

Nanocapsules modify membrane interaction of polymyxin B to reduce its toxicity and enable safe systemic therapy of sepsis.
Abstract
Systemic therapy of Gram-negative sepsis remains challenging. Polymyxin B (PMB) is well suited for sepsis therapy due to the endotoxin affinity and antibacterial activity. However, the dose-limiting toxicity has limited its systemic use in sepsis patients. For safe systemic use of PMB, we have developed a nanoparticulate system, called D-TZP, which selectively reduces the toxicity to mammalian cells but retains the therapeutic activities of PMB. D-TZP consists of an iron-complexed tannic acid nanocapsule containing a vitamin D core, coated with PMB and a chitosan derivative that controls the interaction of PMB with endotoxin, bacteria, and host cells. D-TZP attenuated the membrane toxicity associated with PMB but retained the ability of PMB to inactivate endotoxin and kill Gram-negative bacteria. Upon intravenous injection, D-TZP protected animals from pre-established endotoxemia and polymicrobial sepsis, showing no systemic toxicities inherent to PMB. These results support D-TZP as a safe and effective systemic intervention of sepsis.
INTRODUCTION
Sepsis and septic shock are life-threatening conditions involving severe systemic inflammatory responses to infection. They are frequently encountered in the intensive care unit (ICU) (1), accounting for 4 to 17% of ICU admissions in high-income countries (2), 17 to 26% of hospital mortality (3), and $23.7 billion of hospital expenses (4). Sepsis is caused by the massive invasion of pathogens, which introduce pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) or lipoproteins into the system. The recognition of PAMPs by host immune systems induces the production and release of pro-inflammatory cytokines, chemokines, reactive oxygen species, and nitric oxide, mounting inflammatory responses against the pathogens. In sepsis, the inflammatory responses continue without resolution, taking the host to a vicious cycle of hyperinflammation and compensatory immunosuppression. This leads to a high risk of additional infections and often fatal consequences such as disseminated intravascular coagulation (DIC) and multiple organ dysfunctions (5).
Currently, sepsis is treated by broad-spectrum antibiotics, fluid resuscitation, and vasopressors, aiming to manage early infections and support end-organ functions (6). In addition to the standard care of sepsis, several experimental approaches have been undertaken over the years, resulting in more than 80 clinical trials since 1982 (7). These approaches concern various aspects of sepsis conditions but do not always improve patient outcomes, as evidenced by persistent sepsis mortality (7). The high mortality rate and the lack of success in recent drug development efforts point to a critical unmet need for effective medical intervention in sepsis treatment.
In searching for a new systemic treatment of sepsis, we note an existing drug, polymyxin B (PMB), which is known to address different aspects of Gram-negative sepsis but has not been actively used in patients. PMB is a cationic antimicrobial peptide and potent LPS adsorbent (8). PMB is effective against most Gram-negative bacteria due to the detrimental effect on the membrane (9). Moreover, PMB binds to LPS with high affinity and inhibits the Toll-like receptor 4 (TLR4) signaling to down-regulate acute inflammatory responses (10). PMB was shown to be effective in attenuating LPS-induced endotoxemia in mice as early as 1967 (11) and has been pursued as a potential treatment of Gram-negative sepsis ever since. Gram-negative bacteria account for >60% of severe sepsis (12); thus, PMB would have made an important arsenal for sepsis treatment. However, the nephrotoxicity and neurotoxicity of PMB (13, 14) have limited its clinical application at a dose effective for systemic therapy of sepsis (15). Several attempts have been made to develop less toxic derivatives of PMB only to lose the antibacterial activity (14).
Given the anti-LPS and antibacterial activities of PMB and the significance of Gram-negative sepsis, we aim to develop a new formulation of PMB, which enables its safe systemic use in patients with sepsis. We use nanoparticles as a carrier of PMB. Because of the large surface area relative to the volume, nanoparticles provide a platform for loading therapeutic agents on the surface to help them interact with targets in circulation (16). Moreover, nanoparticles can modify the biodistribution of payloads, thereby reducing the toxicity specific to critical organs (17). On the basis of these features, nanoparticles have been explored in sepsis treatment as a way of sequestering toxins (18), killing bacteria (19), and targeting activated neutrophils (20) or macrophages (21). In this study, we use nanoparticles for attenuating the fatal interaction of PMB with host cells while selectively presenting PMB to endotoxins and bacteria.
We have produced nanoparticles carrying PMB (D-TZP) using a self-assembly of tannic acid (TA)/Fe3+ coordination complex (T) containing vitamin D (D) as a platform and attaching PMB (P) on the surface along with low–molecular weight zwitterionic chitosan (LMZWC; Z), a succinylated chitosan derivative that can attenuate PMB’s undesirable contact with cell membrane (Fig.1A). We evaluated the safety and bioactivity of D-TZP in vitro and in two murine models of sepsis, endotoxemia and cecal ligation and puncture (CLP). The results show that D-TZP maintains antibacterial activities and affinity for LPS while reducing the damaging effects on mammalian cell membrane, thereby increasing the systemic tolerability. Administered intravenously, D-TZP protects animals challenged with pre-established endotoxemia or CLP and reduces their mortality significantly.
RESULTS
Development of D-TZP (Fig. 1)
Fig. 1. Development and characterization of D-TZP.
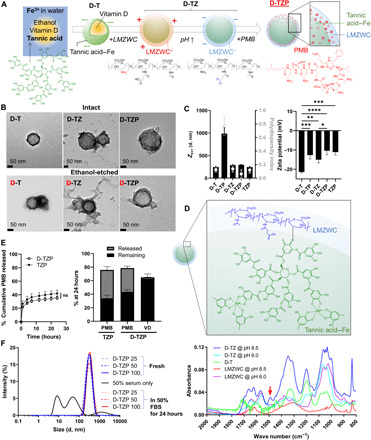
(A) Schematic of D-TZP preparation. D-TZP was formed by encapsulating vitamin D in TA and Fe3+ complexation and then coated with LMZWC and PMB sequentially. D-T, self-assembly of TA/Fe3+ coordination complex containing vitamin D; D-TZ, D-T coated with LMZWC; D-TZP, D-T coated with LMZWC and PMB. (B) Transmission electron micrographs of intact D-T, D-TZ, and D-TZP (top), and ethanol-etched D-T, D-TZ, and D-TZP (bottom). (C) z-average diameter (Zavr), polydispersity index, and zeta potential of nanoparticles at pH 7.4 (10 mM phosphate buffer). The data are shown as means ± SD (n = 3 independently and identically prepared batches). Statistical significance was accessed by Tukey’s multiple comparisons test following ordinary one-way ANOVA (*: p < 0.05; **: p < 0.01; ***: p < 0.001; and ****: p < 0.0001). (D) Schematic of LMZWC binding to TA/Fe3+ assembly (top). Fourier transform infrared spectra of LMZWC at pH 6.0, LMZWC at pH 8.5, D-T and D-TZ at pH 6.0, and D-TZ at pH 8.5 (bottom). (E) In vitro PMB release from D-TZP and TZP in 50% fetal bovine serum (FBS) at 37°C (left). Statistical significance was accessed by Sidak’s multiple comparisons test following two-way analysis of variance (ANOVA) (ns, no significant difference). The data are shown as means ± SD (n = 3 independently and identically prepared batches). Recovery of PMB from D-TZP and TZP after 24-hour incubation in 50% FBS (right). The data are shown as means ± SD (n = 3 independently and identically prepared batches). (F) Size distribution of fresh D-TZP and D-TZP incubated in 50% FBS (at concentrations equivalent to 25 to 100 μg/ml PMB) for 24 hours at 37°C.
Synthesis of TA/Fe nanocapsules encapsulating vitamin D (D-T)
Vitamin D–encapsulated nanocapsules were prepared by interfacial self-assembly of TA-Fe3+ complexes (22). Ethanolic solution containing vitamin D and TA was added to an aqueous Fe3+ solution. TA and Fe3+ form a supramolecular complex instantaneously at the interface between ethanol and water (22) to make spherical capsules encapsulating vitamin D (D-T) (Fig. 1B and fig. S1). According to the dynamic light scattering (DLS) measurement, D-T had an average z diameter of 247 ± 13 nm, a polydispersity index (PdI) of 0.13 ± 0.04, and a surface charge of −21 ± 0.5 mV at pH 7.4 (Fig. 1C). The particle size estimated by transmission electron microscopy (TEM) was 110 ± 24 nm (fig. S1), smaller than DLS measurement, likely due to a moderate degree of aggregation in the buffer in which the DLS was measured. The TA/Fe3+ was clearly visible after ethanol etching, which selectively dissolved vitamin D from D-T (Fig. 1B). The TA/Fe3+ layer was measured to be 15 ± 1 nm (fig. S1).
Primary surface modification of D-T with LMZWC (D-TZ)
We initially attempted to conjugate PMB on the TA/Fe3+ surface using the adhesive properties of the polyphenol (23). However, direct binding of PMB on D-T caused uncontrolled aggregation due to the avid interaction between the cationic drug and highly anionic TA/Fe3+ coating. This PMB-coated D-T had a diameter as large as 987 ± 130 nm and a PdI of 0.77 ± 0.13 (Fig. 1C). The zeta potential of the aggregates was −13 ± 2.0 mV (as compared to −21 ± 0.5 mV of D-T), indicating partial neutralization of the negative charge of D-T by the surface-bound PMB. To prevent the uncontrolled aggregation of PMB-coated particles, D-T was precoated with LMZWC. With 30% succinylation of amine groups (24), LMZWC had an isoelectric point (pI) value of 6.4 (fig. S2). D-T was incubated with LMZWC at pH 6.0 (below pI), where LMZWC was positively charged to attract the polymer via electrostatic interaction. Since the charge density of LMZWC at this pH (+5.5 mV) was lower than that of PMB (+10.4 mV), LMZWC coating did not cause aggregation like PMB did. Then, the pH was slowly raised to pH 8.5 to induce oxidation of the catechol moieties of TA into a quinone form (23), which interacts with the remaining amines of LMZWC via Schiff base reaction (25), thereby stabilizing the LMZWC binding (Fig. 1D). The LMZWC-coated D-T (D-TZ) at pH 8.5, but not the D-TZ at pH 6.0, showed a Fourier transform infrared (FTIR) peak at 1488 cm−1, corresponding to imine (Schiff base, C═N) (26), confirming the covalent binding of LMZWC to TA (Fig. 1D) (27). The surface charge of D-TZ at pH 7.4 was −15 ± 1.6 mV (Fig. 1C), which reflects the surface coverage of LMZWC. D-TZ had a z average of 290 ± 11 nm and a PdI of 0.14 ± 0.05. TEM of ethanol-etched D-TZ visualized the surface layer, which now contained additional LMZWC, with an average thickness of 20 ± 3 nm, thicker than 15 ± 1 nm of D-T (Fig. 1B and fig. S1).
Surface modification of D-TZ with PMB (D-TZP)
PMB was added to D-TZ, forming D-TZP, with an average z diameter of 297 ± 7 nm and a PdI of 0.06 ± 0.06 (Fig. 1C). The inclusion of PMB increased the coating thickness to 27 ± 6 nm, estimated by TEM of ethanol-etched particles (Fig. 1B and fig. S1). The PMB and vitamin D contents in D-TZP were 50 ± 0.9 weight % (wt %) and 23 ± 0.7 wt %, respectively. The balance (27 wt %) is attributable to the TA/Fe3+ assembly and LMZWC. The zeta potential of D-TZP at pH 7.4 was −10 ± 2.1 mV, slightly higher than −15 ± 1.6 mV of D-TZ, indicating the surface loading of PMB (Fig. 1C). To estimate the retention of PMB on D-TZP surface during circulation, D-TZP was incubated in 5% dextrose (D5W) containing 50% fetal bovine serum (FBS) (pH 7.4) for 24 hours at 37°C. Of the total recovered PMB (78.7% of loaded PMB), 44.9% PMB was released from D-TZP over 24 hours in 50% FBS, and 55.1% remained bound on D-TZP (Fig. 1E). In the same condition, most vitamin D remained encapsulated in D-TZP, with 96.5% recovered from D-TZP and 3.5% found in the incubation medium, out of the total recovered vitamin D (65% of loaded vitamin D) (Fig. 1E). D-TZP also maintained colloidal stability in 50% FBS for 24 hours, irrespective of the concentration (Fig. 1F and fig. S3). These results suggest that D-TZP would circulate as nanoparticles with at least 50% of PMB and most of vitamin D on the surface and in the core, respectively, upon systemic administration.
PMB-bound particles without vitamin D (TZP)
TZP were produced to compare with D-TZP in selected studies. Nanoparticulate self-assemblies of TA/Fe3+ were produced without vitamin D and sequentially coated with LMZWC and PMB in the same manner as D-TZP. TZP was slightly smaller than D-TZP, with a z average of 247 ± 10 nm and a PdI of 0.12 ± 0.04 (Fig. 1C). Under TEM, TZP appeared to be clusters of small particles with an average diameter of 34 ± 5 nm (fig. S1). There were no significant differences between D-TZP and TZP in PMB release at any time points (Fig. 1E). Of the total recovered PMB (75.9% of loaded PMB), 55.3% PMB was released from TZP over 24 hours in 50% FBS and 44.7% remained with the particles.
D-TZP reduces toxicity of PMB
D-TZP induces little damage to cell membrane in vitro
PMB causes leakage and damage in the cell membrane by interacting with phospholipids (8, 14). To test whether nanoparticle-bound PMB was less toxic than free PMB, we incubated THP1-XBlue-MD2-CD14 cells, a derivative of the human monocytic leukemia (THP-1) cell line, and J774A.1 murine macrophages with D-TZP, TZP, or free PMB at a concentration equivalent to 50 to 150 μg/ml PMB for 24 hours and stained with propidium iodide (PI), which penetrates damaged cell membrane to intercalate with nucleic acids and emit a fluorescent signal. The cells incubated with free PMB showed dose-dependent increase of PI fluorescence signal in both cell lines, corresponding to the decrease in cell viability measured by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Fig. 2A), indicating membrane damage due to PMB. In contrast, D-TZP induced significantly lower PI fluorescence than free PMB at the same concentrations (Fig. 2A). Consistently, J774A.1 macrophages treated with free PMB or D-TZP for 12 hours showed differential cell membrane morphology. The cells incubated with free PMB showed diffuse signals upon membrane staining with fluorescently labeled wheat germ agglutinin (WGA) in confocal laser scanning microscopy (Fig. 2B). Conversely, those treated with D-TZP showed continuous membrane staining, comparable to the vehicle (D5W)–treated cells. Scanning electron microscopy (SEM) also showed that D-TZP was less damaging than free PMB (Fig. 2C and fig. S4). Free PMB caused notable deformation of cell membrane, whereas D-TZP–treated cells were not different from the vehicle-treated control, showing the characteristic-wrinkled surface of resting macrophages (28), although D-TZP was close to the cell surface (purple in SEM image; Fig. 2C and fig. S4). The fluorescence intensity of PI-stained cells, confocal images of WGA-stained cells, and SEM images of cell morphology indicate that D-TZP causes no membrane damage unlike free PMB at the same dose. TZP omitting the vitamin D core showed the same results as D-TZP in all tests (Fig. 2), supporting the reduced membrane toxicity of nanoparticle-bound PMB. The difference of TZP and D-TZP in the shape and unit size shown in TEM brought no apparent difference in their effects on macrophages. We suspect that the TZP clusters may have interacted with macrophages similarly as D-TZP, based on the comparable hydrodynamic diameters (TZP, 247 ± 10 nm versus D-TZP, 297 ± 7 nm).
Fig. 2. D-TZP reduces toxicity of PMB.
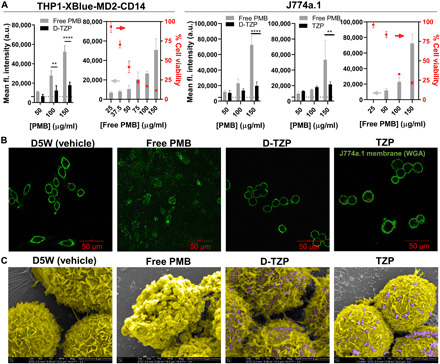
(A) Mean fluorescence intensity of PI-stained THP1-XBlue-MD2-CD14 cells and J774A.1 macrophages after 24-hour incubation with D-TZP, TZP, or free PMB. The dotted lines represent background fluorescence intensity of nontreated control cells. The cell viability measured by MTT assay (dots) was plotted alongside to correlate with PI fluorescence intensity (bars). Statistical significance was accessed by Sidak’s multiple comparisons test following two-way ANOVA (**P < 0.01 and ****P < 0.0001). The data are shown as means ± SD (n = 3 independently and identically prepared batches). (B) Confocal images of J774A.1 macrophages treated with D5W (vehicle), free PMB, TZP, or D-TZP for 12 hours at a concentration equivalent to 150 μg/ml PMB. Green signals indicate the membrane of macrophages stained with wheat germ agglutinin–Alexa Fluor 488 conjugate. (C) Scanning electron micrographs of J774A.1 macrophages treated with D5W, free PMB, TZP, or D-TZP for 12 hours at a concentration equivalent to 150 μg/ml PMB. Images are artificially colored to identify J774A.1 macrophages (yellow) and D-TZP (purple). a.u., arbitrary units.
D-TZP is better tolerated than free PMB in vivo
Because of the damaging effect on cell membrane, PMB is known to induce dose-limiting nephrotoxicity and neurotoxicity in humans (14). To test whether the nanoparticle-bound PMB reduced in vivo toxicity, we administered D-TZP or PMB by intraperitoneal or intravenous injection to male C57BL/6 mice at PMB doses greater than the known LD50 (median lethal dose) values of free PMB [20 mg/kg for intraperitoneal injection; 5 mg/kg for intravenous injection (29)]. While free PMB exceeding its LD50 was fatal (table S1), no mortality was observed with D-TZP at a PMB dose of 40 mg/kg, intraperitoneally, and 10 mg/kg, intravenously, twice the LD50 values of free PMB in respective administration routes. No mice receiving D-TZP lost more than 15% of their original weights, and all regained the weights in 2 days. At each tolerated intravenous dose [free PMB: 5 mg/kg; D-TZP: equivalent to 10 mg/kg PMB, the highest concentration tested], neither free PMB nor D-TZP induced apparent systemic toxicity, and blood chemistry values were within normal ranges (fig. S5). TZP was comparable to D-TZP in blood chemistry. Histological evaluation of major organs had consistent results (fig. S6). In PMB- and D-TZP–treated animals, compared to vehicle-treated controls, mild lymphocyte apoptosis was seen in sections of thymus sampled with the lung in one animal from each group. Similarly, in the TZP group, a mild decrease in lymphocyte cellularity with increased macrophages and mast cells was noted in one animal, but the lesions were of limited severity and are of unlikely significance. There was no other evidence of notable lesions in sections of the brain, lung, liver, spleen, kidney, or heart in all tested groups.
D-TZP retains bioactivities of PMB
Anti-LPS activity
To test whether D-TZP can inactivate LPS, we tested the stimulation of a TLR4 reporter cell line by LPS in the presence of D-TZP. THP1-XBlue-MD2-CD14 cells overexpress MD2 and CD14 to amplify the response to LPS, a TLR4 agonist (30). Upon LPS stimulation of TLR4 via CD14 and MD2, THP1-XBlue-MD2-CD14 cells activate nuclear factor κB and activator protein 1 (31) and subsequently produce secreted embryonic alkaline phosphatase (SEAP) reporter protein, which induces color change of the QUANTI-Blue reagent in proportion to LPS activity. The reporter cells treated with LPS (10 ng/ml) expressed a high level of SEAP as compared to nonchallenged cells. D-TZP equivalent to 9.5 or 18.9 μg/ml PMB (concentrations at which free PMB is not toxic) suppressed the SEAP production, bringing it to the basal level of nontreated cells, similar to free PMB at the same doses (Fig. 3A). This result demonstrates that D-TZP retained the ability of PMB to inactivate LPS. To determine how D-TZP reduced the SEAP production from the LPS-challenged reporter cells, LPS (10 ng/ml) was preincubated with D-TZP or free PMB for 1 hour followed by centrifugation. The supernatant, which contained LPS left unbound to D-TZP or inactivated by free PMB, was incubated with THP1-XBlue-MD2-CD14 cells to detect the LPS activity. The supernatant of LPS preincubated with D-TZP or free PMB showed no significant SEAP production as opposed to nontreated LPS (Fig. 3B), indicating the complete inactivation of free LPS. This result indicates that D-TZP retains the ability of PMB to interact with and neutralize LPS.
Fig. 3. D-TZP retains bioactivities of PMB.
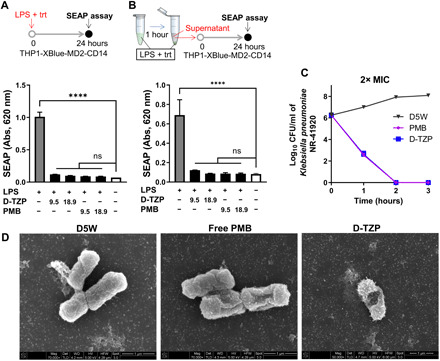
SEAP production from THP1-XBlue-MD2-CD14 TLR4 reporter cells incubated for 24 hours with LPS (10 ng/ml) (A) as a mixture with D-TZP or free PMB at different concentrations (n = 3 independently and identically prepared batches) or (B) precomplexed with D-TZP or free PMB for 1 hour and separated from the complexes. trt, treatment. Numbers in the x axis indicate PMB concentration in micrograms per milliliter (n = 3 independently and identically prepared batches for each test sample; n = 6 replicates for LPS control and no-challenge control). Statistical significance was accessed by Dunnett’s multiple comparisons test following ordinary one-way ANOVA (****P < 0.0001; ns, no significant difference). The data are shown as means ± SD. (C) Time-kill assay of K. pneumoniae NR-41920 treated with free PMB or D-TZP at 2× respective minimum inhibitory concentration (MIC). (D) Scanning electron micrographs of K. pneumoniae NR-41920 treated with D5W, free PMB, or D-TZP (at 5× respective MIC) for 1.5 hours. D-TZP is not visible due to the low concentration.
To investigate which component of D-TZP is responsible for LPS inactivation, control formulations omitting at least one component were tested with THP1-XBlue-MD2-CD14 cells under LPS challenge. D-T, omitting LMZWC and PMB, did not suppress SEAP production from the LPS-challenged reporter cells. D-TZ, omitting PMB, suppressed the response to LPS slightly (fig. S7). The modest effect of D-TZ is attributable to weak affinity of LMZWC for LPS reported previously (32, 33). The lack of significant activity of D-T or D-TZ indicates that the active component of D-TZP in LPS inactivation is PMB, as expected. The omission of vitamin D did not affect the binding and inactivation of LPS, as seen with TZP (fig. S8), further supporting the active role of PMB coating.
Antibacterial activity
To test whether the nanoparticle-bound PMB retains antibacterial activity, the minimum inhibitory concentrations (MICs) of D-TZP, its precursor particles, and free PMB were determined against four clinically isolated Gram-negative bacterial pathogens including Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa (table S2). Both D-TZP and TZP showed antibacterial activity with a two- to fourfold increase in MIC values compared to free PMB (Table 1). D-TZ or D-T, precursor particles without PMB, showed no measurable antibacterial activities, indicating that the other ingredients were not responsible for the bacterial killing. At 2× and 5× respective MICs, both D-TZP and PMB killed K. pneumoniae within 2 hours (Fig. 3C and fig. S9). In addition, K. pneumoniae, incubated with D-TZP or PMB at 5× MIC for 1.5 hours, was observed by SEM. Both D-TZP and PMB induced deformation of bacterial membrane, indicating that D-TZP killed the bacteria in the same mechanism as free PMB (Fig. 3D).
Table 1. Minimum inhibitory concentrations (μg/ml, as PMB equivalent) of PMB, D-TZP, its precursor particles, and control antibiotics.
ATCC, American Type Culture Collection.
| Escherichia coli ATCC 2452 | Escherichia coli ATCC 2469 |
Klebsiella pneumoniae NR-41920 |
Pseudomonas aeruginosa NR-50573 |
|
| PMB | 0.5 | 0.125 | 0.5 | 0.5 |
| D-TZP | 1 | 0.5 | 1 | 1 |
| TZP | 1 | 0.5 | 1 | 1 |
| D-TZ | >64 | >64 | >64 | >64 |
| D-T | >64 | >64 | >64 | >64 |
| Gentamicin | >64 | >64 | 64 | 1 |
| Imipenem | 16 | 16 | 8 | 1 |
D-TZP reduces the mortality of mice with LPS-induced sepsis (endotoxemia model)
On the basis of the safety and anti-LPS activity, therapeutic efficacy of D-TZP was investigated in a mouse model of LPS-induced endotoxemia. Male C57BL/6 mice (6 to 8 weeks old) were challenged with an intraperitoneal injection of LPS, which causes systemic inflammation similar to initial features of sepsis (34). Intraperitoneal injection of LPS (20 mg/kg) was severe enough to kill control mice in less than 60 hours. When D-TZP (equivalent to 40 mg/kg PMB) was administered simultaneously with the LPS, all animals survived (100% survival), while four of five of the control group receiving no treatment but vehicle control (D5W) succumbed to death in 40 hours from the challenge (20% survival) (Fig. 4A and fig. S10). When D-TZP was given intraperitoneally immediately after LPS challenge, the animals treated with D-TZP [PMB (40 mg/kg)] showed 100% survival, while none of the D5W control group survived (0% survival) (Fig. 4B and fig. S11). TZP [PMB (40 mg/kg)] also demonstrated the same effect (100% survival). Since both LPS and D-TZP were administered almost simultaneously to the peritoneal cavity, the anti-LPS effect shown in these two tests may be considered local and not clinically relevant. To mimic a clinical setting where patients present with pre-established septic conditions, mice were challenged with intraperitoneal LPS before the treatment. After intraperitoneally injected LPS was systemically absorbed [2 hours after the injection (35)], D-TZP or TZP was given intravenously at 10 mg/kg PMB equivalent, the known tolerated dose. None of the D5W-treated control survived (0% survival). Free PMB [5 mg/kg, LD50 (29)] was also tested in this model but showed acute toxicity causing early death in two of five mice in 6 hours. Those surviving the acute toxicity of free PMB died in 51 hours (0% survival) (Fig. 4C). In contrast, those treated with D-TZP showed 70% survival, and TZP protected 60% of the animals with no difference between sexes (fig. S12). All surviving animals fully recovered from typical signs of severe endotoxemia, such as hypothermia and weight loss (Fig. 4D), and returned to the normal bright, alert, and responsive (BAR) state. The effectiveness of both D-TZP and TZP as systemic therapy indicates that the surface-bound PMB retained the affinity for LPS in blood.
Fig. 4. D-TZP reduces the mortality of mice with LPS-induced sepsis (endotoxemia model).
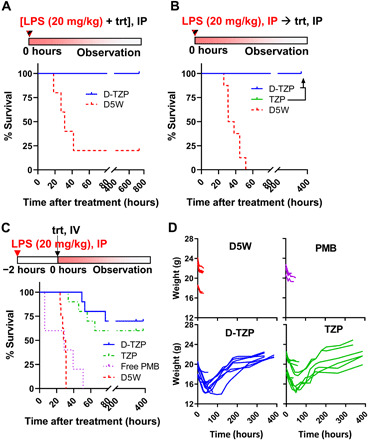
Endotoxemia was induced in C57BL/6 mice by an intraperitoneal (IP) injection of LPS (20 mg/kg). (A) D5W or 40 mg/kg PMB equivalent D-TZP was administered intraperitoneally as a mixture with LPS (n = 5 per group). (B) D5W (n = 8), 40 mg/kg PMB equivalent D-TZP (n = 10), or TZP (n = 8) was administered intraperitoneally, immediately after the LPS challenge. (C) D5W (n = 8), free PMB (5 mg/kg) (n = 5), 10 mg/kg PMB equivalent D-TZP (n = 10), or TZP (n = 10) was administered intravenously 2 hours after the LPS challenge. (D) Body weight change of the mice in (C). trt, treatment.
D-TZP accumulates in the liver and spleen and reduces macrophage response to LPS
We evaluated the pharmacokinetics and biodistribution of PMB in C57BL/6 mice following a single intravenous injection of D-TZP or free PMB (at a dose equivalent to 5 mg/kg PMB). Both D-TZP and free PMB disappeared quickly from the circulation with elimination half-lives of 1.68 and 1 hour, respectively (Fig. 5A). D-TZP–treated mice showed lower PMB levels than free PMB–treated ones in plasma, kidney, and heart at all time points. The high distribution of free PMB in the kidney is consistent with the literature (36). In contrast, D-TZP accumulated predominantly in the liver and spleen, where free PMB showed little distribution except for the first few time points (Fig. 5A).
Fig. 5. D-TZP concentrates PMB in the liver and spleen and reduces macrophage response to LPS.
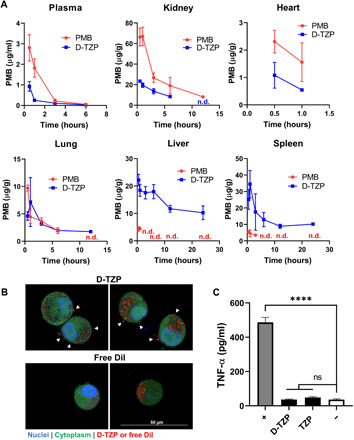
(A) Biodistribution of PMB in C57BL/6 mice after a single intravenous injection of D-TZP or PMB at a dose equivalent to 5 mg/kg. The data are shown as means ± SD (n = 3 mice per group per time point). n.d., not detected. (B) Confocal microscope z-section images of J774A.1 macrophages incubated with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI)–labeled D-TZP or free DiI for 4 hours. White arrowheads: D-TZP on cell surface. (C) Tumor necrosis factor–α (TNF-α) production from J774A.1 macrophages, preincubated with D-TZP or TZP at a concentration equivalent to 25 μg/ml PMB for 24 hours, thoroughly rinsed, and then challenged with LPS (10 ng/ml). +, LPS control; −, no challenge control. Statistical significance was accessed by Dunnett’s multiple comparisons test following ordinary one-way ANOVA (****P < 0.0001; ns, no significant difference). The data are shown as means ± SD (n = 3 independently and identically prepared batches).
To estimate the activity of D-TZP distributing in the liver, we used J774A.1 macrophages as a model of Kupffer cells, which sit on the lumen of the sinusoids and remove most of the circulating nanoparticles through the liver (37) and evaluated their interaction with D-TZP by confocal microscopy. DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate)–labeled D-TZP was located both inside and on the surface of macrophages, unlike free DiI found mostly in the cells. The presence of cell surface–bound D-TZP resonates with the earlier SEM observation, where D-TZP was found on the macrophage surface after 12-hour incubation (Fig. 2C). To examine whether the cell surface–bound D-TZP had anti-LPS activities, we incubated J774A.1 macrophages with the particles (equivalent to 25 μg/ml PMB) for 24 hours, rinsed to remove unbound particles, and challenged the cells with LPS (10 ng/ml) for 12 hours. The J774A.1 macrophages preincubated with D-TZP resisted the LPS challenge and showed basal levels of tumor necrosis factor–α (TNF-α), comparable to that of nonchallenged macrophages (Fig. 5C). These results suggest that D-TZP bound on macrophages can inactivate LPS via the high affinity of the surface-bound PMB for LPS (Fig. 3B and fig. S8b). TZP interaction with macrophages was not observed due to the lack of means to label the particles; however, TZP showed a similar result to D-TZP in the anti-LPS activity following preincubation and rinsing (Fig. 5C), suggesting that TZP has a similar interaction with macrophages to D-TZP.
D-TZP reduces the mortality of mice with CLP (polymicrobial sepsis model) with corresponding reduction of cytokine production and D-dimer level
Given that both D-TZP and TZP were highly effective in treating endotoxemia, we subjected them to the CLP model, which involves polymicrobial infection and thus simulates a clinically relevant septic state (38). D-TZP and TZP were administered intravenously at 10 mg/kg PMB equivalent, 2 hours after CLP, and every 2 subsequent days (a maximum total of three times). Free PMB was given at 5 mg/kg, IV, in the same schedule as D-TZP. The animals receiving vehicle showed 20% survival, and those receiving free PMB had a survival rate of 33% (P = 0.1878 versus vehicle by Log-rank test) (Fig. 6A). Among the animals receiving D-TZP, 75% survived (P = 0.0276 versus vehicle by Log-rank test), recovering from hypothermia and initial weight loss (Fig. 6B) and returning to the BAR state. TZP saved 37.5% (P = 0.0862 versus vehicle by Log-rank test) with weight recovery.
Fig. 6. D-TZP reduces the mortality of mice with CLP (polymicrobial sepsis model) with corresponding reductions of cytokine production and D-dimer level.
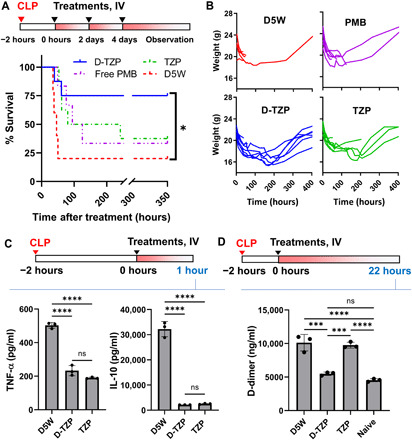
(A) Survival of CLP-operated C57BL/6 mice receiving intravenous injections of D5W (n = 5), PMB (5 mg/kg) (n = 6), D-TZP, or TZP equivalent to 10 mg/kg PMB starting from 2 hours after CLP (q2d × 3) (n = 8 per group). Statistical significance was accessed by Log-rank test (*P < 0.05). (B) Body weight changes of the mice in (B). (C) Plasma levels of TNF-α and IL-10 at 3 hours after CLP surgery (1 hour after treatment). (D) Plasma levels of D-dimer at 24 hours after CLP surgery (22 hours after treatment). Notably, the D-dimer values here are higher than reported levels, which are measured after treatment with heparin (75). Statistical significance for TNF-α, interleukin-10 (IL-10), and D-dimer was accessed by Tukey’s multiple comparisons test following ordinary one-way ANOVA (***P < 0.001; ****P < 0.0001; ns, no significant difference). The data are shown as means ± SD (n = 3 per group).
To evaluate the ability of the particles to promote the resolution of inflammation in CLP-challenged mice, we measured the plasma levels of TNF-α as well as interleukin-10 (IL-10), which is co–up-regulated in the activated neutrophils (39) and monocytes (40) as a secondary effect of TNF-α production, in animals receiving CLP and treatment. D-TZP or TZP (at 10 mg/kg PMB equivalent) was administered intravenously 2 hours after CLP. The production of both cytokines is shown to reach the peak at 2 to 4 hours after CLP (41). Accordingly, the animals were euthanized at 3 hours after CLP (1 hour after the treatment) to determine plasma levels of the cytokines. Both D-TZP and TZP suppressed the production of TNF-α and IL-10 significantly, as compared to the vehicle control (Fig. 6C). This result suggests that the particle-bound PMB (irrespective of the presence of vitamin D in the core) had an immediate effect (≤1 hour).
We also measured the blood level of D-dimer at 24 hours after the CLP surgery (22 hours after the treatment). D-dimer is two covalently bonded D domains of polymerized fibrins and exposed to circulation upon fibrin degradation (42). The elevation of D-dimer level indicates a fibrin clot formation and fibrinolysis, serving as a biomarker of DIC, a common complication of sepsis and the CLP model. When measured at 24 hours after CLP, a time point known to reach the peak D-dimer level in mice (43), the vehicle-treated control group showed 2.2 times higher D-dimer level than naïve animals (Fig. 6D). The TZP group showed no difference from the D5W vehicle control group. In contrast, the D-TZP group showed a significantly lower D-dimer level than both D5W-treated control and TZP groups, comparable to that of naïve (no CLP) control. The differential D-dimer levels suggest greater activity of D-TZP than TZP, consistent with the difference between D-TZP and TZP in the mortality of treated animals in the CLP model.
Differential activities of D-TZP and TZP
D-TZP showed better efficacy than vehicle, but TZP did not, in the CLP model. The D-dimer profile showed a corresponding difference. To determine whether the encapsulated vitamin D was responsible for the difference, we treated male C57BL/6 mice (6 to 8 weeks old) with a single intravenous injection of D-TZP at a dose equivalent to 5 mg/kg vitamin D (the dose used for CLP) and examined whether D-TZP yielded the active metabolite of vitamin D, 25 hydroxyvitamin D (25OHD). Both free vitamin D and D-TZP showed a basal level of 25OHD (fig. S13), indicating that the exogenously supplied vitamin D was not metabolized in mice. Therefore, the superior efficacy of D-TZP in the CLP model may not be explained by biological activity of the encapsulated vitamin D.
We then repeated the in vitro evaluation of anti-LPS activities of D-TZP and TZP, increasing the LPS burden relative to the particle dose than the previous tests. First, THP1-XBlue-MD2-CD14 monocytes were incubated in LPS (10 ng/ml) mixed with D-TZP or TZP at a dose equivalent to 3 to 12 μg/ml PMB, including a concentration range lower than the previous test (Fig. 3A and fig. S8a) for 24 hours. D-TZP outperformed TZP in inhibiting the LPS activity at relatively low PMB concentrations, namely, 3 and 6 μg/ml (fig. S14). Second, J774A.1 macrophages pretreated with a fixed amount of D-TZP and TZP (equivalent of 25 μg/ml PMB) for 24 hours were challenged with LPS for an additional 12 hours, increasing the LPS dose from 25 to 75 ng/ml, as opposed to the previous 10 ng/ml (Fig. 5C). The particles were overall less effective at a relatively higher dose of LPS; nevertheless, D-TZP pretreatment suppressed the TNF-α production from the macrophages significantly better than TZP (fig. S15). These two tests indicate that D-TZP performed better than TZP when challenged with higher LPS burdens relative to the particle dose.
DISCUSSION
Early administration (44) and high-dosage short-term courses of antibiotics therapy (45) help to remove circulating bacteria and endotoxins in patients with sepsis. PMB is well suited for the therapy of Gram-negative sepsis due to the high affinity for endotoxins and antibacterial activities. However, the dose-limiting toxicity has limited its systemic use in patients with sepsis. To overcome the toxicity and exploit the bioactivities of PMB for systemic therapy of sepsis, we have developed a nanoparticle system (D-TZP) that carries PMB on the surface and selectively limits direct exposure of PMB to mammalian cell membrane.
D-TZP was produced by encapsulating vitamin D inside TA/Fe3+ capsules and loading PMB along with LMZWC on the surface of the capsule. TA is a polyphenol from natural origin (46) and forms a supramolecular complex via Fe3+ instantaneously (22) to encapsulate vitamin D. TA can accommodate diverse molecules via hydrogen bonds, hydrophobic interactions, and ionic bonds (47); thus, PMB and LMZWC can be attached on the TA/Fe3+ capsule surface without a coupling agent or chemical modification. LMZWC was added as a hydrophilic coating based on the ability to reduce the interaction of particles with macrophages (24, 48). In addition, the LMZWC layer served two other purposes in PMB loading. First, as a surface-bound polymer, LMZWC helped to maintain the colloidal stability of the particles during the PMB loading, which otherwise would have caused uncontrolled aggregation (Fig. 1C). Second, negatively charged at pH 7.4, LMZWC attracted cationic PMB to the particle surface, mediating the interaction of PMB with the particles. PMB may have noncovalently bonded with LMZWC and/or further migrated toward the TA/Fe3+ surface due to the stronger negative charge of the underlying TA/Fe3+ layer. Despite the presence of cationic PMB, D-TZP assumed a net negative charge, which suggests that PMB may be shielded by the LMZWC layer.
By shielding PMB, LMZWC appears to have served as a “bumper” layer to prevent direct contact of PMB to mammalian cells and help reduce the membrane damage (Fig. 2). As a negatively charged and hydrophilic polymer, LMZWC may swell in water to reduce direct contact of PMB with the cell membrane (fig. S16). However, LMZWC did not negatively affect the affinity of PMB for LPS (Fig. 3, A and B) or the antibacterial activity of PMB (Fig. 3C), indicating that LMZWC did not prevent D-TZP from accessing LPS or bacteria. This differential effect on PMB interaction may be explained by the relatively high negative charge density of LPS (49) and bacterial outer membrane (50), which may have been strong enough to contract PMB overcoming the LMZWC bumper layer. The selective toxicity of D-TZP is also demonstrated by differential membrane damages observed in the SEM images (Figs. 2C and 3D). These results indicate that LMZWC helps to selectively protect mammalian cells from PMB without compromising the ability of PMB to inactivate LPS and kill the bacteria.
The enhanced safety profile of D-TZP enabled systemic administration of PMB at a dose that would have been lethal otherwise. The systemically administered D-TZP protected mice challenged with LPS and CLP. D-TZP was effective in 100% of the animals challenged with endotoxemia, when administered as a mixture with LPS or immediately after LPS (Fig. 3A); in 70%, when administered intravenously 2 hours after LPS challenge (Fig. 3B); and in 75% of the animals challenged with CLP as an intravenous treatment given 2 hours after challenge (Fig. 3C), while the vehicle control resulted in 5% (endotoxemia) or 20% (CLP) survival. All surviving animals fully recovered from the initial signs of morbidity owing to the pre-established septic conditions and restored the BAR state. These survival outcomes compare favorably with other systemic treatments investigated in comparable sepsis models, which show 70 to 85% survival as a simultaneous treatment, 10 to 20% survival as a treatment given at 2 hours after LPS challenge (table S3) (51–53), and 60 to 90% survival as a treatment given at 2 hours after CLP (table S4) (21, 54–56).
The pharmacokinetics and biodistribution profiles suggest that there remains a room for improvement in D-TZP properties. D-TZP accumulated mostly in the organs of the mononuclear phagocyte system (MPS) as evidenced by the PMB levels (Fig. 5). The PMB delivered as D-TZP remained in the MPS organs over 24 hours, much longer than free PMB, with little distribution in the kidneys, which free PMB targets and damages (57). D-TZP accumulating in the liver showed no sign of hepatotoxicity or other abnormalities in blood chemistry and histology (figs. S5 and S6), consistent with the improved safety in the cell-based studies (Fig. 2). The short half-life of D-TZP is attributable to the suboptimal particle size. The hydrodynamic diameter of 297 nm falls in the size range that favors the accumulation in the MPS organs (58), thus interfering with the prolonged circulation. D-TZP with a longer circulation half-life may have a greater exposure to circulating endotoxin and bacteria and affected tissues, as suggested in the literature (59). Future efforts need to be made to reduce the particle size and prolong the circulation half-life.
Despite the high distribution in the MPS organs, D-TZP was effective in two sepsis models. We speculate that D-TZP may have acted mainly on LPS and bacteria entering the MPS organs. Circulating LPS (60) and LPS-lipoprotein complexes (61) are shown to accumulate mostly in the liver and to some extent in the spleen. Blood-borne bacteria follow a similar distribution pattern, accumulating in the liver and spleen (62). In vitro studies with J774A.1 macrophages show that some of the D-TZP remained on the macrophage surface (Figs. 2C and 5B) and suppressed the LPS-induced TNF-α production (Fig. 5C), which suggests that extracellular D-TZP in the MPS organs would have helped inactivate LPS and bacteria. Given that the LMZWC layer reduces the protein adsorption and macrophage uptake of nanoparticles (24, 48), we suspect that LMZWC may have contributed to the partial retention of D-TZP on the cell surface shown in microscope images. On the other hand, we cannot exclude the possibility of D-TZP in the cells playing a role in treating intracellular bacteria residing in macrophages, especially in the polymicrobial sepsis model. Therefore, D-TZP may benefit from further optimization of the LMZWC layer to control the extent of cellular uptake according to the target.
We originally included vitamin D because of its known immunomodulatory activities. The active form of vitamin D, 1,25-dihydroxyviatmin D [1,25(OH)2D], enhances the expression of cathelicidin, an endogenous broad-spectrum antimicrobial peptide (63) and LPS adsorbent (64), from the TLR-activated monocytes and macrophages in humans. Moreover, 1,25(OH)2D down-regulates the synthesis of TLR2 and TLR4 (65) and antagonizes the effect of LPS in proinflammatory cytokine production (66). However, we did not observe the metabolism of vitamin D in mice, with no evidence of 25OHD, the intermediate metabolite (67). This finding may be explained by the low levels of CYP2R1, an enzyme responsible for vitamin D metabolism in mice. We replaced vitamin D with an alternative core with no known biological functions [fluorescein-5-isothiocyanate (FITC)] and tested in endotoxemia model to find that FITC-TZP was as effective as D-TZP (fig. S17). This result supports the idea that the therapeutic activity of D-TZP is mostly attributed to the TZP part of the particles.
Although vitamin D core had no apparent contribution to the bioactivities, D-TZP and TZP show noticeable difference in models involving relatively high insults. D-TZP showed a significant protective effect in the CLP-challenged mice, where the effect of TZP was statistically insignificant (Fig. 6A). D-TZP was also superior to TZP in the reduction of D-dimer, a sign of anticoagulation effect (Fig. 6D), and in the inhibition of monocyte/macrophage activation under relatively high LPS challenge (figs. S14 and S15). In the absence of apparent vitamin D metabolism, we speculate that structural difference between the two particles owing to the vitamin D core may have played a role in their differential activities. D-TZP and TZP showed comparable sizes in DLS measurement (297 ± 7 nm versus 247 ± 10 nm) but appeared smaller in TEM (181 ± 22 nm for D-TZP and 34 ± 5 nm for TZP) (Fig. 1B and fig. S1). The difference between DLS and TEM measurements indicates the aggregation of the particles in aqueous media. With the large difference between the two measurements (247 nm versus 34 nm), we infer that TZP would be present as agglomerates of small particles sequestering a fraction of PMB within the agglomerates, whereas D-TZP with relatively small difference (297 nm versus 181 nm) is less likely so. The agglomeration of TZP would reduce effective exposure of PMB to LPS and bacteria, leading to differential activities between TZP and D-TZP in highly challenging conditions such as the CLP-induced polymicrobial infection. However, we cannot completely rule out the potential contribution of vitamin D, as other studies have shown that vitamin D supplied as dietary supplement had some functions such as bone regeneration (68) or immune homeostasis (69) in mice.
In summary, D-TZP, a nanoparticle formulation of PMB, was developed to reduce toxicity of the drug and enable its systemic use for sepsis therapy at an effective dose. D-TZP carries PMB on the surface of the iron complexed TA capsules, where a chitosan derivative (LMZWC) controls the interaction of PMB with endotoxin, bacteria, and host cells. D-TZP attenuated the membrane toxicity associated with PMB while maintaining the ability of PMB to inactivate LPS and lyse Gram-negative bacteria. Upon intravenous injection, D-TZP inactivated circulating LPS and bacteria and protected animals challenged with endotoxemia and polymicrobial sepsis, showing no systemic toxicities inherent to PMB. These results indicate that D-TZP is a promising systemic therapy of Gram-negative sepsis.
MATERIALS AND METHODS
Chemicals and reagents
Vitamin D (meets United States Pharmacopeia testing specifications), TA (ACS reagent), iron(III) chloride (reagent grade, 97%), PMB sulfate (meets USP testing specifications), colistin sulfate (CS) salt (≥15,000 U/mg), LPSs (from E. coli, O111:B4), hydrogen peroxide 30 wt % solution in water (ACS reagent), hydrochloric acid (≥37%), succinic anhydride (≥99%), sodium phosphate dibasic molecular biology grade, sodium phosphate monobasic, FITC, and Tween 20 (viscous liquid) were purchased from Sigma-Aldrich (St. Louis, MO). Sodium hydroxide (pellets) was purchased from Mallinckrodt. Chitosan (15 kDa; degree of deacetylation, 87%) was purchased from Polysciences Inc. (Warrington, PA). Dialysis bag [with a molecular weight cutoff (MWCO) of 1000 Da] was purchased from Spectrum Laboratories (Rancho Dominguez, CA, USA). Trichloroacetic acid (5%, w/v) was purchased from RICCA Chemical Company (Arlington, TX). Sodium bicarbonate (Certified ACS), sodium sulfate (Certified ACS), acetic acid glacial (≥99.7%), acetonitrile (ACN; meets ACS specifications), Exel International Disposable Safelet I.V. Catheters (20G), hexamethyldisilazane (HMDS), WGA–Alexa Fluor 488 conjugate, Hoechst 33342, an antifade reagent (ProLong Gold Antifade Mountant), formalin (10%, w/v), paraformaldehyde 4% in 0.1 M phosphate-buffered saline (PBS) (pH 7.4), electron microscopy sciences formvar/carbon film 10-nm/1-nm thick on a square 300-mesh copper grid, BD vacutainer plastic blood collection tubes with lithium heparin, 5-0 Perma-Hand Silk Black Braided Suture, occuNomix hot rods warming pack, alcohol prep pad 200/PK, ethyl acetate (certified ACS), and PBS tablets were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Flow cytometry staining buffer (1×) was purchased from R&D Systems (Minneapolis, MN, USA). PI was purchased from VWR International (Radnor, PA). Ethanol (200 proof, anhydrous, meets USP specs) was purchased from Decon Laboratories Inc. (King of Prussia, PA). Fifty percent dextrose (injection, USP) was purchased from Hospira Inc. (Lake Forest, IL) and VetOne (Boise, ID). Yellow-Green Fluor Aminated Particle was purchased from Magsphere Inc. (Pasadena, CA). Deionized (DI) water (18.2 megohms) was obtained from a Milli-Q ultrafiltration system (Millipore, Billerica, MA). Luciferase Cell Culture Lysis 5X Reagent was purchased from Promega (Madison, WI). Nair hair remover lotion was purchased from Church & Dwight Co. Inc. (Ewing, NJ). Enzyme-linked immunosorbent assay (ELISA) kits for TNF-α and IL-10 were purchased from BioLegend (San Diego, CA) and an ELISA kit for D-dimer and 25-hydroxy vitamin D were purchased from MyBioSource Inc. (San Diego, CA). Betadine 5% solution (sterile ophthalmic prep solution) was purchased from Alcon (Geneva, Switzerland). LubriFresh PM was purchased from Major Pharmaceuticals (Livonia, Michigan). Isoflurane, USP was purchased from Akorn (Lake Forest, Illinois). Hoechst 33342, CellTracker Green CMFDA (5-chloromethylfluorescein diacetate), and four-chamber glass bottom dishes were purchased from Thermo Fisher Scientific (Waltham, MA, USA). DiI dye was purchased from Life Technologies Corporation (Carlsbad, CA, USA).
Cell lines, bacterial strains, and reagents
THP1-XBlue-MD2-CD14 cell line, cell culture medium components, SEAP reporter assay (QUANTI-Blue reagent), normocin, zeocin, and G418 were purchased from InvivoGen (San Diego, CA). Gibco Dulbecco’s modified Eagle’s medium [DMEM; d-glucose (4.5 g/liter), l-glutamine (584 mg/liter), and sodium pyruvate (110 mg/liter)], Gibco RPMI 1640 medium, Gibco penicillin-streptomycin (10,000 U/ml), and Gibco Dulbecco’s PBS (without calcium chloride and magnesium chloride) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Sodium pyruvate (100 mM, filter-sterilized) and HEPES solution [1 M (pH 7.0 to 7.6), filter-sterilized] were purchased from Sigma-Aldrich. FBS (premium) was purchased from Atlanta Biologicals (Flowery Branch, GA). J774A.1 macrophage cell line was purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Bacterial strains were obtained from ATCC and BEI Resources (Manassas, VA) (table S1). 0.5 McFarland standard was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cation-adjusted Mueller Hinton broth and tryptone soy agar plates were purchased from BD Biosciences (Franklin Lakes, NJ, USA).
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee at Purdue University, in compliance with the National Institutes of Health guidelines for the care and use of laboratory animals. Six- to 8-week-old C57BL/6 and BALB/c mice were purchased from Envigo (Indianapolis, IN, USA).
Synthesis and characterization of LMZWC
LMZWC was synthesized according to the previously reported method (70). Five grams of chitosan was dissolved in 1% acetic acid (200 ml) and stirred overnight. The solution was centrifuged at 4000 relative centrifugal force (rcf) for 20 min. The supernatant was collected and freeze-dried to obtain an acetate salt form of chitosan. Three grams of chitosan acetate was dissolved in 200 ml of acidified water (pH 3, HCl) and stirred vigorously. For digestion, 30 ml of hydrogen peroxide (30 wt %) was added to the chitosan solution under vigorous stirring for 12 hours. The digestion was quenched by the addition of 50 ml of methanol, and the pH was adjusted to 7 by 1 N NaOH. The digested chitosan [low–molecular weight chitosan (LMWC)] was dialyzed against DI water with an MWCO of 1000 Da for 2 days and freeze-dried. LMWC (400 mg) was dissolved in 1% acetic acid (60 ml). The pH of the chitosan solution was titrated to 6 using 1 N NaHCO3. Subsequently, 60 mg of succinic anhydride was added to achieve an anhydride-to-amine molar feed ratio (An/Am ratio) of 0.3. The pH of the mixture was maintained at 6 with 1 N NaHCO3 for 30 min. The pH was then slowly increased to 8.5 with 1 N NaHCO3 as the reaction proceeded. The reaction mixture was stirred overnight, followed by dialysis against DI water (pH adjusted to 8 to 9 by NaOH) with an MWCO of 1000 Da. The purified LMZWC was freeze-dried and stored at −80°C. For routine quality control, the zeta potential of LMZWC was monitored varying the pH. LMZWC was dissolved to 1 mg/ml in 10 mM NaCl and titrated with 0.1 N HCl bringing the pH from 8 to 3. The zeta potential of LMZWC solution was measured by a Malvern Zetasizer Nano ZS-90 (Worcestershire, UK) at each addition of HCl.
Preparation of core nanoparticles
The core nanoparticles were produced in two forms. First, nanocapsules encapsulating vitamin D (called D-T) were prepared according to a method in the literature (22) with modification. Vitamin D and TA were dissolved in ethanol to 10 and 40 mg/ml, respectively. FeCl3 was dissolved in DI water to 10 mg/ml. The ethanolic vitamin D solution (18 μl) was mixed with ethanolic TA solution (10 μl). Aqueous FeCl3 solution (10 μl) was added to 962 μl of DI water (4°C). The diluted FeCl3 solution was then added to the mixture of vitamin D and TA to initiate interfacial supramolecular assembly of TA and Fe3+ under probe sonication (Sonics Vibracell probe sonicator, Newtown, CT, USA) for 2 min, with 4 s on, 2 s off at 40% amplitude, forming D-T nanocapsules. The formation of the TA-Fe3+ coordination complex was instantly evident from the characteristic blue color (71). Alternatively, nanoparticles omitting vitamin D (called T) were prepared by mixing the ethanolic TA with the aqueous FeCl3 solution under probe sonication. For confocal microscopy, DiI was added to ethanol phase at 0.8 wt % of D-T. The core nanoparticles (D-T or T) were collected by centrifugation at 16,100 rcf for 20 min. Two batches were pooled and used for surface modification.
Surface modification of core nanoparticles
The core nanoparticles (D-T or T) collected from the previous step were incubated in LMZWC solution (4.5 mg in 1.5 ml of DI water, acidified to pH 6.0) for 15 min. The pH was slowly raised to pH 8.5 using NaHCO3 and incubated for an additional 45 min. The LMZWC-coated nanoparticles (D-TZ or TZ) were collected by centrifugation at 16,100 rcf for 20 min. The LMZWC-coated nanoparticles were then incubated with PMB (800 μg in 1.5 ml of DI water) for 1 hour so that the cationic PMB was attracted to the particle surface via the negatively charged LMZWC. The PMB- and LMZWC-coated nanoparticles (D-TZP or TZP) were centrifuged at 16,100 rcf for 20 min, and the supernatant was analyzed by high-performance liquid chromatography (HPLC). The particles were washed with DI water again and freeze-dried in D5W. The freeze-dried particles were reconstituted in D5W. In the rest of this article, the PMB- and LMZWC-coated nanoparticles are referred to as D-TZP or TZP, depending on the inclusion of vitamin D in the core.
Characterization of surface-modified nanoparticles
D-TZP and TZP were suspended in phosphate buffer [10 mM (pH 7.4)], and their sizes and zeta potentials were measured by a Malvern Zetasizer Nano ZS90. Nanoparticle morphology was observed by Tecnai TEM (FEI, Hillsboro, OR, USA) after negative staining with 1% uranyl acetate. To visualize the PMB and LMZWC-bound TA/iron coating, D-TZP was incubated in 100% ethanol for 1 hour, collected by centrifugation at 16,100 rcf for 20 min, and imaged by TEM. To confirm covalent conjugation of ZWC on D-T, D-T and D-TZ were analyzed by FTIR spectroscopy. D-T was dissolved in DI water. LMZWC and D-TZ were dissolved in DI water after adjusting the pH to 6.0 and 8.5, respectively. All formulations were lyophilized and analyzed by the Thermo Nicolet Nexus 470 FTIR (Madison, WI, USA).
HPLC analysis of vitamin D content
D-TZP was precisely weighed and dissolved in ethanol for 1 hour to extract vitamin D. Vitamin D in the supernatant was collected by centrifugation and quantified by the Agilent 1100 HPLC system equipped with Ascentis C18 column (25 cm by 4.6 mm; particle size, 5 μm). The sample injection volume was 50 μl. The mobile phase was a 75:25 volume mixture of methanol and ACN and ran at 1 ml/min. The column temperature was maintained at 40°C. Vitamin D was detected by an ultraviolet detector at a wavelength of 265 nm. Vitamin D was dissolved in ethanol to 50 to 400 μg/ml and analyzed in the same condition to establish a calibration curve. The vitamin D content in D-TZP was calculated as the vitamin D amount divided by the D-TZP mass.
HPLC analysis of PMB content
The PMB content in TZP or D-TZP was indirectly determined by measuring the unbound PMB remaining in the supernatant after 1-hour incubation of PMB with TZ or D-TZ. Briefly, 0.8 ml of supernatant was collected and mixed with 0.2 ml of ACN. HPLC analysis was conducted with an Agilent 1100 HPLC system (Palo Alto, CA, USA) equipped with Ascentis C18 column (25 cm by 4.6 mm; particle size, 5 μm). The sample injection volume was 20 μl. The mobile phase was a 76:24 volume mixture of aqueous Na2SO4 solution [30 mM (pH 2.5)] and ACN and eluted at a flow rate of 1 ml/min. The column temperature was maintained at 35°C. PMB was detected at a wavelength of 215 nm. PMB was dissolved in the mobile phase to 50 to 400 μg/ml and analyzed in the same condition to build a calibration curve. The surface-conjugated PMB was calculated by subtracting the unbound PMB from the PMB feed. The PMB content was calculated as the PMB amount divided by the nanoparticle mass.
In vitro PMB and vitamin D release in 50% FBS
D-TZP or TZP equivalent to 80 μg of PMB was suspended in 0.6 ml of 50% FBS and incubated at 37°C under constant rotation. At regular time points, the suspension was centrifuged at 16,100 rcf for 20 min. The supernatant was collected for analysis. The particles were resuspended in fresh 50% FBS by brief probe sonication and returned to the previous incubation condition. After the final sampling, the pellets were disintegrated in a mixture of 0.1 ml of 2 N HCl, 0.3 ml of DI water, and 0.1 ml of ACN, briefly probe-sonicated and analyzed by HPLC after filtration to quantify the remaining PMB. Vitamin D and PMB in the sampled supernatant were analyzed by HPLC and LC–tandem mass spectrometry (LC-MS/MS), respectively, as described in the following.
HPLC analysis of in vitro released vitamin D
The supernatant was mixed with 1.4 ml of ethyl acetate and vigorously shaken for 1 hour. The organic phase was separated by centrifugation at 16,100 rcf for 20 min and evaporated in a desiccator for 12 hours. The dry mass containing vitamin D was dissolved in 0.2 ml of ethanol and analyzed by HPLC. The pellets were disintegrated in a mixture of 0.16 ml of DI and 0.84 ml of ethanol, briefly probe-sonicated, and analyzed by HPLC.
LC-MS/MS analysis of in vitro released PMB
The sampled release medium was mixed with 5 μl of CS (1 mg/ml, DI water) as an internal standard. To precipitate serum proteins, 0.6 ml of trichloroacetic acid was added to the mixture and vigorously shaken for 1 hour. PMB was separated by centrifugation at 16,100 rcf for 20 min. ACN (0.3 ml) and 8 μl of pyridine were added to the supernatant and analyzed by LC-MS/MS (Agilent 6460 QQQ with the Agilent 1200 Rapid Resolution HPLC). Atlantis T3 column (15 cm by 2.1 mm; particle size, 3 μm) was used for the analysis. Agilent 6460 QQQ equipped with Agilent Jet Stream Electrospray Ionization was operated in the positive ion. The following parameters were used for operation: dwell time, 200 ms; collision energy, 15 V; cell accelerator voltage, 7 V; and fragmentor, 160 V. The precursor ions [mass/charge ratio (m/z)] for PMB and CS were 602.3 and 585.3, respectively. The product ions (m/z) for PMB and CS were 241.1. The injection volume was 10 μl. A gradient chromatography was performed with 0.1% formic acid in DI water (A) and 0.1% formic acid in ACN (B) at a flow rate of 0.3 ml/min. Initially, the column started with the mobile phase consisting of 90% solvent A and 10% solvent B, and a linear gradient was then applied, decreasing solvent A from 90% to 5% over 10 min. Subsequently, solvent A was returned to 90% in 1 min. The mobile phase was maintained as the initial condition to re-equilibrate for an additional 5 min. PMB was dissolved in DI water to 0.39 to 25 μg/ml and analyzed in a similar manner to build a calibration curve.
Nanoparticle size stability in serum
To evaluate the size stability, D-TZP equivalent to 25 to 100 μg of PMB was suspended in 1 ml of 50% FBS and incubated for 24 hours at 37°C. The particle size distribution was measured by a Malvern Zetasizer Nano ZS90.
Cell culture
THP1-XBlue-MD2-CD14 cells were cultured in RPMI 1640, supplemented with 10% FBS, 1% penicillin-streptomycin, 1 mM sodium pyruvate, 10 mM Hepes, normocin (100 μg/ml), zeocin (200 μg/ml), and G418 (250 μg/ml). J774A.1 macrophages were cultured in DMEM, supplemented with 10% FBS and 1% penicillin-streptomycin.
Cytotoxicity of PMB and D-TZP
Cytotoxic concentration range of free PMB was first determined with THP1-XBlue-MD2-CD14 cells and J774A.1 macrophages in a concentration ranging from 25 to 150 μg/ml by the MTT assay. THP1-XBlue-MD2-CD14 cells were seeded in a 96-well plate at a density of 105 cells per well and treated with PMB. After 24-hour incubation, the medium was removed by centrifugation (233 rcf, 5 min), and the cells were resuspended in 115 μl of medium containing MTT (0.75 mg/ml) and incubated for 3.5 hours. One hundred microliters of stop solution was added, and the plate was incubated in 37°C for 24 hours to dissolve the formazan crystal. The absorbance of dissolved formazan was measured at 560 nm by the SpectraMax M3 microplate reader. Cell viability was expressed as the percent of the absorbance of treated samples relative to the absorbance of the D5W treated control. J774A.1 macrophages were suspended in complete medium and plated in a 48-well plate at a density of 105 cells per well. After 24-hour incubation at 37°C in 5% CO2, the cells were treated with PMB for an additional 24 hours and tested by the MTT assay. Next, cytotoxicity of free PMB, TZP, and D-TZP was compared using PI and flow cytometry. THP1-XBlue-MD2-CD14 cells were seeded in a 24-well plate at a density of 5 × 104 cells per well with 0.463 ml of complete medium. After overnight incubation at 37°C in a 5% CO2 atmosphere, the cells were treated with free PMB, TZP, or D-TZP. After 24 hours of incubation, the cells were collected by centrifugation at 233 rcf for 5 min. The cell pellet was dispersed in 300 μl of flow cytometry staining buffer. Twelve microliters of PI (10 μg/ml) was added to cells, and the mixture was incubated in the dark for 1 min. The PI fluorescence of 10,000 cells was measured with the BD Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA) via the FL-2 detector. Nanoparticles and cell debris were excluded by gating, but all cell population (dead or alive) was analyzed. J774A.1 macrophages were plated in a 12-well plate at a density of 1 × 105 cells per well with 0.925 ml of complete medium and treated in the same manner.
Effect of PMB and D-TZP on cell membrane
The effect of D-TZP on J774A.1 cell membrane was monitored by two methods (SEM and confocal microscopy). For SEM, J774A.1 macrophages were plated in a six-well plate at a density of 3 × 105 cells per well with 3 ml of complete medium. After overnight incubation at 37°C in a 5% CO2 atmosphere, the cells were treated with either free PMB, TZP, or D-TZP at a concentration equivalent to 150 μg/ml PMB for 12 hours. The cells were centrifuged at 233 rcf for 3 min. The medium was removed, and 2.5% glutaraldehyde solution was added for fixation overnight at 4°C. The cells were then rinsed three times with water (5 min each) and incubated in 2% osmium tetroxide for 1 hour. After rinsing another three times with water (5 min each), the cells were dehydrated with a series of ethanol gradients (50, 75, and 95%) for 10 min at each step. The dehydrated cells were rinsed with 100% ethanol three times for 10 min each time, stored in a 1:1 mixture of HMDS and ethanol in 1:1 for 30 min, and rinsed with 100% HMDS for 30 min twice. After overnight drying, the cells were placed on a carbon tape attached to an aluminum stud and coated with platinum for SEM analysis. The morphology of treated J774A.1 macrophages was then observed by a FEI NOVA NanoSEM scanning electron microscope. For confocal microscopy, J774A.1 macrophages were plated in a 12-well plate at a density of 105 cells per well with 0.925 ml of complete medium. The cells were treated in the same way as above, collected by centrifugation at 233 rcf for 3 min, and fixed with 4% paraformaldehyde in PBS for 15 min. The fixed cells were stained with WGA (5 μg/ml) for 10 min and Hoechst 33342 (1 μM) for 5 min, mounted on a glass slide with an antifade reagent (ProLong Gold Antifade Mountant), and imaged with a Nikon A1Rsi confocal microscope.
In vivo safety
Male C57BL/6 mice (6 weeks old for weight monitoring; 8 to 10 weeks old for blood chemistry and histology) were used to evaluate the safety of D-TZP and TZP in comparison with free PMB. The treatment was administered by intraperitoneal (2.5 to 40 mg/kg) or intravenous (3 to 10 mg/kg) injection. Animals were monitored over 1 week for signs of toxicity including body weight change. For blood chemistry, blood was collected at 12 hours after intravenous injection of vehicle (D5W), free PMB, D-TZP, or TZP (all equivalent to 10 mg/kg PMB). Animals were euthanized by CO2 asphyxiation. Blood was obtained by cardiac puncture. Major organs were collected at 24 hours after intravenous injection of vehicle (D5W), free PMB (5 mg/kg), D-TZP, or TZP (equivalent to 10 mg/kg PMB), fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) for histological evaluation by a board-certified veterinary pathologist. The samples were imaged with an Olympus BX43 light microscope. All H&E slides were digitized at ×40 magnification using the Aperio VERSA 8-slide scanner. All images were acquired using Aperio ImageScope 12.3 (Leica Biosystems). To evaluate the safety of multiple intravenous injections, PMB (5 mg/kg) or D-TZP equivalent to 10 mg/kg PMB was given every 2 days up to four times total with body weight monitoring over 2 weeks.
In vitro anti-LPS activity
Five microliters of LPS (400 ng/ml) was mixed with 15 μl of free PMB, D-TZP, or TZP at different concentrations in a flat-bottom 96-well plate. THP1-XBlue-MD2-CD14 cell suspension (105 cells in 180 μl) was then added to the mixtures. After 24-hour incubation, cells were centrifuged at 233 rcf for 5 min. Twenty microliters of the cell medium was mixed with the QUANTI-Blue reagent. After 2-hour incubation, the color was measured at 620 nm by a Spectra Max M3 microplate reader (Molecular Devices, Sunnyvale, CA). In another test, 5 μl of LPS was preincubated with 15 μl of free PMB, D-TZP, or TZP for 1 hour. Each mixture was centrifuged at 16,100 rcf for 20 min to separate a supernatant. The supernatant was incubated with THP1-XBlue-MD2-CD14 cell suspension (105 cells in 180 μl) for 24 hours. The medium was analyzed in the same manner as above.
Antimicrobial activities
The MICs of D-TZP and its precursors, PMB, and control antibiotics (gentamicin and imipenem), against E. coli, K. pneumoniae, and P. aeruginosa strains, were determined by the broth microdilution method, according to guidelines outlined by the Clinical and Laboratory Standards Institute (72). Test agents were serially diluted along the plates. A bacterial solution equivalent to 0.5 McFarland standard was prepared and diluted in cation-adjusted Mueller Hinton broth to achieve a bacterial concentration of ~5 × 105 colony-forming units/ml and incubated with serial dilutions of test agents, aerobically at 37°C for 16 to 18 hours. MICs were determined as the minimum concentrations of each agent that completely inhibited the bacterial growth as observed visually.
Effect on bacterial membrane
K. pneumoniae NR-41920 was treated with either vehicle (D5W), free PMB, or D-TZP at 5× MIC for 1.5 hours. The bacteria were collected by centrifugation at 5000 rcf for 10 min, fixed in 2.5% glutaraldehyde solution at 4°C, and placed on poly-l-lysine–coated coverslip. After 1 hour, the coverslip was rinsed three times with water (5 min each) and incubated in 2% osmium tetroxide for 30 min. SEM imaging was performed as described in the previous section.
Therapeutic efficacy in LPS-induced endotoxemia model
Six- to 8-week-old C57BL/6 mice (male and female) were challenged with an intraperitoneal injection of LPS solution (20 mg/kg) (in D5W) via a 20G catheter (Exel International Disposable Safelet I.V.), which causes systemic inflammation similar to initial clinical features of sepsis (34, 73). D-TZP or TZP was administered in three regimens: (i) intraperitoneally at 40 mg/kg PMB equivalent, simultaneously with LPS ([LPS + treatment], IP); (ii) intraperitoneally at 40 mg/kg PMB equivalent, immediately after LPS challenge (LPS, IP ➔ treatment, IP); and (iii) intravenously at 10 mg/kg PMB equivalent, 2 hours after LPS challenge (LPS, IP + 2 hours ➔ treatment, IV). A vehicle control (D5W) or free PMB (5 mg/kg, IV) was given in the same manner for comparison. Buprenorphine (0.05 mg/kg) was injected subcutaneously shortly after the treatment and every 2 to 12 hours as the signs of discomfort were observed. The body weight was recorded at each observation. When an animal was found dead at the time of observation, the time of death was estimated to be halfway between the last two observation times. When animals were lethargic and cold at the time of observation, the animals were euthanized by CO2 asphyxiation. The mouse survival was monitored for 2 weeks.
Pharmacokinetics and biodistribution of PMB
Six- to 8-week-old male C57BL/6 mice were administered with free PMB or D-TZP at a dose equivalent to 5 mg/kg PMB by tail vein injection. At predetermined time points, three mice per group were euthanized for the collection of blood and major organs. Blood was obtained by cardiac puncture and collected in a BD Vacutainer tube (lithium heparin). Plasma was separated from blood by 10 min centrifugation at 1000 rcf and mixed with the same volume of 5% trichloroacetic acid and CS (internal standard, 0.83 μg/ml as a final concentration) by vigorous vortex for 30 min and centrifuged at 16,100 rcf for 20 min. ACN (75 μl) and pyridine (2 μl) were added to the supernatant for analysis. All organs, except for the liver, were incubated in 0.4 ml of 5% trichloroacetic acid, spiked with CS (5 μg/ml), homogenized by an Omni Tissue Master 125 homogenizer (Kennesaw, GA, USA), and centrifuged at 16,100 rcf for 20 min. The supernatant was mixed with ACN in 4:1 v/v ratio for LC-MS/MS analysis. The liver was cut into four pieces, each of which was incubated in 0.4 ml of 5% trichloroacetic acid and spiked with CS (7.5 μg/ml). Each supernatant was collected by centrifugation at 16,100 rcf for 20 min, pooled, and analyzed by LC-MS/MS. Standards with known amounts of PMB were prepared to estimate the concentrations in the samples. The blood and organs from mice with no treatments were spiked with standards and processed in the same manner. The elimination half-lives of PMB in circulation were calculated by the PKSolver using a noncompartmental model (Microsoft Excel, add-in program) (74).
D-TZP interaction with J774A.1 macrophages
J774A.1 macrophages were stained with CellTracker Green CMFDA (5.4 μM) for 30 min and plated in a four-chamber glass bottom dish at a density of 105 cells per well with 0.45 ml of complete medium. After 24.5-hour incubation, the cells were treated with DiI-labeled D-TZP (at 150 μg/ml PMB equivalent) for 4 hours. D-TZP–containing medium was then removed by centrifugation at 233 rcf for 3 min. The cells were stained with Hoechst 33342 (1 μM) in PBS for 5 min and imaged with a Nikon A1Rsi confocal microscope. To exclude the possibility of dye leaching out, another set of macrophages was incubated with an equal amount of free DiI and imaged in the same manner.
TNF-α production by particle-pretreated J774A.1 macrophages
J774A.1 macrophages were suspended in complete medium and plated in a 12-well plate at a density of 105 cells per well. After 24-hour incubation, the cells were treated with either D-TZP or TZP at a concentration equivalent to 25 μg/ml PMB and incubated for an additional 24 hours. The cells were collected, rinsed with PBS two times to remove uninternalized or loosely bound D-TZP or TZP, and incubated in complete medium containing LPS (10 to 75 ng/ml). After 12 hours, the supernatant was collected and analyzed by ELISA to quantify TNF-α production.
Therapeutic efficacy in polymicrobial sepsis model by CLP
The efficacy of D-TZP and TZP was evaluated in a CLP model. The abdomen of 6-week-old male C57BL/6 mice was cleaned with a hair-removal cream (Nair, Church & Dwight Co. Inc). The mice were anesthetized by 2.5% isoflurane. Betadine 0.5% solution and 70% isopropyl alcohol swap were alternately applied three times to disinfect the shaved abdomen. Midline laparotomy was performed under an aseptic condition to exteriorize the cecum. The cecum was ligated below the ileocecal valve with a 5-0 suture (perma-hand silk, Ethicon) and perforated twice with a 20G needle on the same side of the cecum to release feces into the peritoneal cavity. After perforation, the intestines were returned to the abdominal cavity. The abdominal wall and the skin were closed with 5-0 suture. Buprenorphine (0.05 mg/kg) was injected subcutaneously right after the procedure. The animals were kept on top of a warming pack until recovery from anesthesia. D-TZP or TZP (10 mg/kg PMB equivalent) was administered by intravenous injection at 2 hours after CLP and every 2 subsequent days a maximum total of three times (CLP + 2 hours ➔ treatment, IV, q2d × 3). A vehicle control (D5W) or free PMB (5 mg/kg; intravenous LD50) was administered in the same manner. Animals were monitored as described in the endotoxemia model.
Cytokine and D-dimer levels in CLP-challenged mice
The CLP procedure was performed on 6-week-old male C57BL/6 mice, and D-TZP or TZP (10 mg/kg PMB equivalent) was administered intravenously at 2 hours after CLP. D5W was administered as a vehicle control. Three hours (for cytokine measurement) or 24 hours (for D-dimer) after CLP, blood was obtained by cardiac puncture and collected in a BD Vacutainer tube (lithium heparin). Plasma was separated from blood through 10 min centrifugation at 1000 rcf and diluted with assay diluents, 5 times for TNF-α, 20 times for IL-10, and 10 times for D-dimer analysis. TNF-α, IL-10, and D-dimer in the diluted plasma were quantified by ELISA.
Vitamin D metabolism in healthy mice
To evaluate whether the encapsulated vitamin D was metabolized in mice, healthy 8-week-old male C57BL/6 mice were administered with vehicle (D5W), free vitamin D (5 mg/kg) (cholecalciferol, dissolved in 2.5% v/v ethanol and 2.5% v/v propylene glycol), or D-TZP (equivalent to 5 mg/kg vitamin D) by tail-vein injection. Twenty-four hours later, blood was obtained by terminal cardiac puncture and collected in a BD Vacutainer tube (lithium heparin). Plasma was separated from the blood by 10-min centrifugation at 1000 rcf and diluted with assay diluents. The concentration of 25OHD, an intermediate vitamin D metabolite, in the diluted plasma was determined by ELISA.
Statistical analysis
Statistical analysis of all data was performed using GraphPad Prism 9 (La Jolla, CA). Statistical significance was accessed by unpaired t test or one-/two-way analysis of variance (ANOVA) followed by the recommended multiple comparisons test. P < 0.05 was considered statistically significant.
Acknowledgments
Funding: This work was supported by NIH grant R21 AI119479 (to Y.Y.), the Graduate Assistance in Areas of National Need Fellowship (S.A.Y.), and the Purdue University Institute for Drug Discovery (to Y.Y.). Author contributions: Conceptualization: S.A.Y., M.D.T., and Y.Y. Methodology: S.A.Y., H.K., M.S.T., and Y.Y. Investigation: S.A.Y., H.K., and N.S.A. Data curation: S.A.Y. and Y.Y. Visualization: S.A.Y. and A.M.D. Funding acquisition: Y.Y. and S.A.Y. Project administration: Y.Y. Supervision: L.T.L., M.N.S., and Y.Y. Writing—original draft: S.A.Y. and Y.Y. Writing—review and editing: S.A.Y., H.K., N.S.A., A.M.D., M.S.T., M.D.T., L.T.L., M.N.S., and Y.Y. Competing interests: S.A.Y. and Y.Y. are inventors on a patent application (no. PCT/US20/47289) related to this work filed by the Purdue Research Foundation (PRF) on 21 August 2020. Y.Y. is also an inventor on patents (US patents 9,962,401 B2, 9,517,246 B2, and 9,393,262 B2) related to LMZWC, filed by the PRF on 8 November 2016, 18 July 2016, and 27 September 2012 and published on 8 May 2018, 13 December 2016, and 19 July 2016, respectively. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. LMZWC, D-TZP, and TZP can be provided by Y.Y. pending scientific review and a completed material transfer agreement. Requests for LMZWC, D-TZP, and TZP should be submitted to Y.Y. (yyeo@purdue.edu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabj1577/DC1
REFERENCES AND NOTES
- 1.Rhodes A., Evans L. E., Alhazzani W., Levy M. M., Antonelli M., Ferrer R., Kumar A., Sevransky J. E., Sprung C. L., Nunnally M. E., Rochwerg B., Rubenfeld G. D., Angus D. C., Annane D., Beale R. J., Bellinghan G. J., Bernard G. R., Chiche J. D., Coopersmith C., De Backer D. P., French C. J., Fujishima S., Gerlach H., Hidalgo J. L., Hollenberg S. M., Jones A. E., Karnad D. R., Kleinpell R. M., Koh Y., Lisboa T. C., Machado F. R., Marini J. J., Marshall J. C., Mazuski J. E., McIntyre L. A., McLean A. S., Mehta S., Moreno R. P., Myburgh J., Navalesi P., Nishida O., Osborn T. M., Perner A., Plunkett C. M., Ranieri M., Schorr C. A., Seckel M. A., Seymour C. W., Shieh L., Shukri K. A., Simpson S. Q., Singer M., Thompson B. T., Townsend S. R., Van der Poll T., Vincent J. L., Wiersinga W. J., Zimmerman J. L., Dellinger R. P., Surviving sepsis campaign: International guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 43, 304–377 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Esteban A., Frutos-Vivar F., Ferguson N. D., Penuelas O., Lorente J. A., Gordo F., Honrubia T., Algora A., Bustos A., Garcia G., Diaz-Reganon I. R., de Luna R. R., Sepsis incidence and outcome: Contrasting the intensive care unit with the hospital ward. Crit. Care Med. 35, 1284–1289 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Fleischmann C., Scherag A., Adhikari N. K., Hartog C. S., Tsaganos T., Schlattmann P., Angus D. C., Reinhart K., Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am. J. Respir. Crit. Care Med. 193, 259–272 (2016). [DOI] [PubMed] [Google Scholar]
- 4.C. M. Torio, B. J. Moore, in Healthcare Cost and Utilization Project (HCUP) Statistical Briefs (Agency for Healthcare Research and Quality (US), 2016). [PubMed]
- 5.van der Poll T., van de Veerdonk F. L., Scicluna B. P., Netea M. G., The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 17, 407–420 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Gotts J. E., Matthay M. A., Sepsis: Pathophysiology and clinical management. BMJ 353, i1585 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Fink M. P., Warren H. S., Strategies to improve drug development for sepsis. Nat. Rev. Drug Discov. 13, 741–758 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Evans M. E., Feola D. J., Rapp R. P., Polymyxin B sulfate and colistin: Old antibiotics for emerging multiresistant Gram-negative bacteria. Ann. Pharmacother. 33, 960–967 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Trimble M. J., Mlynárčik P., Kolář M., Hancock R. E. W., Polymyxin: Alternative mechanisms of action and resistance. Cold Spring Harb. Perspect. Med. 6, a025288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roger T., Froidevaux C., Le Roy D., Reymond M. K., Chanson A.-L., Mauri D., Burns K., Riederer B. M., Akira S., Calandra T., Protection from lethal Gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc. Natl. Acad. Sci. U.S.A. 106, 2348–2352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rifkind D., Prevention by polymyxin B of endotoxin lethality in mice. J. Bacteriol. 93, 1463–1464 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayr F. B., Yende S., Angus D. C., Epidemiology of severe sepsis. Virulence 5, 4–11 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai C., Xiao X., Li J., Ciccotosto G. D., Cappai R., Tang S., Schneider-Futschik E. K., Hoyer D., Velkov T., Shen J., Molecular mechanisms of neurotoxicity induced by polymyxins and chemoprevention. ACS Chem. Nerosci. 10, 120–131 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Falagas M. E., Kasiakou S. K., Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit. Care 10, R27 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harm S., Gabor F., Hartmann J., Low-dose polymyxin: An option for therapy of Gram-negative sepsis. Innate Immun. 22, 274–283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peer D., Karp J. M., Hong S., Farokhzad O. C., Margalit R., Langer R., Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2, 751–760 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Blanco E., Shen H., Ferrari M., Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 33, 941–951 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henry B. D., Neill D. R., Becker K. A., Gore S., Bricio-Moreno L., Ziobro R., Edwards M. J., Mühlemann K., Steinmann J., Kleuser B., Japtok L., Luginbühl M., Wolfmeier H., Scherag A., Gulbins E., Kadioglu A., Draeger A., Babiychuk E. B., Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 33, 81–88 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Lam S. J., O’Brien-Simpson N. M., Pantarat N., Sulistio A., Wong E. H. H., Chen Y.-Y., Lenzo J. C., Holden J. A., Blencowe A., Reynolds E. C., Qiao G. G., Combating multidrug-resistant Gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nat. Microbiol. 1, 16162 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Wang Z., Li J., Cho J., Malik A. B., Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat. Nanotechnol. 9, 204–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spence S., Greene M. K., Fay F., Hams E., Saunders S. P., Hamid U., Fitzgerald M., Beck J., Bains B. K., Smyth P., Themistou E., Small D. M., Schmid D., O’Kane C. M., Fitzgerald D. C., Abdelghany S. M., Johnston J. A., Fallon P. G., Burrows J. F., McAuley D. F., Kissenpfennig A., Scott C. J., Targeting Siglecs with a sialic acid-decorated nanoparticle abrogates inflammation. Sci. Transl. Med. 7, 303ra140 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Shen G., Xing R., Zhang N., Chen C., Ma G., Yan X., Interfacial cohesion and assembly of bioadhesive molecules for design of long-term stable hydrophobic nanodrugs toward effective anticancer therapy. ACS Nano 10, 5720–5729 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Shin M., Lee H.-A., Lee M., Shin Y., Song J.-J., Kang S.-W., Nam D.-H., Jeon E. J., Cho M., Do M., Park S., Lee M. S., Jang J.-H., Cho S.-W., Kim K.-S., Lee H., Targeting protein and peptide therapeutics to the heart via tannic acid modification. Nat. Biomed. Eng. 2, 304–317 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Xu P., Bajaj G., Shugg T., Van Alstine W. G., Yeo Y., Zwitterionic chitosan derivatives for pH-sensitive stealth coating. Biomacromolecules 11, 2352–2358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin M., Park S.-G., Oh B.-C., Kim K., Jo S., Lee M. S., Oh S. S., Hong S.-H., Shin E.-C., Kim K.-S., Kang S.-W., Lee H., Complete prevention of blood loss with self-sealing haemostatic needles. Nat. Mater. 16, 147–152 (2017). [DOI] [PubMed] [Google Scholar]
- 26.G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts (John Wiley and Sons, ed. 3, 2004), pp. 366. [Google Scholar]
- 27.Zhang Y., Thomas Y., Kim E., Payne G. F., pH- and voltage-responsive chitosan hydrogel through covalent cross-linking with catechol. J. Phys. Chem. B. 116, 1579–1585 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Petty H. R., Hafeman D. G., McConnell H. M., Disappearance of macrophage surface folds after antibody-dependent phagocytosis. J. Cell Biol. 89, 223–229 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sigma-Aldrich (Sigma-Aldrich safety data sheet, version 3.7, 12/13/2016).
- 30.Foit L., Thaxton C. S., Synthetic high-density lipoprotein-like nanoparticles potently inhibit cell signaling and production of inflammatory mediators induced by lipopolysaccharide binding Toll-like receptor 4. Biomaterials 100, 67–75 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Sharif O., Bolshakov V. N., Raines S., Newham P., Perkins N. D., Transcriptional profiling of the LPS induced NF-κB response in macrophages. BMC Immunol. 8, 1 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho E. J., Doh K.-O., Park J., Hyun H., Wilson E. M., Snyder P. W., Tsifansky M. D., Yeo Y., Zwitterionic chitosan for the systemic treatment of sepsis. Sci. Rep. 6, 29739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bajaj G., Van Alstine W. G., Yeo Y., Zwitterionic chitosan derivative, a new biocompatible pharmaceutical excipient, prevents endotoxin-mediated cytokine release. PLOS ONE 7, e30899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buras J. A., Holzmann B., Sitkovsky M., Animal models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 4, 854–865 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Hirano S., Migratory responses of PMN after intraperitoneal and intratracheal administration of lipopolysaccharide. Am. J. Physiol. Lung Cell. Mol. Physiol. 270, L836–L845 (1996). [DOI] [PubMed] [Google Scholar]
- 36.He J., Gao S., Hu M., Chow D. S., Tam V. H., A validated ultra-performance liquid chromatography–tandem mass spectrometry method for the quantification of polymyxin B in mouse serum and epithelial lining fluid: Application to pharmacokinetic studies. J. Antimicrob. Chemother. 68, 1104–1110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouyang B., Poon W., Zhang Y.-N., Lin Z. P., Kingston B. R., Tavares A. J., Zhang Y., Chen J., Valic M. S., Syed A. M., MacMillan P., Couture-Senécal J., Zheng G., Chan W. C. W., The dose threshold for nanoparticle tumour delivery. Nat. Mater. 19, 1362–1371 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Toscano M. G., Ganea D., Gamero A. M., Cecal ligation puncture procedure. JoVE , e2860 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kasten K. R., Muenzer J. T., Caldwell C. C., Neutrophils are significant producers of IL-10 during sepsis. Biochem. Biophys. Res. Commun. 393, 28–31 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foey A. D., Parry S. L., Williams L. M., Feldmann M., Foxwell B. M., Brennan F. M., Regulation of monocyte IL-10 synthesis by endogenous IL-1 and TNF-α: Role of the p38 and p42/44 mitogen-activated protein kinases. J. Immunol. 160, 920–928 (1998). [PubMed] [Google Scholar]
- 41.Restagno D., Venet F., Paquet C., Freyburger L., Allaouchiche B., Monneret G., Bonnet J. M., Louzier V., Mice survival and plasmatic cytokine secretion in a "Two Hit" model of sepsis depend on intratracheal Pseudomonas aeruginosa bacterial load. PLOS ONE 11, e0162109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adam S. S., Key N. S., Greenberg C. S., D-dimer antigen: Current concepts and future prospects. Blood 113, 2878–2887 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Song J., Hu D., He C., Wang T., Liu X., Ma L., Lin Z., Chen Z., Novel biomarkers for early prediction of sepsis-induced disseminated intravascular coagulation in a mouse cecal ligation and puncture model. J. Inflamm. 10, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seymour C. W., Gesten F., Prescott H. C., Friedrich M. E., Iwashyna T. J., Phillips G. S., Lemeshow S., Osborn T., Terry K. M., Levy M. M., Time to treatment and mortality during mandated emergency care for sepsis. N. Engl. J. Med. 376, 2235–2244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pea F., Viale P., Bench-to-bedside review: Appropriate antibiotic therapy in severe sepsis and septic shock—does the dose matter? Crit. Care 13, 214 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin D., Mumper R. J., Plant phenolics: Extraction, analysis and their antioxidant and anticancer properties. Molecules 15, 7313–7352 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan H., Wang J., Zhang Q., Jin Z., Tannic acid-based multifunctional hydrogels with facile adjustable adhesion and cohesion contributed by polyphenol supramolecular chemistry. ACS Omega 2, 6668–6676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park J., Pei Y., Hyun H., Castanares M. A., Collins D. S., Yeo Y., Small molecule delivery to solid tumors with chitosan-coated PLGA particles: A lesson learned from comparative imaging. J. Control. Release 268, 407–415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosenfeld Y., Shai Y., Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta 1758, 1513–1522 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Matsuzaki K., Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 1788, 1687–1692 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Jeong Y.-I., Jung I. D., Lee C.-M., Chang J. H., Chun S. H., Noh K. T., Jeong S. K., Shin Y. K., Lee W. S., Kang M. S., Lee S.-Y., Lee J.-D., Park Y.-M., The novel role of platelet-activating factor in protecting mice against lipopolysaccharide-induced endotoxic shock. PLOS ONE 4, e6503 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soromou L. W., Jiang L., Wei M., Chen N., Huo M., Chu X., Zhong W., Wu Q., Baldé A., Deng X., Feng H., Protection of mice against lipopolysaccharide-induced endotoxic shock by pinocembrin is correlated with regulation of cytokine secretion. J. Immunotoxicol. 11, 56–61 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Berger J.-M., Loza Valdes A., Gromada J., Anderson N., Horton J. D., Inhibition of PCSK9 does not improve lipopolysaccharide-induced mortality in mice. J. Lipid Res. 58, 1661–1669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soh M., Kang D. W., Jeong H.-G., Kim D., Kim D. Y., Yang W., Song C., Baik S., Choi I. Y., Ki S.-K., Kwon H. J., Kim T., Kim C. K., Lee S.-H., Hyeon T., Ceria-zirconia nanoparticles as an enhanced multi-antioxidant for sepsis treatment. Angew. Chem. Int. Ed. 56, 11399–11403 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Miksa M., Wu R., Dong W., Komura H., Amin D., Ji Y., Wang Z., Wang H., Ravikumar T. S., Tracey K. J., Wang P., Immature dendritic cell-derived exosomes rescue septic animals via milk fat globule epidermal growth factor VIII. J. Immunol. 183, 5983–5990 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X., Gu H., Qin D., Yang L., Huang W., Essandoh K., Wang Y., Caldwell C. C., Peng T., Zingarelli B., Fan G.-C., Exosomal miR-223 contributes to mesenchymal stem cell-elicited cardioprotection in polymicrobial sepsis. Sci. Rep. 5, 13721 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zavascki A. P., Nation R. L., Nephrotoxicity of polymyxins: Is there any difference between colistimethate and polymyxin B? Antimicrob. Agents Chemother. 61, e02319-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He C., Hu Y., Yin L., Tang C., Yin C., Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 31, 3657–3666 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Thamphiwatana S., Angsantikul P., Escajadillo T., Zhang Q., Olson J., Luk B. T., Zhang S., Fang R. H., Gao W., Nizet V., Zhang L., Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc. Natl. Acad. Sci. U.S.A. 114, 11488–11493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yao Z., Mates J. M., Cheplowitz A. M., Hammer L. P., Maiseyeu A., Phillips G. S., Wewers M. D., Rajaram M. V. S., Robinson J. M., Anderson C. L., Ganesan L. P., Blood-borne lipopolysaccharide is rapidly eliminated by liver sinusoidal endothelial cells via high-density lipoprotein. J. Immunol. 197, 2390–2399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shao B., Munford R. S., Kitchens R., Varley A. W., Hepatic uptake and deacylation of the LPS in bloodborne LPS-lipoprotein complexes. Innate Immun. 18, 825–833 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Surewaard B. G. J., Deniset J. F., Zemp F. J., Amrein M., Otto M., Conly J., Omri A., Yates R. M., Kubes P., Identification and treatment of the Staphylococcus aureus reservoir in vivo. J. Exp. Med. 213, 1141–1151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gombart A. F., Borregaard N., Koeffler H. P., Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 19, 1067–1077 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Dürr U. H. N., Sudheendra U. S., Ramamoorthy A., LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 1758, 1408–1425 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Sadeghi K., Wessner B., Laggner U., Ploder M., Tamandl D., Friedl J., Zügel U., Steinmeyer A., Pollak A., Roth E., Boltz-Nitulescu G., Spittler A., Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur. J. Immunol. 36, 361–370 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Equils O., Naiki Y., Shapiro A. M., Michelsen K., Lu D., Adams J., Jordan S., 1,25-Dihydroxyvitamin D inhibits lipopolysaccharide-induced immune activation in human endothelial cells. Clin. Exp. Immunol. 143, 58–64 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.G. Jones, D. E. Prosser, M. Kaufmann, in Chapter 5—The Activating Enzymes of Vitamin D Metabolism (25- and 1α-Hydroxylases), Vitamin D (Fourth Edition), D. Feldman, Ed. (Academic Press, 2018), pp. 57–79. [Google Scholar]
- 68.Cignachi N. P., Ribeiro A., Machado G. D. B., Cignachi A. P., Kist L. W., Bogo M. R., Silva R. B. M., Campos M. M., Bone regeneration in a mouse model of type 1 diabetes: Influence of sex, vitamin D3, and insulin. Life Sci. 263, 118593 (2020). [DOI] [PubMed] [Google Scholar]
- 69.An J. H., Cho D. H., Lee G. Y., Kang M. S., Kim S. J., Han S. N., Effects of Vitamin D supplementation on CD4+ T cell subsets and mTOR signaling pathway in high-fat-diet-induced obese mice. Nutrients 13, 796 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amoozgar Z., Park J., Lin Q., Yeo Y., Low molecular-weight chitosan as a pH-sensitive stealth coating for tumor-specific drug delivery. Mol. Pharm. 9, 1262–1270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ejima H., Richardson J. J., Liang K., Best J. P., van Koeverden M. P., Such G. K., Cui J., Caruso F., One-step assembly of coordination complexes for versatile film and particle engineering. Science 341, 154–157 (2013). [DOI] [PubMed] [Google Scholar]
- 72.C. Clinical and Laboratory Standards Institute, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition (Clinical and Laboratory Standards Institute, 2012), pp. CLSI document M07-A09. [Google Scholar]
- 73.Copeland S., Warren H. S., Lowry S. F., Calvano S. E., Remick D.; Inflammation and the Host Response to Injury Investigators , Acute inflammatory response to endotoxin in mice and humans. Clin. Diagn. Lab. Immunol. 12, 60–67 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Y., Huo M., Zhou J., Xie S., PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 99, 306–314 (2010). [DOI] [PubMed] [Google Scholar]
- 75.Weiler H., Lindner V., Kerlin B., Isermann B. H., Hendrickson S. B., Cooley B. C., Meh D. A., Mosesson M. W., Shworak N. W., Post M. J., Conway E. M., Ulfman L. H., von Andrian U. H., Weitz J. I., Characterization of a mouse model for thrombomodulin deficiency. Arterioscler. Thromb. Vasc. Biol. 21, 1531–1537 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabj1577/DC1