

Abstract
COVID-19 is a novel disease caused by SARS-CoV-2 and has made a catastrophic impact on the global economy. As it is, there is no officially FDA approved drug to alleviate the negative impact of SARS-CoV-2 on human health. Numerous drug targets for neutralizing coronavirus infection have been identified, among them is 3-chymotrypsin-like-protease (3CLpro), a viral protease responsible for the viral replication is chosen for this study. This study aimed at finding novel inhibitors of SARS-CoV-2 3C-like protease from the natural library using computational approaches. A total of 69,000 compounds from natural product library were screened to match a minimum of 3 features from the five sites e-pharmacophore model. Compounds with fitness score of 1.00 and above were consequently filtered by executing molecular docking studies via Glide docking algorithm. Qikprop also predicted the compounds drug-likeness and pharmacokinetic features; besides, the QSAR model built from KPLS analysis with radial as binary fingerprint was used to predict the compounds inhibition properties against SARS-CoV-2 3C-like protease. Fifty ns molecular dynamics (MD) simulation was carried out using GROMACS software to understand the dynamics of binding. Nine (9) lead compounds from the natural products library were discovered; seven among them were found to be more potent than lopinavir based on energies of binding. STOCK1N-98687 with docking score of −9.295 kcal/mol had considerable predicted bioactivity (4.427 µM) against SARS-CoV-2 3C-like protease and satisfactory drug-like features than the experimental drug lopinavir. Post-docking analysis by MM-GBSA confirmed the stability of STOCK1N-98687 bound 3CLpro crystal structure. MD simulation of STOCKIN-98687 with 3CLpro at 50 ns showed high stability and low fluctuation of the complex. This study revealed compound STOCK1N-98687 as potential 3CLpro inhibitor; therefore, a wet experiment is worth exploring to confirm the therapeutic potential of STOCK1N-98687 as an antiviral agent.
Keywords: Covid-19, 3-Chymotrypsin-like-protease, Natural compounds, Docking studies, SARS-COV-2
Introduction
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a new coronavirus that emerged in Wuhan, China, in December 2019 and is responsible for the global pandemic previously known as coronavirus disease 2019 (COVID-19) (Zhu et al. 2019). This is to a certain extent, dissimilar from the familiar rampant human coronaviruses HCoV-229E, HCoV-NL63, HCoV-HKU1, and HCoV-OC43; the zoonotic Middle East respiratory syndrome coronavirus (MERS-CoV); and the severe acute respiratory syndrome coronavirus (SARS-CoV) famous with tall mortality (Sharma et al. 2020). Individuals diseased with SARS-CoV-2 are presented with symptoms comprising fever, dry cough, tiredness, loss of speech and difficulty in breathing (Elfiky and Azzam 2020; Pant et al. 2020). Regrettably, there is no available effective drug for COVID-19 (Wu et al. 2020a, b). Different studies have observed the genetic code of SARS-CoV-2 shows 80% similarity with the severe acute respiratory syndrome (SARS), which instigated worldwide outbreak two decades ago (Wu et al. 2020a, b; Chen et al. 2020; Wang et al. 2019).
Based on the available knowledge of the SARS-CoV-2 and closely related coronaviruses, reports on virtual screening of available antiviral drugs (Boopathi et al. 2020; Muralidharan et al. 2020) available databases (Khan et al. 2020), and natural agents breathing (Elfiky and Azzam 2020; Pant et al. 2020; Aanouz et al. 2020) against evolving targets such as viral spike proteins (Hasan et al. 2020), envelop protein (Gupta et al. 2020), proteases (Khan et al. 2020), nucleocapsid protein (Sarma et al. 2020), 2'-O-ribose methyltransferase and 3CL hydrolase is rapidly emerging (Sarma et al. 2020; Elmezayen et al. 2020).
The SARS-CoV-2 coronavirus encodes essential proteases, namely papain-like Protease (PLpro) and 3-chymotrypsin-like Protease (3CLpro) as part of its non-structural protein (nsp)-3 domains. These proteases are attractive antiviral drug targets because they are essential for coronaviral replication. Although the primary function of PLpro and 3CLpro is to process the viral polyprotein in a coordinated manner, they possess the additional function of stripping ubiquitin and ISG15 from host-cell proteins to aid coronaviruses in their evasion of the host innate immune responses (Báez-Santos et al. 2015).
Hence, 3CLpro has been regarded by many scientists across the globe as a drug target against SARS-CoV-2 due to its role in the viral replication cycle (Báez-Santos et al. 2015; Qamar et al. 2020). The 3CLpro is answerable for the catalytic cleavage of eleven conserved sites in polyprotein 1ab (PP1ab) and 1a (PP1a) comprising an enormous hydrophobic residues, a glutamine residues as well as a small number of amino acid residues (Anand et al. 2003). The 3C-like cleavage sites on the polyproteins of coronaviruses are incredibly conserved, and their sequence and substrate specificities are matching (Anand et al. 2003; Wu et al. 2020a, b). This sequential resemblance offers the basis for paralleling SARS-CoV-2 with its prior counterpart leading to the discovery of compounds with great potentials to control or inhibit the replication of SARS-CoV-2. Hence, identification of small molecules with the attribute of inhibiting replication mechanism of SARS-CoV-2 may serve as insight against COVID-19. In this direction, molecular docking and other computational procedures have proved valuable in the initial large-scale screening of several natural compounds and small molecules that directly inhibit essential target proteins (Elekofehinti et al. 2020a, b; Anand et al. 2003).
Using in silico studies, many small molecular weight compounds including the existing FDA-approved drugs have proven to be promising therapeutics against 3CLpro of SARS-CoV-2 (Li et al. 2020a, b; Wei-chung et al. 2021; Johnson et al. 2021; Al-Bustany et al. 2021). Some of these compounds have been validated using experimental studies, and they showed considerable inhibitory prowess (pIC50) against 3CLpro of SARS-CoV-2. However, none of the newly designed compounds has made it to the clinical stage due to the number of years it can take to develop classical antiviral drugs. Nevertheless, few of the repurposed existing drugs (lopinavir, ritonavir, chloroquine, hydroxychloroquine) targeting 3CLpro have shown good outcome in clinical trials (Oscanoa et al. 2020; Horby et al. 2020). Despite the efficacy of the repurposed drugs, no effective therapies exist for treating COVID-19 patients by targeting 3CLpro of SARS-CoV-2 (Mody et al. 2021). Therefore, the medical world are still searching for effective treatments, especially during the early stage of SARS-CoV-2 infection, where no pharmacological intervention has been approved by FDA. The present study aimed to find small molecules from screened natural products with inhibitory attribute against 3CLpro of SARS-CoV-2, thus availing new compounds that can be designed as antiviral agents against the novel pandemic coronavirus disease (COVID-19).
Methods
Schrodinger suite 2018-4 (Windows version) served as the computational tool employed to conduct the in silico analysis devised in this study.
Protein Starting Structure and Ligand Preparation
Crystal structure of SARS-CoV-2 3C-like Protease (3CLPro) (PDB ID: 6W63) was retrieved from Protein Data Bank (http://www.rcsb.org) using the Protein Preparation Wizard of Maestro-v11.2 molecular interface (Iwaloye et al. 2020a). The protein crystal structure was prepared to assign bond orders and add missing hydrogen atoms. The protocol used has been well described in our previous computational studies (Iwaloye et al. 2020b, c). About sixty-nine thousand (69,000) compounds were retrieved from Natural product library (IBS Database, Inter Bio Screen Ltd, http://www.ibscreen.com/natural.shtml) in SDF format, and prepared for docking using Ligprep (Schrödinger suites) and the procedure for preparation is detailed by Iwaloye et al (2020c).
Generation of pharmacophore modelling for database screening and molecular docking study
The e-pharmacophore model of protein–ligand complex was generated by docking the co-ligand with the protein using glide extra precision (XP) docking, a maximum of five pharmacophore features were left as default. The five features include the following:
Hydrogen bond acceptor (A)
Hydrogen bond donor (D)
Aromatic ring (R)
Positive ionizable (P) and
Negative ionizable (N)
The hydrogen bond donor and acceptor features are vector properties and possess a vectorial nature which indicates the direction of electron sharing (Dixon et al. 2006; Salam et al. 2009). Compounds retrieved from natural products database were screened against a minimum of 3 sites from the five generated sites, and compounds with fitness score above the value of 1.0 were further filtered by molecular docking. Docking protocol was carried out by the method described in previous studies (Iwaloye et al. 2020a, b). Since the active site of SARS-CoV-2 3CLpro do not provide a medium of covalent interaction with any compound, all compounds experimented against SARS-CoV-2 3CLpro are non-covalent inhibitors. Therefore, this study explored molecular docking study through non-covalent interactions. Initially, the compounds were docked with the protein using high throughput virtual screening (HTVS) glide docking precision with settings left as default. Subsequently, 10,000 compounds from HTVS screening that appeared as top docked compounds were docked using standard precision (SP). Finally, the best 200 compounds in term of binding affinity were subjected to glide XP docking. Nine compounds were chosen because they had the lowest binding energies with SARS-CoV-2 3CLpro. The antiviral agents ivermectin and lopinavir were compared with the nine natural compounds because of their established therapeutic potential against SARS-CoV-2. To improve the binding affinities of these compounds with the protein crystal structure, a flexible docking procedure, known as induced fit docking (IFD) was employed. This method offers an accurate prediction of the compounds binding affinity to accommodate concomitant structural changes in the receptor upon ligand binding (Sherman et al. 2006).
Calculation of free binding energy
The essence of calculating the free energy of binding is to determine the stability of the protein–ligand complex. The binding free energy was calculated by uploading the docked complex output files to Prime molecular mechanics/generalized born surface area (MM-GBSA), a post-docking analysis embedded in Schrodinger suite. This post-docking module does the calculation by generating a lot of energy properties. These properties report energies for the ligand, receptor, and complex structures as well as energy differences relating to strain and binding, and are broken down into contributions from various terms in the energy expression. The Prime MM-GBSA calculates five fundamental energy which are optimized free receptor (= “ Receptor”), optimized free ligand (= “ Ligand”), optimized complex (= “ Complex”), receptor from minimized/optimized complex and ligand from minimized/optimized complex (Elekofehinti et al. 2020a).
The equations for calculating binding energy are as follow:
| 1 |
| 2 |
where Ecomplex, Eprotein, and Eligand indicate the minimized energies for protein-inhibitor complex, protein, and inhibitor, respectively.
| 3 |
| 4 |
where, ∆GSA is the non-polar contribution to the solvation energy due to the surface area. GSA(complex), GSA(protein) and GSA(ligand) are the surface energies of complex, protein and ligand respectively.
ADME/T properties calculations
The prediction of ADME (Absorption, Distribution, Metabolism, and Excretion) properties of the chemical compounds was calculated by Qikprop module (Qikprop 2018). Parameters such as Lipinski's rule of five (RO5) were evaluated to predict the drug-likeness of the chemical compounds.
Preparation of dataset and generation of automated QSAR
Previously reported compounds identified as 3CLPro inhibitors were identified from two studies (Jin et al. 2020), alongside their inhibition constant (IC50). An online converting tool was employed to convert the compounds IC50 to pIC50 (Selvaraj et al. 2011). A machine-learning algorithm called AutoQSAR was used to build the QSAR model through automation (de Oliveira and Katekawa 2017).
Molecular dynamics (MD) simulation
The MD simulation was performed on 3CLpro in its apo form and bound form (in complex with STOCK1N-98687) using GROMACS 2016.4 software running on Dell (Processor 3.60 GHz Intel Core i5 Memory 4 GB 1600 MHz DDR3) with a GROMOS54A7 force field. Ligand topology was generated using Ac-pype, and the protein–ligand complexes were solvated using SPC water in a dodecahedral box with a minimum of 1.0 nm distance between any protein atoms to the closest box edge. The box was solvated, and NaCl added at a concentration of 150 mM while at the same time neutralizing the system. The system was first energy-minimized, equilibrated in the NVT ensemble (i.e., with a constant number of molecules, volume, and temperature) for 0.1 ns and then simulated for 100 ns in the NPT ensemble at 300 K. Energy minimization was performed using a steepest-descent gradient method for a maximum of 50,000 steps. Each complex was restrained using an isothermal-isochloric (NVT) ensemble and isothermal-isobaric ensemble (NPT) for 200 ps (Elekofehinti et al. 2013). Parrinello-Rahman algorithm was used to couple the temperature and pressure (Shyu et al. 2010). The temperature of 300 K and a pressure of 1.0 atm were maintained. The LINCS algorithm was used to constrain the length of all bonds containing hydrogen atoms. The system was equilibrated for 1 ns with constant temperature and pressure while the production MD run was performed for 50 ns.
Results
The molecular docking results disclosed that nine compounds showed good docking with SARS-CoV-2 3CLPro (Fig. 1a; Table 1). Six of the compounds (STOCK1N-98687, STOCK1N-89003, STOCK1N-84615, STOCK1N-84519, STOCK1N-92347, STOCK1N-94719) possessed better docking score and induced fit score than Lopinavir (− 8.052 kcal/mol and − 633.27 kcal/mol respectively) while three of them (STOCK1N-98135, STOCK1N-80093 and STOCK1N-93501) had better docking score and induced fit score than Ivermectin (− 5.889 kcal/mol and − 634.90 kcal/mol respectively), and the results are shown in Table 1.
Fig. 1.

a Chemical structures of hit compounds. b Screening hypothesis generated by structure based e-pharmacophore model consisting of two hydrogen bond acceptor (A) and three aromatic ring (R)
Table 1.
Molecular Docking score, Induced fit docking score and interacting residues of investigated natural products and reference compounds
| Entry name | Docking score | Induced fit score | No of H-bond | Interacting residues | Hydrogen bond distance (Å) | pIC50 |
|---|---|---|---|---|---|---|
| STOCK1N-98687 | − 9.604 | − 639.47 | 5 | CYS145, CYS44, HIE41, THR26 | CYS145 [2.27] CYS44 [2.23] HIE41 [1.91] THR26 [1.89] | 4.427 |
| STOCK1N-89003 | − 8.869 | − 638.86 | 5 | ASN142, GLU166, HIE163 SER144, THR26 | ASN142 [2.32] GLU166 [1.55] HIE163[1.61] SER144 [2.55] THR26 [2.75] | 3.698 |
| STOCK1N-84615 | − 8.848 | − 634.72 | 5 | HIE41, HIE163, GLU166, THR26, ASN142 | HIE163 [1.83] GLU166 [2.49, 2.03], THR26, ASN142 [1.93] | 4.449 |
| STOCK1N-84519 | − 8.017 | − 637.72 | 3 | GLU166, GLN192, CYS44, HIE41 | GLU166 [1.75], GLN192 [1.76], CYS44 [1.80] | 4.198 |
| STOCK1N-92347 | − 8.664 | − 635.89 | 4 | HIE41, THE190, GLN189, GLU166, CYS44 | THE190 [2.03] GLN189 [2.45], GLU166 [1.81] CYS44 [1.90] | 4.484 |
| STOCK1N-94719 | − 8.053 | − 637.69 | 3 | HIE41, THR190, GLN189, GLU166 | THR190 [1.89] GLN189 [2.11] GLU166 [1.80] | 4.491 |
| Lopinavir | − 8.052 | − 633.27 | 3 | GLU166, ASN142, GLN189 | GLU166 [2.44] ASN142[2.32] GLN189 [1.90] | 5.165 |
| STOCK1N-98135 | − 8.095 | − 638.92 | 4 | HIE41, THR190, GLN189, HIE164 | THR190 [2.42, 2.50] GLN189 [2.72] HIE164 [1.89] | 4.461 |
| STOCK1N-80093 | − 7.439 | − 628.46 | 6 | THR26, PHE140, ASN142, GLU166, HIE163, SER144 | THR26, PHE140 [2.13] ASN142 [1.85] GLU166 [1.99] HIE163 [1.78] SER144 [2.01] | 4.194 |
| STOCK1N-93501 | − 8.090 | − 637.48 | 2 | HIE41, GLN189, THR190 | GLN189 [1.83] THR190[2.50] | 4.280 |
| Ivermectin | − 5.889 | − 634.90 | 3 | GLU166, HIE183, ASN142 | GLU166 [2.13] HIE183 [1.82] ASN142 [2.23] | 4.308 |
Furthermore, the selected hits (Fig. 1b) were further accessed for interacting profiles with 3CLPro of SAR-CoV-2. The predicted pIC50, docking score, induced fit score as well as the number hydrogen bond cum interacting amino acids with the ligands are reported in Table 1. Table 2 showed the result of the binding free energy calculation for the lead compounds and reference compounds, and the results shows that STOCK1N-98687 exhibited the highest calculated binding free energy with 3CLPro of SAR-CoV-2. Figure 2a-i presented the 2D diagram of the lead natural products and reference compounds elucidating their intermolecular interactions with amino acid residues at the catalytic cavity of 3CLPro of SAR-CoV-2. All of the ligands have shown common interactions with amino acid residues such as CYS145, CYS44, HIS41, THR26, ASN142, GLU166, HIS163, GLU166, THR25, GLN192, GLN189, THR190, HIS164 and PHE140 at the binding pocket of SAR-CoV-2 3CLPro (Table 1). The Drug-likeness and ADME/T properties of the lead natural products and reference compounds are shown in Tables 3 and 4 respectively. The parameters corresponding to the best model generated by AutoQSAR was reported in Table 5 and the best model obtained was KPLS_Radial_46 (R2 = 0.8180 and Q2 = 0.5287); this constructed model predicted the bioactivity of both the lead compounds and known antiviral drugs (Tables 1, 5; Fig. 3).
Table 2.
Binding free energy calculation for investigated natural products and reference compounds using Prime MM-GBSA
| s/n | Name | ΔGBinda | ΔGBindb Coulomb | ΔGBindcLipo | ΔGBinddvdW | ΔGBindeH-bond |
|---|---|---|---|---|---|---|
| 1 | STOCK1N-98687 | − 66.44 | − 36.27 | − 17.70 | − 42.97 | − 2.45 |
| 2 | STOCK1N-89003 | − 62.56 | − 14.61 | − 15.35 | − 59.80 | − 1.43 |
| 3 | STOCK1N-84615 | − 52.68 | − 27.89 | − 10.41 | − 43.56 | − 4.32 |
| 4 | STOCK1N-84519 | − 55.37 | − 17.24 | − 12.37 | − 49.34 | − 5.05 |
| 5 | STOCK1N-92347 | − 74.92 | − 19.62 | − 21.04 | − 60.62 | − 6.49 |
| 6 | STOCK1N-94719 | − 68.56 | − 34.85 | − 18.82 | − 51.75 | − 2.61 |
| 7 | Lopinavir | − 54.54 | − 12.88 | − 21.29 | − 59.32 | − 3.73 |
| 8 | STOCK1N-98135 | − 52.17 | − 20.61 | − 13.10 | − 43.70 | − 1.04 |
| 9 | STOCK1N-80093 | − 46.96 | − 15.20 | − 10.19 | − 41.92 | − 4.92 |
| 10 | STOCK1N-93501 | − 48.73 | − 8.15 | − 14.91 | − 57.73 | − 6.24 |
| 11 | Ivermectin | − 36.53 | − 10.98 | − 12.89 | − 54.69 | − 1.10 |
aMM-GBSA free energy (kcal/mol) of binding
bContribution to the MM-GBSA free energy of binding (kcal/mol) from the Coulomb energy
cContribution to the MM-GBSA free energy of binding (kcal/mol) from lipophilic binding
dContribution to the MMGBSA free energy of binding (kcal/mol) from the van der Waals energy
eContribution to the MM-GBSA free energy of binding (kcal/mol) from H-bond
Fig. 2.







a The 2D ligand interaction diagram of STOCK1N-98687. b The 2D ligand interaction diagram of STOCK1N-89003. c The 2D ligand interaction diagram of STOCK1N-84615. d The 2D ligand interaction diagram of STOCK1N-84519. e The 2D ligand interaction diagram of STOCK1N-98135. f The 2D ligand interaction diagram of STOCK1N-92347. g The 2D ligand interaction diagram of STOCK1N-94719
Table 3.
Drug-likeness properties (Lipinski’s rule of five) of investigated natural products and reference compounds as predicted by QikProp
| s/n | Name | mol_MWa | donorHBc | donorHBd | QPlogPo/wb | RuleOfFivee |
|---|---|---|---|---|---|---|
| 1 | STOCK1N-98687 | 452.463 | 4.25 | 9 | 0.946 | 0 |
| 2 | STOCK1N-89003 | 487.465 | 3 | 9.75 | 1.542 | 0 |
| 3 | STOCK1N-84615 | 420.381 | 5 | 8 | 0.35 | 0 |
| 4 | STOCK1N-84519 | 365.342 | 2 | 8.25 | 0.672 | 0 |
| 5 | STOCK1N-92347 | 492.576 | 1 | 5.75 | 6.224 | 1 |
| 6 | STOCK1N-94719 | 489.481 | 4 | 9.7 | 2.135 | 0 |
| 7 | Lopinavir | 628.81 | 4 | 9.45 | 5.608 | 2 |
| 8 | STOCK1N-98135 | 416.473 | 3.25 | 8.25 | 1.469 | 0 |
| 9 | STOCK1N-80093 | 456.451 | 5 | 13.5 | 0.465 | 0 |
| 10 | STOCK1N-93501 | 493.564 | 1 | 6 | 5.894 | 1 |
| 11 | Ivermectin | 730.977 | 1 | 11.75 | 7.484 | 2 |
aMolecular weight of the molecule (Range 130.0 to 725.0)
bPredicted octanol/water partition coefficient. (Range − 2.0 to 6.5)
cNumberof hydrogen bond donors (Range 0.0 to 6.0)
dNumber of hydrogen bond acceptors (Range 2.0 to20.0)
eNumber of violations of Lipinski’s rule of five (Range maximum is 4)
Table 4.
ADME (pharmacokinetic) properties of investigated natural products and reference compounds
| s/n | Name | QPlogKhsaa | QPlogHERGb | QPPMDCKc | PSAd | QPPCacoe |
|---|---|---|---|---|---|---|
| 1 | STOCK1N-98687 | − 0.609 | − 3.186 | 16.625 | 167.116 | 22.313 |
| 2 | STOCK1N-89003 | − 0.162 | − 4.296 | 12.643 | 166.082 | 15.527 |
| 3 | STOCK1N-84615 | − 0.467 | − 4.427 | 4.239 | 165.188 | 5.308 |
| 4 | STOCK1N-84519 | − 0.452 | − 4.413 | 24.074 | 138.286 | 28.196 |
| 5 | STOCK1N-92347 | 1.483 | − 7.136 | 422.331 | 89.895 | 863.86 |
| 6 | STOCK1N-94719 | − 0.315 | − 4.991 | 87.265 | 148.495 | 154.407 |
| 7 | Lopinavir | 0.553 | − 4.417 | 434.943 | 126.261 | 418.358 |
| 8 | STOCK1N-98135 | − 0.551 | − 2.219 | 101.839 | 145.381 | 105.75 |
| 9 | STOCK1N-80093 | − 0.765 | − 6.699 | 14.565 | 163.267 | 38.329 |
| 10 | STOCK1N-93501 | 1.331 | − 6.592 | 505.625 | 99.173 | 1020.398 |
| 11 | Ivermectin | 1.566 | − 4.902 | 645.145 | 111.352 | 1278.435 |
aQPlogKhsa Prediction of binding to human serum albumin. Range from − 1.5 to + 1.5
bQPlogHERG Predicted IC50 value for blockage of HERG K + channels. concern below − 5
cQPPMDCK Predicted apparent MDCK cell permeability in nm/sec. MDCK cells are considered to be a good mimic for the blood–brain barrier. QikProp predictions are for non-active transport. < 25 poor, > 500 great
dPSA:Van der Waals surface area of polar nitrogen and oxygen atoms. Range from 7.0 to 200.0
eQPPCaco:Predicted apparent Caco-2 cell permeability in nm/sec. Caco-2 cells are a model for the gutblood barrier. QikProp predictions are for non-active transport. < 25 poor, > 500 great
Table 5.
Parameters corresponding to best model generated by AutoQSAR
| Model code | S.D | R2 | RMSE | Q2 |
|---|---|---|---|---|
| kpls_radial_46 | 0.5085 | 0.8180 | 0.5685 | 0.5287 |
Fig. 3.

Scatter plot analysis of the best model
Furthermore, Gromacs was deployed to execute the MD simulations for 50 ns for the protein–ligand complex (indicated in red colour) and apo protein (Indicated in black color). The RMSD for both apo 3CLpro and STOCKIN-98687-3CLpro complex is presented in Fig. 4a while the fluctuation in backbone known as RMSF is presented in Fig. 4b. The hydrogen bond number for SARS-CoV-2 3CLpro in apo form (black color) and 3CLpro-STOCKIN-98687 complex (red color) during 50 ns MD simulation is shown in Fig. 4c. The high number of hydrogen bonds in the apoprotein (protein unbound state) is due to intra-molecular hydrogen interactions between the residues within the protein. However, in 3CLpro-STOCKIN-98687 complex, only the number of hydrogen bond interacting with the complex is counted (intermolecular hydrogen interaction).
Fig. 4.

a Time dependence of RMSD values for SARS-CoV-2 3CLpro (black color) and 3CLpro–STOCKIN-98687 complex (red color) during 50 ns MD simulation. b The root mean square fluctuation (RMSF) of C-alpha for SARS-CoV-2 3CLpro (black color) and 3CLpro–STOCKIN-98687 complex (red color) during 50 ns MD simulation. c The hydrogen bond number of SARS-CoV-2 3CLpro in apo form (black color) and 3CLpro-STOCKIN-98687 complex (red color) during 50 ns MD simulation
Discussion
The pharmacophore model generated five active sites exhibiting different scores (Tables 6, 7; Fig. 1b), and the sites comprise two hydrogen acceptors and three aromatic rings. The prepared library of natural compounds that match a minimum of 3 sites exhibiting fitness score ≥ 1.0 was subjected to molecular docking. This screening disclosed that nine compounds (Fig. 5) (STOCK1N-98687, STOCK1N-89003, STOCK1N-84615, STOCK1N-84519, STOCK1N-92347, STOCK1N-94719, STOCK1N-98135, STOCK1N-80093 and STOCK1N-93501) had favorable good docking scores (− 9.295 kcal/mol, − 8.869 kcal/mol, − 8.848 kcal/mol, − 8.017 kcal/mol, − 8.664 kcal/mol, − 8.053 kcal/mol, − 8.095 kcal/mol, − 7.439 kcal/mol and − 8.090 kcal/mol respectively) than lopinavir. Several antiviral agents have been repurposed against COVID-19 to conduct a rapid study of the viral infection, at lower costs and increase the safety profile of drugs (Esakandari et al. 2020; Parlakpinar et al. 2020). Among them are lopinavir and ivermectin; the therapeutic efficiency of these compounds are promising against SARS-CoV-2 in in vitro study. Hence, the present study employed lopinavir and ivermectin as compounds of comparison with the investigated natural products. The favourable binding energies attained by the compounds may denote their inhibitory prowess (Rampogu et al. 2018). The docking protocol was validated by docking native ligand (co-crystal ligand) with the prepared crystal structure of 3CLPro to validate the docking efficiency by determining the root mean square deviation (RMSD). An RMSD value of 1.55 Å showed that the docking procedure is reproducible (Fig. 1b) (Elekofehinti et al. 2018).
Table 6.
Fitness and alignment score of investigated natural products and reference compounds using Energy optimized pharmacophore model (e-pharmacophore)
| s/n | Entry name | Vector score | Align score | Fitness score |
|---|---|---|---|---|
| 1 | STOCK1N-98135 | 0.883 | 0.795 | 1.570 |
| 2 | STOCK1N-98687 | 0.855 | 0.828 | 1.532 |
| 3 | STOCK1N-94719 | 0.625 | 0.717 | 1.485 |
| 4 | STOCK1N-84519 | 0.620 | 0.614 | 1.465 |
| 5 | STOCK1N-84615 | 0.641 | 0.869 | 1.419 |
| 6 | STOCK1N-92347 | 0.625 | 0.775 | 1.319 |
| 7 | STOCK1N-89003 | 0.389 | 0.636 | 1.214 |
| 8 | STOCK1N-93501 | 0.764 | 1.010 | 1.154 |
| 9 | STOCK1N-98135 | 0.887 | 1.004 | 1.034 |
| 10 | Lopinavir | 0.709 | 0.903 | 1.164 |
| 11 | Ivermectin | – | – | – |
Table 7.
Feature score of the pharmacophore sites
| Protein target | No. of possible site | No. of accepted site | Hypotheses | Pharmacophore features with score |
|---|---|---|---|---|
| 6W63 | 6 | 5 | AARRR | A3: − 0.48, A4: − 0.64, R9: − 0.48, R10: − 9.95, R11 − 0.53: |
A, H-bond acceptor; R, aromatic ring
Fig. 5.
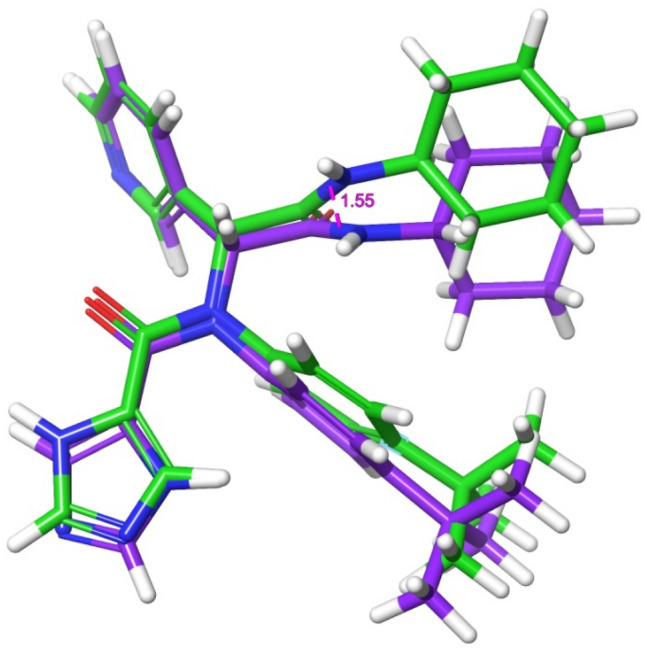
Superposition of the co-crystal ligand with its docked pose with RSMD of 1.55 Å
To further authenticate the docking procedure and determine the stability of protein–ligand complexes, the prime molecular mechanics/generalized Born surface area (MM-GBSA) calculations were engaged. The nine (9) natural products and antiviral compounds exhibited favourable stability with the protein, and the binding free energy (ΔGBind) score were within the range of − 74.92 kcal/mol to − 36.53 kcal/mol. While STOCK1N-92347 formed the most stable complex with the protein, Ivermectin formed the least stability (Table 1). The primary energy contributors to the binding free energy were identified as van der Waals, Lipophilic energy, Coulomb interaction and Hydrogen bond that enhances the binding affinity of the compounds towards the binding pocket of the protein.
A monomer of 3CLpro consists of domain I, domain II, and domain III; and a long loop connects domains II and III. The catalytic site of 3CLpro occupies the gap between domains I and II and has a Cys-His catalytic dyad (Cys145 and His41) (Jo et al. 2020). The enzymatic activity of the 3CLpro resides in the catalytic dyad of Cys145 and His41 (Yang et al. 2003). Several efforts made to inhibit SARS-CoV has led to the identification of covalent molecules capable of targeting the catalytic dyad of the 3CLpro, these covalent inhibitors, however, often come with their disadvantages such as toxic side effects, reduced potency and adverse drug responses (Paasche et al. 2014; Tuley 2018). Figure 2 presented the 3D diagram of the top seven docked natural products elucidating the non-covalent intermolecular interaction at the catalytic cavity of 3CLPro of SAR-CoV-2. The ligands showed common interactions with amino acid residues such as CYS145, CYS44, HIS41, THR26, ASN142, GLU166, HIS163, GLU166, THR25, GLN192, GLN189, THR190, HIS164 and PHE140. The interaction of STOCK1N-98687 with the protein was stabilized by hydrogen bonding and hydrophobic interaction. Interaction profiles showed that five hydrogen bonds contributed to the stability of STOCK1N-98687 in the active site of 3CLpro of SARS-CoV-2 by interacting with CYS145, CYS44, HIS41, and THR26 amino acid residues.
Prompt valuation of ADME properties enormously declines pharmacokinetics-related debacles in the clinical juncture on drug discovery (Daina et al. 2017). Accordingly, the compounds drug-likeness and pharmacokinetics were examined using RO5 and other parameters. RO5 is one of the parameters obligatory before a compound is considered as a drug candidate (Lipinski et al. 1997). Here, it was discovered that the lead compounds except for STOCK1N-92347 and STOCK1N-93501 were in accordance with RO5, and therefore they can be considered as drug candidates. Relevant pharmacokinetic properties of the leads were further carried out; the prediction of human serum albumin binding (QPlogKhsa) showed that lead compounds would bind to human serum albumin during distribution. The predicted IC50 for HERGK+ (QPlogHERG) showed that only STOCK1N-92347 (− 7.136), STOCK1N-80093 (− 6.699) and STOCK1N-93501 (− 6.592) fell within the standard and acceptable range (> -5). The Predicted apparent MDCK cell permeability (a good mimic for blood–brain barrier) (QPPMDCK) shows that a number of the lead compounds possess an adequate capacity of passing through the blood–brain barrier. The predicted Van der Waals surface area of polar nitrogen and oxygen atoms (PSA) for the natural compounds reveals that none was out of range (7.0 – 200.0).
AutoQSAR, a machine-learning algorithm provided by Schrödinger suite computed about 497 physicochemical and topological descriptors, alongside a variety of Canvas fingerprints (de Oliveira and Katekawa 2017), giving out a large pool of independent variables from which to build models. The AutoQSAR splits the experimental compounds randomly into 75% training set, and 25% test set (Table 8). The best predictive model from the manually collected experimental datasets is kpls_radial_46, computed from kernel-based partial least square regression (KPLS) analysis which supports radial binary fingerprint as independent variable. The model parameters include standard deviation (S.D) of 0.5085, R2 of 0.8180; root means square error (RMSE) value of 0.5685 and Q2 of 0.5287. Details of predicted activity of experimental compounds and observed activity by the predictive model are given in Table 4. The inhibitory prowess (pIC50) of the hit and reference compounds are listed in Table 1. Lopinavir had the most satisfactory inhibitory attribute against the protein target with pIC50 value of 5.165 µM. Among the lead compounds, STOCK1N-84615, STOCK1N-92347 and STOCK1N-94719 exhibited relatively moderate pIC50 values.
Table 8.
Details of AutoQSAR predicted activities of investigated compounds compared with the observed activities
| s/n | Pubchem ID | Set | Observed pIC50 | Predicted pIC50 | Residue Error |
|---|---|---|---|---|---|
| 1 | 2540 | Train | 5.5596 | 6.1479 | 0.5883 |
| 2 | 2541 | Test | 5.0246 | 5.5848 | 0.5602 |
| 3 | 2577 | Test | 5.7399 | 5.3475 | − 0.3924 |
| 4 | 2719 | Train | 5.1451 | 5.5285 | 0.3834 |
| 5 | 3108 | Train | 6.2596 | 6.2635 | 0.0039 |
| 6 | 3117 | Train | 5.3307 | 5.1813 | − 0.1494 |
| 7 | 3194 | Test | 6.1739 | 5.1582 | − 1.0157 |
| 8 | 4594 | Test | 4.6786 | 4.5755 | − 0.1031 |
| 9 | 5320 | Train | 4.3010 | 4.4479 | 0.1469 |
| 10 | 5770 | Test | 5.4685 | 5.1246 | − 0.3439 |
| 11 | 10,177 | Test | 3.5331 | 3.9240 | 0.3909 |
| 12 | 10,207 | Train | 3.8794 | 4.1595 | 0.2801 |
| 13 | 10,215 | Train | 3.5229 | 3.9240 | 0.4011 |
| 14 | 56,704 | Train | 4.6922 | 5.0189 | 0.3267 |
| 15 | 72,172 | Train | 5.0491 | 5.0810 | 0.0319 |
| 16 | 72,281 | Train | 4.2218 | 4.0822 | − 0.1396 |
| 17 | 92,769 | Train | 3.9508 | 4.1210 | 0.1702 |
| 18 | 148,192 | Train | 5.1232 | 4.2480 | − 0.8752 |
| 19 | 219,104 | Test | 4.6698 | 4.6437 | − 0.0261 |
| 20 | 222,284 | Train | 3.0393 | 3.7310 | 0.6917 |
| 21 | 479,503 | Train | 4.8027 | 4.3966 | − 0.4061 |
| 22 | 644,241 | Train | 2.4100 | 2.5835 | 0.1735 |
| 23 | 3,000,706 | Train | 6.0706 | 5.2699 | − 0.8007 |
| 24 | 5,281,708 | Test | 3.9788 | 4.8750 | 0.8962 |
| 25 | 5,362,440 | Train | 4.3652 | 4.3110 | − 0.0542 |
| 26 | 5,479,529 | Train | 2.0090 | 2.2547 | 0.2457 |
| 27 | 11,313,622 | Train | 5.8097 | 6.1753 | 0.3656 |
| 28 | 16,129,778 | Train | 7.0000 | 5.7891 | − 1.2109 |
| 29 | 17,755,052 | Train | 5.7351 | 6.4158 | 0.6807 |
| 30 | 23,663,996 | Train | 5.1372 | 4.6559 | − 0.4813 |
| 31 | 23,682,211 | Train | 3.9400 | 4.0372 | 0.0972 |
| 32 | 54,675,779 | Train | 4.8196 | 4.1067 | − 0.7129 |
| 33 | 135,413,534 | Train | 4.7780 | 5.0215 | 0.2435 |
Based on the docking score results and ADME properties, STOCK1N-84615 is the ideal drug candidate against SARS-CoV-2 3CLPro, hence SARS-CoV-2 3CLPro-STOCK1N-84615 complex was subjected to MD simulation for 50 ns. The root mean square deviation (RMSD), which provides information about a protein in respect to its backbone structure, showed that the SARS-CoV-2 3CLPro-STOCK1N-84615 complex was stable throughout the duration of the simulation. The protein in Apo state also achieved stabilization all through the duration of the simulation. The root mean square fluctuation (RMSF) gives a detail information about the dynamic behavior of the amino residues of the protein in both Apo state and bound state. There was atomic positional fluctuation of backbone residues of the protein both in bound state and unbound state, which depicts high degree of flexibility in backbone residues. However, less fluctuation of the backbone residues were observed between 600 and 1300, and between 2200 and 2600 in both unbound and bound state of the protein. Sequel to the dynamics of STOCKIN-98687 with SARS-CoV-2 3CLPro, additional efforts are needed to be made pre-clinically and/or clinically in order to evaluate their therapeutic claim in combating SARS-CoV-2Sequel to the dynamics of STOCKIN-98687 with SARS-CoV-2 3CLPro, additional efforts are needed to be made pre-clinically and/or clinically in order to evaluate their therapeutic claim in combating SARS-CoV-2.
Conclusions
In the present study, the natural product STOCK1N-98687 appears to possess superior criteria than the other compounds screened from a natural product library (IBS Database) as a potential inhibitor of 3CLpro of SARS-CoV-2: it has high Glide docking score, induced fit docking score as well as favourable calculated binding free energy score and good predicted pIC50. MD simulation confirmed the stability of the 3CLpro-STOCKIN-98687 complex. In view of this, supplementary in vitro and in vivo biological research are obligatory to confirm its therapeutic inhibitory effects against SARS-CoV-2 3CLpro in combating the viral infection.
Abbreviations
- SARS-CoV-2
Severe acute respiratory syndrome coronavirus-2
- COVID-19
Coronavirus disease 2019
- PLpro
Papain-like protease
- 3CLpro
3-Chymotrypsin-like protease
- PP1ab
Polyprotein 1ab
- PDB ID
Protein databank identification number
- XP docking
Extra precision docking
- IFD
Induced fit docking
- HTVS
High throughput virtual screening
- MM-GBSA
Molecular mechanics/generalized born surface area
- ADME
Absorption, distribution, metabolism, and excretion
- QSAR
Quantitative structure activity relationship
- RMSD
Root mean square deviation
- ROF
Lipinski rule of five
- QPlogKhsa
Human serum albumin binding
- QPPMDCK
Predicted apparent MDCK cell permeability
- RMSE
Root means square error
- IBS Database
Inter bio screen database
Author contribution
OOE: conceptualization, methodology and reviewing; OI: writing of original draft, reviewing and editing, methodology; ORM: conceptualization, methodology; CDF: writing of original draft. All authors have read and approved the manuscript.
Data availability
We declare that all the data generated are included in this study.
Declarations
Competing interest
The authors declare that they have no completing interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aanouz I, Belhassan A, El Khatabi K, Lakhlifi T, El Idrissi M, Bouachrine M. Medicinal plants as inhibitors of COVID-19: computational investigations. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.17587901-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bustany HA, Ercan S, Ince E, et al. Investigation of angucycline compounds as potential drug candidates against SARS Cov-2 main protease using docking and molecular dynamic approaches. Mol Divers. 2021 doi: 10.1007/s11030-021-10219-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Báez-Santos YM, St. John SE, Mesecar AD. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boopathi S, Poma AB, Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J Biomol Struct Dynam. 2020 doi: 10.1080/07391102.2020.1758788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YW, Yiu CB, Wong K (2020) Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like Protease (3CL (pro)) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. FL000 Res 23:129. 10.12688/f1000research.22457.1 [DOI] [PMC free article] [PubMed]
- Daina A, Michielin O, Zoete V. Swiss ADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira MT, Katekawa E. On the virtues of automated QSAR the new kid on the block. Future Med Chem. 2017 doi: 10.4155/FMC-2017-0170. [DOI] [PubMed] [Google Scholar]
- Dixon SL, Smondyrev AM, Rao SN. PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem Biol Drug Des. 2006;67:370–372. doi: 10.1111/j.1747-0285.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- Du A, Zheng R, Disoma C, Li S, et al. Epigallocatechin-3-gallate, an active ingredient of traditional Chinese medicines, inhibits the 3CLpro activity of SARS-CoV-2. Int J Biol Macromol. 2021;176:1–12. doi: 10.1016/j.ijbiomac.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elekofehinti OO, Aladenika YV, Alli-Smith YR, Ejelonu OC, Lawal AO. Molecular modeling, dynamics simulation and characterization of human inositol hexakisphosphate kinase 1 (IP6K1) related to diabetes. J Appl Sci Enviro Managt. 2013;23:461–467. [Google Scholar]
- Elekofehinti OO, Ejelonu OC, Kamdem JP. Discovery of potential visfatin activators using in silico docking and ADME predictions as therapy for type 2 diabetes. Beni-Suef Univ J Basic Appl Sci. 2018;7:241–249. doi: 10.1016/j.bjbas.2018.02.007. [DOI] [Google Scholar]
- Elekofehinti OO, Iwaloye O, Famusiwa CD, Akinseye O, Rocha BT. Identification of main protease of coronavirus SARS-CoV-2 (Mpro) inhibitors from Melissa officinalis”. Curr Drug Discov Technol. 2020;2020(17):1. doi: 10.2174/157016381799920091810370. [DOI] [PubMed] [Google Scholar]
- Elekofehinti OO, Iwaloye O, Josiah SS, et al. Molecular docking studies, molecular dynamics and ADME/tox reveal therapeutic potentials of STOCK1N-69160 against papain-like protease of SARS-CoV-2. Mol Divers. 2020 doi: 10.1007/s11030-020-10151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfiky AA, Azzam EB. Novel guanosine derivatives against MERS CoV polymerase: an in silico perspective. J Biomol Struct Dyn. 2020;23:1–9. doi: 10.1080/07391102.2020.1758789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmezayen AD, Al-Obaidi A, Şahin AT, Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J Biomol Struct Dyn. 2020;23:1–13. doi: 10.1080/07391102.2020.1798812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakandari H, Nabi-Afjadi M, Fakkari-Afjadi J, Farahmandian N, Miresmaeili SM, Bahreini E. A comprehensive review of COVID-19 characteristics. Biological Procedures Online. 2020;22:1–10. doi: 10.1186/s12575-020-00128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MK, Vemula S, Donde R, Gouda G, Behera L, Vadde R. In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1751300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan A, Paray BA, Hussain A, Qadir FA, Attar F, Aziz FM, et al. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J Biomol Struct Dynam. 2020 doi: 10.1080/07391102.2020.1754293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horby P, Lim WS, Emberson JR. Dexamethasone in hospitalized patients with Covid-19 preliminary report. N Engl J Med. 2020 doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaloye O, Elekofehinti OO, Oluwarotimi EA, et al. Insight into glycogen synthase kinase-3β inhibitory activity of phyto-constituents from Melissa officinalis: in silico studies. Silico Pharmacol. 2020;8:2. doi: 10.1007/s40203-020-00054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaloye O, Elekofehinti OO, Babatomiwa K, Fadipe TM, Akinjiyan MO, et al. Discovery of TCM derived compounds as wild type and mutant Plasmodium falciparum dihydrofolate reductase inhibitors: induced fit docking and ADME studies. Curr Drug Discov Technol. 2020 doi: 10.2174/1570163817999200729122753. [DOI] [PubMed] [Google Scholar]
- Iwaloye O, Elekofehinti OO, Oluwarotimi EA, Babatomiwa K, Momoh IA. In silico molecular studies of natural compounds as possible anti-Alzheimer’s agents: ligand-based design. Net Model Anal Health Inform Bioinform. 2020;9:54. doi: 10.1007/s13721-020-00262-7. [DOI] [Google Scholar]
- Jin Z, Du X, Xu Y. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Jo S, Kim S, Shin DH. Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem. 2020;35:145–151. doi: 10.1080/14756366.2019.1690480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T, Adegboyega A, Iwaloye O, Eseola O, Plass W, Afolabi B, Rotimi D, Ahmed E, Albrakati A, Batiha G, Adeyemi O. Computational study of the therapeutic potentials of a new series of imidazole derivatives against SARS-CoV-2. J Pharmacol Sci. 2021 doi: 10.1016/j.jphs.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Zia K, Ashraf S, Uddin R, Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J Biomol Struct Dynam. 2020 doi: 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- Li JY, You Z, Wang Q, Zhou ZJ, Qiu Y, Luo R, et al. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. 2020;22:80–85. doi: 10.1016/j.micinf.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Li X, Huang YL, Wu Y, Liu R, Zhou L, Lin Y, Wu D, et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc Natl Acad Sci. 2020;117:27381–27387. doi: 10.1073/pnas.2010470117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Mody V, Ho J, Wills S, et al. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun Biol. 2021;4:93. doi: 10.1038/s42003-020-01577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan N, Sakthivel R, Velmurugan D, Gromiha MM. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 Protease against COVID-19. J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- Oscanoa TJ, Romero-Ortuno R, Carvajal J. a pharmacological perspective of chloroquine in SARS-CoV-2 infection: an old drug for the fight against a new coronavirus? Antimicrob Agents. 2020;56:106078. doi: 10.1016/j.ijantimicag.2020.106078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paasche A, Zipper A, Schäfer S, Ziebuhr J, Schirmeister T. Engels B (2014) Evidence for substrate binding-induced zwitterion formation in the catalytic Cys-His dyad of the SARS-CoV main protease. Biochem. 2014;53(37):5930–5946. doi: 10.1186/s12885-018-4050-1. [DOI] [PubMed] [Google Scholar]
- Pant S, Singh M, Ravichandiran V, Murty U, Srivastava HK. Peptide-like and small-molecule inhibitors against Covid-19. J Biomol Struct Dynam. 2020;123:1–10. doi: 10.1080/07391102.2020.1757510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlakpinar H, Polat S, Acet HA. Pharmacological agents under investigation in the treatment of coronavirus disease 2019 and the importance of melatonin. Fundam Clin Pharmacol. 2019 doi: 10.1111/fcp.12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar MT, Alqahtani SM, Alamri MA, Chen LL. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. 2020;10:313–319. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampogu S, Baek A, Zeb A, Lee KW. Exploration for novel inhibitors showing back-to-front approach against VEGFR-2 kinase domain (4AG8) employing molecular docking mechanism and molecular dynamics simulations. BMC Cancer. 2018;18:264. doi: 10.1186/s12885-018-4050-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Release S. 2: Qikprop 2018 Schrödinger. LLC, New York, NY 2018.
- Salam NK, Nuti R, Sherman W. Novel method for generating structure-based pharmacophore using energetic analysis. J Chem Inf Model. 2009;49:2356–2368. doi: 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- Sarma P, Sekhar N, Prajapat M, Avti P, Kaur H, Kumar S, et al. Insilicohomology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain) J Biomol Struct Dyn. 2020 doi: 10.1080/07391102.2020.1753580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj C, Tripathi SK, Reddy KK, Singh SK. Tool development for prediction of pIC50 values from the IC50 values-A pIC50 value calculator. Curr Trends Biotech Pharmacy. 2011;5:1104–1109. [Google Scholar]
- Sharma A, Tiwari S, Deb MK, Marty JL. Severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. Int J Antimicrob Agents. 2020;56:106054. doi: 10.1016/j.ijantimicag.2020.106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Shyu C, Brown CJ, Ytreberg FM. Computational study of evolutionary selection pressure on rainbow trout estrogen receptors. PLoS ONE. 2010;5(3):e9392. doi: 10.1371/journal.pone.0009392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuley A. The taxonomy of covalent inhibitors. Biochem. 2018;57:3326–3337. doi: 10.1021/acs.biochem.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2019;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei-Chung C, Meng-Shiuan H, Yun-Ti C, et al. Repurposing existing drugs: identification of SARS-CoV-2 3C-like protease inhibitors. J Enzyme Inhib Med Chem. 2021;36:147–153. doi: 10.1080/14756366.2020.1850710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, Meng J, Zhu Z, Zhang Z, Wang J, Sheng J. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe. 2020 doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China. New Engl J Med. 2019;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We declare that all the data generated are included in this study.