

Abstract
Asparagine synthetase (ASNS) is a gene on the long arm of chromosome 7 that is copy number amplified in the majority of glioblastomas. ASNS copy number amplification is associated with a significantly decreased survival. Using patient-derived glioma stem cells (GSCs), we showed significant metabolic alterations occur in gliomas when perturbing the expression of asparagine synthetase, which is not merely restricted to amino acid homeostasis. ASNS-high GSCs maintained a slower basal metabolic profile yet readily shifted to a greatly increased capacity for glycolysis and oxidative phosphorylation when needed. This led ASNS-high cells to a greater ability to proliferate and spread into brain tissue. Finally, we demonstrate that these changes confer resistance to cellular stress, notably oxidative stress, through adaptive redox homeostasis which led to radiation resistance. Furthermore, ASNS overexpression led to modifications of the one-carbon metabolism to promote a more antioxidant tumor environment revealing a metabolic vulnerability that may be therapeutically exploited.
Introduction
Glioblastoma (GBM, WHO grade IV gliomas) represents one of the most lethal solid tumors with an overall survival of approximately 12–14 months with standard of care therapy (1). Negligible progress has been made in survival despite advancements in chemotherapy, radiation therapy, and surgical techniques over the past 30 years.
Glioma stem cells (GSCs) have been shown to be responsible for glioma propagation and recurrence through their resistance to multiple modes of therapy. These cells are characterized by in vitro self-renewal capacity, multiple lineage differentiation, neurosphere formation, and neural stem cell marker expression such as Nestin, Sox2, Prom1/CD133, and Nanog (2). GSCs have been shown to be more resistant to both chemotherapy and radiation compared to differentiated tumor cells and studies have shown GSC ability to repopulate a tumor and drive secondary tumor recurrence post-treatment (3–5). Adding another layer of complexity, there’s a great degree of heterogeneity within the GSC pools, consistent with the models of CSC maintenance and propagation (2, 6). Distinct clones even from a single tumor can display variability in gene expression profile and metabolic dependencies (7–10). allowing cancer stem cells to be adaptable and plastic in order to maintain high rates of self-renewal and differentiation (11–13).
The metabolic landscape of the tumor is heterogeneous, and cells will metabolize differently depending on the environment they are in. When most cells would die from radiation, GSCs can remain protected from the stress and enter the cell cycle afterward in order to repopulate the tumor. This plasticity could allow CSCs to respond to fluctuating conditions and survive in unfavorable conditions whether it be stress from treatment or the stress of metastatic sites (14, 15). This also presents a new problem wherein single inhibition of one metabolic pathway may not be effective in vivo despite presumably being effective in vitro, since the true CSC population may be able to modify its metabolic source and pathway depending on what is available to it.
Asparagine synthetase (ASNS) is the ATP dependent enzyme responsible for catalyzing the reaction converting aspartate and glutamine into asparagine. This enzyme is found throughout the body, in all organ systems, and is very tightly regulated since it is an exergonic reaction that also consumes glutamine, thereby being metabolically costly (16). The normal biological need for asparagine does not necessitate constant and elevated levels of ASNS activation, but this is exactly what is observed in many cancers, which suggests this mechanism may in fact play a crucial role in cellular adaptations to nutritional or other stress in cancer cells.
Many studies in recent years have shown ASNS inhibition in solid tumors resulted in diminished cell growth, sometimes cell cycle arrest, and even increased sensitivity to specific stresses (16–19). The exact role ASNS that asparagine plays in the maintenance of growth and survival in solid tumors however has yet to be fully elucidated, although it is apparent that its role goes well beyond simple amino acid biosynthesis. In this study, we show that ASNS is often copy-number amplified in high grade gliomas and corresponds to poor patient survival. Furthermore, the elevated ASNS corresponds to increased metabolic plasticity and resistance to nutrient withdrawal and oxidative stress, via adaptive redox homeostasis. This highlights the extent to which gliomas are capable of hijacking cell intrinsic metabolic pathways to maintain a fitness advantage under stress conditions and continue to support a hyper-proliferative phenotype.
Materials & Methods
Retrospective Analysis of ASNS Copy-Number in Human Gliomas
All database analyses were done using data acquired from cBioportal (MKSCC) and Oncomine. Kaplan Meier plots were created using copy-number data and patient survival data from cBioportal and created using GraphPad Prism 6. ASNS gain was determined as any patients scored with more than two copy of ASNS (diploid), while ASNS diploid was two copy only. All ASNS deletions were excluded from any analyses.
Fluorescence in-situ Hybridization (FISH)
In situ hybridization was carried out by the CSMC Genomics Core using human ASNS probes (Abnova) as per standard CSMC departmental protocols. De-identified patient tumor slides were provided by the obtained under an IRB protocol.
Glioma Stem Cell (GSCs) and Media
GSCs were isolated as previously described and cultured in NBE media or differentiation media and infected with shRNAs as previously described and used in limiting dilution assays, neurosphere formation assays, ELISA, FACs or orthotopic xenograft mouse models. GSC827 and GSC604 were provided by Dr. Lincoln Edwards, previously established at the NCI/NIH Brain Tumor Research Lab (Supp Table 1). All other GSCs mentioned here were procured and developed at Cedars-Sinai Medical Center. All blood, brain tumors and patient derived GSCs were approved by the Cedars-Sinai Medical Center institutional review board (IRB). Informed patient consent was obtained from all patients. All methods were carried out in accordance with the relevant guidelines of the IRB at Cedars-Sinai Medical Center.
qPCR
For all qRT-PCR analyses, RNA extraction was performed using the RNeasy Mini Kit (Qiagen; Valencia, CA) and reverse transcribed to cDNA using the Quantitect Reverse Transcription Kit (Qiagen). For qRT-PCR, 50 ng of cDNA was mixed with the appropriate primers and the iQ SYBR-Green Supermix (BioRad; Hercules, CA), and run on the CFX96 Real-Time System (BioRad). Data were analyzed using the 2−ΔCT method. All mRNA data were normalized to GAPDH expression.
RNA-Seq Analysis
Ultra-low input mRNA sequencing was performed through the CSMC Genomics Core, using the NextSeq 550 platform. Unsupervised PCA analysis was done on 827, 827-shASNS, 604, and 604–48 cell lines, in addition to differential expression analysis. PCA indicated that the two sample types in the comparisons are very different (first dimension: 604 vs. 604–48 = 99.10%; 827 vs. 827-A = 99.24%). A stringent statistic criteria was applied (adjusted p value < 0.01 and log2FC >2) to get a reasonable number of significant DE genes for downstream pathway analysis as well as avoiding more false positives. The heatmap is for the DE genes with adjusted p value < 0.01 and log2FC > 2. Pathway analysis was performed using DAVID (https://david.ncifcrf.gov/home.jsp). Ingenuity Pathway analysis (IPA) was performed on all samples. Data is publicly available under GEO accession # GSE171163.
Lentiviral Transductions
GSCs were either transfected with non-silencing lentiviral particle control, shRNA ASNS lentiviral particles (OriGene, shASNS A, B), or an ASNS overexpression lentiviral particle (GE Dharmacon, Precision LentiORF, Lenti LV-48, LV-23). Transfections were done with lentiviral particles directly onto target cells in a 24-well plate with 500 μl Opti-MEM, calculated particles to have MOI of at least 3, per manufacturer and batch measurements and polybrene (8 μg/ml). Fresh NBE media added after 48 hours, and antibiotic selection for next 7–14 days or until validated by western blot or qPCR.
Western Blot
Whole cell extract was prepared by lysis of the cells in TNN buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 5 mM EDTA pH 8, 1x protease inhibitor cocktail for mammalian cells (Sigma) for 30 minutes at 4°C by rotation and pre-cleared by centrifugation at maximum speed for 10 minutes at 4°C. 50 mg of lysate were denatured in 1x Laemli buffer and separated by SDS-polyacrylamide gel electrophoresis in tris-glycine buffer and transferred onto nitrocellulose membranes (BioRad). The membranes were blocked in 5% milk in PBST for 30 minutes and then incubated with the corresponding primary antibodies (1:1000). After washing with PBST, the membranes were incubated with conjugated goat anti-mouse antibody or goat anti-rabbit antibody (1:5000) for 1 hour at room temperature. After washing the membranes 3 times for 5 minutes, the membranes were scanned and analyzed using an Odyssey infrared system (LI-COR Bioscience).
Tumor Samples
Patient brain tumor samples were classified as GBM based on the World Health Organization (WHO) criteria. Patient information corresponding to GSC604 and GSC827 from the NCI/NIH Brain Tumor Research Lab has been summarized in the supplement (Supp Table 1). All blood, brain tumors and patient derived GSCs were approved by the Cedars-Sinai Medical Center institutional review board (IRB). Informed patient consent was obtained from all patients. All methods were carried out in accordance with the relevant guidelines of the IRB at Cedars-Sinai Medical Center.
Normal Samples
Normal human brain tissues were obtained from two sources: (1) Cedars-Sinai Medical Center. (2) Alzheimer’s Disease Research Center (ADRC) Neuropathology Core at the Department of Pathology in the University of Southern California (USC, Los Angeles, CA; IRB protocol HS-042071). Human tissue collection protocols were approved by institutional committees. Our work at Cedars-Sinai Medical Center was performed under IRB protocols 3636/MOD00001343, Pro00053412 and Pro00019393.
Immunofluorescence for Stem Markers
Cells were grown on coverslips and fixed with 4% paraformaldehyde for 15 minutes, washed with PBS, and permeabilized with ice-cold 100% methanol for 10 minutes at −20°C. The coverslips were then blocked with 5% normal goat serum in PBS for 1 hour at room temperature. This was followed by incubation with primary antibody for 1 hour at 37°C or overnight at 4°C depending on the manufacturer’s recommendation. Antibodies included Nestin (Abcam ab22035) and Sox2 (Abcam ab97959). After washing with PBS, cells were incubated with the appropriate fluorescently conjugated secondary antibody (1:1000 dilution; Invitrogen) for 1 hour at 37°C. The coverslips were mounted on slides using Vectashield mounting medium with DAPI (Vector Labs, Burlingame, CA).
Sphere Formation Assay
Cells were seeded onto 6-well plates at a density of 1000 cells/well and allowed to grow for 7 days, after which point a manual grid was drawn and each well was counted and scored to determine the sphere forming efficiency (SFE) %. SFE was determined as a function of spheres counted per cell seeded.
Cellular Growth Assays
All growth charts were started by seeding 104 cells/well into a 6-well plate and cell counts were taken at days 0, 3, and 7. Any adjustments to media were taken into consideration and all experiments were done at minimum in biological triplicates.
Limiting Dilution Assay
GSCs were dissociated with trypsin and seeded into each well of 96-well ultra-low attachment plate at various densities, such as 1–200 cells per well. After about a week or two, number of well with neurospheres were counted for each density and the data was analyzed by ELDA software (http://bioinf.wehi.edu.au/software/elda/) (20)
CRISPR/Cas9 Knockdown Assays
Two gRNAs targeting exons 13 and 14 of the human ASNS gene designed using the Benchling CRISPR design tool (www.benchling.com). The best gRNA candidates were selected based on the highest on-target and the lowest off-target cleavage scores. A pair of oligonucleotides for each targeting site was designed in forward and reverse orientation containing Bsa1 5’-end extensions were annealed, phosphorylated and ligated into Bsa1 digested px601 Sacas9 U6-Bsa1-scafold AAV plasmid. The ligation mixture was transformed into competent cells and the insertion of the gRNAs was confirmed by Sanger sequencing using U6 primers.
HEK293 cells were cultured in DMEM medium containing 10% FBS and gentamicin (10 μg/ml). One day before transfection, the cells were plated in 6-well plates at a density of 0.3 × 106 cells per well. The next day, the cells were transfected with 2 μg of control px601 plasmid or px601containg ASNS gRNAs using fugene transfection reagent. Eight hours later, media was removed and replaced with fresh media. 48 hours after transfection, the cells were harvested and genomic DNA was isolated from the cells using Nucleospin tissue kit (Macherey-Nagel) according to the manufacturer’s protocol. 300 ng of extracted DNA was subjected to PCR using FailSafe PCR kit and buffer D (Epicentre) under the following PCR conditions: 95°C for 5 minutes, 30 cycles (95°C/30s, 57°C/30s, 72°C/30s), 72°C/7 minutes. The PCR products were resolved in 1% agarose gel and purified using a gel purification kit then cloned into a TA vector (Invitrogen) and analyzed by Sanger sequencing (Genewiz). The sequence alignment for full length and the excision sequences was done using a multiple sequence alignment program (ClustalW2).
GSC827 cells were cultured in serum free stem cell culture medium containing B-27 supplement, pen-strep, 250 μl EGF (50 ng/ml), 400 ul FGF (20 ng/ml) and 40 μl heparin (160 ng/ml). One day before transfection, the cells were plated in 12-well plates at a density of 2 × 106/per well. The next day, the cells were transfected with 2 μg control px601 plasmid or px601 containing ASNS gRNAs using fugene transfection reagent. After 24 hours, media was removed and replaced with fresh media. 48 hours after transfection, the cells were harvested and genomic DNA was isolated from the cells using the Nucleospin tissue kit (Macherey-Nagel) according to the manufacturer’s protocol. 300ng of extracted DNA was subjected to PCR using FailSafe PCR kit and buffer D (Epicentre) under the following PCR conditions: 95°C for 5 minutes, 30 cycles (95°C/30s, 57°C/30s, 72°C/30s), 72°C/7 minutes. The PCR products were resolved in 1% agarose gel and purified using a gel purification kit and cloned into a TA vector (Invitrogen) and analyzed by Sanger sequencing (Genewiz).
Nutrient Restriction Assays
Nutrient restriction assays were done exactly like standard growth curves as described above. Asparagine depletion was done by incubating complete media with 2 IU/ml L-Asparaginase (Prospec) overnight followed by filter dialyzing. Glutamine depleted media was complete NBE media without added GlutaMax (Life Technologies). Glucose free media was complete NBE media without adding standard glucose (4.5 g/l). Any low glucose assays were done with glucose concentration of 0.45 g/l. Cell survival and growth was measure by cell count and normalized to untreated control.
Intracranial Tumor Implantation in Mice
All animal procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Cedars-Sinai Medical Center. We used a stereotactic device (Stoelting Co, Wood Dale, IL) to inject B6.Cg-Foxn1nu/J (approximately 6 weeks old; Jackson labs, Bar Harbor, MI) into the left lateral ventricle with the following GSCs (105 cells per injection; n = 8 mice per group): 0827 infected with non-silencing vector (OriGene), 0827 stably infected with shRNA targeting ASNS (shRNA-ASNS_A, OriGene), 0604 infected with ASNS overexpression vector (GE Dharmacon), or 0604 infected with non-silencing vector. Following injection, the mice were monitored for tumor growth by checking for changes in body weight, lethargy, dehydration, and/or labored breathing. The mice were killed when one or more of the above criteria were met as per the National Cancer Institute Animal Care and Use Committee policy with isoflurane followed by cervical dislocation. Mouse brains were either perfused overnight with 4% paraformaldehyde and washed with PBS, followed by storage overnight at 4°C in 30% sucrose in PBS. The latter tumors were then frozen in isopentane solution (Sigma) that had been cooled in dry ice and serially sectioned on a CM1850 cryostat (Leica, Richmond Hill, ON) into 7-μm (for in situ hybridization) or 10-μm (for immunofluorescence labeling and hematoxylin–eosin staining) sections. Sections were mounted on silanized slides (Superfrost slides; Thermo Fisher Scientific, Pittsburgh, PA). All procedures involving mice were in adherence with NIH Animal Care and Use Committee protocols.
Subcutaneous Tumor Implantation in Mice
Same GSCs described for the intracranial tumor implantation were also used on the same mice, B6.Cg-Foxn1nu/J (approximately 6 weeks old; Jackson labs, Bar Harbor, MI), but with only n = 2/group and 106 cells/mouse injected subcutaneously into lower back left flank. Mice were sacrificed and tumors were collected as described previously once tumors reached a size deemed too burdensome for the mouse or if the tumors began to blister. All procedures involving mice were in adherence with NIH Animal Care and Use Committee protocols.
Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR).
The OCR and ECAR of monolayers vs. neurospheres were determined using the Seahorse XF Extracellular Flux Analyzer (Seahorse Bioscience) through the CSMC Metabolism & Mitochondrial Research Core. The Extracellular Flux Analyzer allows for analyzing OCR and ECAR of a defined number of cells in a defined small volume of culture media in real time and for monitoring their response to drug treatment. Twenty-four-well plates (Seahorse Bioscience) were coated with laminin (Sigma) as described previously to allow the single cells derived from neurosphere cultures to attach for this assay without differentiating. Briefly, each well of the 24-well plate was coated with 50 μl laminin diluted in PBS (10 μg/ml) overnight. The next day the wells were washed three times with PBS, and cells from monolayer or sphere cultures were plated at a density of 100,000 cells per well and allowed to attach overnight in either monolayer or neurosphere media. The following day the adherent cells were washed and fresh media was added. The cartridge was loaded to dispense three metabolic inhibitors sequentially at specific time points: oligomycin (inhibitor of ATP synthase, 1 μM), followed by FCCP (a protonophore and uncoupler of mitochondrial oxidative phosphorylation, 0.5 μM), followed by the addition of a combination of rotenone (mitochondrial complex I inhibitor, 100 nM) and myxothiazol (inhibitor of cytochrome C reductase, 100 nM). Basal OCR and ECAR were measured, as well as the changes in oxygen consumption caused by the addition of the metabolic inhibitors described above.
L-Lactate Production Assay
L-Lactate production was detected using the L-Lactate assay kit (Abcam ab65331). Cell lysates were created from 2 × 106 cells/sample. Protocol was followed as per manufacturer recommendations.
ATP Content Assays
For comparison of ATP levels, the one-step ATPlite Assay (Perkin Elmer) was used as per the manufacturer protocol. Cells were plated in 96-well plate at 5000 cells/well in 100 μl media per well.
ROS Detection Assay
ROS detection was done using the cellular reactive oxygen species detection assay kit (Abcam ab186027). Cells were plated at 104/well in 100 μl/well overnight. Protocol was followed as per manufacturer recommendations. ROS with H2O2 was done at three concentrations, 0.1, 1.0, and 10 μM for 30 minutes at 37°C/5% CO2.
GSSG/GSH Detection Assay
ROS detection was done using the cellular reactive oxygen species detection assay kit (Abcam ab138881). Cell lysates were created from 2 × 106 cells/sample post treatment. Protocol was followed as per manufacturer recommendations. ROS with H2O2 was done at one 1 μM for 30 minutes at 37°C/5% CO2.
Radiation Response Assay
Cells were plated in T-25 flasks with 5 × 104 cells/5 ml media. Radiation was performed using the X-Rad Precision X-ray, care of Dr. Barry Stripp at Cedars-Sinai Medical Center. Cells were exposed to 3, 5, and 10Gy radiation and allowed to grow at 37°C/5% CO2 for 14 days. Neurospheres were counted before dissociating and counting to determine survival.
RNA-Seq for Irradiated GSCs and Data Analysis
Sample preparation:
GSCs were dissociated with trypsin and seeded into T-25 flasks with 5 × 105 cells/5 ml media. Radiation was performed using the Precision X-Ray X-Rad, cells were irradiated at 5Gy or no radiation followed by incubation at 37°C with 5% CO2. On day 0, 3 and 14, cells were harvested and total RNA was extracted using RNeasy Plus Mini Kit (Qiagen).
Library preparation and sequencing:
Total RNA samples were assessed for concentration using a Qubit fluorometer (ThermoFisher Scientific, Waltham, MA) and for quality using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Up to one μg of total RNA per sample was used for library construction using the Illumina TruSeq Stranded mRNA library preparation kit (Illumina, San Diego, CA). Library concentration was measured with a Qubit fluorometer and library size on an Agilent 4200 TapeStation (Agilent Technologies). Libraries were multiplexed and sequenced on a NovaSeq 6000 (Illumina, San Diego, CA) using 75bp single-end sequencing. On average, approximately 30 million reads were generated from each sample.
Data analysis:
Raw sequencing data was demultiplexed and converted to fastq format using bcl2fastq v2.20 (Illumina, San Diego, California). Then reads were aligned to the transcriptome using STAR (version 2.6.1) (Dobin A et al., 2013) / RSEM (version 1.2.28) (Li B and Dewey CN, 2011) with default parameters, using a custom human GRCh38 transcriptome reference downloaded from http://www.gencodegenes.org, containing all protein coding and long non-coding RNA genes based on human GENCODE version 33 annotation. DESeq2 (version 1.26.0) was used for normalization and principal component analysis. Expression of each gene was fitted into a negative binomial generalized linear model, and the likelihood ratio test was applied to assess the differential expressions among all conditions. Benjamini and Hochberg procedure was applied to adjust for multiple hypothesis testing, and differential expression gene candidates were selected with a false discovery rate less than 0.05. For visualization of coordinated gene expression in samples, a two-way hierarchical clustering with Pearson correlation distance matrix was performed with samples and DEG candidates using the Bioconductor g-plots package (version 3.0.3) in R. Genes were extracted from the identified clusters for GO enrichment analysis by using DAVID v6.8 (Huang DW et al., 2009, https://david.ncifcrf.gov).
Methotrexate + L-ASNase Treatment
Set up was similar to growth curve described above. Treated cells with L-Asparaginase (2 IU/ml) for 48 hours before replacing media with fresh L-Asn depleted media as described in the nutrient restriction assay. Methotrexate was given at 1, 5, and 10 μM concentrations (only 10 μM shown in figure). Growth curves and survival analysis was performed. All experiments done in triplicate.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (version 6.0; GraphPad Software). Statistically significant data in in vitro and in vivo assays were assessed by unpaired Student’s t-test unless otherwise noted. Data are expressed as the mean ± SEM. Intergroup differences were considered statistically significant when p < 0.05.
Results
ASNS copy number gain is an indicator of poor glioma patient survival
ASNS gain correlates to poor patient survival in both higher grade and lower grade gliomas. Kaplan-Meier survival plots generated using copy-number data from the 2016 Merged 2016 GBM cohort (cBioPortal, MSKCC) shows a hazard ratio 1.480 in the high-grade cohort and 2.248 in the low-grade cohort (Fig. 1A). ASNS gain appears to enrich in the higher-grade non-G-CIMP brain tumor clusters compared to lower grade G-CIMP tumors, without any particular molecular subtype preference, although there are more classical/mesenchymal cases and more IDH wildtype cases (Fig. 1B, C). ASNS expression is observed in a grade-dependent manner in gliomas and is seen only at scant levels in normal brain tissue (Fig. 1D, E). More recent theories of gliomagenesis suggest that in the context of glioma expression subtypes, there is likely a general shift from a more stem-like precursor representative of the proneural subtype to a more latent mesenchymal profile, with the classical and neural expression subtypes falling somewhere along this continuum. This enrichment in the later stage expression profiles therefore agrees with the idea that ASNS may be positively selected for at a later stage of glioma progression following early focal amplifications leading to chromosome 7 aneuploidy. ASNS copy number does not seem to be correlated to progression free survival (Supplementary Fig. 1B, 1C). Gain of ASNS is enriched in IDH1 wt tumors versus IDH1 mutant (Fig. 1C) and ASNS gain seems almost mutually exclusive of IDH1 mutation (Supplementary Fig. 4). This may also explain the larger survival variance seen between the two groups in the lower grade KM plot, perhaps in part supported by IDH1 mutant derived survival benefit. ASNS copy number gain looks to correlate with poor survival in IDH wt cohorts and does not correlate as well in IDH mutant cohorts, which is reverse of what is observed in the same cohorts when stratifying by CDKN2A deletion status (Supplementary Fig. 2).
Figure. 1. ASNS gain is a poor prognostic indicator in glioma patients.
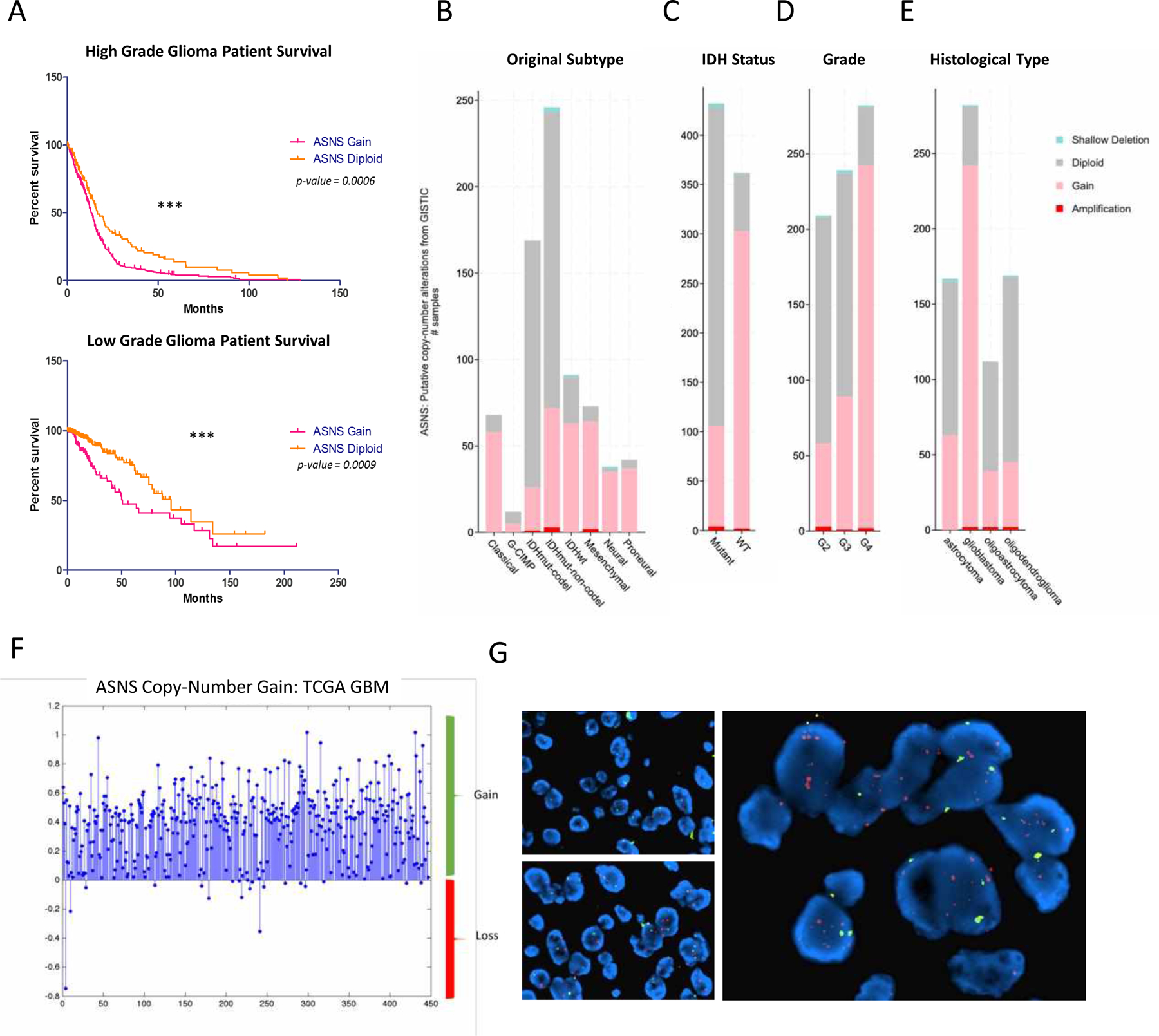
(A) Survival variance is observed in both high grade and lower grade patient cohorts, with hazard ratio of 1.48 and 2.248, respectively. KM plots generated using copy number data from 2016 Merged TCGA Cohort, cBioPortal (MSKCC). (B) more aggressive classifications within gliomas had higher enrichment of ASNS gain, including CL/MES non-GCIMP versus lower grade GCIMP tumors. (C) IDH mutant gliomas had markedly lower levels of ASNS gain compared to IDH wildtype tumors. All charts compiled from cBioPortal (MSKCC), Merged 2016 Cohort. [Gain is defined as 3+ copies of ASNS versus 2 copies for diploid] (D-E) ASNS copy number gain increases in higher grade gliomas compared to normal tissues. ASNS gain far less enriched in lower grade gliomas when compared to high grade gliomas. Grade 4 glioblastoma shows highest enrichment for patients with gain of ASNS. ASNS ‘gain’ defined as 3+ copies of ASNS versus ‘diploid’ (normal 2 gene copies). (F) CNA analysis from TCGA 2009 Provisional database shows gain of ASNS in majority of patient samples. (G) FISH analysis of ASNS copy number in human glioma patient samples.
ASNS catalyzes the ATP-dependent biosynthesis of the non-essential amino acid asparagine from glutamine and aspartate. This process can be antagonized by L-Asparaginase (ASNase) which converts asparagine back into aspartate. The ASNS gene is located on the 7q21.3 loci very near CDK6 and MCM7. In fact, ASNS gain is likely observed as a consequence of chromosome 7 gain which commonly occurs in gliomas accompanied by chromosome 10 loss. Epidermal growth factor receptor (EGFR) is found on chr. 7, and the EGFRvIII mutation is the most common oncogenic alteration found in GBMs (21), and EGFR amplification has often been considered the major driver mutation on chr. 7. Studies have also identified episomal amplification of EGFR on double minute chromosomes as another mechanism of oncogene enrichment in gliomas (22). Although this may function as a unique mechanism of oncogene amplification and perhaps therapeutic escape, EGFR is often still maintained on the chromosome even in the presence of episomal amplicons. ASNS is located far enough from the EGFR loci and is usually not found on any EGFR episomal amplicons. Copy number analysis from the TCGA 2009 provisional GBM dataset shows significant gain of ASNS in patient samples (approx. 6x copies/patient) and this corresponds with fluorescence in situ hybridization (FISH) analysis of ASNS status in glioma patient tumor samples (Fig. 1F, G). ASNS is not a likely driver of chromosome 7 gain in gliomas and is more likely to be a positively selected passenger that promotes the transition from focal amplification on the long arm of chr. 7, to whole chromosome gain that includes the short arm of chr. 7. It should also be noted that these described ASNS elevations mirrored the variance in ASNS elevation observed across our own patient derived glioma stem cell lines (GSCs). Most patient derived GSCs significant levels of ASNS when compared to normal brain, along with other pathological signs of advanced disease (Supplementary Fig. 5A).
ASNS gain drives a hyper-proliferative profile in GSCs
We looked at the effect ASNS gain might have on proliferation in our patient derived glioma stem cells (GSCs). Using two GSC lines with different endogenous levels of ASNS, we used shRNA knockdown and lentiviral overexpression to assess ASNS specific contributions to tumor growth and survival. GSC604 were endogenously low for ASNS, whereas GSC827 were endogenously high. Parental patient information for these cell lines has been described in the supplement (Supp Table 1). Our cell constructs clustered into three groups, ASNS hi (827), ASNS lo (604), and ASNS-mid (604–48, 827-A) (Fig. 2A, B). We will henceforth refer to these cell pairs as 604 ctrl (ASNS low) vs. 604 ASNS-hi (ASNS mid) and 827 ctrl (ASNS high) vs. 827 shASNS (ASNS mid). In addition, CRISPR based removal of the ASNS gene was also shown to be effective in reducing ASNS expression by more than 50% (Supplementary Fig. 6).
Figure 2. ASNS-high cells grow faster and form tumors more rapidly in vivo.
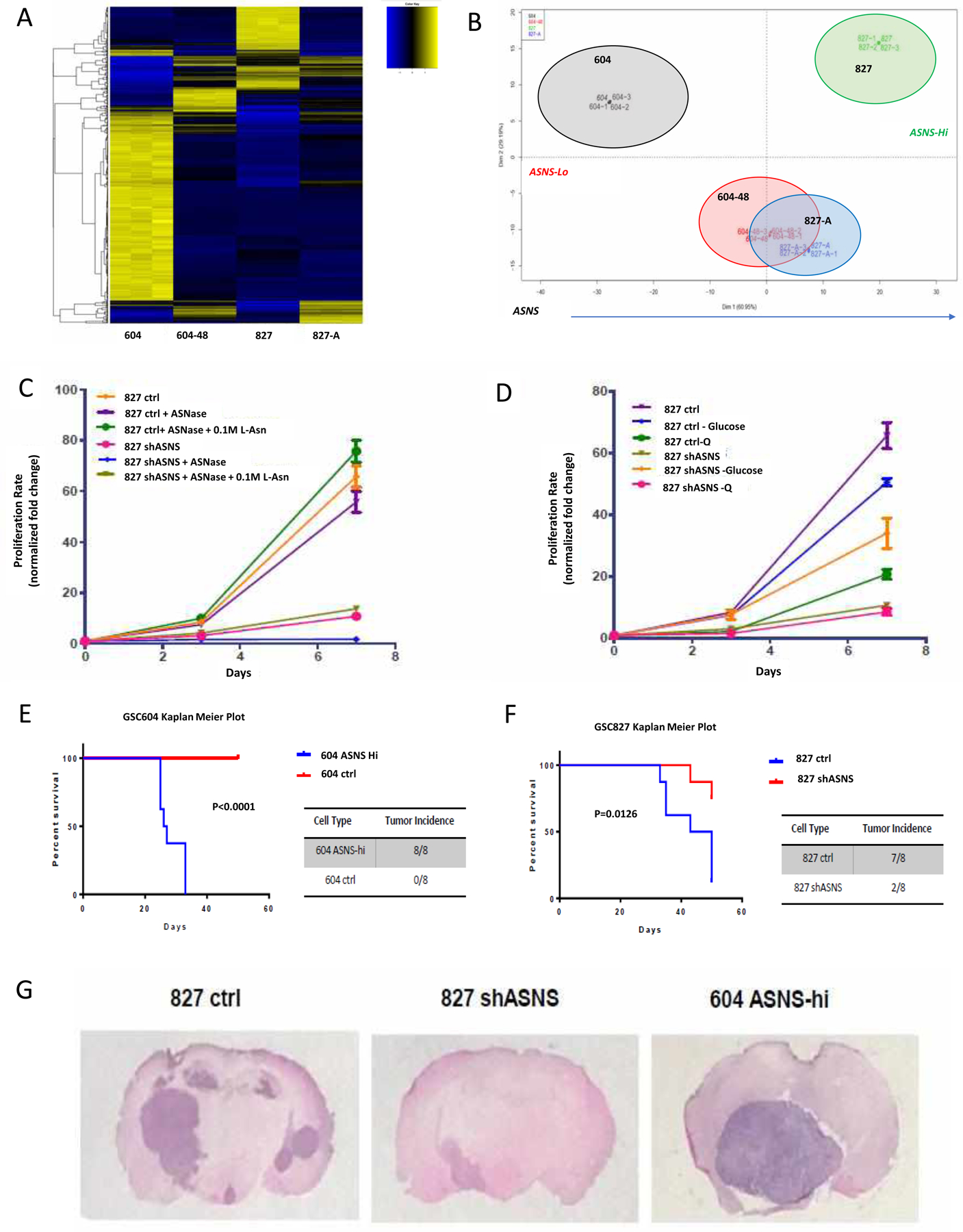
(A, B) RNAseq heatmap shows clustering of cells based on ASNS expression levels, with ASNS hi, mid, and lo. (C) in vitro growth curves for 827 cells with asparagine depletion via ASNase as well as L-Asn supplementation rescue. (D) in vitro growth assays showing glutamine (Q) and glucose withdrawal. ASNS high cells were able to retain robust growth rates in the absence of L-asparagine while this depletion reduced ASNS low growth and induced significant cell death. Regression comparison of matched constructs shows significance (p<0.0001). Glutamine withdrawal had a more significant effect on ASNS high cells (GSC827, 604 ASNS-hi) compared to ASNS low cells (604, 827 shASNS). ASNS low cells were significantly more affected by loss of nutrients within 24 hrs. Rescue of survival could be seen with exogenous supplementation of 0.1 M L-Asn. Regression comparison indicates significant difference in slopes (p<0.0001). Asparagine withdrawal had a more significant effect on ASNS high cells (GSC827, 604 ASNS-hi) compared to ASNS low cells (604, 827 shASNS). Regression comparison indicates significant difference in slopes (p<0.0001). Intracranial tumor implantation model in B6/Foxn1−/− mice shows significant survival difference based on ASNS status. (E) GSC604–48 average survival of 26.5-days (HR=22.92). (F) GSC827 N.S. average survival of 46.5-days (HR=6.44). Cells injected (1×106 cells/mouse) into left flank on B6.Cg-Foxn1 nu/J mice (n=2 mice/group). (G) GSC604–48 tumors were larger and grew faster than 827 control mice, although both are ASNS-high.
Modulation of ASNS expression had an acute effect on GSC morphology and sphere formation efficiency (Supplementary Fig. 5C). Some expression changes were observed in our GSCs with respect to stem maintenance/self-renewal pathways via RNA-seq, and this was further validated using sphere formation assays (Supplementary Fig. 8). ELDA assays confirmed that the efficiency of neurosphere formation decreased significantly in ASNS knocked down GSCs and increased in ASNS overexpressed GSCs (Supplementary Fig. 8F, G). Preliminary data suggested loss of ASNS pushed GSCs into a more differentiated, mesenchymal profile, with decreased expression of certain stem markers such as Sox2, Olig2, Klf4, and EZH2 (Supplementary Fig. 8D, E), perhaps suggesting a latent shift from a more stem to a more mesenchymal phenotype.
RNA sequencing showed a very distinct enrichment for many genes involved in DNA replication, cell cycle regulation, and transition to mitosis, and this is observed in both in vitro and in vivo growth assays. In vitro growth deficiencies as a result of lower ASNS expression could be partially rescued via exogenous supplementation with 0.1 M L-asparagine (Fig. 2C). High ASNS expression also protected growth under certain nutrient restricted conditions. ASNS high cells had a higher baseline proliferation rate which was more tolerant of glucose withdrawal. ASNS high cells were sensitive to glutamine withdrawal, while ASNS low cells were not, consistent with other studies (Fig. 2D). It is unclear however why 827 shASNS cells responded positively to glucose withdrawal. The growth effects were replicated in the CRISPR ASNS knockdowns as well (Supplementary Fig. 7I). In vivo growth was first assessed via a subcutaneous tumor model (data not shown), followed by an intracranial tumor model. Both models showed a significantly more aggressive growth pattern in ASNS high tumors (Fig. 2E, F). In addition to growing significantly larger and faster, 827 ctrl [high ASNS] tumors also appeared to develop more secondary tumors at distant sites when compared to the 827 shASNS tumors (Fig. 2G, Supplementary Fig. 6). Recent studies have implicated ASNS expression with pro-metastatic progression in several solid tumor models, which could be represented in these tumors. GSC604 ASNS-hi tumors grew rapidly albeit without secondary tumors, while GSC604 ctrl cells could not produce detectable tumors. Expectedly, ASNS low cells were very sensitive to asparagine depletion, while ASNS high cells grew very well despite this restriction. ASNS high cells actually seemed to grow better upon ASNase treatment which was puzzling at first but may be explained by the pro-tumorigenic effects of excess aspartate in solid tumors, remembering aspartate is the byproduct of ASNase mediated asparagine depletion.
ASNS facilitates metabolic plasticity and supports a stress adaptive phenotype
We then looked at how ASNS status could alter the metabolic state of our GSCs. Metabolomic analysis of relative abundance of various glycolytic and TCA intermediates under glutamine restricted conditions further revealed a specific inflection in ASNS high cells (827, green/purple) which is absent in ASNS low cells (604 yellow/orange) (Fig. 3). ASNS mid cells were expectedly somewhere between these two groups, but the major metabolites are generally elevated under ASNS high conditions vs. ASNS low conditions. Within the glycolytic pathway, glutamine withdrawal induced increased abundance of intermediates such as G6P-F6P, F16BP, G3P, and P-Serine, while ASNS low and mid cells had the opposite response (Fig. 3A). Nearly all TCA intermediates also increased in ASNS high cells under glutamine withdrawal, while all other cells showed the opposite trend (Fig. 3B). The most pronounced spike was seen in G6P levels, which is also the most NAD+ dependent step within the pathway, suggesting ASNS may be selecting for NAD+ dependent processes downstream. Serine and glycine levels are the only metabolites to show an inverse trend in ASNS high cells when glutamine was withdrawn, suggesting that these metabolites may be used up for some function which is providing a selective advantage under stress.
Figure 3. Modifications in Glycolytic and TCA Pathways under glutamine withdrawal reveal ASNS dependency.
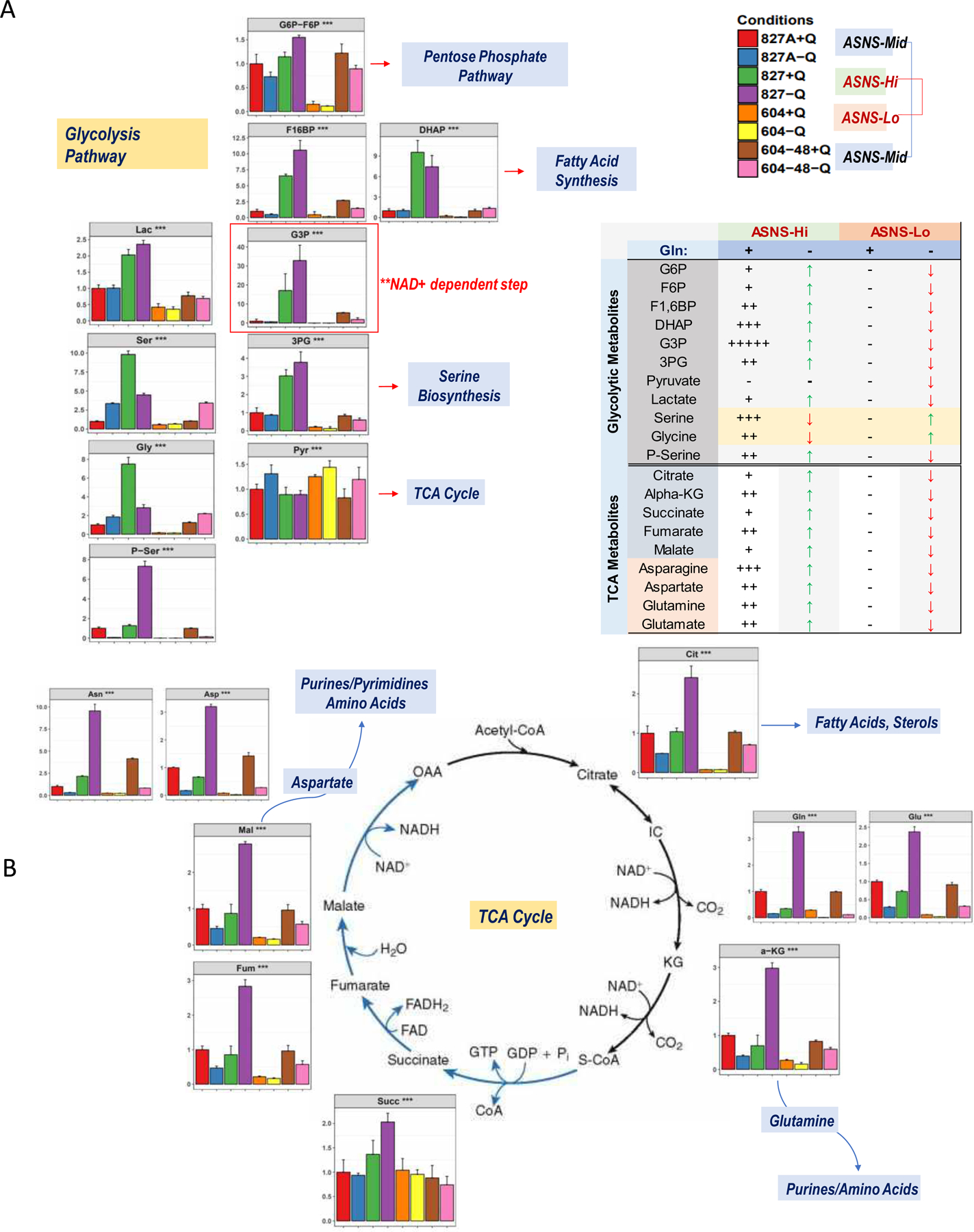
(A) Relative metabolite levels in the glycolysis pathway show significant increases under ASNS high conditions with a strong buildup at NAD+ dependent steps [G3P], (B) increase in all TCA cycle substrates under ASNS high conditions. Strongest glutamine withdrawal related shifts occur in ASNS high cells [827, green/purple], while the weakest shifts occur in ASNS low cells [604, yellow/orange].
Using the Seahorse Bioanalyzer to observe the metabolic phenotype of these cells, we saw that ASNS high cells not only had a higher baseline level of oxidative phosphorylation measured by the oxygen consumption rate (OCR), but also had a higher mitochondrial reserve capacity (Fig. 4E, F). This once again was replicated in the CRISPR knockdown GSCs as well (Supplementary Fig. 7H). In fact, ASNS high cells presented higher baseline levels of glycolysis, suggesting a more activated general metabolic profile. We examined how these metabolic states fluxed, as opposed to steady state profiles. A key hallmark of CSCs is the ability to have metabolic plasticity in order to maintain a degree of adaptability under changing conditions. First, we looked at shifting between oxidative phosphorylation and glycolysis in response to specific inhibitors [oligomycin] and/or drivers [FCCP] of mitochondrial respiration. We observed a much stronger metabolic shift in ASNS high GSCs as well as a greater metabolic range, in terms of the robustness of the oxidative phosphorylation and/or glycolysis (Fig. 4A, B). Another way to look at this was to measure the slope of the extracellular acidification rate (ECAR) spike upon inhibition of mitochondrial respiration via oligomycin, as a measure of shift towards glycolysis. Once again ASNS high GSCs had a higher rate of transition between pathways (Fig. 4C–F). To again address metabolic plasticity, we looked at the effect of single and dual inhibition of metabolism using either oligomycin (OXPHOS inhibitor) or 2-deoxyglucose (2-DG, glycolysis inhibitor), or both. We see that single inhibition of any one metabolic pathway had minimal effect on ASNS high cells but did affect the ATP levels in ASNS low cell, while dual inhibition was required to reduce the ATP levels in ASNS high cells (Fig. 4G). This suggests ASNS high cells more readily metabolically compensate when one metabolic pathway is blocked. Finally, we looked at how fuel flexible these ASNS high cells may be, as preliminary data suggested ASNS expression correlated strongly with elevated fatty acid oxidation in GSCs (data not shown). We see that feeding cells glucose/glutamine deficient media supplemented with palmitate-BSA inhibited ATP levels in GSCs with low levels of ASNS and resulted in cell death, whereas ASNS high GSCs were able to maintain nearly 80% of ATP levels compared to baseline in complete media, indicating the ability of ASNS high cells to utilize fatty acids in lieu of traditional fuel sources (Fig. 4G, H).
Figure 4. ASNS expression corresponds to a more metabolically plastic and stress resistant phenotype.
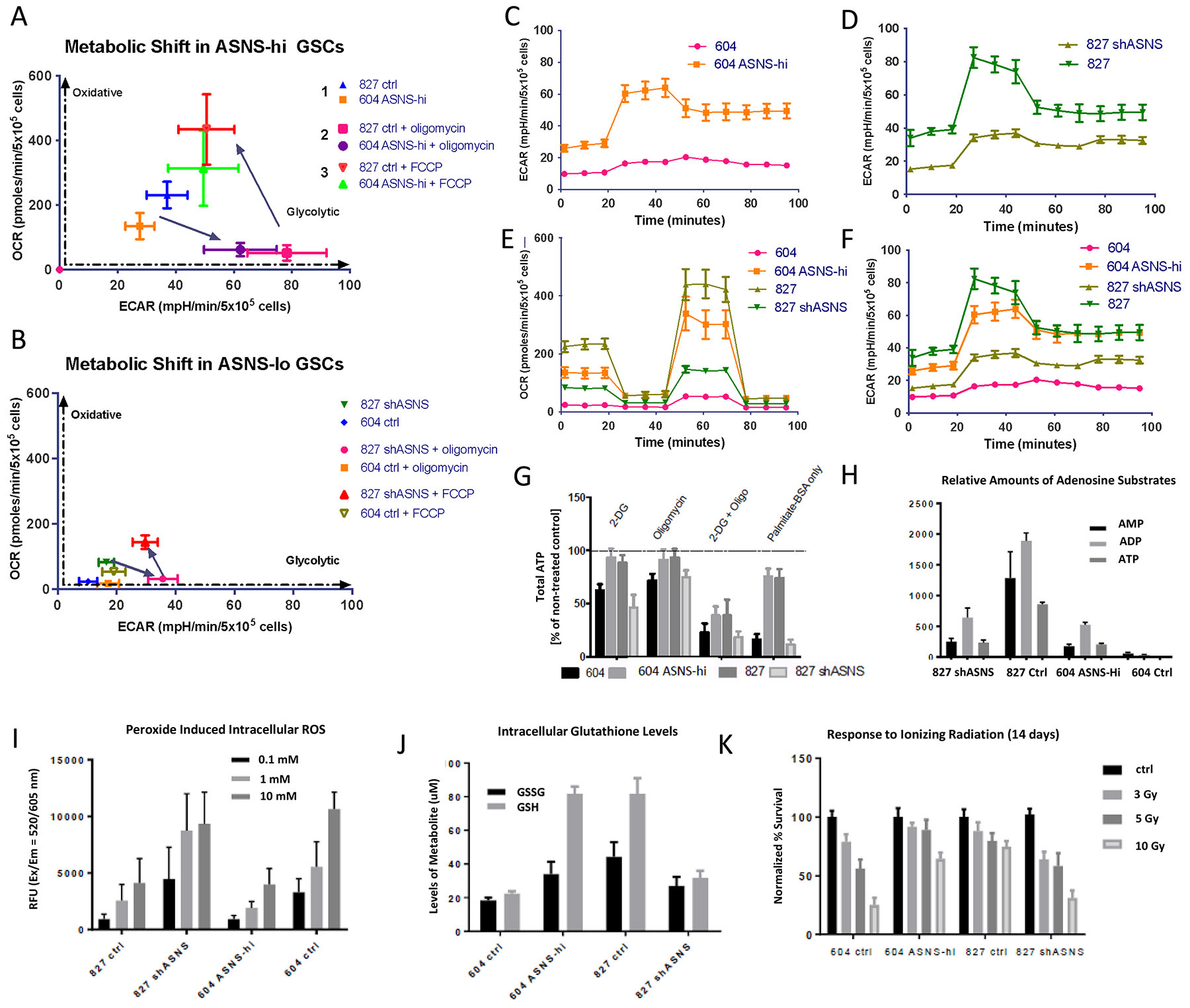
(A, B) ASNS high cells display more robust metabolic plasticity. ASNS high cells can switch between oxidative phosphorylation and glycolysis, bi-directionally more quickly compared to ASNS low cells. These plots express the metabolic transitions in each GSC as seen in the Cell Mito Stress Test, with the application of oligomycin (1 mM), FCCP (1 mM), and rotenone/Antimycin A (0.5 mM). ASNS high cells switch to glycolysis upon treatment with 1mM oligomycin much more quickly than ASNS low cells. (C, D) ECAR plot shows more rapid transition into glycolysis upon mitochondrial inhibition, (E, F) OCR and ECAR more robust in ASNS high GSCs (error bars shown indicate S.E.M). All plots (Fig 4C–F) were normalized in GraphPad, and certain error bars became smaller than the symbols on the data line. Figure 4F represents the combination of plots from Fig 4C and 4D. (G) Total ATP levels under different metabolic inhibitors indicated improved metabolic switching in ASNS high cells, as well as improved fuel flexibility through fatty acid utilization (p<0.0001, student t-test). (H) adenosine substrate levels in cells measured by metabolomics shows higher ATP levels in ASNS high cells versus ASNS low cells. (I) ROS levels in response to hydrogen peroxide treatment (0.1–10 mM) (p<0.001), (J) GSH/GSSG levels in GSCs treated with 1 mM hydrogen peroxide, (K) cell survival post radiation exposure (3–10Gy) for 14 days. Cell survival normalized to control for each GSC based on cell counts at day 14. (p<0.001, student t-test).
ASNS high cells tended to have higher max mitochondrial respiratory capacity and a consequent higher mitochondrial reserve capacity. This excess reserve capacity has been linked to resistance to oxidative stress, so we looked at how ASNS might affect oxidative stress response. First, we saw ASNS low cells produce higher levels of intracellular reactive oxygen species (ROS) in a dose dependent manner in response to peroxide exposure (Fig. 4I). We looked at glutathione response to peroxide stress and saw higher levels of glutathione (GSH) and glutathione disulfide (GSSG), in ASNS high cells (Fig. 4J). Furthermore, ASNS high cells had a lower GSH:GSSG ratio, indicating either decreased oxidation of GSH or improved recycling of GSSG into GSH. GSSG results from two glutathione molecules binding through a peroxide reduction reaction, such as those in response to intracellular ROS, organic hydroperoxides, and hydrogen peroxide.
Clinically, glioma patients often fail treatment once they become radio-resistant, so we looked at GSC response to ionizing radiation in the context of ASNS. GSCs with high levels of ASNS maintained resistance to radiation up to 10Gy, while ASNS low cells consistently showed a dose-dependent cytotoxicity (Fig. 4K). We followed this up by looking by doing transcriptomic profiling of these cells following irradiation to look at what molecular mechanism may be at play (Fig. 5). We found certain genes reversely expressed in two comparisons, which were GSC827-shASNS vs. 827-ctrl and GSC604-ASNS-OE vs. 604-ctrl. 838 genes were upregulated in comparison of GSC827-shASNS vs. 827-ctrl (reversely downregulated in comparison of GSC604-ASNS-OE vs. 604-ctrl), while 485 genes were downregulated in comparison of GSC827-shASNS vs. 827-ctrl (reversely upregulated in comparison of GSC604-ASNS-OE vs. 604-ctrl). These reversely regulated genes were separated into 4–6 clusters depending on expression patterns along with radiation status and time points. Functional annotation analysis of genes in each cluster by DAVID software showed that cell cycle and cell proliferation were upregulated as basic biological processes in GSC827-ctrl (ASNS hi). DNA damage response and repair signals were also activated at the earlier stage of post-radiation culture, decreasing at later time points. In this cell line, Epithelial-Mesenchymal Transition (EMT) seemed moderately activated through entire experiment and more transcription was induced on day 3. FoxO and HIF-1 signaling pathways were activated on day 14. In GSC827-shASNS, Ras/MAPK pathway was activated on day 0 and getting weaker on later time points. Cellular metabolic processes were upregulated at earlier phase and immune response signatures went up on day 3 but no DNA damage response was activated. In GSC604-ctrl, cell growth, MAPK and NFkB signaling were activated with some inflammation reaction on day 0. Wnt signaling and GTPase were activated especially on day 3. On day 14, genes involved in negative regulation of apoptosis and cell proliferation were upregulated. In GSC604-ASNS-OE, translation and DNA damage response were highly activated as default expression of the cell line and increased at early stage of post-radiation response (day 0). When the DNA damage response decreased on day 3, cell division and transcription became more activated. Lastly, regulation of cell growth and EMT increased on day 14.
Figure 5. Changes in transcriptomic profiling and gene ontologies after irradiation.
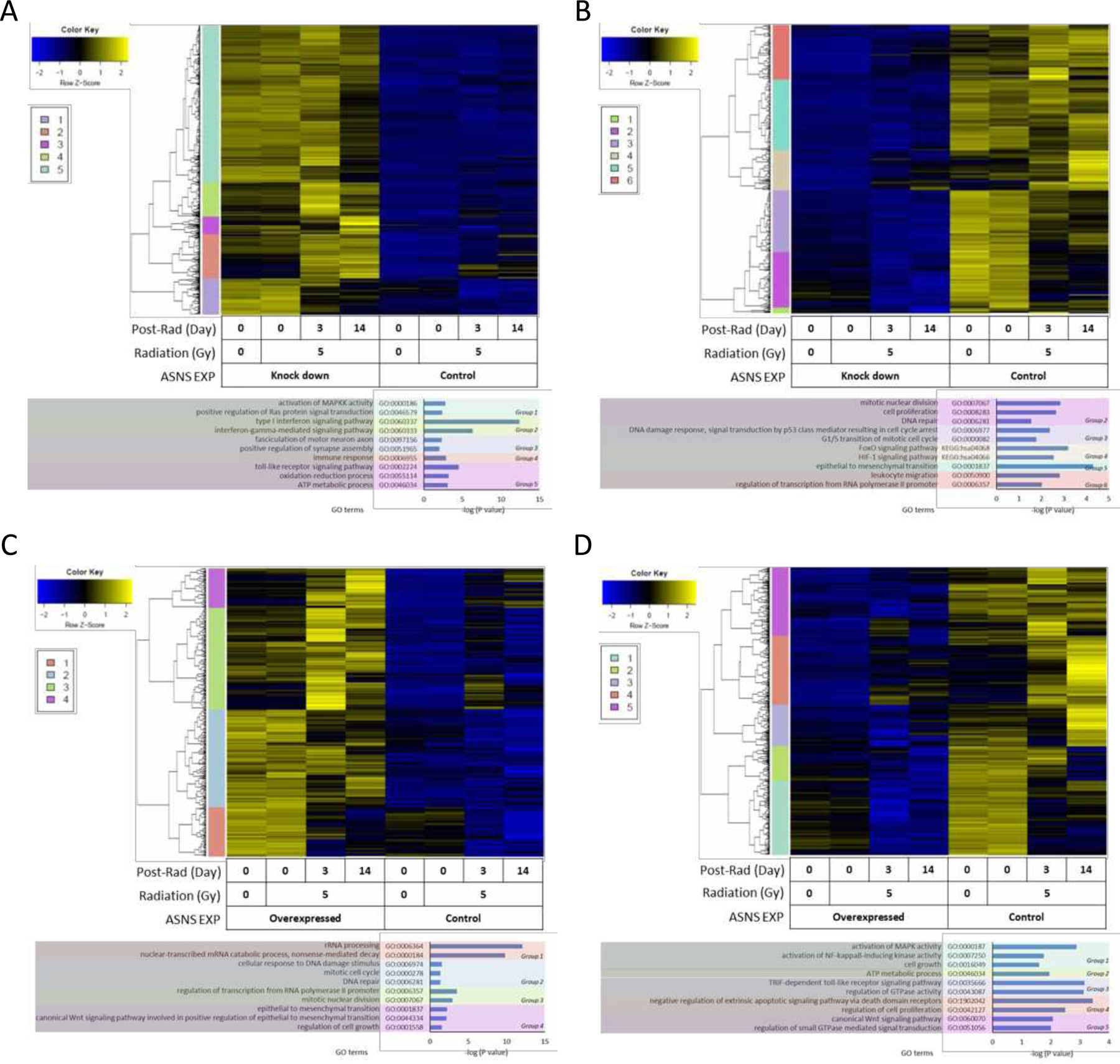
(A) Significantly upregulated genes in 827-shASNS comparing to 827-ctrl, which also showed opposite expression in comparison between 604-ASNS-OE and 604-ctrl. (B) Significantly downregulated genes in 827-shASNS comparing to 827-ctrl, which also showed opposite expression in comparison between 604-ASNS-OE and 604-ctrl. (C) Significantly upregulated genes in 604-ASNS-OE comparing to 604-ctrl, which also showed opposite expression in comparison between 827-shASNS and 827-ctrl. (D) Significantly downregulated genes in 604-ASNS-OE comparing to 604-ctrl, which also showed opposite expression in comparison between 827-shASNS and 827-ctrl. Color bar indicates gene clusters showing expression along with the time point after irradiation and gene ontology (GO) terms and pathways. Color bar indicates gene clusters showing expression along with the time point after irradiation and gene ontology (GO) terms and pathways.
Further metabolomics showed a marked increase in one-carbon metabolites such as methionine, serine, glycine, and homocysteine as well as GSH and GSSG in ASNS high GSCs. Certain metabolites including asparagine, aspartate, glutamine, and a-ketoglutarate responded to glutamine withdrawal, whereas others such as homocysteine, cystathionine, arginine, and lysine was unaffected, and remained elevated in ASNS high GSCs (Supplementary Fig. 9A). Metabolomics again corroborated these findings and showed elevations in GSH biosynthesis, the methionine cycle, and the transsulfuration pathway (Supplementary Fig. 9B). With elevations in homocysteine, we looked at whether elements of the ER stress response pathway upstream of ASNS were involved. ASNS as well as ATF4 and GCN2 are all elevated in the sequencing data, suggesting that the stress response pathway is activated in ASNS high cells, however we see minimal CHOP activation (Supplementary Fig. 9C, D). RNA sequencing data also indicated an enrichment of genes involved in glutathione binding [GSTM1, GSTM2, MGST1], amino acid binding [SHTM1, SHTM2, ASS1], and many amino acid transporters in ASNS high cells (Supplementary Fig. 9E). This indicates an anti-apoptotic adaptive stress response profile which renders cells more flexible under stress (23, 24). Taken together, the ASNS status in GSCs modified the cellular redox homeostasis by promoting an oxidative stress resistant profile that mirrors radio-resistant profiles of glioma patients in the clinic.
Discussion
Advances in our understanding of stem cell biology over the past few decades have opened up new areas of exploration in cellular and molecular biology. The more recent extension of this understanding on stem cells to the pathogenesis of cancer has reframed how many now model early tumorigenesis, tumor progression, and even tumor recurrence post treatment. It can certainly be appreciated that the plasticity of stem cells in many ways mirrors the tumor plasticity & adaptability observed in the clinic, especially as tumors evolve in response to many different selective pressures, therapeutics being chief among them. It is now well established that cancer stem cell models are very good research tools for studying tumor development due to their ability to recapitulate the molecular landscape of actual patient tumors (25–27). In fact, patient derived glioma stem cell cultures, like the ones used in this study, have been able to phenocopy the intertumoral heterogeneity from matched patient tumors in the clinic with reasonably high fidelity (25–27). Current work in this space has begun to move towards even more complex organoid models of tumorigenesis, but GSCs remain a strong platform to study glioma biology in concert with larger OMICs based analyses (28, 29).
The gain of ASNS in glioma patients does not represent a major driver of tumorigenesis in the classical sense, but growing evidence indicates that this alteration is a strong adaptation that is selected for in cancer. ASNS is likely carried as a passenger gain on chromosome 7. In a similar fashion, EGFR focal amplification alone does not initiate gliomas but contributes to progression (21). Many prognostic factors confirm that increased levels of ASNS correspond to worsened clinical outcome, and this is further seen with the mutual exclusivity of ASNS gain and IDH1 mutation. Additionally, enrichment in the classical and mesenchymal expression subtypes suggests a more aggressive clinical presentation and that ASNS gain occurs later in gliomagenesis, since it is less enriched in proneural tumors. Unaddressed in this study is the origin of this ASNS alteration, beyond simply being included in glioma related chromosome 7 aneuploidies. Further work is being conducted to begin to address these and other such alterations that present as common features of aggressive gliomas. In this case, ASNS does in fact demonstrate a sufficient fitness advantage to benefit tumors that acquire this alteration, especially in tumor regions under significant metabolic stress. Metabolic perturbations in cancer are often observed as passenger alterations but retain strong selective pressure as cells develop and contribute to the heterogeneity and plasticity demonstrated in many high-grade tumors, and ASNS appears to follow this general paradigm.
Metabolic perturbations are primarily selected for pro-growth contributions to tumor development, and this is clearly shown with ASNS gain in our glioma cells, both in vitro and in vivo. ASNS high tumors seem to enrich in the classical and mesenchymal expression subtypes and the exhibited growth patterns supportive of an aggressive mesenchymal growth profile. Elevated asparagine bioavailability in these cells provides an extra level of growth support and metabolic maneuverability in response to fluctuating environmental conditions. In vitro asparagine supplementation allowed rescue of growth under nutrient deprived conditions, indicating asparagine is a crucial factor in tumor growth (Fig. 2C, D), and protected against nutrient withdrawal mediated cell death, especially in the context of glutamine withdrawal. Tumor cells are constantly making adjustments to maintain access to key substrates, and often can coordinate a very complicated network of amino acid sensing and sourcing to support the biochemical pathways involved in biomass production and proliferation. Glutamine, a key amino acid and the most abundant metabolite in the body, is an important metabolite involved in cancer growth and progression (30, 31). Many tumors rely on glutamine and glutamine-dependent processes as much or even more than glucose, even in the context of metabolic demand. Asparagine is a unique amino acid to be involved in cancer since it is a non-essential amino acid that is very tightly controlled by stress response mechanisms in the cell, but rarely contributes directly to processes linked to central carbon metabolism. Furthermore, we see invasive and migratory growth patterns in vivo for some ASNS high tumors but never in ASNS low tumors. These data support recent work indicating asparagine supports the EMT process in breast cancer and drives metastatic progression (32).
The most common metabolic perturbation that is observed in the clinic with respect to gliomas is IDH1 mutations. IDH1 is one of the three isozymes of isocitrate dehydrogenase (33). IDH1 reversibly catalyzes oxidative decarboxylation of isocitrate into a-ketoglutarate (a-KG) as part of the TCA cycle, and heterogenous mutations of IDH1 result in dysfunctional enzymatic activity and the elevated production of 2-hydroxyglutarate (2-HG). IDH1 mutation is considered a driver alteration in early tumorigenesis, especially in glioblastomas, often a first hit in the development of diffuse gliomas (33). IDH1 mutant tumors tend to be more amenable to ionizing radiation (34). We see that ASNS gain is almost exclusively enriched in IDH1 wild-type patients, with very few examples of ASNS copy number amplification in patients harboring an IDH mutant (Supplementary Fig. 4). The modulation of the ASNS pathway within aggressive gliomas may not overtake the IDH status as a prognostic indicator, but it does reflect another nuance to high grade gliomas and their ability to mitigate cellular stress and continue to outcompete normal tissue as the tumor expands. This stress mitigation may also represent an important step towards supporting more invasive capabilities when expanding beyond the original tumor niche.
Plasticity and adaptive stress response mechanisms in cancer may well require multiple layers of coordination, which includes not only maintaining growth but also resisting cell death. In our cells, we observed that robust metabolic plasticity was linked to resistance to oxidative stress. This is not surprising when considering the significant cross-over between the metabolic and anti-apoptotic functions of the mitochondria. The metabolic plasticity that we describe in the ASNS high cells focuses on their ability to rapidly switch between glycolysis and oxidative phosphorylation. Baseline shifting in response to the mitochondrial inhibitor oligomycin showed much faster transition from an oxidative profile to a more glycolytic profile (Fig. 4A–F). Cellular plasticity is likely a strong underlying factor that drives resistance to many metabolic interventions in the clinic, and we see in these cells single inhibition is largely ineffective with elevated ASNS, since the robust switching compensates for any detrimental effects on cell survival. Further indication of the plastic nature of ASNS high cell metabolism is seen in the fuel flexibility, where ASNS high cells can readily utilize fatty acid fuel sources to maintain tumor growth and survival. Electron microscopy revealed that in at least one of our cell lines (GSC827), there was evidence of structural differences in mitochondria between ASNS high and low cells (Supplementary Fig. 9G–L). ASNS high mitochondria in this cell line were significantly larger with deformation of the inner cristae structures. Although these differences were not seen in GSC604 cells irrespective of ASNS status, these changes seem to demonstrate previously described cristalysis in GBM cells.
Functional annotation analysis of genes reversely expressed in GSC604 and GSC827 cell lines by DAVID software showed that cell cycle and cell proliferation were upregulated as basic biological processes in GSC827-ctrl (ASNS high). DNA damage response and repair signals were also activated at the earlier stage of post-radiation culture and getting reduced on later time points (Fig. 5B). In this cell line, Epithelial-Mesenchymal Transition (EMT) seemed moderately activated through entire experiment and more transcription was induced on day 3. As demonstrated in earlier studies, cellular quality control and other crucial transcriptional programs involved in cancer biology including angiogenesis, cell survival, and glucose metabolism were activated late, around day 14 (35, 36). By knocking down ASNS (GSC827-shASNS), Ras/MAPK pathway, which is involved in cell growth, division and differentiation, was activated on day 0 and getting weaker on later time points (Fig. 5A). Cellular metabolic processes were upregulated at earlier phase and immune response went up on day 3 but no DNA damage response was activated.
In the cell lines with endogenously lower expression of ASNS (GSC604-ctrl), basic biological processes to maintain cell growth may be indicated by the pro-inflammation signature on day 0 (Fig. 5D). Wnt signaling and GTPase were activated especially on day 3, implying that some type of cytoskeletal organization and polarized cell migration might have been induced (37). On day 14, cells might be trying to minimize radiation-induced cell death and promote cell proliferation by negatively regulating apoptosis. By overexpressing ASNS in GSC604 by transfecting gene (GSC604-ASNS-OE), translation and DNA damage response were highly activated at baseline and increased early on post-radiation (day 0) (Fig. 5C). When the DNA damage response decreased on day 3, cell division and transcription became more activated. Lastly, regulation of cell growth and EMT increased on day 14. Ionizing radiation (IR)-induced EMT is known to promote resistance to radiation (38, 39). Wnt signaling is also important for stem cell maintenance and is associated with the radiation resistance of GBM (40). Overall, activation of DNA damage response and EMT in ASNS high cell lines (GSC827-ctrl and GSC604-ASNS-OE) implies that ASNS confers a radio-protective phenotype in glioma stem cells.
Certain tumors can be incredibly difficult to treat, and this difficulty usually reflects the ability of a tumor to find many pivot points within its biology that can be used to evade stress and potential loss of selective advantage. ASNS CNA is one such pivot point within glioma biology. This metabolic alteration indicates an adoption of an adaptive cellular phenotype, and this is also mirrored in the adaptive stress response profile in the ASNS high cells. The mitochondrial stress tests show that at baseline, ASNS high cells have higher baseline and max OCR, but more importantly ASNS high cells had higher mitochondrial reserve capacity, which has been shown to correlate with resistance to oxidative stress (Fig. 4E, F). The ASNS high GSCs not only had lower levels of intracellular ROS, but also higher levels of glutathione, which was further indicated by elevated one carbon metabolites and gene processes related to GSH binding and antioxidant production. Metabolomic analysis supported the idea that ASNS high cells provided not only increased metabolites that fed into biomass production but also a distinct mechanism that favored one-carbon metabolites and NAD+ dependent intermediates (Fig. 3). Resistance to induced oxidative stress, either by hydrogen peroxide or ionizing radiation exposure, is closely linked to the ASNS mediated hyper-activity of one carbon metabolism and elevated antioxidant activity. As previously noted, ASNS high cells had higher GSH:GSSG ratio, another indication of a resistant phenotype, but it is not clear whether this is due to more efficient reduction of GSSG, rapid elimination of GSSG, decreased oxidation of GSH, or increased production of GSH. It should be noted that we have not yet understood how ASNS directly affects increased one carbon metabolism, but one potential explanation is that increased intracellular asparagine could drive serine/glycine flux into the cells to drive this pathway as described by Krall et al 2016 (41). Metabolomic data shows increased levels of serine and glycine, as well as intracellular asparagine in ASNS high cells, and measurements from the media suggest more asparagine leaving the cells in ASNS high cells, but serine and glycine flux is less clear without more accurate tracing experiments. Additionally, more should and is being done to elucidate the details of what and how elements of the one carbon cycle are directly being altered. A specific ASNS inhibitor can be very effective in combination with other treatments, compared to simple systemic administration of L-Asparaginase, as has been reported in recent years (42, 43). Preliminary data suggested that targeting of folate using methotrexate in combination with L-Asparagine could potentially be one such combination that could target resistant ASNS high cells. Fundamentally, the observed metabolomic shifts indicate ASNS high cells are more readily capable of responding to oxidative stress via a more robust redox response. Additionally, elements of the stress response pathway in the endoplasmic reticulum seem to support oxidative resistance. HERPUD1 elevation in ASNS high cells corresponds to increased levels of homocysteine, and these same cells seem to have lower levels of CHOP activation, indicating a shift towards an adaptive stress response as has been previously described (23, 24). The exact nature of the ER contribution to this resistance profile is unclear but more study is required to fully understand any coordination between the ER and mitochondrial signaling axes that may be mediated by asparagine, directly or indirectly.
We are beginning to see the dynamic role and function that asparagine and its regulation play in brain tumor propagation. The role of ASNS extends well beyond amino acid biosynthesis, and well beyond asparagine. ASNS gain may well reasonably be considered a prognostic indicator of poor response to radiation or any treatment aimed at causing oxidative stress in tumor cells. However, it is more important to understand that the modulation of the asparagine metabolic pathway reflects another example of the weaponization of cell intrinsic mechanisms within gliomagenesis and that these more nuanced modifications should inform therapeutic approaches to this heterogenous disease. The role of ASNS overexpression in clinical radio-resistance requires further study and pharmacological silencing of ASNS by small molecule inhibitors may be a lead into a novel mechanism to overcome one pathway of resistance.
Supplementary Material
Implications:
This study reveals a new role for ASNS in metabolic control and redox homeostasis in glioma stem cells and proposes a new treatment strategy that attempts to exploit one vulnerable metabolic node within the larger multilayered tumor network.
Acknowledgments
Funding for this project was supported by the National Institute for Neurological Disorders & Stroke (NIH; 2R01# NS 048959 to Dr. Yu) and a grant from the FasterCures, a center of the Milken Institute.
Footnotes
Declaration of Interests: The authors declare no competing interests.
References:
- 1.Stupp R, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Ahmed AU, Auffinger B & Lesniak MS Understanding glioma stem cells: rationale, clinical relevance and therapeutic strategies. Expert Rev Neurother 13, 545–555 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balasubramaniyan V, et al. Aberrant mesenchymal differentiation of glioma stem-like cells: implications for therapeutic targeting. Oncotarget 6, 31007–31017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee G, et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model. Mol Cancer Ther 15, 3064–3076 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Codrici E, Enciu AM, Popescu ID, Mihai S & Tanase C Glioma Stem Cells and Their Microenvironments: Providers of Challenging Therapeutic Targets. Stem Cells Int 2016, 5728438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bao S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66, 7843–7848 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Wu J, et al. [Malignant transformation of glioma stromal cells induced by glioma stem cells heterotopically inoculated in host liver]. Zhonghua Yi Xue Za Zhi 94, 2775–2780 (2014). [PubMed] [Google Scholar]
- 8.Dzobo K, et al. Cancer Stem Cell Hypothesis for Therapeutic Innovation in Clinical Oncology? Taking the Root Out, Not Chopping the Leaf. OMICS 20, 681–691 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Rich JN, Matsui WH & Chang JC Cancer stem cells: A nuanced perspective. Medicine (Baltimore) 95, S26–28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rich JN Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine (Baltimore) 95, S2–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baysan M, et al. Micro-environment causes reversible changes in DNA methylation and mRNA expression profiles in patient-derived glioma stem cells. PLoS One 9, e94045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chalmers AJ Radioresistant glioma stem cells--therapeutic obstacle or promising target? DNA Repair (Amst) 6, 1391–1394 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Dong J, et al. Glioma stem/progenitor cells contribute to neovascularization via transdifferentiation. Stem Cell Rev 7, 141–152 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Vlashi E & Pajonk F Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol 31, 28–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vlashi E, McBride WH & Pajonk F Radiation responses of cancer stem cells. J Cell Biochem 108, 339–342 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balasubramanian MN, Butterworth EA & Kilberg MS Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am J Physiol Endocrinol Metab 304, E789–799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell 56, 205–218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toda K, et al. Metabolic Alterations Caused by KRAS Mutations in Colorectal Cancer Contribute to Cell Adaptation to Glutamine Depletion by Upregulation of Asparagine Synthetase. Neoplasia 18, 654–665 (2016). 125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Q, et al. Knockdown of asparagine synthetase (ASNS) suppresses cell proliferation and inhibits tumor growth in gastric cancer cells. Scand J Gastroenterol 51, 1220–1226 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Hu Y, and Smyth GK (2009). ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of Immunological Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- 21.Koga Tomoyuki, Li Bin, Figueroa Javier M, Ren Bing, Chen Clark C, Carter Bob S, Furnari Frank B, Mapping of genomic EGFRvIII deletions in glioblastoma: insight into rearrangement mechanisms and biomarker development, Neuro-Oncology, Volume 20, Issue 10, October 2018, Pages 1310–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka H, & Watanabe T (2020). Mechanisms underlying recurrent genomic amplification in human cancers. Trends in cancer, 6(6), 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishitoh Hideki; CHOP is a multifunctional transcription factor in the ER stress response, The Journal of Biochemistry, Volume 151, Issue 3, 1 March 2012, Pages 217–219, 10.1093/jb/mvr143 [DOI] [PubMed] [Google Scholar]
- 24.Rozpędek W, Pytel D, Mucha B, Leszczyńska H, Diehl JA, & Majsterek I (2016). The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Current Molecular Medicine, 16(6), 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, … & Phillips HS (2010). A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer cell, 17(4), 362–375. [DOI] [PubMed] [Google Scholar]
- 26.Venere M, Fine HA, Dirks PB, & Rich JN (2011). Cancer stem cells in gliomas: identifying and understanding the apex cell in cancer’s hierarchy. Glia, 59(8), 1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fine HA (2009). Glioma stem cells: not all created equal. Cancer cell, 15(4), 247–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, … & Fine HA (2019). Modeling patient-derived glioblastoma with cerebral organoids. Cell reports, 26(12), 3203–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ozawa T, Riester M, Cheng Y-K, Huse JT, Squatrito M, Helmy K, … Holland EC (2014). Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell, 26(2), 288–300. 10.1016/j.ccr.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ratnikov B, et al. Glutamate and asparagine cataplerosis underlie glutamine addiction in melanoma. Oncotarget 6, 7379–7389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang C, et al. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 69, 7986–7993 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knott SR, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, Turgeon MO, Fish L, Erard N, Gable AL and Maceli AR, 2018. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature, 554(7692), p.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimitrov L, Hong CS, Yang C, Zhuang Z, & Heiss JD (2015). New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. International journal of medical sciences, 12(3), 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molenaar RJ, Maciejewski JP, Wilmink JW, & van Noorden CJ (2018). Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene, 37(15), 1949–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webb AE, Brunet A., FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014. April;39(4):159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003. October;3(10):721–32. [DOI] [PubMed] [Google Scholar]
- 37.Lee Y, Lee JK, Ahn SH, Lee J, Nam, DHWNT signaling in glioblastoma and therapeutic opportunities. Lab Invest. 2016. February;96(2):137–50. [DOI] [PubMed] [Google Scholar]
- 38.Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH, Park HG, Han SI, Kang HS., Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. 2017. January 30;16(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng J, Li P, Zhang Q, Yang Z, Fu S, A radiosensitivity gene signature in predicting glioma prognostic via EMT pathway. Oncotarget. 2014. July 15;5(13):4683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwadate Y, Epithelial-mesenchymal transition in glioblastoma progression. Oncol Lett. 2016. March;11(3):1615–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krall AS, Xu S, Graeber TG, Braas D, & Christofk HR (2016). Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nature communications, 7(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panosyan EH, Grigoryan RS, Avramis IA, Seibel NL, Gaynon PS, Siegel SE, … & Avramis VI (2004). Deamination of glutamine is a prerequisite for optimal asparagine deamination by asparaginases in vivo (CCG-1961). Anticancer research, 24(2C), 1121–1126. [PubMed] [Google Scholar]
- 43.Panosyan EH, et al. Clinical aggressiveness of malignant gliomas is linked to augmented metabolism of amino acids. J Neurooncol 128, 57–66 (2016). 126 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.