

Abstract
Abiological enzymes offers new opportunities for sustainable chemistry. Here we report the development of biological catalysts derived from sperm whale myoglobin that exploit a carbene transfer mechanism for the asymmetric synthesis of cyclopropane-fused-δ-lactones, which are key structural motifs found in many biologically active natural products. While hemin, wild-type myoglobin, and other hemoproteins are unable to catalyze this reaction, the myoglobin scaffold could be remodeled by protein engineering to permit the intramolecular cyclopropanation of a broad spectrum of homoallylic diazoacetate substrates in high yields and with up to 99% enantiomeric excess. Via an alternate evolutionary trajectory, a stereodivergent biocatalyst was also obtained for affording mirror-image forms of the desired bicyclic products. In combination with whole-cell transformations, the myoglobin-based biocatalyst was readily applied to enable the asymmetric construction of a cyclopropyl-δ-lactone scaffold at a gram scale, which could be further elaborated to furnish a variety of enantiopure trisubstituted cyclopropanes.
Keywords: intramolecular cyclopropanation, δ-lactones, carbene transfer, myoglobin, biocatalysis
Graphical Abstract
Lords of the Rings: A biocatalytic strategy for the stereodivergent synthesis of cyclopropyl-δ-lactones via the intramolecular cyclopropanation of homoallylic diazoacetates is reported. Myoglobin was re-engineered into two enantiocomplementary biocatalysts capable of producing a range of aryl- and alkyl-substituted cyclopropane-fused δ-lactone scaffolds useful for medicinal chemistry and natural product synthesis.
Cyclopropane-containing scaffolds have attracted significant interest due to their importance for the design of pharmaceuticals, occurrence in biologically active natural products, and value as synthetic intermediates.[1] Fused cyclopropyl-δ-lactones and their tetrahydropyran derivatives are key structural pharmacophores found in numerous biologically active natural products including microphyllandiolide, dinardokanshone B, 2-caren-8,4-olide and chubularisin J (Figure 1).[2] Cyclopropyl-δ-lactones can be accessed through the intramolecular cyclopropanation of homoallylic diazoacetates but synthetic approaches to perform these transformations have been notoriously scarce and essentially limited to rhodium-based catalysts as reported by Doyle and coworkers.[3] In addition, achieving high levels of enantioselectivity using these systems has proven challenging and variable levels of stereocontrol were observed depending on the nature of substituents appended to the olefinic group (Scheme 1).[3] We therefore envisioned that the development of potentially more general and enantioselective iron-based biocatalysts for this transformation would offer an attractive and sustainable alternative for the asymmetric construction of fused cyclopropane-containing bicyclo[4.1.0] scaffolds via intramolecular cyclopropanation.
Figure 1.
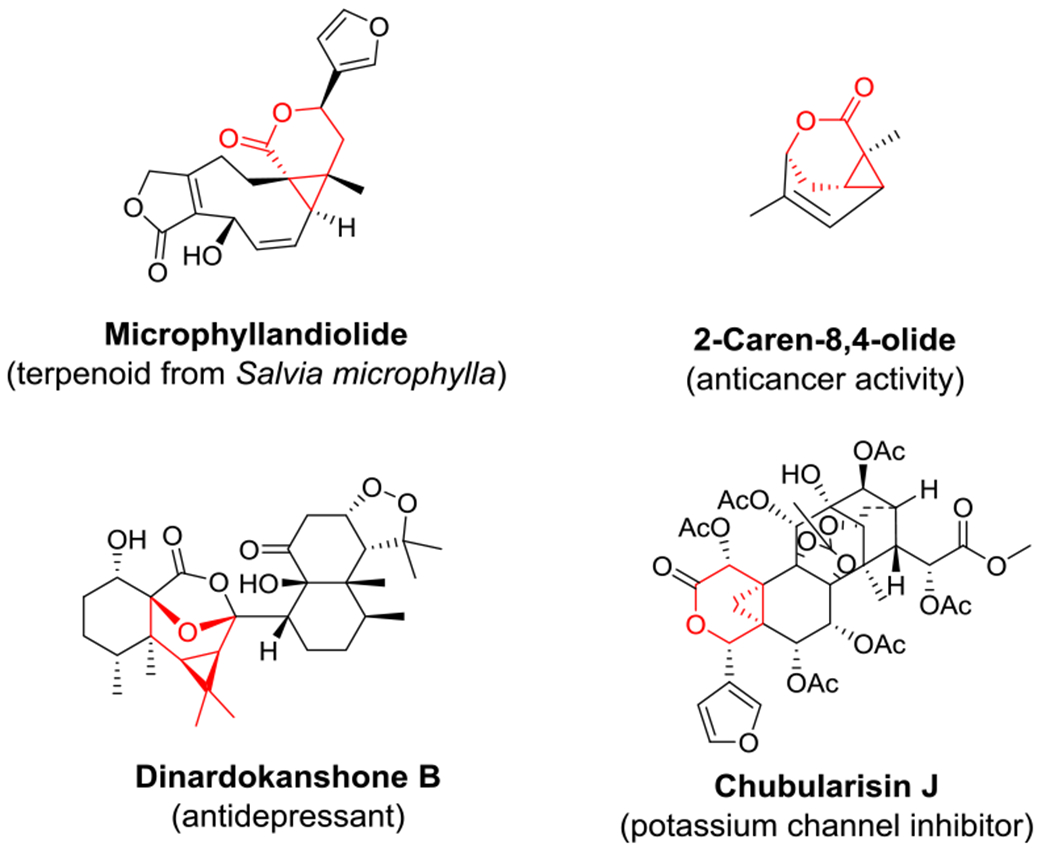
Biologically active compounds containing fused bicyclo[4.1.0] scaffolds
Scheme 1.
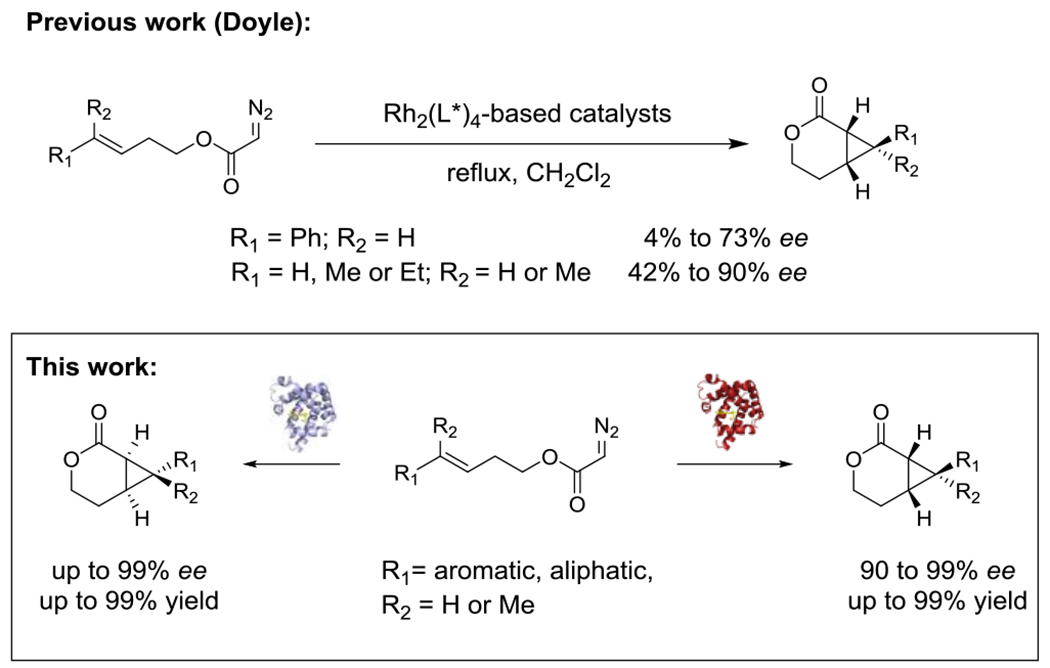
Catalytic strategies for the synthesis of fused bicyclo[4.1.0] scaffolds via intramolecular cyclopropanation
Over the past few years, we and others have demonstrated the potential of engineered hemoproteins (myoglobin, cytochrome P450s, cytochrome c)[4] and artificial metalloenzymes[5] for promoting abiological carbene transfer reactions. Efforts in this area have also expanded the range of diazo compounds amenable to intermolecular cyclopropanation reactions.[6] More recently, the possibility of mediating intramolecular cyclopropanation reactions using engineered hemoprotein-based biocatalysts was demonstrated.[7] Despite this progress, the construction of cyclopropyl-δ-lactones through the cyclization of homoallylic α-diazoacetates has proven elusive,[7a] reflecting the challenge of orchestrating these energetically and sterically demanding ring-closing reactions both with synthetic catalysts and within the active site an enzyme (vide infra). In addition, no iron-based catalysts have so far been reported for promoting these transformations. Here, we report the successful development of myoglobin-based biocatalysts capable of promoting the intramolecular cyclopropanation of homoallylic α-diazoacetates with high enantioselectivity as well as stereodivergent selectivity. These systems are shown to provide an efficient and scalable approach to access enantiomeric pairs of optically active cyclopropyl-δ-lactones as valuable synthons for medicinal chemistry and natural product synthesis (Scheme 1).
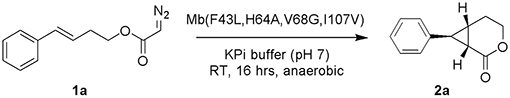
In initial experiments, we evaluated the ability of wild type myoglobin (Mb) and a diverse panel of other hemoproteins, including cytochromes P450 (P450BM3, XplA, BezE), catalase and cytochromes c, to catalyze the cyclization of (E)-4-phenylbutenyl 2-diazoacetate (1a) to give the corresponding intramolecular cyclopropanation products 2a/3a (Tables 1 and S1). However, none of these metalloproteins show any detectable catalytic activity toward this reaction (Table S1). Similar results were obtained with two engineered Mb variants previously developed for the intramolecular cyclopropanation of allyl α-diazoacetates (Mb(H64V,I107S))[7a] and allyl α-diazoacetamides (Mb(F43Y,H64V,V68A,I107V)),[7b] which were also found to be catalytically incompetent toward this transformation (Table 1, Entries 4-5). Considering the high efficiency of Mb(H64V,I107S) toward mediating the cyclization of trans-cinnamyl-2-diazoacetate,[7a] we attributed this result to the inherently higher steric and entropic demands associated with mediating the intramolecular cyclopropanation of 1a to give the cyclopropane-fused-bicyclo[4.1.0] product 2a within the active site of the hemoprotein when compared to the allylic counterpart. Furthermore, neither iron-tetraphenylporphyrin (Fe(TPP)) nor free hemin showed any activity toward conversion of 1a into 2a/3a (Table 1, Entry 1-2), which is in contrast to their previously observed ability to catalyze the cyclopropanation of allyl α-diazoacetates.[7a] This result indicated that the former reaction is energetically unfavorable in the presence of iron-porphyrins as catalysts, even in the absence of steric constraints around the metal active site.
Table 1.
Intramolecular cyclopropanation of (E)-4-phenylbut-3-en-1-yl 2-diazoacetate (1a) with Mb and variants thereof. [a]
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | OD600 | Yield[b] | TON | e.e. |
| 1 | Hemin | - | 0% | 0 | n.d. |
| 2 | Fe(TPP)Cl in DCM | 0% | 0 | n.d. | |
| 3 | Mb (WT) | 0% | 0 | n.d. | |
| 4 | Mb(H64V,I107S) | - | 0% | 0 | n.d. |
| 5 | Mb(F43Y,H64V, V68A,I107V) | - | 0% | 0 | n.d. |
| 6 | Mb(V68G) | - | 1.1% | 1 | 16% |
| 7 | Mb(H64A,V68G) | 1.9% | 2 | 33% | |
| 8 | Mb(H64A,V68G, I107V) | - | 45% | 56 | 61% |
| 9 | Mb(F43L,H64A, V68G, I107V) | - | 63% | 79 | 99% |
| 10 | Mb(F43L,H64A, V68G,I107V) | 20 | >99% | 316 | 99% |
| 11 | Mb(F43L,H64A, V68G,I107V) | 5 | 92% | 1164 | 99% |
| 12[c] | Mb(F43L,H64A, V68G,I107V) | 20 | 66% | 208 | 99% |
Reaction conditions: 2.5 mM (E)-4-phenylbut-3-en-1-yl 2-diazoacetate (1a), 20 μM Mb variant (or C41(DE3) E.coli cells at indicated OD600) in KPi buffer (50 mM, pH 7), 10 mM Na2S2O4 (protein only), RT, 16 hrs in anaerobic chamber.
GC yield based on the calibration curves by authentic standard.
reaction time: 2 min
Next, we screened a diverse ‘in-house’ library of ~80 Mb variants containing up to four amino acid substitutions in their heme pocket but the large majority (98%) of these proteins showed either no activity (0% yield, 59/82 variants) or negligible activity (<1% yield; 21/82 variants) in the reaction with 1a (Table S2). Within this panel of Mb variants, however, Mb(V68G) and Mb(H64V,V68G) showed some appreciable activity toward formation of 2a although both the yields (1-2%) and enantioselectivity of these reactions were low (16% and 4% ee, respectively) (Table 1, Entries 6-7). Interestingly, these Mb variants share a space-creating mutation (Val→Gly) at position 68, whose side-chain is projected toward the meso position between rings A and D of the hemin cofactor (Figure 2a).[8] These results suggested that enlargement of the active site cavity in Mb is beneficial for promoting the cyclization reaction, supporting the notion about the high sensitivity of this reaction to steric factors. This aspect is further highlighted by the lack of activity of Mb(V68A) and Mb(H64A,V68A) (Table S2), which feature only a subtle increase in steric bulk (H→Me) at position 68 compared to Mb(V68G) and Mb(H64A,V68G), respectively.
Figure 2.
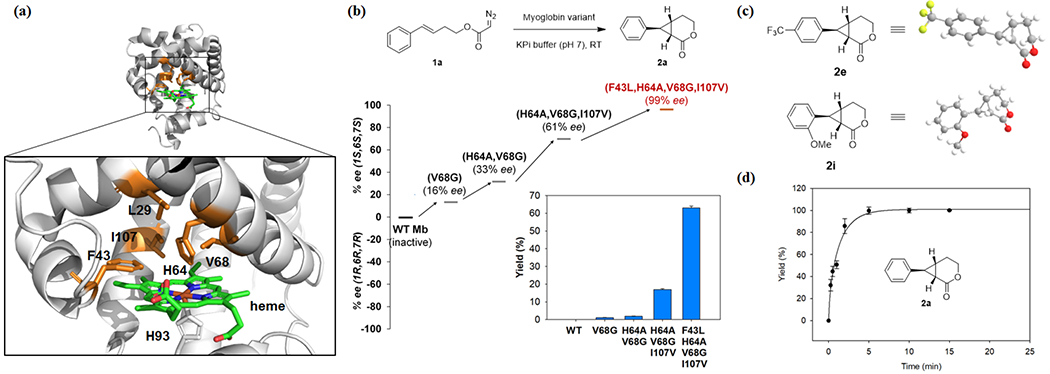
Engineering of myoglobin scaffold for cyclopropyl-δ-lactone synthesis. (a) Crystal structure of sperm whale myoglobin (pdb 1VXA) and close-up view of the heme distal pocket (box). The heme cofactor (green) and the active site residues targeted for mutagenesis (orange) are shown as stick models. (b) Evolutionary path leading to an enantioselective biocatalyst for the intramolecular cyclopropanation of 1a to give 2a. The bar graph reports the GC yields of the reactions with the different variants. See Table S3 for further details. (c) Crystal structures of compound 2e (CCDC:1998584) and 2i (CCDC:1998585). (d) Time course experiments for Mb(F43L,H64A,V68G,I107V)-catalyzed synthesis of 2a using whole cells (C41(DE3) cells, OD600 = 20) and 2.5 mM 1a in oxygen-free potassium phosphate buffer (50 mM, pH 7.0).
Based on the results above, Mb(V68G) was chosen as the starting point for the development of a biocatalyst with improved activity and enantioselectivity for this intramolecular cyclopropanation reaction via protein engineering. To this end, starting from Mb(V68G), iterative rounds of single-site site-saturation mutagenesis (NNK codon) were performed by sequentially targeting each of the active site residues Leu29, Phe43, His64, and Ile107, which surround the heme active center (Figure 2a). The resulting libraries were expressed in multi-well plates and screened as whole cells using 1a as the substrate. The most promising ‘hits’ were validated by characterizing the corresponding Mb variants in purified form prior to the next round of mutagenesis and screening. Using this approach, three beneficial mutations corresponding to H64A, I107V, and F43L (Figure 2b) were accumulated in three consecutive rounds of directed evolution, resulting in a ~60-fold improvement in yield (1→63%) and a dramatic enhancement in enantioselectivity from 16% ee to 99% ee toward formation of 2a (Table 1, Entry 9). The (1S,6S,7S)-configuration of 2a was assigned on the basis of crystallographic analysis of related products 2e and 2i (Figures 2c, Tables S8–S9) described further below.
A whole-cell biotransformation with E. coli cells expressing the Mb(F43L,H64A,V68G,I107V) biocatalyst enabled the quantitative conversion of 1a into 2a with excellent enantioselectivity (99% ee; Table 1, entry 10). A cell density (OD600) of 20 was determined to be sufficient for achieving quantitative yield of the desired product 2a, corresponding to 316 catalytic turnovers (TON) supported by the intracellular enzyme (Table 1, Entry 10; Table S4). By lowering the cell density (OD600 20 → 5), a nearly four-fold higher TON value of 1,160 was obtained while maintaining excellent enantioselectivity (99% ee) and high yields (92%) (Table 1, entry 11). Thus, in addition to offering higher enantioselectivity and involving an iron-based catalyst, the catalytic activity (TON) of this Mb-based system is 10- to 20-fold higher than that previously achieved using Rh-based organometallic catalysts (TON ~ 50-80),[3] highlighting its superior efficiency over synthetic methods previously available for attaining this transformation. Furthermore, time-course experiments showed that the Mb-catalyzed whole-cell reaction reaches ~70% product conversion in 2 min (Table 1, entry 12) and quantitative conversion of 1a to 2a in less than 10 min (Figure 2d), thus exhibiting fast reaction kinetics. Additional experiments (vide infra) demonstrated that the whole-cell biotransformation can be volumetrically scaled up to the gram scale without noticeable loss in yield and enantioselectivity, thus demonstrating the utility of this biocatalytic method for preparative scale synthesis.
To examine the substrate tolerance of Mb(F43L,H64A,V68G,I107V), we then tested its activity in the intramolecular cyclopropanation of a diverse panel of homoallylic α-diazoacetate derivatives under the optimized conditions described above and at a preparative scale (0.2 mmol) (Table 2). To our delight, both electron-withdrawing and electron-donating groups on the aromatic ring were accepted by the Mb variant, which exhibited high to excellent enantioselectivity (90-99% ee) across all of the tested substrates (1b-1i). Substrates carrying a halogen functional group (1c, 1d) useful for further derivatization were all efficiently processed to afford the corresponding bicyclic products (2b, 2d) in good yields (54% to 88%). Similar results were obtained with fluorinated substrates such as 1e, which was transformed into 2e in 55% yield and 99% ee. Encouraged by these results, we then surveyed the effects of substitutions at the para, meta and ortho positions on the aromatic ring using a methyl group as the probe substituent (Table 2, Entries 5-7). Whereas all substrates were readily cyclized by the Mb catalyst, the para-substituted methylated substrate 1f was found to be the least favorable substrate, resulting in the formation of 2f in 36% yield and 90% ee (Table 2, Entry 5). In comparison, the Mb(F43L,H64A,V68G,I107V)-catalyzed cyclization of the meta- and ortho-substituted regioisomers 1g and 1h proceeded with significantly higher efficiency, resulting in their quantitative or near-quantitative conversion to the cyclopropyl-δ-lactones 2g and 2h (95-99%) along with high(er) enantioselectivity (92-94% ee) (Table 2, Entries 6-7). The efficient conversion of substrate 1i into 2i and excellent enantioselectivity of this reaction (99% ee; Table 2, Entry 8) further highlighted the tolerance of the Mb catalyst to steric hindrance in close proximity to the olefinic group. Finally, we extended these studies to an aliphatic diazoacetate substrate containing an unactivated olefinic group such as 4-methylpent-3-en-1-yl 2-diazoacetate (1j). Albeit in moderate yield (41%), this substrate was successfully cyclized by the Mb catalyst to afford 2j in 91% ee. Notably, the Mb(F43L,H64A,V68G,I107V)-catalyzed intramolecular cyclopropanation reaction was determined to exhibit a conserved (1S,6S,7S)-stereoselectivity across this diverse panel of substrates, as evinced from crystallographic analysis of 2e and 2i (Figure 2c) and the similar chromatographic behavior of the other products in chiral GC (Figure S2). Furthermore, in all cases, the desired cyclopropyl-δ-lactone product could be readily isolated from the whole-cell reactions, resulting in up to 84% isolated yields (Table 2).
Table 2.
Substrate scope for Mb(F43L,H64A,V68G,I107V)-catalyzed intramolecular cyclopropanation of homoallylic α-diazoacetats [a]
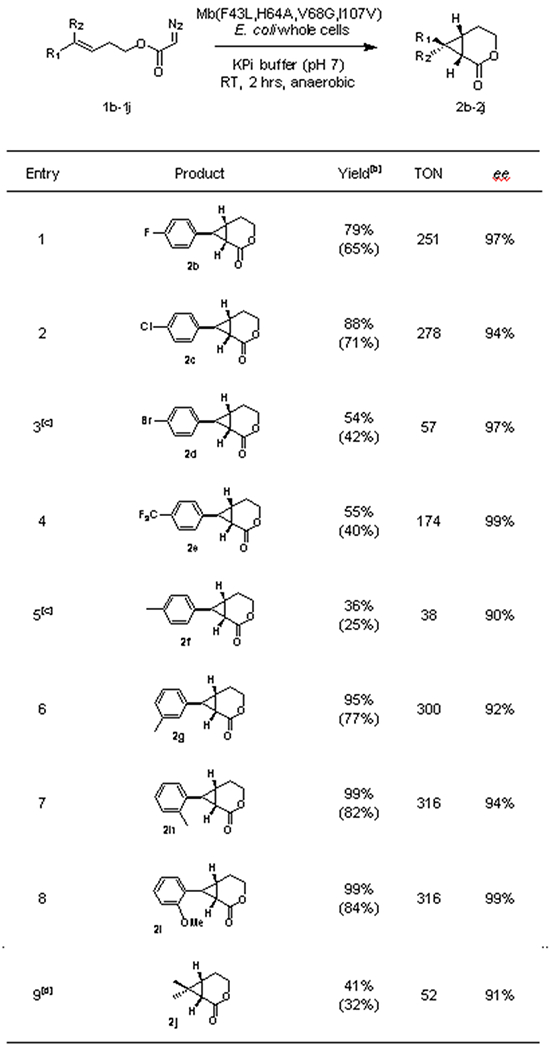 |
Reaction conditions: 2.5 mM substrate, Mb(F43L,H64A,V68G,I107V)-expressing E.coli (OD600 = 20) in KPi buffer (50 mM, pH 7), 80 mL-scale, RT, 2 hrs.
Product conversion as determined by GC. Yields of isolated products are reported in brackets. Errors are within 10%.
Using OD600 = 60.
Using 1 mM substrate, 200 mL-scale.
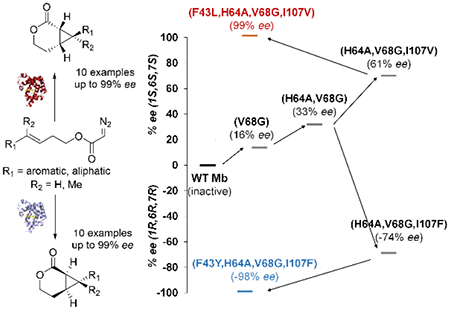
Albeit rare,[9] enantiocomplementary biocatalysts are highly valuable for the synthesis of drugs and complex molecules since mirror-image forms of bioactive molecules often display distinct pharmacological activities. Upon noting that some of the Mb variants favor the formation of the (1R,6R,7R) enantiomer 3a (Table S2), we envisioned the possibility to develop an enantiocomplementary Mb-based catalyst for the synthesis of the cyclopropane-fused δ-lactones. To this end, Mb(V68G) and Mb(H64A,V68G) were chosen as the starting points for these directed evolution campaigns in view of their higher catalytic activity compared to the aforementioned Mb variants, which exhibit only marginal cyclopropanation activity on 1a (<1% yield, Table S2). Gratifyingly, a progressive improvement of the desired (1R,6R,7R)-selectivity was obtained by subjecting both parent enzymes to iterative rounds of site-saturation mutagenesis directed to the yet unmodified active site residues (Figure 3). In both evolutionary pathways, a Ile→Phe mutation at position 107, which is located in the ‘back side’ of the heme distal pocket with respect to its solvent exposed side (Figure 2a), played a critical role in favoring stereoinduction toward formation of enantiomer 3a as evinced from the increase in (1R,6R,7R)-enantioselectivity in the case of Mb(H64L,V68G) (−21 → −89% ee) and the inversion of enantioselectivity in the Mb(H64A,V68G) background (33% ee → −74% ee; Figure 3). Further mutagenesis of these I107F-containing variants led to the identification of biocatalysts with further improved (1R,6R,7R)-enantioselectivity, namely Mb(F43Y,H64A,V68G,I107F) and Mb(F43H,H64L,V68G,I107F), which catalyze the intramolecular cyclopropanation of 1a to give 3a in 98% ee and 99% ee, respectively (Figure 3, Table S3). Notably, the improvement in (1R,6R,7R)-enantioselectivity across these variants is accompanied by an increase in catalytic activity, resulting in significantly improved yields for the reactions catalyzed by Mb(F43Y,H64A,V68G,I107F) and Mb(F43H,H64L,V68G,I107F) (43-69%) compared to the parent enzymes (1-2% yield) (Figure 3). It is also worth noting that the beneficial mutations accumulated in the (1R,6R,7R)-selective variants show a consistent trend with respect to increasing the steric bulk at position 107 (Ile107 →Phe) and introducing aromatic groups with H-bond donor/acceptor capability at position 43 (Phe43 →Tyr/His). These features are in stark contrast with the effects of the mutations beneficial toward inducing (1S,6S,7S) enantioselectivity, which reduce the steric bulk at both the 107 and 43 positions (i.e., Ile107 →Val and Phe43 →Leu; Figure 2b). These structure-selectivity trend highlight the differential demands in terms of active site configuration that are required for steering the enantioselectivity of the Mb-catalyzed intramolecular cyclopropanation reaction in opposite directions.
Figure 3.
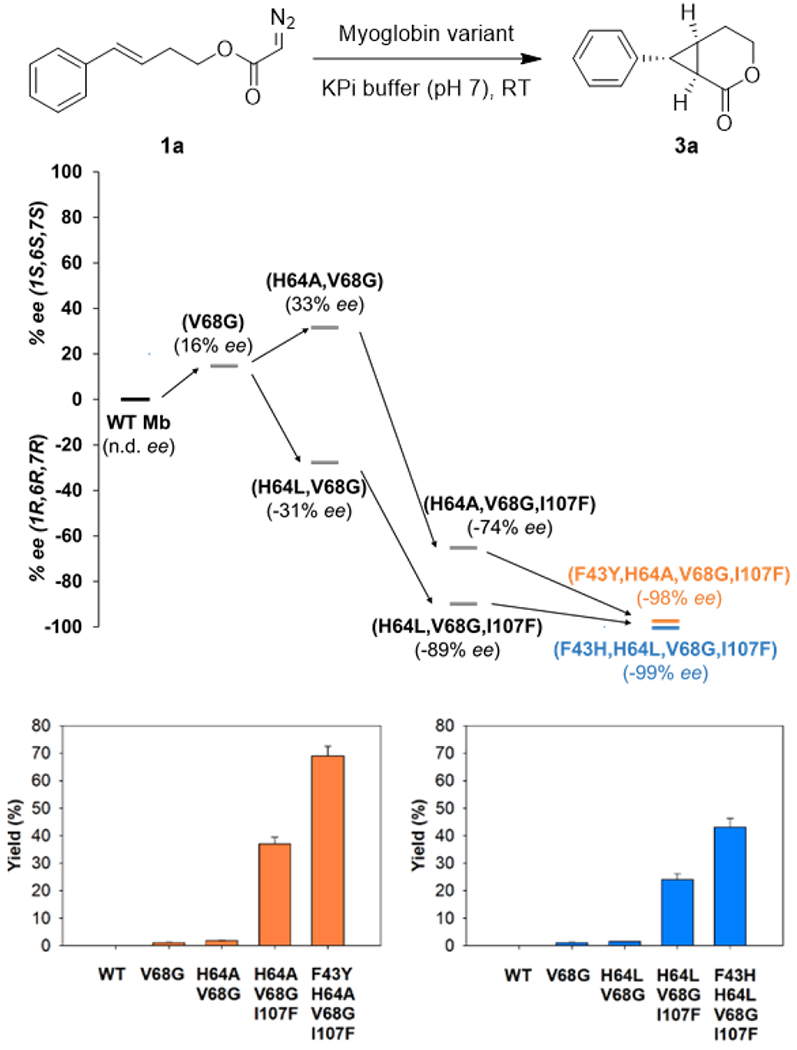
Development of enantiodivergent biocatalysts. The diagram shows the evolutionary paths leading to the (1R,6R,7R)-selective myoglobin variants for the synthesis of 3a from 1a. The bar graphs report the GC yields of the reactions with the different variants. See Table S3 for further details.
The substrate scope of Mb(F43Y,H64A,V68G,I107F) was further investigated by challenging this biocatalyst with the panel of diazoacetate substrates described in Table 2. These experiments show that Mb(F43Y,H64A,V68G,I107F) is able to efficiently process these substrates producing the desired (1R,6R,7R)-configured cyclopropane-δ-lactones in good to excellent enantiomeric excess (73 to >99% ee; Scheme 2), thus exhibiting a broad substrate tolerance along with a consistent, stereodivergent selectivity compared to Mb(F43L,H64A,V68G,I107V). The aliphatic substrate 1j was also efficiently converted by the (1R,6R,7R)-selective catalyst to give 3j albeit with reduced enantioselectivity (51% ee) compared to the other substrates. The Mb variant Mb(F43H,H64A,V68G,I107F) was also found to be a competent and enantioselective catalyst for these reactions, offering superior enantioinduction for the synthesis of 3e and 3g (90-97% ee vs. 50-71% ee) compared to Mb(F43Y,H64A,V68G,I107F).
Scheme 2.
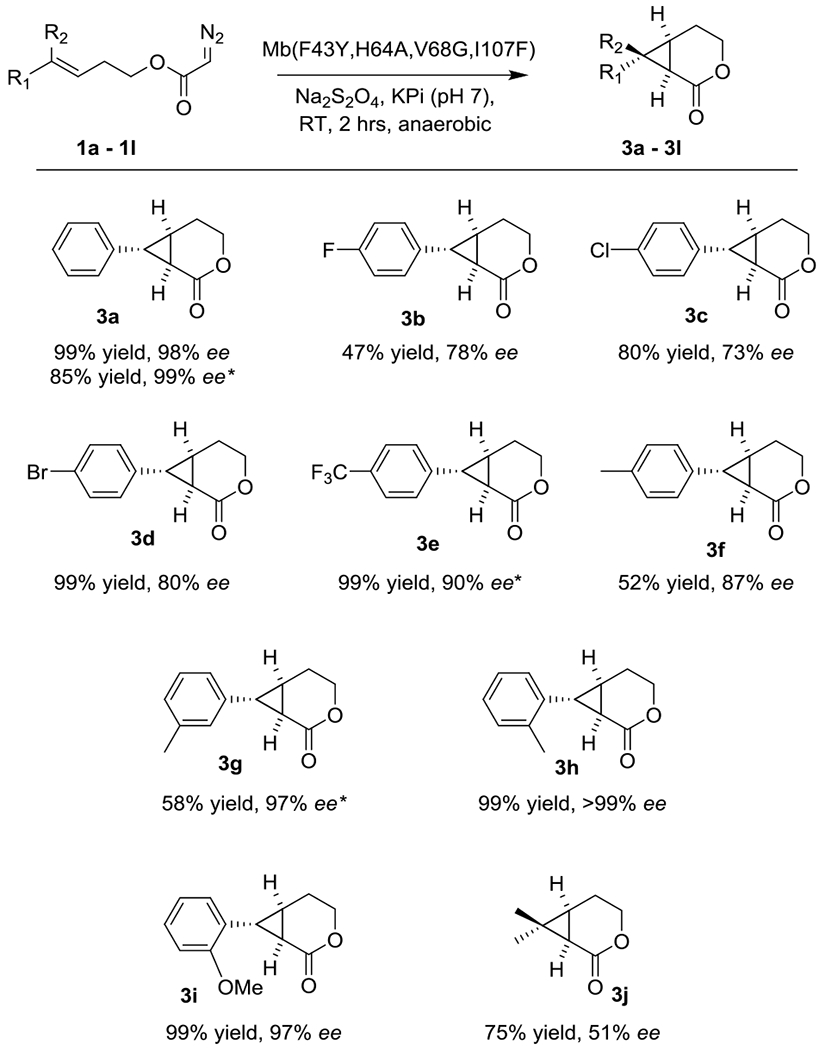
Substrate scope of the (1R,6R,7R)-selective biocatalyst Mb(F43Y,H64A,V68G,I107F). Reaction conditions: 1 mM substrate, 20 μM protein, 10 mM Na2S2O4, in KPi buffer (50 mM, pH 7), RT, anaerobic, 2 hours. Reported yields correspond to GC yields. * Using Mb(F43H,H64L,V68G,I107F).
To further assess the synthetic value of the present strategy, a large-scale biotransformation with Mb(F43L,H64A, V68G,I107V)-expressing E. coli cells was carried out in the presence of 650 mg of 1a (3 mmol). This reaction enabled the isolation of 480 mg of enantiopure 2a (99% ee) in 85% isolated yield (Scheme 3), thus demonstrating the scalability of this biocatalytic method. The enzymatically produced cyclopropane-δ-lactone was then chemically reduced with LiAlH4 to give the cis hydroxymethyl/ethyl-substituted cyclopropane 4 in 89% yield and 99% ee in a single step (Scheme 3). Furthermore, alkaline hydrolysis of 2a afforded the trisubstituted cyclopropane 5 in 94% isolated yield as a single enantiomer. These results thus demonstrate the versatility of these bicyclic enzymatic products toward gaining access to optically active trisubstituted cyclopropanes of value for medicinal chemistry and total synthesis.[1].
Scheme 3.
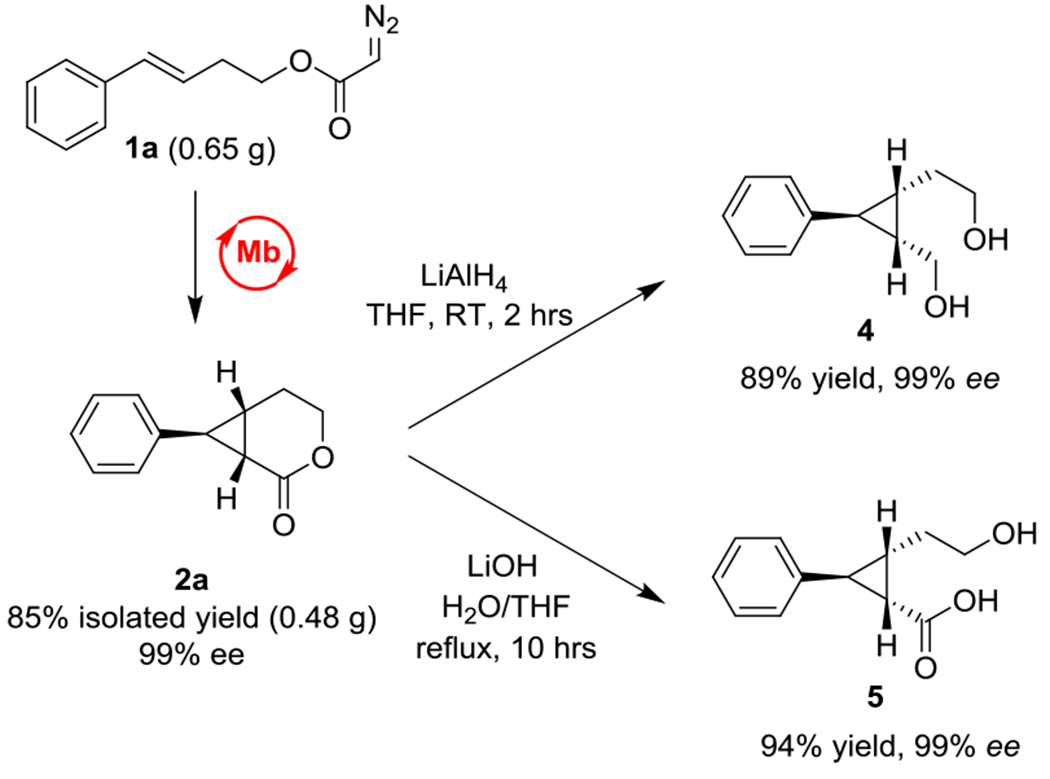
Gram-scale synthesis and chemoenzymatic diversification of cyclopropyl-δ-lactones.
In summary, we have developed an efficient, versatile, and sustainable biocatalytic platform for the enantioselective synthesis of cyclopropyl-δ-lactones, which are key motifs in bioactive molecules (Figure 1) as well as versatile intermediates for the preparation of trisubstituted cyclopropanes (Scheme 3). While neither wild-type myoglobin nor its cofactor (hemin) are able to catalyze this intramolecular cyclopropanation reaction, two biocatalysts capable of executing these reactions with high enantioselectivity as well as stereodivergent selectivity across a broad range of substrates were obtained through re-design of the active site of this hemoprotein. These biocatalytic transformations can be carried out using whole cell systems, which eliminates the need for protein purification, and could be readily performed at the gram scale, which further demonstrates their value for synthetic applications. This study expands the range of synthetically valuable, abiotic transformations achievable via biocatalysis and our findings suggest that a broader spectrum of intramolecular carbene transfer reactions than currently possible[5b, 7] may become accessible through re-engineering of hemoprotein scaffolds.
Supplementary Material
Acknowledgements
This work was supported by the U.S. National Institute of Health grant GM098628. The authors are grateful to Dr. William Brennessel for assistance with crystallographic analyses. MS and X-ray instrumentation are supported by U.S. National Science Foundation grants CHE-0946653 and CHE-1725028.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Talele TT, J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- [2].a) Bautista E, Toscano RA, Ortega A, Org. Lett 2013, 15, 3210–3213; [DOI] [PubMed] [Google Scholar]; b) Liu HB, Zhang H, Li P, Wu Y, Gao ZB, Yue JM, Org. Biomol. Chem 2012, 10, 1448–1458; [DOI] [PubMed] [Google Scholar]; c) Wu HH, Chen YP, Ying SS, Zhang P, Xu YT, Gao XM, Zhu Y, Tetrahedron Lett. 2015, 56, 5851–5854. [Google Scholar]
- [3].Doyle MP, Austin RE, Bailey AS, Dwyer MP, Dyatkin AB, Kalinin AV, Kwan MMY, Liras S, Oalmann CJ, Pieters RJ, Protopopova MN, Raab CE, Roos GHP, Zhou QL, Martin SF, J Am Chem Soc 1995, 117, 5763–5775. [Google Scholar]
- [4].a) Coelho PS, Brustad EM, Kannan A, Arnold FH, Science 2013, 339, 307–310; [DOI] [PubMed] [Google Scholar]; b) Bordeaux M, Tyagi V, Fasan R, Angew. Chem. Int. Ed 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang ZJ, Peck NE, Renata H, Arnold FH, Chem Sci 2014, 5, 598–601; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sreenilayam G, Fasan R, Chem Commun 2015, 51, 1532–1534; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tyagi V, Bonn RB, Fasan R, Chem Sci 2015, 6, 2488–2494; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Kan SBJ, Lewis RD, Chen K, Arnold FH, Science 2016, 354, 1048–1051; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Tyagi V, Fasan R, Angew. Chem. Int. Ed 2016, 55, 2512–2516; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Tyagi V, Sreenilayam G, Bajaj P, Tinoco A, Fasan R, Angew. Chem. Int. Ed 2016, 55, 13562–13566; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Weissenborn MJ, Low SA, Borlinghaus N, Kuhn M, Kummer S, Rami F, Plietker B, Hauer B, Chemcatchem 2016, 8, 1636–1640; [Google Scholar]; j) Kan SBJ, Huang X, Gumulya Y, Chen K, Arnold FH, Nature 2017, 552, 132–136; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Vargas DA, Tinoco A, Tyagi V, Fasan R, Angew. Chem. Int. Ed 2018, 57, 9911–9915; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Chen K, Huang XY, Kan SBJ, Zhang RK, Arnold FH, Science 2018, 360, 71–75; [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Vargas D, Khade R, Zhang Y, Fasan R, Angew. Chem. Int. Ed 2019, 58, 10148–10152; [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Zhang RK, Chen K, Huang X, Wohlschlager L, Renata H, Arnold FH, Nature 2019, 565, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC, Nat. Commun 2015, 6, 7789; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dydio P, Key HM, Nazarenko A, Rha JY, Seyedkazemi V, Clark DS, Hartwig JF, Science 2016, 354, 102–106; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sreenilayam G, Moore EJ, Steck V, Fasan R, Adv. Synth. Cat 2017, 359, 2076–2089; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sreenilayam G, Moore EJ, Steck V, Fasan R, ACS Catal. 2017, 7, 7629–7633; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Moore EJ, Steck V, Bajaj P, Fasan R, J. Org. Chem 2018, 83, 7480–7490; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ohora K, Meichin H, Zhao LM, Wolf MW, Nakayama A, Hasegawa J, Lehnert N, Hayashi T, J Am Chem Soc 2017, 139, 17265–17268; [DOI] [PubMed] [Google Scholar]; g) Villarino L, Splan KE, Reddem E, Alonso-Cotchico L, de Souza CG, Lledos A, Marechal JD, Thunnissen AMWH, Roelfes G, Angew. Chem. Int. Ed 2018, 57, 7785–7789; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Carminati DM, Fasan R, ACS Catal. 2019, 9, 9683–9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Tinoco A, Steck V, Tyagi V, Fasan R, J. Am. Chem. Soc 2017, 139, 5293–5296; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chandgude AL, Fasan R, Angew. Chem. Int. Ed 2018, 57, 15852–15856; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang JE, Huang XY, Zhang RJK, Arnold FH, J Am Chem Soc 2019, 141, 9798–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Chandgude AL, Ren X, Fasan R, J. Am. Chem. Soc 2019, 141, 9145–9150; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ren X, Chandgude AL, Fasan R, ACS Catal. 2020, 10, 2308–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tinoco A, Wei Y, Bacik J-P, Carminati DM, Moore EJ, Ando N, Zhang Y, Fasan R, ACS Catal. 2019, 9 1514–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Mugford PF, Wagner UG, Jiang Y, Faber K, Kazlauskas RJ, Angew. Chem. Int. Ed 2008, 47, 8782–8793; [DOI] [PubMed] [Google Scholar]; b) Wu Q, Soni P, Reetz MT, J Am Chem Soc 2013, 135, 1872–1881; [DOI] [PubMed] [Google Scholar]; c) Koszelewski D, Grischek B, Glueck SM, Kroutil W, Faber K, Chem. Eur. J 2011, 17, 378–383; [DOI] [PubMed] [Google Scholar]; d) Bajaj P, Sreenilayam G, Tyagi V, Fasan R, Angew. Chem. Int. Ed 2016, 55, 16110–16114; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) France SP, Aleku GA, Sharma M, Mangas-Sanchez J, Howard RM, Steflik J, Kumar R, Adams RW, Slabu I, Crook R, Grogan G, Wallace TW, Turner NJ, Angew. Chem. Int. Ed 2017, 56, 15589–15593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.