

Abstract
The outbreak of the triple mutant strain of severe acute respiratory syndrome coronavirus-2 (SARS-COV-2) was more virulent and pathogenic than its original strain. The viral triple mutant strain of SARS-COV-2 is extremely adaptive and increases penetrability into the host. The triple mutant viral strain was first reported in Brazil and South Africa and then communicated to different countries responsible for the second wave of the coronavirus disease (COVID-19) global pandemic with a high mortality rate. The reported genomic mutations are responsible for the alterations in the viral functional and structural proteins, causing the ineffectiveness of the existing antiviral therapy targeting these proteins. Thus, in current research, molecular docking simulation-based virtual screening of a ligand library consisting of FDA-approved existing drugs followed by molecular dynamics simulation-based validation of leads was performed to develop a potent inhibitor molecule for the triple mutant viral strain SARS-CoV-2. Based on the safety profile, tamibarotene was selected as a safe and effective drug candidate for developing therapy against the triple mutant viral spike protein of SARS-CoV-2.
Keywords: Drug repurposing, Drug repositioning, SARS-CoV-2, COVID-19, Corona, Antiviral, Tamibarotene, Resistance, Drug-resistance, Delta variant
Graphical abstract

1. Introduction
Eighteen months before the first confirmed case of Corona Virus Disease (COVID-19) was reported in December 2019, in Wuhan, the Republic of China. The highly contagious and communicable nature of this virus resulted in its vigorous spread among humans, affecting a large population in almost every country around the globe [1]. The lack of any suitable therapeutic regimen and the uncontrolled communication of the COVID-19 disease across international borders led to the declaration of the COVID-19 as a global pandemic by the World Health Organization (WHO) in April 2020. As of July 31, 2021, more than 199 million people were infected by this contagious virus, with more than 4.3 million deaths and around 1.5 million active cases worldwide (https://www.worldometers.info/coronavirus/). The US, India, and Brazil are the most badly affected countries with a maximum number of reported cases as well as deaths [2].
Lack of effective therapy for the COVID-19 pandemic forced social distancing, self-isolation, travel restrictions, and the implementation of either full or partial lockdown to cease human-to-human communication of the pathogenic disease. These situations for such a long duration result in the closure of organizations, communities, and businesses, which has directly or indirectly impacted the livelihood of humans and the global economy. The forced uncoordinated lockdown by the governing authorities has also disrupted the supply chain, leading to the closure of the manufacturing units. This unfavorable situation led to the global recession, causing the loss of millions of jobs and leading to starvation. The worst effect of this global pandemic has been observed in developing as well as underprivileged countries with a dense population, like India, Africa and Brazil [3].
The SARS-CoV-2 infection in humans ranges from mild asymptomatic infection to severe infection leading to respiratory failure associated with multiple organ failure leading to death. High expression of Angiotensin-Converting Enzyme-2 (ACE-2) in the epithelial tissues in the alveolar cavity of the lungs, heart, kidneys, and bladder leads to more complications in these organs because of the COVID-19 infection [4,5].
Coronaviruses are a group of pathogenic viruses having the largest single-stranded RNA (ssRNA) genome, ranging from 24 to 32 kb in length. The viral genome sequence encodes important structural and functional proteins like replicases, proteases, ribonucleases, and spike proteins. With more than 82% genome sequence similarity and more than 90% of essential protein sequence similarity between the SARS-CoV and MERS-CoV, the novel SARS-CoV-2 is found to be more pathogenic and virulent [5].
SARS-CoV-2 is a spherical shape covered with an envelope having spike protein embedded all around it. Spike protein (S), envelop protein (E), nucleocapsid protein (C), and membrane protein (M) are the main characteristic structural proteins of SARS-CoV-2 [6]. The viral life cycle within the host is comprised of a five-step process, including attachment to the host cell, penetration into the cell through endocytosis, multiplication within the nucleus, biosynthesis of the viral proteins, maturation of new viral particles, and release to infect the surrounding cells. Based on the structural similarity, the mechanism of viral pathogenicity has been revealed to develop effective therapeutic strategies.
The spike proteins of SARS-CoV-2 are structurally different from those of SARS-CoV. Spike protein is comprised of a transmembrane trimetric glycoprotein bulging out from the viral surface. The viral spike proteins have two functional subunits, S1 is involved in the binding to the host cell, while S2 has a role in the fusion of the viral and cellular membrane of the host cell. Thus, the interactive role in complexation with the host's ACE-2 and the viral entry is dependent upon this step during cellular entry by the process of endocytosis.
Like other viruses, the SARS-CoV-2 also has one of the most dynamic natures, resulting in the continuous adaptability of mutational changes resulting in the development of resistance to the existing therapeutic options, becoming more sustainable, and an increase in virulence as well as pathogenicity. Because of this dynamic and mutational nature, the viral pathogens become more and more adaptable to their host cells, resulting in more complex pathogenesis, causing increased mortality and morbidity in humans [7]. These mutational changes in the genomic sequence of the pathogen resulting in alterations in most of the viral structural and functional proteins which are supposed to be important drug targets. Structural change in the viral drug targets results in the development of pathogenic resistance to existing as well as upcoming therapies [8].
The 20A.EU1 variant of the SARS-CoV-2 was first reported in Spain, which is characterized by a mutation A222V on the viral spike protein [9]. The B.1.1.7 variant of the original virus was reported in the UK in December 2020 and was characterized by N501Y in the spike protein [9]. It has been found that this wild type of viral strain is 50% more transmissible and binds more tightly to the cellular ACE-2 receptor as compared to the original form. Parallelly, a new variant B.1.351 was reported in South Africa which has been characterized by notable mutations E484K, N501Y, and K417N [10]. These genetic alterations are responsible for the viral evasion of the immune system as well as vaccines. In January 2021, a new viral strain B.1.1.28 was reported in Brazil with a notable mutation E484K similar to that of the UK viral strain B.1.351 [9]. It has been reported that the antibodies generated by the host for the original virus strain were not completely neutralized by the mutated wild-type viral strains [10]. This indicates that the so-far-developed vaccines for the original viral strain will become ineffective after a period because of these mutations.
Drug repurposing is an economical approach to identifying the alternative pharmacological role of existing approved drugs. Computational screening of approved drugs against a specific pathophysiological target involved in a disease condition can be highly beneficial in the identification of potential leads to developing newer therapeutics against a new disease in a shorter time [4,11]. Computational drug repurposing saves a lot of time as we only have to consider the binding affinity of the ligand molecules against a specific receptor as the approved drugs are already established for their pharmacokinetics and toxicity profile [4,8].
Thus, in the current study, a computational drug repurposing approach was utilized to identify potential inhibitors of the triple mutant viral spike protein of the SARS-CoV-2 for developing a novel therapy to control the COVID-19 global pandemic.
2. Material and methods
-
(i)
Macromolecular Antiviral Target
The spike protein of SARS-CoV-2 is found to have a vital role in the viral entry into the host cell by interacting with the human ACE-2 receptor. The analysis of the viral entry revealed the structural and functional involvement of spike protein, ACE-2, as well as transmembrane protease serine-2 (TMPRSS2) protein in the viral pathogenicity [12]. The competitive inhibition of the spike protein can interfere with the complexation of the host's ACE-2 and TMPRSS2 and terminate the viral entry into the host cell, avoiding the chances of viral infection.
The structural model of the viral spike protein was attained from the RCSB protein data bank (PDB i-6M0J). The procured structure model is a macromolecular complex comprised of the S1 subunit of the viral spike protein in complex with the host's ACE-2 receptor with bound N-acetyl glucosamine (NAG) [13].
-
(ii)
Molecular Docking Simulation
The viral spike protein is separated from the macromolecular complex by deleting chain A, i.e., the ACE-2 enzyme of the host. Further, the separated S1 unit of the viral spike protein was prepared for molecular docking simulation by removal of non-interacting redundant water molecules, the addition of hydrogen atoms, and computing Gasteiger charges for each of the atoms followed by assigning an Autodock4 (AD4) type to each atom of the macromolecule [14]. The Autodock tools perform the docking of the macromolecular viral target against the small chemical molecules at a molecular level by simulating the temperature and pressure conditions of the human body to identify their best possible binding conformation to reveal the strength of association between them [4,15,16].
The prepared macromolecular receptor is further utilized for the generation of grid parameters to prepare an imaginary grid box by covering each of the interacting residues involved in the binding interactions with the host's ACE-2 receptor [17,18]. This grid box was utilized by the Autogrid module of the Autodock suite to generate map files for different atoms required to perform molecular docking simulation [15]. The validation of the parameters utilized in the Autodock based docking protocol was performed by docking it with the already reported viral spike protein inhibitor K22. It has been already reported that the viral spike protein inhibitors interfere with the process of complexation of the S1 subunit of the viral spike protein with the host's ACE-2 receptor for entering into the host cell [13].
-
(iii)
Mutations
The wild type of mutant variant of the SARS-CoV-2 was obtained from various geographical locations, like the UK, Spain, Brazil, South Africa, and India. The sequential analysis of the mutant variant has revealed that the important amino acids of the S1 subunit of the viral spike protein which are involved in the binding interactions with the host's TMPRSS2 as well as ACE-2 receptors were found to be mutated. Important mutations reported in most of the virulent types of the wild variants were found to be E484K, N501Y, and K417N [2,10]. These mutations play a major role in the development of pathogenic drug resistance against the existing viral entry inhibitors as these residues are found to have an important interacting role with the inhibitor molecules. Thus, to assure the broad spectrum of action of the designed viral spike protein inhibitors against these wild variants, mutations were created in the original spike protein model of SARS-CoV-2 by using Swiss PDB Viewer (SPDBV) followed by energy minimization by using the AMBER force field. This energy minimized structure model of the wild-type triple mutant variant of the SARS-CoV-2 spike protein was further used to develop potential viral entry inhibitors [8].
-
(iv)
Virtual Screening
The mutated spike protein of SARS-CoV-2 was virtually screened with a ligand library containing 2890 FDA-approved drugs obtained from the ZINC database [19] for exploring potential lead molecules as viral entry inhibitors. The potential lead molecules were shortlisted based upon their observed binding energy and were further evaluated for their structure-activity relationship as well as their safety profile based upon their established pharmacological role, and it was assured that their existing pharmacological action does not interfere with the normal physiological process, resulting in any undesirable effects on the human body during their administration as an antiviral agent to inhibit SARS-CoV-2 virus for the treatment of COVID19. The entry inhibitors are expected to interfere with the viral entry mechanism into the host cell and are proving to be potential antiviral agents against SARS-CoV-2 [16,20,21]. The detailed framework applied in the current research for computational drug repurposing of Tamibarotene against triple mutant viral Spike protein of SARS-CoV-2 was represented in Fig. 1 .
-
(v)
Molecular Dynamic Simulation
Fig. 1.

A schematic representation of the developed framework for computational drug repurposing against the triple mutant spike protein of SARS-CoV-2 to control the severity of the viral pathogenicity in humans via the development of drug resistance.
Based on their safety profile, these shortlisted lead molecules were further screened to shortlist Tamibarotene, Itraconazole, and Irinotecan based on their safety profile, non-involvement in the normal physiological processes of the human body, as well as their high docking score to proceed further to molecular dynamic simulation to observe the stability of their macromolecular complex conformation concerning time. Molecular dynamics simulation of a shorter duration, i.e. 10 ns, was performed for all three shortlisted leads. Based upon the observed results, tamibarotene was found to be the most stabilized molecule against the triple mutant viral spike protein of the SARS-CoV-2, which was further confirmed by amplified simulations for longer durations of 100 ns. The molecular dynamics simulation of the protein-ligand macromolecular complex was performed at a constant temperature condition of 300K for 100 ns by using the Desmond module of Schrodinger [22,23]. The molecular dynamics simulations of the macromolecular complex were performed by solvating them in an explicit water box of size 10 Å by using the OPLS3e force field [24] to model the macromolecular complex of protein and ligand. The water molecules were implied according to the single point charge (SPC) model [25,26]. The use of the OPLS3e force field for macromolecular complexes having organic ligands bound to proteins and the SPC water model was already reported for optimum reproducible results [27]. The macromolecular complex was neutralized by the addition of the ions followed by their energy minimization. The energy minimized macromolecular complex was maintained at a temperature of 300 K by applying the Nose–Hoover thermostatic algorithm [28] while constant pressure was maintained throughout the simulations with the help of the Martina–Tobias–Klein method [29]. The NPT ensemble MD simulation was carried out for 100 ns and the long-range electrostatic interactions between the ligand and the macromolecule were calculated by using the particle-mesh Ewald (PME) [30] method by using 0.8 Å of grid spacing and a cutoff radius of 9.0 Å for Coulomb interactions. The detailed binding interactions of the ligand with the macromolecular viral spike protein were analyzed by using the simulation interaction diagram tool of the Desmond 2019-4 package [22,23].
The Root Mean Square Deviation (RMSD) of the atoms of both receptors as well as the complexed ligand was calculated concerning its reference frame to observe the atomic displacement for a given time frame during the complexation process. The Root Mean Square Fluctuation (RMSF) for the macromolecular residues was calculated during the whole simulation process regarding their initial state present in the crystallized structure. The macromolecular secondary structure elements (SSE) like alpha-helices and beta-strands were monitored throughout the simulation process to generate a plot of SSE distribution by the residue index. The macromolecular interaction of the ligand within its active binding site was monitored throughout the simulation process into four main categories: hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridges. Ligand properties were evaluated by observing their RMSD, radius of gyration (rGyR), molecular surface area (MolSA), solvent accessible surface area (SASA), and polar surface area (PSA). The RMSD value of the ligand molecule was calculated during the whole timeframe of the simulation process concerning its initial frame. The extended length of the ligand equivalent to its principal moment of inertia was measured in terms of rGyr MolSA equivalent to the van der Waals surface area was calculated with a 1.4 Å probe radius, whereas PSA was calculated by considering the contribution of oxygen and nitrogen atoms.
3. Results
-
(i)
Macromolecular Antiviral Target
The structure model of the viral spike protein was revealed by using the X-ray diffraction technique at a resolution of 2.45 Å. The structure model consists of the host's ACE-2, consisting of 603 amino acids, complexed with the viral spike protein S1, consisting of 229 amino acids. Only the S1 subunit of the macromolecular complex is utilized for the development of viral entry inhibitors as it is supposed that the competitive inhibition of the viral S1 subunit terminates its interaction with the host's ACE-2 receptor, failing the viral entry into the host cell. The three-dimensional structural representation of the S1 subunit of the viral spike protein is shown in Fig. 2 .
-
(ii)
Molecular Docking Simulation
Fig. 2.

Crystal structure of the S1 subunit of the Spike Protein: The three-dimensional structure of the S1 subunit of the Spike Protein of SARS-CoV-2 was obtained from the RCSB protein data bank (PDB ID-6M0J) and processed for molecular docking simulation by the addition of polar hydrogens, Gasteiger charge, and removal of redundant water molecules by Autodock software.
All the 80 redundant water molecules and chain A of the macromolecular complex were removed with the help of the software UCSF Chimera to retain the S1 subunit of the viral spike protein. All the non-standard ligands associated with this S1 subunit were removed to allow the nascent viral receptor molecule to proceed further for performing molecular docking simulation. The nascent S1 subunit of the viral spike protein was incorporated with a charge value of 1.99 as per Gasteiger computation and saved in the default Autodock format after assigning the AD4 atom type. The detailed nascent structure of the S1 subunit of the viral spike protein has been represented in Fig. 2 and the coordinates used for the preparation of the grid box are tabulated in Table 1 . The molecular docking results of the reported viral spike protein inhibitor K22 against the prepared macromolecular target are given in Table 2 .
-
(iii)
Mutations
Table 1.
The coordinates of grid box for the viral S1 spike protein of SARS-CoV-2.
| Proteins | x-D | y-D | z-D | Spacing (Ả) | x center | y center | z center |
|---|---|---|---|---|---|---|---|
| 6M0J | 48 | 48 | 48 | 0.492 | −33.692 | 28.069 | 7.071 |
Table 2.
Molecular docking results of ligand K22 with the viral S1 spike protein of SARS-CoV-2.
| Proteins | Ligand | Interacting residues | Binding energy (kcal/mol) | Binding affinity (μM) |
|---|---|---|---|---|
| 6M0J (Non-mutated) | K22 | Asn501, Gly496, Tyr495 | −6.33 | 22.83 |
| 6M0J (Mutated) | K22 | Tyr449, Leu455, Tyr453 | −3.82 | 1590 |
The involvement of the mutated residues of the viral spike protein was validated by comparing the docking results of the spike protein inhibitor molecule against the mutated target protein concerning its non-mutant variant. The involvement of all the three residues, i.e. Lysine-417, Glutamic acid-484, and Aspargine-501. The poor docking score of the mutated spike protein against the established viral protein signifies the involvement of the macromolecular residues 417, 484, and 501. The binding energy of the ligand K22 against the mutated viral spike protein is given in Table 2. The two-dimensional binding interactions of the mutated and non-mutated S1 subunit of the viral spike protein are represented in Fig. 3 .
-
(iv)
Virtual Screening
Fig. 3.

Two-dimensional Binding Interactions: The two-dimensional binding interaction of the reported inhibitor molecule against the mutated S1 subunit of the viral spike protein concerning its original non-mutant variant.
Out of a ligand library containing 2890 FDA-approved drugs, drug molecules having potential binding affinity for the mutated viral spike were selected since they had the lowest binding energy in the predefined range of −5 to −15 kcal/mol. The binding energy obtained for the top ten drug molecules after performing molecular docking simulation-based virtual screening is given in Table 3 .
-
(v)
Molecular Dynamics Simulation
Table 3.
The binding energy of the top ten drug molecules obtained after Autodock based virtual screening of a ligand library containing 2890 FDA-approved drugs against triple mutated viral S1 spike protein of SARS-CoV-2.
| S. No. | ZINC Id | Name | Chemical Structure | Binding Energy (kcal/mol) |
|---|---|---|---|---|
| 1. | ZINC00538415 | Tamibarotene |  |
−7.54 |
| 2 | ZINC00538550 | Ziprasidone |  |
−7.54 |
| 3 | ZINC03876186 | Estrone sulfate |  |
−7.32 |
| 4 | ZINC03830975 | Itraconazole |  |
−7.28 |
| 5 | ZINC01612996 | Irinotecan | 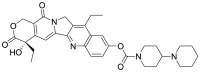 |
−7.22 |
| 6 | ZINC03881958 | Danazol | 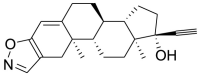 |
−7.13 |
| 7 | ZINC12503133 | Sulindac Sulfoxide | 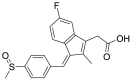 |
−7.13 |
| 8 | ZINC04081771 | Testolactone | 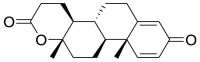 |
−7.06 |
| 9 | ZINC03861599 | Spironolactone | 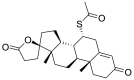 |
−7.05 |
| 10 | ZINC53683151 | Bromocriptine | 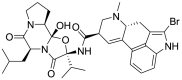 |
−6.98 |
The proposed inhibitor molecule tamibarotene was further validated by performing molecular dynamics simulation for 100 ns using the Desmond module of Schrodinger. The macromolecular receptor has 194 residues while the ligand atom has 26 heavy atoms out of a total of 50 atoms and 4 rotatable bonds. All the macromolecular frames were aligned over the reference frame of the backbone to calculate the RMSD of the macromolecular residues based upon the atomic selection. The RMSD analysis confirms the smooth conduction of the equilibrated simulation process based on the structural confirmation throughout the process. The ligand RMSD indicates the stability of the ligand concerning the macromolecular binding residues during the simulation process by aligning their heavy metals.
The macromolecular RMSD was found to be well within the 3 Å confirms that most of the macromolecular residues are not fluctuating from their initial position during the complexation with the ligand molecule. Despite some initial fluctuations up to 3.0 Å during the ligand adjustment into the macromolecular binding cavity, the ligand molecule has maintained its RMSD value well within 1 Å throughout the simulation process, indicating the potent binding of the ligand within the macromolecular binding cavity during the simulation process. The ligand tamibarotene, after reaching the active binding site of the triple mutant variant of the viral Spike protein of SARS-CoV-2, takes a couple of moves by continuous vibrations to achieve the most stabilized confirmation within the active binding site. Thus, the initial fluctuations in the RMSD value of the ligand in-between ~3-10 ns are because of these continuous vibrations while taking certain moves to achieve the most stabilized confirmation within the active binding site of the triple mutant variant of the viral spike protein. The RMSD of the protein and ligand observed during the molecular dynamic simulation during the 100 ns timeframe was shown in Fig. 4 . The RMSF value of the macromolecular residues was found well within the acceptable range of 3.6 Å. Few residues were found to fluctuate somewhat with an RMSF value of 3.6 Å otherwise, the majority of the residues were found to have fewer fluctuations with an average value of less than 1 Å. The RMSF of the S1 subunit of the viral spike protein and the complex ligand tamibarotene observed during the molecular dynamic simulation during the 100 ns timeframe was shown in Fig. 5 .
Fig. 4.

Root Mean Square Deviation: The RMSD of the protein and ligand observed during the molecular dynamic simulation during the 100 ns timeframe.
Fig. 5.

Root Mean Square Fluctuation: The RMSF of the S1 subunit of the viral spike protein and the complexed ligand tamibarotene were observed during the molecular dynamic simulation during the 100 ns timeframe.
The SSE analysis during the whole simulation process revealed that it has 4.83% of alpha helices and 21.61% of beta-sheets, making their total contributions of 26.44% of SSE, which may remain conserved during most of the simulation process. The macromolecular ligand interaction analysis during the whole 100 ns timeframe of the simulation process has revealed that Lys444 and Tyr449 are found to be interacting with the ligand for 80% of the simulation time by forming a hydrogen bond through a water bridge. While residues Arg346 and Asn450 are found to interact with the ligand for 45% of the simulation time through the formation of hydrogen bonds. During the whole timeframe of the simulation process, more than 6 macromolecular residues were found to be constantly interacting with the complex ligand. The detailed protein-ligand contacts observed during the whole timeframe of the 100 ns molecular dynamic simulation are represented in Fig. 6 . The RMSD value of the ligand was well within the value of 1.5 Å suggesting the minimum fluctuation of the ligand during the simulation process. rGyr value of the ligand was found to be within the range of 4.7–4.9 Å. No intramolecular hydrogen bonds were observed in the ligand during the simulation process. The MolSA of the ligand was found to have been within a range of 336–344 Å2 with an average value of 340 Å2 during the whole simulation process. The SASA of the ligand during the simulation process was found to be around 300 Å2 after some initial fluctuations. The PSA for the complex ligand was found to be within a range of 125–135 Å2 during the simulation process.
Fig. 6.

Protein-Ligand Contacts: The detailed protein-ligand contacts were observed during the whole timeframe of the 100 ns molecular dynamic simulation.
4. Discussion
The viruses are dynamically changing in nature and they generally transform themselves for adaptability as well as better survival. Like other viruses affecting mankind, SARS-CoV-2, responsible for the global pandemic situation of COVID-19, has also started to transform into multiple variants through mutational alterations for better adaptability and increased penetrability in the human host.
After 18 months after the origin of this dreadful virus, at the end of 2019, several mutations were reported in the viral genome, making it a diverse variant from the original type of variant reported initially. These point mutations in the viral genome result in alterations in both functional and structural proteins of the virus. The alterations in the structural protein sequence like spike protein result in a more convenient viral entry into the human host via stronger interaction with the human angiotensin-converting enzyme-2 (ACE-2), which is responsible for viral entry into the human host. Secondly, the alterations in the viral functional proteins which have biochemical involvement in viral physiology, like main protease (Mpro), RNA dependent RNA polymerase (RdRp), and RNA replicase enzyme result in the loss of efficacy of the drugs targeting these viral enzymes for their inhibition. The sequential changes in the viral functional proteins because of genomic mutations cause alterations in their active ligand-binding sites, affecting the binding interactions of the drug inhibitor molecules targeting these viral targets.
The mutated viral variants reported in the recent past are classified based upon the countries in which they were initially reported. The viral variants of SARS-CoV-2 20A.EU1 and B.1.177 were first reported in Spain and were characterized by an A222V point mutation in the viral spike protein. The viral variants of SARS-CoV-2 20I/501Y.V1, VOC202012/01, and B.1.1.7 were reported in the United Kingdom (UK) and were characterized by N501Y point mutations in the viral spike protein. The viral variants 20H/501Y.V2, B.1.351, B.1.1.28, VOC202101/02, and 20J/501Y.V3 were first reported in South Africa as well as Brazil and were characterized by some common mutations E484K, N501Y, and K417N in the viral spike protein. These viral mutations in the SARS-CoV-2 are found to be involved in the increased virulence and pathogenicity of the virus, resulting in the second wave of the COVID-19 pandemic with an increased number of mortalities among the infected patients.
Given the more powerful and pathogenic viral strain of the SARS-CoV-2, in the current study, the author modeled the triple mutant viral spike protein and compared the binding interaction of the existing spike inhibitor with the viral spike protein of the original variant of the SARS-CoV-2. Also, the triple mutant viral spike protein was utilized to perform molecular docking simulation-based in-silico screening of a ligand library consisting of 2890 FDA approved existing drug molecules followed by molecular dynamics simulation-based validation to identify potential inhibitors for the triple mutant viral spike protein of SARS-CoV-2. Tamibarotene is a synthetic analog of retinol and is used as a retinoic acid receptor agonist for the treatment of acute promyelocytic leukemia (APL) and its agonistic activity on the retinoid receptor is supposed to not cause any undesirable effects on the normal human individual. Ziprasidone is an antipsychotic drug used for the treatment of schizophrenia and bipolar disorder by antagonizing 5-HT2A and D2 receptors. Its administration as an antiviral drug can interfere with neurotransmission by antagonizing D2 and 5-HT receptors.
Estrone sulfate is estrogen used for hormonal replacement therapy for the maintenance of hypoestrogenism in females, and the administration of estrone sulfate will result in a hormonal imbalance in normal individuals. Danazol is a synthetic derivative of testosterone with gonadotropic and estrogenic antagonistic activity for the treatment of endometriosis and benign fibrocystic breast disorders. External administration of Danazol will increase the concentration of testosterone in human individuals, leading to a hormonal imbalance and the appearance of male sexual characteristics in women. Sulindac Sulfoxide is a prodrug of sulindac that inhibits the cyclic guanosine monophosphate-phosphodiesterase (cGMP-PDE) enzyme involved in the inhibition of the normal apoptotic process and is used as an anticancer agent. The administration of Sulindac Sulfoxide may inhibit the enzyme associated with the normal apoptotic process, leading to its disturbance, causing certain undesirable effects. Testolactone is a progesterone derivative involved in the treatment of advanced-stage breast cancers and its external administration will again lead to a hormonal imbalance in normal individuals. Spironolactone antagonizes the aldosterone receptor for its antihypertensive effects and its external administration will lead to a disturbance in the blood pressure of normal individuals. Bromocriptine is a D2 receptor agonist used for the treatment of early-stage Parkinsonism disease and its external administration as an antiviral drug may interfere with the normal signaling of the brain, causing certain types of neuropsychological disorders.
Itraconazole is an antifungal agent that targets fungal cytochrome P-450-dependent enzymes involved in the biosynthesis of ergosterol. Irinotecan is a semisynthetic derivative of camptothecin which inhibits the topoisomerase-I enzyme and is used for the treatment of metastatic colorectal cancer. Therefore, Itraconazole, Tamibarotene, and Irinotecan were supposed to not interfere with the normal well-being of humans as their pharmacological role is either associated with foreign pathogens or carcinogenic proteins, and it was assured that their existing pharmacological action does not interfere with the normal physiological process, resulting in any undesirable effects on the human body during its administration as an antiviral agent to inhibit SARS-CoV-2 virus for the treatment of COVID19.
5. Conclusion
The triple mutant variant of SARS-CoV-2 was first reported in South Africa and Brazil. It communicates among the individuals of different countries, like India, and is found to be more virulent and highly pathogenic. All the three mutations in the triple mutations in the strain of SARS-COV-2 make the virus more virulent by increasing its adaptability and vulnerability to the increase in host-pathogen interactions as well as the development of resistance to the existing antiviral drugs. The triple mutant variant of the SARS-CoV-2 was found to be highly communicable in comparison to its original strain and was responsible for an increased number of mortalities because of the ineffectiveness of the existing therapy against this viral strain. Thus, there is an urgent need to control the ever-increasing impact of the COVID19 pandemic around the globe. Therefore, in the current research, a computational framework has been developed for the repurposing of existing drugs with the intent of identifying a potential inhibitor of the spike protein of the triple mutant variant of the SARS-CoV-2 virus. The potential inhibitors of the viral spike protein of the triple mutant variant of the SARS-CoV-2 were further validated for their stability against the viral macromolecular target by performing molecular dynamic simulation for 100 ns. Based on their safety profile, highest docking score as well as stability against the macromolecular target, tamibarotene was selected as a safe and effective drug candidate for developing therapy against triple mutant viral spike protein of SARS-CoV-2.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2021.104748.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y., Xia J., Yu T., Zhang X., Zhang L. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naqvi A.A.T., Fatima K., Mohammad T., Fatima U., Singh I.K., Singh A., Atif S.M., Hariprasad G., Hasan G.M., Hassan M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2020;1866:165878. doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicola M., Alsafi Z., Sohrabi C., Kerwan A., Al-Jabir A., Iosifidis C., Agha M., Agha R. The socio-economic implications of the coronavirus pandemic (COVID-19): a review. Int. J. Surg. 2020;78:185–193. doi: 10.1016/j.ijsu.2020.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jain R., Mujwar S.J.S.C. Vol. 31. 2020. pp. 2487–2499. (Repurposing Metocurine as Main Protease Inhibitor to Develop Novel Antiviral Therapy for COVID-19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuki K., Fujiogi M., Koutsogiannaki S. COVID-19 pathophysiology: a review. Clin. Immunol. 2020;215:108427. doi: 10.1016/j.clim.2020.108427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj C.T.D., Kandaswamy D.K., Danduga R., Rajasabapathy R., James R.A. COVID-19: molecular pathophysiology, genetic evolution and prospective therapeutics-a review. Arch. Microbiol. 2021;203(5):2043–2057. doi: 10.1007/s00203-021-02183-z. 33555378. In this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallamkondu J., John A., Wani W.Y., Ramadevi S.P., Jella K.K., Reddy P.H., Kandimalla R. SARS-CoV-2 pathophysiology and assessment of coronaviruses in CNS diseases with a focus on therapeutic targets. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2020;1866:165889. doi: 10.1016/j.bbadis.2020.165889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mujwar S., Deshmukh R., Harwansh R.K., Gupta J.K., Gour A. Drug repurposing approach for developing novel therapy against mupirocin-resistant Staphylococcus aureus. Assay Drug Dev. Technol. 2019;17:298–309. doi: 10.1089/adt.2019.944. [DOI] [PubMed] [Google Scholar]
- 9.Hodcroft E.B., Zuber M., Nadeau S., Crawford K.H.D., Bloom J.D., Veesler D., Vaughan T.G., Comas I., Candelas F.G., Stadler T., Neher R.A. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature. 2021;595(7869):707–712. doi: 10.1038/s41586-021-03677-y. 34098568. In this issue. [DOI] [PubMed] [Google Scholar]
- 10.Wang P., Nair M.S., Liu L., Iketani S., Luo Y., Guo Y., Wang M., Yu J., Zhang B., Kwong P.D., Graham B.S., Mascola J.R., Chang J.Y., Yin M.T., Sobieszczyk M., Kyratsous C.A., Shapiro L., Sheng Z., Huang Y., Ho D.D. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593(7857):130–135. doi: 10.1038/s41586-021-03398-2. 33684923. In this issue. [DOI] [PubMed] [Google Scholar]
- 11.Mujwar S., Kumar V.J.A., technologies d.d. Computational drug repurposing approach to identify potential fatty acid-binding protein-4 inhibitors to develop novel antiobesity. Therapy. 2020;18:318–327. doi: 10.1089/adt.2020.976. [DOI] [PubMed] [Google Scholar]
- 12.Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 14.Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaushal S.K., Brijendra S., Mujwar S., Prakash B.S. Molecular Docking based analysis to elucidate the DNA Topoisomerase IIbeta as the potential target for the Ganoderic acid, A natural therapeutic agent in cancer therapy. Curr. Comput. Aided Drug Des. 2020;16(2):176–189. doi: 10.2174/1573409915666190820144759. 31429692. [DOI] [PubMed] [Google Scholar]
- 16.Mujwar S P.K. Prediction of Riboswitch as a potential drug target for infectious diseases: an Insilico case study of anthrax. Journal of Medical Imaging and Health Informatics. 2015;5:7–16. [Google Scholar]
- 17.Shah K., Mujwar S., Gupta J.K., Shrivastava S.K., Mishra P. Molecular docking and in silico cogitation validate mefenamic acid prodrugs as human cyclooxygenase-2 inhibitor. Assay Drug Dev. Technol. 2019;17:285–291. doi: 10.1089/adt.2019.943. [DOI] [PubMed] [Google Scholar]
- 18.Shah K., Mujwar S., Krishna G., Gupta J.K.J.A., Technologies D.D. Computational Design and Biological Depiction of Novel Naproxen Derivative. 2020;18:308–317. doi: 10.1089/adt.2020.977. [DOI] [PubMed] [Google Scholar]
- 19.Irwin J.J., Shoichet B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minaz N., Razdan R., Hammock B.D., Mujwar S., Goswami S.K. Impact of diabetes on male sexual function in streptozotocin-induced diabetic rats: protective role of soluble epoxide hydrolase inhibitor. Biomed. Pharmacother. 2019;115:108897. doi: 10.1016/j.biopha.2019.108897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mujwar S P.K. Prediction of riboswitch as a potential drug target and design of its optimal inhibitors for Mycobacterium tuberculosis. Int. J. Comput. Biol. Drug Des. 2015;8:326–347. [Google Scholar]
- 22.S.R.-D.M.D. System . Schrödinger; New York, NY: 2021. Maestro-Desmond Interoperability Tools; p. 2020. D. E. Shaw Research, New York, NY, 2021. [Google Scholar]
- 23.Bowers K.J., Chow D.E., Xu H., Dror R.O., Eastwood M.P., Gregersen B.A., Klepeis J.L., Kolossvary I., Moraes M.A., Sacerdoti F.D. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing. IEEE; 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters, SC'06. 43-43. [Google Scholar]
- 24.Roos K., Wu C., Damm W., Reboul M., Stevenson J.M., Lu C., Dahlgren M.K., Mondal S., Chen W., Wang L., Abel R., Friesner R.A., Harder E.D. OPLS3e: extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theor. Comput. 2019;15:1863–1874. doi: 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
- 25.Berendsen H., Grigera J., T.J.J.o.P.C. Straatsma The missing term in effective pair potentials. 1987;91:6269–6271. [Google Scholar]
- 26.Toukan K., Rahman A. Molecular-dynamics study of atomic motions in water. Phys. Rev. B Condens. Matter. 1985;31:2643–2648. doi: 10.1103/physrevb.31.2643. [DOI] [PubMed] [Google Scholar]
- 27.Gahtori J., Pant S., Srivastava H.K. Modeling antimalarial and antihuman African trypanosomiasis compounds: a ligand- and structure-based approaches. Mol. Divers. 2020;24:1107–1124. doi: 10.1007/s11030-019-10015-y. [DOI] [PubMed] [Google Scholar]
- 28.Posch H.A., Hoover W.G., Vesely F.J. Canonical dynamics of the Nose oscillator: stability, order, and chaos. Phys Rev A Gen Phys. 1986;33:4253–4265. doi: 10.1103/physreva.33.4253. [DOI] [PubMed] [Google Scholar]
- 29.Martyna G.J., Tobias D.J., M.L.J.T.J.o.c.p. Klein Constant pressure molecular dynamics algorithms. 1994;101:4177–4189. [Google Scholar]
- 30.H.G.J.T.J.o.c.p. Petersen Accuracy and efficiency of the particle mesh Ewald method. 1995;103:3668–3679. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.