

Abstract
Protein analysis of small numbers of human cells and single cells is primarily achieved by targeted proteomics with antibody-based immunoassays, whereas they have inherent limitations (e.g., low multiplex and unavailability of antibodies for new proteins). Mass spectrometry (MS)-based targeted proteomics has emerged as an alternative in terms of being antibody-free, high multiplex, high specificity, and quantitation accuracy. Recent advances in MS instrumentation makes MS-based targeted single-cell proteomics possible for multiplexed quantification of highly abundant proteins in single cells. However, there is a technical issue for effective processing of single cells with minimal sample loss for MS analysis. To address this issue we have recently developed a simple protein carrier-assisted one-pot sample preparation coupled with liquid chromatography (LC) – selected reaction monitoring (SRM) termed cLC-SRM for targeted proteomics analysis of small numbers of human cells including single cells. This method capitalizes on using the combined excessive exogenous protein as a carrier and low-volume one-pot processing to greatly reduce surface adsorption losses and high-specificity LC-SRM to effectively address the increased dynamic concentration range resulted from the addition of exogeneous carrier protein. Herein we have provided a detailed protocol for cLC-SRM analysis of small numbers of human cells including cell sorting, cell lysis and digestion, LC-SRM analysis, and data analysis. cLC-SRM was demonstrated to enable multiplexed accurate quantification of most moderately abundant proteins in small numbers of cells (e.g., 10–100 cells) and highly abundant proteins in single cells. Most importantly, this method can be easily implemented in any MS and proteomics laboratories at no additional cost for instrument or reagents. Further improvements in detection sensitivity and sample throughput are needed towards multiplexed targeted single-cell proteomics analysis. We anticipate that eventually it can be broadly applied to biomedical research and systems biology with the potential of facilitating precision medicine.
Keywords: Protein Carrier, Carrier-assisted One-pot Sample Preparation, cLC-SRM, Small Numbers of Cells, Single Cells, Mass-limited Sample, PCR Tube, Targeted Single-cell Proteomics
Summary
We recently developed a protein carrier-assisted one-pot sample preparation coupled to Liquid Chromatography (LC) – Selected Reaction Monitoring (SRM) termed cLC-SRM for multiplexed targeted proteomics analysis of small numbers of cells including single cells. This method capitalizes on using excessive exogenous protein as a carrier and high-specificity LC-SRM for targeted quantification.
Introduction
Recent technological advances in genomics (transcriptomics) allow for precise measurements of whole genome (transcriptome) in single cells1–3. However, single-cell proteomics technologies are lagging far behind but as equally important as genomics (transcriptomics) technologies4–8. Furthermore, protein abundance cannot necessarily be inferred from mRNA abundance9, and proteome is more complex and dynamic than transcriptome10. A large number of mixed populations of cells (i.e., bulk cells) are generally used to generate comprehensive proteome data11–13. However, such bulk measurements average out stochastic variations of individual cells, thus obscure important cell-to-cell variability (i.e., cell heterogeneity)4,14. Limitations of such bulk measurements become even more severe when the cells of interest only account for a small portion of the total populations of cells (e.g., cancer stem cells within tumors at an early-stage cancer). Therefore, there is a huge knowledge gap between single-cell proteomics and genomics (transcriptomics).
Antibody-based immunoassays (e.g., flow or mass cytometry) are predominantly used for targeted proteomics analysis of single cells6,7,15–18, whereas they share common shortcomings with other antibody-based methods (e.g., low multiplex and unavailability of antibodies for new proteins). Mass spectrometry (MS)-based targeted proteomics has emerged as an alternative for accurate protein quantification in terms of being antibody-free, high multiplex (≥200 proteins in a single analysis19), high quantitation accuracy (absolute amounts or concentrations), high specificity and reproducibility (≤10% CV)20–23. However, MS-based single-cell proteomics is still at the early infancy stage. Recent significant progress in sample preparation has made MS-based single-cell proteomics possible for quantitative analysis of highly abundant proteins from single human cells. For example, the most advanced MS platform coupled with ultralow-flow RPLC flow rates can only allow label-free MS detection and quantification of ~670–870 proteins out of the total ≥12,000 proteins in single HeLa cells24,25.
Currently, there are five MS-based single-cell proteomics approaches available, in which three are for global proteomics (nanoPOTS: nanowell-based Preparation in One pot for Trace Samples26; iPAD-1: integrated proteome analysis device for single-cell analysis27; SCoPE-MS: single cell proteomics by mass spectrometry28) and the other two are for targeted proteomics (cLC-SRM: carrier-assisted liquid chromatography (LC) – selected reaction monitoring (SRM)29; cPRISM-SRM: carrier-assisted high pressure, high-resolution separations with intelligent selection and multiplexing coupled to SRM30). However, all these approaches have inherent technical drawbacks. nanoPOTS and iPAD-1, which downscale sample processing volume to 2–200 nL, are not easily adoptable for broad benchtop applications26,27. For SCoPE-MS, a TMT (tandem mass tag) carrier is added after single-cell processing so it cannot effectively prevent surface adsorption losses during sample processing28, resulting in low reproducibility with a correlation coefficient of only ~0.2–0.5 between replicates31. Furthermore, due to the inability to fractionate ultrasmall single-cell samples TMT-based single-cell analysis lacks quantitation accuracy from ratio compression or distortion caused by coeluting interferences32. For cLC-SRM and cPRISM-SRM, using exogenous proteins as a carrier is more suitable for targeted proteomics because peptides from excessive exogenous proteins are frequently sequenced by MS/MS, which greatly reduces the chance for sequencing low abundant endogenous peptides29,30. Unlike global single-cell proteomics with relative quantification, the two targeted proteomics approaches can provide accurate or absolute protein analysis of single cells. Most importantly, they can be easily implemented in any MS and proteomics laboratories at no additional cost for instrument or reagents. In contrast to cPRISM-SRM that requires using high-resolution RPLC for PRISM fractionation, cLC-SRM has a significant advantage in multiplexing (100s of proteins in a single analysis) but with slightly lower detection sensitivity29. Therefore, cLC-SRM is more readily to access and should have more broad utilities for accurate multiplexed protein analysis of small numbers of cells as well as mass-limited precious clinical specimens.
Herein we describe a detailed protocol to perform cLC-SRM for convenient targeted proteomics analysis of small numbers of human cells including single cells. The protocol consists of the following major steps: cell sorting by FACS (fluorescence activated cell sorting), cell lysis and digestion processed in low-volume single PCR tube, LC-SRM data collection, and SRM data analysis using publicly available Skyline software (Figure 1). Its broad utility was demonstrated by absolute targeted quantification of EGFR/MAPK pathway proteins in 1–100 MCF7 or MCF10A cells along with our previously well-established SRM assays and determined pathway protein copies per cell at a wide dynamic range of concentrations29. We anticipate that with the detailed protocol most proteomics researchers can readily implement cLC-SRM in their laboratories for accurate protein analysis of ultrasmall samples (e.g., rare tumor cells) to meet their project needs.
Figure 1: Overview of all the steps in cLC-SRM (carried-assisted one-pot sample preparation coupled with liquid chromatography – selected reaction monitoring).
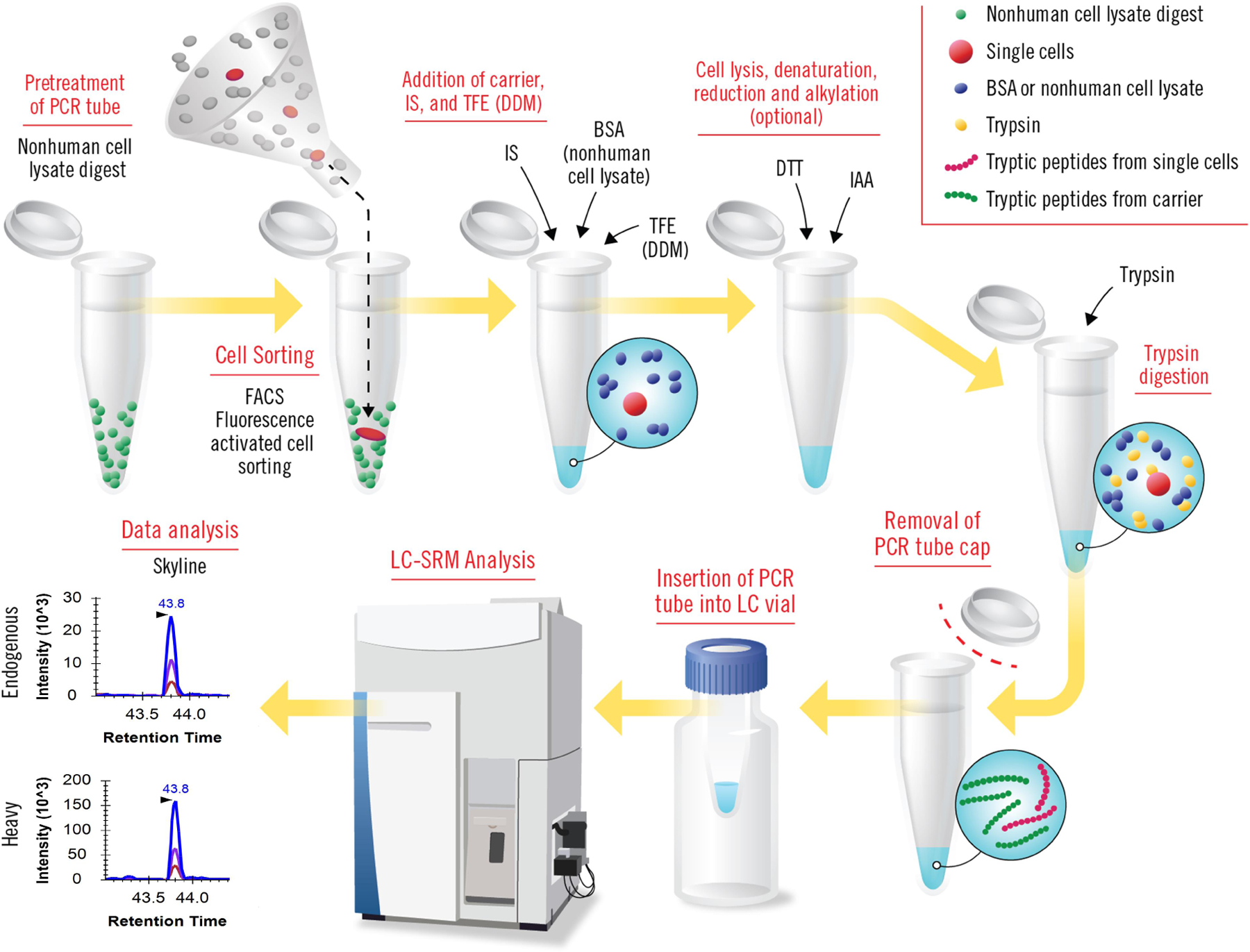
Nonhuman cell lysate digests (e.g., S. oneidensis) is used to pretreat PCR tubes for coating tube surface. Small numbers of human cells or single cells sorted by FACS are collected into pretreated PCR tubes. BSA protein (or nonhuman cell lysate proteome) carrier, heavy internal standards, and TFE (or DDM) are added into sample tubes sequentially for facilitating cell lysis and reducing surface adsorption losses. Conceptually the combined DDM and nonhuman cell lysate proteome carrier will work well for cLC-SRM. Cell lysis is conducted by sonication and protein denaturation is achieved by heating at high temperature. DDT and IAA reagents are used for reduction and alkylation, respectively (the step is optional). Trypsin is added for digestion with much higher ratios of trypsin: protein than that for standard trypsin digestion. The cap of sample tube is removed and then PCR tube is inserted into LC vial for direct LC-SRM analysis. Collected SRM data are analysis using publicly available Skyline software.
Protocol
The step-by-step cLC-SRM analysis is shown in Figure 1. Below is the detailed protocol:
1. Pretreatment of PCR tubes
Add 100 µL of nonhuman (e.g., S. oneidensis) cell lysate digests at 0.2 µg/µL to PCR tubes. Incubate at room temperature for overnight to coat PCR tube surface.
Remove the cell lysate digests, rinse PCR tubes with HPLC-grade water for 3 times, and then airdry PCR tubes in a fume hood.
Store coated PCR tubes into 4 ℃ freezer till further use.
2. FACS sorting
Align a fluorescence-activated cell sorter (FACS) (e.g., BD Influx flow cytometer) into a Hard-Shell 96-Well PCR Plates using fluorescent beads.
Verify that the FACS machine is well aimed for sorting cells into the bottom of PCR plates: 1) Set the angle of the sort stream to the minimum possible, which increases the chance that the cells will be sorted into the bottoms of wells rather than hit the sides; 2) To aim the machine, use the test stream to ‘sort’ droplets of PBS buffer (e.g., 100 droplets) onto the well of empty plate to make sure that the collected droplets are on the bottom. If the droplets do not land onto the right position, recalibrate the cell sorter and repeat until the droplets in the sort stream are deposited correctly.
Place the coated PCR tubes into 96-well PCR tube rack with caps open.
Sort of desired numbers of cells into precoated PCR tubes.
Immediately centrifuge the sorted cells at 850 rpm for 10 min at 4 °C to keep them at the bottom of the tube to avoid potential cell loss.
Store the sorted cells into −80 °C freezer until further analysis.
3. Addition of protein carrier, heavy internal standards, and TFE
Add 4 µL of 25 mM NH4HCO3 into PCR tubes containing collected cells.
Add 1 µL of 10 ng/µL BSA in 25 mM NH4HCO3 into sample tubes.
-
Add 0.3 µL of 100 fmol/µL crude heavy peptide standards (total 30 fmol) into sample tubes.
Note: Dispense of the small volume of crude heavy peptide standards into the middle of the solution and make sure there is no left-over liquid in the pipet tip. High-purity light peptides (>95%) are used to calibrate crude heavy peptide concentrations for absolute quantification.
Add 9 μL of 100% TFE into sample tubes with the final ~60% TFE.
Centrifuge at 5000 rpm for 5 mins and then gently mixed at 850 rpm for 3 mins.
4. Cell lysis and protein denaturation
Sonicate samples on ice for 1 min with 1-min interval for a total of 5 cycles
Incubate samples at incubated at 75–90 °C for 1 h for protein denaturation using the RT-PCR thermocycler with the heated-lid option.
Cool samples to room temperature with centrifugation at 5000 rpm for 3 mins.
5. Reduction and alkylation (optional)
-
Add 0.6 µL of 50 mM DTT with the final concentration of 2 mM.
Note: 10 µL of 500mM DTT is added into 90 µL of 25 mM NH4HCO3 to make 50 mM DTT.
Centrifuge at 5000 rpm for 5 mins and then gently mixed at 850 rpm for 3 mins.
Incubate at 56℃ for 1 h with gentle shaking at 850 rpm.
Cool samples to room temperature and centrifuge at 5000 rpm for 3 mins.
-
Add 0.5 µL of 60 mM IAA was added to the RT-PCR tube with the final concentration of ~2 mM.
Note: 15 µL of 400 mM IAA is added into 85 µL of 25 mM NH4HCO3 to make 60 mM IAA.
Incubate in the dark at room temperature for 30 mins with gentle shaking at 850 rpm.
6. Trypsin digestion
-
Reduce the sample volume down to ~4 µL for greatly reducing TFE volume using a Speed Vac concentrator.
Note: TFE is a volatile solvent with boiling point of 74°C. High percentage TFE inhabits trypsin digestion.
Add 9 µL of 25 mM NH4HCO3 and 1–3 µL of 15 ng/µL trypsin for digestion with the final trypsin concentration 1–3 ng/µL.
Note: When 10 human cells (~1 ng) are digested, the ratio of trypsin enzyme over protein is ≥15:1, 750-fold higher than that in standard trypsin digestion (the ratio of 1:50).
Mix gently at 850 rpm for 3 mins and incubate for overnight (~16 hours) at 37 ℃.
Add 0.5 µL of 5% formic acid to stop enzymatic reaction.
Centrifuge for 1 h at 5000 rpm.
Store in −80 °C freezer until further LC-SRM analysis or in 4 °C freezer for immediate analysis.
7. Preparation for direct LC-SRM analysis
Remove the cap of PCR tube.
Insert PCR tube into LC vial.
-
Close the LC vial for LC-SRM analysis.
Note: For small numbers of cells processed in 96-well PCR plate, the mat cover will be added on the top of plate for direct LC-SRM analysis.
8. LC-SRM analysis
Analyze samples using a nanoACQUITY UPLC® (Waters Corporation, Milford, MA) coupled to a TSQ Vantage triple quadrupole mass spectrometer (Thermo Scientific, San Jose, CA).
The Waters BEH 1.7 µm C18 columns (75 µm i.d. × 20 cm for standard gradient and 100 µm i.d. × 10 cm for short gradient) are connected to a chemically etched 20 μm i.d. fused silica electrospray emitter via a stainless metal union.
Replace 5 µL sample loop with 20 µL sample loop for directly loading all the samples (~15 µL in total) into LC column to maximize detection sensitivity.
Use 0.1% formic acid in water and 0.1% formic acid in 90% acetonitrile as mobile phases A and B for capillary RPLC separation.
The binary LC gradient is used at flow rates of 300 nL/min for standard gradient and 400 nL/min for short gradient. Standard gradient: 5–20% B in 26 mins, 20–25% B in 10 mins, 25–40% B in 8 mins, 40–95% B in 1 min and at 95% B for 7 mins for a total of 52 mins and the analytical column re-equilibrated at 99.5% A for 8 mins. Short gradient for fast separation: 5–95% B in 5 mins and the analytical column re-equilibrated at 99.5% A for 5 mins for a total of 10 mins.
The TSQ Vantage mass spectrometer is operated with ion spray voltages of 2,400 ± 100 V, a capillary offset voltage of 35 V, a skimmer offset voltage of −5 V and a capillary inlet temperature of 220° C.
The tube lens voltages are obtained from automatic tuning and calibration without further optimization.
Before LC-SRM analysis of samples, different concentrations of crude heavy peptide standards are spiked into 20–50 ng/µL of nonhuman cell lysate digests to determine peptide LC retention time (RT) for building scheduled SRM method and potential interference transitions for accurate quantification.
-
The RT scheduled SRM mode is used for multiplexed quantification with the scan window of ≥6 mins.
Note: The scan window will be adjusted accordingly depending on the number of transitions in a certain time window.
The cycle time is set to 1 s, and the dwell time for each transition is automatically adjusted depending on the number of transitions scanned at different retention time windows. A minimal dwell time 10 ms is used for each SRM transition. All target proteins can be simultaneously monitored in a single LC-SRM analysis
9. Data Analysis
Import raw data files into publicly available Skyline software33 for data visualization of each target peptide to determine their detectability.
Use the best transition without matrix interference for SRM quantification.
Use two criteria to determine SRM peak detection and integration: 1) same retention time; 2) approximately the same relative SRM peak intensity ratios across multiple transitions between endogenous (light) peptides and heavy peptide internal standards.
Use multiple technical or biological replicates to obtain the standard deviation (SD) and coefficient of variation (CV).
Inspect all the SRM data manually to ensure correct SRM peak assignment and SRM peak boundary for reliable quantification.
The signal-to-noise ratio (S/N) is calculated by the peak apex intensity over the highest background noise within a retention time region of ±15s for standard gradient and ±6s for short gradient. The limit of detection (LOD) and the limit of quantification (LOQ) are defined as the lowest concentration points of each target protein at which the S/N ratio of surrogate peptides was at least 3 and 7, respectively.
For conservatively determining the LOQ values, in addition to the requirement of S/N ≥7, one additional criteria is applied: surrogate peptide response over different cell numbers must be within the linear dynamic range.
All calibration curves are plotted using Microsoft Excel 2010. The RAW data from TSQ Vantage are loaded into Skyline software to display graphs of extracted ion chromatograms (XICs) of multiple transitions of target proteins monitored.
Representative results
The performance of cLC-SRM was first evaluated by targeted quantification of EGFR/MAPK pathway proteins in 0.5–20 ng of MCF7 cell lysates (equivalent to 5–200 cells). As shown in Figure 2A, extracted ion chromatograms (XICs) clearly shows transitions for ATADDELSFK derived from GRB2 present at ~220,000 copies per MCF7 cell34. cLC-SRM was able to reproducibly quantify endogenous ATADDELSFK down to 5 MCF7 cell equivalents with an S/N ratio of 14 and ~1,800 zmol of quantification sensitivity. The resultant calibration curves displayed excellent linearity over a wide dynamic range of MCF7 cell equivalents with LOQs of ~5–10 cells for high-abundance proteins and ~10–20 cells for moderate-abundance proteins (Figure 2B). The median SRM technical CV for all target peptides across all the data points was ~9%, consistent with the technical reproducibility of well-characterized LC-SRM with CV below 10%.20,23,35–38
Figure 2: Sensitivity and accuracy of cLC-SRM for multiplexed quantification of EGFR/MAPK pathway proteins.
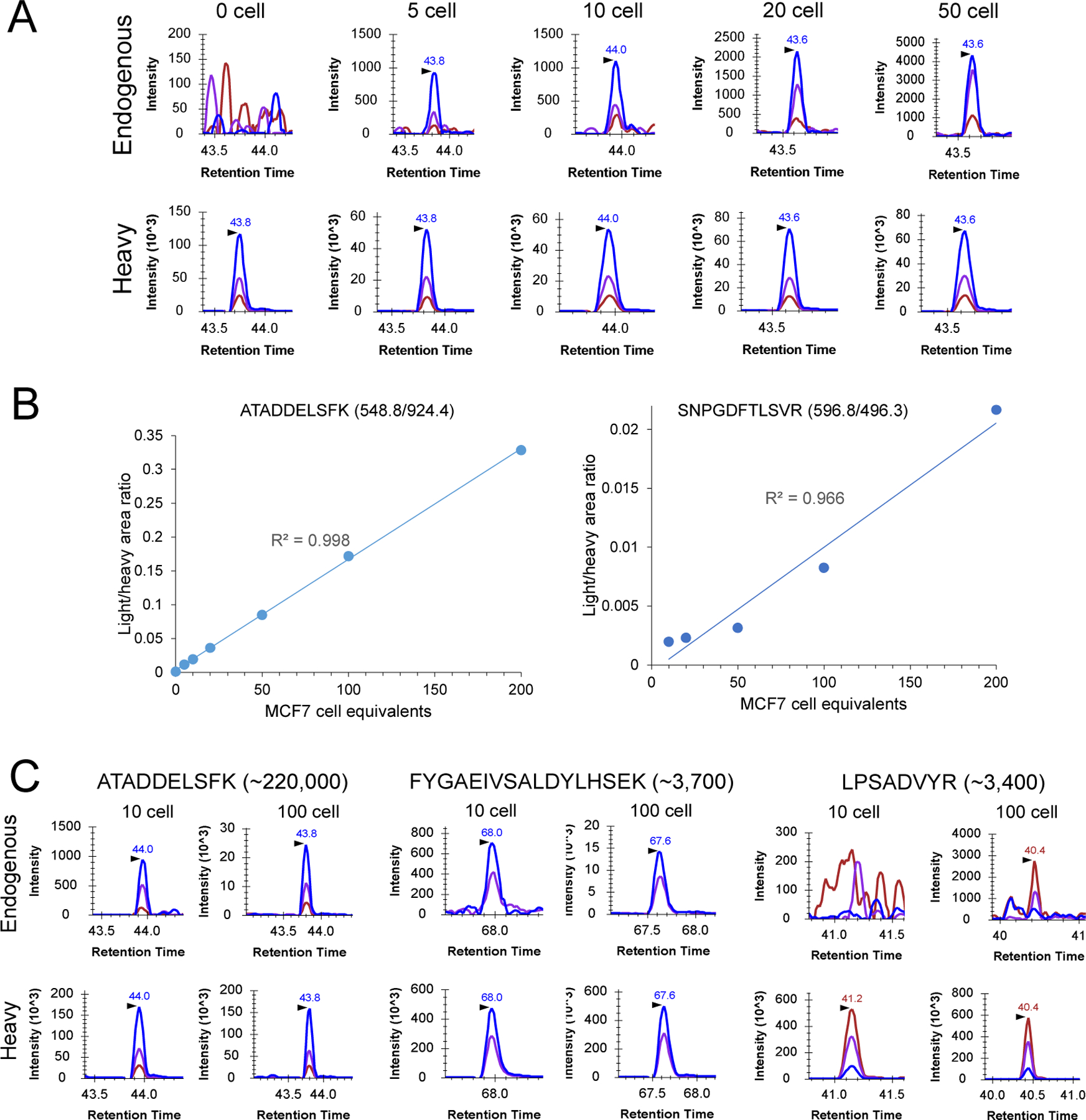
(A) Extracted ion chromatograms (XICs) of transitions monitored for ATADDELSFK derived from GRB2 at different numbers of MCF7 cell equivalents: 548.8/924.4 (blue), 548.8/853.4 (purple), 548.8/738.4 (chestnut). (B) Calibration curves for quantifying high abundance GRB2 and moderate-abundance PTPN11 with the use of the best responsive interference-free transitions, ATADDELSFK (548.8/924.4) for GRB2 and SNPGDFTLSVR (596.9/496.3) for PTPN11. Three and two SRM replicates were performed for 0–10 and 20–200 MCF7 cell equivalents, respectively. (C) Comparison of SRM signal between 10 and 100 MCF7 cells sorted by FACS. Each sample consists of two biological replicates with the addition of ~30 fmol of heavy peptide standards per replicate. XICs of transitions monitored for ATADDELSFK derived from GRB2: 548.8/924.4 (blue), 548.8/853.4 (purple), 548.8/738.4 (chestnut); XICs of transitions monitored for FYGAEIVSALDYLHSEK derived from AKT1: 648.0/897.9 (blue), 648.0/816.4 (purple), 648.0/283.1 (chestnut); XICs of transitions monitored for LPSADVYR derived from SOS1: 460.7/807.4 (blue), 460.7/710.3 (purple), 460.7/404.2 (chestnut).
We next applied cLC-SRM to measure EGFR pathway proteins in 10 and 100 intact MCF7 cells sorted by FACS. cLC-SRM enabled detection of high- and moderate-abundance proteins in 10 intact MCF7 cells (Figure 2C). Even low-abundance AKT1 at ~3700 copies per cell was also detected with the average S/N ratio of 7, suggesting ~60 zmol of absolute sensitivity of cLC-SRM. With the cell number increased to 100, a majority of previously identified important EGFR pathway proteins (22 out of the total 32 proteins)34 were reliably detected and quantified by cLC-SRM. Furthermore, we have recently tested whether a short gradient time (e.g., 5 mins vs standard 45 mins) is sufficient for rapid cLC-SRM analysis. As shown in Figure 3, XICs clearly shows endogenous detection of VLTPTQVK peptide derived from PEBP1 at ~744,000 copies per cell34 in single MCF10A cells sorted by FACS with an S/N ratio of 5 and ~1,240 zmol of quantification sensitivity. As expected, with the cell number increased to 50 and 75 SRM signal became much stronger with detection of all three transitions which have the same pattern as its corresponding heavy internal standard. All these results have shown that cLC-SRM can be used for multiplexed, sensitive, absolute quantification of target proteins in small numbers of human cells including single cells. A short gradient is feasible for cLC-SRM presumably because sample complexity for ≤20 ng of tryptic peptides (10 ng BSA and ≤100 human cells ≈ 10 ng proteins) from carrier-assisted small numbers of cells can be effectively address by high-resolution capillary RPLC separation with high loading capacity of ≥200 ng.
Figure 3: An example of targeted proteomics analysis of small numbers of MCF10A cells including single cells with cLC-SRM at short LC gradient.
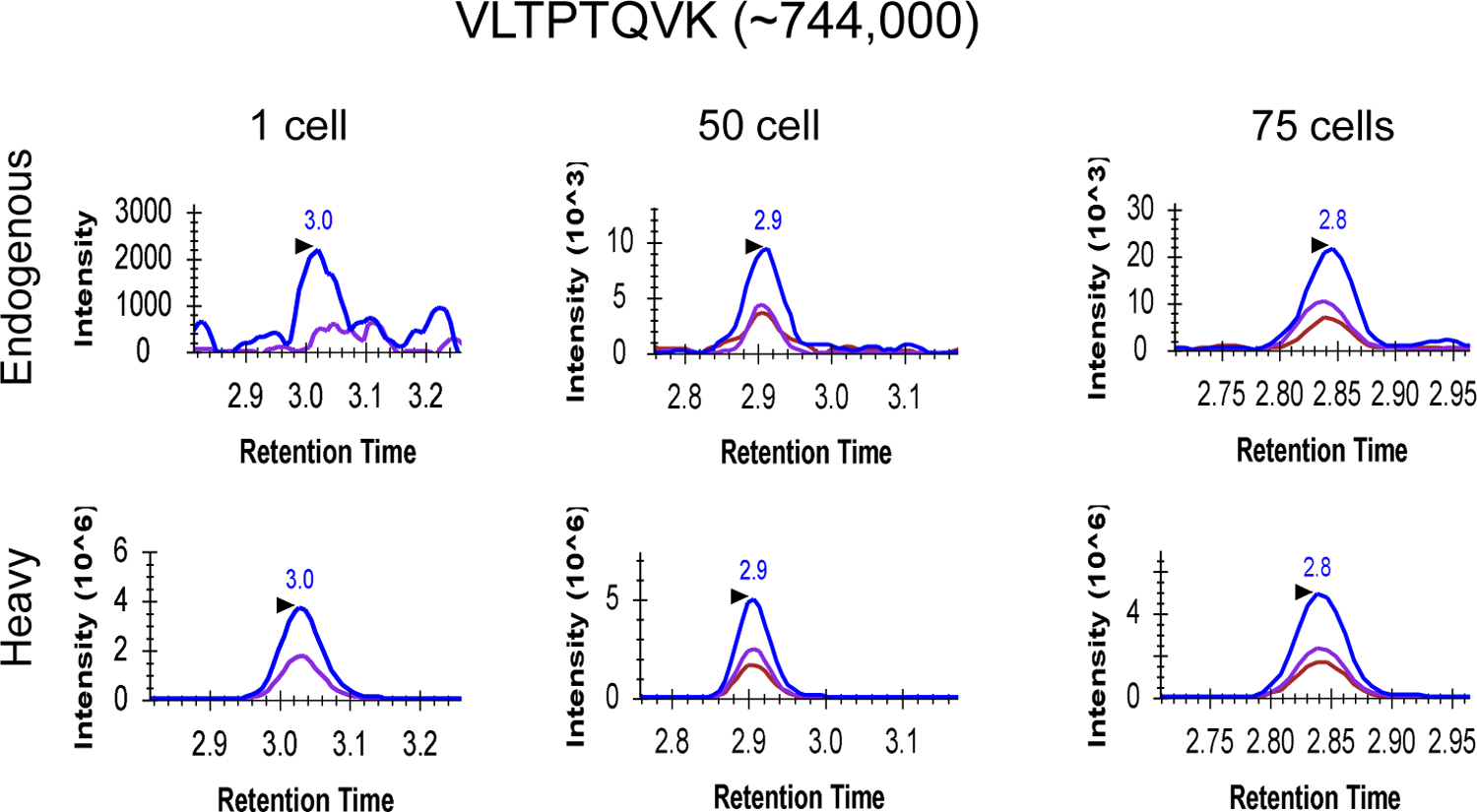
Comparison of SRM signal among 1, 50 and 75 MCF10A cells sorted by FACS. ~30 fmol of internal standard was added to each sample. XICs of transitions monitored for VLTPTQVK derived from PEBP1: 443.3/673.4 (blue), 443.3/572.3 (purple), 443.3/213.2 (chestnut). For single cells, the transition 443.3/213.2 was removed due to severe matrix interference.
Discussion
cLC-SRM is a convenient targeted proteomics method that enables accurate multiplexed protein analysis of small numbers of cells including single cells. This method capitalizes on protein carrier-assisted one-pot sample preparation that combines all steps into one pot (e.g., single tube or single well) including cell collection, multistep cell lysis and digestion, transfer of peptide digests to capillary LC column for MS analysis (Figure 1). This ‘all-in-one’ low-volume one-pot processing presumably maximizes recovery of small numbers of cells for quantitative targeted proteomics analysis by greatly reducing possible surface absorption losses.
There are two critical steps for cLC-SRM analysis: 1) after FACS sorting immediate centrifugation is required to ensure collected cells at the bottom of tubes or wells because low volume (~15 µL) is used to process small numbers of cells; 2) prior to the addition of trypsin, it was necessary to reduce the TFE levels to ≤10% for effective trypsin digestion. To avoid drying out samples with unrecoverable loss, it was necessary to frequently check the sample volume (~4 µL of the remaining volume) during Speed Vac concentrating. One-pot sample preparation can be further simplified by removal of reduction and alkylation steps with neglectable effect on digestion efficiency (Figure 3). One drawback is that cysteine containing peptides cannot be selected as surrogate peptides for target proteins because of instability of free cysteine. To avoid Speed Vac concentrating step for TFE removal we consider replacing TFE with MS-compatible surfactants (e.g., DDM: n-Dodecyl β-D-maltoside) for cell lysis because frequent checking sample volume severely prevents automation of sample processing with low throughput. In addition, single carrier protein (e.g., BSA) may not be sufficient to unbiasedly reduce surface adsorption losses for every protein. This was evidenced by a few undetected proteins at >30,000 copies per cell (e.g., MAP2K1) in 10 MCF7 cells that should have been detected29. Nonhuman cell lysates presumably can be used as a more effective proteome carrier in contrast to our current BSA carrier because there are many different types of nonhuman proteins with less interference for SRM detection of human proteins.
When compared to other available MS-based single-cell proteomics methods that require specific devices and only provide relative quantification26,27, cLC-SRM is readily adopted and implemented in any MS proteomics laboratory without additional costs and can provide accurate or absolute quantification of target proteins in small numbers of cells including single cells. But unlike other global single-cell proteomics methods, cLC-SRM is not suitable for global proteome profiling. Furthermore, cLC-SRM has significant advantages over conventional targeted single-cell proteomics using antibody-based immunoassays in terms of multiplexing (≥200 proteins in a single analysis) and quantitation specificity and accuracy. Future developments will focus on significant improvements in detection sensitivity and sample throughput for enabling rapid absolute quantification of most proteins (e.g., important negative feedback regulators) in single human cells. Automation of cLC-SRM is another direction to improve its robustness, reproducibility, and the overall sample throughput. We anticipate that cLC-SRM can be broadly used for multiplexed accurate quantification of target proteins in small subpopulations of cells and rare tumor cells as well as ultrasmall clinical specimens. In turn, it will be greatly beneficial to current biomedical research and systems biology.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| 2 mL glass LC vial | Microsolv | 9502S-WCV | Vessel to hold PCR tube for autosample injection |
| BSA | Sigma-Aldrich | P0834-10×1mL | Carrier protein for greatly reducing surface adsorption losses |
| DTT | Thermo Scientific | A39255 | Reagent for reduction |
| Formic acid | Thermo Scientific | 28905 | Reagent for stopping enzyme reaction |
| IAA | Thermo Scientific | A39271 | Reagent for alkylation |
| Peptide internal standards | New England peptide | Targeted quantification of EGFR/ MAPK pathway proteins | |
| RT-PCR tube | GeneMate Bioexpress | T-3035-1 | 0.2 mL PCR tube for one-pot sample preparation |
| Skyline software | University of Washington | Publicly available for SRM data analysis | |
| Sonicator | Hielscher Ultrasound Technology | UTR200 | Sonication on ice for cell lysis |
| Speed Vac concentrator | Thermo Scientific | Reduction of the percentage of TFE for effective trypsin digestion | |
| TFE | Sigma-Aldrich | 18370-10×1mL | 60% TFE for cell lysis |
| Thermocycler w/ heated lid | Peltier Thermal Cycler | PTC-200 | Heating for protein denaturation |
| Trypsin Gold | Promega | V528A | Enzyme for protein digestion |
| Waters BEH C18 column | Waters | C18 column for peptide separation |
Acknowledgements
This work was supported by NIH Grant R21CA223715 (TS). Dr. Pengfei Zhang acknowledges the fellowship support from China Scholarship Council. The experimental work described herein was performed in the Environmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, a national scientific user facility sponsored by the United States of America Department of Energy under Contract DE-AC05-76RL0 1830.
Footnotes
Disclosures
The authors have nothing to disclose.
References
- 1.Wang Z, Gerstein M & Snyder M RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10 (1), 57–63, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyamoto DT et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 349 (6254), 1351–1356, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y & Navin NE Advances and applications of single-cell sequencing technologies. Mol Cell 58 (4), 598–609, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macaulay IC, Ponting CP & Voet T Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet 33 (2), 155–168, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gavasso S, Gullaksen SE, Skavland J & Gjertsen BT Single-cell proteomics: potential implications for cancer diagnostics. Expert Rev Mol Diagn 16 (5), 579–589, (2016). [DOI] [PubMed] [Google Scholar]
- 6.Wu M & Singh AK Single-cell protein analysis. Curr Opin Biotechnol 23 (1), 83–88, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes AJ et al. Single-cell western blotting. Nature Methods 11 (7), 749–U794, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 10.1038/nbt.3973, (2017). [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Beyer A & Aebersold R On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165 (3), 535–550, (2016). [DOI] [PubMed] [Google Scholar]
- 10.Choudhary C & Mann M Decoding signalling networks by mass spectrometry-based proteomics. Nat Rev Mol Cell Biol 11 (6), 427–439, (2010). [DOI] [PubMed] [Google Scholar]
- 11.Mertins P et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534 (7605), 55–62, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166 (3), 755–765, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B et al. Proteogenomic characterization of human colon and rectal cancer. Nature 513 (7518), 382–387, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaudet S & Miller-Jensen K Redefining Signaling Pathways with an Expanding Single-Cell Toolbox. Trends Biotechnol 34 (6), 458–469, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods 11 (4), 417–422, (2014). [DOI] [PubMed] [Google Scholar]
- 16.Di Palma S & Bodenmiller B Unraveling cell populations in tumors by single-cell mass cytometry. Curr Opin Biotechnol 31 122–129, (2015). [DOI] [PubMed] [Google Scholar]
- 17.Schulz KR, Danna EA, Krutzik PO & Nolan GP Single-cell phospho-protein analysis by flow cytometry. Curr Protoc Immunol Chapter 8 Unit 8 17 11–20, (2012). [DOI] [PubMed] [Google Scholar]
- 18.Willison KR & Klug DR Quantitative single cell and single molecule proteomics for clinical studies. Curr Opin Biotechnol 24 (4), 745–751, (2013). [DOI] [PubMed] [Google Scholar]
- 19.Lee JY et al. Detection of Head and Neck Cancer Based on Longitudinal Changes in Serum Protein Abundance. Cancer Epidemiol Biomarkers Prev 10.1038/msb4100014 doi: ., (2020). [DOI] [PubMed] [Google Scholar]
- 20.Shi T et al. Advancing the sensitivity of selected reaction monitoring-based targeted quantitative proteomics. Proteomics 12 (8), 1074–1092, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi T et al. Advances in targeted proteomics and applications to biomedical research. Proteomics 16 (15–16), 2160–2182, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lange V, Picotti P, Domon B & Aebersold R Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol 4 222, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picotti P & Aebersold R Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Meth 9 (6), 555–566, (2012). [DOI] [PubMed] [Google Scholar]
- 24.Zhu Y et al. Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angew Chem Int Ed Engl 57 (38), 12370–12374, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong Y et al. Improved Single-Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal Chem 92 (3), 2665–2671, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Y et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat Commun 9 (1), 882, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao X et al. Integrated Proteome Analysis Device for Fast Single-Cell Protein Profiling. Anal Chem 90 (23), 14003–14010, (2018). [DOI] [PubMed] [Google Scholar]
- 28.Budnik B, Levy E, Harmange G & Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol 19 (1), 161, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang P et al. Carrier-Assisted Single-Tube Processing Approach for Targeted Proteomics Analysis of Low Numbers of Mammalian Cells. Anal Chem 91 (2), 1441–1451, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi T et al. Facile carrier-assisted targeted mass spectrometric approach for proteomic analysis of low numbers of mammalian cells. Commun Biol 1 103, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vitrinel B, Iannitelli DE, Mazzoni EO, Christiaen L & Vogel C A simple method to quantify proteins from one thousand cells. bioRxiv doi: 10.1101/753582 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rauniyar N & Yates JR 3rd. Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res 13 (12), 5293–5309, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacLean B et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26 (7), 966–968, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi T et al. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci Signal 9 (436), rs6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Addona TA et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol 27 (7), 633–641, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He J et al. Analytical platform evaluation for quantification of ERG in prostate cancer using protein and mRNA detection methods. J Transl Med 13 54, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi T et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc Natl Acad Sci U S A 109 (38), 15395–15400, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi T et al. Targeted quantification of low ng/mL level proteins in human serum without immunoaffinity depletion. J Proteome Res 12 (7), 3353–3361, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]