

Abstract
Background
Pulmonary vascular changes in postoperative pulmonary artery hypertension (PAH) are similar to those seen in idiopathic PAH. Data are sparse on direct comparative midterm outcomes for these 2 high-risk populations.
Methods
Patients with idiopathic or postoperative PAH referred to a large tertiary hospital between June 2005 and July 2019 were retrospectively evaluated.
Results
A total of 364 consecutive patients were studied, including 201 postoperative PAH patients and 163 patients with idiopathic PAH, with a median age of 18.7 (interquartile range 10.0, 31.5) and 7.3 (IQR: 2.9, 18.3) years, respectively. PAH-specific drugs were used in 77.7% of patients; 31.4% received combination therapy. Patients with idiopathic PAH had a shorter 6-mintue walk distance, lower percutaneous oxygen saturation, and higher B-type natriuretic peptide levels than those with postoperative PAH at diagnosis (all P < 0.001), During a median follow-up time of 3.4 (interquartile range: 2.1, 5.8) years, 56 patients (15.4%) died, and one underwent bilateral lung transplantation. Patients with postoperative PAH had better survival than those with idiopathic PAH, according to age (hazard ratio [HR] 0.128, 95% confidence interval [CI]: 0.07-0.22, P < 0.0001); Kaplan–Meier survival estimates at 5 years for idiopathic and postoperative PAH patients were 74.3% and 92.6%, respectively. Patients in New York Heart Association functional class III–IV had an over 4-fold increased risk of death (HR 4.85, 95% CI: 2.61-9.00, P < 0.0001). Patients < 18 years of age at idiopathic PAH diagnosis had a worse survival compared to adult patients (HR 6.90, 95% CI: 4.19-15.56, P = 0.040).
Conclusions
Postoperative-PAH patients had better midterm survival compared to patients with idiopathic PAH. Mortality was significant in both PAH groups, reinforcing the need for early diagnosis and optimal individualized management to improve outcomes.
Résumé
Contexte
Les changements vasculaires pulmonaires associés à l'hypertension artérielle pulmonaire (HAP) postopératoire sont semblables à ceux observés dans l'HAP idiopathique. On dispose de peu de données comparatives directes sur les résultats à moyen terme dans ces deux populations à risque élevé.
Méthodologie
Les patients présentant une HAP idiopathique ou postopératoire qui avaient été dirigés vers un important centre hospitalier de soins tertiaires entre juin 2005 et juillet 2019 ont fait l'objet d'une évaluation rétrospective.
Résultats
Au total, 364 patients consécutifs ont été étudiés, dont 201 cas d'HAP postopératoire et 163 cas d'HAP idiopathique, dont l'âge médian était de 18,7 (intervalle interquartile : de 10,0 à 31,5) et de 7,3 (intervalle interquartile : de 2,9 à 18,3) ans, respectivement. Des médicaments traitant précisément l'HAP avaient été utilisés chez 77,7 % des patients; 31,4 % des patients avaient reçu un traitement en association. Chez les patients présentant une HAP idiopathique, la distance parcourue au test de marche de six minutes était inférieure, la saturation percutanée en oxygène était inférieure et les taux de peptide natriurétique de type B étaient supérieurs aux valeurs affichées par les patients présentant une HAP postopératoire au diagnostic (toutes les valeurs p < 0,001). Pendant une période de suivi d'une durée médiane de 3,4 (intervalle interquartile : de 2,1 à 5,8) ans, 56 patients (15,4 %) étaient décédés, et un patient avait subi une transplantation pulmonaire bilatérale. La survie des patients présentant une HAP postopératoire était supérieure à celle des patients avec une HAP idiopathique, en fonction de l'âge (rapport des risques instantanés [RRI] 0,128, intervalle de confiance à 95 % [IC] : de 0,07 à 0,22, p < 0,0001); la survie à cinq ans des patients atteints d'HAP idiopathique et postopératoire, estimée selon la méthode de Kaplan-Meier, était de 74,3 % et de 92,6 %, respectivement. Chez les patients appartenant aux classes fonctionnelles III et IV de la New York Heart Association, le facteur multiplicatif du risque de décès était plus de quatre fois supérieur (RRI 4,85, IC à 95 % : de 2,61 à 9,00, p < 0,0001). Les patients âgés de moins de 18 ans au moment du diagnostic d'HAP idiopathique avaient un taux de survie inférieur à celui des patients adultes (RRI 6,90, IC à 95 % : de 4,19 à 15,56, p = 0,040).
Conclusions
La survie à moyen terme des patients atteints d'une HAP postopératoire était supérieure à celle des patients dont l'HAP était idiopathique. La mortalité était importante dans les deux groupes d'HAP, ce qui met en lumière la nécessité d'un diagnostic précoce et d'une prise en charge individualisée optimale afin d'améliorer les résultats.
Pulmonary arterial hypertension (PAH) is characterized by increased pulmonary arterial pressure (PAP) and pulmonary vascular resistance (PVR) that ultimately lead to right heart failure and premature death.1 Postoperative PAH refers to PAH that either persists immediately after repair of congenital heart disease (CHD) or recurs or develops months or years after repair in the absence of significant postoperative residual congenital lesions or defects.2 Idiopathic PAH, on the other hand, is a sporadic form of PAH encountered in patients without family history or known triggering factors for PAH.1
Pulmonary vascular disease prior to CHD surgery or intervention may be due to persistently elevated PVR from birth, or to excessive pressure and/or volume load to the pulmonary circulation due to a significant systemic-to-pulmonary shunt.3,4 Early changes to the pulmonary vasculature are likely to be reversible if the cardiac defect is repaired early in life (at a few months of age).5 However, the potential for morbidity and/or mortality benefit will be lost beyond a certain point—repairing intracardiac lesions at this stage may not be helpful, and may even accelerate the onset of right ventricular failure due to the loss of “decompression” function in the later stages of the disease.3, 4, 5 In this case, postoperative and idiopathic PAH may have similar clinical features, with medial hypertrophy and intimal proliferation in pulmonary arterioles having been noted in both.6,7 Despite similarities in pathophysiology and hemodynamics between the 2 groups, midterm prognosis varies from study to study, with reports of similar survival rates in postoperative and idiopathic PAH patients,8 or marginally better midterm survival for the former.9
Patients living in regions with limited economic and health care resources may be subject to late diagnosis and repair for CHD. With increased access to CHD surgery and improved perioperative care, we have experienced a surge in the number of patients with postoperative PAH. With limited resources and reduced awareness of the disease, idiopathic PAH is often diagnosed late, with a significant impact on prognosis.
We assessed the clinical, functional, and hemodynamic characteristics of a contemporary cohort with postoperative PAH patients and compared these, as well as midterm survival, to those for patients with idiopathic PAH.
Methods
Study population
Data from consecutive patients (children and adults, with or without Down syndrome) with postoperative or idiopathic PAH referred to Beijing Anzhen Hospital (one of the largest PAH centres in China with standardized treatment strategies and referrals from all over China) between June 2005 and July 2018 were studied retrospectively. All patients met the clinical definition of PAH2,10,11 with mean PAP ≥ 25 mm Hg, pulmonary artery wedge pressure ≤ 15 mm Hg, and PVR > 3 Wood units measured by right heart catheterization (RHC), and/or invasive PAP monitoring. Acute vasodilator testing was performed with inhaled iloprost during cardiac catheterization, and a positive response was defined according to current guidelines as a decrease in mean PAP of at least 10 mm Hg to < 40 mm Hg, with a stable cardiac output.12 For CHD-PAH patients, a positive response was defined as a > 20% fall in the PVR index, and PVR index/systemic vascular resistance index with respective final values of < 6 Wood units and < 0.3.13
Patients presenting with PAH deemed too ill to undergo full RHC were also included. Postoperative PAH was a subtype of CHD-PAH that was defined as PAH diagnosed at any point after CHD repair.2 Idiopathic PAH was defined as PAH without a familial history of PAH or known triggering factor. Patients were classified according to cardiac anatomy into those with a mild or moderate defect.14 Mild and moderate defects included isolated atrial septal defects, ventricular septal defects, patent ductus arteriosus, and the combination of these defects.14 Patients with univentricular hearts were not included.
The date of PAH diagnosis was defined as the date of the first confirmatory RHC and also was used as baseline in both patients with idiopathic PAH and postoperative PAH. For patients who had PAH definitively diagnosed by echocardiography prior to RHC, the date of definitive echo study was used as a baseline. Baseline clinical data of selected patients were collected from the electronic medical record system, including demographic data (age, biological sex), percutaneous oxygen saturation, New York Heart Association cardiac function classification (NYHA-FC), 6-minute walking distance, and brain natriuretic peptide. Systolic pulmonary arterial pressure and right ventricle/left ventricle basal diameter ratio estimated by echocardiography and mean PAP, right atrial pressure, cardiac index, and PVR estimated by RHC were collected. Medications, including approved PAH-specific drugs, were collected. The results of regular follow-up of all selected patients were collected from June 2005 to July 2018, focusing on the endpoints of lung transplantation and death. Causes of death were also collected.
Study procedures were approved by the Research Ethics Committee of Beijing Anzhen Hospital.
Statistical analysis
Statistical analysis was performed using SPSS version 24 (IBM Corporation, Armonk, NY). Normally distributed continuous data were summarized as mean ± standard deviation; median (interquartile range [IQR]) was reported when the distribution was not normal. Categorical variables were presented as a number (percentage). Comparisons between groups were performed using the Wilcoxon rank sum test or the χ2 test, as appropriate.
Univariate Cox proportional hazards regression analysis was used to compare all-cause mortality between diagnostic groups; a multivariate Cox proportional hazards model was also used to determine the variables associated with an increased risk of death. Composite and subgroup Kaplan–Meier curves were plotted and analyzed using a log-rank test between groups. A 2-sided P-value of 0.05 was considered to indicate statistical significance.
Results
Patient characteristics
A total of 364 consecutive patients were included, referred from at least 27 regions in China.
Postoperative PAH
Postoperative PAH was present in 201 (55.2%) patients. Median age at diagnosis was 18.7 (10.0, 31.5) years, and 68.2% were female. A total of 172 (85.6%) patients were diagnosed with PAH before operation, including 142 patients by RHC, and 30 patients by echocardiography only. Preoperative PAP was unknown in 29 patients. A ventricular septal defect was the most common defect, present in 60 (29.9%) patients, followed by combined defects (54 patients; 26.9%), atrial septal defects (48 patients; 23.9%), and patent ductus arteriosus (39 patients; 19.4%). Pre-tricuspid shunts were most frequent in adult patients (42 in 102 patients; 41.2%), and post-tricuspid shunts were most frequent in pediatric patients (65 in 99 patients; 65.7%). There were significant differences in hemodynamic characteristics between patients with post-tricuspid vs pre-tricuspid defects: systolic PAP by echo (82.9 ± 25.6 vs 73.3 ± 18.2 mm Hg, P = 0.007), mean PAP by RHC (72.4 ± 23.5 mm Hg vs 49.3 ± 16.8 mm Hg, P < 0.001), right atrial pressure (10.1 ± 3.2 mm Hg vs 8.4 ± 2.9 mm Hg, P = 0.044), and PVR index (25.5 ± 13.8 Wood units·m2 vs 14.9 ± 12.9 Wood units·m2, P = 0.007). Median age at repair was 15.1 (IQR: 7.9, 28.7) years, whereas median time from repair to diagnosis of postoperative-PAH was 1.5 (IQR: 0, 2.9) years.
A total of 142 patients underwent pre-repair cardiac catheterization examinations, with acute vasodilator testing performed simultaneously. Forty-eight patients had a positive vasodilator response before CHD surgery; their mean age and defect locations are shown in Figure 1. There is no statistical difference in age at diagnosis between groups. Among these patients, 11 patients had a positive vasodilator response after surgery (Table 1), and only 3 patients showed a positive response both pre- and postoperatively. These patients were 1 adult with a ventricular septal defect and an atrial septal defect, and 2 children with post-tricuspid CHD.
Figure 1.
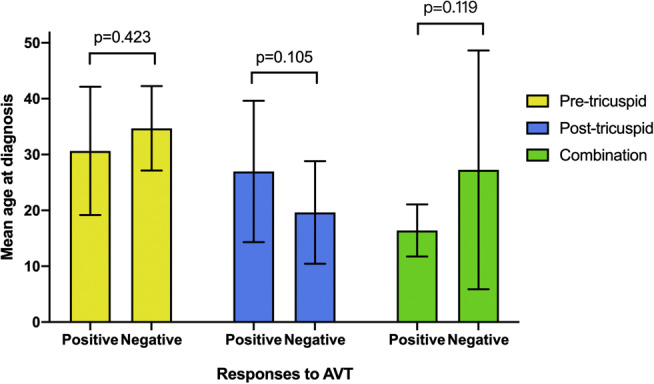
Pre-repair postoperative–pulmonary arterial hypertension patients’ responses to acute vasodilator testing (AVT) by right heart catheterization, and their mean age at diagnosis (years), as well as defect locations.
Table 1.
Demographic and clinical patient characteristics and treatment strategies
| Characteristic/treatment strategy | Postoperative PAH | Idiopathic PAH | P |
|---|---|---|---|
| Number of patients | 201 (55.2) | 163 (44.8) | N/A |
| Age at diagnosis, y | 18.7 (10.0, 31.5) | 7.3 (2.9, 18.3) | < 0.001 |
| Follow-up time, y | 4.2 ± 2.8 | 3.7 ± 2.6 | 0.069 |
| Female | 137 (68.2) | 92 (56.4) | 0.021 |
| Down syndrome | 5 (2.5) | 2 (1.2) | 0.384 |
| NYHA-FC III–IV | 28 (13.9) | 63 (38.7) | < 0.001 |
| 6MWD, m | 489.6 ± 86.9 | 423.8 ± 83.3 | < 0.001 |
| Room air SpO2, % | 97.4 ± 3.0 | 94.8 ± 5.8 | < 0.001 |
| BNP, pg/ml | 76.0 (35.8, 213.0) | 373.0 (93.0, 930.0) | 0.001 |
| SPAP, mm Hg | 71.5 ± 21.3 | 79.7 ± 20.0 | < 0.001 |
| RV/LV diameter ratio | 0.53 ± 0.18 | 0.87 ± 0.41 | < 0.001 |
| RHC parameters | 173 (86.1) | 83 (50.9) | N/A |
| mRAP, mm Hg | 9.0 ± 3.4 | 8.4 ± 3.4 | 0.243 |
| mPAP, mm Hg | 58.1 ± 22.4 | 61.7 ± 17.6 | 0.217 |
| PVRI, Wood units · m2 | 13.9 (9.2, 27.9) | 16.8 (10.0, 23.4) | 0.405 |
| Cardiac index, L/min/m2 | 3.2 ± 1.1 | 3.5 ± 1.5 | 0.119 |
| AVT | 11/173 (6.4) | 14/83 (16.9) | 0.008 |
| CCBs | 0/11 | 6/14 (42.9) | 0.030 |
| PAH Rx at last visit | 160 (79.6) | 123 (75.5) | 0.345 |
| ERA | 51 (31.9) | 47 (38.2) | 0.267 |
| PDE-5 inhibitor | 60 (37.5) | 21 (17.1) | < 0.001 |
| Prostacyclin | 9 (5.6) | 6 (4.9) | 0.781 |
| Combination therapy | 40 (25.0) | 49 (39.8) | 0.008 |
Values are n (%), median (interquartile range), or mean (±standard deviation).
AVT, acute pulmonary vasodilatation test; BNP, brain natriuretic peptide; CCB, calcium channel blockers; ERA, endothelin receptor antagonist; mPAP, mean pulmonary artery pressure; mRAP, mean right atrial pressure; N/A, not applicable; NYHA-FC, New York Heart Association functional class; PAH, pulmonary arterial hypertension; PDE, phosphodiesterase; PVRI, pulmonary vascular resistance index; RHC, right heart catheterization; RV/LV, right ventricle/left ventricle basal diameter ratio; Rx, treatment; SPAP, systolic pulmonary artery pressure; SpO2, percutaneous oxygen saturation; 6MWD, 6-minute walk distance.
Idiopathic pulmonary arterial hypertension
A total of 163 patients with idiopathic PAH were included. Of these, 123 (75.5%) were children, with a median age of 7.3 (IQR: 2.9, 18.3) years, a much younger cohort compared with that with postoperative PAH (Table 1). Female sex predominance was also observed in the idiopathic PAH cohort (92 female patients; 56.4%). Two patients had Down syndrome, and one had Noonan syndrome. Eighty-three idiopathic PAH patients completed acute vasodilator testing during RHC. Fourteen patients were positive, 10 of them were children.
Differentiation between postoperative and idiopathic PAH
Baseline characteristics
The baseline demographic and clinical characteristics of the 2 etiologic groups are shown in Table 1. Twenty percent of the patients with PAH were considered too ill to undergo RHC. A total of 9.7% of patients underwent RHC in other centers in China with incomplete data. Postoperative PAH patients were more likely to be female. Percutaneous oxygen saturation was also lower in idiopathic PAH patients (Table 1).
Patients with idiopathic PAH had worse exercise capacity, and functional capacity at the time of diagnosis. A total of 154 patients carried out a 6-minute walking distance test, and completed a shorter distance compared to patients with postoperative PAH (423.8 ± 83.3 m vs 489.6 ± 86.9 m, respectively, P < 0.001). More idiopathic PAH patients were in NYHA-FC III-IV at both diagnosis and enrollment, compared with postoperative PAH patients (P < 0.001). The idiopathic PAH cohort had higher brain natriuretic peptide levels compared to the postoperative PAH cohort (Table 1).
The idiopathic PAH patients had a higher right ventricle to left ventricle diameter ratio compared to patients with postoperative PAH (Table 1). Baseline RHC findings revealed moderate-to-severe PAH in both groups of patients. No significant differences were identified between the 2 cohorts in mean PAP, PVR index, or right atrial pressure.
Treatment
A total of 283 patients (77.7%) received treatment with pulmonary vasodilators during follow-up. Idiopathic PAH patients received more treatment with an endothelin receptor antagonist (n = 47; 38.2%) and were more often on combination therapy (n = 49; 39.8%) compared to patients with postoperative PAH. Only 4 (1.4%) patients were treated with subcutaneous/intravenous prostacyclin. Six idiopathic PAH patients with positive acute vasodilator testing were treated with calcium channel blockers.
Survival analysis
During a median follow-up of 3.4 (IQR: 2.1, 5.8) years, 56 (15.4%) patients died—13 (23.2%) with postoperative PAH, and 43 (76.8%) with idiopathic PAH, and one patient with idiopathic PAH underwent bilateral lung transplantation. The causes of death were not statistically significantly different in the 2 cohorts (all P > 0.05; Table 2). The majority of patients (17; 30.3%) died due to heart failure.
Table 2.
Characteristics of patients who died during follow-up
| Characteristic | Postoperative PAH | Idiopathic PAH | P |
|---|---|---|---|
| Died | 13 (6.5) | 43 (26.4) | < 0.001 |
| Time from diagnosis to death, y | 2.1 (0.7, 4.4) | 2.6 (0.2, 4.1) | 0.687 |
| Median age at death, y | 21.7 (9.7, 32,2) | 9.4 (6.0, 12.9) | 0.001 |
| Children | 5 (38.5) | 39 (90.7) | < 0.001 |
| Cause of death | |||
| Heart failure | 3 (23.1) | 14 (32.6) | 0.515 |
| Sudden death∗ | 3 (23.1) | 9 (20.9) | 0.869 |
| Hemoptysis | 0 | 4 (9.3) | 0.072 |
| PHC | 3 (23.1) | 5 (11.6) | 0.301 |
| Cerebral embolism | 0 | 1 (2.3) | 0.579 |
| Respiratory distress | 1 (7.7) | 0 | 0.066 |
| Non-disease-related | 0 | 4 (9.3) | 0.254 |
| Unknown | 3 (23.1) | 6 (14.0) | 0.433 |
| On PAH therapies at death | 7 (53.8) | 31 (72.1) | 0.217 |
Values are n (%) or median (interquartile range).
PAH, pulmonary arterial hypertension; PHC: pulmonary hypertensive crisis.
“Sudden death” was defined as the sudden, natural unexpected death of unknown or cardiac cause outside the hospital—last seen alive and functioning normally 24 hours before being found.38
Freedom from death or transplantation for the whole study population at 1-, 3-, and 5-year follow-up was 94.3%, 87.9%, and 82.0%, respectively. Patients with postoperative PAH had better survival compared to those with idiopathic PAH (hazard ratio [HR] 0.320, 95% confidence interval [CI]: 0.17-0.48, P < 0.0001; Fig. 2A); survival at 1, 3, and 5 years was 96.9%, 95.4%, and 92.6%, respectively, for patients with postoperative PAH, and 91.4%, 81.4%, and 74.3%, respectively, for patients with idiopathic PAH. To avoid the unavoidable time bias, survival analysis based on patient age was also conducted (Fig. 2B). There was no difference in survival based on the location of the defect in patients with postoperative PAH (P = 0.251).
Figure 2.

Kaplan–Meier curve in each subgroup of idiopathic pulmonary arterial hypertension (IPAH) and postoperative-PAH patients. Kaplan–Meier curve in patients with idiopathic PAH and postoperative PAH according to (A) follow-up time and (B) age, in PAH patients with New York Heart Association functional class in (C) the 2 cohorts (survival rate IPAH I-II vs postoperative PAH, P < 0.0001), and (D) age-associated mortality in the IPAH cohort.
The characteristics of patients with postoperative vs idiopathic PAH who died were further compared (Table 2). NYHA-FC III-IV (HR 6.16, 95% CI: 2.03-18.66, P = 0.001) was the only independent predictor of mortality in patients with postoperative PAH (Table 3). In the overall population, patients with NYHA-FC III–IV had more than a 4-fold increased risk of death compared with those with NYHA-FC I–II (HR 4.85, 95% CI: 2.61-9.00, P < 0.0001). Survival of the 2 cohorts affected by NYHA-FC is further illustrated in Figure 2C.
Table 3.
Univariate and multivariate Cox proportional hazards model estimates of risk factors for mortality
| Variable | Univariate analysis |
Multivariate analysis |
||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
| Postoperative PAH | ||||||
| NYHA-FC III-IV | 6.62 | 2.20-19.73 | 0.001 | 6.16 | 2.03-18.66 | 0.001 |
| Room air SpO2 ≥ 95% | 0.31 | 0.07-1.40 | 0.126 | 0.44 | 0.09-2.08 | 0.297 |
| Idiopathic PAH | ||||||
| Age < 18 y | 3.84 | 1.36-10.82 | 0.011 | 6.90 | 4.19-15.56 | 0.040 |
| Female | 0.51 | 0.28-0.94 | 0.03 | |||
| RAP < 14 mm Hg | 0.31 | 0.09-1.11 | 0.07 | |||
Univariate analysis P values < 0.2 are listed. P values < 0.05 are in bold.
CI, confidence interval; HR, hazard ratio; NYHA-FC, New York Heart Association functional class; PAH, pulmonary arterial hypertension; RAP, right atrial pressure; SpO2, percutaneous oxygen saturation.
Among patients who were positive on acute vasodilator testing, only one child with idiopathic PAH died, after 1.84 years of follow-up, due to heart failure.
Influence of age
No difference in survival between adult and pediatric patients was found in the postoperative-PAH groups (P = 0.071), whereas being <18 years old (HR 6.90, 95% CI: 4.19-15.56, P = 0.040) was the only independent predictor of outcome in the idiopathic PAH cohort, and there was a statistically significant difference in survival (Fig. 2D; Table 3). The median age of death was 21.7 (9.6, 32.2) years in the postoperative-PAH group, and 9.4 (6.0, 12.9) years in idiopathic PAH patients (P = 0.001). Eighteen patients, including 17 pediatric patients (94.4%), died within 6 months from diagnosis (12 in the idiopathic PAH group, 6 in the postoperative-PAH group), 13 of whom were aged <10 years at PAH onset. Time from diagnosis to death was 2.6 (0.2, 4.1) years in idiopathic-PAH patients; it was 2.1 (0.7, 4.4) years in postoperative-PAH patients (P = 0.687; Table 2).
Influence of biological sex
Although biological sex was not independently associated with mortality in our study, there was a significant difference between the 2 etiologic groups in the survival rates of male patients. Male idiopathic-PAH patients had a higher risk of death (HR 7.20, 95% CI: 3.29-15.77, P < 0.0001). In fact, male patients had higher mortality compared to female patients in the idiopathic-PAH cohort (HR 2.23, 95% CI: 1.19-4.19, P = 0.012), whereas no difference in survival was observed between biological sexes in the postoperative-PAH cohort. However, in the entire cohort, there was no significant difference in survival between male and female patients (P = 0.10).
Discussion
We report considerable mortality in a contemporary cohort of children and adults with postoperative or idiopathic PAH treated at a single tertiary centre, despite the majority of patients receiving PAH therapies. Mortality was worse in the group with idiopathic PAH compared with those with postoperative PAH. Mortality was related to baseline functional class in both groups, and a younger age at diagnosis in patients with idiopathic PAH.
With major advances in the care of children and adults with congenital and acquired heart disease globally, the detection, treatment, and successful outcomes of these patients continue to improve. The burden of CHD in China is substantial, as is the challenge to optimize clinical care and outcomes. According to data from the National Bureau of Statistics of China,15 the birth rate in 2017 was 1.24%, whereas the incidence of fetal CHD was 0.74% per year, similar to levels reported in national studies in Western countries.15,16 The total surgical and interventional volume for repair of CHD is approximately 100,000 cases per year. With a population base of more than 1 billion, the number of tertiary centres, and physicians with the expertise to achieve a timely diagnosis and repair of CHD, remains relatively low. Limited access to advanced CHD care in less-developed regions is likely to contribute to CHD-related complications, including PAH, either through late repair (postoperative PAH) or no repair of a cardiac defect, which can allow the development of Eisenmenger syndrome.
The widespread application of PAH-specific therapies is a key factor in the overall improvement of patient prognosis in recent years. In our study, 20%-25% of patients did not receive targeted treatment. Monotherapy remained the mainstay for the patients. This may have contributed to the relatively lower survival rates. The reasons behind this situation are affordability, compliance, and logistics. PAH-targeted drugs were not covered by health insurance, which rendered this therapy unaffordable to the general population. Secondly, some patients with mild clinical symptoms take medication irregularly or ceased drug use upon experiencing mild side effects. Lastly, due to the limited number of physicians, many patients face challenges in scheduling follow-up visits and long-term management. However, as an increasing number of targeted drug therapies are being covered by national health insurance since 2020, new guidelines have been released, and PAH therapy has entered a new era in China.
Previous reports have suggested that patients with postoperative PAH have a similar8,17 or even worse9 prognosis compared to patients with idiopathic PAH, based on smaller cohort studies.18,19 In the current study, our 2 cohorts shared significant similarities in clinical features, especially invasive hemodynamics, but their midterm prognosis differed significantly. Patients with postoperative PAH had better cardiac performance compared to patients with idiopathic PAH,20 with longer 6-minute walking test distances and a lower proportion of deaths caused by heart failure. Although the RV faced similar afterload in the 2 cohorts, RV adaptation to the chronic pressure overload appears to be superior in the postoperative-PAH group.21, 22, 23 Indeed, the ratio of RV to left ventricle diameter was significantly greater in the patients with idiopathic PAH (Table 1), suggesting a greater tendency for heterometric adaptation, that is, RV dilatation and dysfunction, compared with patients with postoperative PAH. The difference in cardiac adaptation between these 2 PAH groups is also supported by the significantly greater brain natriuretic peptide concentration found in idiopathic-PAH patients,9 and may be related to RV conditioning from early childhood in CHD patients.19,22, 23, 24 RV adaptation is a major determinant of survival in PAH, and optimizing RV function is a major target for PAH therapies.25, 26, 27
Our data confirmed that pediatric patients with idiopathic PAH have a significantly greater mortality rate compared to adults, as previously reported in the Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Disease Management (REVEAL) in a smaller population.28 Indeed, many pediatric patients with idiopathic PAH do not survive to adulthood, and aggressive treatment is warranted. The survival of children with postoperative PAH was better than that for children with idiopathic PAH in our study, which may also reflect better RV adaptation, or differences in the pathogenesis and rate of progression of the disease compared to that in idiopathic PAH. Previous studies have shown that genetic factors play an important role in the pathogenesis and progression of idiopathic PAH, suggesting that we should conduct genetic testing in idiopathic PAH, especially in children.29 Further investment is required to enhance our knowledge of the various, often very aggressive, forms of pediatric PAH.30
Our findings have significant clinical implications. Postoperative PAH had a very significant impact on the outcomes of our congenital heart population, supporting the need for caution when deciding on the operability of patients with CHD, especially in patients with abnormal preoperative hemodynamics. Current pulmonary hypertension guidelines support this cautious approach, proposing a cutoff for PVR around 3-5 Wood units for defining operability, even though these criteria are the result of expert consensus or small sample studies rather than being evidence based.31, 32, 33 Moreover, the fairly poor correlation between invasive hemodynamics and histology from lung biopsies in previous studies requires clinicians to use their clinical judgment to predict the outcome of CHD repair, using a constellation of information.34
Once postoperative or idiopathic PAH is diagnosed, an aggressive therapeutic approach is warranted.35 PAH therapies have been shown to alter prognosis in the adult and pediatric populations,35,36; upfront or early sequential therapy also has relatively recently entered routine clinical practice, especially in patients with adverse hemodynamics or risk factors for an adverse prognosis.12,29,37 Our study population was treated with PAH-targeted therapies in similar proportions to those in Western populations. For instance, the REHAP registry (Registro Español de Hipertensión Arterial Pulmonar [Spanish Registry of Pulmonary Arterial Hypertension]) study showed that 74.6% of patients with PAH received PAH-targeted therapies, which is similar to the percentage in our study population.8 A major gap remains between PAH patients treated in China vs Western countries, driven in part by financial constraints and inadequate health insurance.
Limitations
This was a single-centre, retrospective, observational study, which included both children and adults, who are known to have different rates of PAH progression. This study bears the burden of all the limitations of a retrospective study. We lacked serial hemodynamic data in the postoperative-PAH group, as well as more comprehensive serial assessment of right ventricular function by magnetic resonance imaging, which is currently the reference method. There may have been genetic influences on outcome—not all patients were examined in regard to genetic predisposition. There were only 56 deaths total; the validity of the Cox multivariate regression model was insufficient to a certain extent. Our patients were selected by referral to our centre, thus, a selection bias toward patients with more severe PAH is possible. However, such a bias would affect both groups, and therefore our survival comparison remains valid.
Conclusions
In contrast to prior reports, we have identified better midterm survival for postoperative-PAH patients compared to patients with idiopathic PAH. However, despite significant advances in management, mortality remains significant in both idiopathic-PAH and postoperative-PAH patients, reinforcing the need for early diagnosis and optimal use of PAH therapies.
Acknowledgements
We are grateful to the patients and their families, whose generosity and cooperation have made this study possible.
Funding Sources
This work was supported by funding from the National Natural Science Foundation of China (82070243).
Disclosures
The authors have no conflicts of interest to disclose.
Footnotes
Ethics Statement: Study procedures were approved by the Research Ethics Committee of Beijing Anzhen Hospital.
See page 878 for disclosure information.
References
- 1.Galie N., Hoeper M., Humbert M., et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) Eur Heart J. 2009;30:2493–2537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- 2.Galie N., Humbert M., Vachiery J.L., et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Respir J. 2015;46:903–975. doi: 10.1183/13993003.01032-2015. [DOI] [PubMed] [Google Scholar]
- 3.D'Alto M., Mahadevan V.S. Pulmonary arterial hypertension associated with congenital heart disease. Eur Respir Rev. 2012;21:328–337. doi: 10.1183/09059180.00004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Feen D.E., Bartelds B., de Boer R.A., Berger R.M.F. Assessment of reversibility in pulmonary arterial hypertension and congenital heart disease. Heart. 2019;105:276–282. doi: 10.1136/heartjnl-2018-314025. [DOI] [PubMed] [Google Scholar]
- 5.Haworth S.G. Pulmonary hypertension in the young. Heart. 2002;88:658–664. doi: 10.1136/heart.88.6.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaki S., Mohri H., Haneda K., Endo M., Akimoto H. Indications for surgery based on lung biopsy in cases of ventricular septal defect and/or patent ductus arteriosus with severe pulmonary hypertension. Chest. 1989;96:31–39. doi: 10.1378/chest.96.1.31. [DOI] [PubMed] [Google Scholar]
- 7.Rich J.D. Pulmonary hypertension and the right ventricle-thinking outside the box (Third International Right Heart Failure Summit, part 1) Pulm Circ. 2014;4:696–704. doi: 10.1086/678543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alonso-Gonzalez R., Lopez-Guarch C.J., Subirana-Domenech M.T., et al. Pulmonary hypertension and congenital heart disease: an insight from the REHAP National Registry. Int J Cardiol. 2015;184:717–723. doi: 10.1016/j.ijcard.2015.02.031. [DOI] [PubMed] [Google Scholar]
- 9.Barst R.J., Ivy D.D., Foreman A.J., McGoon M.D., Rosenzweig E.B. Four- and seven-year outcomes of patients with congenital heart disease-associated pulmonary arterial hypertension (from the REVEAL Registry) Am J Cardiol. 2014;113:147–155. doi: 10.1016/j.amjcard.2013.09.032. [DOI] [PubMed] [Google Scholar]
- 10.Subias P.E. SEC Working Group for ESC/ERS 2015 Guidelines for Diagnosis and Treatment of Pulmonary Hypertension; Expert Reviewers for ESC/ERS 2015 Guidelines for Diagnosis and Treatment of Pulmonary Hypertension. SEC Guidelines Committee. Rev Esp Cardiol (Engl Ed) 2016;69:102–108. doi: 10.1016/j.rec.2015.11.030. [DOI] [PubMed] [Google Scholar]
- 11.Abman S.H., Hansmann G., Archer S.L., et al. Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation. 2015;132:2037–2099. doi: 10.1161/CIR.0000000000000329. [DOI] [PubMed] [Google Scholar]
- 12.Sitbon O., Humbert M., Jais X., et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–3111. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 13.Georg H., Christian A., Hashim A.K., et al. Executive summary. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart. 2016;102:ii86–ii100. doi: 10.1136/heartjnl-2015-309132. [DOI] [PubMed] [Google Scholar]
- 14.Stout K.K., Daniels C.J., Aboulhosn J.A., et al. 2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e637–e697. doi: 10.1161/CIR.0000000000000602. [DOI] [PubMed] [Google Scholar]
- 15.China Statistics Press; 2017. National Bureau of Statistics of China. New Statistical Yearbook.http://www.stats.gov.cn/tjsj/ndsj/2017/indexeh.htm Available at: [Google Scholar]
- 16.Gatzoulis M.A., Beghetti M., Landzberg M.J., Galie N. Pulmonary arterial hypertension associated with congenital heart disease: recent advances and future directions. Int J Cardiol. 2014;177:340–347. doi: 10.1016/j.ijcard.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 17.Zijlstra W.M.H., Douwes J.M., Rosenzweig E.B., et al. Survival differences in pediatric pulmonary arterial hypertension: clues to a better understanding of outcome and optimal treatment strategies. J Am Coll Cardiol. 2014;63:2159–2169. doi: 10.1016/j.jacc.2014.02.575. [DOI] [PubMed] [Google Scholar]
- 18.Zhang D., Zhang C., Zhao W.G., Gu H. Follow up study on patients with pulmonary arterial hypertension after CHD repair. Chinese J Thorac Cardiovasc Surg. 2017;33:21–25. [Google Scholar]
- 19.Li Q., Dimopoulos K., Zhang C., et al. Acute effect of inhaled iloprost in children with pulmonary arterial hypertension associated with simple congenital heart defects. Pediatr Cardiol. 2018;39:757–762. doi: 10.1007/s00246-018-1818-7. [DOI] [PubMed] [Google Scholar]
- 20.Righini F.M., Apostolo A., Heck P.B., et al. Exercise physiology in pulmonary hypertension patients with and without congenital heart disease. Eur J Prev Cardiol. 2019;26:86–93. doi: 10.1177/2047487318809479. [DOI] [PubMed] [Google Scholar]
- 21.Hopkins W.E. The remarkable right ventricle of patients with Eisenmenger syndrome. Coron Artery Dis. 2005;16:19–25. doi: 10.1097/00019501-200502000-00004. [DOI] [PubMed] [Google Scholar]
- 22.William E.H., Alan D.W. Severe pulmonary hypertension without right ventricular failure: the unique hearts of patients with Eisenmenger syndrome. Am J Cardiol. 2002;89:34–38. doi: 10.1016/s0002-9149(01)02159-2. [DOI] [PubMed] [Google Scholar]
- 23.Chin K.M., Kim N.H., Rubin L.J. The right ventricle in pulmonary hypertension. Coron Artery Dis. 2005;16:13–18. doi: 10.1097/00019501-200502000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Dias C.A., Assad R.S., Caneo L.F., et al. Reversible pulmonary trunk banding. II. An experimental model for rapid pulmonary ventricular hypertrophy. J Thorac Cardiovasc Surg. 2002;124:999–1006. doi: 10.1067/mtc.2002.124234. [DOI] [PubMed] [Google Scholar]
- 25.Beghetti M., Tissot C. Pulmonary hypertension in congenital shunts. Rev Esp Cardiol. 2010;63:1179–1193. doi: 10.1016/s1885-5857(10)70232-2. [DOI] [PubMed] [Google Scholar]
- 26.D'Alonzo G.E., Barst R.J., Ayres S.M., et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 27.Sandoval J., Bauerle O., Palomar A., et al. Survival in primary pulmonary hypertension. Validation of a prognostic equation. Circulation. 1994;89:1733–1744. doi: 10.1161/01.cir.89.4.1733. [DOI] [PubMed] [Google Scholar]
- 28.Barst R.J., McGoon M.D., Elliott C.G., et al. Survival in childhood pulmonary arterial hypertension: insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation. 2012;125:113–122. doi: 10.1161/CIRCULATIONAHA.111.026591. [DOI] [PubMed] [Google Scholar]
- 29.Chida A., Shintani M., Yagi H., et al. Outcomes of childhood pulmonary arterial hypertension in BMPR2 and ALK1 mutation carriers. Am J Cardiol. 2012;110:586–593. doi: 10.1016/j.amjcard.2012.04.035. [DOI] [PubMed] [Google Scholar]
- 30.Favoccia C., Kempny A., Yorke J., et al. EmPHasis-10 score for the assessment of quality of life in various types of pulmonary hypertension and its relation to outcome. Eur J Prev Cardiol. 2018;26:1338–1340. doi: 10.1177/2047487318819161. [DOI] [PubMed] [Google Scholar]
- 31.Baumgartner H., De Backer J., Babu-Narayan S.V., et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur Heart J. 2021;42:563–645. doi: 10.1093/eurheartj/ehaa554. [DOI] [PubMed] [Google Scholar]
- 32.Attie F., Rosas M., Granados N., et al. Surgical treatment for secundum atrial septal defects in patients > 40 years old. A randomized clinical trial. J Am Coll Cardiol. 2001;38:2035–2042. doi: 10.1016/s0735-1097(01)01635-7. [DOI] [PubMed] [Google Scholar]
- 33.Oster M., Bhatt A.B., Zaragoza-Macias E., Dendukuri N., Marelli A. Interventional therapy versus medical therapy for secundum atrial septal defect: a systematic review (part 2) for the 2018 AHA/ACC Guideline for the management of adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e814–e830. doi: 10.1161/CIR.0000000000000605. [DOI] [PubMed] [Google Scholar]
- 34.Ramjug S., Hussain N., Hurdman J., et al. Pulmonary arterial hypertension associated with congenital heart disease: comparison of clinical and anatomic-pathophysiologic classification. J Heart Lung Transplant. 2016;35:610–618. doi: 10.1016/j.healun.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Li Q., Dimopoulos K., Liu T., et al. Peripartum outcomes in a large population of women with pulmonary arterial hypertension associated with congenital heart disease. Eur J Prev Cardiol. 2019;26:1067–1076. doi: 10.1177/2047487318821246. [DOI] [PubMed] [Google Scholar]
- 36.Engelfriet P.M., Duffels M.G., Moller T., et al. Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease. Heart. 2007;93:682–687. doi: 10.1136/hrt.2006.098848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Loon R.L., Roofthooft M.T., Delhaas T., et al. Outcome of pediatric patients with pulmonary arterial hypertension in the era of new medical therapies. Am J Cardiol. 2010;106:117–124. doi: 10.1016/j.amjcard.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 38.Winkel B.G., Holst A.G., Theilade J., et al. Nationwide study of sudden cardiac death in persons aged 1-35 years. Eur Heart J. 2011;32:983–990. doi: 10.1093/eurheartj/ehq428. [DOI] [PubMed] [Google Scholar]