

Summary:
Age-related clonal hematopoiesis (CH) is a risk factor for malignancy, cardiovascular disease and all-cause mortality. Somatic mutations in DNMT3A are drivers of CH, but decades may elapse between acquisition of a mutation and CH, suggesting that environmental factors contribute to clonal expansion. We tested whether infection provides selective pressure favoring expansion of Dnmt3a-mutant hematopoietic stem cells (HSCs) in mouse chimeras. We created Dnmt3a-mosaic mice by transplanting Dnmt3a−/− and WT HSCs into WT mice and observed substantial expansion of Dnmt3a−/− HSCs during chronic mycobacterial infection. Injection of recombinant IFNγ alone was sufficient to phenocopy CH by Dnmt3a−/− HSCs upon infection. Transcriptional and epigenetic profiling and functional studies indicate reduced differentiation associated with widespread methylation alterations and reduced secondary stress-induced apoptosis account for Dnmt3a−/− clonal expansion during infection. DNMT3A-mutant human HSCs similarly exhibit defective IFNγ-induced differentiation. We thus demonstrate that IFNγ signaling induced during chronic infection can drive DNMT3A-loss of function CH.
Keywords: clonal hematopoiesis, CH, infection, interferon gamma, clonal competition, mycobacterial infection, DNMT3A, epigenetic regulation
One Sentence Summary:
Hormaechea-Agulla et al. demonstrate that infection drives CH by promoting expansion of Dnmt3a-loss of function hematopoietic stem cells.
eTOC Blurb
King and colleagues demonstrate that chronic mycobacterial infection drives expansion of hematopoietic stem cells deficient in Dnmt3a, the gene most commonly mutated in clonal hematopoiesis. Interferon gamma signaling is sufficient to promote Dnmt3a−/− clonal expansion, which is associated with broad changes in epigenetic regulation of a pro-differentiation gene program.
Graphical Abstract
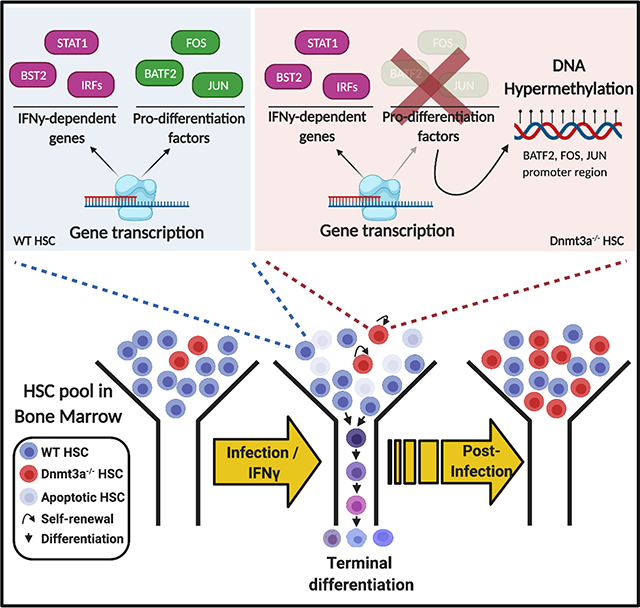
Introduction:
Humans have tens of thousands of hematopoietic stem cells (HSCs) at birth that collectively sustain lifelong blood production through self-renewal and differentiation. In 2014, three independent groups reported a higher than expected frequency of genetically variant cells in the peripheral blood of elderly individuals, reflective of expanded HSC clones (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). This phenomenon, referred to as clonal hematopoiesis (CH), occurs increasingly with age such that approximately 15% of individuals in their seventies are affected (Genovese et al., 2014; Steensma, 2018). Strikingly, CH is associated with a 13-fold increased risk of myeloid hematologic malignancy and an increased risk of all-cause mortality, largely due to cardiovascular disease and stroke (Jaiswal and Libby, 2019). Defined as the presence of a clonal population of cells comprising >4% of the peripheral blood as identified by a specific variant gene allele (DeZern et al., 2019), CH represents the acquisition of genetic mutations combined with selective pressures that result in advantages for the overrepresented clone (Bowman et al., 2018). Clarifying the mechanisms by which CH emerges may aid in identifying key interventions for this widespread phenomenon with tremendous public health implications.
Mutations in several genes are associated with CH, and the incidence of CH correlates directly with age, reaching approximately 50% by the tenth decade (Zink et al., 2017). However, genes alone are unlikely to provide a complete explanation for CH. By some estimates, mutations in DNMT3A, the gene most commonly mutated in CH, are nearly universal in individuals by the age of 50 (Young et al., 2016), suggesting that for most people, decades elapse between acquisition of a CH-associated mutation and CH itself. In addition, many have noted that DNMT3A mutations confer little phenotype on steady state hematopoiesis (Buscarlet et al., 2017); thus, environmental factors likely play a major role in CH emergence. Indeed, epidemiologic studies have demonstrated that CH is associated with smoking and smoking-associated conditions such as chronic lung disease (Zink et al., 2017). Chemotherapy has been shown to promote expansion of PPM1D-mutant clones in treatment-related leukemias (Bolton et al., 2020b; Hsu et al., 2018; Kahn et al., 2018), highlighting a direct link between an environmental selection pressure and a specific mutation well-suited to that environment.
Similarly, inflammation has been shown to be a driver of Tet2-mutant CH. Microbial signals, TNFα, and IL-6 stimulation promote expansion of Tet2 clones in murine models through their superior resistance to inflammation-mediated toxicity compared to WT cells (Abegunde et al., 2018; Cai et al., 2018; Fuster et al., 2017; Meisel et al., 2018). A new study indicates IL-1 exposure can promote expansion of Cebpa-deficient hematopoietic progenitors (Higa et al., 2020). Thus, prior studies have identified biological adaptations that allow HSCs with certain mutations to gain selective advantage in specific environmental conditions (King et al., 2019).
Despite the fact that mutations in DNMT3A are approximately six times more common among CH cases than any other gene, specific environmental drivers of DNMT3A-mutant CH have not been determined. Given the strong epidemiologic links of CH with age and smoking, and given that age and smoking are both associated with inflammation, we reasoned that inflammation may drive DNMT3A clonal hematopoiesis.
In prior work, we established a murine model of chronic infection by Mycobacterium avium, an intracellular pathogen that induces a sustained interferon gamma (IFNγ)-mediated immune response. We demonstrated that systemic inflammation promotes HSC depletion during chronic infection in these mice (Matatall et al., 2016), and that many of the HSC responses to systemic M. avium infection can be recapitulated by administration of IFNγ alone (Baldridge et al., 2010). We found that HSC loss was attributable to a loss of quiescence and self-renewal among HSCs in wild type mice, with excessive differentiation and increased susceptibility to secondary stress-induced apoptosis ultimately leading to exhaustion of the HSC reserve (Matatall et al., 2016). Such a loss of self-renewal in favor of differentiation events has long been recognized as a potential avenue for HSC depletion (Holstege et al., 2014; Morrison and Kimble, 2006). Since prior studies showed that Dnmt3a−/− HSCs demonstrate defective differentiation and enhanced self-renewal (Challen et al., 2011; Jeong et al., 2018), we used a mouse model to test the hypothesis that Dnmt3a loss of function mutations, which comprise the majority of human CH-associated mutations (Russler-Germain et al., 2014), alter the biological responses of HSCs to inflammation, and thus that infection provides an environmental selection pressure favoring the expansion of Dnmt3a-loss of function clones.
Results:
Chronic infection promotes expansion of Dnmt3a-loss of function clones in mice.
To test how infection affects the self-renewal and differentiation of HSCs with Dnmt3a mutations, we used a Mx1-Cre+ Dnmt3af/f mouse model that approximates DNMT3A loss of function mutations commonly found in human CH (Sandoval et al., 2019). We next created mosaic mice to model CH by transplanting a mixture of whole bone marrow from PIPC-treated CD45.2 Mx1-Cre+ Dnmt3af/f (“Dnmt3a−/−”) and CD45.1 WT rescue marrow into lethally irradiated CD45.1 WT mice. This approach yields a starting engraftment of approximately 10% 45.2 Dnmt3a−/− cells as detected in peripheral blood (Figure S1A). Mice transplanted with a mixture of Mx1-Cre− Dnmt3af/f (“WT”) and CD45.1 WT marrow were used as controls (Figure 1A). Two months after transplantation, engrafted mice were systemically infected with M. avium by IV injection. After 16 weeks, 2 months from the start of M. avium infection, we examined bone marrow by flow cytometry (Figure S1B). Of note, we omitted Sca1 from the phenotypic definition of HSCs in these experiments because it has been shown to be nonspecifically upregulated in the setting of infections that induce an interferon response (Baldridge et al., 2011). Mx1-Cre− control “WT” HSCs and Dnmt3a−/− HSCs remained at a low frequency in the absence of infection. However, Dnmt3a−/− HSCs expanded significantly in the marrow in the setting of infection (Figure 1B). This increase was evident regardless of whether Dnmt3a−/− HSCs were analyzed as a percentage of WBM or as absolute number, indicating that the relative expansion was not simply a result of depletion of the WT competitor marrow (Figure S1C).
Figure 1. M. avium infection promotes Dnmt3a-mutant clonal expansion in mice.
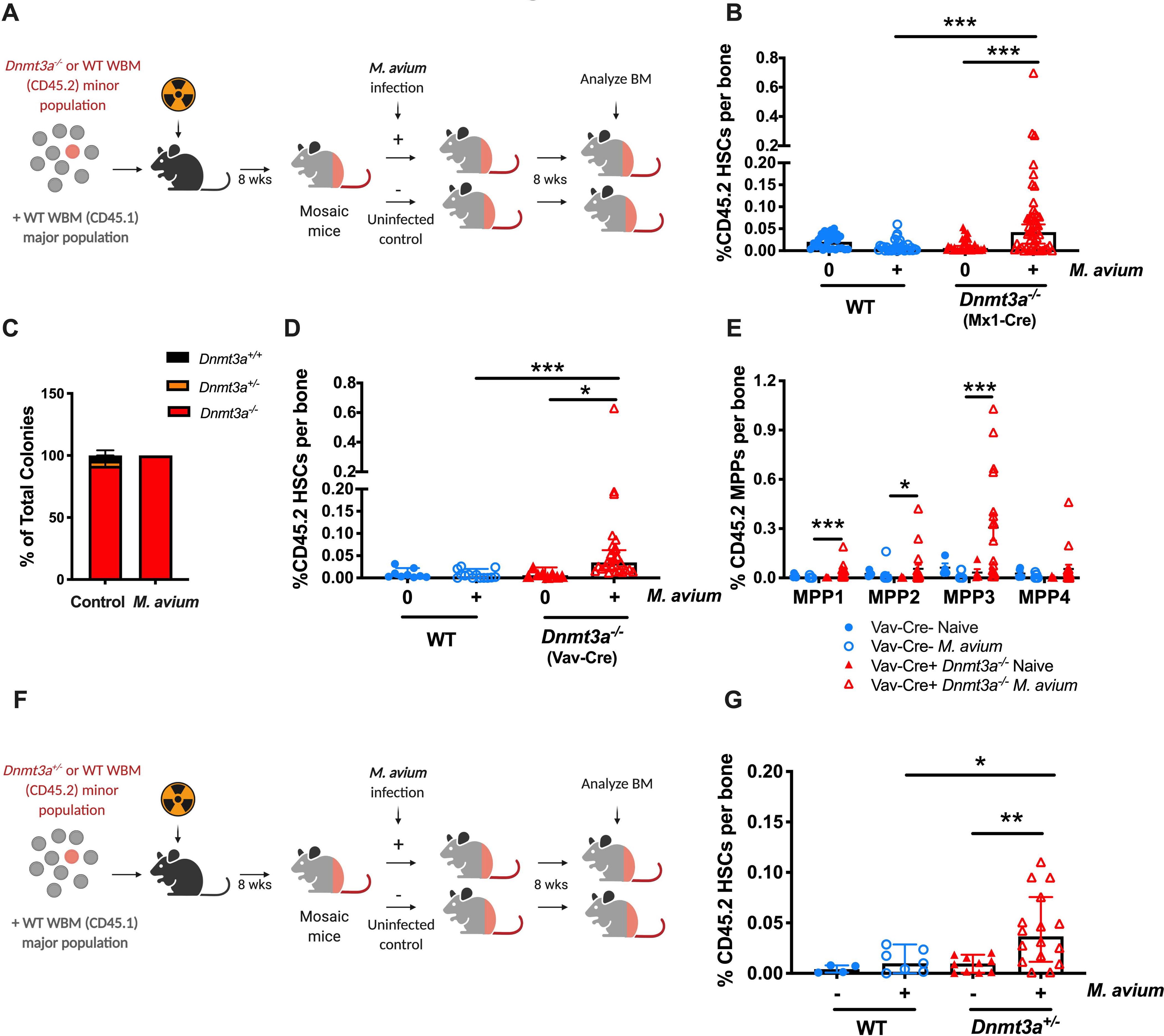
(A) Mosaic mouse model to compare Dnmt3afl/fl vs Dnmt3a−/− HSCs upon infection. WT recipient mice were transplanted with a minor population of “test” Dnmt3afl/fl or Dnmt3a−/− whole bone marrow and a major population of WT competitor. Two months after transplant, half of the mice were infected with M. avium, and the percent of test (Dnmt3a−/− or WT) CD45.2 HSCs measured eight weeks later. (B) CD45.2 HSCs (KL CD150+ CD48− CD34−) shown as a percentage of WBM in mosaic mice. Dnmt3a−/− cells were from Mx1-Cre Dnmt3afl/fl donors treated with PIPC 4 weeks before transplant. Data represent 4 independent experiments with n=20–40 per group. Median with 95% CI. P values calculated by Kruskal-Wallis test. (C) Genotyping of single-cell derived CD45.2+ colonies from bone marrow of mosaic mice at the end of the experiment; n=12–20 per group. (D) CD45.2 HSCs (KL CD150+ CD48− CD34−) shown as percentage of WBM in mosaic mice. Here, Dnmt3a−/− cells were from Vav-Cre Dnmt3afl/fl donors. n=5–20 per group. Median with 95% CI. P values calculated by Kruskal-Wallis test. (E) CD45.2 MPPs as percentage of total WBM in mosaic mice (corresponding to Figure 1D). (F). Mosaic mouse model to compare WT vs Dnmt3a+/− HSCs upon 2-month of infection with M. avium. (G). CD45.2 HSCs (KL CD150+ CD48− CD34−) shown as a percentage of WBM in mosaic mice model to compare Dnmt3afl/fl vs Dnmt3a+/− HSCs upon infection. Median with 95% CI. P values calculated by Kruskal-Wallis test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To confirm that Dnmt3a was efficiently knocked out in the Mx1-Cre+ Dnmt3af/f donors, CD45.2 HSCs were sorted from the recipient mosaic mice at the time of bone marrow evaluation and colonies were cultivated in liquid media. After 2 weeks of expansion, we genotyped the colonies by PCR and found high efficiency of KO in both the uninfected and infected mosaic mice (Figure 1C). Thus, expansion of Dnmt3a−/− HSCs occurred only in the setting of M. avium infection and was not related to a difference in gene deletion frequency.
In the experiments described above, a PIPC-dependent interferon response induced Dnmt3a mutation in the donor mice. To test the hypothesis that this inflammatory signaling altered the bone marrow to provide a selective advantage upon subsequent infectious stress, we repeated our experiments using Vav-Cre Dnmt3af/f mice as donors. These mice do not require PIPC treatment to induce the deletion within the hematopoietic compartment. Expansion of Dnmt3a−/− HSCs was also seen upon infection of mosaics created with Vav-Cre Dnmt3af/f donors (Figure 1D, S1D), indicating that the expansion effect was not dependent on prior PIPC exposure.
We analyzed other bone marrow populations and detected expansion of Dnmt3a−/− cells among multipotent progenitors (MPPs) (Figure 1E). This finding was similar regardless of the Cre-driver used to generate the Dnmt3a-mutant mice. Notably, we did not see expansion among more downstream populations such as granulocyte-monocyte progenitors, megakaryocyte progenitors, or B and T cells, macrophage/monocytes, or granulocytes (Figures S1E–L), suggesting a differentiation block in the Dnmt3a-mutant population at the level of the MPP.
Given that most mutations in DNMT3A in humans are heterozygous, we next tested whether Dnmt3a+/− HSCs expand in our mosaic mouse infection model. Here, we created mosaic mice using WBM from CD45.2 mice with a germline heterozygous Dnmt3a deletion and CD45.1 rescue marrow, or CD45.2 WT mice and CD45.1 rescue marrow as controls (Figure 1F). After 8 weeks of M. avium infection, we were able to detect an expansion of Dnmt3a+/− HSCs in the marrow (Figure 1G). Predictably, the effect size was smaller than that seen for Dnmt3a−/− HSCs.
To confirm the observed phenotypic expansion of HSCs, we sorted CD45.2 WBM from mosaic mice at the end of the experiment and performed colony forming assays in methylcellulose. Colony formation was increased in the infected Dnmt3a−/− mosaics compared to uninfected and WT controls, indicating that there were more functionally defined stem and progenitor cells in the 45.2 compartment of infected Dnmt3a−/− mosaic mice (Figure S1M). Indeed, the number of colonies observed in methylcellulose culture correlated with the number of phenotypically defined HSCs as determined by flow cytometry (Figure S1N, Pearson correlation coefficient 0.76).
To ascertain whether the degree of HSC expansion correlated with the bacterial load during infection, we isolated the spleen and HSCs from two Dnmt3a−/− mosaic mice that showed very high levels of Dnmt3a−/− expansion and the spleen and HSCs from two Dnmt3a−/− mosaic mice that showed little Dnmt3a−/− expansion. In both cases, no bacterial DNA was detected by PCR in the HSCs themselves, but equally high levels of bacterial DNA were detected in the spleen (Figure S1O). These results indicate that the HSCs are not themselves infected, and that the systemic bacterial burden does not account for differences in Dnmt3a−/− expansion.
Chronic infection promotes Dnmt3a−/− clonal hematopoiesis in the peripheral blood of mice.
In humans, CH is defined by variant allele frequency detectible in peripheral blood. In order to determine whether chronic infection promotes expansion of Dnmt3a−/− clones in the peripheral blood, we retained Dnmt3a−/−:WT mosaic mice and monitored the peripheral blood over a period of 12 months (Figure 2A, Figure S2A). A potential limitation of this study is that we did not perform the same 12-month analysis of the WT:WT mosaics, which showed not expansion of HSCs at 2 months post infection. Whereas the percentage of Dnmt3a−/− CD45.2 peripheral blood mononuclear cells (PBMCs) expanded to an insignificant degree in this time frame for naïve mice, the percentage increased significantly for infected mosaic mice at both 8- and 12-months post infection (Figure 2B–G). In the bone marrow, the percentage of total HSCs that were Dnmt3a−/− reached an average of 82.8% in the infected mosaic mice by 12 months after infection, compared to 19.5% for the uninfected controls (Figure 2H–I, S2B).
Figure 2. M. avium infection promotes late Dnmt3a-mutant clonal peripheral blood expansion.
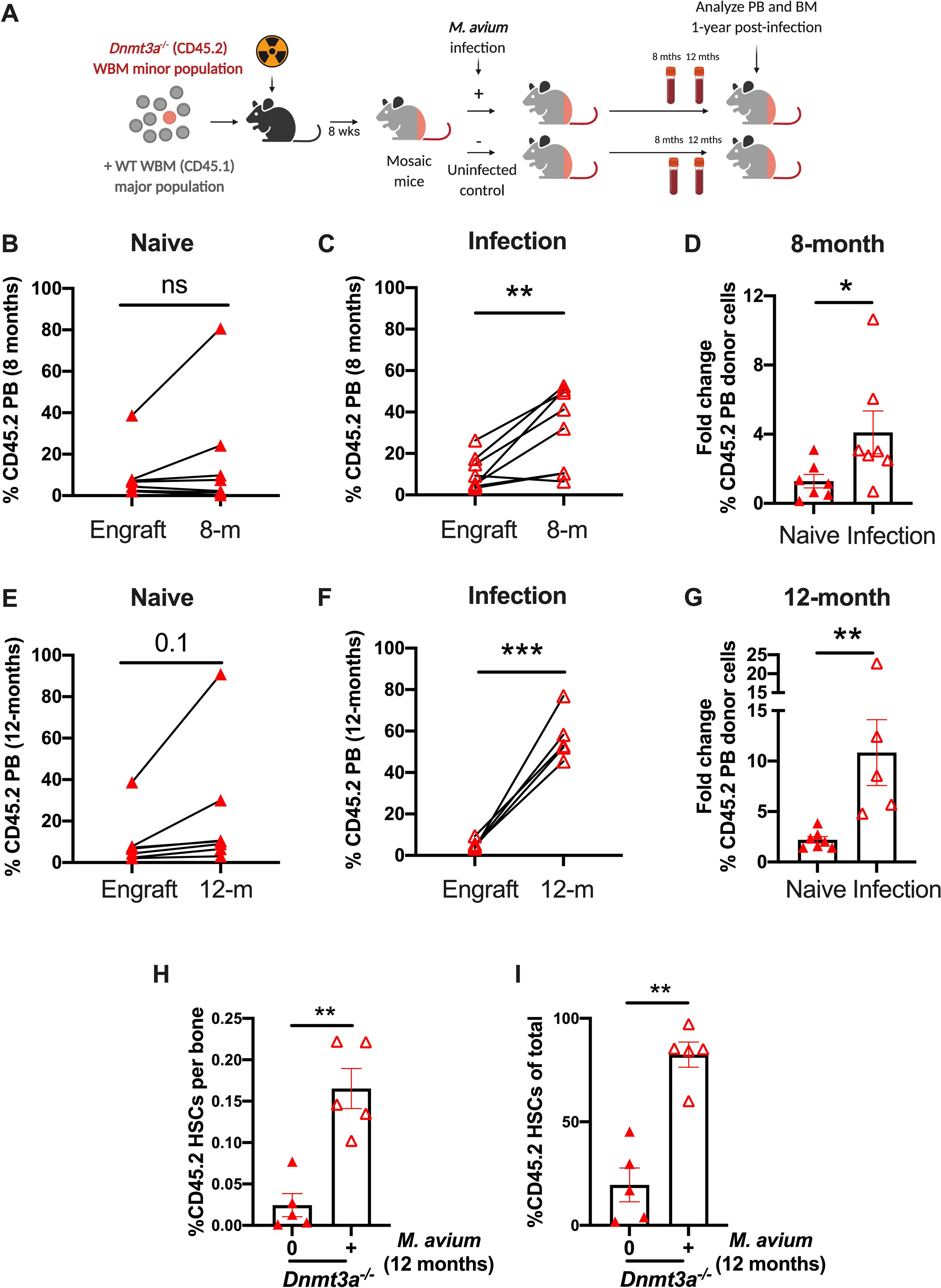
(A) WT recipient mice were transplanted with a minor population of “test” Dnmt3afl/fl or Dnmt3a−/− CD45.2 WBM and a major population of CD45.1 WT competitor. Two months after transplant, half of the mice were infected with M. avium, and the percent of CD45.2 PB leucocytes was measured eight and twelve months later likewise the percent of test CD45.2 HSCs in the BM after twelve months of infection. (B) Paired analysis of CD45.2 population of individual mice at engraftment and after 8-months of treatment with PBS (naïve). (C) Paired analysis of percentage CD45.2 population of individual mice at engraftment and after 8-months of infection with M. avium. (D) Fold change from starting engraftment of CD45.2 population in naïve and M. avium infected mice after 8-months of PBS treatment or infection. (E) Paired analysis of CD45.2 population of individual mice at engraftment and after 12-months of treatment with PBS (naïve). (F) Paired analysis of percentage CD45.2 population of individual mice at engraftment and after 12-months of infection with M. avium. (G) Fold change from starting engraftment of CD45.2 population in naïve and M. avium infected mice after 12-months of PBS treatment or infection. CD45.2 HSCs (KL CD150+ CD48− CD34−) shown as a percentage of WBM (H) or total HSCs (I) in mosaic mice after 12-month of infection. (I) Paired analysis were performed by Paired t-test (Figures 2B-C and 2E-F). Statistical analysis of Figures 2G and 2H-I were performed by using Mann-Whitney test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Dnmt3a−/− HSCs show reduced differentiation and secondary stress-induced apoptosis in response to infection.
In order to understand the mechanism of infection-induced Dnmt3a-loss of function clonal expansion, we next compared the responses of WT versus Dnmt3a−/− HSCs to infectious stress. We previously showed that HSCs divide during chronic infection with M. avium, but these divisions are accompanied by a loss of self-renewal and an increase in differentiation, ultimately depleting the HSC compartment (Baldridge et al., 2010; Matatall et al., 2016). Prior studies have also demonstrated a defect in post-transplant differentiation by Dnmt3a−/− HSCs (Challen et al., 2011), but their capacity to respond to inflammatory stress has not been measured. To ascertain whether Dnmt3a loss of function affects hematologic responses to infection, we infected with M. avium Mx1-Cre Dnmt3af/f mice in which Dnmt3a-deletion had been induced by PIPC treatment 4 weeks prior. After 4 weeks of systemic infection, there were few differences in the bone marrow of Dnmt3a−/− mice compared to PIPC-treated Cre-negative controls. Specifically, neither the number of HSCs nor apoptosis rate of Dnmt3a−/− HSCs were different compared to either näive or infected control mice (Figure 3A and B, S3A and B). The increase in cell division upon infection was similar in Dnmt3a−/− HSCs compared to controls (Figure 3C).
Figure 3: Hematological responses of Dnmt3a−/− mice to infection with M. avium.
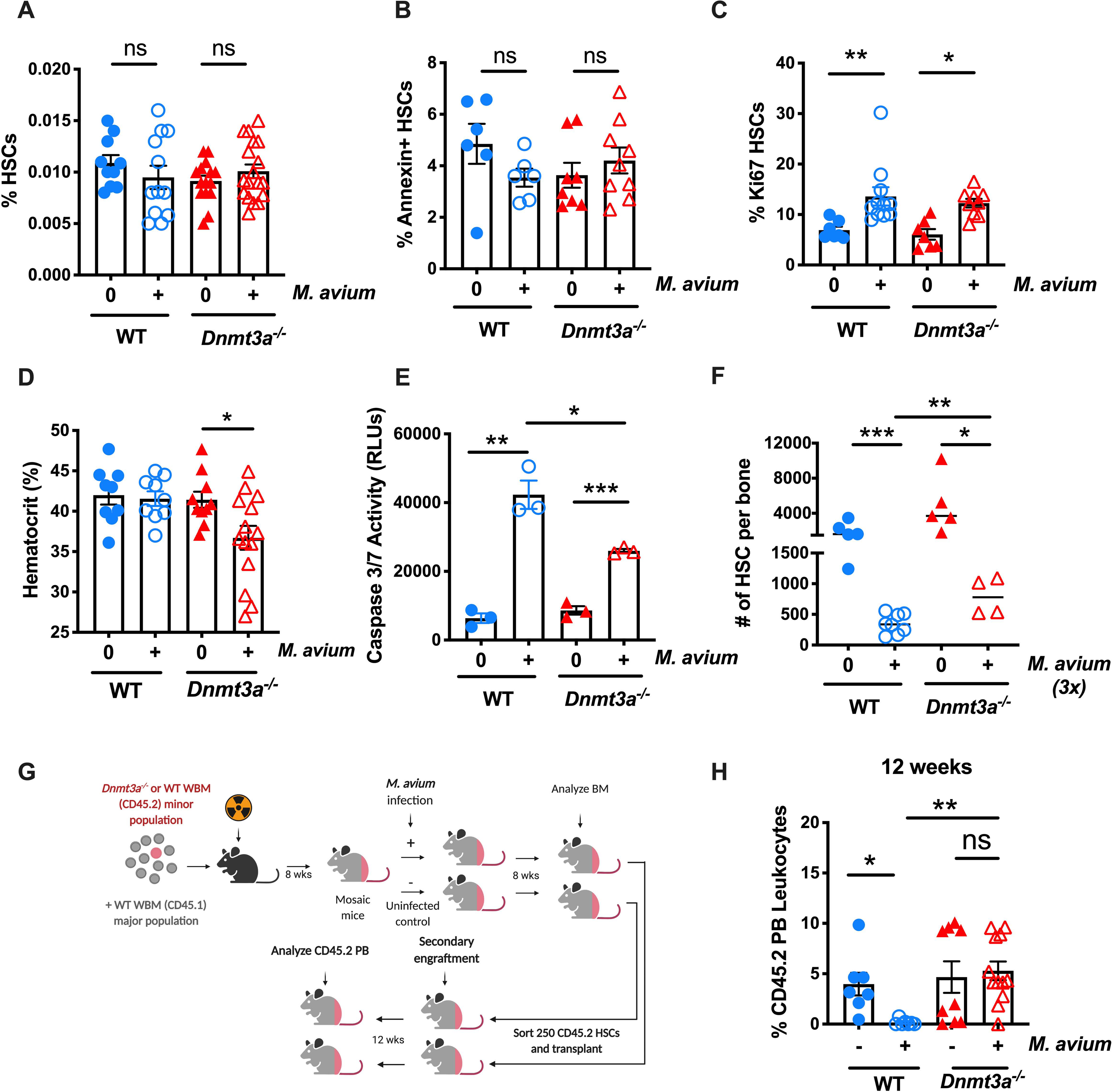
(A-C) WT and Dnmt3a−/− mice were infected with M. avium for 1 month. (A) Percentage of HSCs (KL CD150+ CD48− CD34−) of total WBM. n=10–20 per group; data are representative of 5 independent experiments. Error bars, mean ± SEM. P values calculated by ordinary one-way ANOVA. (B) Percentage of Annexin + HSCs. n=6–10 per group; data are representative of two independent experiments. Error bars, mean ± SEM. P values calculated by ordinary one-way ANOVA. (C) Percentage of Ki67+ HSCs. n=10–15 per group; data are representative of two independent experiments. Error bars, mean ± SEM. P values calculated by ordinary one-way ANOVA. (D) Hematocrit in WT and Dnmt3a−/− mice infected with M. avium for 1 month. n=10–20 per group. Error bars, mean ± SEM. P values calculated by ordinary one-way ANOVA. (E) 400 HSCs (KSL CD150+ CD48− CD34−) were sorted, treated for 12h with IFN-y in vitro prior to Caspase 3/7 assay. n=2–3 per group. Error bars, mean ± SEM. P values calculated by ordinary one-way ANOVA. (F) WT and Dnmt3a−/− mice were infected with M. avium for 3 consecutive months before we analyzed the absolute number of HSCs (KL CD150+ CD48− CD34−) of total WBM. (G) Mouse model of secondary transplant from primary mosaic mice (Figure 1A). 250 sorted donor CD45.2 HSCs (LK CD150+ CD48− CD34−) from previously transplanted animals were co-transplanted with 2.5 × 105 CD45.1 rescue marrow into lethally irradiated mice. PB was assessed 12 weeks post-transplant, and engraftment is shown as percentage of CD45.2 cells in blood (H). Data are presented as mean ± SEM; n = 7–14 per group. P values calculated Kruskal-Wallis test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Functionally, HSCs from WT or Dnmt3a−/− mice were equally able to engraft in WT recipients, and infection did not significantly alter the total engraftment. However, we noted that unlike WT mice Dnmt3a−/− mice became anemic upon M. avium infection, possibly indicating impaired adaptive responses in the blood system (Figure 3D).
IFNγ exposure confers sensitivity to secondary stress-induced apoptosis, demonstrated by an increased propensity of HSCs from M. avium-infected or IFNγ-treated mice to undergo apoptosis after ex vivo culture for 24 hours compared to PBS-treated controls (Matatall et al., 2016; Pietras et al., 2014). In other words, whereas M. avium infection alone is not sufficient to trigger apoptosis in HSCs, it does leave HSCs in a vulnerable state in which any additional stress activates apoptosis pathways. Thus, we compared secondary stress-induced apoptosis in HSCs from infected WT and Dnmt3a−/− mice. Whereas secondary stress-induced apoptosis was elevated in both WT and Dnmt3a−/− HSCs after infection, the degree of apoptosis was less in Dnmt3a−/− HSCs (Figure 3E), indicating that loss of DNMT3A partially protects HSCs against this avenue of depletion.
We previously used differential equations to model the dynamics of HSCs during chronic infection (Matatall et al., 2016), describing the loss of HSCs over time as a function of their starting number (N), division rate (λ), and the proportion of daughter cells lost from the HSC pool to events such as death, differentiation or mobilization (d) (see Supplementary Information). Based on that model, and the reduced differentiation capacity and rate of secondary stress-induced apoptosis of Dnmt3a−/− HSCs (Challen et al., 2011), we predicted that the inflammatory stress of chronic infection would serve as a selection pressure favoring the expansion of Dnmt3a−/− HSC clones compared to WT. Indeed, when we applied the observed differences in differentiation and stress-induced apoptosis to our mathematical modeling, we could predict that a minor population of Dnmt3a−/− HSCs would overtake a major population of WT HSCs in the presence of chronic infection (Figures S3C–F).
To test whether Dnmt3a−/− mice are more resistant to HSC exhaustion upon chronic infection, as indicated by our mathematical model, we infected WT versus Dnmt3a−/− mice with M. avium over the course of three months. Whereas HSCs were depleted in both WT and Dnmt3a−/− mice after three months of infection, the final number of HSCs at the end of the observation period was significantly greater in Dnmt3a−/− mice, indicating a relative protection against the depleting effects of infection (Figure 3F). Further, in order to test whether the Dnmt3a−/− HSCs that expanded after infection (Figure 1B) were bona fide HSCs, we performed a secondary transplant of sorted CD45.2 HSCs collected from mosaic naïve or infected mice (Figure 3G). When 250 sorted donor CD45.2 HSCs from mosaic mice were transplanted into naïve CD45.1 recipients, the Dnmt3a−/− HSCs from both naïve and infected mosaics showed strong long-term engraftment (Figure 3H). By contrast, the WT HSCs from infected mosaics had reduced long-term engraftment, consistent with a known impairment in self-renewal after infection (Figure 3H). Collectively, these studies indicate that the phenotypically defined Dnmt3a−/− HSCs that expand upon infection remain functional in long-term reconstitution studies, are more resistant to loss by secondary stress-induced apoptosis and differentiation, and retain their self-renewal capacity better than WT HSCs when stressed by chronic infection with M. avium.
Human and murine Dnmt3a-mutant HSPCs show decreased differentiation and increased serial replating capacity under inflammatory stress.
To further examine the differentiation capacity of WT versus Dnmt3a−/− HSCs in response to inflammatory stress, we conducted in vitro differentiation assays. LT-HSCs were sorted from WT or Dnmt3a−/− mice and incubated in methylcellulose in the presence or absence of recombinant murine IFNγ (Figure S4A). At high concentrations (100 ng/mL), IFNγ resulted in a loss of total WT colonies, whereas Dnmt3a−/− HSCs were relatively resistant to these toxic effects (Figure 4A, S4B). At low concentrations (1 ng/mL), IFNγ stimulated increased WT colony formation, whereas this increase was diminished for Dnmt3a−/− HSCs (Figure 4B, S4C). Characterization of the colonies formed after co-incubation with 1ng/mL IFNγ revealed few undifferentiated WT CFU-GEMM colonies and an abundance of CFU-GM, CFU-G, and CFU-M colonies, consistent with differentiation. In contrast, the Dnmt3a−/− HSCs generated more CFU-GEMM colonies and fewer CFU-GM, CFU-G, and CFU-M colonies, consistent with a differentiation defect in the Dnmt3a−/− background (Figure 4C, S4D). Collectively, these experiments show that Dnmt3a−/− HSCs have improved survival at high IFNγ concentrations and reduced differentiation at low IFNγ concentrations, both of which may contribute to a survival advantage.
Figure 4: Dnmt3a-mutant HSCs show defective differentiation and increased self-renewal in response to inflammatory insults.
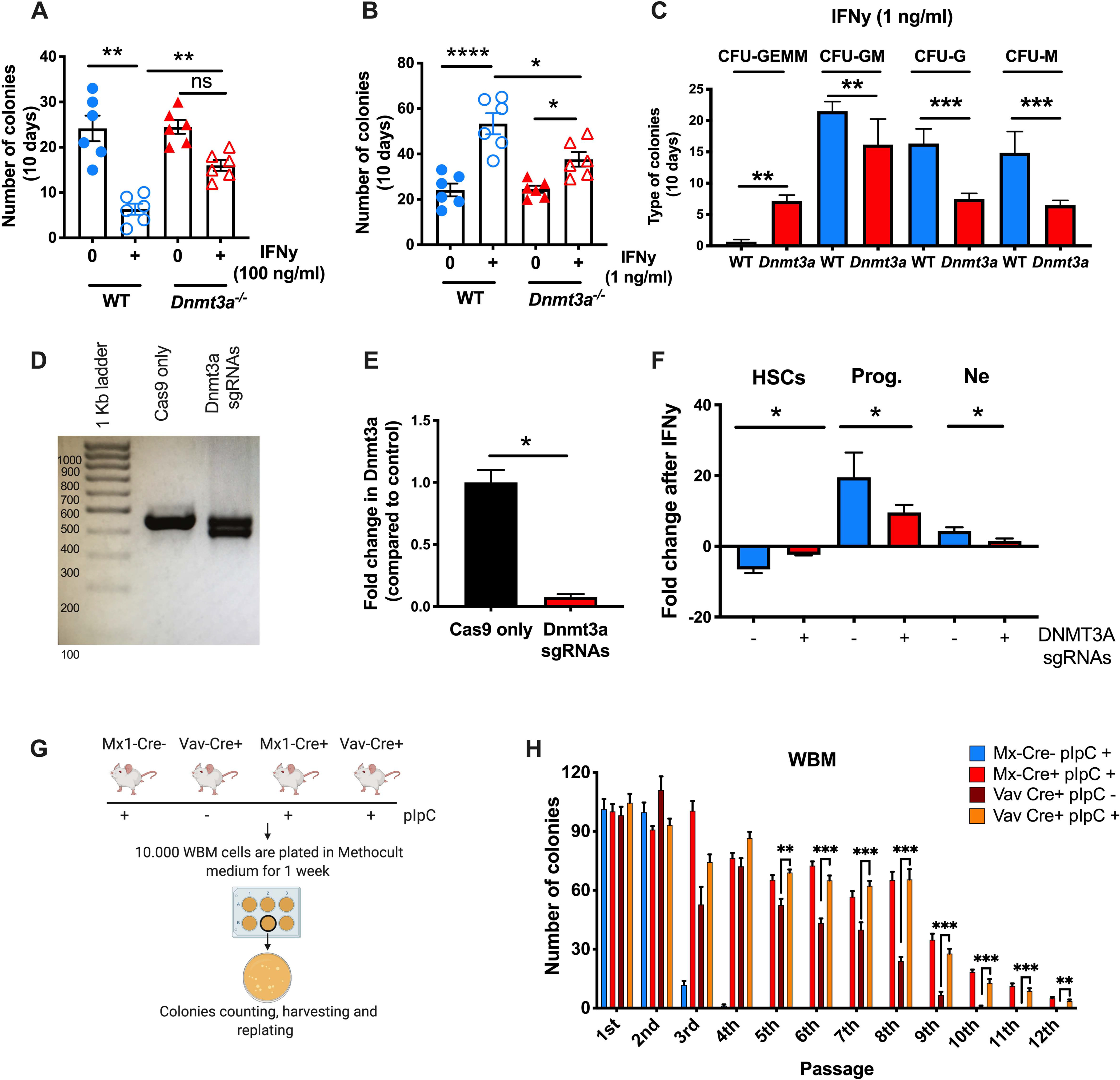
(A-C) 80 LT-HSCs (LSK CD150+ CD48-) from the pool of 3 Dnmt3afl/fl and 3 Dnmt3a−/− mice were sorted and cultured in 2 ml of complete Methocult medium with the presence of absence of rm-IFNy. Two different doses of rm-IFNy were used: 100 ng/ml (Figure 4A) or 1 ng/ml (Figure 4B-C). After 10d of culture, colonies were counted and scored. (A) Number of colonies after 10d of culture with 100 ng/ml of rm-IFNy. Data are representative of 2 independent experiments. Error bars, mean ± SEM. P values calculated by ordinary one-way ANOVA. (B) Number of colonies after 10d of culture with 1 ng/ml of rm-IFNy. Data are representative of 2 independent experiments. Error bars, mean ± SEM. P values calculated by ordinary one-way ANOVA. (C) Morphology of colonies after 10d of culture with 1 ng/ml of rm-IFNy. Data are representative of 2 independent experiments. Error bars, mean ± SEM. P values calculated by Mann Whitney test. (D) Representative gel of Dnmt3a exon 13 region. Expected size of band from WT allele is 450 bp. (E) hDnmt3a RNA expression. P values calculated by two-sided t-test. Error bars, mean ± SEM. Data are representative of two independent experiments performed in triplicate. (F) Differentiation assay in human CD34+ cells treated for 72h with rhIFN-y. Fold change is relative to time 0. Error bars, mean ± SEM. P values calculated by two-sided t-test. Data are representative of three independent experiments. (D-E) Serial replating assay to compare the self-renew capacity of WBM cells from Mx1-Cre and Vav-Cre+ mice with/without pIpC treatment. Data are representative of two independent experiments. Error bars, mean ± SEM. P values calculated by two-way ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To test these observations in human CD34+ HSPCs, we used CRISPR/Cas9 to delete DNMT3A from CD34+ cells isolated from human buffy coats and validated efficient knock out (KO) of the gene by quantitative PCR (Figure 4D–E). Typically, WT CD34+ HSPCs differentiate in response to in vitro exposure to IFNγ, as evidenced by loss of HSC markers (e.g., CD34) and gain of markers of more differentiated cells (e.g., CD38, CD66b) (Matatall et al., 2016). We carried out differentiation assays with the KO cells versus controls that were subjected to the same editing procedures but without the sgRNAs (“Cas9 only”). Upon IFNγ stimulation, control CD34+ cells differentiated at a high rate, as indicated by a loss of CD34+ CD38− HSCs and a significant increase in CD38+ progenitors and CD66b+ neutrophils. In contrast, DNMT3A−/− human CD34+ cells did not differentiate normally, showing little change in either their HSC or neutrophil numbers after three days in culture with IFNγ (Figure 4F). These data demonstrate DNMT3A-mutant HSCs have a striking defect in IFNγ-induced differentiation.
To test the impact of Dnmt3a mutation on HSC self-renewal in the setting of inflammatory stress, we performed serial replating assays of murine bone marrow from PIPC-treated Mx1-Cre−Dnmt3af/f controls, Mx1-Cre+ Dnmt3af/f mice, and Vav1-Cre+ Dnmt3af/f (Figure 4G). We found that whole bone marrow from Dnmt3a−/− mice persisted for 8 to 9 rounds of serial replating as opposed to 2–3 rounds for WT controls, regardless of the Cre driver (Figure 4H, compare red and maroon to blue). Furthermore, to test the effect of PIPC induction on Dnmt3a−/− marrow, we serially replated WBM from Vav1-Cre+ Dnmt3af/f mice that had been previously treated with PIPC or PBS alone. As shown in Figure 4H, PIPC treatment slightly augmented the serial replating capacity of Dnmt3a-mutant WBM (compare maroon to orange), extending the potential for replating from 9 to 12 passages. A similar increase was also noted for Dnmt3a−/− cells upon culture of cKit+ enriched cells, indicating that increased replating capacity was not simply a result of increased progenitor numbers (Figure S4E). Altogether, these serial replating studies indicate that inflammatory signals actually reinforce the self-renewal Dnmt3a−/− HSCs.
Dnmt3a loss of function blunts HSC response to infection.
To investigate the mechanisms underlying differential responses to infectious stimulation by WT versus Dnmt3a−/− HSCs, we conducted cytokine profiling of WT and Dnmt3a−/− mice in the presence or absence of infection. In prior studies we noted that many of the effects of M. avium infection are potentiated by the inflammatory cytokine IFNγ which is abundantly expressed throughout chronic mycobacterial infection. Indeed, administration of recombinant IFNγ alone is sufficient to interrupt HSC quiescence and promote myeloid differentiation (Matatall et al., 2014); whereas HSCs lacking the IFNγ receptor do not increase their cell cycle rate in response to M. avium infection (Baldridge et al., 2010). Here, cytokine profiling demonstrated that Dnmt3a−/− mice produce similar levels of IFNγ in response to M. avium infection as WT mice, as well as other inflammatory cytokines such as IL6 and TNFα (Figure 5A). However, some differences were noted; specifically, IL1α and IL9 were significantly suppressed, whereas IL1β and IL12 were elevated in Dnmt3a−/− mice compared to WT (Figure 5A, S5A).
Figure 5. IFN-y signaling is necessary for Dnmt3a-mutant clonal expansion in M. avium infection.
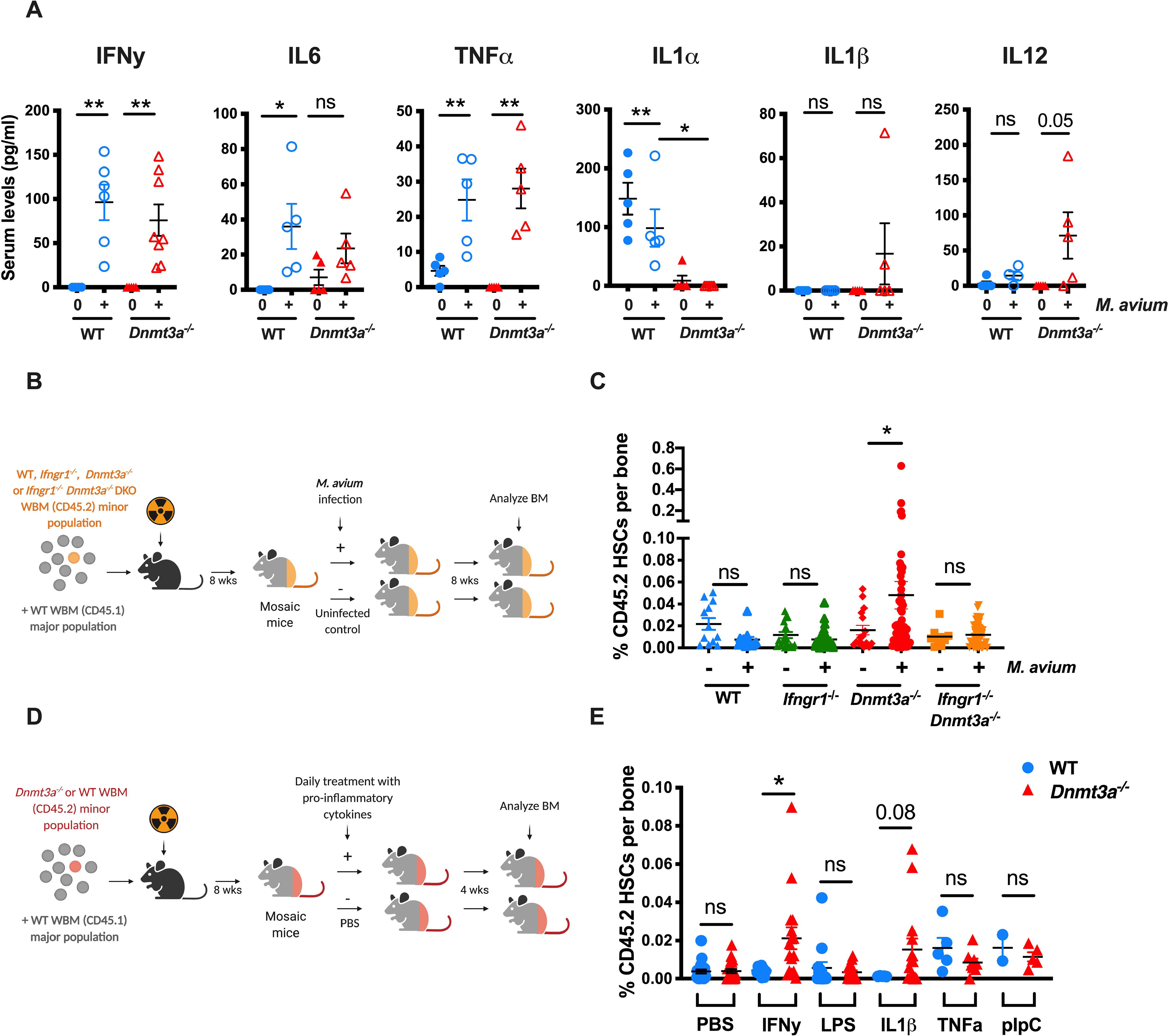
(A) Serum cytokine levels of IFNy, IL6, TNFa, IL1a, IL1b and IL12 in WT and Dnmt3a−/− (Mx1-Cre) mice that were naïve or infected for one month with M. avium. n=5 per group. P values calculated by ordinary one-way ANOVA or Kruskal-Wallis test. (B) Mosaic mouse model to compare Ifngr1−/− Dnmt3a−/− double KO (DKO) vs Dnmt3a−/− HSCs upon infection. WT recipient mice were transplanted with a minor population of “test” WT, Ifngr1−/−, Dnmt3a−/− or Ifngr1−/− Dnmt3a−/− DKO WBM and a major population of WT competitor. Two months after transplant, half of the mice were infected with M. avium, and the percent of test CD45.2 HSCs measured eight weeks later. (C) Percentage of CD45.2 HSCs of WBM in mosaic mice. Data are representative of two independent experiments; n=10–20 per group. Error bars, mean ± SEM. P values calculated by Kruskal-Wallis test. (D) Mosaic mouse model to compare Dnmt3afl/fl vs Dnmt3a−/− HSCs upon daily treatment with pro-inflammatory cytokines for 1-month. (E) Mosaic mice were injected daily with IFNy, LPS, IL1b, TNFa or pIpC for one month prior to determination of percentage of CD45.2 HSCs in WBM. Error bars, mean ± SEM. P values calculated by two-sided t-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To assess the role of IFNγ signaling in Dnmt3a−/− clonal expansion during M. avium infection, we infected mosaic mice containing a minor population of Dnmt3a−/− Ifngr1−/− DKO HSCs (Figure 5B). As shown in Figure 5C, Dnmt3a−/− clonal expansion occurred as expected upon M. avium infection, but no clonal expansion was evident among mosaics with the DKO minor population or in mosaic mice with a minor population of Ifngr1−/− HSCs. These data indicate that IFNγ signaling is required for M. avium-induced Dnmt3a−/− clonal expansion. Furthermore, our findings reinforce the observation that inflammation-mediated depletion of the major WT population is insufficient to yield clonal expansion of Dnmt3a−/− HSCs and indicate that cell-autonomous IFNγ signaling is essential for their expansion.
In order to test if a different type of infection similarly induces Dnmt3a−/− HSC expansion, we created mosaic mice and replaced M. avium infection with four rounds of acute lymphochoriomeningitis virus (Armstrong strain), administered IP every 10 days (Figure S5B). This strain of LCMV induces a brief 2-day spike of IFNα followed by a lower level of IFNγ over the course of 7–10 days (Matatall et al., 2014). In this experiment, we saw no statistically significant expansion of Dnmt3a−/− HSCs (S5C–D), indicating not all types of infections are sufficient to drive clonal expansion.
To test whether inflammatory stimulation is sufficient to drive CH, we attempted to induce clonal expansion of Dnmt3a−/− HSCs by administering a selection of recombinant inflammatory cytokines alone (Figure 5D). Strikingly, daily injection of recombinant IFNγ for one month was sufficient to induce expansion of the Dnmt3a−/− clones (Figure 5E). Furthermore, daily injection of IL1β induced a clear trend toward expansion, whereas no trend was observed in the setting of daily LPS, TNFa, or polyinosinic-polycytidylic acid (pIpC) treatment (Figure 5E). Collectively, these experiments indicate that sustained IFNγ signaling is necessary and sufficient to induce infection-associated Dnmt3a−/− HSC expansion.
To identify the mechanisms underlying Dnmt3a−/− HSC expansion during infection, we conducted RNAseq analysis of WT and Dnmt3a−/− HSCs in the presence or absence of M. avium infection. Whereas infection significantly altered gene expression in WT HSCs, there were fewer differences in the Dnmt3a−/− background (Figure 6A, 6B, Supplemental Tables 2–3). Indeed, blinded analysis of the RNAseq data confirmed that the differences between WT infected and naive samples were greater than those between the Dnmt3a−/− infected and naïve samples (Figure S6A). HSCs from both genotypes appeared to respond to IFNγ signaling during the infection, as evidenced by increased expression of interferon response genes and similar expression of the IFNγ receptor Ifngr1 (Figure S6B–C) as well as similar induction of common IFNγ-regulated genes such as Stat1 and Bst2, as validated by qPCR (Figure 6C). However, the total number of differentially expressed genes was lower in the Dnmt3a−/− background than WT (Figure 6D), with 155 genes upregulated in WT but not Dnmt3a−/− HSCs. Gene ontogeny analysis revealed that genes involved in cell differentiation were particularly enriched among those induced in WT but not Dnmt3a−/− mice (Figure 6E). Indeed, an interacting network of AP1 family transcription factors involved in differentiation, including Fos, Jun, and Batf2, were upregulated in the WT but not Dnmt3a−/− HSCs (Figure 6F). We used qPCR to verify that these interrelated genes were upregulated during M. avium infection in WT but not Dnmt3a−/− HSCs (Figure 6C). Also notable was a lack of induction of autoregulatory genes Socs3 and Nr4a1 in the Dnmt3a−/− background (Figure 6C). These genes provide feedback regulation of interferon signaling (Freire and Conneely, 2018; Kim et al., 2017). Collectively, these findings highlight a series of differentiation-related genes whose expression is induced in WT mice during infection, but not in the Dnmt3a−/− background.
Figure 6. Responses to infection are blunted in Dnmt3a-mutant HSCs.
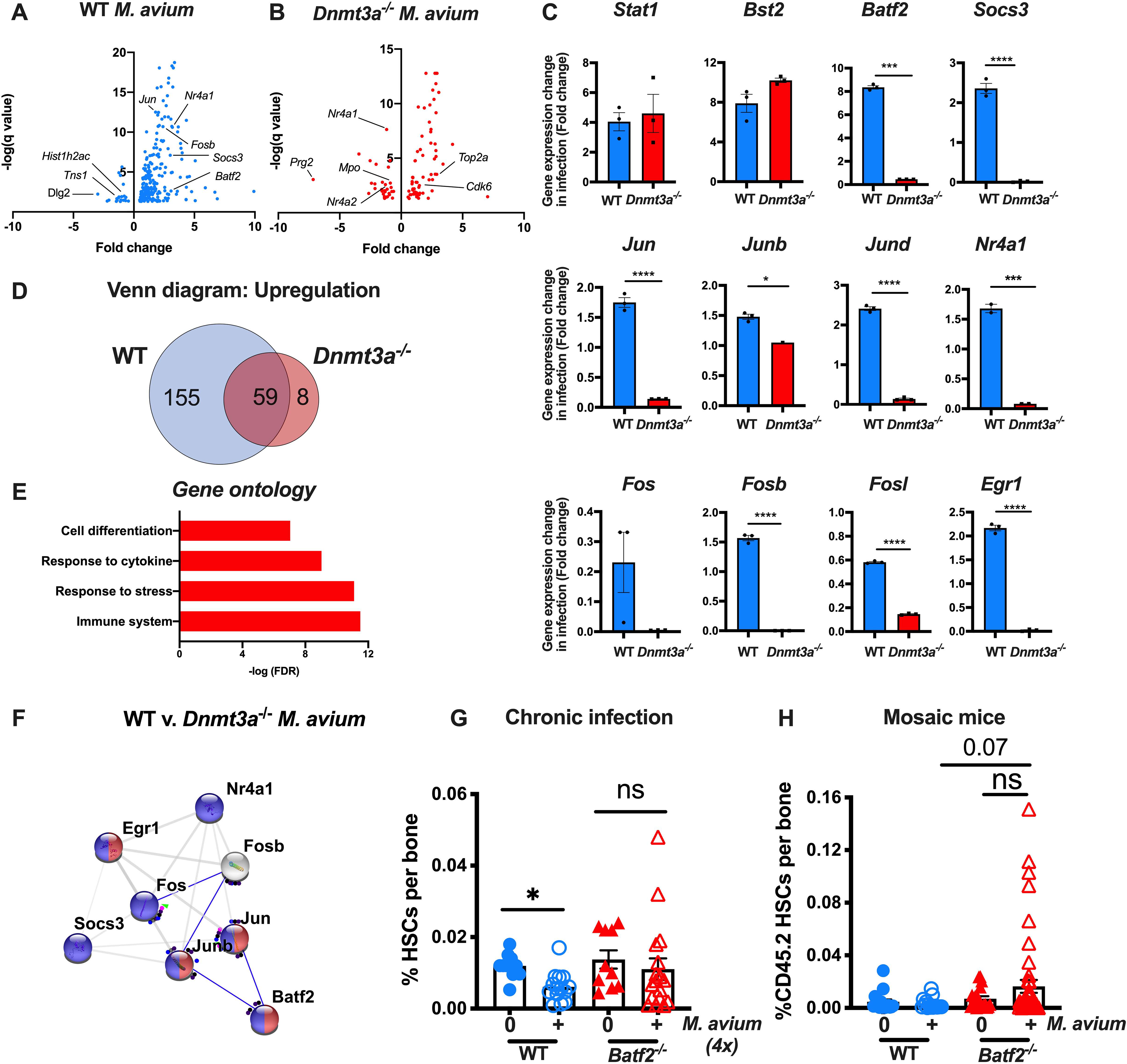
(A) RNA-seq volcano plot showing genes with a significant change (q value<0.05) between HSCs from infected and naïve WT mice. (B) RNA-seq volcano plot showing genes with a significant change (q value<0.05) between HSCs from infected and naïve Dnmt3a−/− mice. (C) qPCR validation of genes from RNA seq data. n=3 per group. P values calculated by two-sided t-test. (D) Venn diagram with number of genes upregulated during infection in WT only (blue), Dnmt3a−/− only (light red), or both (dark red). (E) Gene ontology pathways enriched among genes upregulated in HSCs from WT but not Dnmt3a−/− mice during infection. (F) String interactions of validated genes with altered expression during infection in WT but not Dnmt3a−/− HSCs shows a network of transcription factors, including Batf2. (G) Percentage of HSCs (LK CD150+ CD48− CD34−) of total WBM in WT and Batf2−/− mice infected with M. avium for 4 months. Data are representative of two experiments, each with n=5–10 per group. Error bars, mean ± SEM. P values calculated by Kruskal-Wallis test. (H) CD45.2 HSCs (KL CD150+ CD48− CD34−) shown as a percentage of WBM in mosaic mice after two-month infection with M. avium. Error bars, mean ± SEM. P values calculated by Kruskal-Wallis test. *p < 0.05, **p < 0.01, ***p < 0.001.
We noted significant mouse-to-mouse variability in the expansion of Dnmt3a−/− HSCs in infected mosaic mice (Figure 1B). In order to assess whether this variability might be explained by acquisition of secondary mutations or alterations in the transcriptional response to infection, we performed RNAseq on CD45.2 HSPC populations from mice with large clonal expansion versus those without. Although RNAseq is less sensitive than whole exome or whole genome sequencing, these analyses yielded little to no differences in the transcriptional responses by HSPCs in mosaic mice that did or did not expand during infection, suggesting that secondary mutations were not responsible for the variability (Figures S6D–E).
We previously reported IFNγ-dependent induction of the differentiation factor BATF2 in HSCs upon IFNγ stimulation or M. avium infection and further showed that BATF2-deficient human CD34+ HSPCs display disrupted myeloid differentiation upon IFNγ stimulation in vitro (Matatall et al., 2016). To determine whether knockdown of Batf2 phenocopies Dnmt3a deficiency to give HSCs a selection advantage during chronic inflammatory stress, we created Batf2 KO mice in a C57Bl6 background by CRISPR/Cas9 deletion of mouse embryos (Figure S6F) and infected them with M. avium repeatedly over the course of four months. As noted in Figure 6G, whereas HSCs were depleted upon chronic repeated infection of WT mice, the number of HSCs remained relatively intact in Batf2 KO mice, similar to the protection observed in chronically infected Dnmt3a−/− mice (Figure 3F). These findings indicate that, similar to Dnmt3a−/− HSCs, Batf2-mutant HSCs are more resilient in the setting of chronic inflammatory stress than WT.
We further created mosaic mice with a minor population of Batf2−/− HSCs and a major population of WT HSCs, and infected these mice with M. avium. Just as Dnmt3a−/− HSCs expanded in mosaic mice upon infection, Batf2−/− HSCs expanded but to a lesser degree than seen in Dnmt3a−/− HSCs (Figure 6H). These findings indicate that loss of the pro-differentiation factor Batf2 partially phenocopies the clonal expansion effects of Dnmt3a loss during chronic infection.
Prodifferentiation factors are hypermethylated in Dnmt3a−/− HSCs
Since DNMT3A is a de novo DNA methyltransferase, transcriptional differences in WT versus Dnmt3a−/− HSCs upon infection suggest a possible role for epigenetic regulation in the HSC transcriptional response to inflammation. We therefore performed whole genome bisulfite sequencing (WGBS) of HSCs isolated from WT or Dnmt3a−/− mice that were naïve or infected with M. avium. Hierarchical clustering of WGBS indicated that the global methylation of Dnmt3a−/− cells were more divergent upon infection compared to WT (Figure 7A). Indeed, there were vastly more hyper- and hypo-methylated regions in infected versus naive Dnmt3a−/− HSCs compared to WT (Figure 7B). Among the altered regions, both WT and Dnmt3a−/− HSCs showed slightly more hypomethylated rather than hypermethylated regions, although changes in both directions were present (Figure 7C). In the WT background, regions of hyper- and hypomethylation were mostly restricted to CpG islands (Figure 7D). By contrast, highly significant differences in hypomethylation were found in the Dnmt3a−/− HSCs in CpG islands, shores, enhancers, and promoters (Figure 7D). Collectively, these findings indicate that DNMT3A is highly active in HSCs during infection, and that loss of DNMT3A function results in significant global changes in methylation, including both hyper- and hypo-methylation, in genome regions that likely affect gene transcription.
Figure 7. Profound changes in the methylation landscape of Dnmt3a−/− HSCs in infection.
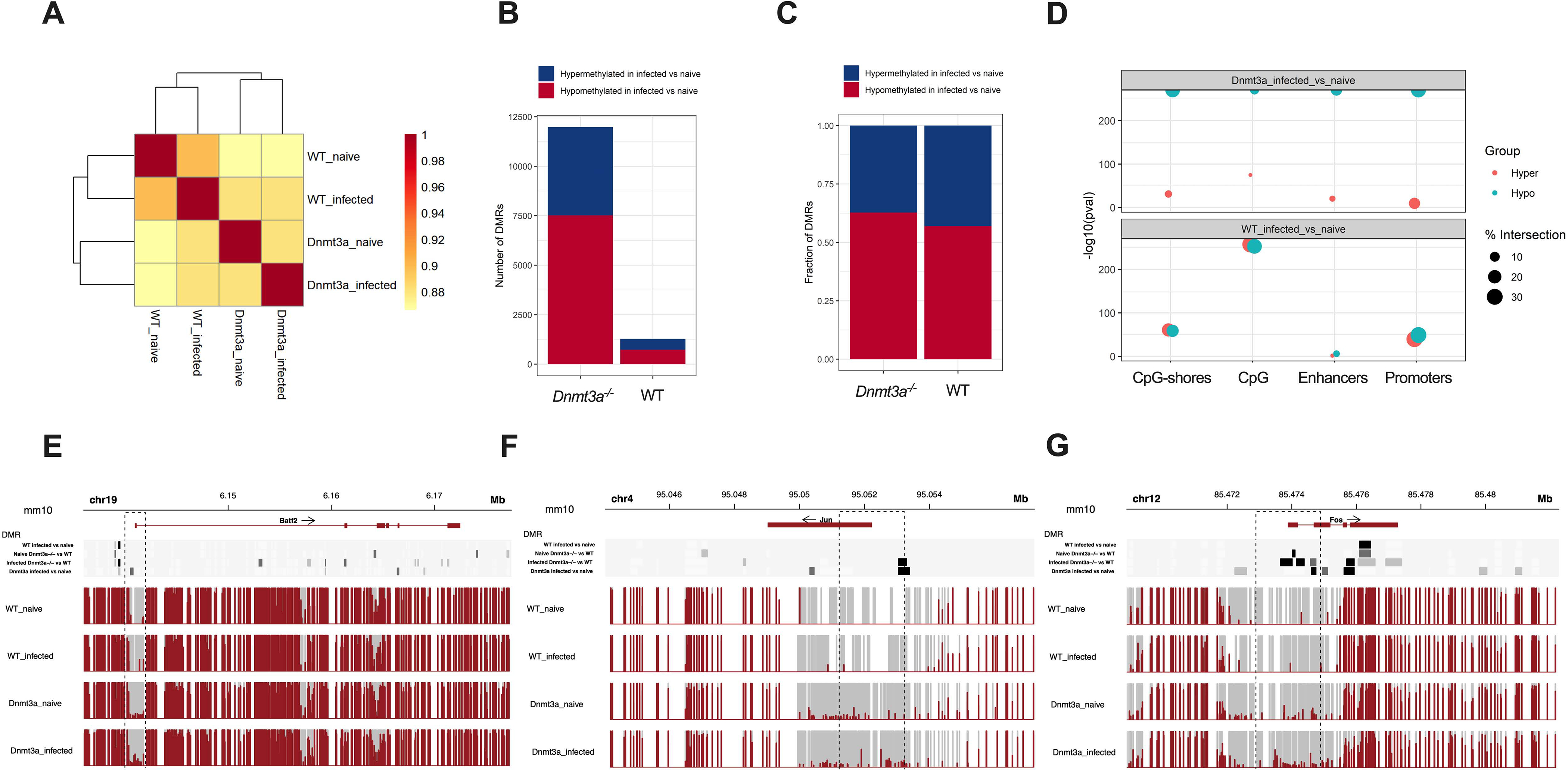
(A) Hierarchical clustering based on CpG methylation correlation in WT and Dnmt3a−/− HSCs naïve and after 1-month of M. avium infection (B) Number of hyper- or hypo-methylated DMRs in WT and Dnmt3a−/− HSCs naïve and after 1-month of M. avium infection. (C) Fraction of hyper- or hypo-methylated DMRs in WT and Dnmt3a−/− HSCs naïve and after 1-month of M. avium infection. (D) Enrichment analysis. Size of data points represents the overlap percentage with the size of the corresponding regulatory regions in the denominator. (E) DNA methylation profile of Batf2 in WT or Dnmt3a−/− HSCs naïve or after 1-month of infection. (F) DNA methylation profile of Jun in WT or Dnmt3a−/− HSCs naïve or after 1-month of infection. (G) DNA methylation profile of Fos in WT or Dnmt3a−/− HSCs naïve or after 1-month of infection. For Figures E-G, significance is shown at the top with light gray denoting p<0.05; medium gray denoting p<0.01, and black denoting p<0.001.
To assess the relationship between DNA methylation and specific differentially expressed genes identified through RNAseq analysis, we examined the methylation of Batf2, Jun, and Fos. All three of these prodifferentiation genes showed increased methylation in the promoter region in the Dnmt3a−/− background as compared to the WT background (Figure 7E–G, S7A–C). Hypermethylation of these promoter regions is consistent with their transcriptional repression.
Discussion:
Here we present the first evidence of an environmental factor that drives Dnmt3a-loss of function CH. Our controlled biological experiments provide proof of concept that an infection can promote Dnmt3a-loss of function CH through increased resistance to stress-induced apoptosis and a defect in differentiation, driven by impaired expression of an IFNγ-dependent transcriptional network (Figure S7). We find that inflammatory IFNγ signaling is both necessary and sufficient for Dnmt3a-loss of function CH. Importantly, these data demonstrate that DNMT3A is a critical participant in the normal HSC response to infection and inflammation.
In prior work we showed that infection significantly impairs HSC quiescence and self-renewal (Baldridge et al., 2010). Indeed, months-long exposure to systemic M. avium infection drives extensive HSC differentiation and depletes the number of bone marrow resident HSCs by over 90% (Matatall et al., 2016). The baseline rate of HSC differentiation is impaired in Dnmt3a−/− HSCs (Challen et al., 2011), and a modestly higher rate of self-renewal is thought to underlie the natural age-associated expansion in HSC number that is observed in Dnmt3a−/− mice (Challen et al., 2011). We extended these findings to test the prediction that the differentiation defect in Dnmt3a−/− HSCs allows them to overtake WT HSCs in the setting of infection. The fact that HSC−/− clones expand in both percentage and absolute number suggests that their expansion is not simply a reflection of decay of the competing WT compartment (Lee-Six and Kent, 2020). Furthermore, since the mosaic system enables a comparison of the behavior of genetically diverse HSCs in the same WT environment, clonal expansion must be attributable at least in part to a cell autonomous difference in the behavior of the HSCs and is not dependent on loss of Dnmt3a in nonhematopoietic niche cells. We find after inflammatory stress Dnmt3a−/− HSCs are significantly more resistant to both secondary stress-induced apoptosis and differentiation than WT, which together confer a selection advantage under inflammatory conditions.
Competition between HSC clones can be thought of in evolutionary terms (Hsu et al., 2018; Rozhok and DeGregori, 2015); and, similar to the evolution of species, competitive selection of HSCs depends on both genetic variation and environmental pressure. A recent study showed that intestinal permeability and elevated inflammatory signaling are necessary for CH in a mouse model of somatic Tet2 mutation (Jaiswal et al., 2017; Meisel et al., 2018). Furthermore, these studies highlight the fact that the environmental cues that promote CH (e.g. microbial exposure) may be closely tied to the biological consequence of the corresponding mutation (e.g. altered inflammatory signaling by Tet2-deficient macrophages (Jaiswal et al., 2017)), thus demonstrating that interactions between genes and environment drive CH (King et al., 2019).
Immune signaling pathways are known to contribute to DNMT3A-associated hematologic conditions including aplastic anemia, myeloproliferative neoplasms, myelodysplastic syndrome, and acute myelogenous leukemia (Azrakhsh et al., 2019; Jacquelin et al., 2018; Yoshizato et al., 2015). Our data provide evidence that the measurable differences in response to infection and inflammation by Dnmt3a−/− HSCs are sufficient to support their competitive advantage against WT cells. The rapid rate of cell division in our post-transplant mosaic mice exaggerates competitive differences that would be expected to take a longer period of time in non-transplant conditions. Aging itself is associated with increased inflammation (Piber et al., 2019), and variability in inflammation in individuals over decades may drive the variable presentation of CH. Our study provides definitive evidence that inflammation can drive Dnmt3a loss of function CH, thus supporting the concept that management of acute and chronic inflammatory conditions including infections may modify risk for clonal expansion and subsequent malignancy (Binder et al., 2018; Mantovani et al., 2018).
Our transcriptional profiling data introduce the novel concept that Dnmt3a−/− clones outlast WT clones by remaining agnostic to the differentiation pressures of inflammation. Since many CH-associated mutations themselves contribute to increased inflammatory signaling, including mutations in TET2, FLT3, JAK2, STAT1, CBL, and RAS (Azrakhsh et al., 2019; Cull et al., 2017; Jaiswal et al., 2017), resistance to the damaging effects of inflammation may be a common theme in CH.
Transcriptional and epigenetic differences in Dnmt3a−/− clones upon infection provide direct evidence that an epigenetic regulator plays a key role in the responses of HSCs to inflammation. An array of pro-differentiation factors, including members of the Fos and Jun family of transcription factors, are relatively suppressed in the Dnmt3a−/− background. Consistent with this observation, we found that the promoters of these genes were hypermethylated in the Dnmt3a−/− background. While hypermethylation in the setting of a Dnmt3a deletion seems counterintuitive, it has also been reported in other studies, and may be related to compensatory effects of alternative methyltransferases (Jeong et al., 2018). Strikingly, loss of the histone demethylase Kdm6b leads to opposite effects on Fos and Jun, suggesting an interrelated regulatory mechanism (Mallaney et al., 2019). In a recently published study examining gene expression differences between serially transplanted Dnmt3a−/− HSCs and WT HSCs, these genes are also differentially suppressed in the Dnmt3a−/− cells, suggesting that infection is likely not the only chronic stress or source of chronic inflammation that contributes to their regulation (Jeong et al., 2018). Our findings lend further support to the emerging theme that inflammation is a major driver of both aging and age-associated CH, and cells that resist the depleting effects of inflammation attain dominance over time.
As a case in point, we previously identified BATF2 to be a critical IFNγ-induced transcription factor that drives myeloid differentiation of HSCs. Here we show that, unlike their WT counterparts, Batf2-deficient stem cells resist depletion upon chronic infection. One can surmise that this survival advantage comes at the expense of immunological responsiveness; it will be interesting to determine the extent to which age-related deficits in immunity are attributable to reduced inflammatory responses in the HSC compartment.
Of note, two autoregulatory genes, Nr4a1 and Socs3, were induced in WT but not Dnmt3a−/− HSCs upon infection. Nr4a1 has been reported to be reduced in Dnmt3a-mutant hematopoietic progenitors, leading to suppression of apoptosis (Kramer et al., 2017). Both NR4A1 and SOCS3 have been noted to play a role in supporting HSC homeostasis by dampening inflammatory responses (Freire and Conneely, 2018; Kim et al., 2017). The fact that the gene signature in Dnmt3a−/− HSCs during infection shows a dampening of inflammatory signaling suggests that reduced expression of these two genes in the Dnmt3a−/− background are passenger rather than driver effects. We also note that PIPC treatment enhanced the self-renewal of Dnmt3a−/− HSCs in serial replating assays and, consistent with that observation, many alterations in genome methylation occurred in bivalent canyon sites that have been associated previously with developmentally important genes (Jeong et al., 2014).
In summary, we demonstrated that chronic IFNγ signaling is sufficient to drive expansion of Dnmt3a-loss of function HSC clones in a mouse model of HSC clonal competition. Clonal expansion occurred as a result of differences in secondary stress-induced apoptosis and inflammation-induced differentiation, normally driven by DNMT3A-dependent induction of Fos/Jun family pro-differentiation factors. Our studies suggest that an array of inflammatory cytokines including IFNγ, IL1β, and IL12 may be sufficient to promote these effects, and they provide the first evidence that DNMT3A participates actively in HSC-based responses to infection.
Limitations of the Study:
Our study has a number of limitations, the most significant of which is a lack of validating human data. The challenge of gathering longitudinal data spanning decades in humans is not trivial but we hope this will be the subject of future work. We also show that Dnmt3a−/− mice become anemic with infection, but this is not a common feature of humans with CH although increased red cell distribution width (RDW) has been reported (Jaiswal et al., 2014). It will be interesting to investigate to what extent CH may contribute to conditions such as anemia of chronic disease. Of note, there have been recent reports of human epidemiologic data correlating CH with severe infections (Bolton et al., 2020a), and it will be important to assess the degree to which these phenomena are interrelated in humans. The phenotypes reported in this study largely focus on KO mice, and as shown in Figure 1 give a stronger effect than heterozygous loss of function mutations commonly found in humans and do not accurately model gain of function mutations.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Katherine Y. King (kyk@bcm.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Online resources:
RNAseq and WGBS data are publicly available through Mendeley as follows: http://dx.doi.org/10.17632/zbc66krbv4.1 (RNAseq 1), http://dx.doi.org/10.17632/wj5knck449.1 (RNAseq 2), http://dx.doi.org/10.17632/t5szbhjwdc.1 (WGBS).
Code availability:
Formulas used to calculate mathematical predictions in Supplemental Figures 2C–F are available in Supplementary Table 1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
We used wild-type C57BL/6 (CD45.2) and C57BL/6.SJL (CD45.1) mice 6–12 weeks of age. Dnmt3afl/fl mice, a kind gift from Dr. Margaret Goodell, were crossed to either Mx1-Cre mice (Stock No: 003556) or Vav-iCre mice (Stock No: 008610) obtained from Jackson Labs. Dnmt3afl/fl Ifngr1 DKO mice were produced by mating Mx1-Cre Dnmt3afl/fl with Ifngr1−/− mice (Stock No: 025394) all on C57BL/6 background. Dnmt3a+/− germline heterozygous mice were also received from the Goodell lab. Batf2-KO mice were generated in conjunction with the genetically engineered mouse core facility at BCM by the use of CRISPR gene editing to remove exons 1 and 2 of Batf2 in a C57BL/6 background. Heterozygous mice from the initial transfer were outcrossed to WT mice and F1 pups were then crossed to generate homozygous mice. Genotyping was determined by PCR and sequencing. The following CRISPR single guide RNAs were used: GGTGCACTCACTCGCACTCGCTC and GGTCTCACTCTTGGTTCAAAAGG. All mice were maintained at an AALAC-accredited, specific pathogen-free animal facility at Baylor College of Medicine. All experiments were approved by the institutional review board of Baylor College of Medicine.
Microbial infections
Mice were infected with 2 × 106 colony-forming units of Mycobacterium avium intravenous (IV) as described (Feng et al., 2008). For chronic infections, mice were repeatedly infected every four weeks for either 3 or 4 months, as indicated. M. avium was detected by growth on Middlebrook agar and by PCR (Park et al., 2000). LCMV infections were performed with 1 × 105 plaque-forming units (PFUs) with four injections every 10 days.
METHOD DETAILS
Bone marrow transplantation
Whole bone marrow transplants were performed by IV injection of 2 × 105 CD45.2 donor WBM cells from infected mice or naive controls with 2 × 105 CD45.1 competitor into CD45.1 WT recipients following a split dose of 10.5 Gy of irradiation. For mosaic mice, lethally irradiated CD45.1 naïve recipient mice were transplanted with 1. 5 × 105 unfractionated CD45.2 donor WBM (Dnmt3a−/−, Dnmt3a−/− Ifngr1−/−, Batf2−/−, or Cre-negative/WT) competed against 6 × 105 unfractionated WBM cells from age-matched CD45.1 mice. Deletion of floxed alleles in Mx1-Cre Dnmt3af/f donor WBM was carried out 5–6 weeks before transplantation in primary recipients by six intraperitoneal injections (300 μg per mouse) of pIpC (Sigma) in PBS every other day. Eight weeks after transplantation, mosaic mice were infected with M. avium (or mock-infected with PBS) for eight additional weeks prior to bone marrow evaluation. To check the efficiency of deletion at the end of the experiment, CD45.2+ HSCs from mock-infected and infected mosaic mice were sorted into StemSpan media (Stem Cell Technologies), incubated for 2 weeks, and then the colonies were genotyped.
Flow cytometry
Peripheral blood was analyzed with a Hemavet 950. WBM cells were isolated from femurs, tibias, and pelvises. For antibody staining, cells were suspended at a concentration of 108 cells/mL and incubated at 4°C for 15 minutes with the desired antibodies. Magnetic enrichment was performed with anti-c-Kit microbeads (eBioscience, San Diego, CA) on an AutoMACS (Miltenyi Biotec, Germany). Post-enrichment, the positive cell fraction was labeled with antibodies to identify HSCs (Lineage- c-Kit+ CD150+ CD48− CD34−). Cell sorting was performed on an Aria II (BD Biosciences, San Jose, CA) and analysis performed on a LSRII (BD Biosciences, San Jose, CA). List of antibodies used for Flow cytometry and cell sorting are indicated in Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD45.1 (PB conjugated, clone A20) | Biolegend | Cat#110721 |
| Anti-Mouse CD45.1 (APC conjugated, clone A20) | eBioscience | Cat#17–0453–81 |
| Anti-Mouse CD45.2 (BV 605 conjugated, clone 104) | Biolegend | Cat#109841 |
| Anti-Mouse CD45.2 (PE conjugated, clone A20) | eBioscience | Cat#12–0453–81 |
| Anti-Mouse c-kit/CD117 (APC-Cy7 conjugated, clone 2B8) | eBioscience | Cat#25–1171–82 |
| Anti-Mouse CD4 (PE-Cy5 conjugated, clone GK 1.5) | eBioscience | Cat#15–0041–82 |
| Anti-Mouse CD4 (FITC conjugated, clone GK1.5) | eBioscience | Cat#11–0041-82 |
| Anti-Mouse CD8 (PE-Cy5 conjugated, clone 53–6.7) | eBioscience | Cat#15–0081–82 |
| Anti-Mouse CD8 (FITC conjugated, clone 53–6.7) | eBioscience | Cat#11–0081–82 |
| Anti-Mouse CD45R/B220 (PE-Cy5 conjugated, clone RA3–682) | eBioscience | Cat#15–0452–82 |
| Anti-Mouse CD45R/B220 (PE-Cy7 conjugated, clone RA3–6B2) | eBioscience | Cat#25–0452–82 |
| Anti-Mouse/Human CD45R/B220 (FITC conjugated, clone RA3–6B2) | eBioscience | Cat#11–0452–82 |
| Anti-Mouse Ly-6G/Gr1 (PE-Cy5 conjugated, clone RB6–8C5) | eBioscience | Cat#15–5931–82 |
| Anti-Mouse Ly-6G/Gr1 (PE-Cy7 conjugated, clone RB6–8C5) | eBioscience | Cat#25–5931–82 |
| Anti-Mouse CD11b/Mac1 (PE-Cy5 conjugated, clone M1/70) | eBioscience | Cat#15–0112–82 |
| Anti-Mouse CD11b/Mac1 (PE-Cy7 conjugated, M1/70) | eBioscience | Cat#25–0112–81 |
| Anti-Mouse Ter119 (PE-Cy5 conjugated, clone M1/70) | eBioscience | Cat#15–5921–82 |
| Anti-Mouse CD150/SLAM (PE-Cy7 conjugated, clone TC15–12F12.2) | Biolegend | Cat#115914 |
| Anti-Mouse CD48 (APC conjugated, clone HM48–1) | eBioscience | Cat#17–0481–82 |
| Anti-Mouse CD48 (FITC conjugated, clone HM48–1) | eBioscience | Cat#11–0481–82 |
| Anti-Mouse CD34 (FITC conjugated, clone RAM34) | eBioscience | 11–0341–82 |
| Anti-Mouse Flk2/Flt3/CD135 (PE conjugated, clone A2F10) | eBioscience | Cat#12–1351–82 |
| Anti-Mouse Ki67 (PE conjugated, clone SolA15) | eBioscience | Cat#12–5698–82 |
| Anti-Mouse Annexin V (PE conjugated) | BD Pharmingen | Cat#556422 |
| Anti-Human CD38 (PE conjugated, clone HIT2) | BD Bioscience | Cat#555460 |
| Anti-Human CD45 (APC conjugated, clone HI30) | Biolegend | Cat#304012 |
| Anti-Human CD66b (PE-Cy7 conjugated, clone G10F5) | eBioscience | Cat#25–0666-42 |
| Anti-Human CD15 (PB conjugated, clone W6D3) | Biolegend | Cat#B2557908 |
| DAPI | Life technologies | Cat#D1306 |
| Fc block | MACS Miltenyi Biotec | Cat#130–046–702 |
| Bacterial and Virus Strains | ||
| Mycobacterium avium | N/A | SmT 2151 |
| Lymphocytic choriomeningitis virus (LCMV) | N/A | Armstrong |
| Biological Samples | ||
| Human Buffy coat samples | Gulf Coast Regional Blood Center | https://www.gulfcoastconsortia.org |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Polyinosinic-polycytidylic acid (pIpC) | Invivogen | Cat#TLRLPIC5 |
| Recombinant Mouse IFNy | eBioscience | Cat#BMS326 |
| Recombinant Mouse IL1b | eBioscience | Cat#B215819 |
| Recombinant Mouse LPS | Sigma | Cat#L8643–5MG |
| Recombinant Mouse TNFa | Gibco | PMC3016 |
| Recombinant Human Flt3-Ligand | Preprotech | Cat#300–19 |
| Recombinant Human IL-3 | Preprotech | Cat#200–03 |
| Recombinant Human SCF | Preprotech | Cat#300–07 |
| Recombinant Human TPO | Preprotech | Cat#300–18 |
| Recombinant Human IFNy | RD Biosystems | Cat#285-IF-100 |
| Mouse CD117 Micro Beads | MACS Miltenyi Biotec | Cat#130–091-224 |
| Human CD34 Micro Beads | MACS Miltenyi Biotec | Cat#130–046-702 |
| Critical Commercial Assays | ||
| mouse IFN alpha platinum ELISA kit | eBioscience | Cat#BMS6027 |
| CaspaseGlo 3/7 | Promega | Cat#G8090 |
| NucleoSpin RNA Plus XS kit | Macherey-Nagel | Cat# 740990 |
| SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian | Takara | Cat# 634411 |
| RNAqueous Kit | Ambion | Cat#AM1912 |
| SuperScript III First-Strand Synthesis Supermix | Invitrogen | Cat#18080051 |
| iTaq Universal SYBR Green Supermix | BioRad | Cat#172–5121 |
| Annexin V Binding Buffer, 10X concentrate | BD Pharmingen | Cat#556422 |
| Deposited Data | ||
| RNAseq_1 | Mendeley | http://dx.doi.org/10.17632/zbc66krbv4.1 |
| RNAseq_2 | Mendeley | http://dx.doi.org/10.17632/wj5knck449.1 |
| WGBS | Mendeley | http://dx.doi.org/10.17632/t5szbhjwdc.1 |
| Experimental Models: Organisms/Strains | ||
| Mice: CD45.1 | CCM core - BCM | NA |
| Mice: CD45.2 | CCM core - BCM | NA |
| Mice: Dnmt3a fl/fl | Provided by Margaret Goodell | NA |
| Mice: Mx1-Cre | The Jackson Laboratory | Stock No: 003556 |
| Mice: Vav-iCre | The Jackson Laboratory | Stock No: 008610 |
| Mice: Ifngr1−/− | The Jackson Laboratory | Stock No: 025394 |
| Mice: Batf2−/− mice | BCM Mouse ES Cell Core | NA |
| Oligonucleotides | ||
| CRISPRevolution sgRNA EZ Kit (3 nmol) - h3A_ex13_sg78 AGGUGGCCAGCAGCCGCGCG - Modified |
Synthego | NA |
| CRISPRevolution sgRNA EZ Kit (3 nmol) - h3A_ex13_sg75 UGACACUGCCAAGGCCGUGG - Modified |
Synthego | NA |
| List of primers used for qPCR are in Supplemental Table 4 | ||
| Software and Algorithms | ||
| Prism 7 | N/A | https://www.graphpad.com/scientific-software/prism/ |
| Flow Jo | N/A | https://www.flowjo.com/solutions/flowjo |
| R Project for Statistical Computing | N/A | https://www.r-project.org |
| R studio | N/A | https://www.rstudio.com/products/rstudio/download/#download |
| HTStream | N/A | https://github.com/ibest/HTStream |
| STAR | N/A | https://github.com/alexdobin/STAR |
| Biomart –Genome annotations- | N/A | http://uswest.ensembl.org/biomart/martview/f7089c3f574fcc3ee405faed118b166e |
| Differential Expression with Limma-Voom | N/A | https://ucdavis-bioinformatics-training.github.io/2018-June-RNA-Seq-Workshop/thursday/DE.html |
| Samtools | N/A | https://samtools.github.io/samtools/samtools.html |
| Biorender | N/A | https://biorender.com/ |
| Other | ||
| MethoCultTM | Stemcell Technologies | Cat#M3434 |
| StemSpan SFEM | Stemcell Technologies | Cat#9600 |
Quantitative real-time PCR.
RNA was isolated using the RNAqueous kit (Ambion) and reverse transcribed with random hexamer primers using SuperScript III (Invitrogen). We standardized cDNA input and performed RT-PCR using iTaq Universal SYBR Green Supermix (BioRad) with 18S rRNA probe (VIC-MGB) and a gene-specific probe (FAM-MGB) for 40 cycles with an Applied Biosystems StepOnePlus Real-Time PCR System. Samples were normalized to 18S rRNA levels and fold change determined by the ΔΔCT method. Primers used in RT-PCR reactions are in Table S1. M. avium was detected by PCR as previously described (Park et al., 2000).
DNA extraction and amplification of 16S bacterial gene
50000 LT-HSCs (LK CD150+ CD48− CD34−) were sorted from the pool of naive or 1-month infected WT/Dnmt3a−/− mice (n=6–8 per group) and DNA was isolated with the AllPrep DNA/RNA Mini Kit DNeasy blood & Tissue kit. DNA from spleen of the same mice was extracted with the DNeasy blood & Tissue kit. Pre-amplification of DNA was performed with Pre-Amp Mastermix (Fluidigm, Cat# 100–5581). PCR was run for 40 cycles with an annealing temperature of 60 C and PCR product was run in an 1.5% agarose gel.
RNA purification and RNA-Seq
We sorted 5000–10000 HSCs (KL CD150+ CD48− CD34−) into lysis buffer from the pools of naive or 1-month infected WT/Dnmt3a−/− mice (n=10–12 per group). RNA was isolated with the NucleoSpin ® RNA Plus XS kit (Macherey Nagel). RNA-seq libraries were prepared by using SMARTer® Stranded Total RNA-Seq Kit v2 - Pico Input Mammalian (Takara Bio Usa). Illumina NovaSeq SP was used for sequencing with a paired-end sequencing length of 10bp. FASTQ files were preprocessed using HTS stream (https://github.com/ibest/HTStream) and the clean FASTQ file were aligned using STAR. Differential expression (DE) analysis of gene expression was performed using Limma-Voom. False discovery rate (FDR)<0.05 was considered statistically significant. We performed gene ontology analysis for differentially expressed genes with q value <0.05. Statistically significant results are listed in Table S2: RNAseq WT infected vs WT naïve (q value<0.05), and Table S3: RNAseq Dnmt3a−/− infected vs Dnmt3a−/− naïve, both related to Figure 6.
We conducted Between Group Analysis (BGA) (Culhane et al. 2002) using the made4 Bioconductor package based on regularized log-transformed reads. The plots in Figure S3 show relative distance between samples obtained by projecting them into two-dimensional space using correspondence analysis (CoA).
Whole Genome Bisulfite Sequence Analysis
100000 LT-HSCs (LK CD150+ CD48− were sorted into lysis buffer from the pools of naive or 1-month (M. avium) infected WT/Dnmt3a−/− mice (n=7–8 per group). Around 300 ng of DNA per group was extracted with AllPrep DNA/RNA Mini Kit. NEBNext Ultra II DNA library prep kit was used for library preparation. Bisulfite conversion step was added after “methylated” adaptor ligation and USER excision. Then, methylated adaptor ligated DNA fragment was treated with bisulfite conversion using EZ DNA methylation-lightning kit. These modified DNA fragments were then amplified with index primers with Kapa HiFi urasil+ specialized polymerase for bisulfite converted DNA. For WGBS library sequencing, 150-high output kit from illumine was used. Samples (400 million reads/sample (15–20X coverage)) were run in NovaSeq S1 FC (2, lanes 1,600 Million reads).
Quality control was conducted using FastQC (Andrews, 2010) and CGmapTools (Guo et al., 2018). Adapter and quality trimmed reads, processed using trim_galore, were aligned to GRCm38 reference genome using bs_seeker2 (Guo et al., 2013), based on bowtie read aligner (Langmead et al., 2009). The average alignment rate for all samples was 48%. Duplicate reads were removed using MarkDuplicates from the Picard tool set (Picard Tools. Broad Institute 2018; Available from: http://broadinstitute.github.io/picard/), resulting in an average of 11.5x coverage across the entire genome for all of the samples (minimum 10.7x maximum 12.5x for individual samples). Raw methylation calls were extracted from pre-processed reads using bs_seeker2 (Guo et al., 2013). Differentially methylated regions (DMRs) were identified with CGmapTools (Guo et al., 2018) using all CG sites with a minimum of 4x and maximum of 100x coverage and Benjamini & Hochberg correction for multiple testing. We assumed 0.01 significance level and retained only sites that showed at least 0.1 methylation level difference. Visualizations of DMRs were created with Gviz (Hahne and Ivanek, 2016), using GENCODE mouse gene annotation database v24 (Frankish et al., 2019).
DNMT3A deletion and human HSPC differentiation assay
DNMT3A deletion was conducted on human CD34+ cells according to the method of Gundry et al (Gundry et al., 2017) by using two separate single guide RNAs (sgRNAs) targeted to exon thirteen of human DNMT3A. Guide sequences were: AGGUGGCCAGCAGCCGCGCG - modified and UGACACUGCCAAGGCCGUGG - modified. Mixed guide RNAs were used at 0.5 ug/ul final concentration for each guide, mixed with 1 ug Cas9 protein (PNA Bio) and transduced into CD34+ cells by electroporation. Gene deletion was confirmed by PCR and realtime using the following primers; forward, GATGGAATCGCTACAGGGCT and reverse: CCCCCAATCACCAGATCGAAT.
Human CD34+ cells were isolated from buffy coat samples by enrichment using anti-CD34 magnetic microbeads (Macs, Miltenyi). CD34+ blood cells were expanded in StemSpan SFEM (Stem Cell Technologies) supplemented with 100ng/ml rhFLT-3L, 100ng/ml rhSCF, 100ng/ml rhTPO and 20ng/ml IL-3 (Preprotech). Differentiation of human cells was measured by incubation of CD34+ cells in the above media with IFNy (1000 U/mL) for 3 days. Cells were characterized by flow cytometry using anti CD45, CD34, CD38 and CD66b antibodies (eBioscience, Biolegend, and BD Pharmingen) (Yang et al., 2005). Each experimental replicate was conducted with cells from a different biological sample, representing a different individual.
LT-HSC mouse differentiation assay on Methocult
Mouse differentiation assay was conducted according to the LT-HSC Methylcellulose Assay of Kerenyi et al (Kerenyi et al. 2013 and 2014). LT-HSCs (LSK CD150+ CD48-) were sorted from 3 Mx1-Cre+ Dnmt3a−/− mice and 3 Mx1-Cre− Dnmt3afl/fl mice. A total of 80 LT-HSCs were suspended in 2mL of methyl cellulose semisolid medium (MethoCult GF M3434 methylcellulose-based medium) and seeded into a 12-well culture plate in which IFN-γ was added at 100 ng/mL in the indicated groups. The cells were incubated at 37 °C in 5% CO2 for 10 days. After that time, colonies were counted and scored according to the instructions found in Miller et al. (2008).
Serial Replating
10000 WBM cells or 500 ckit+ cells from pIpC-treated Mx1-Cre− and Mx1-Cre+ Dnmt3af/f mice and pIpC untreated and treated Vav-Cre+ Dnmt3af/f mice were plated in Methocult medium for one week. After that time, colonies were counted, harvested and 5000 cells were replated every passage until no colonies were observed.
Annexin staining
After surface antibody staining, murine HSCs were incubated with 5μl of Annexin-PE antibody (BD Pharmingen, Cat#556422) in 100μl of 1X binding buffer for 15min at RT.
Ki67 measurement
Murine cells stained with cell surface markers were fixed, permeabilized and refixed using buffers from BD Biosciences BrdU Flow Kit (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. After fixation, cells were stained for 30 mins at RT with Ki67 PE (eBioSciences) and Alexa 488 goat anti-rabbit secondary antibody (Abcam) for 20 mins at RT.
Apoptosis assay
Apoptosis within murine HSCs was determined using Promega’s Caspase-Glo 3/7 Assay (Promega) according to the manufacturers protocol. Briefly, 400 HSCs were sorted directly into white-walled 96 well plates containing StemPro+ media (Stem Cell Technologies). Caspase Glo reagent was then added after 12 hr incubation at 37°C. We recounted cells after 12 hr incubation to ensure uniform cell numbers. We read luciferase output after 1hr at room temperature. We heat shocked cells at 60°C for 5 minutes to use as a positive control. Plates were read on a Cytation 5 Imaging Reader (BioTek).
IFNy, LPS, and IL1β chronic treatment
Recipient mice were transplanted with 1.5 × 105 Cre− Dnmt3afl/fl CD45.2 WBM or 1.5 × 105 Cre+ Dnmt3a−/− CD45.2 WBM + 6 × 105 CD45.1 WBM. At 8 weeks post-transplantation, mice were then treated with daily IP injections of either 1μg of IFNy, 6 μg of LPS or 0.5μg of IL1β for 4 weeks.
Cytokine bead array
Peripheral blood was collected from naïve and M. avium infected WT and Dnmt3a−/− mice via cardiac puncture. Serum was isolated from the blood through centrifugation using BD microtainers and frozen at –80 C until further use. Serum was thawed on ice and vortexed thoroughly prior to being diluted 1:2 in Assay Buffer provided in the Millipore-Sigma Mouse Cytokine/Chemokine Magnetic Bead Panel 96 well plate assay kit. Preparation of reagents for the immunoassay including antibody-immobilized beads, quality controls, wash buffer, and serum matrix were completed according to the manufacturer’s protocol. Serum samples and antibody-immobilized beads were incubated together in the dark overnight at 4°C with agitation. Detection antibodies and secondary streptavidin-phycoerythrin were added the following day for the appropriate time per kit instructions. Control and experimental serum samples were run in triplicate on a Bio-Rad Bio-Plex 200 using Bioplex manager 6.1 software. Cytokine and chemokine concentrations were calculated using the median fluorescent intensity (MFI) calculated using a spline curve-fitting method.
QUANTIFICATION AND STATISTICAL ANALYSIS
The basis for predicting dynamics of two competing populations of HSCs is the differential equation of the form N(t)=−λN(t) + 2(1-d)λN(t) where N(t) is the HSC count at time (t), λ is the division rate of the HSC, and d is the fraction of HSC progeny that are not HSC (i.e., that constitutes HSC “loss”) (Matatall et al., 2016). We calculated λ and N(t) values from Ki67 (Figure 3C) and flow cytometric analysis of HSC counts in WT or Dnmt3a−/− mice with or without chronic M. avium infection (Figure S3B) and used these values to calculate d for infected versus uninfected states. These λ and d values were then used to predict N(t) from a starting set of 10 “minor population” HSCs and 20 “major population” (competitor) HSCs in post-transplant mice with or without infection introduced at 60 days post-transplant. Parameters used in these predictions are listed in Table S4. Further details on modeling can be found in the Methods S1. For all data figures, mean values ± SEM are shown. Student’s t test, Kruskal-Wallis test or ordinary one-way ANOVA were used for comparisons (GraphPad Prism v.8.0). Statistical details relevant to each experiment including n and number of times each experiment was repeated are listed in the figure legends.
Supplementary Material
Highlights.
Dnmt3a−/− and Dnmt3a+/− hematopoietic stem cells outcompete WT during infection.
Recombinant interferon gamma is sufficient to drive expansion of Dnmt3a−/− clones.
DNA methylation silences differentiation programs in Dnmt3a−/− HSCs upon infection.
Inflammatory signaling during infection drives Dnmt3a-mutant clonal hematopoiesis.
Acknowledgments:
The authors would like to thank Catherine Gillespie and members of the King Lab for useful discussions. We thank Claudine S. Kadmon, Jack Toups, and Meghan Kisiel for technical assistance and Margaret Goodell for scientific advice and sharing the Mx1-Cre Dnmt3af/f mice. This project depended on the support of Joel Sederstrom and the BCM Cytometry and Cell Sorting Core with funding from the NIH (NCRR grant S10RR024574, NIAID AI036211 and NCI P30CA125123), NIH S10 OD020066, and the Dan L. Duncan Cancer Center and help from Lisa White and the genomic and RNA profiling core at Baylor College of Medicine, with funding from the NIH (NIDDK-DK56338 and NCI-CA125123). Development of the Batf2 KO mice was done by the BCM Mouse ES Cell Core supported by the Dan L Duncan Cancer Center Grant P30 CA125123. RJ and PK were supported by the Polish National Science Centre grants No. 2016/23/D/ST7/03665 and 2018/29/B/ST7/02550, respectively. KYK, KAM, DHA, DL, and BK were supported by the NIH grants R01HL136333 and R01HL134880 (KYK), T32DK060445 (KAM, BK), T32AI053831(BK), and T32HL092332 (DL).
Footnotes
Declaration of Interests: The authors have no conflicts of interest to disclose.
Inclusion and Diversity: We worked to ensure sex balance in the selection of non-human subjects. One or more authors from this paper self-identifies as an underrepresented ethnic minority in science, one or more authors of this paper received support from a program designed to increase minority representation in science.
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abegunde SO, Buckstein R, Wells RA, and Rauh MJ (2018). An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Experimental Hematology 59, 60–65. [DOI] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data.
- Azrakhsh NA, Mensah-Glanowska P, Sand K, and Kittang AO (2019). Targeting Immune Signaling Pathways in Clonal Hematopoiesis. Current medicinal chemistry 26. [DOI] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, King KY, and Goodell MA (2011). Inflammatory signals regulate hematopoietic stem cells. Trends in Immunology 32, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder S, Luciano M, and Horejs-Hoeck J (2018). The cytokine network in acute myeloid leukemia (AML): A focus on pro- and anti-inflammatory mediators. Cytokine and Growth Factor Reviews 43, 8–15. [DOI] [PubMed] [Google Scholar]
- Bolton KL, Koh Y, Foote MB, Im H, Jee J, Sun CH, Safonov A, Ptashkin R, Moon JH, Lee JY, et al. (2020a). Clonal hematopoiesis is associated with risk of severe Covid-19. medRxiv, 2020.2011.2025.20233163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, Patel M, Berthon A, Syed A, Yabe M, et al. (2020b). Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet 52, 1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Busque L, and Levine RL (2018). Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Stem Cell 22, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lepine G, Mollica L, Szuber N, Dube M-P, and Busque L (2017). DNMT3Aand TET2dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 130, 753–762. [DOI] [PubMed] [Google Scholar]
- Cai Z, Kotzin JJ, Ramdas B, Chen S, Nelanuthala S, Palam LR, Pandey R, Mali RS, Liu Y, Kelley MR, et al. (2018). Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 23, 833–849.e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. (2011). Dnmt3a is essential for hematopoietic stem cell differentiation. Nature Genetics 44, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull AH, Snetsinger B, Buckstein R, Wells RA, and Rauh MJ (2017). Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol 55, 56–70 e13. [DOI] [PubMed] [Google Scholar]
- DeZern AE, Malcovati L, and Ebert BL (2019). CHIP, CCUS, and Other Acronyms: Definition, Implications, and Impact on Practice. American Society of Clinical Oncology educational book American Society of Clinical Oncology Annual Meeting 39, 400–410. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic acids research 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire PR, and Conneely OM (2018). NR4A1 and NR4A3 restrict HSC proliferation via reciprocal regulation of C/EBPalpha and inflammatory signaling. Blood 131, 1081–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu C-L, Sano S, Muralidharan S, Rius C, et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. New England Journal of Medicine 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen PY, and Pellegrini M (2013). BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. Bmc Genomics 14, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Zhu P, Pellegrini M, Zhang MQ, Wang X, and Ni Z (2018). CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 34, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol Biol 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Higa KC, Goodspeed A, Chavez JS, Zaberezhnyy V, Rabe JL, Tenen DG, Pietras EM, and DeGregori J (2020). Chronic Interleukin-1 exposure triggers selective expansion of Cebpa-deficient multipotent hematopoietic progenitors. bioRxiv, 2020.2003.2025.008250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege H, Pfeiffer W, Sie D, Hulsman M, Nicholas TJ, Lee CC, Ross T, Lin J, Miller MA, Ylstra B, et al. (2014). Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res 24, 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JI, Dayaram T, Tovy A, De Braekeleer E, Jeong M, Wang F, Zhang J, Heffernan TP, Gera S, Kovacs JJ, et al. (2018). PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 23, 700–713.e706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquelin S, Straube J, Cooper L, Vu T, Song A, Bywater M, Baxter E, Heidecker M, Wackrow B, Porter A, et al. (2018). Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 132, 2707–2721. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. New England Journal of Medicine 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, and Libby P (2019). Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nature reviews Cardiology 355, 2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. (2017). Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. The New England journal of medicine 377, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M, Park HJ, Celik H, Ostrander EL, Reyes JM, Guzman A, Rodriguez B, Lei Y, Lee Y, Ding L, et al. (2018). Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Reports 23, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong M, Sun D, Luo M, Huang Y, Challen GA, Rodriguez B, Zhang X, Chavez L, Wang H, Hannah R, et al. (2014). Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat Genet 46, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn JD, Miller PG, Silver AJ, Sellar RS, Bhatt S, Gibson C, McConkey M, Adams D, Mar B, Mertins P, et al. (2018). PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 132, 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Cheng Y, Bolton-Gillespie E, Cai X, Ma C, Tarangelo A, Le L, Jambhekar M, Raman P, Hayer KE, et al. (2017). Rb family proteins enforce the homeostasis of quiescent hematopoietic stem cells by repressing Socs3 expression. The Journal of Experimental Medicine 214, 1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KY, Huang Y, Nakada D, and Goodell MA (2019). Environmental influences on clonal hematopoiesis. Exp Hematol. 83:66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer AC, Kothari A, Wilson WC, Celik H, Nikitas J, Mallaney C, Ostrander EL, Eultgen E, Martens A, Valentine MC, et al. (2017). Dnmt3a regulates T-cell development and suppresses T-ALL transformation. Leukemia 31, 2479–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H, and Kent DG (2020). Tracking hematopoietic stem cells and their progeny using whole-genome sequencing. Exp Hematol 83, 12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallaney C, Ostrander EL, Celik H, Kramer AC, Martens A, Kothari A, Koh WK, Haussler E, Iwamori N, Gontarz P, et al. (2019). Kdm6b regulates context-dependent hematopoietic stem cell self-renewal and leukemogenesis. Leukemia 33, 2506–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Barajon I, and Garlanda C (2018). IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunological Reviews 281, 57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, and King KY (2016). Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Reports 17, 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall KA, Shen C-C, Challen GA, and King KY (2014). Type II Interferon Promotes Differentiation of Myeloid-Biased Hematopoietic Stem Cells. Stem Cells 32, 3023–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, Pierre JF, Ernest JD, Galipeau HJ, Thuille N, et al. (2018). Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, and Kimble J (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068–1074. [DOI] [PubMed] [Google Scholar]
- Park H, Jang H, Kim C, Chung B, Chang CL, Park SK, and Song S (2000). Detection and identification of mycobacteria by amplification of the internal transcribed spacer regions with genus- and species-specific PCR primers. J Clin Microbiol 38, 4080–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piber D, Olmstead R, Cho JH-J, Witarama T, Perez C, Dietz N, Seeman TE, Breen EC, Cole SW, and Irwin MR (2019). Inflammaging: Age and Systemic, Cellular, and Nuclear Inflammatory Biology in Older Adults. The journals of gerontology Series A, Biological sciences and medical sciences 74, 1716–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, and Passegué E (2014). Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. The Journal of Experimental Medicine 211, 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhok AI, and DeGregori J (2015). Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proc Natl Acad Sci U S A 112, 8914–8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P, Townsend RR, Wilson RK, et al. (2014). The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 25, 442–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval JE, Huang YH, Muise A, Goodell MA, and Reich NO (2019). Mutations in the DNMT3A DNA methyltransferase in acute myeloid leukemia patients cause both loss and gain of function and differential regulation by protein partners. J Biol Chem 294, 4898–4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steensma DP (2018). Clinical Implications of Clonal Hematopoiesis. Mayo Clinic proceedings 93, 1122–1130. [DOI] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nature Medicine 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, Sato-Otsubo A, Sato Y, Liu D, Suzuki H, et al. (2015). Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med 373, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AL, Challen GA, Birmann BM, and Druley TE (2016). Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nature Communications 7, 12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, et al. (2017). Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Online resources:
RNAseq and WGBS data are publicly available through Mendeley as follows: http://dx.doi.org/10.17632/zbc66krbv4.1 (RNAseq 1), http://dx.doi.org/10.17632/wj5knck449.1 (RNAseq 2), http://dx.doi.org/10.17632/t5szbhjwdc.1 (WGBS).
Code availability:
Formulas used to calculate mathematical predictions in Supplemental Figures 2C–F are available in Supplementary Table 1.