

Time-lapse imaging delineates the mitochondrial life cycle in neurons regulated by PINK1 and Parkin.
Abstract
Altered mitochondrial quality control and dynamics may contribute to neurodegenerative diseases, including Parkinson’s disease, but we understand little about these processes in neurons. We combined time-lapse microscopy and correlative light and electron microscopy to track individual mitochondria in neurons lacking the fission-promoting protein dynamin-related protein 1 (Drp1) and delineate the kinetics of PINK1-dependent pathways of mitochondrial quality control. Depolarized mitochondria recruit Parkin to the outer mitochondrial membrane, triggering autophagosome formation, rapid lysosomal fusion, and Parkin redistribution. Unexpectedly, these mitolysosomes are dynamic and persist for hours. Some are engulfed by healthy mitochondria, and others are deacidified before bursting. In other cases, Parkin is directly recruited to the matrix of polarized mitochondria. Loss of PINK1 blocks Parkin recruitment, causes LC3 accumulation within mitochondria, and exacerbates Drp1KO toxicity to dopamine neurons. These results define a distinct neuronal mitochondrial life cycle, revealing potential mechanisms of mitochondrial recycling and signaling relevant to neurodegeneration.
INTRODUCTION
Changes in mitochondrial quality control and dynamics, the balance of mitochondrial fission and fusion, are implicated in the pathogenesis of several neurodegenerative diseases, especially Parkinson’s disease (PD) (1). The dopamine (DA) neurons in the substantia nigra (SN) that degenerate in PD are sensitive to changes in mitochondrial fission (2) and fusion (3), and altered expression or mutation of proteins implicated in familial forms of PD (e.g., synuclein, DJ-1, Parkin, PINK1, LRRK2, and VPS35) influences mitochondrial morphology and quality control (1, 4–6).
These findings raise the possibility that familial PD genes produce toxicity by altering mitochondrial fission and quality control and call into question how the level of fission and quality control may converge to affect cytotoxicity. In Drosophila, PINK1 compensates for the loss of the central mitochondrial fission protein dynamin-related protein 1 (Drp1) to maintain mitochondrial morphology and survival (7), and loss of PINK1 leads to neurodegeneration and death (8). Moreover, when mitochondria are depolarized, the mitochondrial fusion proteins Mfn1 and Mfn2 are targeted for degradation by Parkin in a PINK1-dependent manner (9, 10), suggesting that PINK1 increases the fission-fusion balance. However, it is unclear whether the effects of losing PINK1 on mitochondrial morphology are important to the toxicity of Drp1 loss. Instead, mitochondrial fission and PINK1 may converge at mitochondrial turnover, and the extent of fission may influence the toxicity of PINK1 loss by affecting the number of mitochondria needing degradation.
Drp1-dependent fission may be critical to mitochondrial turnover. In INS1 and COS cells, fission isolates mitochondria with lower mitochondrial membrane potential, and the depolarized mitochondria are degraded by autophagy (11). Fission may be required to produce mitochondria small enough to be engulfed by autophagosomes. Supporting this possibility, cardiomyocytes and mouse embryonic fibroblasts (MEFs) lacking Drp1 have decreased Parkin-dependent mitophagy after depolarization by the protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (9). They may also have fewer mitochondria targeted to lysosomes (12, 13). However, if fission is truly required for mitochondrial turnover, then the mitochondrial content of neurons would be expected to increase (rather than decrease) when fission is compromised (2), and several studies have failed to find a critical role for Drp1 in mitochondrial turnover (14–16). Moreover, the physiology of mitochondrial turnover is different in neurons from that in other cell types. In many cell lines, depolarization with CCCP causes rapid Parkin accumulation on the outer mitochondrial membrane (OM) and the complete removal of all mitochondria, while in neurons, the induction of Parkin-based mitophagy is far less robust, and there is no clear effect on mitochondrial content (17–19), suggesting that neuronal mitochondria may be more resistant to degradation.
To better understand how mitochondrial fission and PINK1/Parkin promote mitochondrial quality control in neurons, we developed neuronal culture and mouse models with deletion of Drp1 in DA neurons, with and without PINK1. We took advantage of the individual, large, and swollen Drp1KO mitochondria to track the fates and functions of individual mitochondria and delineate new mechanisms of mitochondrial quality control.
RESULTS
Midbrain DA neurons require PINK1 to survive when fission is compromised
Midbrain DA neurons lacking Drp1 have reduced mitochondrial mass, although the size of individual mitochondria is increased (2). We hypothesized that this reflects the ongoing degradation of Drp1KO mitochondria that is critical to DA neuron health. To determine whether disrupting turnover would enhance the toxicity of losing Drp1, we assessed the impact of deleting PINK1 (6, 20, 21). If Drp1KO mitochondria are turned over through a PINK1-dependent mechanism, loss of PINK1 would increase the toxicity of Drp1 loss, although PINK1 loss alone does not produce degeneration (22). Conversely, if Drp1KO mitochondria can no longer be turned over by conventional mitophagy, loss of PINK1 might have no effect.
We generated mice with targeted deletion of Drp1 in DA neurons on a PINK1 KO (knockout) background (DATcre-Drp1KO;PINK1 KO). In our prior report, loss of Drp1 alone in DA neurons led to the death of ~25% of mice by day 50 [(2); data reproduced in Fig. 1A, left]. As expected, loss of PINK1 alone had no effect on survival. However, loss of PINK1 in mice with Drp1KO DA neurons markedly impaired survival; ~85% of DATcre-Drp1KO;PINK1KO mice died by 50 days of age, while ~40% of mice with heterozygous loss of PINK1 died over the same period (Fig. 1A).
Fig. 1. Midbrain DA neurons require PINK1 to survive when fission is compromised.
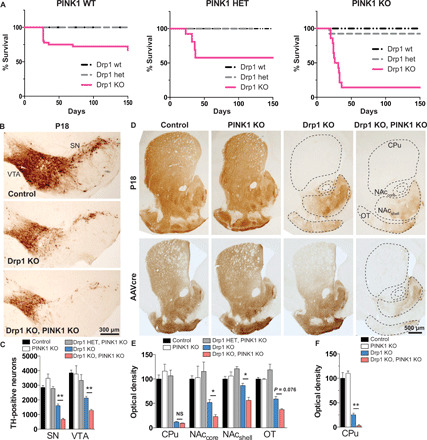
(A) Kaplan-Meier survival curve of Drp1 wt, heterozygous (het), and Drp1KO mice on a PINK1 wt background (left), PINK1 het background (middle), and PINK1 KO background (right). Drp1KO-PINK1KO mice were significantly more likely to die than either Drp1KO-PINK1 wt [hazard ratio (HR) 14.4, 95% confidence interval (CI): 4.67 to 44.6, P < 0.001 by log-rank (Mantel-Cox) test] or Drp1KO-PINK1 het mice (HR 4.21, 95% CI: 1.50 to 11.8, P < 0.01). n = 18 to 36 PINK1 wt mice per group, 5 to 13 PINK1 het mice per group, and 13 to 15 PINK1 KO mice per group. Data in left (PINK1 WT) were published (2) and reproduced here with permission from The Journal of Neuroscience. (B and C) Targeted deletion of Drp1 in DA neurons causes loss of DA cell bodies in the SN and VTA by P18 (assessed by TH staining), which is exacerbated by concurrent PINK1 loss. Data show means ± SEM, n = 4 mice per group with 5 to 6 fields per mouse. In (D) (top) and (E), the loss of cell bodies is preceded by early loss of DA terminals projecting to the caudate putamen (CPu) by P18. Although DA projections to the nucleus accumbens (NAc) core and shell and to the olfactory tubercule (OT) are relatively spared in Drp1KO, concurrent loss of PINK1 markedly increases their susceptibility. n = 4 mice per group, 14 to 20 fields per mouse. In (D) (bottom) and (F), AAVcre was delivered to the SNc of 6- to 7-month-old Drp1lox/lox and Drp1lox/lox;PINK1KO mice. Two months later, mice lacking PINK1 were more susceptible to Drp1 loss, indicating that the synergistic effect also occurs in adult animals. n = 3 to 5 mice per group with 4 to 5 fields per mouse. NS, not significant; *P < 0.05 and **P < 0.01 by one-way ANOVA with Games-Howell post hoc test.
We used stereology to examine survival of midbrain DA neurons from postnatal day 18 (P18) mice. We found that concurrent loss of PINK1 decreased the survival of Drp1KO DA neurons in the SN and ventral tegmental area (VTA) of Drp1KO mice, although PINK1 KO alone had no detectable impact (Fig. 1, B and C).
The terminals of Drp1KO DA neurons degenerate before the cell bodies (2). To determine whether PINK1 KO exacerbates this effect, we measured the optical density of TH (tyrosine hydroxylase) immunostaining. Drp1KO alone caused almost complete loss of DA terminals in the caudate and putamen by P18 but spared most terminals in the nucleus accumbens and olfactory tubercule (Fig. 1, D, top row, and E). On the PINK1 KO background, many of these otherwise resistant Drp1KO terminals were lost.
To determine whether PINK1 KO also causes degeneration when Drp1 is lost in adult animals, we injected AAVcre into 6- to 8-month-old Drp1lox/lox;PINK1KO and Drp1lox/lox mice. Mice were sacrificed after 2 months, and we assessed their striatal denervation. Deletion of Drp1 alone in adult mice produced severe loss of terminals in the caudate putamen, with only ~25% spared. However, many of those resistant terminals were lost when Drp1 was deleted on the PINK1 KO background (Fig. 1, D, bottom row, and F).
The increased toxicity of Drp1KO when PINK1 is absent could indicate that PINK1-dependent turnover of Drp1KO mitochondria enables neurons to survive or that PINK1 protects cells from loss of Drp1 (7) by increasing the fission/fusion balance, perhaps by degrading the fusion proteins Mfn1 and Mfn2 (9, 10). However, we failed to find evidence for the latter model, as PINK1 KO alone did not affect the morphology of mitochondria in DA neurons, and concurrent loss of PINK1 did not increase the proportion of Drp1KO DA neurons with swollen mitochondria at the cell body (fig. S1, A to D). Loss of either PINK1 or Drp1 can also increase oxidative stress (23, 24). However, neither Drp1KO, PINK1 KO, or the combination increased oxidative stress, as assessed by mitochondria-targeted redox-sensitive green fluorescent protein (roGFP) (mito-roGFP) (fig. S1, E and F). Thus, changes in mitochondrial morphology or oxidative stress are unlikely to underlie the toxicity of PINK1 KO to Drp1KO DA neurons.
Drp1KO increases mitochondrial targeting to lysosomes
To investigate the fate of Drp1KO mitochondria, we evaluated their fusion with lysosomes. In the absence of Drp1, an increased percentage of mitochondria colocalized with the lysosomal marker lysosomal associated membrane protein 1 (LAMP1), both in cultured hippocampal neurons where LAMP1 fluorescence ringed mitochondria and in DA neurons in vivo where LAMP1 fluorescence overlapped with mitochondria (fig. S2, A and B). These differential patterns of colocalization might reflect different stages of mitochondrial degradation or result from technical differences.
To quantify the impact of Drp1KO on mitochondrial pH, we used mitochondria-targeted mKeima (mitoKeima), a pH-sensitive reporter that changes fluorescence as mitochondria are delivered to lysosomes [peak excitation, 586 nm (acidic pH) and 440 nm (neutral pH)] (25). Drp1KO increased the ratio of red (acidic) to green (neutral) fluorescence, indicating that Drp1KO mitochondria are at a lower pH (Fig. 2, A to C). The increased red/green ratio in the Drp1KO group was driven by an upward shift in the mitoKeima signal in >50% of cells, while other Drp1KO cells remained at control levels (fig. S2, C and D). The increase was quenched by both ammonium chloride (NH4Cl), to alkalinize cells, and bafilomycin, which blocks the acidification of lysosomes and fusion of autophagosomes with lysosomes (26). To confirm that Drp1KO mitochondria fuse with lysosomes, we performed a Magic Red assay and observed fluorescence in some Drp1KO mitochondria (Fig. 2D and fig. S2E), indicating cleavage of the substrate by the lysosomal enzyme cathepsin B.
Fig. 2. Drp1KO increases mitochondrial targeting to lysosomes in a process that is facilitated by PINK1 and Parkin.
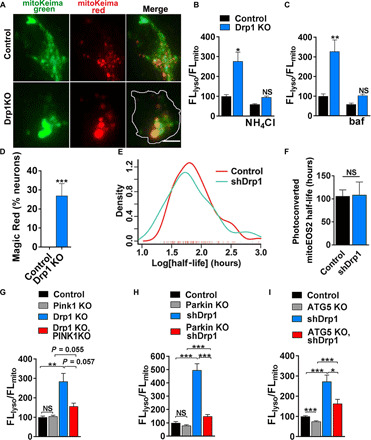
(A) Images show control and Drp1KO hippocampal neurons expressing mitoKeima. Scale bar, 4 μm. (B) Quantification of Drp1KO hippocampal neurons expressing mitoKeima. Drp1KO mitochondria have proportionally increased fluorescence in the acidic, lysosomal (FLlyso) versus mitochondrial (FLmito) channel, and this signal is decreased by NH4Cl. Data show means ± SEM, n = 7 to 8 coverslips per group, 6 to 12 cells per coverslip, compilation of two experiments. (C) Bafilomycin (baf), which blocks lysosome acidification, also decreased the FLlyso/FLmito ratio. n = 3 coverslips per group, 5 to 17 cells per coverslip. (D) Neurons were incubated with Magic Red for 1 hour. Magic Red signal accumulates in Drp1KO but not control neurons. n = 9 to 10 coverslips per group, 4 to 12 cells per coverslip, compilation of two experiments. (E and F) Estimate of mitochondrial turnover in neurons expressing Drp1 shRNA or scramble shRNA assessed on the basis of the rate of loss of photoconverted mitoEOS2 from the cell body. Individual neurons expressing mitoEOS2 were photoconverted and longitudinally imaged using robotic microscopy. Loss of Drp1 (shRNA) did not influence the rate of dissipation of red (photoconverted) fluorescence despite producing characteristic swollen mitochondria (fig. S2F). Data are means ± SEM, not significant versus scramble shRNA by two-sided Mann-Whitney test, n = 109 neurons in total from two experiments. (G and H) Increased lysosomal targeting in Drp1KO neurons is inhibited by PINK1 KO or Parkin KO. (G) n = 10 to 11 coverslips per group, n = 3 to 15 cells per coverslip, compilation of two experiments; (H) n = 16 coverslips per group, n = 3 to 9 cells per coverslip from three experiments. (I) Increased lysosomal targeting in neurons treated with shDrp1 is also largely inhibited by ATG5 KO. n = 25 to 32 coverslips per group, n = 3 to 5 cells per coverslip from six experiments; *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA with Games-Howell post hoc test (B, G, H, and I) or Tukey’s multiple comparisons test (C) or t test (D).
To test whether the increased mitochondrial acidification and lysosomal fusion indicated a faster turnover rate, we examined the dissipation of a photoconvertible mitochondria-targeted fluorophore as a surrogate for mitochondrial turnover. Neurons were cotransfected with mitochondrial matrix–targeted EOS2 (mito-EOS2) and Drp1 short hairpin RNA (shRNA) or scrambled shRNA. EOS2 was irreversibly photoconverted from green to red with a single pulse of 405-nm light and longitudinally imaged by robotic microscopy at 12-hour intervals for 30 to 40 hours to measure clearance of the red fluorescence. Single-cell red fluorescence intensities were normalized to the baseline, and the half-life of mito-EOS2 was calculated for each cell (Fig. 2, E and F, and fig. S2F). Unexpectedly, we found no difference in mean mito-EOS2 half-life or the distribution of mito-EOS2 half-lives in neurons with Drp1 knockdown. Combined with the mitoKeima data, this suggests either that most Drp1KO mitochondria are not degraded despite being acidified or that the acidified mitochondria are degraded, but at least a subset of mitochondrial proteins (including the photoconverted red mito-EOS2 reporter) remain intact in the resulting acidified structures for long periods.
Parkin is targeted across the OM of polarized mitochondria
The synthetic lethality of PINK1 and Drp1KO suggests that PINK1 may be required for quality control of dysfunctional Drp1KO mitochondria. Consistent with this, the increase in acidified mitoKeima signal in Drp1KO neurons was largely blocked when PINK1 was also absent (Fig. 2G). Parkin KO also markedly decreased the acidified mitoKeima signal (Fig. 2H) in the setting of Drp1 loss. We next tested the requirement for the core autophagy protein ATG5. Hippocampal neurons from ATG5lox/lox mice (27) were cotransfected with mitoKeima, Cre (to delete ATG5), or vector control and with Drp1 or scrambled shRNA. Similar to PINK1 KO and Parkin KO, deletion of ATG5 decreased the acidification of Drp1-deficient mitochondria (Fig. 2I and fig. S2, G and H).
To further assess how PINK1 and Parkin contribute to mitochondrial quality control in neurons, we next asked whether Parkin is recruited to Drp1KO mitochondria by examining the colocalization of Parkin [visualized with green fluorescent protein (GFP)–Parkin] with mitochondria (mito-BFP). In many Drp1KO neurons, Parkin was dispersed evenly throughout the cytosol (Fig. 3, A and B). However, in ~5% of Drp1KO neurons, GFP-Parkin encircled a single mitochondrion. In others, punctae of GFP-Parkin were adjacent to mitochondria, perhaps representing an earlier stage before Parkin fully encircles mitochondria. Of interest, ~30% of neurons contained one or more mitochondria where the GFP-Parkin signal overlapped with the mitochondria, suggesting that Parkin had transited through the OM and either localized to the inner mitochondrial membrane or entered the mitochondrial matrix. Confocal imaging of 0.4-μm z-stacks of live and fixed neurons revealed an overlapping pattern consistent with GFP-Parkin in the matrix or inner mitochondrial membrane and confirmed that the circling pattern is GFP-Parkin at the OM (hereafter referred to as OM-Parkin; Fig. 3C and fig. S3, A and B). Live imaging of GFP targeted to the OM versus the matrix and immunostaining against Tom20 (an OM protein) versus pyruvate dehydrogenase (PDH; a matrix protein) confirmed our ability to distinguish these compartments. Although the OM pattern of GFP-Parkin fluorescence could be explained by increased acidity in the matrix of mitochondria, quenching the GFP fluorescence in the matrix, adding NH4Cl to alkalinize the mitochondrial contents (28) failed to restore GFP fluorescence within the matrix of mitochondria with OM-Parkin (fig. S3C), further suggesting that these are distinct patterns of GFP-Parkin recruitment. In the absence of both PINK1 and Drp1, all three forms of Parkin configuration around the mitochondria were absent (Fig. 3B). Moreover, although most Drp1KO mitochondria are polarized (Fig. 3D) (2), the mean mitochondrial membrane potential was markedly decreased in Drp1KO;PINK1KO neurons, consistent with PINK1’s role in protecting against Drp1KO toxicity.
Fig. 3. Parkin is recruited into polarized Drp1KO mitochondria.
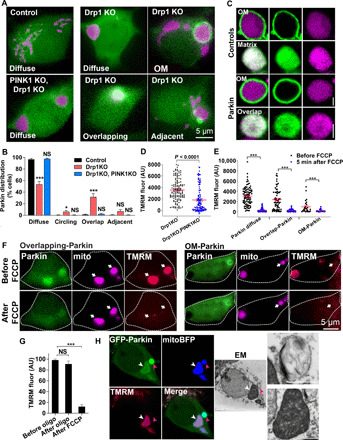
(A) Neurons expressing GFP-Parkin (green) and mitoBFP (purple, mitochondrial matrix–targeted BFP). Parkin is diffusely distributed in control neurons. A subset of Drp1KO neurons contain mitochondria with either adjacent Parkin punctae, overlapping Parkin, or Parkin ringing (circling). Drp1KO;PINK1KO blocks all Parkin recruitment. (B) Quantification of Parkin distribution patterns. Data show means ± SEM; n = 12 coverslips per group, 8 to 20 neurons per coverslip from three experiments. (C) Confocal microscopy shows single mitochondria in Drp1KO neurons expressing a mitochondrial matrix marker (mitoFarRed, purple) and either a mitochondrial outer membrane marker (OM-GFP) or mitochondrial matrix marker (matrix-GFP) as controls or GFP-Parkin recruited either in a mitochondrial OM or overlapping pattern. Scale bars, 1 μm. (D) Scattergraph of mitochondrial membrane potential (TMRM) of individual Drp1KO and Drp1KO;PINK1KO mitochondria. n = 128 to 130 mitochondria per group from two experiments. P < 0.0001 by unpaired t test. (E and F) Scattergraphs (E) and images (F) of mitochondrial membrane potential (TMRM, red) of mitochondria with different Parkin patterns in Drp1KO neurons expressing mitoBFP and GFP-Parkin, before and after FCCP treatment (1.25 μM). n = 26 to 87 mitochondria per group from three experiments. (G) Mitochondrial membrane potential (TMRM) of mitochondria with overlapping-Parkin in Drp1KO neurons before and after oligomycin (5 μM, 5 min) and FCCP (1.25 μM, 5 min). n = 17 cells from two experiments. *P < 0.05 and ***P ≤ 0.001 by one-way ANOVA with Tukey’s multiple comparisons test (G) or Games-Howell post hoc test (B and E). (H) CLEM showing GFP-Parkin accumulation in polarized (white arrowheads) and depolarized (pink arrowheads) mitochondria. Corresponding ultrastructure shows that the polarized mitochondrion with internalized Parkin has intact membranes and cristae (bottom inset), while the depolarized mitochondria (top inset) is a vesicular structure containing degraded mitochondrial membranes. Scale bars, 2 μm (main panels) and 1 μm (inset). AU, arbitrary units.
Notably, this overlapping pattern of Parkin accumulation was not unique to neurons. In immortalized Drp1KO MEFs (4), a subset of cells contained one or more mitochondria with either OM, overlapping, or adjacent Parkin patterns (fig. S3, D to F). In contrast, treatment of wild-type (wt) MEFs with carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) for 2 hours led to Parkin recruitment on the OM but not to the overlapping pattern (fig. S3G).
We next aimed to characterize the mitochondria with overlapping Parkin. Only one-third of mitochondria with overlapping Parkin had normal Tom20 signal, while the remaining had either decreased or no detectable Tom20 immunofluorescence (fig. S4, A and B). This raises the possibility of partial or complete degradation of the OM in a subset of mitochondria with overlapping Parkin. In contrast, the inner mitochondrial membrane markers MTCO1 and COX7B were fully intact in mitochondria with the overlapping-Parkin pattern (fig. S4, C to E), suggesting that the subset of mitochondria with overlapping Parkin that lack Tom20 staining have features of mitoplasts, where the inner mitochondrial membrane is preserved despite degradation of the OM (29). Parkin has been shown to mediate OM rupture, independent of mitophagy, leaving mitoplast-like structures behind (30). Alternatively, the subset of mitochondria with disrupted Tom20 immunofluorescence may actually be degrading mitochondria that retain normal immunoreactivity to mitochondrial matrix markers.
To confirm that Parkin did indeed enter mitochondria, we also performed immunostaining against Parkin. Using two separate Parkin antibodies, we first confirmed that Parkin immunofluorescence directly overlapped with GFP fluorescence ringing OM-Parkin mitochondria (fig. S5, A to C). Moreover, we confirmed that Parkin immunofluorescence also localized to the matrix of many overlapping-Parkin mitochondria. Notably, the Parkin immunofluorescence failed to localize to a subset of overlapping-Parkin mitochondria, although the frequency of this differed depending on the antibody used, suggesting that Parkin either has a different conformation or has been partially degraded in a subset of the overlapping-Parkin mitochondria. In addition, the cytosolic GFP-Parkin fluorescence intensity was independent of the observed Parkin pattern (fig. S5D), indicating that the different patterns of GFP-Parkin distribution are not artifacts of expression level. Some cells contained mitochondria with all three patterns of Parkin distribution (OM, overlapping, and diffuse; fig. S5E).
We hypothesized that Parkin localizes to the OM of depolarized mitochondria, which might precede targeting to the matrix or the inner mitochondrial membrane, and hence that both OM and overlapping-Parkin mitochondria would be depolarized. To assess this, we examined mitochondrial membrane potential by tetramethyl-rhodamine methyl ester (TMRM) fluorescence before and after treatment with FCCP (2). Although most mitochondria with OM-localized Parkin were depolarized, ~25% were polarized (Fig. 3, E and F), consistent with Parkin being recruited to these mitochondria secondary to mitochondrial stress that occurs independent of depolarization (16, 31). In contrast, although some mitochondria with overlapping-Parkin were depolarized, many others were polarized and functional, as they maintained their membrane potential even after treatment with oligomycin, indicating that membrane potential was not being supported by reverse pumping of adenosine triphosphate (ATP) synthase with ATP hydrolysis (Fig. 3G).
These findings on mitochondrial membrane integrity, Parkin localization, and mitochondrial membrane potential all indicate that there are two subsets of mitochondria with overlapping-Parkin patterns. Consistent with this, analysis of mitoKeima signals revealed a bimodal distribution; some overlapping-Parkin mitochondria had very high mitoKeima signals, while others had relatively low (but still elevated) mitoKeima signals consistent with mild acidification. In contrast, most mitochondria with OM-Parkin had low (neutral) mitoKeima signals (fig. S5, E and F). Despite these differences, in all populations of mitochondria with overlapping Parkin or OM-Parkin, the mitoKeima signal was present throughout the matrix, consistent with homogeneous pH throughout the matrix.
To define the ultrastructure of polarized and depolarized mitochondria with overlapping Parkin, we performed correlative light and electron microscopy (CLEM). Using this approach, we confirmed that polarized mitochondria containing overlapping Parkin are intact mitochondria with cristae with enhanced electron density and honeycomb or concentric onion patterns (Fig. 3H and fig. S5G), as can be observed in mitochondria from patients with mitochondrial disorders (32). In contrast, depolarized overlapping-Parkin mitochondria are actually degrading mitochondrial remnants that we hypothesized had been degraded by autophagy to form a mitolysosome, presumably explaining why some mitochondria with overlapping Parkin failed to exhibit immunoreactivity to the Parkin antibody.
Although GFP-Parkin is commonly used to study Parkin localization, its expression could potentially artificially amplify Parkin-dependent mitophagy. To verify that GFP-Parkin patterns are independent of overexpression, we also performed CLEM in neurons with only endogenous Parkin expression, using Parkin antibodies to detect recruitment patterns. With this approach, we were able to observe mitochondria with overlapping endogenous Parkin and intact cristae (fig. S5H). No mitochondria with overlapping endogenous Parkin were found to be degrading mitochondria structures, consistent with a disruption of Parkin conformation or degradation of Parkin in these structures. Therefore, although the depolarized overlapping-Parkin mitochondria are indistinguishable from other mitochondria by typical mitochondrial markers (e.g., mito-BFP), they are actually in the process of degradation.
Parkin dynamically accumulates in functional and degrading mitochondria through distinct pathways
We next used time-lapse imaging to define the steps of Parkin recruitment to neuronal mitochondria. First, to delineate when lysosomal fusion occurs in relation to Parkin recruitment to the OM and matrix, we simultaneously tracked Parkin distribution with GFP-Parkin and mitochondrial acidity using mitoKeima. We found that Parkin accumulates on the OM of Drp1KO mitochondria with normal pH (Fig. 4, A and B). This pattern of OM-Parkin accumulation was maintained for 3.6 ± 0.4 hours (n = 11 mitochondria, means ± SEM; fig. S5I), before Parkin began to dissipate from the OM. Parkin dissipation occurred simultaneously with a marked increase in the acidity of the mitochondrial contents (Fig. 4, C and D). Following mitochondrial acidification, there was a rapid decrease in mitochondrial size, followed by the redistribution of Parkin to an overlapping pattern within ~80 min of OM-Parkin dissipation (Fig. 4D). Loss of OM-Parkin was invariably followed by the accumulation of overlapping Parkin in the acidic, shrunken mitochondrial structures (hereafter referred to as “indirect” overlapping-Parkin recruitment), previously identified by CLEM as degrading mitochondrial structures (Fig. 3H). These data suggest that conversion to the overlapping-Parkin pattern via the indirect pathway marks the fusion with lysosomes and the associated transition to a mitolysosome.
Fig. 4. Time-lapse imaging reveals distinct Parkin-dependent quality control pathways.
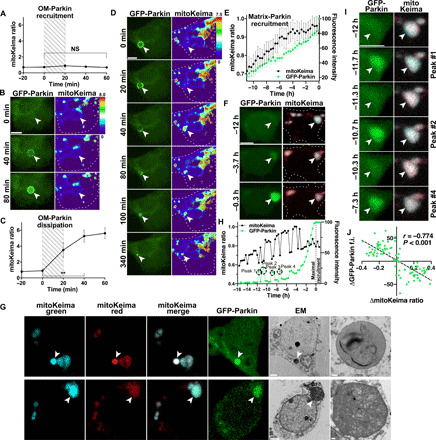
Drp1KO neurons coexpressing GFP-Parkin and mitoKeima were imaged every 20 min for up to 21 hours. (A) Relative mitochondrial acidity during OM-Parkin recruitment. Hatched area represents period of GFP-Parkin OM accumulation. Data represent means ± SEM. n = 10 mitochondria from 9 neurons, 1 to 4 neurons per dish, three experiments. (B) GFP-Parkin accumulates at the OM of a mitochondrion with low acidity. (C) Mitochondrial acidity during OM GFP-Parkin dissipation. Hatched area delineates period when GFP-Parkin begins to dissipate. n = 10 mitochondria from 9 neurons, 1 to 4 neurons per dish, two experiments. **P < 0.01 by one-way ANOVA with repeated measures. (D) OM-Parkin dissipates concomitantly with mitochondrial acidification. Parkin then accumulates inside the shrunken mitochondrion. (E) Direct matrix-Parkin accumulation. GFP-Parkin (green) slowly accumulates in the mitochondrial matrix as acidity (black) increases. GFP-Parkin fluorescence was normalized such that the lower and upper bounds were defined by the nuclear and maximum mitochondrial intensities, respectively. Traces are aligned so maximum matrix-Parkin intensity is at 0 hours. n = 23 mitochondria from 22 neurons, 1 to 9 neurons per dish, seven experiments. (F) GFP-Parkin directly accumulates in mitochondria, without prior OM recruitment. (G) CLEM shows that acidified overlapping-Parkin mitochondria are degrading mitochondrial contents within lysosomes (mitolysosomes) (top). Overlapping-Parkin mitochondria with slightly acidic pH (fig. S5F) are intact mitochondria with electron-dense cristae (bottom). Scale bars, 2 μm (cell overviews) and 0.2 μm (insets). (H and I) Corresponding repetitive single spikes of Parkin accumulation and decreased mitochondrial acidity in a Drp1KO mitochondrion during direct matrix-Parkin recruitment. (B, D, F, and I) Scale bars, 5 μm. (J) Inverse relationship between changes in GFP-Parkin intensity and mitoKeima ratio during Parkin spikes. The change in GFP-Parkin and mitoKeima was measured between time points before, during, and after the peak. n = 90 measurements, 40 individual spikes in 6 neurons, 5 to 9 spikes per neuron, 1 mitochondrion per neuron; Pearson r = −0.774, P < 0.001. f.i., fluorescence intensity.
Notably, Parkin also accumulated in the matrix of mitochondria directly, without passing through a stage of OM-Parkin (Fig. 4, E and F). In these instances, Parkin began accumulating in the matrix of mitochondria with normal pH. However, direct Parkin recruitment to the matrix occurred more slowly than indirect recruitment to the overlapping pattern via the OM, taking 14.5 ± 0.6 hours (n = 23 mitochondria, means ± SEM) to reach maximal recruitment, during which time the mitochondria also gradually became mildly acidic. This suggests that the mildly acidified overlapping-Parkin mitochondria are the larger intact mitochondria with preserved mitochondrial membrane potential (Fig. 3E) that we visualized by CLEM (Fig. 3H and fig. S5G). To further confirm this, we performed CLEM on mitochondria expressing mitoKeima and showed that small highly acidic mitochondria with overlapping Parkin are mitolysosomes, while larger mitochondria with lower acidity are intact mitochondria (Fig. 4G). Preserved mitochondrial membrane potential in the context of mild acidification may suggest partial lysosomal fusion or ion channel–driven changes (33). In contrast, when Parkin recruitment to the OM precedes mitochondrial acidification, Parkin triggers mitochondrial acidification by initiating lysosomal fusion, ultimately leading to mitochondrial degradation.
During direct recruitment of matrix-Parkin, we also observed large, repetitive fluctuations in GFP-Parkin fluorescence (hereafter called “Parkin spikes”), suggesting rapid and repetitive bursts of Parkin entry and exit from the mitochondrial matrix (Fig. 4, H and I). These fluctuations were observed in ~26% (6 of 23) of cases of direct matrix-Parkin recruitment, with the individual spikes occurring on average every 3.2 ± 0.3 hours (n = 6 mitochondria, means ± SEM) and lasting less than 40 min each (Fig. 4, H and I, and fig. S6). The spikes of Parkin entry were strongly correlated with small but sharp decreases in mitochondrial acidity (r = −0.77, P < 0.001; Fig. 4J), indicating that Parkin accumulation is intimately associated with mitochondrial pH, although it remains unclear whether matrix-Parkin triggers or resolves the acidification. Notably, loss of GFP-Parkin fluorescence was not due to quenching of GFP fluorescence at low pH, as we observed far higher mitoKeima signals indicative of low mitochondrial pH without any loss of GFP fluorescence in other mitochondria. The rapid fluctuations in acidity may be mediated by direct fusion with lysosomes at points of transient contact between lysosomes and mitochondria (34), although we cannot exclude the possibility of fusion with already acidified mitochondria (see below) or other ion channel–driven changes (33). Regardless of the mechanism, the existence of Parkin spikes establishes that both Parkin recruitment to mitochondria and mitochondrial acidification are not always irreversible processes.
Mitochondria with indirect Parkin recruitment are degraded by autophagy
We next assessed whether mitochondria with indirect overlapping-Parkin accumulation undergo canonical macroautophagy in which they are engulfed by autophagosomes to produce mitophagosomes, before fusing with lysosomes to form mitolysosomes. To visualize and define the kinetics of autophagosome formation and fusion with lysosomes, we examined GFP-LC3 localization at different stages following indirect overlapping-Parkin recruitment. No GFP-LC3 accumulation on Drp1KO mitochondria was visible at baseline, indicating either that Drp1KO mitochondria are degraded independent of microtubule-associated protein 1A/1B-light chain 3 (LC3) or that degradation occurs too rapidly for detection. In support of the latter, after a 4-hour treatment with the autophagy inhibitor bafilomycin, GFP-LC3 accumulated on a subset of LAMP1-circled Drp1KO mitochondria (Fig. 5A). In addition, of all GFP-LC3–positive mitochondria, most (~90%) were also encircled by LAMP1 (Fig. 5B), suggesting that lysosomal fusion occurs rapidly once autophagosomes are formed. Conversely, only ~30% of mitochondria encircled by LAMP1 were also encircled by GFP-LC3 (Fig. 5C), suggesting that LC3 on the inner autophagosome membrane is rapidly degraded following lysosomal fusion (35, 36). Subsequent time-lapse imaging showed that LC3 accumulates in a discrete spot on the OM of OM-Parkin mitochondria, presumably representing the autophagosome formation site (37), before forming a ring around the organelle to seal the mitophagosome (Fig. 5D). After Parkin transitions to the overlapping pattern (i.e., after lysosomal fusion to form the mitolysosome), GFP-LC3 again rapidly clustered into a discrete punctum on the outer membrane of the mitophagosome before dissipating.
Fig. 5. PINK1-dependent formation of LC3-positive autophagosomes precedes rapid lysosome fusion.
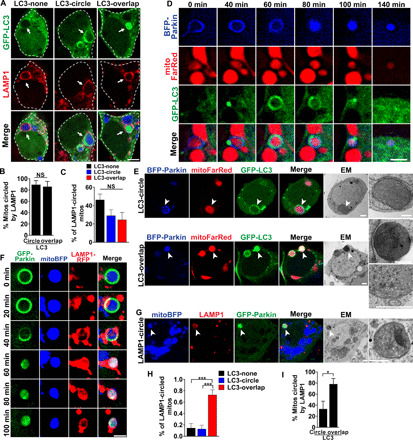
(A) Drp1KO neurons expressing GFP-LC3 (green) and mitoBFP (blue) treated with 100 nM bafilomycin for 4 hours and stained for LAMP1 (lysosomes, red). Scale bar, 4 μm. (B) Most mitochondria with circling or overlapping patterns of GFP-LC3 were circled by LAMP1. n = 8 coverslips per group, 4 to 7 neurons per coverslip, three experiments. (C) Among LAMP1-circled mitochondria, ≈50% did not have LC3 accumulation (diffuse LC3); others were circled by GFP-LC3 (LC3-circle) or had GFP-LC3 overlapping the mitochondrial matrix (LC3-overlap). n = 8 coverslips per group, n = 6 to 11 cells per coverslip, three independent experiments. (D) Drp1KO neurons coexpressing GFP-LC3, mitoFarRed, and BFP-Parkin. GFP-LC3 accumulates in a distinct punctum on the mitochondrion with OM-Parkin, before dispersing to encircle the mitochondrion. BFP-Parkin then transitions to the overlapping pattern, and LC3 reaccumulates into a punctum. Scale bar, 2 μm. (E) CLEM shows ultrastructure of Parkin-positive mitochondria with distinct patterns of GFP-LC3. Mitochondria with LC3-circle are surrounded by an isolation membrane (top). LC3-overlap mitochondrion (bottom) is electron-dense but intact, with packed cristae (see insets). Bottom inset is the same structure from a different section. Scale bars, 1 μm. (F) Drp1KO neurons coexpressing GFP-Parkin, LAMP1-RFP, and mitoBFP. Images show formation of a LAMP1 ring, indicating lysosomal fusion, concurrent with Parkin shifting from OM to overlapping. Scale bar, 2 μm. (G) Overlapping-Parkin mitochondria circled by LAMP1 are mitolysosomes by ultrastructure. Scale bars, 1 μm. (H) Drp1KO;PINK1KO neurons expressing GFP-LC3 and mitoBFP treated with bafilomycin for 4 hours and stained for LAMP1. Among LAMP1-circled mitochondria, ≈80% had LC3-overlap, and a few were either circled or lacked GFP-LC3 accumulation. n = 9 coverslips per group, 5 to 12 neurons per coverslip, two experiments. (I) Most mitochondria with LC3-overlap were circled by LAMP1. n = 6 coverslips per group, 5 to 12 neurons per coverslip, three experiments. *P < 0.05 and ***P < 0.001 by t test (B and I) or one-way ANOVA with Tukey’s multiple comparisons test (C and H).
Notably, an additional ~20% of swollen Drp1KO mitochondria had a distinct overlapping pattern of LC3, suggestive of LC3 accumulation within mitochondria, while ~50% of LAMP1-circled mitochondria did not have any GFP-LC3 accumulation (Fig. 5C). These different patterns of GFP-LC3 distribution occurred independently of the level of GFP-LC3 in the cytosol (fig. S7A), indicating that the overlapping pattern is not an artifact of LC3 overexpression. To gain insight into the differences between mitochondria with distinct patterns of LC3 and Parkin accumulation, we again used CLEM. This confirmed that the mitochondria with ringing patterns of LC3 accumulation are indeed mitochondria encircled by autophagosomes (i.e., mitophagosomes; Fig. 5E, top). Therefore, degrading Drp1KO mitochondria up to at least 2 to 3 μm are engulfed by autophagosomes, but the structures are very short-lived. Time-lapse imaging confirmed that LAMP1 recruitment occurs rapidly and coincides with the transition from OM to overlapping Parkin after autophagosome formation (Fig. 5, D and F), while CLEM showed LAMP1 circling a degrading mitochondrion with overlapping Parkin (Fig. 5G). Thus, the acidic degrading mitochondrial structures that emerge from the indirect pathway are LAMP1 positive and depolarized, further confirming that they are indeed mitolysosomes. Conversely, and of particular interest, overlapping-Parkin mitochondria with the overlapping LC3 and LAMP1-circled patterns appear to be intact mitochondria packed with electron-dense cristae (Fig. 5E, bottom), perhaps suggestive of either stalled degradation or an alternate degradation pathway. To assess the requirement for PINK1 in this LC3 accumulation, we examined GFP-LC3 in Drp1KO;PINK1KO neurons (Fig. 5, H and I). Without PINK1, most LAMP1-circled mitochondria had the overlapping pattern of GFP-LC3, and few had the LC3-circling or no-LC3 pattern, indicating that simultaneous accumulation of LAMP1 and the circling pattern of LC3 on the OM requires PINK1. It remains unclear how LC3 is recruited to the overlapping pattern, but this LC3 recruitment in the absence of PINK1 suggests that the overlapping pattern of GFP-LC3 may represent an as-yet undefined secondary quality control pathway. Notably, follow-up CLEM experiments to visualize the ultrastructure of mitochondria circled by endogenous LC3 revealed short stretches of membranes adjacent to the mitochondria rather than the intact autophagosome membranes visualized using GFP-LC3 (fig. S7B). This may be due to autophagosome membrane damage incurred during membrane permeabilization for antibody staining. However, we cannot exclude the possibility that GFP-LC3 promotes the formation of autophagosome membranes that are distinct from those formed by endogenous LC3. We similarly failed to visualize either Drp1KO or Drp1KO-PINK1KO mitochondria with the overlapping pattern of endogenous LC3. Hence, further characterization of LC3 action using endogenous markers is required.
Parkin is targeted into mitochondria in wt neurons
We also tested whether Parkin dynamics and mitochondrial acidification occur similarly in wt neurons. At baseline, most mitochondria in wt neurons had diffuse Parkin, while only occasional mitochondria had either OM or overlapping-Parkin patterns. However, treatment with either the oxidative stressor paraquat or deferoxamine (DFO), an iron chelator that can trigger mitophagy (38), caused robust Parkin recruitment in both the OM and the overlapping patterns (fig. S8, A and B). Moreover, consistent with the Drp1KO mitochondria, ≈2/3 of wt mitochondria with overlapping Parkin had either decreased or absent Tom20 immunofluorescence (fig. S8C), while the intensity of the inner mitochondrial marker MTCO1 was similar in mitochondria with and without Parkin accumulation (fig. S8D). Similar to Drp1KO mitochondria, time-lapse imaging showed that, following treatment with DFO, Parkin moved from the OM to an overlapping pattern in a subset of wt mitochondria, with a concomitant increase in acidity (fig. S8E). Notably, although much harder to detect, we also observed direct Parkin recruitment in wt neurons in response to DFO (fig. S8F), and the overlapping pattern of Parkin recruitment was also visualized with endogenous Parkin (fig. S8G).
A subset of mitolysosomes is engulfed by polarized mitochondria
The fate of mitolysosomes is poorly understood, especially in neurons. To delineate their fates, we tracked individual mitolysosomes over time. Mitochondria with indirect Parkin recruitment were identified on the basis of direct visualization of the OM-Parkin to overlapping-Parkin pattern transition and followed for an additional 2.3 to 19.7 hours following conversion into mitolysosomes. Unexpectedly, during this period, the mitolysosomes not only made repeated contacts with nonacidified mitochondria (visualized in 55 ± 7.2% of images, n = 12 mitochondria, means ± SEM) but also fused with and were periodically engulfed by seemingly healthy mitochondria with neutral pH (5 of 12 mitochondria engulfed; Fig. 6A). In some cases, these interactions were temporary, as mitolysosomes were observed to emerge from mitochondria after being engulfed (fig. S9A). Although the function of these interactions remains unclear, they may represent a mechanism to recycle mitochondrial components. If so, recipient mitochondria would presumably be functional. To test this, we used TMRM to track mitochondrial membrane potential during engulfment of overlapping-Parkin mitochondria. We observed engulfment of a depolarized overlapping-Parkin mitochondrion by a polarized, mitoBFP-positive mitochondrion (Fig. 6B). This indicates that mitochondria with overlapping Parkin can be engulfed by functional mitochondria, although the short half-life of TMRM dye limited our ability to quantify this phenomenon.
Fig. 6. Acidified overlapping-Parkin mitochondria are engulfed by mitochondria with normal pH.
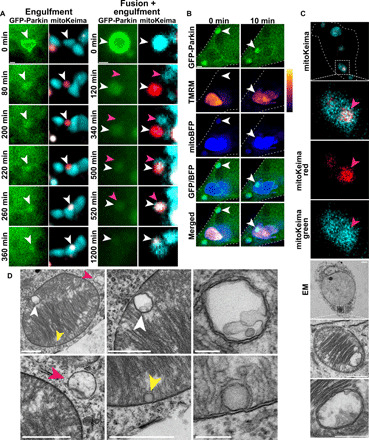
Mitochondria in Drp1KO hippocampal neurons coexpressing GFP-Parkin and mitoKeima were tracked individually to determine their fates. (A) Time-lapse images show the fates of single mitochondria that were acidified following OM-Parkin disruption (white arrowheads). Some mitolysosomes are engulfed by nonacidified mitochondria (left). The mitolysosome on the right first fuses with a nonacidified mitochondrion (pink arrowheads) before being engulfed by another. Scale bars, 1 μm. (B) Drp1KO neurons expressing GFP-Parkin and mitoBFP were incubated with TMRM and imaged every 10 min for several hours. Representative images show overlapping-Parkin mitochondria being engulfed by polarized mitochondria (white arrowheads). Overlapping-Parkin colocalizes with mito-BFP before and after engulfment. Scale bar, 1 μm. (C) CLEM images show a healthy mitochondrion with intact cristae that contains a discrete acidified structure (by mitoKeima) that corresponds to a membrane-bound structure enclosing mitochondrial materials. Scale bars, 2 μm (cell overview), 1 μm (mitochondria overview), and 0.5 μm (inset). (D) EM images show a healthy mitochondrion engulfing two structures (white and yellow arrowheads) similar to what was described in (C). A lysosome (pink arrowheads) was also observed interacting with the same mitochondrion. Scale bars, 1 μm for images containing arrowheads and 0.2 μm for insets.
We next used CLEM to visualize the ultrastructure of internalized mitochondria. Analysis of a mitochondrion containing a region of increased acidity (based on mitoKeima signal) revealed an internal double-membrane structure enclosing mitochondrial remnants (Fig. 6C). Using EM, we observed healthy-appearing mitochondria (based on intact cristae morphology) containing membrane-bound structures with degraded contents resembling mitolysosomes (Fig. 6D), which we hypothesize may have been engulfed. We also observed a depolarized mitochondrion with GFP-Parkin–positive, membrane-bound structures (fig. S9B). The presence of the GFP reporter suggests that these membrane-bound structures underwent Parkin-mediated degradation before being engulfed by the recipient mitochondria. Notably, we also observed mitochondrial engulfment in wt neurons, where mitochondria with acidified overlapping-Parkin patterns contacted and were engulfed by other nonacidified mitochondria (fig. S9, C and D). However, only a limited number of mitochondria could be definitively tracked in wt neurons because of their small size and greater number and mobility.
Mitolysosomes persist and dynamically interact with other mitochondria
We hypothesized that mitochondrial engulfment of mitolysosomes enables the recycling of mitochondrial materials. Moreover, some materials may no longer be useful to mitochondria but still useful to other cellular functions, while other materials may need complete degradation. However, we found that mitolysosomes are stable structures, making it difficult to determine their fates. None of the definitively identified indirect overlapping-Parkin mitolysosomes tracked for at least 5 hours (and up to 19.7) underwent notable change in morphology or acidity or loss of expression of the targeted fluorescent probes, following the OM- to overlapping-Parkin transition over the duration of the imaging period (0 of 11), indicating that mitochondrial degradation in neurons is often a slow process.
To better assess their fates, we tracked mitolysosomes at a later stage by selecting mitochondria that already had overlapping-Parkin localization at the start of the live imaging session, instead of trying to capture both the recruitment and fate in a single session. Although we did not directly visualize whether Parkin onset was direct or indirect in these mitochondria, most were both acidified (high mitoKeima signal) and small in size, suggesting that they were predominantly mitolysosomes in the indirect pathway (Fig. 7A). In ~41% of the cases, GFP-Parkin remained localized in the mitolysosomes for the entire imaging session (12 to 21 hours), and the mitochondrial pH was stable, showing only a small decrease in acidity (Fig. 7, B and C), suggesting that mitolysosomes can maintain a stable status for many hours. In another ~43% of cases, the mitolysosomes underwent a sudden, large decrease in signal of both mitoKeima channels and of GFP-Parkin intensity, which coincided with a decrease in size (Fig. 7, B, D, and E), presumably reflecting lysosomal membrane permeabilization or rupture and diffusion of degrading materials into the cytosol. This phenomenon was not unique to Drp1KO mitochondria, as we also observed it in wt neurons (fig. S10A). If this resulted from bursting of mitolysosomes, we reasoned that it should be accompanied by an increase of emitted fluorescent signal in the cytosol. To test this, we measured cytosolic Keima signals, given that they are low at baseline and would therefore allow for the detection of even a small change in signal. We observed a sharp increase in cytosolic Keima fluorescence in both channels (Fig. 7, F and G) that occurred simultaneously with the decrease in mitolysosome size and signal, strongly supporting that mitolysosomes do indeed release partially degraded materials into the cytosol.
Fig. 7. Long-lived mitolysosomes burst and release contents.
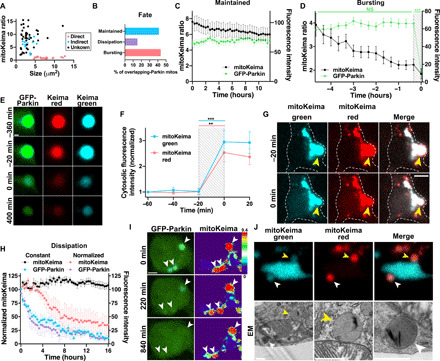
(A) Mitochondria with confirmed indirect OM-Parkin (blue, n = 12) are smaller and more acidic than those with direct Parkin recruitment (pink, n = 23). Most with overlapping-Parkin from t = 0 (unknown origin, black, n = 46) are similar to those from the indirect pathway. (B) Fates of these 46 mitolysosomes. (C) Constant acidity levels in mitolysosomes that maintain GFP-Parkin signal (n = 19 mitochondria from 18 neurons, 1 to 4 neurons per dish, 10 experiments; not significant for interaction between GFP-Parkin and mitoKeima ratio by two-way ANOVA with repeated measures). (D) GFP-Parkin sharply decreases after bursting. Acidity decreases over hours preceding degradation (P < 0.05). Traces are aligned so bursting occurs at 0 hours. n = 19 mitochondria from 17 neurons, 1 to 5 neurons per dish, seven experiments. ***P < 0.001 by two-way ANOVA with repeated measures. (E) Post-burst mitolysosome structure tracked for 400 min. Scale bar, 1 μm. (F) Cytosolic mitoKeima increases when mitolysosomes burst. Traces are aligned so bursting occurs at 0 min. n = 20 mitochondria from 18 neurons, 1 to 5 neurons per dish, seven experiments. **P < 0.01 and ***P < 0.001 by two-way ANOVA with repeated measures and Bonferroni’s multiple comparison test. (G) Mitolysosome bursting (yellow arrowheads) is accompanied by increased cytosolic mitoKeima. Scale bar, 5 μm. (H) In some mitolysosomes, GFP-Parkin dissipates followed by pH normalization (pink, n = 3 mitochondria from 3 neurons, 1 neuron per dish, three experiments), while others remain acidic (black, n = 4 mitochondria from 3 neurons, 1 to 2 neurons per dish, two experiments). GFP-Parkin intensity similarly decreases in both (constant acidity, blue; normalized acidity, purple). (I) In three mitochondria with overlapping-Parkin, GFP-Parkin signal fades by 220 min and is at cytosolic levels by 840 min. By then, the mitolysosome in the lower left corner has normalized acidity. Scale bar, 3 μm. (J) CLEM shows close contacts between a mitochondrion with intact cristae and two acidified mitolysosomes (yellow and white arrowheads). Scale bars, 1 μm.
In ~50% of these bursting cases, the mitolysosomes vanished following bursting (fig. S10B), suggesting complete degradation, while in the others, a remnant structure persisted for the duration of the imaging session (up to 15.3 hours) (Fig. 7E). In most cases, the mitolysosome pH partially normalized in the few hours preceding bursting (Fig. 7D), indicative of lysosomal deacidification that may prevent the release of potentially harmful acidic contents into the cytosol. Thus, some mitochondrial proteins can withstand the acidity of the lysosome for several hours before getting released to the cytosol.
In the remaining ~15% of mitolysosomes, mitoKeima fluorescence persisted, but the Parkin signal gradually dissipated (Fig. 7, B, H, and I). Unexpectedly, a subset of these structures partially or fully normalized their pH following Parkin dissipation. The mechanism of this pH normalization remains to be defined and could involve increased proton leakage and inhibition of lysosomal acidification (39). However, live imaging showed that the mitolysosomes normalized their pH as they made repeated contacts with both adjacent mitolysosomes and neutral mitochondria (Fig. 7J and fig. S10C). This raises the possibility that these contacts may also contribute to the deacidification by enabling the transfer of partially degraded mitochondrial contents (fig. S10, D and E).
DISCUSSION
Disruption in mitochondrial quality control is implicated in the pathophysiology of PD and other neurodegenerative diseases, but little is known about the fate of degrading mitochondrial contents in neurons. Here, we simultaneously tracked the acidity and fate of individual mitochondria for up to 21 hours, taking advantage of the enlarged, segregated, and relatively immobile nature of Drp1KO mitochondria in neurons. We identified distinct patterns of Parkin recruitment to mitochondria and delineated the specific sequence and timing by which mitochondria are degraded in Drp1KO and wt neurons. We found that following fusion of mitophagosomes with lysosomes, the resulting acidic mitolysosomes continue to actively interact with other healthy mitochondria for many hours and are sometimes engulfed by functional mitochondria. Moreover, a subset eventually normalize their pH, burst, and release their contents into the cytosol (Fig. 8).
Fig. 8. Indirect and direct pathways of mitochondrial Parkin recruitment.
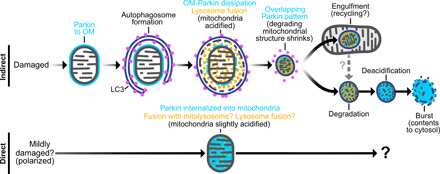
Schema shows indirect and direct mechanisms by which Parkin accumulates in mitochondria and the fates of mitochondria in each of these pathways. In the indirect pathway, Parkin accumulates on the OM of depolarized mitochondria (OM-Parkin). These mitochondria are engulfed by autophagosomes and then rapidly fuse with lysosomes, which occurs concurrently with Parkin dissipation from the OM. These degrading mitochondrial structures then shrink to form mitolysosomes with an overlapping-Parkin pattern (and which are indistinguishable from healthy mitochondria when visualized by either light microscopy with mitochondria-targeted reporters or immunofluorescence against mitochondrial matrix proteins). Mitolysosomes are stable for many hours. A subset is engulfed by mitochondria, and others eventually undergo deacidification before bursting and releasing contents into the cytosol. In the direct pathway, Parkin is gradually recruited directly into mitochondria over many hours, as mitochondria concurrently undergo mild acidification through undefined mechanisms.
Loss of mitochondrial fission promotes PINK1-dependent mitochondrial quality control in DA neurons
Fission is thought to be required for mitochondrial turnover. However, we found that Drp1KO increases the Parkin- and PINK1-dependent engulfment of even very large mitochondria by autophagosomes to produce mitophagosomes, as well as their subsequent fusion and acidification by lysosomes to form mitolysosomes. Therefore, in contrast to previous reports in other cell types (13, 40), Drp1KO increases mitophagy in neurons. Our finding that Drp1KO mitochondria continue to be degraded is in line with the finding that Drp1KO DA neurons and MEFs have decreased (rather than increased) mitochondrial content (2, 14) and that Drp1KO increases the number of acidified mitochondria in HeLa cells (16). Moreover, ongoing turnover indicates that primary mitochondrial dysfunction is the cause of increased toxicity in Drp1KO neurons and suggests that the toxicity of Drp1KO is not due to a primary deficit in mitochondrial degradation.
The findings that PINK1 protects against Drp1KO toxicity and that loss of PINK1 decreases Drp1KO-induced mitochondrial acidification and membrane potential suggest that PINK1-mediated mitochondrial quality control is critical in protecting against toxic Drp1KO mitochondria. However, the mechanism by which Drp1KO triggers mitochondrial dysfunction and increased turnover is unknown. Previous work suggested that Drp1 may protect against unchecked PINK1-Parkin activity (16); however, this is seemingly inconsistent with our finding that concurrent loss of Drp1 and PINK1 is more toxic than loss of Drp1 alone.
Our data also clarify the epistatic relationship between Drp1KO and PINK1 KO. The synergistic toxicity of PINK1 and Drp1 loss in Drosophila (7, 41) is preserved in mice, but the effect is independent of mitochondrial morphology. This likely explains the variable effects of PINK1 KO on mitochondrial morphology in mammalian cell culture (9, 10, 41–43). Our in vivo system is among the most robust examples of PINK1 KO toxicity in a mouse model. Although PINK1 KO alone fails to produce neurodegeneration or affect survival in mice (22, 42), it increases toxicity of A53T synuclein in vivo (44) and the size of mitochondrial inclusions (45, 46). Notably, inhibiting Drp1 with the small molecule mdivi-1 or expression of a dominant negative Drp1 normalizes decreased striatal DA release in PINK1 KO mice (47). Several important differences in experimental design likely explain these contrasting results, including that Rappold et al. assessed partial inhibition of Drp1 function. Mdivi-1 also has potent antioxidant effects that may be more prominent than its effects on mitochondrial morphology, and the effects of mdivi-1 may differ from Drp1KO (48). In addition, we assayed neurodegeneration of DA neurons, whereas Rappold et al. assessed changes in DA homeostasis, which may represent compensatory changes rather than toxicity.
Parkin accumulates in mitochondria through distinct pathways
We found that Parkin is targeted into mitochondria through two distinct pathways (Fig. 8). In the indirect pathway, depolarized mitochondria first accumulate Parkin on their OM. This is followed by engulfment by an autophagosome, rapid fusion with lysosomes, and acidification to form a mitolysosome, with concurrent redistribution of Parkin across the OM into the degrading mitochondria. Although hypothesized based on work in cell lines, this temporal sequence of events has not previously been directly visualized and correlated with the cellular ultrastructure in individual mitochondria. We found that the indirect pathway occurs both in Drp1KO mitochondria and in wt mitochondria exposed to stressors. In the “direct” pathway, polarized mitochondria directly accumulate Parkin, without any apparent preceding accumulation on the OM. Notably, we also tried to track the fates of these intact, mildly acidic, and polarized mitochondria with direct overlapping-Parkin mitochondria. However, the long duration of direct Parkin recruitment precluded us from following the fates of mitochondria with proven direct Parkin recruitment. Understanding the fates and function of this subset of mitochondria will be an important subject for future studies. We also found that direct overlapping-Parkin accumulation occurs in wt neurons, supporting that the direct pathway is not only a unique response to loss of mitochondrial fission but also occurs normally and in response to other stressors in wt neurons.
Mitochondria with direct Parkin recruitment have an intact OM, supporting that Parkin internalization into mitochondria is not contingent on membrane rupture. Once inside mitochondria, Parkin may accumulate on the outer surface of the inner mitochondrial membrane. Alternatively, Parkin may be imported into the mitochondrial matrix, as demonstrated in proliferating cells using biochemical approaches and immunoEM (49). We found that many mitochondria with the overlapping-Parkin pattern were polarized, indicating an intact membrane potential (50). Although Parkin is not predicted to have a mitochondrial targeting sequence, mitochondrial import might be mediated through interactions with other mitochondrial-targeted proteins, analogous to peroxisomal targeting receptors that import cytosolic proteins into the peroxisome matrix (51). Such a function is proposed for Klokin1, a splice variant of human chondroitin polymerizing factor (ChPF), that was found to transport Parkin to the mitochondrial matrix during the cell cycle (52). Other proteins may also contribute, for instance, F-box protein 7, which both binds and translocates Parkin to mitochondria and also enters and forms aggregates within mitochondria (53, 54). However, we speculate that some mitochondria may internalize Parkin as a by-product of engulfing or fusing with mitolysosomes, perhaps explaining why mitochondria in the direct pathway are far less acidic than the degrading mitolysosomes in the indirect pathway. If true, mitochondria in the “direct pathway” may be a downstream consequence of mitolysosomes formed by canonical mitophagy in the “indirect pathway.” Notably, regardless of their origin, overlapping-Parkin mitochondria in the direct pathway retain polarization and at least partial function even after mild acidification, consistent with the finding that mitochondria can retain function in acidic environments (55, 56).
Mitolysosomes are dynamic structures with distinct fates
Although the machinery driving PINK1/Parkin-based mitophagy has been extensively studied, there is very little known regarding the kinetics of this process, especially in neurons, presumably because there has been no means to track individual mitochondria for prolonged periods of time. Hence, studies have primarily relied on snapshots of population-based changes following the administration of mitochondrial toxins or other stressors, as well as short imaging sessions visualizing individual steps such as Parkin recruitment, which have limited ability to determine the timing and sequence of events in individual neurons. Here, we took advantage of the decreased mobility and large size of Drp1KO mitochondria, combined with the decreased total number of mitochondria in Drp1KO neurons, to track individual mitochondria for up to 21 hours, which allowed us to visualize key steps in the mitochondrial life cycle and define their kinetics. Moreover, we used a genetic model (Drp1KO) that produces a nonlethal stressor and drives mitophagy in only a fraction of mitochondria in a fraction of cells at any given time, presumably better recapitulating mitochondrial turnover in normal biology and in neurodegeneration in PD. These studies provided several important insights. First, we confirmed the proposed sequence of events in which depolarized mitochondria destined for degradation are first coated with OM-Parkin before being engulfed by an autophagosome and then acidified following lysosomal fusion. In addition, we defined the precise kinetics of this pathway, showing that autophagosome recruitment is followed rapidly (≈30 min) by lysosomal recruitment and fusion leading to acidification, Parkin redistribution, and formation of the mitolysosome. Notably, these findings differ from those of a recent study that used LysoTracker, superecliptic pHluorin (SEP)–LAMP1-RFP, and mitochondria-targeted EGFP-mCherry reporters to conclude that mitochondrial acidification is a very slow process that takes several hours following mitophagosome formation and LAMP1 accumulation (57). We hypothesize that these discrepant findings result from differences in the sensitivity of methodologies used to detect acidification, as we found that GFP and other mitochondrial-targeted fluorescent reporters continued to fluoresce even after acidification (as detected by mitoKeima and the circling of mitochondria by LAMP1) occurred due to formation of mitolysosomes. Tracking the time course of acidification in individual mitochondria, as opposed to snapshots at discrete time points, may also have increased sensitivity, but further experiments will be required to determine whether these or other differences in the experimental paradigms underlie the discrepant results.
Our studies also used longitudinal tracking to provide some of the first insights into the fates of mitolysosomes. Fusion of mitochondria with lysosomes has been generally considered to inevitably lead to rapid and complete degradation. However, we found that ≈40% of mitolysosomes survived throughout the imaging period (≈12 to 21 hours), during which time they dynamically contacted other mitochondria. In some cases, these contacts may regulate mitochondrial motility (58), while in others, they may lead to transfer of contents, perhaps as a means of recycling mitochondrial components. A subset of mitolysosomes were fully engulfed by other mitochondria, an intriguing but perhaps not unexpected finding given the evolutionary origin of mitochondria as an endosymbiotic bacterium integrated into cells (59). We hypothesize that such events represent a mechanism to enable the recycling of mitochondrial contents, which may be a more efficient process than fully degrading mitochondria before rebuilding them.
Another subset of mitolysosomes suddenly burst and released their contents into the cytosol, which could enable some contents to be reused by other organelles and others to be degraded further by the proteosome. Notably, although lysosomes are known to release recycled amino acids and other nutrients (60), we show that at least some proteins (i.e., the mitochondrial-targeted fluorescent reporters) are not fully broken down but rather released in a functional state. Understanding which mitochondrial contents are degraded and to what extent, as well as what triggers the release of mitochondrial contents, will be of great interest. Notably, the pH of these mitolysosomes partially normalized before bursting, potentially representing a mechanism to avoid damaging cellular structures when the contents are released. Moreover, the cytosolic release of mitochondrial contents may also represent a key mechanism by which mitochondrial contents enter the cytosol to trigger cell signaling pathways, including those involved in innate immunity and cell death (61–65). Recent evidence shows that chemically induced lysosomal rupture recruits the PD protein LRRK2 to lysosomes, further suggesting that a disruption in mitolysosome processing may be relevant to PD pathophysiology (66).
Last, a subset of mitolysosomes persisted for hours even after GFP-Parkin fluorescence dissipated and partially normalized their pH over time. The significance of this pH normalization is unclear, but we speculate that it reflects compensatory mechanisms to decrease lysosomal catabolism in the face of chronic mitochondrial dysfunction, to preserve residual mitochondria that are needed to sustain energy levels (67). It remains unclear whether these mitolysosomes eventually release their contents as well or if they have distinct fates from those mitolysosomes that burst.
In contrast, mitochondria in the direct pathway were larger and less acidic than those that first recruit Parkin to the OM, suggesting that these are distinct pathways. Mitochondria in the direct pathway may be healthy or mildly damaged mitochondria that have either engulfed or transiently fused with mitolysosomes. We speculate that these mitochondria have undergone targeted repair, consistent with prior work suggesting that the incorporation of lysosomal proteins within mitochondria may promote the elimination of oxidized mitochondrial proteins (68, 69). However, their slow accumulation of Parkin over many hours precluded us from determining their fates, which will require longer-term analysis.
Conclusions and questions
Critical questions raised by our findings include understanding how and why mitolysosomes fuse with and are engulfed by mitochondria, including the extent to which mitochondrial contents are recycled through these processes. It will also be important to better define the process by which mitolysosomes burst and whether this facilitates recycling of degraded mitochondrial contents and degradation of nonrecyclable components. Particularly important will be understanding how and when these processes are required for neuronal health and survival. PINK1 and Parkin do not appear necessary for mitochondrial turnover in neurons, since loss of either Parkin or PINK1 does not affect mitochondrial content, and both PINK1 and Parkin KO mice are viable, indicating that alternative pathways compensate when they are lost. However, our data with PINK1 KO mice, and other findings with Parkin KO mice (70), suggest that these compensatory processes are insufficient when mitochondria are damaged and require increased turnover. The engulfment of mitolysosomes by mitochondria may represent a mechanism to recycle mitochondrial contents when mitochondria cannot be repaired and an opportunity to strengthen or repair the recipient mitochondria. Moreover, Parkin and PINK1 may also have specific functions in the matrix such as enhancing mitochondrial transcription to promote biogenesis (49, 52) or initiating quality control pathways by ubiquitinating specific matrix proteins. Such a mechanism might explain how Parkin KO differentially increases the half-life of mitochondrial proteins in Drosophila heads, including proteins in the matrix (71, 72).
In summary, our results not only define a distinct mitochondrial life cycle in neurons wherein mitolysosomes remain dynamically active for long periods but also reveal mechanisms that may enable cells to salvage and reuse components of damaged mitochondria. At the same time, our findings also show that at least a subset of proteins can remain functional in mitolysosomes despite being acidified, and the presence of these mitochondrial proteins and reporters may lead to the misidentification of mitolysosomes as intact mitochondria. Further research is required to determine whether the mechanisms of mitochondrial quality control we delineate here, including the release of mitochondrial contents into the cytosol, may initiate immune and other signaling pathways and contribute to the selective vulnerability of neurons to PINK1 and Parkin mutations.
MATERIALS AND METHODS
Molecular biology
AAV2/1.CMV.PI.Cre.RBG was purchased from Penn Vector Core (Philadelphia, PA). Drp1 shRNA-smCBA-mTagBFP2 was cloned into the pTR-CBA eGFP vector to replace eGFP [Drp1-targeting shRNA was a gift of S. Strack, University of Iowa (73)]. The following constructs were previously cloned into the pCAGGS vector downstream of the chicken actin promoter: ATP-YEMK (gift of H. Noji, Osaka University) (74), Cre recombinase (2), EGFP-LC3 (Addgene), mitoGFP (4), and mitoTagBFP (2). mitoFarRed was generated by fusing TagRFP657 [gift from V. Verkhusha (Albert Einstein)] to the mitochondria-targeting sequence, cytochrome C oxidase subunit VIII (75, 76), and mitoKeima was generated by cloning mito-mKeima from pIND-mito-mKeima (gift of A. Miyawaki) into pCAGGs. OM-GFP was made by fusing the OM-targeting sequence of rat Tom20 to GFP (77). GFP-Parkin was made by fusing GFP to Parkin (gift from A. Abeliovich, Columbia) and Matrix-roGFP from Addgene. pGW1-mitoEOS2 was cloned into the pGW1 from pEOS2-mito.
Mice
Following procedures previously described in (2), mice were group-housed in a colony maintained with a standard 12-hour light/dark cycle and given food and water ad libitum. All mice received food on the cage floor. Experiments were performed on age-matched mice of either sex, with the specific number of males and females matched between groups in each experiment (typically divided 50:50 between males and females). No differences between sexes were noted in any of the experiments. Experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, as adopted by the National Institutes of Health (NIH), and with approval of the University of California, San Francisco Institutional Animal Care and Use Committee. Drp1lox/lox and ATG5lox/lox mice were maintained on the original mixed C57Bl/6 and 129/SvEv background. All other mouse lines were maintained on a pure C57Bl/6 background (the Jackson laboratory; RRID:IMSR_JAX:000664). Floxed Drp1KO mice (78) and PINK1 KO mice (46) have been described. DATcre mice (79) and Parkin KO mice (Stock No: 006582) were obtained from the Jackson laboratory.
Primary neuronal cultures
Primary hippocampal cultures were prepared from early postnatal (P0 to P1) mouse or rat pups of both sexes as described (2, 80). Neurons were transfected via electroporation (Amaxa) with the relevant constructs and cultured in minimum essential medium (MEM) Eagle’s with Earle’s balanced salt solution medium (UCSF Cell Culture Facility, CCFAC001) supplemented with 5% fetal bovine serum (HyClone, SH3007103), 21 mM glucose, 1% Glutamax (Gibco, 175007), 2% B27 supplement (Gibco, 17504-044), and 0.1% serum extender (Thermo Fisher Scientific, 355006). Neurons were plated at 600 to 900 cells/mm2, and uridine (10 μM, Sigma-Aldrich, U3750) was added on day 4 to minimize glial overgrowth. Neurons were cultured at 37°C and 5% CO2 for 9 to 11 days before live imaging or fixation.
Cell line
Immortalized Drp1KO MEFs were cultured in DMEM-H (UCSF Cell Culture Facility) containing 10% fetal bovine serum (JR Scientific) and penicillin-streptomycin at 37°C and 5% CO2 before live imaging or analysis.
Stereotaxic recombinant adeno-associated virus injection
For stereotaxic injection of AAV2/1.CMV.PI.Cre.RBG, 6- to 8-month-old Drp1wt/wt mice, Drp1lox/lox mice, and PINK1 KO and Drp1lox/lox PINK1 KO mice were used. Mice were anesthetized with ketamine/xylazine, and their skulls were immobilized with a stereotaxic frame (Kopf). A 0.5-μl volume of AAVcre (2.16 × 1013 genome copies/ml) was injected using a Hamilton syringe and a cannula (33 gauge) unilaterally into the SN/VTA (anteroposterior, −3.0 mm from bregma; mediolateral, 1.1 mm; dorsoventral, 4.3 mm) at a rate of 0.2 μl/min (2, 81). Animals were sacrificed 2 months after injection.
Histology
Mice were anesthetized and perfused with phosphate-buffered saline (PBS) and then 4% paraformaldehyde (PFA). Brains were removed, postfixed, cryoprotected in 30% sucrose, and frozen in isopentane. Coronal sections of 40-μm thickness were cut on a cryostat throughout the brain, including the SNc/VTA, and every sixth section was analyzed. Cells were fixed for 15 min in medium with 4% PFA.
For immunofluorescence, brain section and cells were rinsed with PBS and transferred in blocking solution containing either 0.2% Triton X-100 or 0.01% for CLEM, with either 3 to 5% bovine serum albumin or 5 to 10% goat serum for 1 hour. Samples were then incubated overnight at room temperature (RT) or for 24 to 48 hours at 4°C, with the appropriate primary antibody in blocking solution.
The following primary antibodies were used: rabbit anti-Tom20 (1:1000 to 1500; Santa Cruz Biotechnology, catalog no. SC-11415, RRID:AB_2207533), rabbit anti-COX7B (1:100; Abcam, catalog no. ab137094), mouse anti-PDH (1:200 to 1500; Abcam, catalog no. ab110333, RRID:AB_10862029) (24), mouse anti-MTCO1 (1:1000; Abcam, catalog no. ab14705, RRID:AB_2084810), rat anti-Lamp1 (1:400; BD Pharmingen, catalog no. 553792, RRID:AB_2134499), or anti-TH (mouse; 1:20,000; EMD Millipore, catalog no. MAB318, RRID:AB_2201528; or rabbit, 1:1000; Millipore, catalog no. AB152, RRID:AB_390204). Sections were rinsed and incubated for 2 hours with the appropriate secondary antibodies as follows: Alexa Fluor 488, 594, 568, or 647 anti-mouse, anti-rat, or anti-rabbit immunoglobulin G (IgG) (1:400; Invitrogen). Samples were rinsed in PBS, and coverslips were mounted on glass slides immerged in ProLong Gold Antifade Mountant (Thermo Fisher Scientific, #P36934) and stored at 4°C.
Immunofluorescence samples were imaged and analyzed with the examiner blind to the genotype using either a laser scanning confocal microscope (Zeiss LSM 880 with Airyscan or LSM510-Meta, Carl Zeiss) equipped with 63× [1.4 numerical aperture (NA)] and 100× (1.3 NA) PlanApo objectives or with 10× (0.3 NA) and 100× oil immersion (1.4 NA) objectives, respectively, on a Nikon Ti-E inverted microscope with an Andor iXon EMCCD camera. Confocal z-stacks of individual mitochondria were acquired using the optimal step size (0.25 or 0.4 μm) along the mitochondrial thickness (2 to 5 μm). The exposure time to acquire images was calculated to make the z-stack duration to be less than 5 s, and the images were reconstructed with Zen Lite software (Zeiss).
For peroxidase experiments, sections were incubated with rabbit polyclonal anti-TH and visualized with biotinylated goat anti-rabbit IgG (1:300, Vector Laboratories, Burlingame, CA; BA-1000, RRID:AB_2313606), followed by streptavidin-conjugated horseradish peroxidase (1:300; Vectastain ABC kit, Vector Laboratories). Positive immunostaining was visualized with 3,3′-diaminobenzidine (Sigma-Aldrich) after reaction with hydrogen peroxide.
Stereology
Total numbers of TH-positive neurons in the SNc and VTA were quantified with the examiner blind to the genotype using a computer-assisted image analysis system, consisting of an Olympus BX-51 microscope equipped with an XYZ computer-controlled motorized stage and a SIA-L9C digital camera (Scientific Instruments and Applications, Duluth, GA), and counted using the Optical Fractionator probe of Stereo Investigator software (MicroBrightfield, Williston, VT; RRID:nif-0000-00110).The numbers of TH-stained cells were counted at high power (100× oil; NA 1.4) using a 75 mm by 75 mm counting frame as described (2).
Correlative light and electron microscopy
Primary neurons cultured on gridded 35-mm MatTek glass-bottom dishes (MatTek Corp., Ashland, MA, USA) were imaged using a fluorescence microscope and then fixed in 2% glutaraldehyde and 2% PFA in phosphate buffer (pH 7.4) (EMS, Hatfield, PA, USA). Samples were washed (3×; 5 min, RT) in 0.1 M sodium cacodylate buffer (pH 7.2) and covered in 1% osmium tetroxide with 1.6% potassium ferricyanide in 0.1 M sodium cacodylate buffer for 30 min. Samples were washed (3×; 5 min, RT) in buffer and then in distilled water (1×; 5 min, RT). Samples were then put through an ascending ethanol gradient (7 min; 35, 50, 70, 80, and 90%) followed by pure ethanol (2×; 5 min, RT). Samples were incubated with Epon resin (EMS, Hatfield, PA, USA) and polymerized at 60°C for 24 hours. The glass coverslips were then removed using ultrathin Personna razor blades (EMS, Hatfield, PA, USA) and liquid nitrogen exposure as needed. The gridded alphanumerical labeling was used to identify regions of interest that were dissected out and mounted for sectioning using cyanoacrylate glue. A Leica UC6 Ultramicrotome (Leica, Wetzlar, Germany) was used to cut thin sections (100 nm) from the surface of the block until reaching a depth of 4 to 5 μm to ensure full cell coverage. Formvar-coated 50-mesh copper grids were used to collect serial sections and were poststained with 2% uranyl acetate followed by Reynold’s lead citrate, for 5 min each. Imaging was performed using a Tecnai 12 120-kV transmission electron microscope (FEI, Hillsboro, OR, USA), and data were recorded using UltraScan 1000 with Digital Micrograph 3 software (Gatan Inc., Pleasanton, CA, USA).
Live imaging
Magic Red
For cathepsin B activity assays, neurons were incubated with Magic Red (a cathepsin B substrate, ImmunoChemistry, #937) for 1 hour at 37°C before live imaging (572/35 excitation and 632/60 emission).
mito-roGFP
For mito-roGFP experiments, sequential images were acquired at 402/15 excitation and 535/50 emission and 490/20 excitation and 535/50 emission. Background was subtracted, and images were created on the basis of the ratio of the 402-nm/490-nm images on a pixel-per-pixel basis. To calibrate the assay, 2 mM dithiothreitol was used to fully reduce the mitochondria and determine the minimum fluorescence ratio and 100 μM Aldrithiol (DPS) to oxidize the mitochondria for the maximum value (82).
Mitochondrial membrane potential
Neurons were pretreated for 60 min with TMRM (20 nM) in neuron culture medium and then transferred to Tyrode’s medium with 20 nM TMRM for live imaging. Images were captured before and 5 min after FCCP (1.25 μM) treatment. TMRM fluorescence of the mitochondria (an estimate of the extent of polarization of these mitochondria) was calculated as the average fluorescent intensity within the individual mitochondria subtracted by the average fluorescent intensity in the nucleus of the same cell.
mitoKeima
For mitoKeima experiments, images were acquired at 572/35 excitation and 632/60 emission and 430/24 excitation and 632/60 emission, or 561 excitation and 602 to 706 emission and 458 excitation and 602 to 706 emission on a laser scanning confocal microscope, for time-lapse imaging at 561 excitation and 607/36 emission and 440 excitation and 607/36 emission.
Time-lapse imaging of mitochondrial turnover
Live imaging of Drp1KO neurons coexpressing mitoKeima and GFP-Parkin was performed on a Nikon Ti-E microscope with Flash4.0 camera (Hamamatsu) controlled by NIS Elements 4.2 (Nikon Instruments). All components were encased in an environmental chamber (In Vivo Scientific) maintained at ~37°C and ~5% CO2. Z-stacks of single neurons were acquired every 20 min for up to 21 hours. Live imaging of Drp1KO and wt neurons incubated with TMRM or expressing GFP-LC3 or LAMP1-RFP was performed on a DeltaVision OMX SR imaging system with an environmental control module (~37°C and ~5% CO2) and controlled by AcquireSRsoftWoRx (GE Healthcare). Z-stacks were acquired every 10 to 20 min for up to 12 hours. For all time-lapse runs, the z section offering the best focus for the mitochondria of interest was selected at each time point. Images were then aligned using MultiStackReg in ImageJ (NIH). MitoKeima ratio was calculated after background subtraction using the ratio of red over green mitoKeima channels. Regions of interest were drawn around tracked mitochondria at every time point. Quantitative data were obtained from images using MetaMorph software (Universal Imaging), ImageJ (NIH), or ZEN 2.3 lite (Zeiss).
Automated fluorescence microscopy
Experiments involving neuronal survival analysis and optical pulse imaging used an automated microscopy platform as described (83). Briefly, images were obtained with an inverted microscope (Nikon TE2000) equipped with the PerfectFocus system, a 20× Plan Fluor S 0.45 NA ELWD objective, and a 16-bit Andor Clara digital camera with a cooled charge-coupled device. Illumination was provided by a Lambda XL Xenon lamp (Sutter). All of the components were encased in a custom-designed, climate-controlled environmental chamber (In Vivo Scientific), kept at ~37°C and ~5% CO2. The Semrock BrightLine full-multiband filter set [4′,6-diamidino-2-phenylindole (DAPI), fluorescein isothiocyanate (FITC), tetramethyl rhodamine isothiocyanate (TRITC), and cyanine 5 (Cy5)] was used for fluorophore photoactivation (DAPI) and excitation and detection (FITC and TRITC). The illumination, filter wheels, focusing, stage movements, and image acquisitions were fully automated and coordinated with publicly available (ImageJ and μManager) software.
Neurons expressing mitoEOS2 were photoconverted with a 4-s pulse of 405-nm light and longitudinally imaged using robotic microscopy, which can return to the same cell repeatedly over time (84). Single-cell red fluorescence intensities were normalized to the red fluorescence immediately after photoconversion, and the half-life of the photoconverted red mito-EOS2 reporter was calculated for each cell using the standard half-life equation.
Relevant data were extracted from the raw, digital images in a sequential process using an original script developed in PipelinePilot (Accelrys, San Diego, CA). Briefly, the median background fluorescence from a portion of all images was calculated and subtracted from each individual image. The images were then assembled into montages representing each well at each time point. The montages were temporally sequenced and aligned automatically, and neuron cell bodies were segmented on the basis of intensity and morphology. Neurons with photoswitched mitoEOS2 values below 10 arbitrary units, neurons that survived <~30 hours after photoswitching, and those with a calculated half-life of >1000 hours were excluded from half-life analyses as this value was outside of the range of half-lives that the OPL technique is realistically able to detect. Statistical analyses and the generation of cumulative hazard plots and density plots were accomplished using custom-designed algorithms and the survival package within R, while scatterplots and bar graphs were created using Prism (GraphPad).
Statistical analysis
Data were compiled and processed using Microsoft Excel. GraphPad Prism (GraphPad Software) and SPSS (IBM) were used for statistical analyses, and GraphPad Prism was used to generate graphs. All statistical analyses, including the n, what n represents, description of error bars, statistical tests used, and level of significance, are found in the figure legends. Data were processed using two-tailed unpaired t tests with Welch’s correction or by analysis of variance (ANOVA). Before ANOVA, the homogeneity of variance between experimental groups was assessed by Levene’s test. If homogeneous (P > 0.05), data were subjected to one- or two-way ANOVA followed by Bonferroni, Sidak, or Tukey post hoc test. If not homogeneous, data were processed by one-way ANOVA followed by Games-Howell post hoc test.
Acknowledgments
We thank M. Calvert, B. Ndjamen, and the Gladstone Histology and Light Microscopy core as well as D. Larsen and the UCSF Nikon Imaging Center, R. Zalpuri, and the University of California, Berkeley Electron Microscope Laboratory for advice and assistance in EM sample preparation and data collection and G. Howard and K. Claiborn for helping edit the manuscript. Funding: This work was supported by the Joan and David Traitel Family Trust and Betty Brown’s Family. K.N., H.L., Z.D., A.B., and M.K.N. were also supported by a Burroughs-Wellcome Medical Scientist Fund Career Award; NIH RO1NS091902 to K.N.; and NIH 5P30 NS069496 to L. Mucke (Gladstone). Z.D. was also supported by a Hillblom Fellowship and a Berkelhammer Award for Excellence in Neuroscience. I.H. and E.H. were supported by NIH R01 NS098516 and VA Merit Award BX001108 to E.H. J.M. and S.F. were supported by the Taube/Koret Center for Neurodegenerative disease; the Michael J. Fox Foundation; and NIH R37NS101995, RF1 AG058476, and RF1 AG065151. This work was also supported by NIH RR18928 to the Gladstone Institutes. Author contributions: H.L., Z.D., A.B., M.K.N., J.M., E.H., and K.N. designed the research. H.L., Z.D., A.B., D.M.J., M.K.N., I.H., J.M., and R.F. performed research. D.M.J., J.D., H.S., S.F., and E.H. provided guidance and reagents for critical techniques. H.L., Z.D., A.B., M.K.N., J.M., R.F., E.H., and K.N. analyzed data. K.N. and Z.D. wrote the paper with assistance from co-authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabf6580/DC1
REFERENCES AND NOTES
- 1.Itoh K., Nakamura K., Iijima M., Sesaki H., Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 23, 64–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berthet A., Margolis E. B., Zhang J., Hsieh I., Zhang J., Hnasko T. S., Ahmad J., Edwards R. H., Sesaki H., Huang E. J., Nakamura K., Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 34, 14304–14317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pham A. H., Meng S., Chu Q. N., Chan D. C., Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 21, 4817–4826 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakamura K., Nemani V. M., Azarbal F., Skibinski G., Levy J. M., Egami K., Munishkina L., Zhang J., Gardner B., Wakabayashi J., Sesaki H., Cheng Y., Finkbeiner S., Nussbaum R. L., Masliah E., Edwards R. H., Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 286, 20710–20726 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang W., Wang X., Fujioka H., Hoppel C., Whone A. L., Caldwell M. A., Cullen P. J., Liu J., Zhu X., Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 22, 54–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., Youle R. J., PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLOS Biol. 8, e1000298 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poole A. C., Thomas R. E., Andrews L. A., McBride H. M., Whitworth A. J., Pallanck L. J., The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 105, 1638–1643 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., Guo M., Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D. F., Karbowski M., Youle R. J., Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziviani E., Tao R. N., Whitworth A. J., Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. U.S.A. 107, 5018–5023 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G., Alroy J., Wu M., Py B. F., Yuan J., Deeney J. T., Corkey B. E., Shirihai O. S., Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kageyama Y., Hoshijima M., Seo K., Bedja D., Sysa-Shah P., Andrabi S. A., Chen W., Hoke A., Dawson V. L., Dawson T. M., Gabrielson K., Kass D. A., Iijima M., Sesaki H., Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 33, 2798–2813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikeda Y., Shirakabe A., Maejima Y., Zhai P., Sciarretta S., Toli J., Nomura M., Mihara K., Egashira K., Ohishi M., Abdellatif M., Sadoshima J., Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116, 264–278 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Song M., Mihara K., Chen Y., Scorrano L., Dorn G. W. II, Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita S. I., Jin X., Furukawa K., Hamasaki M., Nezu A., Otera H., Saigusa T., Yoshimori T., Sakai Y., Mihara K., Kanki T., Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J. Cell Biol. 215, 649–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burman J. L., Pickles S., Wang C., Sekine S., Vargas J. N. S., Zhang Z., Youle A. M., Nezich C. L., Wu X., Hammer J. A., Youle R. J., Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 216, 3231–3247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai Q., Zakaria H. M., Simone A., Sheng Z. H., Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 22, 545–552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Laar V. S., Arnold B., Cassady S. J., Chu C. T., Burton E. A., Berman S. B., Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 20, 927–940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grenier K., McLelland G. L., Fon E. A., Parkin- and PINK1-dependent mitophagy in neurons: Will the real pathway please stand up? Front. Neurol. 4, 100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vives-Bauza C., Zhou C., Huang Y., Cui M., de Vries R. L., Kim J., May J., Tocilescu M. A., Liu W., Ko H. S., Magrane J., Moore D. J., Dawson V. L., Grailhe R., Dawson T. M., Li C., Tieu K., Przedborski S., PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lazarou M., Sliter D. A., Kane L. A., Sarraf S. A., Wang C., Burman J. L., Sideris D. P., Fogel A. I., Youle R. J., The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitada T., Pisani A., Porter D. R., Yamaguchi H., Tscherter A., Martella G., Bonsi P., Zhang C., Pothos E. N., Shen J., Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11441–11446 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dagda R. K., Cherra S. J. III, Kulich S. M., Tandon A., Park D., Chu C. T., Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kageyama Y., Zhang Z., Roda R., Fukaya M., Wakabayashi J., Wakabayashi N., Kensler T. W., Reddy P. H., Iijima M., Sesaki H., Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 197, 535–551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katayama H., Kogure T., Mizushima N., Yoshimori T., Miyawaki A., A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 18, 1042–1052 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Mauvezin C., Nagy P., Juhasz G., Neufeld T. P., Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 6, 7007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N., Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Mellman I., Fuchs R., Helenius A., Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 55, 663–700 (1986). [DOI] [PubMed] [Google Scholar]
- 29.Pronevich L. A., Mirzabekov T. A., Rozhdestvenskaya Z. E., Mitochondrial porin regulates the sensitivity of anion carriers to inhibitors. FEBS Lett. 247, 330–332 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Yoshii S. R., Kishi C., Ishihara N., Mizushima N., Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 286, 19630–19640 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin S. M., Youle R. J., The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vincent A. E., Ng Y. S., White K., Davey T., Mannella C., Falkous G., Feeney C., Schaefer A. M., McFarland R., Gorman G. S., Taylor R. W., Turnbull D. M., Picard M., The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci. Rep. 6, 30610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poburko D., Santo-Domingo J., Demaurex N., Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 286, 11672–11684 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong Y. C., Ysselstein D., Krainc D., Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshii S. R., Mizushima N., Monitoring and measuring autophagy. Int. J. Mol. Sci. 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura S., Noda T., Yoshimori T., Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Itakura E., Mizushima N., Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 764–776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen G. F., Toth R., James J., Ganley I. G., Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 14, 1127–1135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson D. E., Ostrowski P., Jaumouille V., Grinstein S., The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 212, 677–692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Favaro G., Romanello V., Varanita T., Andrea Desbats M., Morbidoni V., Tezze C., Albiero M., Canato M., Gherardi G., De Stefani D., Mammucari C., Blaauw B., Boncompagni S., Protasi F., Reggiani C., Scorrano L., Salviati L., Sandri M., DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 10, 2576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y., Ouyang Y., Yang L., Beal M. F., McQuibban A., Vogel H., Lu B., Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. U.S.A. 105, 7070–7075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gispert S., Ricciardi F., Kurz A., Azizov M., Hoepken H. H., Becker D., Voos W., Leuner K., Muller W. E., Kudin A. P., Kunz W. S., Zimmermann A., Roeper J., Wenzel D., Jendrach M., Garcia-Arencibia M., Fernandez-Ruiz J., Huber L., Rohrer H., Barrera M., Reichert A. S., Rub U., Chen A., Nussbaum R. L., Auburger G., Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLOS ONE 4, e5777 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lutz A. K., Exner N., Fett M. E., Schlehe J. S., Kloos K., Lammermann K., Brunner B., Kurz-Drexler A., Vogel F., Reichert A. S., Bouman L., Vogt-Weisenhorn D., Wurst W., Tatzelt J., Haass C., Winklhofer K. F., Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 284, 22938–22951 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gispert S., Brehm N., Weil J., Seidel K., Rub U., Kern B., Walter M., Roeper J., Auburger G., Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 24, 1061–1076 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song S., Jang S., Park J., Bang S., Choi S., Kwon K. Y., Zhuang X., Kim E., Chung J., Characterization of PINK1 (PTEN-induced putative kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila. J. Biol. Chem. 288, 5660–5672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L., Xie Z., Turkson S., Zhuang X., A53T human α-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci. 35, 890–905 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rappold P. M., Cui M., Grima J. C., Fan R. Z., de Mesy-Bentley K. L., Chen L., Zhuang X., Bowers W. J., Tieu K., Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 5, 5244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bordt E. A., Clerc P., Roelofs B. A., Saladino A. J., Tretter L., Adam-Vizi V., Cherok E., Khalil A., Yadava N., Ge S. X., Francis T. C., Kennedy N. W., Picton L. K., Kumar T., Uppuluri S., Miller A. M., Itoh K., Karbowski M., Sesaki H., Hill R. B., Polster B. M., The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 40, 583–594.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuroda Y., Mitsui T., Kunishige M., Shono M., Akaike M., Azuma H., Matsumoto T., Parkin enhances mitochondrial biogenesis in proliferating cells. Hum. Mol. Genet. 15, 883–895 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Harbauer A. B., Zahedi R. P., Sickmann A., Pfanner N., Meisinger C., The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 19, 357–372 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Kim P. K., Hettema E. H., Multiple pathways for protein transport to peroxisomes. J. Mol. Biol. 427, 1176–1190 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuroda Y., Sako W., Goto S., Sawada T., Uchida D., Izumi Y., Takahashi T., Kagawa N., Matsumoto M., Matsumoto M., Takahashi R., Kaji R., Mitsui T., Parkin interacts with Klokin1 for mitochondrial import and maintenance of membrane potential. Hum. Mol. Genet. 21, 991–1003 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Burchell V. S., Nelson D. E., Sanchez-Martinez A., Delgado-Camprubi M., Ivatt R. M., Pogson J. H., Randle S. J., Wray S., Lewis P. A., Houlden H., Abramov A. Y., Hardy J., Wood N. W., Whitworth A. J., Laman H., Plun-Favreau H., The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 16, 1257–1265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Z. D., Xie S. P., Sathiyamoorthy S., Saw W. T., Sing T. Y., Ng S. H., Chua H. P., Tang A. M., Shaffra F., Li Z., Wang H., Ho P. G., Lai M. K., Angeles D. C., Lim T. M., Tan E. K., F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 24, 6314–6330 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Selivanov V. A., Zeak J. A., Roca J., Cascante M., Trucco M., Votyakova T. V., The role of external and matrix pH in mitochondrial reactive oxygen species generation. J. Biol. Chem. 283, 29292–29300 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khacho M., Tarabay M., Patten D., Khacho P., MacLaurin J. G., Guadagno J., Bergeron R., Cregan S. P., Harper M. E., Park D. S., Slack R. S., Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat. Commun. 5, 3550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans C. S., Holzbaur E. L., Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. eLife 9, e50260 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong Y. C., Peng W., Krainc D., Lysosomal regulation of inter-mitochondrial contact fate and motility in Charcot-Marie-Tooth type 2. Dev. Cell 50, 339–354.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roger A. J., Munoz-Gomez S. A., Kamikawa R., The origin and diversification of mitochondria. Curr. Biol. 27, R1177–R1192 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Mony V. K., Benjamin S., O’Rourke E. J., A lysosome-centered view of nutrient homeostasis. Autophagy 12, 619–631 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C. J., Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.West A. P., Khoury-Hanold W., Staron M., Tal M. C., Pineda C. M., Lang S. M., Bestwick M., Duguay B. A., Raimundo N., MacDuff D. A., Kaech S. M., Smiley J. R., Means R. E., Iwasaki A., Shadel G. S., Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.West A. P., Shadel G. S., Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17, 363–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sliter D. A., Martinez J., Hao L., Chen X., Sun N., Fischer T. D., Burman J. L., Li Y., Zhang Z., Narendra D. P., Cai H., Borsche M., Klein C., Youle R. J., Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rongvaux A., Jackson R., Harman C. C., Li T., West A. P., de Zoete M. R., Wu Y., Yordy B., Lakhani S. A., Kuan C. Y., Taniguchi T., Shadel G. S., Chen Z. J., Iwasaki A., Flavell R. A., Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bonet-Ponce L., Beilina A., Williamson C. D., Lindberg E., Kluss J. H., Saez-Atienzar S., Landeck N., Kumaran R., Mamais A., Bleck C. K. E., Li Y., Cookson M. R., LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 6, eabb2454 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fernandez-Mosquera L., Yambire K. F., Couto R., Pereyra L., Pabis K., Ponsford A. H., Diogo C. V., Stagi M., Milosevic I., Raimundo N., Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy 15, 1572–1591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miyamoto Y., Kitamura N., Nakamura Y., Futamura M., Miyamoto T., Yoshida M., Ono M., Ichinose S., Arakawa H., Possible existence of lysosome-like organella within mitochondria and its role in mitochondrial quality control. PLOS ONE 6, e16054 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miyamoto T., Kitamura N., Ono M., Nakamura Y., Yoshida M., Kamino H., Murai R., Yamada T., Arakawa H., Identification of 14-3-3γ as a Mieap-interacting protein and its role in mitochondrial quality control. Sci. Rep. 2, 379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pickrell A. M., Huang C. H., Kennedy S. R., Ordureau A., Sideris D. P., Hoekstra J. G., Harper J. W., Youle R. J., Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87, 371–381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vincow E. S., Merrihew G., Thomas R. E., Shulman N. J., Beyer R. P., MacCoss M. J., Pallanck L. J., The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. U.S.A. 110, 6400–6405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vincow E. S., Thomas R. E., Merrihew G. E., Shulman N. J., Bammler T. K., MacDonald J. W., MacCoss M. J., Pallanck L. J., Autophagy accounts for approximately one-third of mitochondrial protein turnover and is protein selective. Autophagy 15, 1592–1605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cribbs J. T., Strack S., Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Imamura H., Huynh Nhat K. P., Togawa H., Saito K., Iino R., Kato-Yamada Y., Nagai T., Noji H., Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. U.S.A. 106, 15651–15656 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morozova K. S., Piatkevich K. D., Gould T. J., Zhang J., Bewersdorf J., Verkhusha V. V., Far-red fluorescent protein excitable with red lasers for flow cytometry and superresolution STED nanoscopy. Biophys. J. 99, L13–L15 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Subach O. M., Gundorov I. S., Yoshimura M., Subach F. V., Zhang J., Gruenwald D., Souslova E. A., Chudakov D. M., Verkhusha V. V., Conversion of red fluorescent protein into a bright blue probe. Chem. Biol. 15, 1116–1124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kanaji S., Iwahashi J., Kida Y., Sakaguchi M., Mihara K., Characterization of the signal that directs Tom20 to the mitochondrial outer membrane. J. Cell Biol. 151, 277–288 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wakabayashi J., Zhang Z., Wakabayashi N., Tamura Y., Fukaya M., Kensler T. W., Iijima M., Sesaki H., The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Backman C. M., Malik N., Zhang Y., Shan L., Grinberg A., Hoffer B. J., Westphal H., Tomac A. C., Characterization of a mouse strain expressing Cre recombinase from the 3′ untranslated region of the dopamine transporter locus. Genesis 44, 383–390 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Shields L. Y., Kim H., Zhu L., Haddad D., Berthet A., Pathak D., Lam M., Ponnusamy R., Diaz-Ramirez L. G., Gill T. M., Sesaki H., Mucke L., Nakamura K., Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis. 6, e1725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shin J. H., Ko H. S., Kang H., Lee Y., Lee Y. I., Pletinkova O., Troconso J. C., Dawson V. L., Dawson T. M., PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Lith M., Tiwari S., Pediani J., Milligan G., Bulleid N. J., Real-time monitoring of redox changes in the mammalian endoplasmic reticulum. J. Cell Sci. 124, 2349–2356 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barmada S. J., Serio A., Arjun A., Bilican B., Daub A., Ando D. M., Tsvetkov A., Pleiss M., Li X., Peisach D., Shaw C., Chandran S., Finkbeiner S., Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 10, 677–685 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Arrasate M., Finkbeiner S., Automated microscope system for determining factors that predict neuronal fate. Proc. Natl. Acad. Sci. U.S.A. 102, 3840–3845 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/32/eabf6580/DC1