

Abstract
Taxol® is widely regarded as amongst the most famed natural isolates ever discovered, and has been the subject of innumerable studies in both basic and applied science. Its documented success as an anticancer agent, coupled with early concerns over supply, stimulated a furious worldwide effort from chemists to provide a solution for its preparation through total synthesis. Those pioneering studies proved the feasibility of retrosynthetically-guided access to synthetic Taxol, albeit in minute quantities and with enormous effort. In practice, all medicinal chemistry efforts and eventual commercialization have relied upon natural- (plant material) or biosynthetically-derived (synthetic biology) supplies. Here we show how a complementary divergent synthetic approach that is holistically patterned off of biosynthetic machinery for terpene synthesis can be used to arrive at Taxol®.
GRAPHICAL ABSTRACT
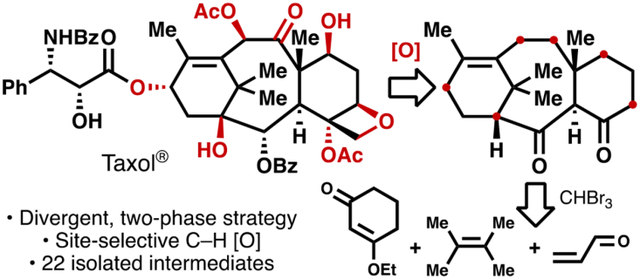
Introduction
Taxol® (1, Figure 1) stands as amongst the most famous terpene-based natural products to be used in a clinical setting.1 Registering >$9 billion in sales between 1993 and 20022 for use as an anticancer agent, Taxol is still prescribed today in generic form, and alternative formulations such as Abraxane® (albumin-bound) have generated >$5.5 billion in revenue as of January 2020.3 Numerous other taxanes have either been approved by the FDA, or are in various stages of clinical development.4–6 The storied history of Taxol in society began in ancient times and has been narrated in numerous books and reviews.1,7–10 After the molecule’s iconic structure was fully elucidated in 1971,11 and clarity regarding its unique mechanism of action was revealed in 1979,12 numbers of clinicians, formulations experts, biologists, and chemists contributed to its eventual use as a life-saving medicine in 1992.13 Taxol has stimulated research in nearly all branches of chemical science, including medicinal, natural product, engineering, chemical biology, biochemistry, and last but not least, synthetic organic. Historically, the clinical use of 1 relied exclusively on semisynthesis,14,15 both for the preparation of analogs (from 2), and as the initial commercial source before the advent of a synthetic biology-based route using plant-cell cultures.16,17
Figure 1.
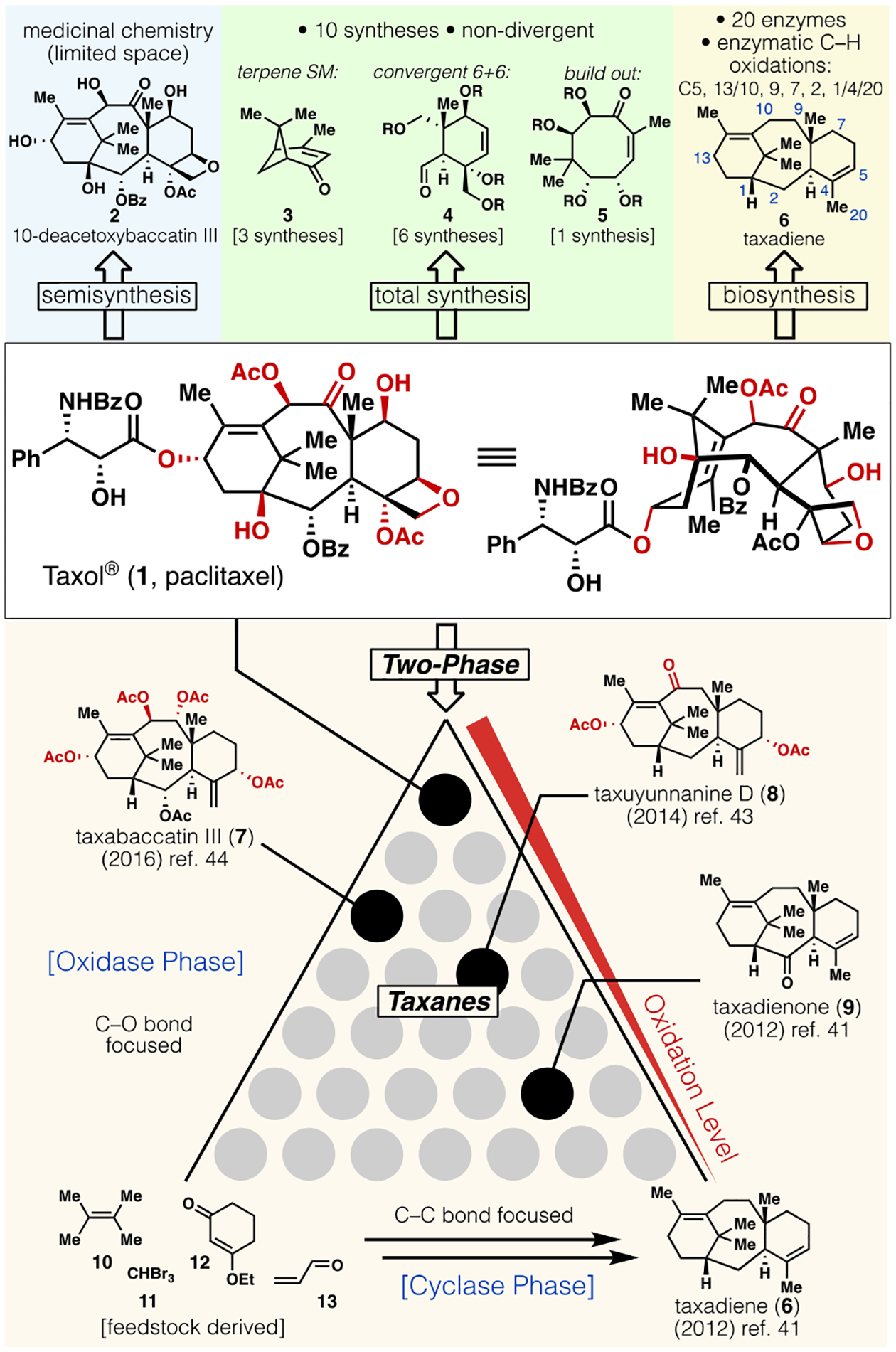
Approaches to the synthesis of Taxol (1): Semi-synthesis, canonical total synthesis, biosynthesis, and two-phase synthesis (this work).
Structurally, this heavily-oxygenated polycyclic diterpenoid presents a monumental challenge for total synthesis due to the dense orientation of functionality that render precise and predictable manipulations difficult. Any route to such a molecule must address the issue of a conformationally flexible central eight-membered ring system, which makes the neighboring reactivity of various functional groups unpredictable and substrate-dependent. In addition, the congested array of similarly reactive secondary alcohols creates a chemoselectivity puzzle of the highest magnitude. In the early 1990’s, at least 30 teams18 competed for finishing the synthesis first, and all completed syntheses, regardless of date, have been deservingly heralded as major (even “herculean”)1 accomplishments in the annals of organic chemistry. A relatively concise totally synthetic approach to 1 has been widely viewed as impossible, with one textbook (published in 2007) declaring that “…even an academic-type synthesis of 1 poses a major challenge, unlikely to be solved by a preparation under 30 or 40 steps in length”.9 This prediction had so far proven true, as ten distinct syntheses have been disclosed (from 1994 to 2015).19–31 Of those, seven have reported totally synthetic approaches and three have described formal syntheses that intercepted known intermediates. From a strategic perspective, three of these syntheses used naturally-occurring terpene starting materials (e.g. 3),19,20, 23,24,27 six of them fused two chemically synthesized six-membered ring systems to form the central eight-membered ring (e.g. 4),21,22, 25,27–31 and one built out by double annulation onto an already-constructed eight-membered ring (e.g. 5).26 Previous approaches to 1 showed that it was feasible to create such a molecule in the lab and paved a foundation for future approaches to taxanes. However, these pioneering studies remained within the realm of academic research as they did not form a basis for scalable chemical production of 1 or a viable medicinal chemistry campaign to explore taxane bioactivity. Obviously, the quantity of chemically synthesized 1 by these efforts (or ours) can never match the metric ton quantities prepared yearly through plant-derived sources. Aside from efficiency, another salient feature of taxane biosynthesis is its inherent divergency and the formation of hundreds of differentially oxidized analogs. It is this feature of biosynthesis that we sought to mimic in the two-phase retrosynthetic approach. Since prior approaches followed the canonical rules of retrosynthetic analysis (e.g. maximizing convergency) to establish the feasibility of accessing specifically 1, actual taxanes (compounds containing the full carbon skeleton) are not accessed in those syntheses until late in the sequence.
The goal of this study, initiated in 2007, was to answer to a fundamental question of whether an approach to terpene synthesis loosely patterned on the logic of biosynthesis, would enable a hypothetical medicinal chemistry approach to taxanes in the absence of a viable semisynthetic option. Herein we present the realization of our ultimate goal: a two-phase synthesis of Taxol®(1).
Nature’s route to complex terpenes is well-documented and enlists two conceptually different types of enzymes32,33. The first class of enzymes (cyclases) forges C–C bonds to generate complex hydrocarbon-based frameworks in what is known as a “cyclase-phase”. These minimally oxidized terpenes are then processed by a second set of enzymes that install C–O bonds in what is known as an “oxidase-phase”. This ingenious synthetic plan, despite being linear rather than convergent, benefits strategically from near-perfect redox economy and ideality. Divergent access to analogs is encoded into this logic, as each oxidative event during the oxidase-phase potentially gives rise to different bioactivity; thus, the oxidase-phase represents a superb medicinal chemistry machinery from an evolutionary standpoint. It therefore stands to reason that designing a chemical synthesis patterned off this biosynthetic route for structures as complex as 1 could harbor similar divergence, in contrast to conventional abiotic approaches to 1 that have been reported to date.
In 2009, we disclosed a proof-of-concept to efficiently access highly oxidized terpenes that holistically mimics this underlying logic of biosynthesis.34 Two-phase terpene synthesis logic, despite lacking convergency, could be applied to concise syntheses of complex systems such as ingenol,35 phorbol,36 thapsigargin,37 and others.38,39 Application of this strategy to the non-skeletally rearranged taxanes,40 of which 1 resides at the apex of structural complexity, remained aspirational for the past decade. This can be visualized in Figure 1, with a hypothetical retrosynthetic pyramid for the two-phase synthesis of 1 requiring a chemist to navigate no less than nine different C–O bond forming steps from a minimally oxidized taxane precursor such as taxadienone (9). Initial progress towards this goal was registered in 2012 with the scalable synthesis of enantiopure 9 and taxadiene (6) through a concise cyclase-phase.41 Although bioengineering approaches to 6 are known,42 our laboratory has been a primary supplier of this molecule to numerous groups around the world (nine as of this writing). Extensive studies were then performed to decipher the innate reactivity of 6 towards allylic oxidation (installation of C–5, 10, and 13) culminating in the total synthesis of taxuyunnanine D (8).43 Translating those lessons to the total synthesis of taxabaccatin III (7) via 9 was successful.44 As aforementioned, the vexing arrangement of oxygenation required more than a consideration of chemo-, regio-, and stereoselectivity which Nature mastered using eons of evolution with enzymes. Scheme 1 outlines a route to 1 that overcomes such challenges.
Scheme 1. Two-phase synthesis of 1.
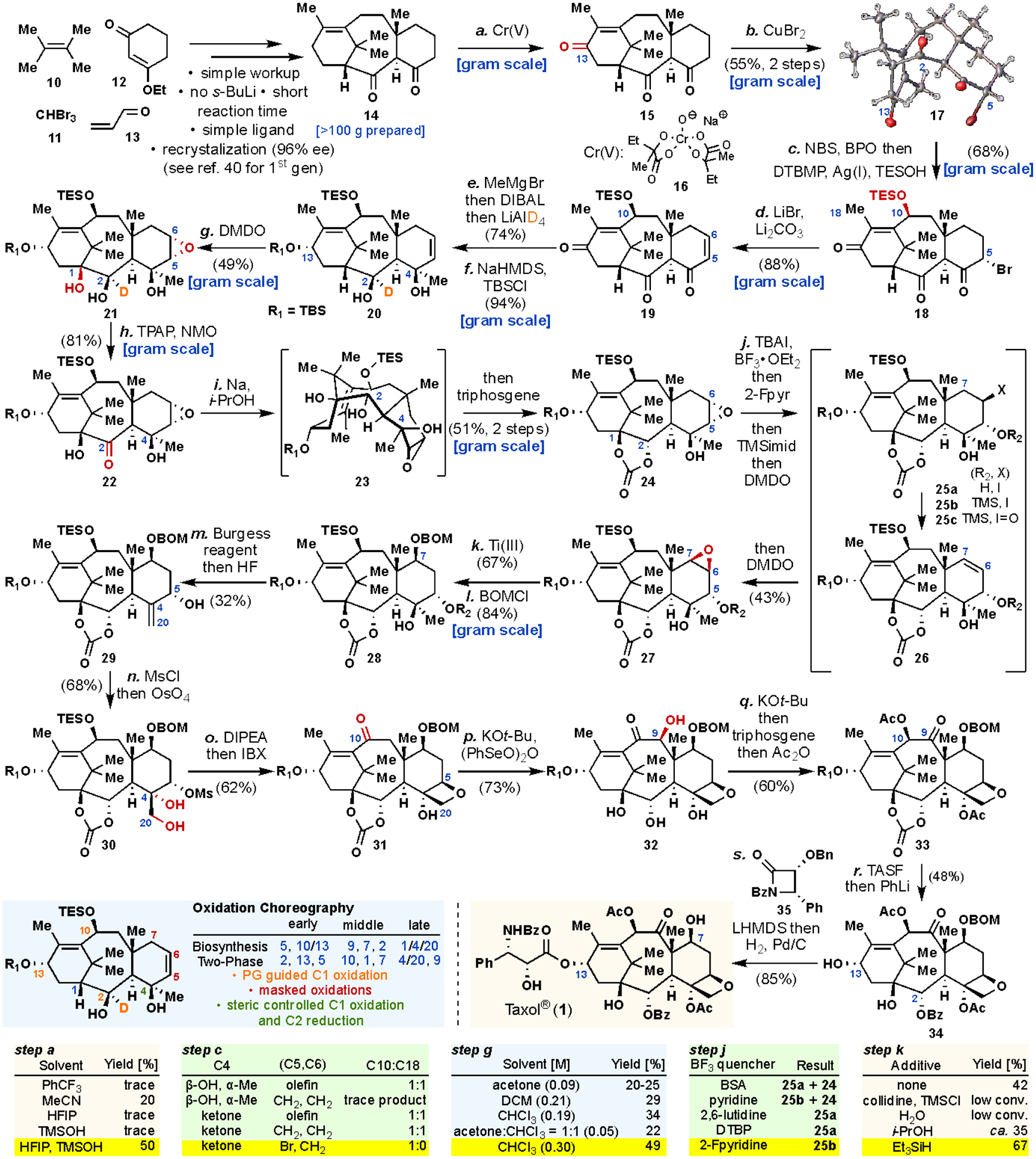
Reagents and conditions are as follows. (a) Cr(V) (16), HFIP, TMSOH, t-BuOH, 80 °C. (b) CuBr2, THF, rt, 55%, over 2 steps. (c) NBS, BPO, CCl4, then DTBMP, AgOTf, TESOH, 4Å MS, PhMe, rt, 68%. (d) LiBr, Li2CO3, DMF, 100°C, 88%. (e) MeMgBr, DCM, 0 °C to rt, then DIBAL, −78 °C, DCM, then LiAlD4, THF, 60 °C, 74%. (f) NaHMDS, TBSCl, THF, 0 °C to rt, 94%. (g) DMDO, 0 °C, 49%. (h) TPAP, NMO, DCM, rt, 81%. (h) Na, i-PrOH, Et2O, rt, then triphosgene, pyr, DMAP, DCM, −78 to 0 °C. (j) TBAI, BF3•OEt2, DCM, −78 °C, then 2-Fpyr, −78 °C, then TMSIm, rt, then DMDO, −78 °C to rt, 43%. (k) Cp2TiCl, Et3SiH, THF, −78 °C to rt, 67%. (l) BOMCl, TBAI, DIPEA, DCE, 45 °C, 84%. (m) Burgess reagent, PhMe, dioxane, then HF, H2O, pyr, MeCN, rt, 32%. (n) MsCl, pyr, 0 °C to rt, then OsO4, THF, pyr, 0 °C to rt, 68%. (o) DIPEA, PhMe, then IBX, DMSO, H2O, 80 °C, 62%. (p) KOt-Bu, (PhSeO)2O, THF, H2O, −78 to 0 °C, 73%. (q) KOt-Bu, THF, then triphosgene, pyr, DMAP, DCM, −78 to 0 °C, then Ac2O, DMAP, pyr, rt, 60%. (r) TASF, THF, rt, then PhLi, THF, −78 °C, 48%. (s) β-lactam (35), LHMDS, THF, −78 °C to rt, then Pd/C, H2, EtOH, 80 °C, 85%.; HFIP, hexafluoroisopropanol; TMSOH, trimethylsilanol; NBS, N-bromosuccinimide; BPO, benzoyl peroxide; DTBMP, 2,6-di-tert-butyl-4-methylpyridine; TESOH, triethylsilanol; DIBAL, diisobutylaluminium hydride; HMDS, hexamethyldisilazane; TBSCl, t-butyldimethylsilyl chloride; DMDO, dimethyldioxirane; TPAP, tetrapropylammonium perruthenate; NMO, N-methylmorpholine N-oxide; pyr, pyridine; DMAP, 4-dimethylaminopyridine; TBAI, tetra-n-butylammonium iodide; 2-Fpyr, 2-fluoropyridine; TMSIm, N-(trimethylsilyl)imidazole; Cp2TiCl, bis(cyclopentadienyl)titanium(III) chloride; BOMCl, benzyl chloromethyl ether; DIPEA, N,N-diisopropylethylamine; DCE, 1,2-dichloroethane; MsCl, methanesulfonyl chloride; IBX, 2-iodoxybenzoic acid; TASF, tris(dimethylamino)sulfonium difluorotrimethylsilicate.
Two-phase synthesis of Taxol®
Starting from simple feedstock-derived building blocks 10-13, the cyclase-phase published in 201241 underwent significant optimization and was initially accomplished using flow chemistry by Albany Molecular Research Inc. (AMRI) to deliver multigram quantities of 14.45 Subsequently, our collaborators (Chemveda) devised a semi-process scale 2nd generation cyclase-phase route through the following modifications (see SI for full summary and graphical procedure): (i) use of alternative solvents for extracting volatile intermediates to simplify workup; (ii) replacement of s-BuLi with less expensive n-BuLi; (iii) use of a more easily-accessible ligand to set the stereochemistry; (iv) development of an operationally- convenient and reproducible enolate trapping sequence; (v) shorter reaction time; and (vi) development of a simple recrystallization protocol for purification. In this way, ca. 110 grams of 14 were prepared in a simple batch protocol. Lessons from previous studies on the selective C–H functionalization of allylic C–H bonds in the taxane core suggested that oxidation at C–13 would be feasible. It was found that the solvent played a decisive role in controlling the regioselectivity (as three allylic positions can be oxidized) and conversion to enone 15 using the Cr(V)-based oxidant 16.43,46 As shown in the inset table for step a, a unique protic and non-nucleophilic mixture of HFIP/TMSOH (2:1) exhibited critical synergy to achieve the high C–13 selectivity over C–11 to afford 15, which could be carried on directly to the next reaction with minimal purification. It has been speculated that the mechanism of this oxidation putatively involves a cationic intermediate.47 This tri-carbonyl-bearing scaffold was selectively brominated at C–5 rather than C–1, C–3, C–10, or C–14 using CuBr2, delivering 17 (whose structure was undoubtably confirmed by X-ray crystallography) in 55% isolated yield (19-gram scale). This high selectivity stemmed from the fact C–1 and 2 are virtually non-enolizable, and the C–14 enolate would form a strained bridgehead diene. Bromination at this stage was essential, as it served as a placeholder for the eventual C–5 and C–7 oxidations and also drove subtle conformational shifts that enabled complete selectivity in the ensuing C–H allylic oxidation at C–10 (see inset table for step c, and SI for discussion). Radical-based oxidation of C–10 took place smoothly following our 2014 protocol,43 proceeding through a dibromide (C–5/10) that could be selectively displaced at C–10 with TESOH in the presence of Ag(I) to deliver 18 in 68% isolated yield (gram-scale). Elimination of the C–5 bromide under standard conditions (LiBr/Li2CO3) afforded di-enone 19 in 88% isolated yield on gram-scale. To set the stage for the pivotal C–1 oxidation, the last carbon atom of the taxane core was installed, the C–13 stereochemistry was set, and a deuterium atom was strategically placed onto C–2; that is, taxane 20 was produced from 19 by sequential addition of MeMgBr and DIBAL, and then LiAlD4 (relative reactivity dictated by C–19, 11 and 20 methyl groups, respectively) followed by TBS installation in 67% yield over two transformations (gram-scale). The remarkable site-selectivity in functionalizing 19 was enabled by exploiting the unique steric and electronic features associated with each carbonyl group. A complex matrix of experiments across multiple taxanes (see SI) revealed substrate 20 to be ideally suited to chemo- and stereoselective oxidation events using DMDO in step g to deliver epoxy-triol 21 in 49% yield as a single diastereomer (gram-scale).48 The Δ11,12-olefin remained intact because of the steric shielding by C–10 and 13 substituents as well as the nature of this hyperstable olefin.49,50 The stereoselectivity of the Δ5,6-olefin epoxidation was guided by the C–19 methyl group. The solvent (CHCl3, which presumably enhances the reactivity of DMDO through H-bonding) and concentration choice were essential (see inset table for step g)51,52 as was the precise functionality expressed in taxane 20. Thus, if C–2 bearing alcohol did not have the β-stereochemistry (presumably prevents C–2 from directing oxidation elsewhere)53 and α-deuterium incorporation (9% with C–2 α–H, see SI),54–57 or if C–4 did not bear full substitution, rapid oxidation back to the C–2 ketone took place with little to no observable hydroxylation at C–1 (see SI).
The second half of the synthesis began with TPAP-mediated58 oxidation of C–2 (22, 85% isolated yield, gram-scale). Thermodynamic reduction of the ketone using Na/i-PrOH59 followed by addition of triphosgene to epoxy-triol 23 furnished carbonate 24 in 51% isolated yield (gram-scale) with the C–5/6 epoxide left completely unscathed. As with the oxidation in step g, the stereochemistry and right balance of sterics at C–4 again played an important role in enabling this outcome, presumably due to its staggered conformation relative to the C–2 α-keto radical intermediate during the reduction. The stage was now set for a redox-relay event to stereoselectively install the C–7 oxygen atom. This maneuver was achieved without isolation of unstable intermediates in one-flask through a carefully designed sequence involving the addition of orthogonally reactive and volatile reagents. In the event, taxane 24 was exposed to TBAI/BF3•OEt2 to regioselectively (via stereoelectronic guidance) afford the corresponding iodohydrin 25a, followed by addition of 2-fluoropyridine to sequester boron-salts. As indicated in the inset table, the use of other additives and alternative bases led to low yields, stemming from incomplete reaction (26) and/or reformation of 24. Immediate capping at C–5 with a TMS group (TMS-imidazole), followed by addition of DMDO, led to iodine-oxidation and spontaneous syn-elimination of iodoso 25c to liberate olefin 26, which could be further stereoselectively oxidized to epoxy-taxane 27 in 43% isolated yield.60 Regioselective elimination through 25c was essential, as E2 elimination of the corresponding iodide 25b did not take place due to steric hindrance around C–7 and C–5 syn hydrogen was conformationally less accessible. It is important to note that this sequential addition design proved to be essential in practice, as 25a was found to revert to 24 upon isolation. Ti-mediated, sterically-guided, regioselective reductive lysis of the epoxide in the presence of Et3SiH followed by BOM installation proceeded in 56% isolated yield to furnish 28 (gram-scale).61 The use of triethyl silane as reductive quenching agent in Ti-mediated epoxide reduction is, to the best of our knowledge, without precedent, and increased both the yield and robustness of the transformation (see inset table for step k). At this juncture, 28 structurally resembled one of Holton’s intermediates and the following transformations were thus heavily inspired by their findings.20 The tertiary alcohol at C–4, which played a pivotal role in numerous transformations, was dismantled through Burgess dehydration, followed by addition of HF•py to afford allylic alcohol 29 in 32% yield, accompanied by a C–5 ketone side product (see SI, compound SI-13). Next, the C–5 hydroxyl group was activated with MsCl and the resulting allylic mesylate (which could not be isolated due to its instability) was immediately subjected to dihydroxylation using OsO4 to deliver oxetane-precursor diol 30 in 68% isolated yield. Oxetane formation was achieved using a hindered amine base (DIPEA), and subsequent addition of IBX directly oxidized the C–10 allylic -OTES group to the C–10 enone, forming 31 in 62% isolated yield. C–9 oxidation took place first, using a modification of Holton’s conditions (KOt-Bu and water/(PhSeO)2O) to deliver 32 (with the cyclic carbonate cleaved) in 73% yield. Redox-isomerization (KOt-Bu) and quadruple acylation to reinstall the cyclic carbonate and acetylate C–4/10 proceeded smoothly to afford taxane 33. Tandem desilylation at C–13 and addition of PhLi gave rise to baccatin III bearing a BOM group at C–7 (34) in 48% isolated yield.24 The total synthesis of 1 could be completed by Ojima-acylation62 using β-lactam 35 and the Li-alkoxide of 34, followed by hydrogenation in 85% isolated yield. The use of 35, which has not been utilized in the context of taxane synthesis before,63 enabled more direct access to 1. This sequence could be performed with high reproducibility through the isolation and purification of 22 intermediates and has been used to prepare 35.2 mg of 1 in multi-batch fashion.
Discussion
It is worth contrasting the strategy employed in Nature with the pioneering syntheses that preceded this work. As aforementioned, the biological two-phase approach to 1 is rather linear, requiring no fewer than 20 different enzymes (2 in the cyclase-phase and 18 in the oxidase-phase).32,33,64 Each transformation takes place in high yield and intermediates are individually sequestered leading to incredible efficiency and scalability. Conventional retrosynthetic analysis would teach that a more convergent approach to 1 should be a better solution for procurement in the laboratory. Yet, as history has proven, such designs so far did not succeed in meaningfully adding to the supply of 1. This issue stems from the sheer number of manipulations required by all syntheses (including our work). A potential approach for medicinal chemistry explorations was uncharted because prior retrosynthetic designs accessed taxanes (compounds bearing the complete carbon skeleton) late into the syntheses, a challenge the present approach overcomes. To be sure, of the 10 syntheses reported, actual taxanes are accessed half-way21 or near the end of the sequence.26 Such issues cannot be resolved by creative engineering solutions or sophisticated analytical chemistry, as Takahashi has shown with the fully automated formal synthesis of 1 that delivered 0.0001 g of baccatin III after 44 machine-automated transformations.27 While the current synthesis offers no benefit in terms of overall yield, and clearly was not developed in a vacuum and benefitted from the many pioneering syntheses that came before, it does however provide a conceptual blueprint for how a viable medicinal chemistry evaluation could take place without semi-synthetic approaches. Furthermore, since multigram quantities of the full taxane skeleton are accessed at a very early stage (step e), this could serve as a new platform for further analogue synthesis combined with the known structure-activity relationships of 1 (as demonstrated with 6, 7, and 8).65,66 Since taxanes only exhibit useful bioactivity when a C–13 sidechain is present, it is notable that our route mirrors biosynthesis in that the proper oxidation is installed at C–13 very early in the synthesis (step a) rather than at a late-stage as in most of the past routes (see Scheme 1 oxidation choreography for a summary). The strategic advance of this two-phase retrosynthesis is reinforced by the fact that all of the methods employed to access 1 were, in principle, known and available to past practitioners. The robust nature of these reactions ensured that two-thirds of the transformations in this synthesis could be conducted on a gram-scale.
Conclusion and Outlook
It is worth recounting some of the key lessons, tactics, and maneuvers that enabled the current route to 1. As with the biosynthetic two-phase approach, this synthesis focuses mostly on the oxidase-phase and thus the precise choreography of O-installation was critical to access the target. C–2 was chosen as the first oxygenation not only due to the retrosynthetic simplification it enabled during the cyclase phase but also because of its critical role in bioactivity.66 Oxidation of C–13, 5 and 10 gave the best material throughput in this order. These oxidations were rapidly completed based on our previous work43,44 to set the stage for the key C–1 oxidation. The challenge of installing C–1 and C–7 could be addressed in unison through careful selection of neighboring functionality and redox-relay through a C–5/6 epoxide group. The C–7 oxidation state was installed through a carefully designed redox relay originating from the C–4 ketone, the C–5 bromide, the C–5/6 epoxide, the C–6 iodide, the Δ6,7-olefin, the C–6/7-epoxide, and finally to the C–7 alcohol. As the supporting information detail, multiple generations of strategies were evaluated before finalizing this pathway. This is clearly a weakness of the two-phase design with regards to one of the most densely functionalized terpenes known. In biosynthesis, enzymatic hydroxylation events accomplished through eons of evolution don’t necessitate protecting groups or functional group manipulations and are largely impervious to subtle substrate modifications. C–4 and C–20 benefited from simple dihydroxylation of an olefin, and the pioneering work of Holton20 guided the final C–9 oxidation (and stereochemistry thereof).
While this synthesis does employ protecting groups (C–5, C–7, C–10, and C–13) their effect was maximized by strategic selections and allocations so that analogous functional groups were well differentiated throughout the synthesis – e.g. simultaneous installation of an –OTES group upon C–H oxidation (C–10) or tandem removal during oxidation (C–10) in the presence of –OTBS (C–13). The silyl groups at C–13 and C–10 did play a pivotal role in shielding the bridgehead olefin from oxidation. Unconventional means of protecting functionality and directing reactivity were also employed including the use of deuterium (kinetic isotope effect) at C–2,67 a bromine atom at C–5, a tertiary alcohol at C–4, and an epoxide at C–5/6. The dense functionality innates to 1 enabled unique positionally selective reactions such as the reactivity of a tricarbonyl (step e), the oxidation at C–1 (step g), and the stability and reactivity of epoxide intermediates (steps h-j). Reactive intermediates (c, j, n, and q) could also be utilized through consecutive operations in order to achieve challenging transformations with minimal labor.
Through this unique opportunity of implementing the “pyramid-climbing” approach to one of the most densely functionalized terpenes (arguably, the most iconic known), we could reevaluate the way one approaches disconnections to other terpenes. The two-phase approach is a synthetically high-risk/high-reward strategy as we have demonstrated with other syntheses (such as ingenol,35 phorbol,36 and thapsigargin37). As a carbon skeleton of a target terpene is built in early stage, all oxidase-phase reactions are essentially a late-stage functionalization of a complex natural product. As the oxidation level ascends, there are more and more (similar) functional groups in a molecule; thereby, protecting group manipulations and choice of substrate compatible reagents and conditions become progressively more challenging. However, all late-stage intermediates can in principle be elaborated to bioactive candidates, since a carbon skeleton is already present (akin to Nature’s route). Such a route might pass through a low yielding transformation, but that is typically a minor issue when it comes to an initial medicinal chemistry campaign. This leads to the second point that the two-phase synthesis is a useful strategy depending on the context. For example, the overall yield of Nicolaou’s convergent Taxol synthesis was 0.0078%,21 while our linear two-phase approach ended up yielding only 0.0014%. Employing either approach for a target-oriented synthesis (e.g. process chemistry) would be inappropriate and result in poor material throughput. On the other hand, under a circumstance where multiple non-specific analogues were required (e.g. medicinal chemistry), the two-phase strategy would be an extremely powerful way of achieving maximal analog output. Lastly, we felt this aspect was enabling for navigating the dead-ends and detours often encountered in complex natural product synthesis.68 Arrays of substrates possessing the same carbon skeleton but slightly different reactivity due to subtle differences in functional group arrangement enable facile substrate screening. In contrast, a traditional convergent synthesis might require a redesigned substrate that needs to be prepared when a particular transformation fails.
As aforementioned, we began our campaign to access Taxol® (1) to answer a fundamental thought-experiment: Would a holistically-biomimetic approach enable an unified medicinal synthetic campaign involving divergent access to natural and non-natural taxanes at various oxidation patterns? In one sense, if 1 were pursued by a large pharma company in the absence of a natural supply, this route would indeed enable such a study (where mg quantities of each analog are needed). On the other hand, the blueprint laid out herein, required deeply-choreographed empirically derived manipulations unique to each substrate’s reactivity. Thus, extensive effort would be required and process chemists might choose a different path to the final target. At the end of the day, chemical synthesis to specifically access 1 will likely never be competitive with a fully enzymatic approach. To avoid unnecessary concession steps that beleaguer all syntheses of 1, we predict that far more improved medicinal chemistry-based approaches to novel taxanes could be accomplished using a hybrid approach that employs synthesis to generate carbon frameworks (for example containing unnatural motifs) and enlists synthetic biology for chemo- and site-specific oxidations.69 In the meantime, the present synthesis demonstrates that two-phase synthesis can open the possibility of divergent access to taxanes, in contrast to classical synthetic strategies.
Supplementary Material
ACKNOWLEDGMENT
We are especially grateful to Yerram Reddy, V. K. R. and Kokkirala, Y. V. of Chemveda for providing ample quantities of compound 14. Financial support for this work was provided by NIH (GM-118176), Funai overseas scholarship (predoctoral fellowship to Y. K.), Honjo international scholarship (predoctoral fellowship to Y. K.) and JSPS (postdoctoral fellowship to H. N.). The early stages of this program were funded in part by grants from TEVA, LEO Pharma, Bristol-Myers Squibb, and The Scripps Research Institute. We thank D.-H. Huang and L. Pasternack (Scripps Research Institute) for assistance with NMR spectroscopy; M. Gembicky (University of California, San Diego) for assistance with X-ray crystallography; J. S. Chen, B. B. Sanchez and E. Sturgell (Scripps Research Institute) for assistance with LCMS analysis and HRMS analysis. We thank the NOSAMS facility for conducting the isotopic analysis (NSF Cooperative Agreement #OCE-1239667).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures and analytical data (PDF)
REFERENCES
- 1).Nicolaou KC; Montagnon T Molecules That Changed the World: A Brief History of the Art and Science of Synthesis and Its Impact on Society, 1st ed.; Wiley-VCH: Weinheim, 2008. [Google Scholar]
- 2).Brickley P NIH Censured for Taxol Deal. Genome Biol. 2003, 4 (1). [Google Scholar]
- 3).Celgene. Press Release Archive. 2020, https://ir.celgene.com/press-releases-archive/defa.
- 4).Commerçon A; Bézard D; Bernard F; Bourzat JD Improved Protection and Esterification of a Precursor of the Taxotere® and Taxol Side Chains. Tetrahedron Lett. 1992, 33 (36), 5185–5188. [Google Scholar]
- 5).Drug Approval Package: Taxotere (Docetaxel) #020449s028 https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/20-449s028_Taxotere.cfm (accessed Feb 11, 2020).
- 6).Drug Approval Package: Cabazitaxel (Jevtana) #201023 https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/201023s000TOC.cfm (accessed Feb 11, 2020).
- 7).Nicolaou KC; Sorensen EJ Classics in Total Synthesis, 6th ed.; Wiley-VCG: Weinheim, 2014. [Google Scholar]
- 8).Suffness M TAXOL: Science and Applications; CRC Press: Boca Raton, 1995. [Google Scholar]
- 9).Hudlicky T; Reed WJ The Way of Synthesis: Evolution of Design and Methods for Natural Products, 1st ed.; Wiley-VCH: Weinheim, 2007. [Google Scholar]
- 10).Wang YF; Shi QW; Dong M; Kiyota H; Gu YC; Cong B Natural Taxanes: Developments since 1828. Chem. Rev 2011, 111 (12), 7652–7709. [DOI] [PubMed] [Google Scholar]
- 11).Wani MC; Taylor HL; Wall ME; Coggon P; Mcphail AT Plant Antitumor Agents.VI.The Isolation and Structure of Taxol, a Novel Antileukemic and Antitumor Agent from Taxus Brevifolia. J. Am. Chem. Soc 1971, 93 (9), 2325–2327. [DOI] [PubMed] [Google Scholar]
- 12).Schiff PB; Fant J; Horwitz SB Promotion of Microtubule Assembly in Vitro by Taxol. Nature 1979, 277 (5698), 665–667. [DOI] [PubMed] [Google Scholar]
- 13).Drugs@FDA: FDA-Approved Drugs: Taxol (Paclitaxel) #020262 https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=020262 (accessed Feb 12, 2020).
- 14).Ojima I; Habus I; Zhao M; Zucco M; Park YH; Sun CM; Brigaud T New and Efficient Approaches to the Semisynthesis of Taxol and Its C-13 Side Chain Analogs by Means of β-Lactam Synthon Method. Tetrahedron 1992, 48 (34), 6985–7012. [Google Scholar]
- 15).Holton RA Method for Preparation of Taxol. EP0400971 (A2), 1990.
- 16).Cino PM; Schwarz SR; Cazzulino DL Callus Cell Induction from Partially Submerged Explant Tissue in Liquid Medium for Preparation of Taxanes. US5527702A, 1996.
- 17).Bringi V; Kadkade PG; Prince CL; Roach BL Enhanced Production of Taxol and Taxanes by Cell Cultures of Taxus Species. US7264951B1, 2007.
- 18).Wender PA; Mucciaro TP A New and Practical Approach to the Synthesis of Taxol and Taxol Analogues: The Pinene Path. J. Am. Chem. Soc 1992, 114 (14), 5878–5879. [Google Scholar]
- 19).Holton RA; Somoza C; Kim H; Liang F; Biediger RJ; Boatman PD; Shindo M; Smith CC; Kim S; Nadizadeh H; Suzuki Y; Tao C; Vu P; Tang S; Zhang P; Murthi KK; Gentile LN; and Jyanwei H Liu. First Total Synthesis of Taxol. 1. Functionalisation of B Ring. J. Am. Chem. Soc 1994, 116 (2), 1597–1598. [Google Scholar]
- 20).Holton RA; Kim HB; Somoza C; Liang F; Biediger RJ; Boatman PD; Shindo M; Smith CC; Kim S; Nadizadeh H; Nadizadeh H; Suzuki Y; Tao C; Vu P; Tang S; Zhang P; Murthi KK; Gentile LN; Liu JH First Total Synthesis of Taxol. 2. Completion of the C and D Rings. J. Am. Chem. Soc 1994, 116 (4), 1599–1600. [Google Scholar]
- 21).Nicolaou KC; Yang Z; Liu JJ; Ueno H; Nantermet PG; Guy RK; Claiborne CF; Renaud J; Couladouros EA; Paulvannan K; Sorensen EJ. Total Synthesis of Taxol. Nature 1994, 367 (6464), 630–634. [DOI] [PubMed] [Google Scholar]
- 22).Masters JJ; Link JT; Snyder LB; Young WB; Danishefsky SJ A Total Synthesis of Taxol. Angew. Chemie Int. Ed. English 1995, 34 (16), 1723–1726. [Google Scholar]
- 23).Wender PA; Badham NF; Conway SP; Floreancig PE; Glass TE; Granicher C; Houze JB; Jänichen J; Lee D; Marquess DG; McGrane PL; Meng W; Mucciaro TP; Mühlebach M; Natchus MG; Paulsen H; Rawlins DB; Satkofsky J; Shuker AJ; Sutton JC; Taylor RE; Tomooka K. The Pinene Path to Taxanes. 5. Stereocontrolled Synthesis of a Versatile Taxane Precursor. J. Am. Chem. Soc 1997, 119 (11), 2755–2756. [Google Scholar]
- 24).Wender PA; Badham NF; Conway SP; Floreancig PE; Glass TE; Houze JB; Krauss NE; Lee D; Marquess DG; McGrane PL; Meng W; Natchus MG; Shuker AJ; Sutton JC; Taylor RE The Pinene Path to Taxanes. 6. A Concise Stereocontrolled Synthesis of Taxol. J. Am. Chem. Soc 1997, 119 (11), 2757–2758. [Google Scholar]
- 25).Morihira K; Hara R; Kawahara S; Nishimori T; Nakamura N; Kusama H; Kuwajima I Enantioselective Total Synthesis of Taxol. J. Am. Chem. Soc 1998, 120 (49), 12980–12981. [Google Scholar]
- 26).Mukaiyama T; Shiina I; Iwadare H; Sakoh H; Tani Y; Hasegawa M; Satoh K Asymmetric Total Synthesis of Taxol. Chem. Eur. J 1998, 5 (1), 121–161. [Google Scholar]
- 27).Doi T; Fuse S; Miyamoto S; Nakai K; Sasuga D; Takahashi T A Formal Total Synthesis of Taxol Aided by an Automated Synthesizer. Chem. - An Asian J 2006, 1 (3), 370–383. [DOI] [PubMed] [Google Scholar]
- 28).Fukaya K; Tanaka Y; Sato AC; Kodama K; Yamazaki H; Ishimoto T; Nozaki Y; Iwaki YM; Yuki Y; Umei K; Sugai T; Yamaguchi Y; Watanabe A; Oishi T; Sato T; Chida N Synthesis of Paclitaxel. 1. Synthesis of the ABC Ring of Paclitaxel by SmI2-Mediated Cyclization. Org. Lett 2015, 17 (11), 2570–2573. [DOI] [PubMed] [Google Scholar]
- 29).Fukaya K; Kodama K; Tanaka Y; Yamazaki H; Sugai T; Yamaguchi Y; Watanabe A; Oishi T; Sato T; Chida N Synthesis of Paclitaxel. 2. Construction of the ABCD Ring and Formal Synthesis. Org. Lett 2015, 17 (11), 2574–2577. [DOI] [PubMed] [Google Scholar]
- 30).Hirai S; Utsugi M; Iwamoto M; Nakada M Formal Total Synthesis of (–)-Taxol through Pd-Catalyzed Eight-Membered Carbocyclic Ring Formation. Chem. - A Eur. J 2015, 21 (1), 355–359. [DOI] [PubMed] [Google Scholar]
- 31).Lim J A Total Synthesis of Taxol, Harvard, 2000.
- 32).Croteau R; Ketchum REB; Long RM; Kaspera R; Wildung MR Taxol Biosynthesis and Molecular Genetics. Phytochem. Rev 2006, 5 (1), 75–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Jennewein S; Wildung MR; Chau MD; Walker K; Croteau R Random Sequencing of an Induced Taxus Cell CDNA Library for Identification of Clones Involved in Taxol Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (24), 9149–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Chen K; Baran PS Total Synthesis of Eudesmane Terpenes by Site-Selective C-H Oxidations. Nature 2009, 459 (7248), 824–828. [DOI] [PubMed] [Google Scholar]
- 35).Jørgensen L; McKerrall SJ; Kuttruff CA; Ungeheuer F; Felding J; Baran PS 14-Step Synthesis of (+)-Ingenol from (+)-3-Carene. Science 2013, 341, 878–882. [DOI] [PubMed] [Google Scholar]
- 36).Kawamura S; Chu H; Felding J; Baran PS Nineteen-Step Total Synthesis of (+)-Phorbol. Nature 2016, 532 (7597), 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Chu H; Smith JM; Felding J; Baran PS Scalable Synthesis of (−)-Thapsigargin. ACS Cent. Sci 2017, 3 (1), 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Condakes ML; Hung K; Harwood SJ; Maimone TJ Total Syntheses of (–)-Majucin and (–)-Jiadifenoxolane A, Complex Majucin-Type Illicium Sesquiterpenes. J. Am. Chem. Soc 2017, 139 (49), 17783–17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Kuroda Y; Nicacio KJ; da Silva-Jr IA; Leger PR; Chang S; Gubiani JR; Deflon VM; Nagashima N; Rode A; Blackford K; Ferreira AG; Sette LD; Williams DE; Andersen RJ; Jancar S; Berlinck RGS; Sarpong R Isolation, Synthesis and Bioactivity Studies of Phomactin Terpenoids. Nat. Chem 2018, 10 (9), 938–945. [DOI] [PubMed] [Google Scholar]
- 40).For a beautiful synthesis of the rearranged taxane, see:; Schneider F; Samarin K; Zanella S; Gaich T Total Synthesis of the Complex Taxane Diterpene Canataxpropellane. Science 2020, 367 (6478), 676–681. [DOI] [PubMed] [Google Scholar]
- 41).Mendoza A; Ishihara Y; Baran PS Scalable Enantioselective Total Synthesis of Taxanes. Nat. Chem 2012, 4 (1), 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Ajikumar PK; Xiao WH; Tyo KEJ; Wang Y; Simeon F; Leonard E; Mucha O; Phon TH; Pfeifer B; Stephanopoulos G Isoprenoid Pathway Optimization for Taxol Precursor Overproduction in Escherichia Coli. Science 2010, 330 (6000), 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Wilde NC; Isomura M; Mendoza A; Baran PS Two-Phase Synthesis of (–)-Taxuyunnanine D. J. Am. Chem. Soc 2014, 136 (13), 4909–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Yuan C; Jin Y; Wilde NC; Baran PS Short, Enantioselective Total Synthesis of Highly Oxidized Taxanes. Angew. Chemie - Int. Ed 2016, 55 (29), 8280–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Krasutsky SG; Jacobo SH; Tweedie SR; Krishnamoorthy R; Filatov AS Route Optimization and Synthesis of Taxadienone. Org. Process Res. Dev 2015, 19 (1), 284–289. [Google Scholar]
- 46).Kende AS; Johnson S; Sanfilippo P; Hodges JC; Junheim LN Synthesis of a Taxane Triene. J. Am. Chem. Soc 1986, 108 (12), 3513–3515. [Google Scholar]
- 47).Wilde NC The Two-Phase Synthesis of Taxane Natural Products: Developing the Tools for the Oxidase Phase, The Scripps Research Institute, 2016. [Google Scholar]
- 48).Horiguchi T; Nagura M; Cheng Q; Oritani T; Kudo T Chemical Oxidation of Taxoids with M-CPBA and Dimethyl Dioxirane: Regioselective Epoxidation of Taxinine J Derivatives. Heterocycles 2000, 53 (12), 2629–2642. [Google Scholar]
- 49).Maier WF; von Ragué Schleyer P Evaluation and Prediction of the Stability of Bridgehead Olefins. J. Am. Chem. Soc 1981, 103 (8), 1891–1900. [Google Scholar]
- 50).McEwen AB; von Ragué Schleyer P Hyperstable Olefins: Further Calculational Explorations and Predictions. J. Am. Chem. Soc 1986, 108 (14), 3951–3960. [Google Scholar]
- 51).Murray RW; Gu D Dioxirane Chemistry. Part 23. The Effect of Solvent on the Dimethyldioxirane Epoxidation Reaction. J. Chem. Soc. Perkin Trans 2 1993, No. 11, 2203–2207. [Google Scholar]
- 52).Gibert M; Ferrer M; Sänchez-Baeza F; Messeguer A Availability and Reactivity of Concentrated Dimethyldioxirane Solutions in Solvents Other Than Acetone. Tetrahedron 1997, 53 (25), 8643–8650. [Google Scholar]
- 53).Murray RW; Gu H Dimethyldioxirane Reactions: Rate Acceleration Due to Intramolecular H-Bonding. J. Phys. Org. Chem 1996, 9 (11), 751–758. [Google Scholar]
- 54).Clive DLJ; Cantin M; Khodabocus A; Kong X; Tao Y Protecting Group Improvement by Isotopic Substitution: Synthesis of the Quinone System of Fredericamycin A. Tetrahedron 1993, 49 (36), 7917–7930. [Google Scholar]
- 55).Vedejs E; Little J Aziridinomitosenes by Anionic Cyclization: Deuterium as a Removable Blocking Group. J. Am. Chem. Soc 2002, 124 (5), 748–749. [DOI] [PubMed] [Google Scholar]
- 56).Miyashita M; Sasaki M; Hattori I; Sakai M; Tanino K Total Synthesis of Norzoanthamine. Science 2004, 305, 495–499. [DOI] [PubMed] [Google Scholar]
- 57).Quasdorf KW; Huters AD; Lodewyk MW; Tantillo DJ; Garg NK Total Synthesis of Oxidized Welwitindolinones and (−)- N -Methylwelwitindolinone C Isonitrile. J. Am. Chem. Soc 2012, 134 (3), 1396–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58).Griffith WP; Ley SV; Whitcombe GP; White AD Preparation and Use of Tetra-n-Butylammonium per-Ruthenate (TBAP Reagent) and Tetra-n-Propylammonium per-Ruthenate (TPAP Reagent) as New Catalytic Oxidants for Alcohols. J. Chem. Soc. Chem. Commun 1987, No. 21, 1625–1627. [Google Scholar]
- 59).Bouveault L; Blanc GL Préparation Des Alcools Primaires Au Moyen Des Acides Correspondants. Bull. Soc. Chim. Fr 1903, 136, 1676–1678. [Google Scholar]
- 60).Liang X; Kingston DGI; Lin CM; Mamel E Synthesis and Biological Evaluation of Paclitaxel Analogs Modified in Ring C. Tetrahedron Lett. 1995, 36 (17), 2901–2904. [Google Scholar]
- 61).Nugent WA; RajanBabu TV Transition-Metal-Centered Radicals in Organic Synthesis. Titanium(III)-Induced Cyclization of Epoxy Olefins. J. Am. Chem. Soc 1988, 110 (25), 8561–8562. [Google Scholar]
- 62).Ojima I; Sun CM; Zucco M; Park YH; Duclos O; Kuduk S A Highly Efficient Route to Taxotere by the β-Lactam Synthon Method. Tetrahedron Lett. 1993, 34 (26), 4149–4152. [Google Scholar]
- 63).Fuji K; Watanabe Y; Ohtsubo T; Nuruzzaman M; Hamajima Y; Kohno M Synthesis of Extremely Simplified Compounds Possessing the Key Pharmacophore Units of Taxol, Phenylisoserine and Oxetane Moieties. Chem. Pharm. Bull 1999, 47 (9), 1334–1337. [DOI] [PubMed] [Google Scholar]
- 64).Davis EM; Croteau R Cyclization Enzymes in the Biosynthesis of Monoterpenes, Sesquiterpenes, and Diterpenes. In Biosynthesis Aromatic Polyketides, Isoprenoids, Alkaloids; Springer: New York, 2000; pp 53–96. [Google Scholar]
- 65).Kingston DGI Taxol: The Chemistry and Structure-Activity Relationships of a Novel Anticancer Agent. Trends Biotechnol. 1994, 12 (6), 222–227. [DOI] [PubMed] [Google Scholar]
- 66).Fang W; Liang X. Recent Progress in Structure Activity Relationship and Mechanistic Studies of Taxol Analogues. Mini-Reviews Med. Chem 2012, 5 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- 67).Angelis YS; Hatzakis NS; Smonou I; Orfanopoulos M Oxidation of Benzyl Alcohols by Dimethyldioxirane. The Question of Concerted versus Stepwise Mechanisms Probed by Kinetic Isotope Effects. Tetrahedron Lett. 2001, 42 (22), 3753–3756. [Google Scholar]
- 68).Sierra MA; de la Torre MC; Nicolaou KC Dead Ends and Detours: Direct Ways to Successful Total Synthesis; Wiley-VCH: Weinheim, 2004. [Google Scholar]
- 69).Zwick C; Sosa M; Renata H Concise Chemoenzymatic Total Synthesis of GE81112 B1. ChemRxiv. 2019, 10.26434/chemrxiv.9684554.v1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.