCR was performed using 2× M5 HiPer SYBR Premix (MF787-01, Mei5) on a Q1000 Real-Time PCR System (LongGene). Primers for amplifying nanog sgRNA target sites and dmd genomic regions are listed in table S1. The 2-ΔΔCt method was used to estimate ploidy at these loci. The average copy number of these amplicons in wild-type embryos was normalized as two.
Acknowledgments
We thank B. Zhang and W. Chen for providing zcas9 and pU6x:sgRNA plasmids. We thank H. Yu, X. Zhao, C. Liu, and S. Wang from the State Key Laboratory of Microbial Technology for assistance with confocal imaging. Funding: This work was supported by the National Natural Science Foundation of China (grant numbers 31871451 and 32070813), the National Key R&D Program of China (grant number 2018YFA0801000), and the Program of Shandong University Qilu Young Scholars. Author contributions: C.Z. and M.S. conceived the idea. C.Z., T.L., Y.-Z.Z., J.L., I.T., F.W., A.C., J.W., Z.Z., and M.S. performed the experiments. C.Z., Y.-J.Z., D.-L.S., and M.S. analyzed and validated the data. M.S. and D.-L.S. acquired funding. M.S. supervised the work and wrote the original draft. M.S., D.-L.S., and C.Z. reviewed and edited the manuscript.Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabg4243/DC1
REFERENCES AND NOTES
- 1.White R. J., Collins J. E., Sealy I. M., Wali N., Dooley C. M., Digby Z., Stemple D. L., Murphy D. N., Billis K., Hourlier T., Füllgrabe A., Davis M. P., Enright A. J., Busch-Nentwich E. M., A high-resolution mRNA expression time course of embryonic development in zebrafish. eLife 6, e30860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuentes R., Tajer B., Kobayashi M., Pelliccia J. L., Langdon Y., Abrams E. W., Mullins M. C., The maternal coordinate system: Molecular-genetics of embryonic axis formation and patterning in the zebrafish. Curr. Top. Dev. Biol. 140, 341–389 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Yan L., Chen J., Zhu X., Sun J., Wu X., Shen W., Zhang W., Tao Q., Meng A., Maternal Huluwa dictates the embryonic body axis through β-catenin in vertebrates. Science 362, eaat1045 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Abrams E. W., Fuentes R., Marlow F. L., Kobayashi M., Zhang H., Lu S., Kapp L., Joseph S. R., Kugath A., Gupta T., Lemon V., Runke G., Amodeo A. A., Vastenhouw N. L., Mullins M. C., Molecular genetics of maternally-controlled cell divisions. PLOS Genet. 16, e1008652 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Escobar-Aguirre M., Elkouby Y. M., Mullins M. C., Localization in oogenesis of maternal regulators of embryonic development. Adv. Exp. Med. Biol. 953, 173–207 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Lindeman R. E., Pelegri F., Vertebrate maternal-effect genes: Insights into fertilization, early cleavage divisions, and germ cell determinant localization from studies in the zebrafish. Mol. Reprod. Dev. 77, 299–313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing Y. Y., Cheng X. N., Li Y. L., Zhang C., Saquet A., Liu Y. Y., Shao M., Shi D. L., Mutational analysis of dishevelled genes in zebrafish reveals distinct functions in embryonic patterning and gastrulation cell movements. PLOS Genet. 14, e1007551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gritsman K., Zhang J., Cheng S., Heckscher E., Talbot W. S., Schier A. F., The EGF-CFC protein one-eyed pinhead is essential for nodal signaling. Cell 97, 121–132 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Hino H., Nakanishi A., Seki R., Aoki T., Yamaha E., Kawahara A., Shimizu T., Hibi M., Roles of maternal wnt8a transcripts in axis formation in zebrafish. Dev. Biol. 434, 96–107 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Ciruna B., Weidinger G., Knaut H., Thisse B., Thisse C., Raz E., Schier A. F., Production of maternal-zygotic mutant zebrafish by germ-line replacement. Proc. Natl. Acad. Sci. U.S.A. 99, 14919–14924 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang F., Li X., He M., Ye D., Xiong F., Amin G., Zhu Z., Sun Y., Efficient generation of zebrafish maternal-zygotic mutants through transplantation of ectopically induced and Cas9/gRNA targeted primordial germ cells. J. Genet. Genomics 47, 37–47 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Shao M., Cheng X. N., Liu Y. Y., Li J. T., Shi D. L., Transplantation of zebrafish cells by conventional pneumatic microinjector. Zebrafish 15, 73–76 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Wu X., Shen W., Zhang B., Meng A., The genetic program of oocytes can be modified in vivo in the zebrafish ovary. J. Mol. Cell Biol. 10, 479–493 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Liu Y., Zhu Z., Ho I. H. T., Shi Y., Xie Y., Li J., Zhang Y., Chan M. T. V., Cheng C. H. K., Germline-specific dgcr8 knockout in zebrafish using a BACK approach. Cell. Mol. Life Sci. 74, 2503–2511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M., RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Y., Zhang B., Luo L., Shi D.-L., Wang H., Cui Z., Huang H., Cao Y., Shu X., Zhang W., Zhou J., Li Y., Du J., Zhao Q., Chen J., Zhong H., Zhong T. P., Li L., Xiong J.-W., Peng J., Xiao W., Zhang J., Yao J., Yin Z., Mo X., Peng G., Zhu J., Chen Y., Zhou Y., Liu D., Pan W., Zhang Y., Ruan H., Liu F., Zhu Z., Meng A.; ZAKOC Consortium , Systematic genome editing of the genes on zebrafish chromosome 1 by CRISPR/Cas9. Genome Res. 30, 118–126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin L., Maddison L. A., Li M., Kara N., LaFave M. C., Varshney G. K., Burgess S. M., Patton J. G., Chen W., Multiplex conditional mutagenesis using transgenic expression of Cas9 and sgRNAs. Genetics 200, 431–441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ablain J., Durand E. M., Yang S., Zhou Y., Zon L. I., A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev. Cell 32, 756–764 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai M., Liang D., Wang Y., Li Q., Wu Y., Li J., Spermatogenic cell-specific gene mutation in mice via CRISPR-Cas9. J. Genet. Genomics 43, 289–296 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Li M., Bui M., Yang T., Bowman C. S., White B. J., Akbari O. S., Germline Cas9 expression yields highly efficient genome engineering in a major worldwide disease vector, Aedes aegypti. Proc. Natl. Acad. Sci. U.S.A 114, E10540–E10549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X., Gays D., Milia C., Santoro M. M., Cilia control vascular mural cell recruitment in vertebrates. Cell Rep. 18, 1033–1047 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reade A., Motta-Mena L. B., Gardner K. H., Stainier D. Y., Weiner O. D., Woo S., TAEL: A zebrafish-optimized optogenetic gene expression system with fine spatial and temporal control. Development 144, 345–355 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ablain J., Xu M., Rothschild H., Jordan R. C., Mito J. K., Daniels B. H., Bell C. F., Joseph N. M., Wu H., Bastian B. C., Zon L. I., Yeh I., Human tumor genomics and zebrafish modeling identify SPRED1 loss as a driver of mucosal melanoma. Science 362, 1055–1060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou W., Cao L., Jeffries J., Zhu X., Staiger C. J., Deng Q., Neutrophil-specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. Dis. Model. Mech. 11, dmm033027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y. L., Xu C. F., Li H. J., Cao Z. T., Liu J., Wang J. L., du X. J., Yang X. Z., Gu Z., Wang J., Macrophage-specific in vivo gene editing using cationic lipid-assisted polymeric nanoparticles. ACS Nano 12, 994–1005 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Grainger S., Nguyen N., Richter J., Setayesh J., Lonquich B., Oon C. H., Wozniak J. M., Barahona R., Kamei C. N., Houston J., Carrillo-Terrazas M., Drummond I. A., Gonzalez D., Willert K., Traver D., EGFR is required for Wnt9a-Fzd9b signalling specificity in haematopoietic stem cells. Nat. Cell Biol. 21, 721–730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y. C., Wang I. J., Heat-shock-induced tyrosinase gene ablation with CRISPR in zebrafish. Mol. Genet. Genomics 295, 911–922 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Cox A. G., Tsomides A., Yimlamai D., Hwang K. L., Miesfeld J., Galli G. G., Fowl B. H., Fort M., Ma K. Y., Sullivan M. R., Hosios A. M., Snay E., Yuan M., Brown K. K., Lien E. C., Chhangawala S., Steinhauser M. L., Asara J. M., Houvras Y., Link B., Heiden M. G. V., Camargo F. D., Goessling W., Yap regulates glucose utilization and sustains nucleotide synthesis to enable organ growth. EMBO J. 37, e100294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehravar M., Shirazi A., Nazari M., Banan M., Mosaicism in CRISPR/Cas9-mediated genome editing. Dev. Biol. 445, 156–162 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Di Donato V., De Santis F., Auer T. O., Testa N., Sánchez-Iranzo H., Mercader N., Concordet J.-P., Del Bene F., 2C-Cas9: A versatile tool for clonal analysis of gene function. Genome Res. 26, 681–692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Onichtchouk D., Aduroja K., Belting H. G., Gnugge L., Driever W., Transgene driving GFP expression from the promoter of the zona pellucida gene zpc is expressed in oocytes and provides an early marker for gonad differentiation in zebrafish. Dev. Dyn. 228, 393–404 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Liu D., Wang Z., Xiao A., Zhang Y., Li W., Zu Y., Yao S., Lin S., Zhang B., Efficient gene targeting in zebrafish mediated by a zebrafish-codon-optimized cas9 and evaluation of off-targeting effect. J. Genet. Genomics 41, 43–46 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Liu Y., Zhang C., Zhang Y., Lin S., Shi D. L., Shao M., Highly efficient genome editing using oocyte-specific zcas9 transgenic zebrafish. J. Genet. Genomics 45, 509–512 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Wu R. S., Lam I. I., Clay H., Duong D. N., Deo R. C., Coughlin S. R., A rapid method for directed gene knockout for screening in G0 zebrafish. Dev. Cell 46, 112–125.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Thermes V., Grabher C., Ristoratore F., Bourrat F., Choulika A., Wittbrodt J., Joly J. S., I-Sce I meganuclease mediates highly efficient transgenesis in fish. Mech. Dev. 118, 91–98 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Gagnon J. A., Obbad K., Schier A. F., The primary role of zebrafish nanog is in extra-embryonic tissue. Development 145, dev147793 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veil M., Schaechtle M. A., Gao M., Kirner V., Buryanova L., Grethen R., Onichtchouk D., Maternal Nanog is required for zebrafish embryo architecture and for cell viability during gastrulation. Development 145, dev155366 (2018). [DOI] [PubMed] [Google Scholar]
- 39.He M., Zhang R., Jiao S., Zhang F., Ye D., Wang H., Sun Y., Nanog safeguards early embryogenesis against global activation of maternal β-catenin activity by interfering with TCF factors. PLOS Biol. 18, e3000561 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moreno-Mateos M. A., Vejnar C. E., Beaudoin J. D., Fernandez J. P., Mis E. K., Khokha M. K., Giraldez A. J., CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 12, 982–988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruce A. E., Howley C., Dixon Fox M., Ho R. K., T-box gene eomesodermin and the homeobox-containing Mix/Bix gene mtx2 regulate epiboly movements in the zebrafish. Dev. Dyn. 233, 105–114 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly C., Chin A. J., Leatherman J. L., Kozlowski D. J., Weinberg E. S., Maternally controlled (beta)-catenin-mediated signaling is required for organizer formation in the zebrafish. Development 127, 3899–3911 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Bellipanni G., Varga M., Maegawa S., Imai Y., Kelly C., Myers A. P., Chu F., Talbot W. S., Weinberg E. S., Essential and opposing roles of zebrafish β-catenins in the formation of dorsal axial structures and neurectoderm. Development 133, 1299–1309 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Kishimoto Y., Lee K. H., Zon L., Hammerschmidt M., Schulte-Merker S., The molecular nature of zebrafish swirl: BMP2 function is essential during early dorsoventral patterning. Development 124, 4457–4466 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Shao M., Wang M., Liu Y. Y., Ge Y. W., Zhang Y. J., Shi D. L., Vegetally localised Vrtn functions as a novel repressor to modulate bmp2b transcription during dorsoventral patterning in zebrafish. Development 144, 3361–3374 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Houwing S., Kamminga L. M., Berezikov E., Cronembold D., Girard A., van den Elst H., Filippov D. V., Blaser H., Raz E., Moens C. B., Plasterk R. H. A., Hannon G. J., Draper B. W., Ketting R. F., A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 129, 69–82 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Hartung O., Forbes M. M., Marlow F. L., Zebrafish vasa is required for germ-cell differentiation and maintenance. Mol. Reprod. Dev. 81, 946–961 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bertho S., Clapp M., Banisch T. U., Bandemer J., Raz E., Marlow F. L., Zebrafish dazl regulates cystogenesis and germline stem cell specification during the primordial germ cell to germline stem cell transition. Development , dev.187773 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta A., Hall V. L., Kok F. O., Shin M., McNulty J. C., Lawson N. D., Wolfe S. A., Targeted chromosomal deletions and inversions in zebrafish. Genome Res. 23, 1008–1017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim S., Wang Y., Yu X., Huang Y., Featherstone M. S., Sampath K., A simple strategy for heritable chromosomal deletions in zebrafish via the combinatorial action of targeting nucleases. Genome Biol. 14, R69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao A., Wang Z., Hu Y., Wu Y., Luo Z., Yang Z., Zu Y., Li W., Huang P., Tong X., Zhu Z., Lin S., Zhang B., Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 41, e141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varshney G. K., Pei W., LaFave M. C., Idol J., Xu L., Gallardo V., Carrington B., Bishop K., Jones M. P., Li M., Harper U., Huang S. C., Prakash A., Chen W., Sood R., Ledin J., Burgess S. M., High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 25, 1030–1042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoshijima K., Jurynec M. J., Shaw D. K., Jacobi A. M., Behlke M. A., Grunwald D. J., Highly efficient CRISPR-Cas9-based methods for generating deletion mutations and F0 embryos that lack gene function in zebrafish. Dev. Cell 51, 645–657.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim B. H., Zhang G., Generating stable knockout zebrafish lines by deleting large chromosomal fragments using multiple gRNAs. G3 10, 1029–1037 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El-Brolosy M. A., Kontarakis Z., Rossi A., Kuenne C., Günther S., Fukuda N., Kikhi K., Boezio G. L. M., Takacs C. M., Lai S.-L., Fukuda R., Gerri C., Giraldez A. J., Stainier D. Y. R., Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma Z., Zhu P., Shi H., Guo L., Zhang Q., Chen Y., Chen S., Zhang Z., Peng J., Chen J., PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Kim S., Peterson S. E., Jasin M., Keeney S., Mechanisms of germ line genome instability. Semin. Cell Dev. Biol. 54, 177–187 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Symington L. S., Gautier J., Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Owens D. D. G., Caulder A., Frontera V., Harman J. R., Allan A. J., Bucakci A., Greder L., Codner G. F., Hublitz P., McHugh P. J., Teboul L., de Bruijn M. F. T. R., Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic Acids Res. 47, 7402–7417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weisheit I., Kroeger J. A., Malik R., Klimmt J., Crusius D., Dannert A., Dichgans M., Paquet D., Detection of deleterious on-target effects after HDR-mediated CRISPR editing. Cell Rep. 31, 107689 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Shin H. Y., Wang C., Lee H. K., Yoo K. H., Zeng X., Kuhns T., Yang C. M., Mohr T., Liu C., Hennighausen L., CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 8, 15464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zuccaro M. V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R. T., Palmerola K. L., Smith M. E., Tsang S. H., Goland R., Jasin M., Lobo R., Treff N., Egli D., Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell 183, 1650–1664.e15 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Korablev A., Lukyanchikova V., Serova I., Battulin N., On-target CRISPR/Cas9 activity can cause undesigned large deletion in mouse zygotes. Int. J. Mol. Sci. 21, 3604 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adikusuma F., Piltz S., Corbett M. A., Turvey M., McColl S. R., Helbig K. J., Beard M. R., Hughes J., Pomerantz R. T., Thomas P. Q., Large deletions induced by Cas9 cleavage. Nature 560, E8–E9 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Kosicki M., Tomberg K., Bradley A., Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanenbaum M. E., Gilbert L. A., Qi L. S., Weissman J. S., Vale R. D., A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang J., Fei F., Berberoglu M. A., Sun S., Wang L., Dong Z., Wang X., Csy4-based vector system enables conditional chimeric gene editing in zebrafish without interrupting embryogenesis. J. Mol. Cell Biol. 10, 586–588 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Lee R. T., Ng A. S., Ingham P. W., Ribozyme mediated gRNA generation for in vitro and in vivo CRISPR/Cas9 mutagenesis. PLOS ONE 11, e0166020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shiraki T., Kawakami K., A tRNA-based multiplex sgRNA expression system in zebrafish and its application to generation of transgenic albino fish. Sci. Rep. 8, 13366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shao M., Lu T., Zhang C., Zhang Y. Z., Kong S. H., Shi D. L., Rbm24 controls poly(A) tail length and translation efficiency of crystallinm RNAs in the lens via cytoplasmic polyadenylation. Proc. Natl. Acad. Sci. U.S.A. 117, 7245–7254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabg4243/DC1