

Abstract
Despite the large number of promising neuroprotective agents identified in experimental traumatic brain injury (TBI) studies, none has yet shown meaningful improvements in long-term outcome in clinical trials. To develop recommendations and guidelines for pre-clinical testing of pharmacological or biological therapies for TBI, the Moody Project for Translational Traumatic Brain Injury Research hosted a symposium attended by investigators with extensive experience in pre-clinical TBI testing. The symposium participants discussed issues related to pre-clinical TBI testing including experimental models, therapy and outcome selection, study design, data analysis, and dissemination. Consensus recommendations included the creation of a manual of standard operating procedures with sufficiently detailed descriptions of modeling and outcome measurement procedures to permit replication. The importance of the selection of clinically relevant outcome variables, especially related to behavior testing, was noted. Considering the heterogeneous nature of human TBI, evidence of therapeutic efficacy in multiple, diverse (e.g., diffuse vs. focused) rodent models and a species with a gyrencephalic brain prior to clinical testing was encouraged. Basing drug doses, times, and routes of administration on pharmacokinetic and pharmacodynamic data in the test species was recommended. Symposium participants agreed that the publication of negative results would reduce costly and unnecessary duplication of unsuccessful experiments. Although some of the recommendations are more relevant to multi-center, multi-investigator collaborations, most are applicable to pre-clinical therapy testing in general. The goal of these consensus guidelines is to increase the likelihood that therapies that improve outcomes in pre-clinical studies will also improve outcomes in TBI patients.
Keywords: : guidelines, pre-clinical, traumatic brain injury
Introduction
The Centers for Disease Control and Prevention estimates that there were about 1.7 million new traumatic brain injuries (TBIs) per year in the United States from 2002 to 2006.1 These TBIs resulted in 1.4 million emergency department visits, 275,000 hospitalizations, and 52,000 fatalities (for a review, see Roozenbeek and colleague2). In addition to new TBIs, there are about 5.3 million people in the United States3 and 7.7 million people in Europe4 living with disabilities from TBI. Unfortunately, many of these individuals suffer from the long-term consequences of TBI for many years post-injury.5 Although estimates of the economic costs of TBI vary (for a review, see Humphries and associates6), the Centers for Disease Control and Prevention estimates medical costs of more than $76 billion annually in the United States alone.7 In view of the staggering economic and psychosocial costs of TBI, it is not surprising that considerable amounts of time and money have been devoted to finding treatments for TBI.
To adequately model the heterogeneous nature of TBI in humans, pre-clinical studies have been conducted using experimental models that replicate the important pathophysiological features of different types of human TBI.8 Despite the large number of promising neuroprotective agents identified in experimental TBI studies, none have shown meaningful improvements in long-term outcome in clinical trials.8–10 A number of factors may contribute to these translational failures. For example, Agoston and colleagues9 observed that the types of data collected (e.g., histopathological, behavioral, imaging) and the collection times (hours, days, or weeks post-TBI) differ markedly between clinical and experimental TBI studies. It is also apparent that pre-clinical testing should involve multiple experimental TBI models,11 preferably in multiple species.12
To develop recommendations and guidelines for the pre-clinical testing of pharmacological/biological therapies for TBI, the Moody Project for Translational Traumatic Brain Injury Research sponsored a symposium attended by investigators with extensive experience in pre-clinical TBI testing to identify potential limitations of current experimental models and protocols. This group also suggested strategies to improve the likelihood of future pre-clinical studies identifying therapies that rapidly and successfully translate to clinical trials.
State of the Art in TBI Clinical Trials
Burke and co-workers13 identified 142 randomized, controlled trials (RCTs) as of October 2013 and Bragge and colleagues14 reported 207 RCTs in moderate-to-severe TBI patients through 2015. Although many of these RCTs tested non-pharmacological therapies (e.g., cerebral perfusion pressure or intracranial pressure [ICP] control), Kabadi and Faden7 reported more than 30 trials of “neuroprotective strategies.” As of 2006, 130 therapies had proved successful in pre-clinical testing15,16 and it is likely that the number now is much higher.
Were “positive” trials really positive?
Unfortunately, most of the beneficial effects observed in clinical trials to date have been transient, not translated into a desirable clinical benefit (e.g., they reduced mortality with a corresponding increase in the vegetative state rate), not reproducible, or offset by negative effects of the therapy. Yet, as summarized in Table 1, there have been several “statistically significant positive results.”17
Table 1.
Randomized Clinical Trials with Positive Results in TBI Patients, 1990–2012
| Therapy | N | Type | Results | References |
|---|---|---|---|---|
| Pharmacological therapies | ||||
| Nimodipine | 123 TBI w SAH |
RCT, multi | Significantly fewer unfavorable GOS scores at 6 months in nimodipine group | Harders et al.28 |
| Progesterone | 159 sev TBI | RCT | Significantly more favorable outcomes (GOS & FIM) and lower mortality in progesterone-treated pts 3 and 6 months post-TBI | Xiao et al.30 |
| Amantadine | 184 sev TBI | RCT, multi | Significantly faster rate of functional recovery (DRS) 4 wks post-TBI in amantadine-treated pts | Giacino et al.27 |
| Phenytoin | 404 sev TBI | RCT, single | Significantly fewer seizures in phenytoin-treated pts 1-7 days post-TBI | Temkin et al.26 |
| Surgical/Management therapies | ||||
| Hypothermia | 215 sev TBI | RCT, multi | Significantly lower ICP and more favorable GOS in sev TBI pts with 5 days vs. 2 days of hypothermia | Jiang et al.20 |
| 47 sev TBI | RCT, single | Significant reduction in number of seizures in hypothermic patients | Clifton et al.23 | |
| 396 sev TBI | RCT, single | Significantly lower mortality and more favorable outcome in hypothermia group | Zhi et al.21 | |
| 82 sev TBI | RCT, single | hypothermia hastened neurological recovery; trend toward improved outcome | Marion et al.24 | |
| Craniectomy | 486 sev TBI | RCT, multi | Significantly more favorable GOS at 6 months in pts with larger (15x15 cm) vs. smaller (6x8 cm) craniectomy | Jiang et al.156 |
| Craniectomy | 408 sev TBI | RCT, multi | Significantly lower mortality in decompressive craniectomy group | Hutchinson et al.157 |
| CBF targeted therapy | 189 sev TBI | RCT, single | Jugular venous desaturation significantly lower in CBF- vs. ICP-targeted therapy | Robertson et al.18 |
| Hyperbaric O2 | 168 sev TBI | RCT, single | Mortality rate was significantly lower in pts treated with hyperbaric O2 | Rockswold et al.19 |
CBF, cerebral blood flow; DRS, disability rating scale; FIM, functional independence measure; GOS, Glasgow Outcome Scale; multi, multi-center RCT; pts, patients; RCT, randomized clinical trial; SAH, subarachnoid hemorrhage; single, single-center RCT; sev, severe; TBI, traumatic brain injury.
Robertson and associates reported that although cerebral blood flow (CBF)-targeted therapy reduced the incidence of jugular venous desaturation substantially compared with ICP-targeted therapy, neurological outcome was the same in the two groups, perhaps because the frequency of adult respiratory distress syndrome was significantly higher with CBF-targeted therapy.18 Hyperbaric oxygen (HBO) therapy in severe TBI patients was associated with significantly lower mortality, but the number of favorable outcomes was no higher in the HBO patients than in the controls.19 In a three-center study of severe TBI patients with intracranial hypertension, Jiang and colleagues20 observed that 5 days of mild hypothermia (33–35°C) resulted in significantly more favorable outcomes than 2 days of hypothermia, but the study design did not include a normothermic control group.
One initially promising technique was reported by Zhi and associates,21 who compared mortality and 6-month Glasgow Outcome Scale (GOS) scores in 198 normothermic patients with severe TBI, and an equal number in whom rectal temperature was maintained at 32–35°C for 1 to 7 days. Mortality was significantly lower and the rate of “good” recovery was significantly higher in the hypothermia group. Additionally, CBF and jugular venous O2 saturation were higher during hypothermia in a small (15) subset of patients in whom those variables were measured prior to and during the period of hypothermia. But in contrast to Zhi and associates, Clifton and colleagues,22 in a multi-center study of 392 patients with severe TBI, observed no significant effect of hypothermia (33°C) on mortality or GOS scores (6 months). Further, the complication rate and length of hospitalization were significantly higher in the hypothermic group. In addition to the study by Zhi and associates,21 the results of Clifton and colleagues22 differ from the results of two earlier studies, at least to some extent. In a single-center RCT of 46 patients with severe TBI, Clifton and collegaues23 observed that hypothermia (32–33°C) was associated with a statistically significant reduction in seizures and a trend toward better outcome (GOS, 3 months) that was not statistically significant. Based on another single-center RCT of 82 patients with severe TBI, Marion and co-workers24 reported that hypothermia (32–33°C) resulted in a significant improvement in GOS (3 and 6 months). Unfortunately, there were no significant differences in GOS scores in the hypothermic compared with the normothermic groups at 12 months post-injury.
Despite the encouraging results of these positive studies, other research failed to confirm these effects. In a multi-center study, Clifton and colleagues22 reported no significant improvement in outcome in patients treated with hypothermia (33°C). But they also noted that,22 although hypothermia may not be beneficial in patients who are normothermic on admission, maintaining hypothermia may be neuroprotective in patients who are hypothermic on admission. In another multi-center RCT of hypothermia (32–35°C) in 384 TBI patients with intracranial hypertension (ICP >20 mm Hg) from 18 trauma centers, Andrews and colleagues25 reported no difference in outcome (6 months, GOS-Extended) between the normothermic and hypothermic patients, and the study was stopped early because of concerns about patient safety in the hypothermic group. Together, these results indicate that hypothermia does not improve long-term outcome and may be associated with a higher rate of complications.
Temkin and associates26 reported significantly fewer seizures in the first 8 days post-injury among patients with severe TBI treated with phenytoin compared with a placebo-treated group. Unfortunately, these positive therapeutic effects were transient, and there were no significant differences in the number of post-traumatic seizures between the two groups from 8 days to 2 years post-TBI. Giacino and colleagues, in a RCT of 184 patients with severe TBI,27 reported that amantadine improved the rate of functional recovery 2 weeks after severe TBI. Although the effect was transient and there were no significant differences between the treated and placebo groups 6 weeks post-TBI, even transient accelerated recovery is a somewhat positive effect. Harders and co-workers28 reported that a group of TBI patients with subarachnoid hemorrhage treated with nimodipine (intravenous and oral for 3 weeks) had significantly fewer unfavorable outcomes (GOS, 6 months) than matched patients treated with placebo. Although these results were encouraging, subsequent studies failed to confirm the positive findings.29 The remaining positive RCT was a single-center trial of progesterone in patients with severe TBI.30 Xiao and colleagues30 reported that progesterone, administered within 8 hours of injury, resulted in significantly better dichotomized GOS scores 3 and 6 months post-injury. Functional Independence Measure (FIM) scores also were significantly better in the progesterone-treated compared with placebo-treated patients 3 and 6 months after injury. Although subsequent studies have failed to confirm the positive results of Xiao and colleagues30 (for a review, see Ma and colleagues,31 and Schumachera and associates32), there is an ongoing debate about the limitations of some of these studies (see Goldstein and associates,33 and Stein and co-workers34). In summary, few, if any, RCTs have demonstrated unequivocal, lasting benefits of surgical/management or pharmacological therapies for TBI.
Why Have Promising Therapies Failed in Clinical Trials?
Potential reasons for the failure of promising candidate therapies to translate to successful RCTs and clinical benefits in TBI patients are legion. A thorough discussion of these issues is beyond the scope of this article, but they have been addressed in detail in several excellent reviews.9,10,13–15,32,35,36 Briefly, the reasons why promising therapies may have failed in clinical testing can be divided into the limitations inherent in pre-clinical testing, and those associated with the clinical trials themselves.
Limitations in pre-clinical testing
TBI in humans is complex and heterogeneous and patients with many different types of TBI (e.g., epidural or subdural hemorrhage, focal contusions, diffuse swelling, diffuse axonal injury [DAI]) may have similar Glasgow Coma Scale (GCS) scores on admission.8,31 It is likely and reasonable that therapies that show pre-clinical efficacy in experimental models replicating focal TBI might not necessarily prove equally effective in patients with, for example, diffuse TBI. For this reason, it would seem necessary that pre-clinical studies be conducted in models that replicate diffuse TBI (e.g., impact acceleration), focal TBI (e.g., controlled cortical impact, [CCI]), and other models that produce a more mixed phenotype (e.g., fluid percussion injury, [FPI]). (For a review of experimental TBI models, see Johnson; Xiong; Crnak; DeWitt; and Morales,and colleagues, respectively37–41.) The STAIR (Stroke Therapy Academic Industry Roundtable) criteria recommend that therapies be tested in pre-clinical stroke studies in multiple animal models, including non-human primates, as well as in both sexes.42,43 However, despite representing a seemingly logical strategy, implementation of the STAIR criteria in pre-clinical stroke research has not led to translational success in clinical pharmacological neuroprotection.
Doppenberg and colleagues44 suggested the following requirements for RCTs for therapies for TBI:
-
1.
The mechanism on which the drug or procedure acts should be demonstrated in animal models of TBI.
-
2.
This mechanism should be blocked in these models by the studied drug or intervention.
-
3.
The mechanism should be demonstrated in human TBI.
-
4.
The brain penetration of the drug must be adequate to affect the mechanism under study.
-
5.
The safety and tolerability of the drug must be shown in human head injury.
To meet the first three requirements, pre-clinical and clinical studies must be designed to include measurements to confirm that the same targets of therapy are present, and the same mechanisms are active. Unfortunately, this is rarely the case in either clinical or experimental studies (see Tables 1 and 2 in Agoston and colleagues,9 and Box 1 in Stocchetti and colleagues36).
Table 2.
Potential Peasons for the Failure of Clinical Trials for Therapies for TBI: Reasons Related to Pre-Clinical Testing
| Reason | Summary | References |
|---|---|---|
| Translational issues | Inadequate understanding of differences in therapeutic window, pharmacokinetics, dosages, etc., between humans and experimental animals | Hawryluk et al.15; Margulies and Hicks121 |
| Multiple injury models | Limited evidence of efficacy in different species, genders, ages, etc. | Hawryluk et al.15; Margulies and Hicks121 |
| Injury mechanisms | Mismatch between primary injury mechanisms in humans and experimental animals (e.g., diffuse vs. focal, DAI) | Hawryluk et al.15; Margulies and Hicks121; Saatman et al.8 |
| End-points | Differences in types of end-points in clinical vs. experimental studies | Agoston et al.9 |
| Sample size/power | Inadequate sample size to produce adequate statistical power | Button et al.77 |
| Time intervals | Experimental studies measure acute effects (<1 month); RCTs measure long-term (>1 month) outcomes | Agoston et al.9 |
| Inadequate scientific rigor/Inadvertent bias/Confounders | Inadequate blinding, selective analyses, reporting bias, randomization, attrition | Holman et al.158; Fanelli et al.159; Vogt et al.160; Tsilidis et al.161; O'Collins et al.162 |
In addition to these limitations related to mechanisms and pharmacological properties, there are large discrepancies between the clinical and pre-clinical outcomes of the studies. Imaging (e.g., computed tomography [CT] and, to a lesser extent, magnetic resonance imaging [MRI]) are widely used in clinical studies as a source of diagnostic information but, until recently, imaging has been relatively rare in experimental TBI studies.9 However, an increasing number of pre-clinical studies have successfully implemented serial in vivo imaging to track progressive changes in rats after TBI with or without treatment.45,46
In human TBI studies, injury severity is defined by the GCS, a scale of neurological function based on levels of consciousness.47,48 Neurological function scales for rodents49,50 are used in some experimental studies but, more frequently, injury level is defined by the impact parameters (e.g., pressure pulse magnitude, in atmospheres, for FPI; impact depth and velocity for CCI). In addition to basic neurological function, clinical studies typically involve assessments of neuropsychological/ cognitive/behavioral outcomes. Cognitive function (typically spatial memory) testing has become relatively common in experimental TBI studies. Other behavioral changes relevant to TBI, including anxiety, sociability, impulsivity, and depression, that were once seldom measured in pre-clinical TBI studies now are recognized as important outcomes. However, there is still a large gap in the frequency of behavioral testing between human and animal TBI studies.9 Post-traumatic epilepsy is another common and serious consequence of TBI that is rarely incorporated into pre-clinical TBI studies, which is likely related to the long-term recovery and extensive electrophysiological monitoring required for the study of post-traumatic epilepsy.51–53
Moreover, in most RCTs, behavioral outcomes are measured 3 to 12 months post-TBI, whereas long-term (> 1 month) assessments of behavioral or cognitive function are seldom included in experimental TBI studies (Table 2).9 The effects of TBI on memory function have been studied for up to 1 year after TBI in rats54–56 (for a review of chronic behavioral studies, see Osier and associates57). Bramlett and Dietrich58–61 and Smith and colleagues62 reported histopathological changes up to 12 months after FPI, and Dixon and colleagues54 for up to 12 months post-CCI. Kochanek and associates reported changes in CBF up to 12 months after CCI.63 These expensive and labor-intensive studies contributed important information about the long-term changes associated with experimental TBI, but they are unusual because most pre-clinical research focuses on the acute, short-term effects of TBI.9 Considering the recognition that even mild TBI often has long-term consequences,5 it is important that pre-clinical TBI studies include evaluations of the chronic behavioral and histopathological effects.
Finally, another important potential factor that may contribute to failure of translation is insufficient rigor in pre-clinical studies. This can contribute to overstated effects and has been shown in other fields such as cancer research to generate irreproducible results and failed clinical translation.64,65 Estimates of the numbers of irreproducible pre-clinical studies have ranged from ∼40% to as high as 80%.66
Limitations in clinical testing
One of the potential reasons for the failure of RCTs is inadvertent bias/confounders (Table 3). Corrigan and colleagues67 observed that follow-up bias can occur in patient populations with lower socioeconomic status or in those patients who sustained a TBI due to violence or had a history of drug abuse. Additionally, based on a survey of 24 meta-analyses of RCTs, Page and co-workers68 observed that the effects of interventions may be exaggerated by the lack of adequate blinding of investigators, especially those assessing outcomes, and inadequate randomization sequence generation. However, this would not explain the failure of translation—rather, it would produce false-positive clinical trials. It is important to note that inadvertent bias is a significant potential contributor to inaccuracy in pre-clinical studies as well.
Table 3.
Potential Reasons for the Failure of Clinical Trials for Therapies for TBI: Reasons Related to Clinical Trial Design and Methods
| Reason | Summary | References |
|---|---|---|
| Single center | RCTs conducted in single centers may be less reliable than multiple center RCTs | Bafeta et al.69; Bellomo et al.70 |
| Follow-up bias | Patient selection bias may occur due to loss of follow-up in some patient populations (low socioeconomic status, substance abuse, violent injury mechanisms) | Corrigan et al.67 |
| Randomization/Blinding | Intervention effect estimates may be exaggerated due to inadequate or unclear random sequence generation or blinding, especially with subjective end-points | Bragge et al.14; Page et al.68 |
| Sample size | Inadequate sample size to produce adequate statistical power | Burke et al.13; Bragge et al.14; Button et al.77 |
| Therapy dose selection | Translation of doses used in pre-clinical studies should be based on physiological, pharmacokinetic, and toxicology data | Blanchard and Smoliga79 |
| Patient selection | Select patient populations most likely to be helped by therapy | Hawryluk et al.15; Narayan et al.35 |
| Outcome errors | Misclassification of outcomes (e.g., GOS score) can significantly reduce effect size | Lu et al.85 |
| Inadvertent bias | Pressures to publish, industry bias, positive results, government vs. industry support | Fanelli et al.159; Dwan et al.163; Fanelli et al.164; Easterbrook et al.165 |
Another potential explanation for the disappointing results of TBI RCTs is the frequency of single-center trials. In a report of the results of meta-analysis of RCTs of therapies in patients with moderate to severe TBI up to 2015, Bragge and colleagues14 observed that three quarters of the 207 RCTs analyzed were single-center trials. Although single-center RCTs are less expensive and easier to organize and coordinate than multi-center trials, and data collection and analysis are more straightforward, the results often prove hard to replicate.14,69,70 Maas and associates71 observed that most of the trials that reported beneficial effects of the therapy tested were single-center RCTs. One example is the single-center RCTs that found hypothermia to be beneficial in patients with severe TBI.21,23,24 Although these studies appeared to be appropriately designed, randomized, and blinded, the results could not be confirmed in follow-on, multi-center RCTs.22,25 Similarly, two single-center RCTs showed decreased mortality and improved functional outcomes with progesterone,30,72 results that were not confirmed in large multi-center clinical trials.73,74
A related issue is that many RCTs may be underpowered. A clinical trial with fewer than 100 patients may overestimate75 or underestimate76 treatment effects. Although the direction of the effect may vary, these and other reports13,77 indicate that small RCTs may yield different results than larger studies of the same therapies.
Therapy dose selection is another important issue. Scaling drug doses from small experimental animals to humans is not as simple as multiplying the mg/kg dose in an animal by the patient's body weight. The U.S. Food and Drug Administration (FDA) recommends that drug doses for humans be scaled using a calculation based on body surface area (BSA)78:
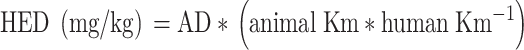 |
where HED = human equivalent dose, AD = animal dose (mg/kg), Km = weight (kg) divided by BSA (m2). The use of a simple body-weight conversion would result in a dose increase of 200-fold for a 60-kg human, whereas dose conversion using BSA would increase the dose by about 65-fold. Although the BSA conversion has been widely used for many years, Blanchard and Smoliga79 recently reviewed the evidence that scaling based on BSA also is inadequate due to the evidence that body structures and function that affect drug outcome do not scale only by BSA or body mass. Use of computational modeling of the key factors affecting drug response (known as allometric scaling) across more than one species is the more accurate method for estimating human equivalent dosing.80,81 Although there is added expense in the assessment of multiple species, such methods improve the accuracy of estimation of key factors of drug disposition.
Granted the majority of RCTs have been conducted in patients with moderate to severe TBI,14 as of 2013 there have been at least 71 RCTs testing therapies for concussion (i.e., mild TBI)13 and several RCTs included patients with mild TBI along with moderate and severe cases.25,82,83 The inclusion of patients with multiple levels of TBI severity facilitates recruitment and increases the likelihood of acquiring a large patient population. However, one of the postulated reasons for RCT failure is the inclusion of patients who are “too good” or “too bad” to be helped by the therapy tested.15,35 In some cases, post hoc sub-group analysis in negative RCTs revealed sub-populations of patients that appeared to have benefited from the therapy tested. As noted above, Clifton and colleagues22 suggested that maintaining hypothermia may be beneficial in TBI patients who were hypothermic on admission. To date, this hypothesis has not been tested.
One of the most important aspects of clinical trial design and execution is the selection and accurate assessment of outcomes. A widely used outcome in TBI RCTs is the GOS.14 The GOS divides outcomes into five categories: Good Recovery (GR); Moderate Disability (MD); Severe Disability (SD); Vegetative; and Death.47 To more accurately define recovery, Jennett and colleagues48 expanded the original five-category GOS to the eight-category GOS-Extended (GOSE or GOS-E) by adding two levels each to GR, MD, and SD for conscious patients (e.g., MD ≥ MD upper, MD lower; GR ≥ GR upper, GR lower, etc.). The GOS-E provides better differentiation of recovery levels, especially in patients with mild or moderate TBI. Even though the GOS-E may be more subjective, it correlates more closely with other disability ratings, neuropsychological tests, etc., than the original GOS.84 However, there have been significant inter-rater differences and misclassifications with the GOS and GOS-E.84–87 Lu and associates85 observed that errors in classifying patients with the GOS could reduce the power of detecting a treatment effect, thereby underestimating the true efficacy of therapy. Further, neither of these scales is adequate to assess more subtle symptoms associated with mild TBI. These limitations inspired the Traumatic Brain Injury Endpoints Development (TED) project that aims to develop and validate novel biomarkers and outcome measures for clinical TBI monitoring.88
Operation Brain Trauma Therapy (OBTT): A Novel Approach to Pre-Clinical Testing
OBTT is a consortium of investigators at the Safar Center at the University of Pittsburgh, the Walter Reed Army Institute of Research (WRAIR), the Miami Project to Cure Paralysis at the University of Miami, Virginia Commonwealth University, the University of Florida, Messina University, and Banyan Biomarkers that is supported by the U.S. Department of Defense.11,89,90 Although OBTT is novel in pre-clinical TBI research, the Multi-center Animal Spinal Cord Injury Study (MASCIS) was a similar multi-center collaboration decades earlier to test the efficacy of therapies for spinal cord injury.91 Financial support for MASCIS was provided by the U.S. National Institute of Neurological Diseases and Stroke (NINDS). Whereas personnel from each of the participating MASCIS centers were trained to use the same weight-drop spinal cord injury model and the Basso, Beattie, Bresnahan (BBB) locomotor function scale,92 the OBTT centers used different TBI models, each center using the model with which it had the most experience (i.e., FPI, Miami; CCI, Pittsburgh; penetrating ballistic-like brain injury [PBBI], WRAIR)11,89 for the express purpose of addressing the marked anatomical heterogeneity of clinical TBI. All of the OBTT centers used the same drug doses and treatment regimen, assessed the cognitive effects of therapy using the Morris water maze (MWM), collected serial blood samples for biomarker measurements, and harvested the brains for histopathological analyses 21 days post-injury. However, there were some differences in procedures among the OBTT centers. For example, vestibular/motor function was measured using cylinder and grid walk tasks by the Miami group, beam balance and walking tasks by Pittsburgh, and a rotating rod task by WRAIR.89 Another significant difference among centers was the level of injury. Pittsburgh and WRAIR used severe levels of CCI and PBBI, respectively, whereas the Miami group used a moderate level of FPI. Details of these and other similarities and differences among the OBTT center have been published.11,89,90
To date, the five therapies that have been tested by OBTT are erythropoietin,93 cyclosporine,94 levetiracetam,95 simvastatin,96 and nicotinamide.97 All of the drugs tested by OBTT were FDA-approved for human use, and all had been effective in experimental TBI models in which therapy was initiated post-injury. Of the five drugs tested, only levetiracetam significantly improved outcome in more than one model. Both doses of levetiracetam improved MWM performance after FPI, whereas the lower dose (54 mg/kg) improved MWM performance after CCI. In the CCI model, the higher dose (170 mg/kg) improved performance on the beam balance task and reduced hemispheric tissue loss. Levetiracetam therapy had no significant effect after PBBI, the most severe TBI model used by OBTT.90 Based on this demonstration of efficacy in two TBI models, levetiracetam is being tested in a micro-pig FPI model with the second round of therapy testing now in progress. Among other therapies, OBTT is testing glibenclamide and Kollidon VA64.90 Glibenclaide (a.k.a. glyburide), a sulfonylurea used to treat diabetes, improved spatial memory and reduced hippocampal injury after CCI in rats.98 Kollidon VA64, a poloaxmer that aids in cell membrane resealing, reduced neuronal degeneration, BBB damage, and brain edema and improved motor function in mice after CCI.99 These studies have been completed but the results have not yet been published. Other ongoing studies by OBTT include testing of amantadine, the aquaporin antagonist AER-271, minocycline, the protease inhibitor E64d, and the nicotinamide phosphoribosyltransferase activator P7C3-A20.
Alhough the results of the first set of therapy testing are somewhat disappointing, they do not diminish the remarkable achievement of the OBTT investigators, including unique elucidation, characterization, and comparison of multiple, gold standard pre-clinical TBI models using conventional outcome metrics and novel candidate biomarkers of brain injury as well as setting standards for methodological rigor.90,94,97,100
Despite its substantive strengths, the OBTT approach has some limitations that should be acknowledged. The time of drug administration (15 min post-TBI) would be impractical in TBI patients. Also, the drug doses and times and routes of administration may not have been optimal, and the OBTT studies weren't designed to test mechanisms or to confirm that the therapies acted on their intended targets, if known. These and other potential flaws have been thoughtfully considered by the OBTT investigators90 The work of OBTT also suggests that some of the reasons for failure of translation of therapies may lay within the pre-clinical realm, further emphasizing the need for rigorous, multi-center, pre-clinical approaches to therapy development. OBTT is an excellent example of the type of pre-clinical testing using multiple TBI models and clinically relevant outcome variables that is likely to identify therapies that will prove effective in clinical trials in patients with different types of TBIs.
Moody Project TBI Symposium
The Moody Project TBI Symposium, held in Galveston, TX, in September 2016, was intended to identify potential limitations and gaps in current experimental TBI therapy testing and suggest guidelines to overcome some of these limitations and improve the translation of promising therapies to successful clinical trials in TBI patients. Investigators with extensive experience with pre-clinical testing were invited to participate and address a series of questions (Table 4) related to model and therapy selection, outcomes, and data collection and analysis. The invitees included physicians, pharmacologists, physiologists, biomedical engineers, and animal behaviorists from the United States, Europe, and Australia.
Table 4.
Moody Project Symposium Discussion Topics and Questions
| Topic | Questions |
|---|---|
| Model and therapy selection | Models |
| 1. Should multiple models be used for therapy testing? If so, how many and which ones? | |
| 2. Should models be standardized across sites? | |
| 3. Injury severity? Mild, moderate, severe? Repetitive mild? | |
| 4. Should studies include both genders? | |
| 5. Should anesthetics be constant across sites? | |
| 6. Should secondary (post-traumatic) insults (e.g., hypotension, hypoxemia) be included? | |
| Therapies | |
| 1. Therapy selection? FDA approved? Human use approval? BBB permeability? | |
| 2. Should PD/PK be established prior to efficacy testing? | |
| 3. Time/route of administration? How many doses? | |
| 4. Should the same doses and routes and times of administration be used in all models? | |
| Outcomes | 1. Which ones? |
| 2. How should outcomes be weighted in “efficacy score”? | |
| 3. Should outcomes be standardized across sites? | |
| 4. At what time after injury should outcomes be assessed? | |
| 5. Long-term (> 21 days) outcomes? | |
| 6. Serum biomarkers? Which ones? Should biomarker data be included in the efficacy score? | |
| 7. Should mechanistic outcomes be included to confirm/refute theoretical mechanisms(s) of action? | |
| 8. Should outcomes be standardized and/or normalized across sites? | |
| 9. Which outcomes must be positive to recommend a therapy for clinical testing? | |
| Data collection, analysis, sharing and intersite communication | 1. Should common data elements be “common” across all sites? |
| 2. How should the outcome data be analyzed? | |
| 3. Should the data be shared outside of the consortium? | |
| 4. Should data be analyzed by each site or at a data analysis core? | |
| 5. Should studies include Go/No-Go preliminary analyses or short-term outcome screening? | |
| 6. Should studies include an interim analysis? | |
| 7. Should data be shared among sites while collection is in progress? | |
| 8. What types of intersite communication should be permitted/encouraged? |
BBB, blood–brain barrier; FDA, Food and Drug Administration; PD/PK, pharmacodynamics /pharmacokinetics.
The participants were divided into three groups and each group discussed one of the broad topic areas at each of the three topic discussion sessions, thereby providing all groups the opportunity to discuss all of the topics and questions (Table 4). Although overall study design was not one of the topics, it was discussed by all of the groups and so it is included as a separate section in this consensus summary. Many of the study design recommendations are specific to multi-model, multi-center studies such as OBTT, whereas others would be applicable to pre-clinical therapy testing in general.
Study Design Recommendations
Large, multi-center pre-clinical studies could be patterned after RCTs, with Stage 1 studies designed for relatively simple, efficient screening using multiple, well-established TBI models. Stage 1 studies would be relatively short (2–3 weeks) with simple behavioral and histopathological outcomes. Stage 1 studies would be preceded by preliminary measurements of pharmacokinetics/pharmacodynamics (PD/PK) and brain penetrance of the therapy to aid in dose selection and route and time of administration. If reliable, species-specific information was available from previous studies, this step could be omitted.
Therapies that improved outcome in Stage 1 screening could advance to Stage 2 trials that might include both sexes, young and old animals, and more numerous, mechanistic outcomes. Stage 2 studies also could include physiological measurements (e.g., CBF, ICP, BBB permeability, microdialysis measurements of transmitters, metabolites), detailed assessments of PD/PK and toxicity, and post-traumatic insults such as hypotension, hypoxia, anemia, and/or polytrauma. An alternative approach would limit the Stage 1 screening studies to single sexes, ages, and injuries, and encourage the formation of additional multi-center consortia that would focus on more complex issues such as the chronic effects of TBI, TBI model development and characterization, mild TBI and/or repetitive mild TBI, combination therapies, etc.
One important requirement of OBTT is the use of a detailed manual of standard operating procedures (MSOP).89 The symposium participants agreed unanimously about the importance of the creation and constant updating and revising of the MSOP. The diligent use of accurate, detailed MSOPs ensures that all methods and procedures are consistently and rigorously used among all participating centers and, more importantly, facilitates the replication of the research by other investigators. This is especially important in view of the doubt about the reliability of much scientific data.77,101–103 When the biotechnology firm Amgen tried to replicate 53 “landmark” pre-clinical cancer studies, it was able to confirm the results in only six (11%).64 Begley and Ellis64 noted that the authors of the studies that were confirmed provided detailed information about controls, reagents, blinding, and investigator bias.
Additional reasons for unreliable data or questionable results include inadequate sample size77 and errors in data collection and analysis.102 In a study that supported the importance of a detailed MSOP, Carp reported103 that, of the 241 functional MRI studies examined, fewer than half provided important information about data collection (e.g., number of subjects not analyzed, the reasons for rejection, whether or how the subjects were compensated, etc.) or methods (e.g., corrections for differences in slice acquisition timing or co-registration to high-resolution scans, whether temporal filtering was conducted, etc.). For a pre-clinical therapy study to be appropriately replicated, transparent reporting and accurate details about the TBI models, surgical procedures, outcomes and data collection, and analysis are required, and those details would be available only with a meticulous, up-to-date MSOP. Further, pre-clinical screening studies, either individual or multi-center, often take months to years to complete and during that interval personnel and/or facilities may change, increasing the likelihood that minor procedural details will vary, either intentionally or inadvertently. Regular, routine updates of the MSOPs will help to minimize the effects of these variations on data reproducibility. These and other recommendations about study design and conduct are included in the RIGOR guidelines from the NINDS.104
Go/No-Go decision points
Go/No-Go decision points are built into many screening studies to limit futile testing of therapies that prove unlikely to produce positive results. The Go/No-Go decision point in the OBTT studies occurred at the end of first-stage testing when levetiracetam, which improved MWM performance in two TBI models, was approved for further study (Go) but erythropoietin, cyclosporine, simvastatin, and nicotinamide were not (No-Go) because they failed to improve outcomes in more than a single TBI model.90 Because the survival interval for the OBTT experiments was 3 weeks, the screening of the five agents took years. A test that predicted the 3-week outcomes but could have been conducted 24 h, or even a week post-injury, would have saved a considerable amount of time, money, and effort. To date, no early test (or method, procedure, task, etc.) that accurately predicts outcome exists but there are some that show promise.
Fluoro-Jade (FJ) is a fluorescent stain that identifies degenerating cells as early as 4 h after injury,105–107 although there is evidence that FJ-positive cells may be injured but not necessarily dying,108 and FJ may stain non-neural cells (e.g., activated microglia, astrocytes) under some circumstances.109 FJ is a potentially valuable screening tool to assess the neuroprotective effects of therapies soon after injury. However, it is unclear whether cell death represents the key target for therapies even in patients with severe TBI, or should be seen as the “one target” for therapies. For example, mechanisms such as axonal injury, cerebral edema, or even cell signaling, among others could be critically important. The key target may also depend on the TBI phenotype, injury severity, timing, and/or presence of secondary extracerebral insults.
Perhaps the most promising early indicators of injury and therapeutic efficacy are serum biomarkers. A component of the OBTT consortium, Mondello and colleagues100 demonstrated that serum levels of glial fibrillary acidic protein (GFAP) sampled 4 h after all three OBTT TBI models (i.e., FPI, CCI, PBBI) correlated with lesion volume and cortical tissue loss 21 days after injury. Four-hour serum levels of GFAP also correlated significantly with MWM latency to goal platform after FPI but only weakly with MWM performance after CCI or PBBI.100 These data provided clear evidence that serum GFAP level measurements sampled 4 h after TBI could represent a valuable surrogate histological and/or theranostic end-point that might also serve an ideal tool for screening of therapies and, perhaps, Go/No-Go decisions very soon after injury. That approach (or other biomarkers) clearly warrants future studies of higher throughput screening in pre-clinical TBI models. GFAP was recently “approved” by the FDA as a biomarker for mild TBI.
Despite the promise of methods such as FJ and serum biomarkers for early decisions about therapeutic efficacy, the desirability of very early screening is unclear. As noted above, the time courses of the pathophysiological mechanisms of TBI vary widely over hours to days post-TBI. Early screening would markedly underestimate the efficacy of agents that required repeated or delayed administration. Restorative therapies would not be expected to act within hours or even days or weeks post-TBI. The inclusion of Go/No-Go decision points and the timing and methods used for those decisions should include consideration of the time courses of the pathophysiological processes of the study injury models and the PD/PK and putative mechanisms of action of the test therapies in the test species.
Intersite communications
The OBTT consortium design includes monthly teleconferences among the site investigators. These calls provide opportunities for progress updates and discussions about problems and/or questions about models (e.g., excess mortality, unexpected morbidity, outcomes [unexpected behaviors or injury or drug effects]) and other procedural issues. To minimize the risk of inadvertent bias, results are not discussed until all centers have completed outcome data collection. Although questions and concerns of the laboratory personnel are addressed in the monthly calls, the personnel conducting the experiments are not included to preserve blinding.
Experimental Models of TBI
Evaluations of the pro and cons of myriad existing TBI models and recommendations for the “best” TBI models were beyond the scope of the symposium discussions, and also of this review article. These issues have been thoroughly covered in numerous other review articles.37–41,110 It is worth noting that all animal models have limitations and only replicate certain features of clinical TBI, which is heterogeneous in nature.
Chronic traumatic encephalopathy (CTE)
Although TBI model development and characterization were not goals of OBTT and Stage 1 therapy testing studies are not well suited to these tasks, the need for an experimental TBI model that reliably replicated significant histopathological and behavioral features of CTE was recognized. The correlation between repeated (usually mild) TBI and chronic or late-developing cognitive dysfunction has been recognized for decades. Punch drunk and dementia pugilistica were terms used in the 1920s and 1930s to describe the long-term effects of TBI.111,112 The dementia related to repeated mTBI (i.e., CTE) is believed to be due to an excessive accumulation of the microtubule-associated protein tau.113–115 Goldstein and colleagues116 observed histopathological evidence of phosphorylated tau deposition in the brains of military personnel who had sustained blast-induced TBI, suggesting that blast injury may contribute to CTE. Currently, there is no single experimental model that produces all CTE-like behavioral and pathological effects, but some of these effects can be replicated in rodent TBI models. Goldstein and colleagues116 reported histological evidence of CTE-like neuropathology including tau protein–linked immunoreactivity, persistent perivascular pathology, degeneration of cortical and hippocampal neurons, and chronic inflammation (i.e., astrocytosis and microgliosis) in the brains of mice 2 weeks after simulated blast (compressed gas-driven shock tube). Shock-wave–exposed mice also exhibited deficits in spatial learning and memory, as measured using the Barnes maze.116 However, it is important to note that most of the CTE-like histopathological and behavioral effects of shock-wave–exposure in mice were prevented by immobilizing the animals' heads, thereby preventing the rapid oscillations produced by the shock wave. Thus, rather than a model of blast-induced CTE, Goldstein and colleagues116 may have created a model of CTE produced by rapid, repetitive head acceleration/deceleration.
Certain CTE-like neuropathological changes also can be replicated by CCI. Glushakova and associates117 reported BBB damage, perivascular microbleeds, and white-matter damage that progressed for at least 3 months after CCI. BBB damage,118 perivascular microbleeds, and deep white-matter damage often are observed in humans after repeated, mild TBI.113 Briggs and co-workers119 reported CTE-like neuropathological changes (e.g., increases in phosphorylated tau, TDP-43, neuroinflammation, reductions in white-matter volume) 53 days after the last of 30 mild, weight-drop TBIs (over 6 weeks) in mice. They also observed deficits in spatial memory acquisition (Barnes maze) and rotarod performance. The authors stated that the TBI levels used were mild, based on duration of righting reflex suppression, but they reported mortality rates of about 20%, a degree of lethality that would not be expected in mild TBI models. In an excellent recent review of CTE and tau, Ojo and colleagues,120 observed that, of the 29 studies of tau levels after TBI in rodents, 23 reported increased tau and six reported no change in tau. Of the 16 tau-positive murine studies, eight used genetically modified mice. The most substantive limitation of most current rodent models of CTE is that they fail to replicate typical neuropathological characteristics of CTE in humans such as neurofibrillary tangles, astroglial tangles, neuropil threads, or peri-vascular or peri-ventricular p-tau.120 An important consideration in the development of rodent models of mild TBI and CTE is the potentially confounding effects of anesthetics on neuronal injury, CBF, neurotransmitter signaling, etc.110
Multiple rodent models
The OBTT consortium used moderate FPI, severe CCI, and severe PBBI TBI models because these models replicated many important features of diffuse, focal, and penetrating TBI, respectively, in humans.11,89 An alternative would be the use of the same model by multiple centers, an approach attempted in the MASCIS spinal cord injury study.90,91 One of the primary advantages of the MASCIS approach was inherent intersite replication of results. However, because human TBI is a heterogeneous insult,8 testing of therapies in rodent models that replicate multiple features of different types of human TBI increases the likelihood that therapies that show promise in different types of experimental TBI models also will do so in heterogeneous human TBI. This is the rationale for the previous recommendations for therapy testing in multiple models in stroke (i.e., STAIR criteria)42 and TBI.9,15,121
Large animal models
Compared with rodents, large-animal TBI studies are resource-intensive in terms of money, space, personnel, etc. Despite these considerations, large-animal studies provide unique contributions to TBI research. The cerebral cortices of rats are lissencephalic, lacking gyri and sulci, whereas those of humans, pigs, and other higher mammals are gyrencephalic and divided into gyri by sulci. The use of experimental animals with sulci may be especially important in modeling CTE because the tau pathologies appear to be preferentially localized in the depths of sulci in human CTE.113,122 There also are significant differences in brain weights, volumes, and surface areas between rats,123 pigs,124 and humans.125,126 The ratios of white to gray matter differ considerably among species and, in general, the volume of white matter increases with the size of the brain due to the need for longer fibers with increasing brain size.127 There are major differences in the anatomy of the skull between pigs and rats.128 These differences in the anatomy of the brains and skulls in the different species used in research should be considered in the interpretation of experimental results, especially in studies of blast TBI.
The OBTT design included plans to test therapies successful in multiple rodent models using the FPI model in micro-pigs.11,89 To date, only levetiracetam has improved cognitive outcome in multiple rodent models (FPI and CCI), and further evaluations of levetiracetam in FPI in micro-pigs are ongoing.95 As noted above, the importance of large animals in therapy testing is well recognized.8,15,42,110,121 but an additional consideration is that FDA approval for a new drug (i.e., Investigational New Drug; IND) or repurposing a previously approved agent may require proof of efficacy in large animals as well as rodents. Cell-based therapies almost invariably require data from large-animal studies (W. Dalton Dietrich, personal communication). Symposium participants who serve or had served on clinical trial study sections for the National Institutes of Health (NIH) commented that proof of efficacy in large-animal studies was crucial for NIH support for clinical trials. Finally, there is an expectation that, in view of the numerous failures of clinical trials for TBI therapies, the NIH is reassessing guidelines for NIH-supported clinical trials with a goal of aligning NIH guidelines with those of the FDA.
Therapies
Therapy selection
The therapies selected by OBTT were FDA-approved agents that had been shown to improve behavioral and/or histopathological outcomes in experimental TBI studies.89 Although a higher priority was assigned to therapies supported by a large body of experimental evidence or those that demonstrated very impressive results in a few or even a single study, novel therapies with significant theoretical potential but limited track records also were considered.11 Several other considerations were suggested by symposium participants (Table 5).
Table 5.
Potential Therapy Selection Criteria
| Criterion | Rationale |
|---|---|
| FDA approval | Therapies approved for human use more readily translate to clinical studies |
| Efficacy | Therapies with efficacy in previous experimental TBI studies are more likely to prove successful in further pre-clinical testing |
| PD/PK in species | Facilitates dose and time and route of administration selection |
| BBB permeability | Increases likelihood of achieving therapeutic levels in brain |
| Human use | Therapies with long histories of use in humans are less likely to have significant negative side effects |
| Known targets/Mechanisms | Facilitates monitoring of dose and time/route of administration efficacy; therapies with multiple targets/mechanisms may act on multiple deleterious pathways |
| Novelty | High risk/high reward |
BBB, blood–brain barrier; FDA, Food and Drug Administration; PD/PK, pharmacodynamics/pharmacokinetics.
Doses and routes of administration
Ideally, pre-clinical studies would use a range of drug doses/concentrations to generate complete dose/response information. This approach often is impractical for in vivo studies, especially in a large, multi-model consortium such as OBTT. Most investigators use a single dose or, like OBTT, low and high doses based on information from previous studies in the same species and consultation with faculty experts in PD/PK.11 Similarly, decisions about route of administration typically are based on previous research and/or the BBB permeability of the therapeutic agent. Pre-clinical screening studies should use routes of administration that translate easily to patient studies (e.g., intravenous). In the case of agents with low brain penetrance, direct injections into the cerebral ventricles might be possible in patients with moderate/severe TBI with intraventricular ICP monitors/drains but not in those without such devices in place. Information about PD/PK in more than one study species is particularly valuable in determining doses, routes of administration, and times and durations of administration (see below). Such evaluations allow for estimation of the relationship between dose and both blood and brain tissue concentrations as they relate to observed neuroprotection and potential toxicity. Further, the relationship between concentration and a biomarker of target engagement with respect to the resultant measure of neuroprotection should be evaluated. Such studies should also evaluate different degrees of brain injury severity to ultimately determine the therapeutic window as a function of the severity of brain injury. However, these relationships are complex and may vary depending on myriad factors such as the therapy, the amount of BBB damage, and toxicity, among others.
Time and duration of administration
Decisions about the post-injury time of therapy initiation primarily are based on two considerations. Initial administration several hours post-TBI is more readily translatable to human TBI because of the time required for patient transport, admission, early diagnostic testing (e.g., examination; imaging, usually CT) and the acquisition of consent from the patient or the patient's family. In theory, safe, stable, easily administered therapies could be started by first responders but rarely is this feasible. In contrast, administration as soon as possible after injury is better suited to interrupt pathophysiological mechanisms that begin soon after injury. For example, poly(ADP-ribose) polymerase (PARP) activation increased as early as 30 min after FPI in rats129 and excitatory amino acid levels increased within minutes after TBI in rats.130 Neuronal injury due to these and numerous other secondary injury processes could be interrupted only by therapies administered within minutes of TBI. To increase the likelihood of detecting a positive effect, therapy was initiated in the OBTT trials 15 min after injury. The symposium consensus opinion was that therapies that showed promise when administered early could then be re-tested with delayed initiation of therapy. However, some deleterious secondary injury mechanisms may only operate in delayed periods after the injury, and thus the timing of therapy administration should be tailored to the mechanistic target. For example, in OBTT, testing of the drug simvastatin used a more delayed and chronic approach to dosing96 based on prior literature and targeting of mechanisms such as regeneration and repair.131,132 Indeed, a therapy that targets regeneration or rehabilitation might optimally be administered days, weeks, or even months after the injury.
Decisions about duration of therapy are best based on PD/PK information about the therapy in the study species and the time course of the pathophysiological mechanism(s) targeted. Therapies with short plasma or tissue half-lives may have to be administered repeatedly to be effective. Even if administered early, drugs with slow BBB penetrance might not reach therapeutic brain tissue concentrations for hours after administration. Knowledge of the time course of targeted mechanisms also is important to ensure adequate drug concentrations during the relevant time intervals. For example, lipid peroxidation and mitochondrial oxidative damage started at 24 h and peaked 72 h after CCI in rats.133 Therapies started immediately after injury might be ineffective unless adequate tissue concentrations persisted for days after administration. Additionally, the duration of therapy should be related to injury processes in each experimental model. Both FPI62 and CCI54 result in progressive neuronal damage for at least a year post-injury. Early treatment might interrupt the processes that result in chronic neurodegeneration, but it is also possible that long-term or delayed administration would be necessary.
Outcomes
As in every other aspect of experimental TBI research, perhaps the most important consideration in outcome assessments is the need to document all outcome measurement methods/procedures in meticulous detail. As evidenced by the OBTT consortium, there are different ways to conduct and analyze outcome measures, particularly behavioral tasks. The Miami group assessed spatial memory performance on post-injury days (PID) 13–16, and tested for retention of the hidden platform location with a probe (missing platform) trial on PID17 and for working memory on PID20–21, whereas the Pittsburgh group conducted spatial memory hidden platform testing on PID14–18, a visible platform test on PID19–20, and a probe trial on PID20.89 A cursory examination of the experimental TBI literature revealed substantive differences in MWM procedures. Smith and colleagues134 measured spatial memory function using an elegant system of overlapping ovals to measure the time to the goal platform in 20 trials over 2 days. Dash and colleagues135 assessed spatial memory using the MWM with 2 days of training followed by a single day of probe trial testing. Dixon and colleagues54 measured spatial memory acquisition and retention with r days of hidden platform testing, a probe trial to measure place memory, and 2 days of visible platform trials. The MWM procedures in these studies were described in appropriate detail, but without such careful descriptions, it would be impossible to replicate these studies. As noted above, scientists doing TBI research, as do all investigators, have a responsibility to document all methods and procedures, especially those related to models and outcomes, in sufficient detail to permit accurate replication.
Primary outcomes selection
The OBTT organizers elected to allow each center to use the outcome measures with which they had the most experience. All centers included primary outcomes that assessed biomarkers, motor and cognitive functions, and neuropathology, but the specific tasks in each category varied among centers.89 Outcome variables that were the same across centers were the timing of blood sampling for biomarker analysis (4 and 24 h post-injury and at euthanasia), time of euthanasia (21 days), and an analysis of time to the goal platform and a probe trial in the MWM task.89 The standardized procedures enabled comparisons across models and centers, whereas the individual methods provided the opportunity to detect treatment effects specific to the method of analysis. The primary consensus recommendation related to outcome selection was that the clinical relevance of the outcome be established. For example, the GCS is the primary determinant of state of consciousness and level of injury in TBI patients. The duration of righting reflex suppression, a reasonable measure of the level of consciousness in experimental animals,110 is a method currently used by several TBI laboratories.40 The righting reflex is a mesencephalic reflex that returns before thalamocortical function during recovery from unconsciousness due to anesthesia or brain injury.136 The return of the righting reflex is considered to be analogous to the return of consciousness in humans.137–139 Spatial and working memory are aspects of cognitive function that are frequently tested in experimental TBI studies. Additionally, other types of higher cortical functioning that are assessed in TBI patients (e.g., inhibition/disinhibition, attention, executive functioning, problem solving) can be measured in rodents.40 (For an excellent review of human outcomes and their animal equivalents, see Shultz and colleagues.110)
Secondary/supplemental outcomes
For pre-clinical therapy screening studies, secondary outcomes were defined as those that provide additional information but wouldn't be given high priority in decisions about the efficacy of the test therapies. The consensus recommendation was that screening studies should be as “stripped down” as possible to provide as much rigorous therapy testing as financial resources permitted. In-depth explorations of mechanisms, pathways, etc., would be reserved for further studies in successful agents. Imaging is frequently a component of clinical TBI studies but has been relatively rare in past experimental TBI research.9 However, as noted above, imaging is becoming more common in experimental TBI research.45,53 Although MRI and other types of imaging may be a component of TBI clinical research studies, imaging typically is limited to CT in the acute care of TBI patients. Imaging may have a role in Stage 2 studies but was not considered to be especially useful in the determination of the success or failure of test drugs.
Common data elements (CDEs)
Recently, the NINDS undertook the development of a set of CDEs for TBI research that were intended to facilitate reporting, data sharing, comparison of results, and collaboration by standardizing the definitions and protocols for clinical TBI research.140 This was followed by the formation of a set of CDEs for experimental TBI research.141 Symposium participants recommended the consideration of the NINDS CDEs in the selection of primary and supplemental outcome variables. This recommendation was made with the understanding that the current CDE lists141 should not be interpreted as excluding potential outcome measures. The CDE lists are evolving and variables not currently included can be added later.
Outcome weighting
The goal of pre-clinical screening is to determine whether a therapy warrants further study and/or translation to clinical studies. It is conceivable that screening studies, especially multi-center, multi-model studies, would yield mixed results with some outcome measures showing positive results but others showing no evidence of efficacy. OBTT uses a point system in which outcome categories contributed part of the total score. The motor (4), cognitive (10), neuropathology (4), and serum biomarker (4) categories are assigned scores that, if the test therapy improves outcome in all categories, total 22 points.89 When designed, this scoring system provided a mechanism to assign priorities for further testing if all five of the therapies in the initial round of screening proved efficacious. Alas, prioritizing proved unnecessary but the system was felt to be reasonable. The consensus opinion was that assigning cognitive outcome a disproportionately high value is appropriate. In fact, the consensus opinion was that improvements in cognitive outcome, even in the absence of positive effects in any other outcome, would be sufficient to qualify a therapy for further testing.
Long-term outcomes
In experimental animals, TBI resulted in long-term deficits in histopathological and behavioral outcomes.11,54,62 (For a review, see Osier and colleagues57 and Bramlett and Dietrich. 60) Patients sustaining a TBI, even a mild TBI, may suffer from neurological and behavior effects for years after injury142,143 as well as a reduced life expectancy.144 (For a review, see Masel and DeWitt.5) Despite this recognition of the lasting consequences of TBI, relatively few experimental studies include long-term end-points. An analysis of 193 “long-term” experimental TBI studies57 yielded only 18, nine, and one with durations of 6 months or more, 1 year or more, and more than 1 year, respectively. These data suggest that less than 10% of all “long-term” experimental TBI studies lasted longer than 6 months and the percentage of all experimental studies with long-term outcome assessments is likely a fraction of a percent. Although there is a clear need for more experimental research on the long-term effects of TBI, assessments of the chronic effects of TBI are ill-suited for Stage 1 screening studies because of the extensive time, money, and personnel required. The consensus opinion is that the best approach is multi-center consortium studies designed specifically to characterize the chronic effects of TBI and identify potential therapies that might prove effective when administered weeks to months post-injury.
Data Analysis and Sharing
Data analysis
The consensus recommendation is to involve statisticians in all aspects of study design and data collection, collation, analysis, and sharing. A central data core is desirable in any multi-center effort. Individual center investigators and their statistician colleagues are capable of analyzing their own data, but a central core is well suited for data analysis across all sites. A relatively autonomous data core reduces the likelihood of the inadvertent bias that could occur if the overall data analysis was conducted by one of the participating centers. A central data core facilitates blinding during the across-center analysis. However, central data cores are an additional expense that might be viewed initially by the source of research support as an unnecessary luxury, making it necessary to establish a compelling rationale that justifies the expenditure.
Data sharing/dissemination
Data dissemination is one of the bedrock principles of research. In the neurotrauma field, data repositories have been created to facilitate the sharing of the results from multiple TBI studies. The International Mission for Prognosis and Analysis of Clinical Trials in TBI (IMPACT) database, which was supported by the NIH, included data from eight RCTs and three observational studies from three TBI centers (later expended to four) in Europe and the United States.145,146 The Federal Interagency TBI Research Informatics System (FITBIR) is a database created by the U.S. Department of Defense and the NIH that comprises TBI patient data.147,148 Presently, FITBIR is limited to clinical data but eventually will expand to include CDEs from pre-clinical studies.141 When FITBIR has the capability of accepting pre-clinical information, data from individual studies as well as multi-center consortia can be added. However, data sharing and dissemination might be delayed by the reluctance of investigators to share unpublished data. Typically, data are sequestered for 6 to 12 months before being widely disseminated.
In an important recent article about data sharing, Ferguson and colleagues,149 observed that in addition to “big data” datasets, there exists a great deal of data collected in small datasets that the authors refer to as “ long tail” data, usually collected by individual investigators as small datasets. Although these small datasets are difficult to collect and disseminate, there is a clear need to do so because the long tail data together represent the majority of data collected by the neuroscience research community. Widespread access to long tail data is useful for discovery purposes but also for issues related to transparency and study replication. Reasons for not disseminating long tail data include concerns about the quality of these data and their use by non-experts, the cost of collecting and maintaining the database, and the potential negative impact of sharing unpublished data on the researchers' careers,149 as well as the commercial interests of pharmaceutical companies. Whether these perceived limitations can be overcome remains to be seen.
A second and perhaps more challenging component of long tail data is so-called dark data from failed pre-clinical studies as well as ancillary data such as animal care records.149 Virtually all TBI researchers have data from studies with negative results and most remain unpublished because publishing negative results is difficult. Researchers have a finite amount of time and most prefer to focus that time on data that are more readily reported. Although this is a reasonable preference, unpublished negative experiments might be repeated by others, thereby wasting the time and money of other researchers. In addition, publication bias toward positive studies, particularly in the absence of adequate rigor, can mislead other investigators to pursue unfruitful paths.150,151 Because many of the negative studies and their unnecessary duplication were supported by government grants, this represents potentially billions of dollars of wasted public funds.152 Ferguson and colleagues153 offer several options to facilitate the collection and dissemination of long tail data and their dark component. There was consensus agreement with Dr. Ferguson's observations, concerns, and recommendations that mechanisms be developed to collect and share all pre-clinical TBI data.
To facilitate data sharing, Ferguson and colleagues proposed that data should be discoverable, accessible, intelligible, assessable, and usable.149 Discoverable accessible data can be readily located through a search and then easily interrogated. Intelligible data use standard nomenclature that can be read and understood by humans and machines. Datasets should be complete with sufficiently detailed information to allow assessments of reliability. Usable data would be formatted to facilitate use without extensive conversion. (For details, see Box 2 in Ferguson and colleagues.149) A similar set of recommendations was incorporated in the FAIR (Findable, Accessible, Interoperable, and Reusable)154,155 guidelines. Among other features, findable includes the assignment of a unique, persistent identifier and the registration of data in searchable resource. Accessible refers to the data being retrievable by their identifier using a standardized communications protocol that is open, free, and universally implementable. Interoperable means that the data use a formal, accessible, shared, and broadly applicable language for knowledge representation. The reusable principle means that the data are described with accurate and relevant attributes. (For details, see Table 2 in Wilkinson and associates.154)
Concluding Statement
The major conclusion from the workshop was that we have identified and drawn attention to several needs, issues, and biases that plague the current experimental TBI research and literature, and we suggested potential steps to deal with them. We hope that the development of multi-institutional networks and consortia, similar to OBTT, will minimize many of these biases while optimizing resources and exploiting the different expertise. Such collaborations would allow easy, standardized, and harmonized data collection and access to information on experimental TBI studies. Further, rigor in designing and performing scientific research will ensure reproducibility and validated results facilitating unbiased evaluation of tested therapies, fewer false-positives, and, hopefully, more rapid identification of effective drugs. Support from funding agencies, regulatory bodies, and journal editors is essential and would greatly improve the chances for success.
Consensus Summary
Below are the consensus recommendations of the participants of the Moody Project TBI Symposium for pre-clinical testing of therapies for TBI.
Study design
Multi-center pre-clinical screening could be categorized as Stage 1 or Stage 2 studies. Stage 1 studies would use a defined species, age, and sex and one or two doses of test drugs.
Therapies that improved outcome in more than one TBI model would advance to Stage 2 testing that could include multiple drug doses and/or times and durations of administration, combined injury models such as TBI and hemorrhage, male and female animals, and mechanistic variables such as imaging, electrophysiology, etc.
All experimental procedures, methods, outcomes, etc., should be described in detail in Manuals of Standard Operating Procedures (MSOP), which is updated regularly. This recommendation is applicable to single-center and single-investigator studies as well as multi-center, multi-investigator pre-clinical studies of therapies for TBI.
Appropriate randomization of drug administration and blinding of key research personnel is recommended to ensure robust and unbiased experimental design. The inclusion of complete details of models and outcome assessments, randomization, blinding, etc., in the operating procedures and resultant publications would facilitate replication.
If included in the study design, Go/No-Go decisions should take into account the time-courses of the pathophysiological processes of the injury models, the PD/PK, and putative mechanisms of action of the test therapies in the test species.
Intersite communications provide the opportunity for progress updates and discussions of unexpected events related to models or outcomes. Questions or concerns of laboratory personnel conducting the experiments would be included in these discussions.
Models
The use of well-characterized experimental TBI models that replicate important features of TBI in humans increases clinical relevance and limiting Stage 1 studies to one level of injury in each model enhances efficiency and feasibility. Additional injury levels could be investigated in Stage 2 experiments.
Therapies that show positive results in Stage 1 and 2 rodent studies should be tested in TBI studies in large animals, such as pigs, with gyrencephalic brains.
Therapies
Therapy selection
Optimally, the selection of potential test therapies for pre-clinical screening studies would be based on the likelihood of eventual use in humans by careful vetting by expert collaborators in pharmacology to ensure that they have potential druggability (low toxicity, BBB permeability, etc.). Ideally, test therapies would have a wide therapeutic window between neuroprotective efficacy and side effects/toxicity to increase the likelihood of eventual translation to patient studies.
Additional criteria for therapy selection could be positive results in previous TBI or other central nervous system (CNS) injury models or novel and/or multiple targets known to be important in the pathophysiology of TBI.
Given the experience of OBTT—where the positive effects of therapies were less than anticipated from the published literature—there would appear to be value in defining therapies showing cross-model efficacy, or potent efficacy in a single model, even if that specific drug could not be advanced. Such findings could help direct the field toward a key mechanism and/or other derivatives that might be able to be synthesized.
Particularly for repurposed drugs, doses used in pre-clinical animal models should be evaluated for the likelihood of human equivalent dosing in future clinical studies through dose estimation methods.
Doses and route of administration
Symposium participants recommended that doses and routes of administration be selected based on known PD/PK and brain penetrance information in the test species, efficacy against a defined mechanistic target viewed as important to TBI, and/or by preliminary studies in the test species. It is especially important to include studies to confirm that the therapy produced the desired effect on the target mechanism, if known.
It is important to include considerations of the feasibility of route in the target patient population in the selection of routes of administration.
Time and duration of administration
If available, PD/PK information would guide the decisions about the most effective time of therapy initiation and duration.
Although an early time of initiation of therapy may reduce the clinical relevance of the study, early administration is appropriate for some Stage 1 screening, depending on the therapy and therapeutic target. Later times of administration could be tested in either Stage 1 or Stage 2 studies of successful therapies.
Considerations of the time courses of the pathophysiological targets of the test therapy in time-of-administration decisions would increase the likelihood of positive results.
Outcomes
Primary outcomes selection
The clinical relevance of the primary outcomes is an important consideration in outcome selection. Outcomes that have no human equivalent are of limited value in pre-clinical therapy screening.
For multi-center screening studies, the inclusion of standardized outcome measures would facilitate comparisons among models and centers.
The selection of a limited number of measurements directly relevant to behavioral or pathological outcomes would improve the efficiency of Stage 1 studies. Supplemental/secondary outcomes such as imaging, physiology (e.g., CBF, ICP) or those intended for detailed explorations of pathophysiological pathways could be reserved for Stage 2 studies. However, in outcome selection, consideration should be given to special cases, such as drugs targeting a reduction in ICP or mechanisms relevant to the link between TBI and chronic neurodegenerative diseases.
Common data elements
The NINDS CDEs should be considered in primary outcome selection with the inclusion of outcomes not currently specified in the CDE list.
Long-term outcomes
Although the importance of the measurements of the chronic effects of TBI is recognized, long-term outcomes are not considered to be practical in Stage 1 pre-clinical screening studies.
Data analysis and sharing
If funding permits, a central data core is an asset to a multi-center pre-clinical screening study. Multi-center data analysis by a data core is more easily blinded and less susceptible to inadvertent bias than data analysis at individual sites.
Rapid dissemination of information generated by any TBI study is important and data moratoria should be as short possible. If available and appropriate, data sharing via large international TBI databases such as IMPACT and FITBIR would facilitate dissemination.
Reporting data and results from unsuccessful/negative pre-clinical screening studies in peer-reviewed journals, and/or disseminated using a widely available database is recommended to reduce unnecessary duplication.
Acknowledgments
We thank Andrew Hall for editorial assistance with this manuscript. The Moody Project Symposium and manuscript submission fees were supported by funds from The Moody Project for Translational Traumatic Brain Injury Research. We would also like to thank the important work performed by pre-clinical and clinical team science efforts such as The Multi-center Animal Spinal Cord Injury Study (MASCIS), Operation Brain Trauma Therapy (OBTT), Transforming Research and Clinical Knowledge in TBI (TRACK-TBI), International Initiative for Traumatic Brain Injury Research (InTBIR), Collaborative European NeuroTrauma Effectiveness Research in TBI (CENTER-TBI), International Mission for Prognosis and Analysis of Clinical Trials in TBI (IMPACT), and Federal Interagency Traumatic Brain Injury Research (FITBIR) of which we collectively amassed previous experience contributing to these discussions.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Faul M., Xu L., Wald M.M., and Coronado V.G. (2010). Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Contr (ed). Available at: http://www.cdc.gov/traumaticbraininjury/tbi_ed.html. Accessed April18, 2017 [Google Scholar]
- 2.Roozenbeek B., Maas A.I., and Menon D.K. (2013). Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 9, 231–236 [DOI] [PubMed] [Google Scholar]
- 3.Tagliaferri F., Compagnone C., Korsic M., Servadei F., and Kraus J. (2006). A systematic review of brain injury epidemiology in Europe. Acta Neurochir. (Wien) 148, 255–268 [DOI] [PubMed] [Google Scholar]
- 4.Langlois J.A., Rutland-Brown W., and Wald M.M. (2006). The epidemiology and impact of traumatic brain injury: a brief overview. J. Head Trauma Rehabil. 21, 375–378 [DOI] [PubMed] [Google Scholar]
- 5.Masel B.E., and DeWitt D.S. (2010). Traumatic brain injury: a disease process, not an event. J. Neurotrauma 27, 1529–1540 [DOI] [PubMed] [Google Scholar]
- 6.Humphreys I., Wood R.L., Phillips C.J., and Macey S. (2013). The costs of traumatic brain injury: a literature review. Clinicoeconom. Outcomes Res. 5, 281–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kabadi S.V., and Faden A.I. (2014). Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int. J. Mol. Sci. 15, 1216–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saatman K.E., Duhaime A.C., Bullock R., Maas A.I., Valadka A., Manley G.T., and Workshop Scientific Team and Advisory Panel Members (2008). Classification of traumatic brain injury for targeted therapies. J. Neurotrauma 25, 719–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agoston D.V., Risling M., and Bellander B.-M. (2012). Bench-to-bedside and bedside back to the bench; coordinating clinical and experimental traumatic brain injury studies. Front. Neurol. 3, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menon D.K. (2009). Unique challenges in clinical trials in traumatic brain injury. Crit. Care Med. 37, S129–S135 [DOI] [PubMed] [Google Scholar]
- 11.Kochanek P.M., Bramlett H., Dietrich W.D., Dixon C.E., Hayes R.L., Povlishock J., Tortella F.C., and Wang K.K.W. (2011). A novel multicenter preclinical drug screening and biomarker consortium for experimental traumatic brain injury: Operation brain trauma therapy. J. Trauma 71, S15–S24 [DOI] [PubMed] [Google Scholar]
- 12.Loane D.J., and Faden A.I. (2010). Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 31, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke M.J., Fralick M., Nejatbakhsh N., Tartaglia M.C., and Tator C.H. (2015). In search of evidence-based treatment for concussion: characteristics of current clinical trials. Brain Inj. 29, 300–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bragge P., Synnot A., Maas A.I., Menon D.K., Cooper D.J., Rosenfeld J.V., and Gruen R.L. (2016). A state-of-the-science overview of randomized controlled trials evaluating acute management of moderate-to-severe traumatic brain injury. J. Neurotrauma 33, 1461–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawryluk G.W.J., and Bullock M.R. (2015). Design of acute neuroprotection studies, in: Handbook of Clinical Neurology, Traumatic Brain Injury, Part II, Volume 128. Grafman J., and Salazar A.M. (eds). Elsevier: Amsterdam: pps. 761–778 [DOI] [PubMed] [Google Scholar]
- 16.Marklund N., Bakshi A., Castelbuono D.J., Conte V., and McIntosh T.K. (2006). Evaluation of pharmacological treatment strategies in traumatic brain injury. Curr. Pharm. Design 12, 1645–1680 [DOI] [PubMed] [Google Scholar]
- 17.Maas A.I., Murray G.D., Roozenbeek B., Lingsma H.F., Butcher I., McHugh G.S., Weir J., Lu J., Steyerberg E.W., and International Mission on Prognosis Analysis of Clinical Trials in Traumatic Brain Injury (IMPACT) Study Group. (2013). Advancing care for traumatic brain injury: findings from the IMPACT studies and perspectives on future research. Lancet Neurol. 12, 1200–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson C.S., Valadka A.B., Hannay H.J., Contant C.F., Gopinath S.P., Cormio M., Uzura M., and Grossman R.G. (1999). Prevention of secondary ischemic insults after severe head injury. Crit. Care Med. 27, 2086–2095 [DOI] [PubMed] [Google Scholar]
- 19.Rockswold G.L., Ford S.E., Anderson D.C., Bergman T.A., and Sherman R.E. (1992). Results of a prospective randomized trial for treatment of severely brain-injured patients with hyperbaric oxygen. J. Neurosurg. 76, 929–934 [DOI] [PubMed] [Google Scholar]
- 20.Jiang J.Y., Xu W., Li W.P., Gao R.Y., Bao Y.H., Liang Y.M., and Luo Q.Z. (2006). Effect of long-term mild hypothermia or short-term mild hypothermia on outcome of patients with severe traumatic brain injury. J. Cereb. Blood Flow Metab. 26, 771–776 [DOI] [PubMed] [Google Scholar]
- 21.Zhi D., Zhang S., and Lin X. (2003). Study on therapeutic mechanism and clinical effect of mild hypothermia in patients with severe head injury. Surg. Neurol. 59, 381–385 [DOI] [PubMed] [Google Scholar]
- 22.Clifton G.L., Miller E.R., Choi S.C., Levin H.S., McCauley S., Smith K.R., Muizelaar J.P., Wagner F.C., Marion D.W., Luerssen T.G., Chesnut R.M., and Schwartz M. (2001). Lack of effect of induction of hypothermia after acute brain injury. N. Engl. J. Med. 344, 556–563 [DOI] [PubMed] [Google Scholar]
- 23.Clifton G.L., Allen S., Barrodale P., Plenger P., Berry J., Koch S., Fletcher J., Hayes R.L., and Choi S.C. (1993). A phase II study of moderate hypothermia in severe brain injury. J. Neurotrauma 10, 263–271 [DOI] [PubMed] [Google Scholar]
- 24.Marion D.W., Penrod L.E., Kelsey S.F., Obrist W.D., Kochanek P.M., Palmer A.M., Wisniewski S.R., and DeKosky S.T. (1997). Treatment of traumatic brain injury with moderate hypothermia. N. Engl. J. Med. 336, 540–546 [DOI] [PubMed] [Google Scholar]
- 25.Andrews P.J., Sinclair H.L., Rodriguez A., Harris B.A., Battison C.G., Rhodes J.K., Murray G.D., and Eurotherm3235 Trial Collaborators. (2015). Hypothermia for intracranial hypertension after traumatic brain injury. N. Engl. J.Med. 373, 2403–2412 [DOI] [PubMed] [Google Scholar]
- 26.Temkin N.R., Dikmen S.S., Wilensky A.J., Keihm J., Chabel S., and Winn H.R. (1990). A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N. Engl. J. Med. 323, 497–502 [DOI] [PubMed] [Google Scholar]
- 27.Giacino J.T., Whyte J., Bagiella E., Kalmar K., Childs N., Khademi A., Eifert B., Long D., Katz D.I., Cho S., Yabion S.A., Luther M., Hammond F.M., Nodenbo A., Novak P., Mercer W., Maurer-Karattup P., and Sherer M. (2012). Placebo-controlled trial of amantadine for severe traumatic brain injury. N. Engl. J. Med. 366, 819–826 [DOI] [PubMed] [Google Scholar]
- 28.Harders A., Kakarieka A., Braakman R., and German tSAH Study Group. (1996). Traumatic subarachnoid hemorrhage and its treatment with nimodipine. J. Neurosurg. 85, 82–89 [DOI] [PubMed] [Google Scholar]
- 29.Vergouwen M.D., Vermeulen M., and Roos Y.B. (2006). Effect of nimodipine on outcome in patients with traumatic subarachnoid haemorrhage: a systematic review. Lancet Neurol. 5, 1029–1032 [DOI] [PubMed] [Google Scholar]
- 30.Xiao G., Wei J., Yan W., Wang W., and Lu Z. (2008). Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit. Care 12, R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J., Huang S., Qin S., You C., and Zeng Y. (2016). Progesterone for acute traumatic brain injury. Cochrane Database Syst. Rev. 2016December22;12:CD008409. Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schumachera M., Deniera C., Oudineta J.P., Adams D., and Guennouna R. (2016). Progesterone neuroprotection: the background of clinical trial failure J. Steroid Biochem. Mol. Biol. 160, 53–66 [DOI] [PubMed] [Google Scholar]
- 33.Goldstein F.C., Caveney A.F., Hertzberg V.S., Silbergleit R., Yeatts S.D., Palesch Y.Y., Levin H.S., and Wright D.W. (2017). Very early administration of progesterone does not improve neuropsychological outcomes in subjects with moderate to severe traumatic brain injury. J. Neurotrauma 34, 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein D.G., Sayeed I., Espinosa-Garcia C., Atif F., and Sergeeva E.G. (2017). Goldstein et al.'s Secondary analysis of progesterone clinical trial for traumatic brain injury can only reflect the same trial design flaws: a response to ‘‘Very early administration of progesterone does not improve neuropsychological outcomes in subjects with moderate to severetraumatic brain injury.’’ J. Neurotrauma 34, 2192–2193 [DOI] [PubMed] [Google Scholar]
- 35.Narayan R.K., Michel M.E., Ansell B., and The Clinical Trials in Head Injury Study Group. (2002). Clinical trials in head injury. J. Neurotrauma 19, 503–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stocchetti N., Taccone F.S., Citerio G., Pepe P.E., Le Roux P.D., Oddo M., Polderman K.H., Stevens R.D., Barsan W., Maas A.I.R., Meyfroidt G., Bell M.J., Silbergleit R., Vespa P.M., Faden A.I., Helbok R., Tisherman S., Zanier E.R., Valenzuela T., Wendon J., Menon D.K., and Vincent J.-L. (2015). Neuroprotection in acute brain injury: an up-to-date review. Crit. Care 19, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson V.E., Meaney D.F., Cullen D.K., and Smith D.H. (2015). Animal models of traumatic brain injury, in: Handbook of Clinical Neurology: Traumatic Brain Injury, Part I, Volume 127. Grafman J., and Salazar A.M. (eds). Elsevier: Amsterdam, pps. 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiong Y., Mahmood A., and Chopp M. (2013). Animal models of traumatic brain injury. Nat. Rev. Neurosci. 14, 128–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cernak I. (2005). Animal models of head trauma. NeuroRx. 2, 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeWitt D.S., Perez-Polo J.R., Hulsebosch C.E., Dash P.K., and Robertson C.S. (2013). Challenges in the development of rodent models of mild traumatic brain injury. J. Neurotrauma 30, 688–701 [DOI] [PubMed] [Google Scholar]
- 41.Morales D.M., Marklund N., Lebold D., Thompson H.J., Pitkanen A., Maxwell W.L., Longhi L., Laurer H., Maegele M., Neugebauer E., Graham D.I., Stocchetti N., and McIntosh T.K. (2005). Experimental models of traumatic brain injury: do we really need a better mousetrap? Neuroscience 136, 971–989 [DOI] [PubMed] [Google Scholar]
- 42.Cook D.J., and Tymianski M. (2012). Nonhuman primate models of stroke for translational neuroprotection research. Neurotherapeutics 9, 371–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stroke Therapy Academic Industry Roundtable (STAIR). (1999). Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 30, 371–379 [DOI] [PubMed] [Google Scholar]
- 44.Doppenberg E.M., Choi S.C., and Bullock R. (2004). Clinical trials in traumatic brain injury: lessons for the future. J. Neurosurg. Anesthesiol. 16, 87–94 [DOI] [PubMed] [Google Scholar]
- 45.Wright D.K., Liu S., van der Poe C., McDonald S.J., Brady R.D., Taylor L., Yang L., Gardner A.J., Ordidge R., O'Brien T.J., Johnston L.A., and Shultz S.R. (2017). Traumatic brain injury results in cellular, structural and functional changes resembling motor neuron disease. Cereb. Cortex 27, 4503–4515 [DOI] [PubMed] [Google Scholar]
- 46.Shultz S.R., Wright D.K., Zheng P., Stuchbery R., Liu S.-J., Sashindranath M., Medcalf R.L., Johnston L.A., Hovens C.M., Jones N.C., and O'Brien T.J. (2015). Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 138, 1297–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jennett B., and Bond M. (1975). Assessment of outcome after severe brain damage. Lancet 1, 480–484 [DOI] [PubMed] [Google Scholar]
- 48.Jennett B., Snoek J., Bond M.R., and Brooks M. (1981). Disability after severe head injury: observations on the use of the Glasgow Outcome Scale. J. Neurol. Neurosurg. Psychiatry 44, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McIntosh T.K., Vink R., Noble L., Yamakami I., Fernyak S., and Faden A.I. (1989). Traumatic brain injury in the rat: characterization of a lateral fluid percussion model. Neuroscience 28, 233–244 [DOI] [PubMed] [Google Scholar]
- 50.Crawley J.N. (2003). Behavioral phenotyping of rodents. Comp. Med. 53, 140–146 [PubMed] [Google Scholar]
- 51.Kharatishvili I., Nissinen J.P., McIntosh T.K., and Pitkänen A. (2006). A model of postttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 140, 685–697 [DOI] [PubMed] [Google Scholar]
- 52.Liu S.-J., Zheng P., Wright D.K., Dezsi G., Braine E., Nguyen T., Corcoran N.M., Johnston L.A., Hovens C.M., Mayo J.N., Hudson M., Shultz S.R., Jones N.C., and O'Brien T.J. (2016). Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 139, 1919–1938 [DOI] [PubMed] [Google Scholar]
- 53.Shultz S.R., Cardamone L., Liu Y.R., Hogan R.E., Maccotta L., Wright D.K., Zheng P., Koe A., Gregoire M.-C., Williams J.P., Hicks R.J., Jones N.C., Myers D.E., O'Brien T.J., and Bouilleret V. (2013). Can structural or functional changes following traumatic brain injury in the rat predict the epileptic outcome? Epilepsia 54, 1240–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dixon C.E., Kochanek P.M., Yan H.Q., Schiding J.K., Griffith R.G., Baum E., Marion D.W., and DeKosky S.T. (1999). One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J. Neurotrauma 16, 109–122 [DOI] [PubMed] [Google Scholar]
- 55.Sell S.L., Johnson K., DeWitt D.S., and Prough D.S. (2017). Persistent behavioral deficits in rats after parasagittal fluid percussion injury. J. Neurotrauma 34, 1086–1096 [DOI] [PubMed] [Google Scholar]
- 56.Pierce J.E.S., Smith D.H., Trojanowski J.Q., and McIntosh T.K. (1998). Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience 87, 359–369 [DOI] [PubMed] [Google Scholar]
- 57.Osier N.D., Carlson S.W., DeSana A., and Dixon C.E. (2015). Chronic histopathological and behavioral outcomes of experimental traumatic brain injury in adult male animals. J. Neurotrauma 32, 1861–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bramlett H.M., and Dietrich W.D. (2002). Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 103, 607–614 [DOI] [PubMed] [Google Scholar]
- 59.Bramlett H.M., and Dietrich W.D. (2007). Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog. Brain Res. 161, 125–141 [DOI] [PubMed] [Google Scholar]
- 60.Bramlett H.M., and Dietrich W.D. (2015). Long-term consequences of traumatic brain injury: current status of potential mechanisms of injury and neurological outcomes. J. Neurotrauma 32, 1834–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bramlett H.M., Dietrich W.D., Green E.J., and Busto R. (1997). Chronic histopathological consequences of fluid percussion brain injury in rats: effects of post-traumatic hypothermia. Acta Neuropathol. 93, 190–199 [DOI] [PubMed] [Google Scholar]
- 62.Smith D.H., Chen X.H., Pierce J.E., Wolf J.A., Trojanowski J.Q., Graham D.I., and McIntosh T.K. (1997). Progressive atrophy and neuron death for one year following brain trauma in the rat. J. Neurotrauma 14, 715–727 [DOI] [PubMed] [Google Scholar]
- 63.Kochanek P.M., Hendrich K.S., Dixon C.E., Schiding J.K., Williams D.S., and Ho C. (2002). Cerebral blood flow at one year after controlled cortical impact in rats: assessment by magnetic resonance imaging. J. Neurotrauma 19, 1029–1037 [DOI] [PubMed] [Google Scholar]
- 64.Begley C.G., and Ellis L. (2012). Drug development: raise standards for preclinical cancer research. Nature 483, 531–533 [DOI] [PubMed] [Google Scholar]
- 65.Begley C.G., and Ioannidis J.P.A. (2015). Reproducibility in Science: Improving the standard for basic and preclinical research. Circ. Res. 116, 116–126 [DOI] [PubMed] [Google Scholar]
- 66.Freedman L.P., Cockburn I.M., and Simcoe T.S. (2015). the economics of reproducibility in preclinical research. PLoS Biol. 13, e1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corrigan J.D., Harrison-Felix C., Bogner J., Dijkers M., Terrill M.S., and Whiteneck G. (2003). Systematic bias in traumatic brain injury outcome studies because of loss to follow-up. Arch. Phys. Med. Rehabil. 84, 153–160 [DOI] [PubMed] [Google Scholar]
- 68.Page M.J., Higgins J.P., Clayton G., Sterne J.A., Hróbjartsson A., and Savović J. (2016). empirical evidence of study design biases in randomized trials: systematic review of meta-epidemiological studies. PLoS One 11, e0159267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bafeta A., Dechartres A., Trinquart L., Yavchitz A., Boutron I., and Ravaud P. (2012). Impact of single centre status on estimates of intervention effects in trials with continuous outcomes: meta-epidemiological study. BMJ 344, e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bellomo R., Warrillow S.J., and Reade M.C. (2009). Why we should be wary of single-center trials. Crit. Care Med. 37, 3114–3119 [DOI] [PubMed] [Google Scholar]
- 71.Maas A.I., Roozenbeek B., and Manley G.T. (2010). Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics 7, 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wright D.W., Kellerman A.L., Hertzberg V.S., Clark P.L., Goldstein F.C., Salomone J.P., Dent L.L., Harris O.A., Ander D.S., Lowery D.W., Patel M.M., Denson D.D., Gordon A.B., Wald M.M., Gupta S., Hoffman S.W., and Stein D.G. (2007). ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann. Emerg. Med. 49, 391–402 [DOI] [PubMed] [Google Scholar]
- 73.Wright D.W., Yeatts S.D., Silbergleit R., Palesch Y.Y., Hertzberg V.S., Frankel M., Goldstein F.C., Caveney A.F., Howlett-Smith H., Bengelink E.M., Manley G.T., Merck L.H., Janis L.S., Barson W.G., and NETT Investigators. (2014). Very early administration of progesterone for acutetraumatic brain injury. N. Engl. J. Med. 371, 2457–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Skolnick B.E., Maas A.I., Narayan R.K., van der Hoop R.G., MacAllister T., Ward J.D., Nelson N.R., Stocchetti N., and SYNAPSE Trial Investigators. (2014). A clinical trial of progesterone for severe traumatic brain injury. N. Engl. J. Med. 371, 2467–2476 [DOI] [PubMed] [Google Scholar]
- 75.Egger M., Davey-Smith G., Schneider M., and Minder C. (1997). Bias in meta-analysis detected by a simple, graphical test. BMJ 315, 629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilkes M.M., and Navickis R.J. (2001). Patient survival after human albumin administration: a meta-analysis of randomized, controlled trials. Ann. Int. Med. 135, 149–164 [DOI] [PubMed] [Google Scholar]
- 77.Button K.S., Ioannidis J.P.A., Mokrysz C., Nosek B.A., Flint J., Robinson E.S.J., and Munafò M.R. (2013). Power failure: why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 14, 365–376 [DOI] [PubMed] [Google Scholar]
- 78.Reagan-Shaw S., Nihal M., and Ahmad N. (2008). Dose translation from animal to human studies revisited. FASEB J. 22, 659–661 [DOI] [PubMed] [Google Scholar]
- 79.Blanchard O.L., and Smoliga J.M. (2015). Translating dosages from animal models to human clinical trials: revisiting body surface area scaling. FASEB J. 29, 1629–1634 [DOI] [PubMed] [Google Scholar]
- 80.Zhao J., Cao Y., and Jusko W.J. (2015). Across-species scaling of monoclonal antibody pharmacokinetics using a minimal PBPK model. Pharma. Res. 32, 3269–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Buck S.S., Sinha V.K., Fenu L.A., Nijsen M.J., Mackle C.E., and Gilissen R.A.H.J. (2007). Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab. Dispo. 35, 1766–1780 [DOI] [PubMed] [Google Scholar]
- 82.Edwards P., Arango M., Balica L., Cottingham R., El-Sayed H., Farrell B., Fernandes J., Gogichaisvili T., Golden N., Hartzenberg B., Husain M., Ulloa M.I., Jerbi Z., Khamis H., Komolafe E., Laloe V., Lomas G., Ludwig S., Mazairac G., Munoz Sanchez Mde L., Nasi L., Olldashi F., Plunkett P., Roberts I., Sandercock P., Shakur H., Soler C., Stocker R., Svoboda P., Trenkler S., Venkataramana N.K., Wasserberg J., Yates D., and Yutthakasemsunt S. (2005). Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet 365, 1957–1959 [DOI] [PubMed] [Google Scholar]
- 83.Mendelow A.D., Gregson B.A., Rowan E.N., Francis R., McColl E., McNamee P., Chambers I., Unterberg A.W., Boyers D., and Mitchell P. (2015). Early surgery versus initial conservative treatment in patients with traumatic intracerebral haemorrhage [STITCH (Trauma)]: the first randomized trial. J. Neurotrauma 32, 1312–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levin H.S., Boake C., Song J., McCauley S., Contant C., Diaz-Marchan P., Brundage S., Goodman H., and Kotrla K.J. (2001). Validity and sensitivity to change of the extended Glasgow Outcome Scale in mild to moderate traumatic brain injury. J. Neurotrauma 18, 575–584 [DOI] [PubMed] [Google Scholar]
- 85.Lu J., Murray G.D., Steverberg E.W., Butcher I., Mchugh G.S., Lingsma H., Mushkudiani N., Choi S., Maas A.I.R., and Marmarou A. (2008). Effects of Glasgow Outcome Scale misclassification on traumatic brain injury clinical trials. J. Neurotrauma 25, 641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maas A.I., Braakman R., Schouten H.J., Minderhoud J.M., and van Zomeren A.H. (1983). Agreement between physicians on assessment of outcome following severe head injury. J. Neurosurg. 58, 321–325 [DOI] [PubMed] [Google Scholar]
- 87.Wilson J.T., Slieker F.J., Legrand V., Murray G., Stocchetti N., and Maas A.I. (2007). Observer variation in the assessment of outcome in traumatic brain injury: experience from a multicenter, international randomized clinical trial. Neurosurgery 61, 123–129 [DOI] [PubMed] [Google Scholar]
- 88.Manley G.T., MacDonald C.L., Markowitz A.J., Stephenson D., Robbins A., Gardner R.C., Winkler E., Bodien Y.G., Taylor S.R., Yue J.K., Kannan L., Kumar A., McCrea M.A., and Wang K.K. (2017). The Traumatic Brain Injury Endpoints Development (TED) Initiative: progress on a public-private regulatory collaboration to accelerate diagnosis and treatment of traumatic brain injury. J. Neurotrauma 34, 2721–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kochanek P.M., Bramlett H.M., Dixon C.E., Shear D.A., Dietrich W.D., Schmid K.E., Mondello S., Wang K.K.W., Hayes R.L., Povlishock J.T., and Tortella F.C. (2016). Approach to modeling, therapy evaluation, drug selection, and biomarker assessments for a multicenter pre-clinical drug screening consortium for acute therapies in severe traumatic brain injury: Operation Brain Trauma Therapy. J. Neurotrauma 33, 513–522 [DOI] [PubMed] [Google Scholar]
- 90.Kochanek P.M., Bramlett H.M., Shear D.A., Dixon C.E., Mondello S., Dietrich W.D., Hayes R.L., Wang K.K.W., Poloyac S.M., Empey P.F., Povlishock J.T., Mountney A., Browning M., Deng-Bryant Y., Yan H.Q., Jackson T.C., Cantania M., Glushakova O., Richieri S.P., and Tortella F.C. (2016). Synthesis of findings, current investigations, and future directions: Operation Brain Trauma Therapy. J. Neurotrauma 33, 606–614 [DOI] [PubMed] [Google Scholar]
- 91.Basso D.M., Beattie M.S., Bresnahan J.C., Anderson D.K., Faden A.I., Gruner J.A., Holford T.R., Hsu C.Y., Noble L.J., Nockels R., Perot P.L., Salzman S.K., and Young W. (1996). MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. J. Neurotrauma 13, 343–359 [DOI] [PubMed] [Google Scholar]
- 92.Basso D.M., Beattie M.S., and Bresnahan J.C. (1995). A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma 12, 1–21 [DOI] [PubMed] [Google Scholar]
- 93.Bramlett H.M., Dietrich W.D., Dixon C.E., Shear D.A., Schmid K.E., Mondello S., Wang K.K., Hayes R.L., Povlishock J.T., Tortella F.C., and Kochanek P.M. (2016). Erythropoietin treatment in traumatic brain injury: Operation Brain Trauma Therapy. J. Neurotrauma 33 538–552 [DOI] [PubMed] [Google Scholar]
- 94.Dixon C.E., Bramlett H.M., Dietrich W.D., Shear D.A., Deng-Bryant Y., Mondello S., Wang K.K., Hayes R.L., Empey P.E., Povlishock J.T., Tortella F.C., and Kochanek P.M. (2016). Cyclosporine treatment in traumatic brain injury: Operation Brain Trauma Therapy. J. Neurotrauma 33, 553–566 [DOI] [PubMed] [Google Scholar]
- 95.Browning M., Shear D.A., Bramlett H.M., Dixon C.E., Mondello S., Schmid K.E., Poloyac S.M., Dietrich W.D., Hayes R.L., Wang K.K.W., Povlischock J.T., Tortella F.C., and Kochanek P.M. (2016). Levetiracetam treatment in traumatic brain injury: Operation Brain Trauma Therapy. J. Neurotrauma 33, 581–594 [DOI] [PubMed] [Google Scholar]
- 96.Mountney A., Bramlett H.M., Dixon C.E., Mondello S., Dietrich W.D., Wang K.K., Caudle K., Empey P.E., Poloyac S.M., Hayes R.L., Povlishock J.T., Tortella F.C., Kochanek P.M., and Shear D.A. (2016). Simvastatin treatment in traumatic brain injury: Operation Brain Trauma Therapy. J. Neurotrauma 33, 567–580 [DOI] [PubMed] [Google Scholar]
- 97.Shear D.A., Dixon C.E., Bramlett H.M., Mondello S., Dietrich W.D., Deng-Bryant Y., Schmid K.E., Wang K.K., Hayes R.L., Povlishock J.T., Kochanek P.M., and Tortella F.C. (2016). Nicotinamide treatment in traumatic brain injury: Operation Brain Trauma Therapy. J. Neurotrauma 33, 523–537 [DOI] [PubMed] [Google Scholar]
- 98.Patel A.D., Gerzanich V., Geng Z., and Simard J.M. (2010). Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. J. Neuropathol. Exp. Neurol. 69, 1177–1190 [DOI] [PubMed] [Google Scholar]
- 99.Mbye L.H., Keles E., Tao L., Zhang J., Chung J., Larvie M., Koppula R., Lo E.H., and Whalen M.J. (2012). Kollidon VA64, a membrane-resealing agent, reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metabol. 32, 515–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mondello S., Shear D.A., Bramlett H.M., Dixon C.E., Schmid K.E., Dietrich W.D., Wang K.K.W., Hayes R.L., Glushakova O., Catania M., Richieri S.P., Povlishock J.T., Tortella F.C., and Kochanek P.M. (2016). Insight into pre-clinical models of traumatic brain injury using circulating brain damage biomarkers: Operation Brain Trauma Therapy. J. Neurotrauma 33, 595–605 [DOI] [PubMed] [Google Scholar]
- 101.Miguel E., Camerer C., Casey K., Cohen J., Esterling K.M., Gerber A., Glennerster R., Green D.P., Humphreys M., Imbens G., Laitin D., Madon T., Nelson L., Nosek B.A., Petersen M., Sedlmayr R., Simmons J.P., Simonsohn U., and Van der Laan M. (2014). Promoting transparency in social science research. Science 343, 30–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simmons J.P., Nelson L.D., and Simonsohn U. (2011). False-positive psychology: undisclosed flexibility in data collection and analysis allows presenting anything as significant. Psychol. Sci. 22, 1359–1366 [DOI] [PubMed] [Google Scholar]
- 103.Carp J. (2012). The secret lives of experiments: methods reporting in the fMRI literature. Neuroimage 63, 289–300 [DOI] [PubMed] [Google Scholar]
- 104.Lapchak P.A., Zhang J.H., and Noble-Haeusslein L.J. (2013). RIGOR Guidelines: Escalating STAIR and STEPS for effective translational research. Transl. Stroke Res. 4, 279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hellmich H.L., Capra B., Eidson K., Garcia J., Kennedy D., Uchida T., Parsley M., Cowart J., DeWitt D.S., and Prough D.S. (2005). Dose-dependent neuronal injury after traumatic brain injury. Brain Res. 1044, 144–154 [DOI] [PubMed] [Google Scholar]
- 106.Sato M., Chang E., Igarashi T., and Noble L.J. (2001). Neuronal injury and loss after traumatic brain injury: time course and regional variability. Brain Res. 917, 45–54 [DOI] [PubMed] [Google Scholar]
- 107.Schmued L.C., Albertson C., and Slikker W. (1997). Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 751, 37–46 [DOI] [PubMed] [Google Scholar]
- 108.Wang J., Jahn-Eimermacherb A., Brucknera M., Werner C., Engelhard K., and Thala S.C. (2015). Comparison of different quantification methods to determine hippocampal damage after cerebral ischemia. J. Neurosci. Methods 240, 67–78 [DOI] [PubMed] [Google Scholar]
- 109.Damjanaca M., Rioux Bilan A., Barrier L., Pontcharraud R., Anne C., Hugon J., and Page G. (2007). Fluoro-Jade® B staining as useful tool to identify activated microglia and astrocytes in a mouse transgenic model of Alzheimer's disease. Brain Res. 1128, 40–49 [DOI] [PubMed] [Google Scholar]
- 110.Shultz S.R., McDonald S.J., Vonder Haar C., Meconi A., Vink R., van Donkelaar P., Taneja C., Iverson G.L., and Christie B.R. (2017). The potential for animal models to provide insight into mild traumatic brain injury: translational challenges and strategies. Neurosci. Biobehav. Rev. 76, 396–414 [DOI] [PubMed] [Google Scholar]
- 111.Millspaugh J.A. (1937). Dementia pugilistica. US Naval Med. Bull. 35, 297–303 [Google Scholar]
- 112.Martland H.S. (1928). Punch drunk. JAMA 91, 1103–1107 [Google Scholar]
- 113.McKee A.C., Cantu R.C., Nowinski C.J., Hedley-Whyte E.T., Gavett B.E., Budson A.E., Santini V.E., Lee H.-S., Kubilus C.A., and Stern R.A. (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McKee A.C., Gavett B.E., Stern R.A., Nowinski C.J., Cantu R.C., Kowall N.W., Perl D.P., Hedley-White T., Price B., Sullivan C., Morin P., Lee H.-S., Kubilus C.A., Daneshvar D.H., Wulff M., and Budson A.E. (2010). TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 69, 918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peskind E.R., Brody D., Cernak I., McKee A., and Ruff R.L. (2013). Military- and sports-related mild traumatic brain injury: clinical presentation, management, and long-term consequences. J. Clin. Psychiatry 74, 180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goldstein L.E., Fisher A.M., Tagge C.A., Zhang X.-L., Velisek L., Sullivan J.A., Upreti C., Karcht J.M., Ericsson M., Wojnarowicz M.W., Goletiani C.J., Maglakelidze G.M., Casey N., Moncaster J.A., Minaeva O., Moir R.B., Nowinski C.J., Stern R.A., Cantu R.C., Geiling J., Blusztajn J.K., Wolozin B.L., Ikezu T., Stein T.D., Budson A.E., Kowall N.W., Chargin D., Sharon A., Saman S., Hall G.F., Moss W.C., Cleveland R.O., Tanzi R.E., Stanton P.K., and McKee A.C. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4, 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Glushakova O.Y., Johnson D., and Hayes R.L. (2014). Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood–brain barrier disruption, and progressive white matter damage. J. Neurotrauma 31, 1180–1193 [DOI] [PubMed] [Google Scholar]
- 118.Doherty C.P., O'Keefe E., Wallace E., Loftus T., Keaney J., Kealy J., Humphries M.M., Molloy M.G., Meaney J.F., Farrell M., and Campbell M. (2016). Blood-brain barrier dysfunction as a hallmark pathology in chronic traumatic encephalopathy. J. Neuropathol. Exp. Neurol. 75, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Briggs D.I., Angoa-Pérez M., and Kuhn D.M. (2016). Prolonged repetitive head trauma induces a singular chronic traumatic encephalopathy-like pathology in white matter despite transient behavioral abnormalities. Am. J. Pathol. 186, 2869–2886 [DOI] [PubMed] [Google Scholar]
- 120.Ojo J.O., Mouzon B.C., and Crawford F. (2016). Repetitive head trauma, chronic traumatic encephalopathy and tau: challenges in translating from mice to men. Exp. Neurol. 275, 389–404 [DOI] [PubMed] [Google Scholar]
- 121.Margulies S., and Hicks R. (2009). Combination therapies for traumatic brain injury: prospective considerations. J. Neurotrauma 26, 925–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McKee A.C., and Robinson M.E. (2014). Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 10, S242–S253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sahin B., Aslan H., Unal B., Canan S., Bilgic S., Kaplan S., and Tumkaya L. (2001). Brain volumes of the lamb, rat and bird do not show hemispheric asymmetry: a stereological study. Image Analysis Stereol. 20, 9–13 [Google Scholar]
- 124.Mayhew T.M., Mwamengele G.L.M., and Dantzer V. (1990). Comparative morphometry of the mammalian brain: estimates of cerebral volumes and cortical surface areas obtained from macroscopic slices. J. Anat. 172, 191–200 [PMC free article] [PubMed] [Google Scholar]
- 125.Cosgrove K.P., Mazure C.M., and Staley J.K. (2007). Evolving knowledge of sex differences in brain structure, function, and chemistry. Biol. Psychiatry 62, 847–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sisodiya S., Free S., Fish D., and Shorvon S. (1990). MRI-based surface area estimates in the normal adult human brain: evidence for structural organization. J. Anat. 188, 425–438 [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang K., and Sejnowski T.J. (2000). A universal scaling law between gray matter and white matter of cerebral cortex. PNAS 97, 5621–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bauman R.A., Ling G., Tong L., Januszkiewicz A., Agoston D., Delanerolle N., Kim Y., Ritzel D., Bell R., Ecklund J., Armonda R., Bandak F., and Parks S. (2009). An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J. Neurotrauma 26, 841–860 [DOI] [PubMed] [Google Scholar]
- 129.LaPlaca M.C., Raghupathi R., Verma A., Pieper A.A., Saatman K.E., Snyder S.H., and McIntosh T.K. (1999). Temporal patterns of poly(ADP-ribose) polymerase activation in the cortex following experimental brain injury in the rat. J. Neurochem. 73, 205–213 [DOI] [PubMed] [Google Scholar]
- 130.Faden A.I., Demediuk P., Panter S.S., and Vink R. (1989). The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800 [DOI] [PubMed] [Google Scholar]
- 131.Mahmood A., Goussev A., Kazmi H., Qu C., Lu D., and Chopp M. (2009). Long-term benefits after treatment of traumatic brain injury with simvastatin in rats. Neurosurgery 65, 187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wu H., Mahmood A., Qu C., Xiong Y., and Chopp M. (2012). Simvastatin attenuates axonal injury after experimental traumatic brain injury and promotes neurite outgrowth of primary cortical neurons. Brain Res. 1486, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hill R.L., Singh I.N., Wang J.A., and Hall E.D. (2017). Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem. Int. 111, 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Smith D.H., Okiyama K., Thomas M.J., Claussen B., and McIntosh T.K. (1991). Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J. Neurotrauma 8, 259–269 [DOI] [PubMed] [Google Scholar]
- 135.Dash P.K., Orsi S.A., and Moore A.N. (2006). Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex–mammalian target of rapamycin pathway. J. Neurosci. 26, 8048–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bignall K.E. (1974). Ontogeny of levels of neural organization: the righting reflex as a model. Exp. Neurol. 42, 566–573 [DOI] [PubMed] [Google Scholar]
- 137.Alkire M.T., McReynolds J.R., E.L. H., and Trivedi A.N. (2007). Thalamic microinjection of nicotine reverses sevoflurane induced loss of righting reflex in the rat. Anesthesiology 107, 264–272 [DOI] [PubMed] [Google Scholar]
- 138.Nguyen H.T., Li K.Y., daGraca R.L., Delphin E., Xiong M., and Ye J.H. (2009). Behavior and cellular evidence form propofol-induced hypnosis involving brain glycine receptors. Anesthesiology 110, 326–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sukhotinsky I., Zalkind V., Lu J., Hopkins D.A., Saper C.B., and Devor M. (2007). Neural pathways associated with loss of consciousness caused by intracerebral microinjection of GABAA-active anesthetics. Eur. J. Neurosci. 25, 1417–1436 [DOI] [PubMed] [Google Scholar]
- 140.Hicks R., Giacino J., Harrison-Felix C., Manley G., Valadka A., and Wilde E.A. (2013). Progress in developing common data elements for traumatic brain injury research: version two—the end of the beginning. J. Neurotrauma 30, 1852–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Smith D.H., Hicks R.R., Johnson V.E., Bergstrom D.A., Cummings D.M., Noble L.J., Hovda D., Whalen M., Ahlers S.T., LaPlaca M., Tortella F.C., Duhaime A.-C., and Dixon C.E. (2015). Pre-clinical traumatic brain injury common data elements: toward a common language across laboratories. J. Neurotrauma 32, 1225–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Levin H.S., Grossman R.G., Rose J.E., and Teasdale G. (1979). Long-term neuropsychological outcome of closed head injury. J. Neurosurg. 50, 412–422 [DOI] [PubMed] [Google Scholar]
- 143.Ponsford J., Draper K., and Schonberger M. (2008). Functional outcome 10 years after traumatic brain injury: its relationship with demographic, injury severity, and cognitive and emotional status. J. Int. Neuropsychol. Soc. 14, 233–242 [DOI] [PubMed] [Google Scholar]
- 144.Brown A.W., Leibson C.L., Malec J.F., Perkins P.K., Diehl N.N., and Larson D.R. (2004). Long-term survival after traumatic brain injury: a population-based analysis. NeuroRehabilitation 19, 37–43 [PubMed] [Google Scholar]
- 145.Maas A.I.R., Marmarou A., Murray G.D., Teasdale G.M., and Steyerberg E.W. (2007). Prognosis and clinical trial design in traumatic brain injury: the IMPACT Study. J. Neurotrauma 24, 232–238 [DOI] [PubMed] [Google Scholar]
- 146.Maas A.I.R., Steyerberg E.W., Marmarou A., McHugh G.S., Lingsma H.F., Butcher I., Lu J., Weir J., Roozenbeek B., and Murray G.D. (2010). IMPACT recommendations for improving the design and analysis of clinical trials in moderate to severe traumatic brain injury. Neurotherapeutics 7, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.FITBIR. Federal Interagency Traumatic Brain Injury Research Informatics System. Available at: https://fitbir.nih.gov. Accessed July6, 2017
- 148.Thompson H.J., Vavilala M.S., and Rivara F.P. (2015). Chapter 1 Common Data Elements and Federal Interagency Traumatic Brain Injury Research Informatics System for TBI Research. Annu. Rev. Nurs. Res. 33, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ferguson A.R., Nielson J.L., Cragin M.H., Bandrowski A.E., and Martone M.E. (2014). Big data from small data: data-sharing in the ‘long tail’ of neuroscience. Nat. Neurosci. 17, 1442–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Begley C.G., and Ellis L.M. (2012). Drug development: raise standards for preclinical cancer research. Nature 483, 531–533 [DOI] [PubMed] [Google Scholar]
- 151.Begley C.G., and Ioannidis J.P. (2015). Reproducibility in science: improving the standard for basic and preclinical research. Circ. Res. 116, 116–126 [DOI] [PubMed] [Google Scholar]
- 152.Chan A.W., Song F., Vickers A., Jefferson T., Dickersin K., Getzsche P.C., Krumholz H.M., Ghersi D., and van der Worp H.B. (2014). Increasing value and reducing waste: addressing inaccessible research. Lancet 383, 257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ferguson A.R., Irvine K.-A., Gensel J.C., Nielson J.L., Lin A., Ly J., Segal M.R., Ratan R.R., Bresnahan J.C., and Beattie M.S. (2013). Derivation of multivariate syndromic outcome metrics for consistent testing across multiple models of cervical spinal cord injury in rats. PLoS One 8, e59712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wilkinson M.D., Dumontier M., Aalbersberg I.J., Appleton G., Axton M., Baak A., Blomberg N., Boiten J.W., da Silva Santos L.B., Bourne P.E., Bouwman J., Brookes A.J., Clark T., Crosas M., Dillo I., Dumon O., Edmunds S., Evelo C.T., Finkers R., Gonzalez-Beltran A., Gray A.J., Groth P., Goble C., Grethe J.S., Heringa J.2, 't Hoen P.A., Hooft R., Kuhn T., Kok R., Kok J., Lusher S.J., Martone M.E., Mons A., Packer A.L., Persson B., Rocca-Serra P., Roos M., van Schaik R., Sansone S.A., Schultes E., Sengstag T., Slater T., Strawn G, Swertz M.A., Thompson M., van der Lei J., van Mulligen E., Velterop J., Waagmeester A., Wittenburg P., Wolstencroft K., Zhao J., and Mons B. (2016). The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mons B., Neylon C., Velterop J., Dumontier M., da Silva Santos L.O.B., and Wilkinson M.D. (2017). Cloudy, increasingly FAIR; revisiting the FAIR Data guiding principles for the European Open Science Cloud. Inf. Serv. Use 37, 49–56 [Google Scholar]
- 156.Jiang J.Y., Xu W., Li W.P., Xu W.H., Zhang J., Bao Y.H., Ying Y.H., and Luo Q.Z. (2005). Efficacy of standard trauma craniectomy for refractory intracranial hypertension with severe traumatic brain injury: a multicenter, prospective, randomized controlled study. J. Neurotrauma 22, 623–628 [DOI] [PubMed] [Google Scholar]
- 157.Hutchinson P.J., Kolias A.G., Timofeev I.S., Corteen E.A., Czosnyka M., Timothy J., Anderson I., Bulters D.O., Bell A., Eynon C.A., Wadley J., Mendelow A.D., Mitchell P.M., Wilson M.H., Critchley G., Sahuquillo J., Unterberg A., Servadei F., Teasdale G.M., Pickard J.D., Menon D.K., Murray G.D., Kirkpatrick P.J. and RESCUEicp Trial Collaborators. (2016). Trial of decompressive craniectomy for traumatic intracranial hypertension. N. Engl. J. Med. 375, 1119–1130 [DOI] [PubMed] [Google Scholar]
- 158.Holman C., Piper S.K., Grittner U., Diamantaras A.A., Kimmelman J., Siegerink B., and Dirnagl U. (2016). Where have all the rodents gone? The effects of attrition in experimental research on cancer and stroke. PLoS Biol. 14, e1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fanelli D., Costas R., and Ioannidia P.A. (2016). Meta-assessment of bias in science. PNAS 114, 3714–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vogt L., Reichlin T.S., Nathues C., and Würbel H. (2016). authorization of animal experiments is based on confidence rather than evidence of scientific rigor. PLoS Biol. 14, e2000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tsilidis K.K., Panagiotou O.A., Sena E.S., Aretouli E., Evangelou E., Howells D.W., Salman R.A.-I., Macleod M.R., and Ioannidis J.P.A. (2013). Evaluation of excess significance bias in animal studies of neurological diseases. PLoS Biol. 11, e1001609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.O'Collins V.E., Macleod M.R., Donnan G.A., Horky L.L., van der Worp B.H., and Howells D.W. (2006). 1,026 experimental treatments in acute stroke. Ann. Neurol. 59, 467–477 [DOI] [PubMed] [Google Scholar]
- 163.Dwan K., Altman D.G., Amalz J.A., Bloom J., Chan A.-W., Cronin E., Decullier E., Easterbrook P.J., Von Elm E., Gamble C., Ghersi D., Ionannidis J.P.A., Simes J., and Williamson P.R. (2008). Systematic review of the empirical evidence of study publication bias and outcome reporting bias. PLOS One 3, e3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fanelli D. (2010). “Positive” results increase down the hierarchy of the sciences. PLoS One 5, e10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Easterbrook P.J., Berlin J.A., Gopalan R., and Matthews D.R. (1991). Publication bias in clinical research. Lancet 337, 867–872 [DOI] [PubMed] [Google Scholar]