

Abstract
The clinical success of cancer immunotherapy is providing exciting opportunities for the development of new methods to detect and treat cancer more effectively. A new generation of biomaterials is being developed to interface with molecular and cellular features of immunity and ultimately shape or control anti-tumor responses. This review focuses on recent advances that are supporting the advancement of engineered T cells. This class of cancer therapy has the potential to cure disease in subsets of patients, yet there remain challenges such as the need to improve response rates and safety while lowering costs to expand their use. To provide a focused overview, we highlight recent strategies in three areas of biomaterials research: low-cost cell manufacturing to broaden patient access, noninvasive diagnostics for predictive monitoring of immune responses, and strategies for in vivo control that enhance anti-tumor immunity. These research efforts shed light on some of the challenges associated with T cell immunotherapy and how engineered biomaterials that interface with synthetic immunity are gaining traction to solve these challenges.
Keywords: biomaterials, adoptive T cell therapy, cancer immunotherapy, cell manufacturing, immune biomarkers, remote control, synthetic immunity, immunoengineering
Graphical Abstract
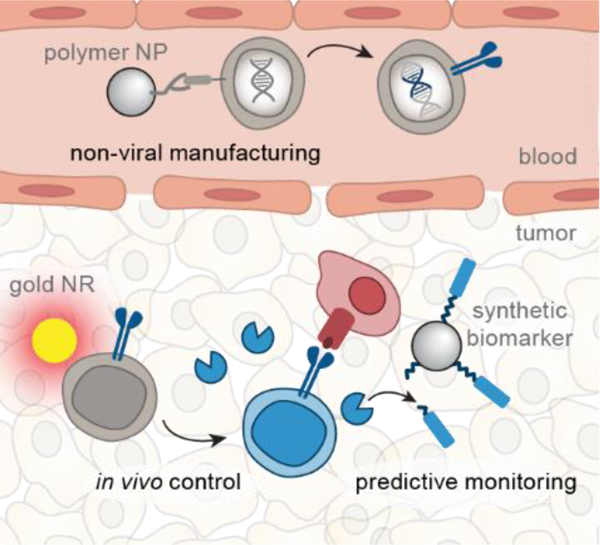
Summary
Biomaterials that interface with synthetic T cell immunity have the potential to detect and treat cancer more effectively. This review highlights recent advances in three areas of biomaterials research to improve engineered T cell therapies: low-cost manufacturing, predictive monitoring, and in vivo control.
1. Introduction
Advances in biomaterials will continue to play a fundamental role in shaping the future of cancer therapies toward more effective and safer treatments. The ability to engineer key properties of biomaterials such as size, charge, and shape contributes to the control of cellular and molecular interactions that ultimately affect therapeutic responses[1]. Biomaterials like lipids, polymers, hydrogels, protein conjugates, and nanoparticles have demonstrated safety and use as U.S. Food and Drug Administration (FDA)-approved cancer therapies to enhance anti-tumor activity and reduce toxicity in healthy tissues[1, 2]. For instance, Gliadel®, a biodegradable polymer wafer loaded with the chemotherapeutic drug carmustine, was developed to be implanted after surgical resection of brain tumors to destroy remaining tumor cells by localized drug delivery[3]. Beyond chemotherapy, biomaterials are generating promising new strategies to enhance cancer immunotherapies as they emerge as the next pillar of cancer treatment. The success of cancer immunotherapy largely depends on the ability to control key steps in the cancer immunity cycle, which includes tumor antigen presentation, immune cell activation, lymphocyte trafficking and infiltration to tumor sites, and targeted killing of tumor cells[4]. At each step, engineered biomaterials have the potential to enhance and boost anti-tumor immune responses while mitigating off-target effects. For example, interleukin-2 (IL-2), the first FDA-approved cytokine for cancer immunotherapy, had modest clinical success due to its short half-life and dose-limiting systemic toxicities[5]. This motivated the development of polyethylene glycol (PEG)-modified IL-2, which significantly extended its circulation half-life and reduced the required dosage while retaining its anti-tumor immune activity[6]. The success of PEGylation has since been extended to additional immunomodulatory cytokines like tumor necrosis factor alpha (TNF-α) and interferon alpha (IFN-α)[7] that have been approved for use in humans by regulatory agencies.
The rapid growth and clinical success of cell-based immunotherapies have led to new opportunities for biomaterials to enhance synthetic T cell immunity. Treatments like chimeric antigen receptor (CAR) T cell therapy are achieving unprecedented patient responses in hematological cancers with objective response rates as high as ~90% in B cell malignancies[8]. Yet major challenges continue to impede the broad clinical benefit of engineered T cell therapies across patient populations and tumor types especially for solid tumors (Figure 1). For example, engineered T cells are personalized for each patient and requires a multistep manufacturing process[9, 10], which includes isolation of T cells, viral transduction to introduce tumor targeting receptors, T cell expansion, and autologous infusion[11]. This complex pipeline precludes broad patient access as a single infusion of CAR T cell therapy costs between $350k–$450k and requires 3–5 weeks to manufacture, during which disease progression and mortality can occur[8, 11, 12]. For solid tumors, clinical response rates remain low compared to hematological cancers because of barriers such as immunosuppression by the tumor microenvironment (TME), chronic receptor activation leading to T cell exhaustion[13, 14] and severe immune-related toxicities from on-target, off-tumor cytotoxicity[15]. Moreover, potent immunomodulators like cytokines that are co-delivered systemically to support engineered T cells can lead to activation of endogenous immune cells and off-target toxicity[16]. These challenges are motiving the development of new approaches to realize the full potential of synthetic T cell immunity.
Figure 1. Opportunities for biomaterials to enhance engineered T cell therapies.
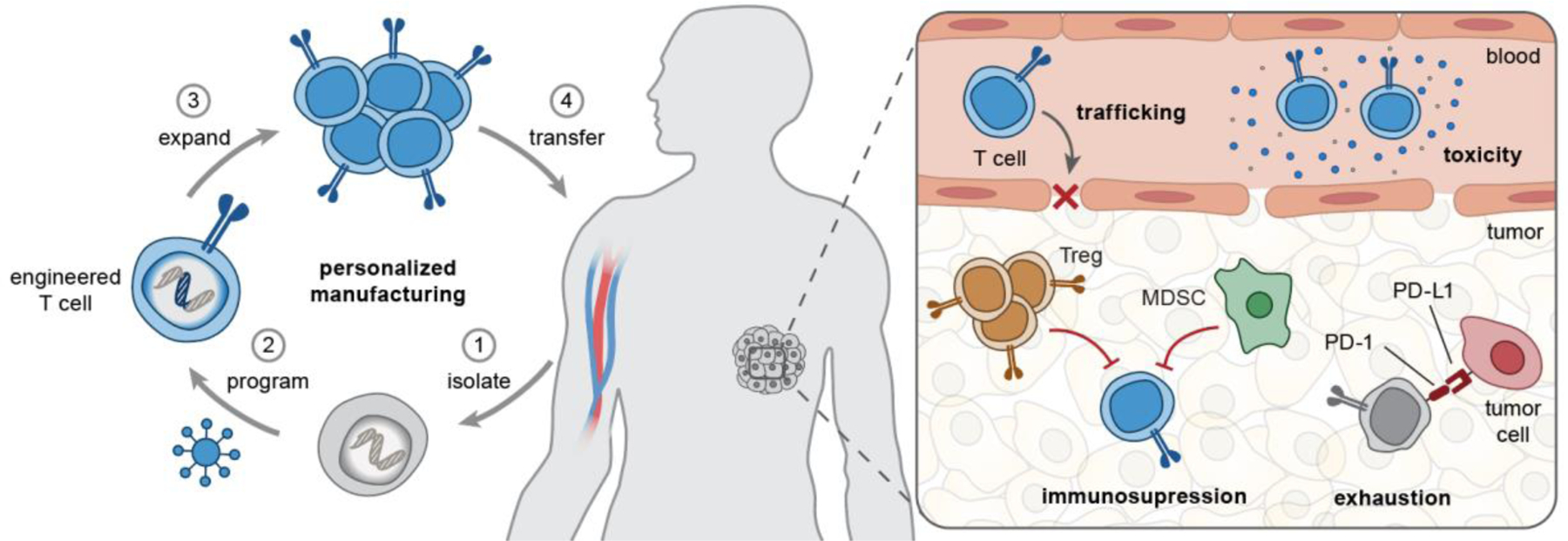
(Left) T cell manufacturing is a personalized, multi-step process that includes isolation of patient T cells, genetic programming using viral vectors, and ex vivo T cell expansion before autologous infusion. (Right) In vivo, engineered T cells need to overcome several challenges associated with T cell trafficking, tumor immunosuppression (e.g., by Treg, MDSCs), exhaustion by chronic antigen stimulation, and immune-related toxicities. MDSC, myeloid-derived suppressor cells; Treg, regulatory T cells; PD-1/PD-L1, programmed death-1/ligand-1; TME, tumor microenvironment.
The overall objective of this review is to summarize recent advances at the interface of biomaterials and engineered T cells. Given the breadth of ongoing research, we will focus our review on three key research areas: low-cost cell manufacturing, predictive response monitoring, and enhancing in vivo control (Table 1). We will discuss opportunities for biomaterials to support the translation of engineered T cell therapies and provide our perspective on future directions of this burgeoning field.
Table 1.
Current progress of biomaterials and technologies to improve engineered T cell therapies
| Application | Material & Approach | Advantages & Caveats | |
|---|---|---|---|
| Cell manufacturing |
Ex vivo nonviral CAR production |
Cationic polymers[17] / Lipid nanoparticles[18] | (+) Easier to manufacture than virus (+) Higher cell visibility than electroporation (–) Limited transfection efficiency |
| In situ CAR production | PBAE polymer nanoparticles loaded with CAR transposon[19, 20] | (+) Lower time and cost than ex vivo production (–) Off-target CAR delivery |
|
| Nonviral transgene insertion |
Transposon system[21–24] | (+) Extended transgene expression (–) Semi-random gene insertion |
|
| CRISPR-Cas9[25] | (+) Extended transgene expression (+) Site-specific gene knock-in (–) Potential immunogenicity |
||
| Predictive monitoring | Multiplexed phenotyping | Combinatorial staining[26, 27] | (+) Expand the multiplexing capacity (–) Complex staining and analysis |
| Mass barcoding (CYTOF)[28–33] | (+) Low background (+) Minimal overlap between mass labels (–) Lower sensitivity than bright fluorophores (–) Samples cannot be recovered |
||
| DNA-barcoded mAb, pMHC[34, 35] | (+) High sensitivity (+) Absolute quantification (+) Theoretically limitless multiplexing capacity (–) Complicated barcode sequence design |
||
| High throughput serial analysis |
Micro-engraved arrays[36–39] | (+) Analyze ~ 104 T cells simultaneously (–) Difficult to analyze cell-cell interaction |
|
| Single cell barcoding chip[40–45] | (+) Spatial encoding increases multiplexing (+) Valves for fluidic control (+) Capable of analyzing intracellular proteins (–) Difficult to analyze cell-cell interaction |
||
| Cell pairing by hydrodynamic traps[46–48] | (+) Precise control of cell-cell interaction (–) Low throughput |
||
| In vivo PET imaging | Radiolabeled mAb[49] | (+) Spatial and temporal analysis (+) Long circulation extends monitoring time (–) Poor tumor penetration (–) Risks of radiation-induced toxicity |
|
| Radiolabeled mAb fragments & peptides[50–53] | (+) Spatial temporal analysis (+) Good tumor penetration (+) Rapid clearance lowers risks of toxicity (–) Require repeated probe injections |
||
| In vivo activity monitoring | Synthetic biomarkers[54–63] | (+) Amplification of detection signals (+) High multiplexing capacity (+) Rapid, cost-effective workflow (–) No spatial resolution |
|
| In vivo control | TME modulation | Viral peptides[64] | (+) Easy to manufacture at GMP facilities (+) Stimulate both innate and adoptive immunity (–) Rely on intra-tumoral injection (–) Require existing antiviral immunity |
| Redirection of antiviral T cells to cancer | Tumor-targeting Ab-peptide conjugates[65, 66] | (+) Deliverable by systemic injections (–) Require existing antiviral immunity |
|
| pMHC-IgG fusion protein[67] | (+) Deliverable by systemic injections (+) No chemical conjugation needed (–) Require existing antiviral immunity |
||
| Targeted modulation | T cell backpack[68, 69] | (+) Selective drug release near T cells or in TME (–) One-time dosing only |
|
| T cell-targeting nanomaterials[70–72] | (+) Allow repeated dosing (+) Broad range of cargo types (–) Off-target delivery |
||
| Remote control | Antibody-based adaptors[73–78] | (+) Modular antigen specificity (–) Lack of spatial resolution |
|
| Microbubbles + ultrasound[79] | (+) Spatial and temporal control (–) Unproven in vivo utility |
||
| Gold nanorods + thermal gene switches[80, 81] | (+) Spatial and temporal control (–) Thermal tolerance |
Abbreviations: PBAE, poly (β-amino ester); CRISPR-Cas9, clustered regularly interspaced short palindromic repeats–CRISPR-associated protein 9; TME, tumor microenvironment; mAb, monoclonal antibody; pMHC, peptide major histocompatibility complex; IgG, Immunoglobulin G; GMP, good manufacturing practice; CYTOF, cytometry by time-of-flight; PET, positron emission tomography.
2. Genetic programming of T cells by nanomaterials
CAR T-cell therapy has resulted in durable responses in cancer patients; however, the complex and costly manufacturing pipeline remains a major obstacle for implementing CAR T cell therapy as standard of care for cancer treatment[11, 12]. One primary driver is the use of viral vectors to genetically engineer CAR T cells, which remains the gold standard[12]. Batch production of clinical-grade viral vectors is time-consuming (3–4 weeks)[12], which delays treatments for patients, increases the potential for mortality, and prevents rapid iteration to optimize CAR designs. Additionally, maintaining quality and safety of current good manufacturing practice (cGMP) viral vectors involves costly extensive manufacturing and testing. Therefore, cGMP-grade, clinical-scale viral production can account for as high as ∼30% of the total production cost[82]. Apart from manufacturing limitations, safety concerns associated with viral transduction, such as insertional mutagenesis and genotoxicity, have led the FDA to restrict the number of integrated viral vectors to 5 copies per T cell[83, 84]. This limits the multiplicity of infection (MOI) that can be used for transduction, resulting in transfection efficiencies as low as 5–10%[85, 86]. Moreover, regulatory agencies recommend monitoring patients for up to 15 years post-treatment for the absence of replication-competent virus in patients, which adds additional burden and cost[12]. These challenges are spurring on the development of nonviral technologies to enable rapid and cost-efficient production of CAR T cells to broaden patient access.
Nonviral gene delivery – such as electroporation, mechanical disruption, and chemical transfection – has been a recent focus due to the potential to reduce costs, shorten manufacturing time, and improve safety profiles compared to viral vectors. Electroporation has been explored as a nonviral alternative for CAR T cell manufacturing, but it leads to lower cell viability and gene transfer efficiency than viral vectors and allows nonspecific transport of molecules into and out of cells[87, 88]. Mechanical disruption, such as by squeezing cells through microfluidic channels to create transient pores in the membrane, has been reported to effectively deliver nucleic acids to the cytosol of T cells[89]. This method requires integrated microfluidic devices to apply both mechanical forces and electrical fields to disrupt cell and nuclear membranes for DNA transfection[90]. Chemical transfection with agents such as lipofectamine is easy-to-use, cost-effective, and can result in lower cytotoxicity compared to electroporation. However, its use for CAR transgene delivery has so far been limited by low gene transfection efficiency in T cells.
2.1. Nonviral gene modification ex vivo
Polymer- and lipid-based biomaterials are emerging as promising agents for T cell transfection (Figure 2a)[17–19, 91]. Constituent lipids and polymers that are positively charged condense negatively charged DNA and mRNA into nanocomplexes by ionic interactions. These nanocomplexes typically carry a net positive surface charge that facilitates cellular uptake through ionic interactions with negatively charged surface proteoglycans[92]. Once nanocomplexes are inside cells, the constituent lipids or polymers are designed to induce translocation of the transgenes from endosomes to the cytosol through fusion with the endosomal membrane or osmotic disruption[93]. Olden and colleagues screened a panel of cationic polymers with a variety of structures (e.g., linear, linear-branched, cyclic-branched) for plasmid DNA delivery to immortalized Jurkat T cells. After optimizing transfection conditions in Jurkat cells, a linear branched polymer transfected primary human T cells with 20% transfection efficiency for mRNA and 10% for plasmid DNA while maintaining cell viability above 75%[17].
Figure 2. Nanomaterial design for T cell manufacturing.
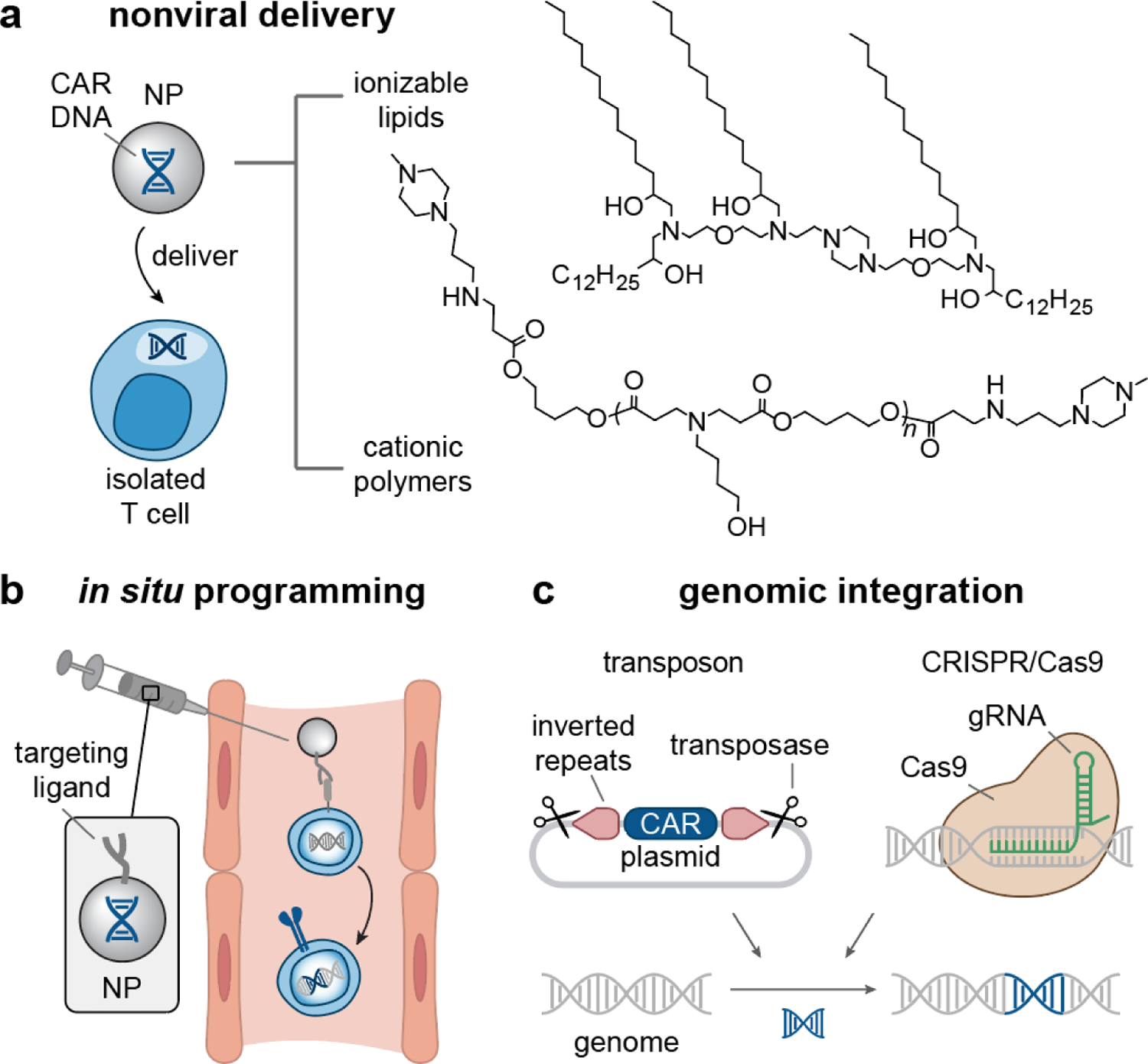
(a) (Left) Lipid/polymer-based Nanoparticles (NPs) can be used as nonviral vectors to deliver CAR-encoded DNA to isolated T cells ex vivo[17–20, 91]. (Right) Examples of lipids and polymers that have shown to successfully transfect T cells with CAR transgene[18–20]. (b) Systemically administered NPs carrying CAR DNA and displaying T cell targeting ligands can reprogram endogenous T cells for CAR expression in situ[19]. (c) Transposon[21–23] and CRISPR/Cas9[25, 95] systems present as nonviral approaches that can integrate CAR transgene into the T cell genome. NP, nanoparticles; CAR, chimeric antigen receptor; CRISPR/Cas9, clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9.
Apart from polymer-based delivery systems, Billingsley et al. formulated a library of ionizable lipid nanoparticles (LNPs) for CAR mRNA delivery to primary T cells[18]. Out of the 24 formulations tested, the top LNP transfected primary T cells with CAR mRNA with efficiency similar to electroporation. Of note, LNP-based transfection of CAR T cells resulted in higher viability than electroporation (>75% vs. 30%) with comparable T cell cytotoxicity. While this LNP system achieved comparable mRNA transfection in primary T cells as the linear polymer system reported by Olden et al.[17], LNPs are more clinically advanced as the first siRNA drug (Alnylam’s Onpattro®, 2018) approved by the FDA is based on LNPs. Moreover, LNPs are also used in the first two FDA-approved, mRNA-based vaccines for COVID-19 (BioNtech/Pfizer, Moderna), further highlighting the translational feasibility of LNP-based transfection systems[94].
Several recent studies have shed light on the properties of T cells that contribute to the limited transfection efficacy of nanoparticle-based transfection agents[96, 97]. Many types of cells express membrane-bound heparan sulfate proteoglycans (HSPG), which are negatively charged molecules that electrostatically bind to positively charged gene carriers to facilitate subsequent cellular uptake[97]. T cells express HSPG at low levels[98] and therefore, are poor at uptake of positively charged carriers. The reduced uptake efficiency by human T cells was recently reported using 2-dimethylaminoethyl methacrylate (DMAEMA) polymers as a nonviral transfection agent compared to HeLa cells [96]. One approach to improve uptake is through ligand-receptor interactions that actively trigger receptor-mediated endocytosis. Indeed, nanoparticles decorated with T cell targeting ligands (e.g., CD3 or CD8 antibodies) have shown greater uptake and transfection efficiency than non-targeting counterparts[91, 99, 100]. Another T cell property hindering efficient transfection is that the acidification gradient in the endosomal pathway is delayed in primary T cells[96]. Using dextran polymers labeled with pH-sensitive fluorophores, one study showed that the endo-lysosomal pH of primary T cells was higher than HeLa cells throughout a 4-hour incubation[96]. The lowest pH was observed at the 4-hour timepoint, with pH 6 for primary T cells and pH 5 for HeLa cells. Delayed endosomal acidification hinder pH-responsive carriers that are formulated to trigger endosomal escape of transgenes in response to the acidic endo-lysosomal condition. Based on these studies, reduced uptake by HSPG and delayed endosomal acidification should be considered in future designs of nonviral vectors for CAR-T cell manufacturing.
2.2. Redirecting T cells in situ
The ability to genetically engineer T cells inside the body has the potential to lower costs and accelerate turnaround times by circumventing the need for a multistep ex vivo manufacturing pipeline (Figure 1 left). In situ CAR production requires delivery of CAR transgenes to T cells in circulation. Early studies focused on the use of viral vectors in vivo but resulted in low transfection efficiencies (~7.5% at two weeks post-administration) and did not improve overall survival in xenograft mouse models[101–103]. Importantly, pre-existing or treatment-induced antiviral immunity are barriers to achieving high transduction efficiency as rapid inactivation and clearance of viral vectors limits the number of doses that can be administered to patients[104]. By contrast, synthetic nanoparticles can be formulated with reagents that are less immunogenic[105] and cheaper to manufacture by cGMP-grade production than viruses. Stephan and colleagues demonstrated synthetic nanoparticles for in situ CAR manufacturing in preclinical studies using polymeric nanoparticles that encapsulated a CAR transgene in the form of plasmid DNA or mRNA[19, 20] (Figure 3). They formulated these nanoparticles using poly (glutamic acid) (PGA) and poly (β-amino ester) (PBAE). PGA was coupled to anti-CD3e f(ab’)2 fragments to achieve T cell targeting and enhance uptake of nanoparticles (Figure 2a and b). To enhance the translocation of CAR-encoded DNA to the nucleus for CAR expression, PBAE polymers were conjugated to synthetic peptides containing microtubule-associated sequences and nuclear localization signals to direct the CAR transgene from the cytosol to the nucleus for CAR expression[19]. In addition, the inclusion of transposons flanking the CAR transgene and a separate plasmid encoding a hyperactive transposase enabled the efficient integration of the CAR vector into chromosomes for persistent CAR expression. In leukemia-bearing mice, five sequential nanoparticle doses resulted in 5.8% of peripheral CD3+ T cells expressing anti-CD19 CAR two days after the last injection. This relatively low transfection efficiency still resulted in tumor regression in a mouse model of leukemia comparably to adoptively transferred CAR T cells that were virally transduced (70% vs. 80% survival respectively). Building on this success, this group further applied the PBAE polymeric nanoparticle to program circulating T cells with CAR/TCR-encoded mRNA[20]. Encoding CAR/TCR transgenes in mRNA offers higher transfection rates and faster CAR expression than plasmid DNA, as mRNA molecules are translated into target proteins in the cytosol without the need to enter the nucleus. The CAR/TCR mRNA loaded PBAE nanoparticles induced potent disease regression, comparable to ex vivo engineered T cells, in murine models of prostate cancer, leukemia, and hepatitis B-induced hepatocellular carcinoma. Collectively, these studies demonstrate the promising potential of using synthetic nanoparticles for in situ production of therapeutic T cells.
Figure 3. In situ T cell programming with engineered, disease-specific CARs or TCRs using polymeric nanocarriers.
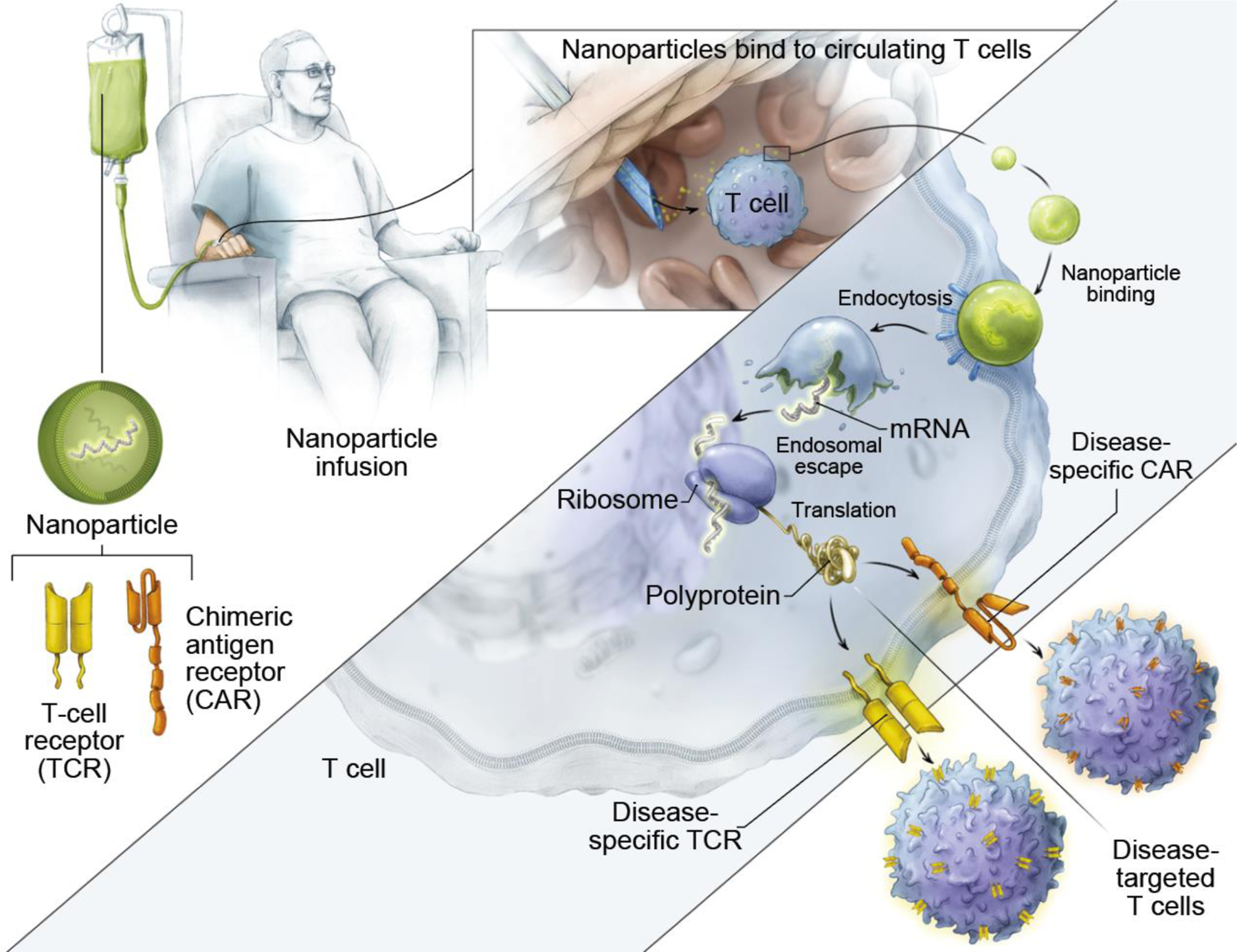
Nanocarriers encapsulated with TCR/CAR-encoded mRNA are modified with antibodies (e.g., anti-CD3 and CD8) that target the nanocarriers to circulating T cells in patients. Upon binding to T cells, the nanocarriers are taken up by T cells through endocytosis. The constituent polymer facilitates endosomal escape of the CAR-encoded mRNA for surface CAR expression through the translational machinery (i.e., ribosomes). Reproduced under the terms of the CCA 4.0 International License.[20] Copyright 2020, Springer Nature.
To achieve durable anti-cancer efficacy of CAR T cells, development of nonviral gene delivery requires a mechanism for genomic integration of CAR transgenes (Figure 2c), as nanoparticles and electroporation lack viral machinery to insert transgenes into host DNA. To address this, strategies include the use of transposons, which have been reported for nonviral CAR production in preclinical and clinical studies[21–23]. Transposon systems comprise two essential components: the transposase enzyme and the transposon DNA that contains the transgene flanked by specific DNA end sequences[106]. The transposase enzyme performs excision at the DNA end sequences and inserts the transgene into the genome of target cells for stable gene expression[106]. Both the transposase enzyme and the transposon DNA have been encoded and delivered as conventional plasmid DNA[21–23]. Transposon systems can also be expressed by DNA minicircles[24, 107], a minimal expression cassette devoid of a bacterial plasmid DNA backbone[108]. Of note, minicircle plasmids have superior transfection efficiency than conventional plasmid DNA – minicircle plasmids delivered by electroporation resulted in 64.3% CAR transgene expression 14 days after transfection, in contrast to 10.6% expression with conventional plasmids[24]. This superior transgene expression afforded by minicircles has been attributed to their reduced size (4k base pairs for minicircles vs. 6k base pairs for plasmids)[24, 109], which could be beneficial to improve the limited transfection efficiency seen in many nanoparticle-based transfection agents for CAR T cell production.
In addition to transposon systems, CRISPR-Cas9 has received increasing attention in T cell engineering for site-specific integration, which avoids potential mutagenesis and malignant transformation resulting from random gene integration by virus or semi-random integration by some transposon systems[25, 110]. CRISPR-Cas9 gene knock-in induces DNA double-strand breaks and homology-directed repair with a donor DNA template containing the desired insert sequence. This approach has been used to program primary T cells with engineered T cell receptors (TCR) in the endogenous TCR locus by delivering CRISPR-Cas9 and CAR/TCR transgenes through electroporation or adeno-associated virus (AAV) vectors[25, 95]. A recent study showed that TCR-engineered T cells produced by CRISPR-Cas9 knock-in specifically recognized tumor antigens and mounted productive anti-tumor T cell responses in vitro and in vivo[95]. Notably, care must be taken when using CRISPR-Cas9 systems for clinical translation due to their potential immunogenicity[111]. To date, studies using CRISPR-Cas9 for CAR/TCR T cell production largely rely on electroporation or AAV vectors, which could potentially be replaced in the future by synthetic nanoparticles[112, 113] for in situ CAR production.
Altogether, nonviral transfection approaches that combine the advantages of nonviral delivery vectors and gene-integrating systems are promising for engineering therapeutic cells ex vivo and for manufacturing of CAR T cells in situ. Nonviral vectors have the potential to reduce manufacturing costs and lead times of engineered T cell therapies and potentially improve safety of CAR T cell therapies. The potential of synthetic nanoparticles to achieve in situ delivery of CAR transgene to T cells without induction of anti-viral immunity could lessen the challenging logistics associated with patient-specific cell manufacturing, thereby lowering costs and improving accessibility. Specific delivery of CAR to T cells is important for in situ CAR production to be realized in the clinic, as off-target delivery of CAR to cancer cells has led to treatment failure and patient relapse[114]. Consequently, restricting CAR expression to T cells is among the forefront goals to ensure clinical potency. This could potentially be achieved by using nanocarriers with preferential delivery at both organ[115] and cell[116] levels or by implementing cell-specific promoters to the transgene design.
3. Identifying predictive biomarkers of T cell response
Despite the clinical success of engineered T cell therapies in hematological malignancies, there remains a need for technologies to assess treatment responses due to modest response rates in solid tumors and development of resistance[13, 14]. Radiographic evaluation of tumor morphology is the standard assessment method for cancer immunotherapy, yet atypical kinetics and patterns of immune-related response can pose a challenge to clinical interpretation[117]. Therefore, there is a significant interest in the development of biomaterials-based diagnostic technologies to identify noninvasive and predictive biomarkers of T cell response including early on-treatment phenotype and functionality of patient T cells [117–119] (Figure 4). In this setting, progress in multiplexed cytometry such as the development of new labeling reagents and detection methods are enabling comprehensive characterization of peripheral T cell populations associated with patient response. Additionally, microfluidic immunoassays implement advances in microfabrication and surface-functionalized materials to serially assess tumor reactivity of patient T cells. Furthermore, image-based and synthetic biomarkers integrate emergent bioconjugation strategies with engineered nanomaterials to provide the ability to monitor key features of the antitumor response such as T cell infiltration and cytotoxicity in vivo. These emerging biomarkers of T cell response can play an important role in predicting tumor responses to improve clinical decision-making during engineered T cell therapy.
Figure 4. Engineering biomarkers of T cell response.
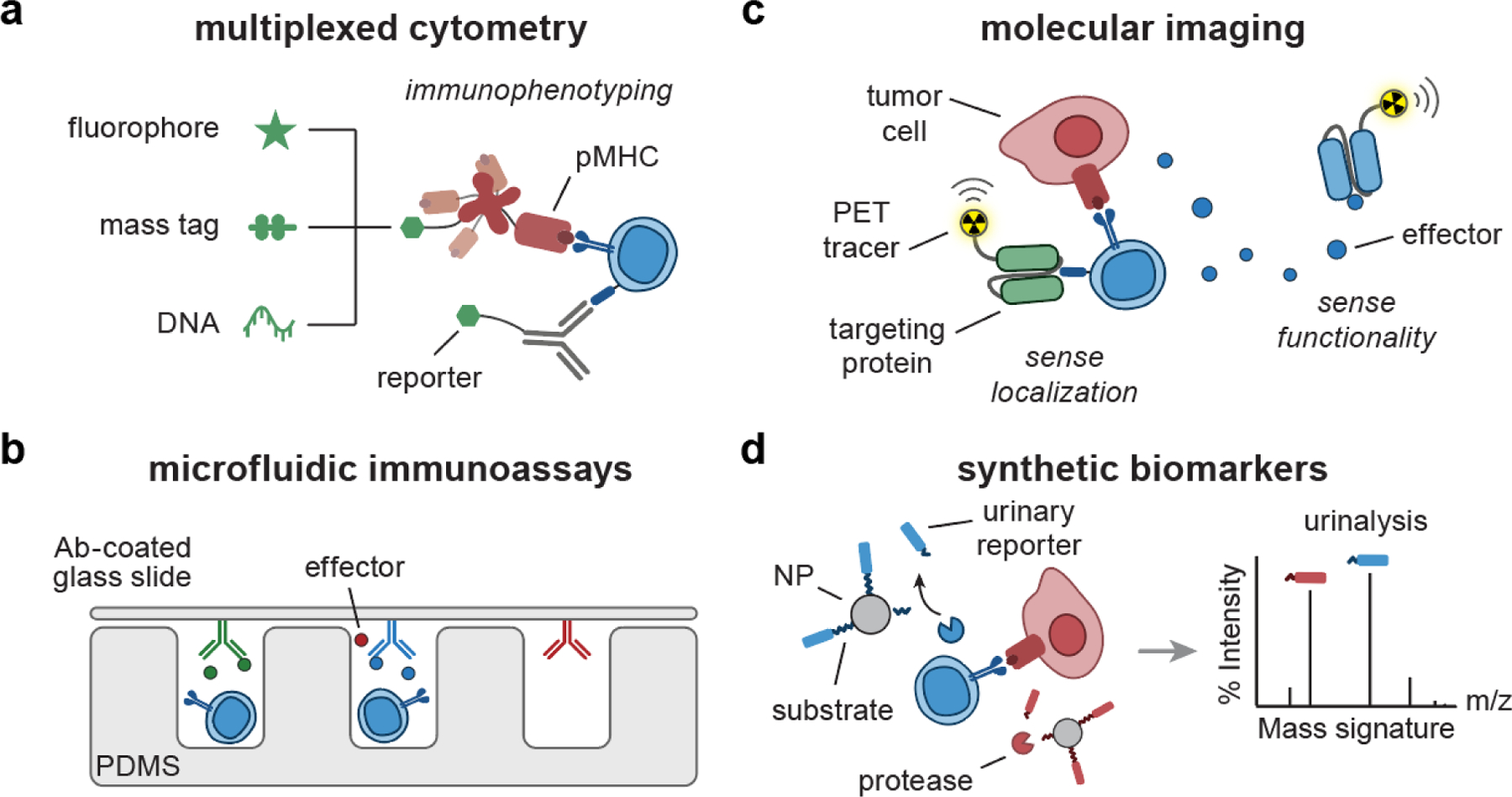
(a) pMHC multimer or Ab can be labeled with combinations of fluorophores[26, 27], mass-encoded peptides[32, 33], and DNA barcodes[120–122] for multiplexed T cell phenotyping. (b) Microfluidic immunoassays[36, 44] are comprised of micro-sized channels and structures fabricated with PDMS to support analysis of secretory effector molecules using a cover glass slide coated with detection Ab. (c) Targeting proteins (e.g., Ab, pMHC) can be conjugated with radionuclides, facilitating PET imaging of T cell localization[51, 123] and functionality[124–126]. (d) Synthetic biomarkers consist of peptide-based protease substrates coupled to NP scaffolds. Upon sensing proteases, the substrates are cleaved, releasing mass-encoded reporters into urine for multiplexed analysis by LC/MS-MS[55, 61, 62]. Ab, antibody; pMHC, peptide-major histocompatibility complex; PDMS, polydimethylsiloxane; PET, positron emission tomography; NP, nanoparticle; LC/MS-MS, liquid chromatography and tandem mass spectrometry.
3.1. Multiplexed cytometry for cell phenotyping
Multi-parameter analysis by flow cytometry remains the gold standard for analysis of single cells[127]. This technique relies on labeling extracellular or intracellular markers with fluorophore-conjugated antibodies or other affinity agents to allow analysis or isolation of target cell populations by fluorescence. Dozens of surface markers are differentially expressed across naïve, memory, and effector T cells, and cells sharing the same lineage can be polyfunctional (e.g., tumor reactive CD8+ or CD4+ T cells expressing different cytokine profiles)[128, 129]. Additionally, the TCR repertoire is incredibly diverse (105–108 unique clones), and in adoptive cell therapy using tumor infiltrating lymphocytes (TILs), hundreds of neoantigen-specific T cell clones contribute to the anti-tumor T cell response[130, 131]. Therefore, the ability to perform densely multiplexed cytometry is important to understand the phenotype and functionality of manufactured T cells during the course of adoptive therapy. As the number or parameters that flow cytometry can analyze simultaneously is limited by the number of fluorophores with nonoverlapping emission spectra[132], this has motivated the development of new labeling agents and methods for multiplexed cytometry (Figure 4a).
Combinatorial staining by labeling a single cell with a unique combination of fluorophores has been developed to expand the multiplexing capacity of fluorescence-based cytometry beyond the limited number of spectrally distinct fluorophores. This strategy is often employed for labelling T cells with peptide-major histocompatibility complex (pMHC), whose multivalent variants (e.g., pMHC tetramer, octamer) have increased avidity for cognate TCRs and are the gold standard reagent for detection and isolation of antigen-specific T cells[133]. A combinatorial encoding strategy using 8 fluorophores to label pMHC multimers enabled parallel detection of up to 25 antigen-specific T cell populations from patient blood samples[26, 27]. To further increase multiplexing density, mass cytometry uses rare earth (lanthanide) heavy metal isotopes as labeling agents whose atomic masses are discriminated by time-of-flight mass spectrometry[134]. Compared to fluorescence-based cytometry, cytometry by time-of-flight (CyTOF) has lower background due to the absence of lanthanides in biological samples and limited overlapping signals between isotopic labels, allowing simultaneous detection of up to 60 cellular markers[135]. CyTOF has enabled comprehensive investigation of complex aspects of the anti-tumor T cell response, including T cell exhaustion[28, 29], characterization of neoantigen-specific TILs[30, 31], and tracking the evolution of T cell subsets during engineered T cell therapy[32, 33].
The large clonal diversity of tumor antigen-specific T cells has motivated the development of DNA-barcoded methods to track T cell responses[120, 121, 136–138]. DNA barcoding significantly expands the degree of multiplexed analysis relative to fluorescence and mass cytometry since the number of unique DNA barcodes scales exponentially with the number of bases. Dahotre and colleagues reported a programmable cytometry platform, called DNA-gated sorting (DGS), for cell detection and isolation[120] (Figure 5). This approach relied on the use of orthogonal DNA gates that function analogously to fluorescence gates; whereas in FACS, cells are sorted based a threshold fluorescent intensity (i.e., gate), in DGS, target cells labeled with DNA gates are sorted by barcode-specific strand displacement reactions. Therefore, DGS allows sequential sorting of target subpopulations by the use of orthogonal DNA strands without fluorescent labels. The team demonstrated this by isolation of different immune subsets from an endogenous antiviral immune response. For massively multiplexed analysis of antigen-specific T cells, Dahotre and team also showed that DNA barcoded pMHC tetramers retain their ability to bind to antigen-specific T cells to allow detection at the single cell level by droplet digital PCR (ddPCR)[138]. For high-purity cell sorting, Kacherovsky et. al. developed a strategy for traceless isolation of CD8+ T cells by first capturing cells with DNA aptamers followed by strand displacement reactions to release bound cells. They showed that CAR T cells generated from aptamer-based isolation retained antitumor activity in vivo[139]. The authors noted that the use of DNA could allow for multiplexed sorting applications upon discovery of aptamers for different cell subsets. For multiplexed analysis of functional neoantigen-specific T cells, Bentzen et. al. generated DNA-barcoded libraries of pMHC multimers and screened > 1,000 antigen specific T cells in a single clinical sample by sequencing DNA barcodes[122]. This approach enabled tracking dynamic changes in melanoma-specific T cells before and after adoptive TIL transfer. Zhang and colleagues further expanded DNA-barcoded pMHC technology with TCR sequencing for high throughput determination of T cell antigen specificities[121]. By binding a library of DNA-barcoded pMHC tetramers to T cells and sequencing barcodes and T cell receptors from single T cells, they identified neoantigen-specific T cell receptors that induce anti-tumor cytotoxicity without cross-reactivity to wild-type antigens. Based on these studies, the ability to use DNA for multiplexed sorting and analysis has potential for identifying functional neoantigen-specific T cells and predicting their antitumor activity.
Figure 5. DNA-gated sorting (DGS) for highly multiplexed detection and isolation of T cells.
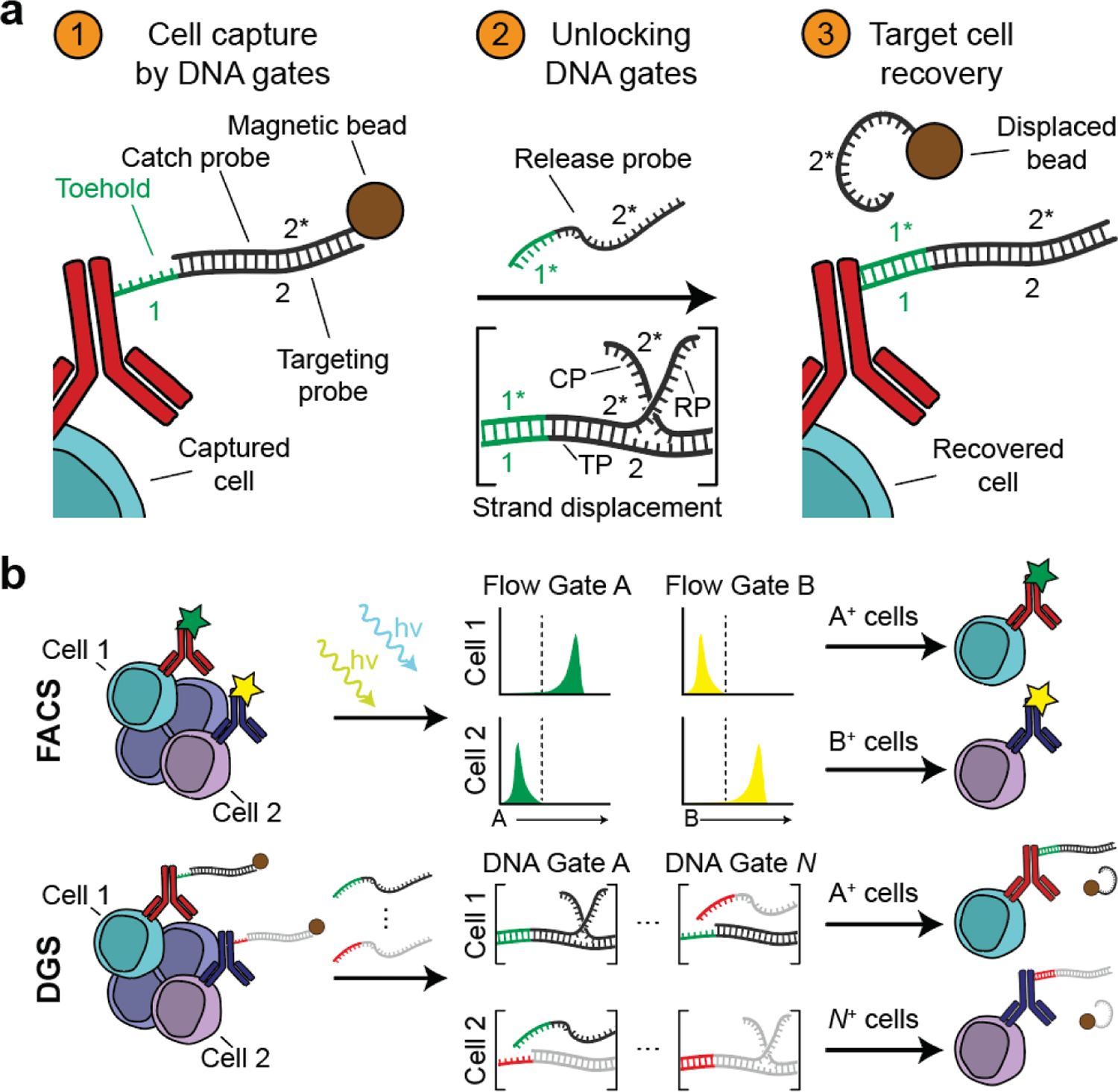
(a) In DGS, T cells are labeled with DNA-barcoded antibodies targeting cell surface markers. The annealing of the DNA barcode, labeled as targeting probe (TP), to a partially complementary catch probe (CP) facilitates magnetic capture of the target cells. Addition of a release probe (RP) fully complementary to the DNA barcode displaces the catch probe via toehold-mediated strand displacement, removing the magnetic label and allowing the labeled cells to be recovered. (b) DGS is analogous to FACS. In FACS, labeled cells are sorted based on fluorescence in a flow gate exceeding a preset threshold. In DGS, labeled cells are sequentially sorted by strand displacement in a given DNA gate. Reproduced with permission.[120] Copyright 2018, National Academy of Sciences. CP, catch probe; TP, targeting probe; RP, release probe; FACS, fluorescence-activated cell sorting.
3.2. Microfluidic immunoassays for serial analysis
Advances in microfabrication and biomaterials are enabling the development of microfluidic immunoassays for high throughput, serial analyses of T cell functionality at single cell resolution (Figure 4b). Kinetic measurements of cytokines and effector molecules secreted during T cell activation have the ability to discriminate functional T cell subsets (e.g., T cells with proliferative and cytotoxic potential)[38, 132]. Microfluidic immunoassays use antibodies for detection of secretory proteins and are composed of micro-sized channels and structures fabricated with a biocompatible material (e.g., polydimethylsiloxane (PDMS)) for single cell culture and analysis[140]. The surface of these microdevices can be functionalized with adhesion molecules (e.g., ICAM-1) or stimulatory ligands (e.g., αCD3, pMHC) to mimic the cellular environment and T cell activation conditions. By handling single cells or groups of cells in microliter- or picoliter-scale systems, microfluidic immunoassays enable analysis of small, valuable clinical samples such as blood and tissue biopsies. Additionally, they offer the throughput necessary for detection and analysis of rare cell populations, such as neoantigen-specific T cells.
Technologies such as microengraved arrays and single-cell barcoding chips (SCBCs) facilitate multiplexed and high throughput analyses of secretory proteins (e.g., cytokines, effector molecules) from activated T cells to evaluate their cytotoxic potential. Microengraved arrays consist of tens of thousands of subnanoliter microwells to isolate single T cells and capture secreted proteins by antibodies conjugated to a glass slide used to cover these wells[36–39] (Figure 6a). The glass slide is then exposed to a series of fluorescently labeled secondary antibodies, and colorimetric discrimination is used to analyze single cell protein secretion. Using this technology, researchers performed longitudinal analyses of proinflammatory cytokines (e.g., IL-2, IFNγ) from arrays containing single T cells to identify antigen-specific CD8+ T cell clones from patient blood samples[36] and to reveal the kinetics of cytokine secretion across states of T cell differentiation[37, 38]. To mimic the surface of antigen presenting cells (APCs), the microwells were coated with supported lipid bilayers, which maintain the stability and fluidity of a cellular membrane. The lipid bilayers were then tethered with recombinant ligands, allowing uniform antigen presentation for assessment of T cell activation by pan (αCD3) or antigen-specific (pMHC) ligands[39].
Figure 6. Microfluidic immunoassays for single-cell analysis of T cell effector functions.
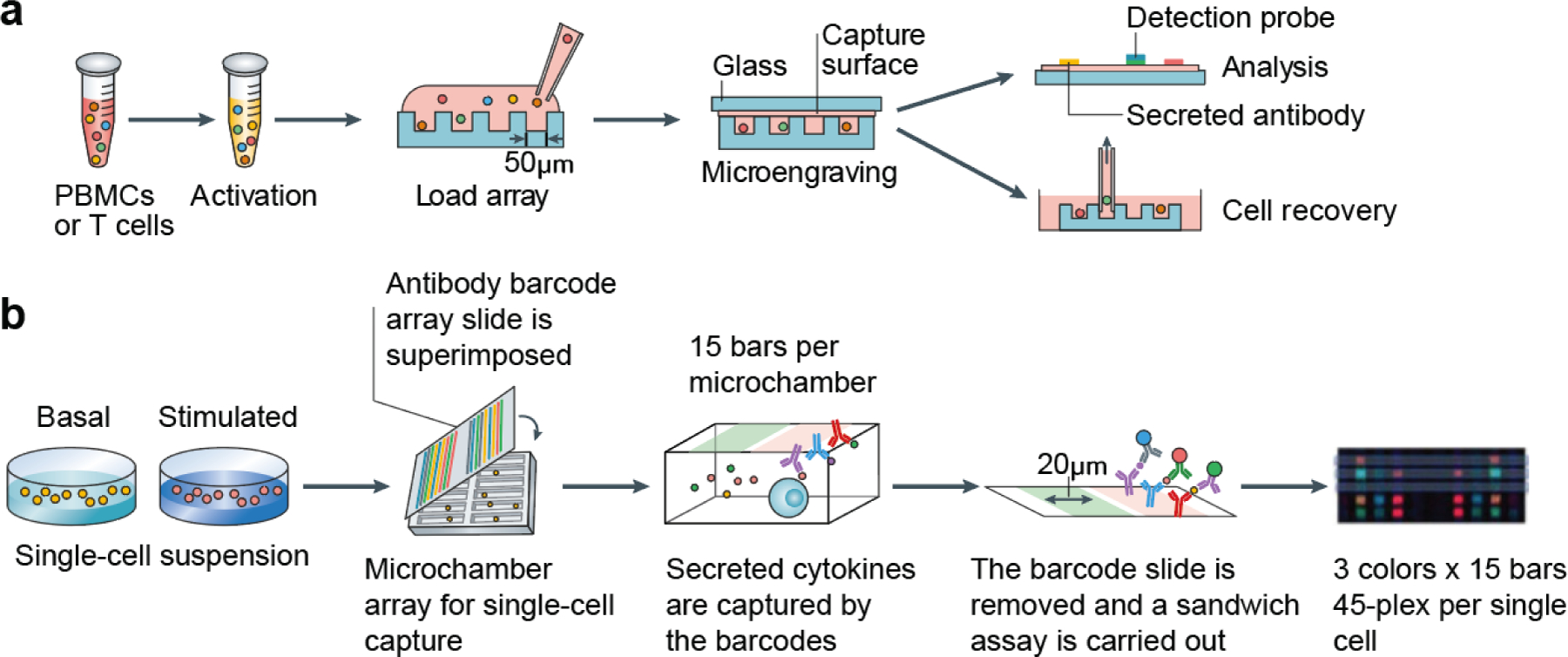
(a) Schematic for microengraved arrays showing the loading of single cells into arrays containing thousands of microwells. Secreted cytokines are measured by capture antibodies tethered to a cover glass slide. (b) Schematic for single cell barcoding chips (SCBCs) showing the use of precisely patterned antibody barcode arrays and microchamber arrays for high throughput and multiplexed analysis of secreted cytokines. Adapted with permission.[141] Copyright 2020, Springer Nature. PBMC, peripheral blood mononuclear cell.
Whereas microengraved arrays achieve multiplexed detection through fluorescent encoding, SCBCs spatially encode capture antibodies in patterns on the cover glass slides, facilitating detection of a greater diversity of secretory proteins[40–45] (Figure 6b). Moreover, the addition of programmable control valves enables isolation of single cells and manipulation of fluidic microenvironments. Fluidic control also allows introduction of a lysis buffer to facilitate measurements of intracellular proteins (e.g., cytokines, transcription factors), which can potentially capture the early kinetics of T cell activation and effector function[42]. SCBCs have been used extensively for multiplexed functional profiling of antigen-specific T cells. Of note, Ma et. al. used this technology to conduct a time-course analysis of T cells from melanoma patients treated by adoptive transfer of T cells specific for the melanoma antigen MART-1. By analyzing 19 proteins secreted from single T cells, they characterized the expansion of effector phenotypes in transferred T cells associated with patient response[44].
In addition to analyzing single T cells in isolation, microfluidic arrays can characterize T cells in engagement with tumor cells or other immune cells. Both microengraved arrays and SCBCs can be used to study intercellular interactions[140], but the generation of cell-cell contacts is governed by stochastic events, resulting in limited control in the number of paired events, cell ratio, and timing of contact formation. By contrast, cell pairing by hydrodynamic traps exploits fluid dynamics and precise microwell patterning to spatially and temporally control cell-cell contacts[46–48]. This technique has been utilized to characterize early activation kinetics and cytokine secretion by T cells and NK cells in respective cocultures with antigen-presenting cells (APCs) or tumor cells[46–48].
By providing spatial and temporal dimensions of the T cell response, microfluidic arrays offer complementary data to those obtained by conventional cellular assays (e.g., ELISA, flow cytometry). Their utility for on-chip characterization of engineered T cells can be further enhanced with biomaterials. For example, metallic nanomaterials display unique properties (e.g., localized surface plasmon resonance (SPR), metal enhanced fluorescence) that can significantly improve the limit of detection for cytokines in antibody-based assays[142]. Additionally, tumor 3D scaffolds embedded in microfluidic chips are emerging to assess the tumoricidal activity of T cells. These models are established by seeding or encapsulating tumor cells in biomaterial-based scaffolds that resemble the extracellular matrix of tumors. Advances in biomaterials have led to the development of natural (e.g., collagen, Matrigel) and synthetic materials (e.g., polyacrylamide, polystyrene) that mimic the biochemical and physical properties of the TME[143]. When coupled with microfluidics, these systems enable high throughput cellular analysis in a controlled fluidic microenvironment[143, 144]. Such systems have been used to investigate the cytotoxic potential of engineered T cells, in addition to various aspects of the TME that could impact T cell activity[145–147]. Overall, the integration of microfluidic technology and biomaterials offers exciting opportunities for sensitive, comprehensive, and high throughput functional assessment of manufactured T cells in engineered T cell therapy.
3.3. Molecular probes for imaging immunity
Radiographic imaging is the standard assessment for tracking patient responses to cancer immunotherapy based on changes in tumor burden. However, distinct immune-related patterns of response – such as pseudoprogression (i.e., transient increase in tumor volume before tumor shrinkage) and variable response kinetics across patients – confound interpretation and have prompted active debate to improve response assessment[117]. This has motivated continual refinement to evaluation criteria – including immune-related response criteria to optimize the timing and frequency of radiographic assessment – and has led to increased interest in the development of molecular imaging probes to monitor T cell responses with modalities like positron emission tomography (PET), molecular magnetic resonance imaging (MRI), and single photon emission computer tomography (SPECT)[119]. ImmunoPET is gaining interest as a molecular imaging strategy for sensitive, quantitative, and noninvasive analysis of T cell responses to immunotherapy[148]. In this strategy, affinity agents (e.g., monoclonal antibodies (mAb), pMHC, binding peptides) are chemically conjugated with a radioactive nuclide to target and label T cell biomarkers for detection by PET imaging (Figure 4c).
Tumor infiltration is a key early step in anti-tumor T cell responses, motivating the development of probes that track T cell trafficking and infiltration. To monitor the infiltration of T cells, Larimer et. al. engineered PET probes targeting the pan-T cell marker CD3 by conjugating anti-CD3 mAbs to 89Zr, a long-lived radionuclide (half-life ~ 78.4h)[49]. PET imaging predicted mice responsive to anti-cytotoxic T lymphocyte antigen 4 (αCTLA-4) checkpoint blockade before detectable differences in tumor burden. To selectively target CD8+ T cells that mediate anti-tumor cytotoxicity, researchers designed CD8-targeted PET probes based on antibody fragments, such as minibodies[123, 149], diabodies[50, 52], single domain antibody (VHH)[51], and single chain variable fragments (scFv)[53]. For instance, Rashidian et. al. developed 89Zr-labeled anti-CD8 VHH probes and showed that homogeneous CD8+ T cell infiltration throughout the tumor is associated with strong responses to immunotherapy across three tumor models[51]. The use of antibody fragments in these probes also improved tumor penetration, increased clearance kinetics, and reduced required radiation doses[50]. To track infiltration of antigen-specific T cells that drive anti-tumor immunity, pMHC multimers are required. However, conventional pMHC multimers use foreign proteins such as streptavidin to form a multivalent construct, which is immunogenic and precludes in vivo use[133]. To address this, Woodham et. al. engineered Fc-based pMHC dimers called SynTacs and site specifically labeled these agents with 64Cu by enzyme-based conjugation chemistry[123]. PET imaging of 64Cu-labeled SynTacs selectively tracked antigen-specific T cell populations in mouse models of cancer and viral infection.
Furthermore, factors like tumor immunosuppression, T cell exhaustion, and resistance can limit T cell activity even after infiltration, and therefore there is growing interest in monitoring downstream effectors of T cell-mediated immunity. As activated T cells secrete molecules like granzyme B (GzmB) and IFNγ during cytotoxic killing, targeting these molecules facilitates evaluation of in vivo T cell activity. To develop PET probes against the cytotoxic protease GzmB, Larimer et. al. radiolabeled a peptide-based inhibitor, which contains a substrate for GzmB and an electrophilic trap that binds irreversibly to the protease, for noninvasive assessment of ICB therapeutic responses. PET imaging with this probe stratified responding and non-responding mice on combination immunotherapy before differences in tumor burden[124, 125]. In addition, Gibson et. al. developed an IFNγ-PET probe to monitor responses to cancer vaccination[126]. In a model of T cell exhaustion, probe uptake did not increase despite T cell infiltration, indicating the ability to distinguish active from immunosuppressed T cell infiltrates. These studies underscore the need for biomarkers of T cell effector functions to assess tumor responses.
3.4. Synthetic biomarkers of immunity
Longitudinal monitoring is necessary to track immune responses over the course of immunotherapy and predict therapeutic efficacy early on-treatment. This has driven work on developing noninvasive biomarkers based on shed content of tumor cells (e.g., circulating tumor DNA (ctDNA)) or immune cells (e.g., circulating cytokines) for the evaluation of anti-tumor T cell responses during treatment with cancer immunotherapy[117]. In contrast to endogenous biomarkers, synthetic biomarkers are an emerging class of activatable biosensors designed to query sites of disease for dysregulated protease activity and release cleaved reporters in urine for noninvasive detection (Figure 7)[61, 63]. Synthetic biomarkers consist of peptide substrates that are labeled with a reporter (e.g., fluorophore, mass barcode, or DNA barcode) and conjugated to a carrier (e.g., nanoparticles, polymers, proteins) (Figure 4d) [61]. The use of the carrier extends the half-life of free peptides that would otherwise be cleared rapidly into urine and facilitates delivery of the peptides to the disease sites[62]. There, dysregulated proteases cleave the peptide substrates, releasing reporters that are filtered by the kidneys into urine for noninvasive detection. Synthetic biomarkers utilize two biological processes to improve detection sensitivity. First, these activity-based sensors rely on enzymatic turnover to generate detection signal. Since one copy of protease can cleave thousands of synthetic substrates, proteases serve as molecular amplifiers for endogenous disease signals (>1,000-fold)[150]. Second, instead of probing endogenous signals that are diluted in blood, synthetic biomarkers generate reporter signals that are concentrated in urine (>50–100-fold relative to blood concentration[151, 152]) to further increase signal-to-noise. This opportunity to challenge current limits of detection has enabled the use of synthetic biomarkers for early disease detection in cancer and other pathological conditions[54–63].
Figure 7. Synthetic biomarkers for noninvasive detection of protease activity.
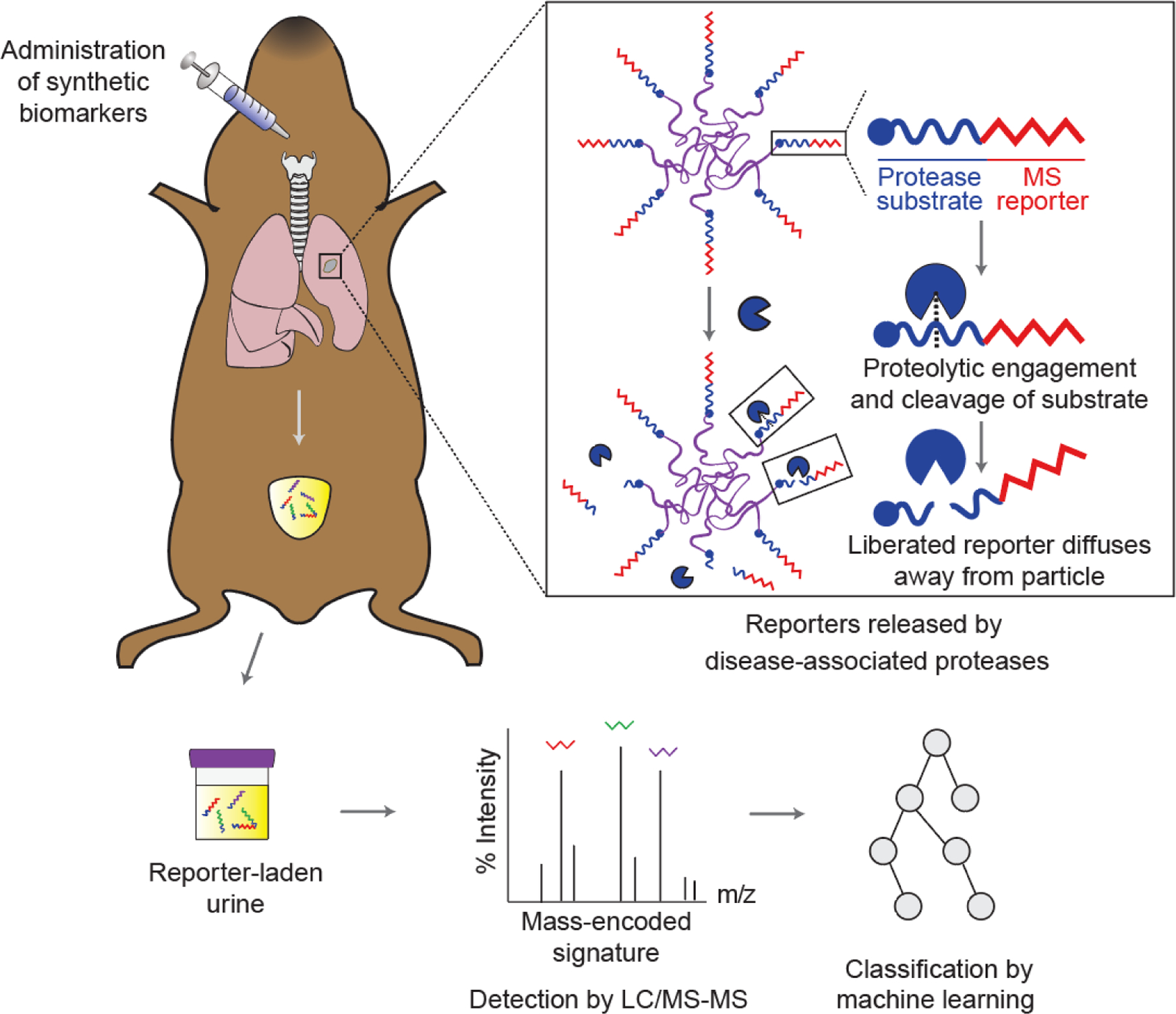
Synthetic biomarkers consist of reporter-labeled peptide substrates conjugated to a nanoparticle or protein carrier. Upon administration, these biomarkers accumulate at the disease sites where dysregulated proteases cleave substrates on the surface of the carriers, releasing the reporters into urine. Multiplexed quantification of mass-barcoded reporters in urine by LC-MS/MS enables diagnostic classification by machine learning. Adapted with permission.[63] Copyright 2020, AAAS. LC-MS/MS, liquid chromatography and tandem mass spectrometry.
During target cell killing, cytotoxic CD8+ T cells release granules containing effector molecules including the protease GzmB to initiate apoptosis in target cells[153]. Therefore, the development of synthetic biomarkers to sense GzmB activity in vivo has the potential for early detection of T cell-mediated conditions such as transplant rejection and tumor responses to cancer immunotherapy[55, 62]. To develop GzmB nanosensors for detection of T cell activity at the onset of acute cellular rejection, Mac et. al. decorated iron oxide nanoparticles with peptide substrates specific for GzmB[62]. In mice bearing allogeneic skin grafts, nanosensors passively accumulated in inflamed grafts and sensed GzmB activity during alloreactive T cell killing of donor cells. This led to production of reporter signals in urine several days before observable graft failure, allowing noninvasive and early detection of acute cellular rejection with high accuracy (area under receiver-operating curve (AUROC) = 0.98). Moreover, antibody-mediated depletion of CD8+ T cells diminished this increase in urinary reporter signals, indicating that GzmB+ CD8+ T cells at the onset of rejection are responsible for nanosensor activation. Synthetic biomarkers monitoring GzmB activity have also been extended for early on-treatment assessment of T cell responses to ICB therapy. In this approach, GzmB peptide substrates were directly coupled to therapeutic checkpoint antibodies (e.g., αPD1, αCTLA4) to harness the biological functions of therapeutic antibodies while sensing anti-tumor T cell activity at the same time points of ICB treatment[55]. In two syngeneic tumor models, ICB antibody-GzmB sensor conjugates produced elevated reporter signals in urine of mice responding to therapy, allowing noninvasive detection of therapeutic responses as early as the second dose of treatment (AUROC = 0.92–1.00) and before changes in tumor burden were detected. The increases in reporter signal correlated with observed increases in GzmB+ CD8+ TILs by flow cytometry, suggesting that GzmB sensor conjugates could detect anti-tumor T cell activity at the onset of therapy response through production of urinary reporters.
In addition to lowering the limits of detection, synthetic biomarkers offer the potential to improve detection specificity through multiplexed activity measurement. Multiplexed detection can be achieved by employing a library of mass- or DNA-barcoded biomarkers to monitor multiple proteases simultaneously[54, 58, 154]. Additionally, recent advances in machine learning enable the training of diagnostic classifiers that accurately differentiate experimental groups based on multiplexed urinary outputs. Mac et. al. extended ICB response assessment by using a mass-barcoded library of synthetic biomarkers to monitor both immune- and tumor-associated proteases for classification of refractory tumors based on resistance mechanisms[55]. In mouse models of resistance, gene knockout of B2m or Jak1 allowed for evasion of CD8+ T cell-mediated tumor control, leading to resistance to checkpoint inhibitors. Administration of a multiplexed library of synthetic biomarkers enabled development of machine learning classifiers based on urinary signatures that stratified B2m from Jak1 resistance with high accuracy (AUROC ≥ 0.9). This finding was consistent with observed differences in proteases expression in these resistance models and highlighted the potential of synthetic biomarkers to identify T cell- and tumor-intrinsic mechanisms leading to ineffective anti-tumor responses.
Synthetic biomarkers have shown promise as noninvasive biomarkers of T cell activity, allowing for early and accurate detection of several T-cell mediated conditions. Strategies to localize protease activation of synthetic biomarkers to the tumor sites can further improve assessment of anti-tumor T cell activity. To increase tumor-targeting, these biomarkers have been conjugated with tumor-penetrating peptides or targeting ligands to enhance on-target signals from CD8+ TILs[54–56]. Alternatively, biomarkers masked with photolabile protecting groups have been unmasked in the TME using a light trigger, facilitating selective activation by local proteases[57]. Furthermore, protease activity has been integrated in synthetic logic circuits, which could further enhance detection specificity of synthetic biomarkers[155–158]. Overall, the results discussed in this section have demonstrated the potential of synthetic biomarkers for assessing anti-tumor T cell responses during engineered T cell therapy.
4. Augmenting T cell responses by biomaterials
Biomaterials-based strategies have the potential to improve treatment outcomes by addressing important roadblocks such as inefficient T cell infiltration into tumors, limited T cell persistence/expansion in the TME, and severe systemic toxicity due to hyperactive T cells[13, 159–166]. Biomaterials are well positioned to address these challenges and others, as they can be programmed to respond to environmental and user-defined cues for improved control of anti-tumor immunity. For example, biomaterials can be functionalized with affinity agents (e.g., antibodies) or stimuli-responsive moieties (e.g., pH sensitive bonds, protease-cleavable peptides) to selectively interact with the TME or specific cell populations. In this section, we summarize recent advances of biomaterials that enhance engineered T cell therapies by controlling the infiltration and effector functions of anti-tumor T cells in the TME (Figure 8).
Figure 8. Enhancing T cell immunity against cancer.
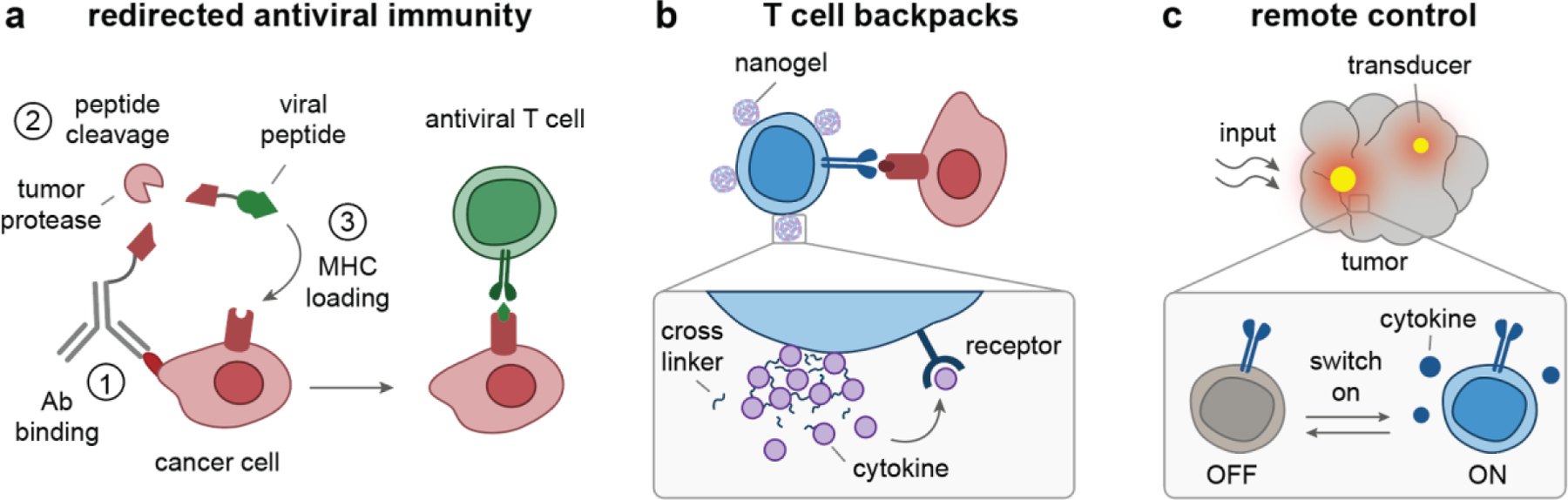
(a) Mechanism of action for antibody-viral epitope conjugates to redirect antiviral immunity against cancer: (1) Antibodies bind to tumor antigens, (2) tumor proteases cleave peptide linkers to release viral peptides, and (3) the released peptides load into empty MHC molecules on cancer cell surfaces for recognition by antiviral T cells[167]. (b) Nanogels can be backpacked on the surfaces of T cells ex vivo, so that T cells migrate through the body carrying their own agonists, which are released in a pseudo-autocrine manner[68, 69]. (c) Nanomaterials can be used as transducers to locally convert external inputs (e.g., light) into signals (e.g., heat) that activate engineered T cells to release effector molecules, such as immunostimulatory cytokines[80, 81]. Ab, antibody; MHC, major histocompatibility complex.
4.1. Redirecting anti-viral immunity against tumor
An emerging strategy to circumvent the scarcity of anti-tumor T cells in the TME is to redirect endogenous virus-specific T cells in the host against cancer cells. Antiviral T cells specific for previously encountered viral infections (e.g., cytomegalovirus [CMV], Epstein-Barr virus [EBV], influenza virus) circulate in the blood and surveil human tissues, including tumors, as “bystander T cells” [64, 168]. T cells against persistent herpesviruses such as CMV are especially widespread in healthy individuals as greater than 60% of the global population has been infected by CMV[169]. Moreover, the frequency of CMV-specific T cells expands with age[170, 171] and can be as high as 85% of total CD8+ T cells[167]. Viral-specific T cells maintain memory phenotypes, which respond quickly to reactivation, are capable of cytotoxicity, and have better persistence and proliferation potential then effector T cells[64, 172, 173]. These features are in stark contrast to tumor-specific T cells, which often have exhausted phenotypes due to chronic receptor activation and the immunosuppressive TME[174].
Although cancer cells are not recognizable by antiviral CD8+ T cells, recent strategies have been reported that recruit antiviral CD8+ T cells to trigger antitumor immunity[64, 66, 167]. For example, intra-tumoral injection of viral peptides turned immunosuppressive solid tumors into immune-activating environments by simulating a local reinfection that broadly activated innate and adaptive immunity[64]. Intra-tumoral injection of viral peptides was found to upregulate MHC I expression on tumor cells, promote the accumulation of CD8+ T cells, natural killer (NK) cells, and DCs within tumors, activate DCs within draining LNs, and upregulate cytotoxic molecules (e.g., GzmB) by CD8+ T cells and NK cells[64]. This peptide therapy potently delayed tumor growth and improved survival rates of B16, MC38, and 4-OHT tumor models in mice pre-infected with a model virus, vesicular stomatitis virus expressing ovalbumin (OVA) antigen.
Extending these viral peptide therapies, alternative administration methods (e.g., systemic injection) have been investigated to circumvent the need for intra-tumoral injection in less accessible tumors and mitigate the fast clearance of peptide epitopes[66, 67, 167]. One approach is to functionalize viral peptides on tumor-targeting antibodies through cleavable linkers that conditionally release peptides in the TME. This approach leverages antibodies to enhance the half-life, biodistribution, and delivery of cargo to the TME[65, 175]. For example, a recent study delivered CMV peptide antigens to tumor cell surfaces by antibody–peptide epitope conjugates (APECs) to reprogram surface antigenicity[167](Figure 8a). In the TME, tumor-expressed proteases (e.g., matrix metalloproteases) cleaved the peptide linkers, liberating viral peptide epitopes from APECs. The epitopes were then loaded onto HLA-I molecules on the tumor cell surfaces, stimulating antiviral T cells. Notably, injection of APECs even without adjuvant improved the survival rate in multiple mouse models, such as human breast cancer (MDA-MB-231) and human liver cancer (SNU-475). A similar antibody-peptide conjugate was published separately by Sefrin et al. to sensitize tumors to attack by virus-specific T cells[66], which further supports the feasibility of this approach. Rather than reprogramming cancer cell surfaces, another approach is to use a fusion protein composed of a tumor-targeting antibody (immunoglobulin G, IgG) and a pMHC targeting CMV-specific T cells, facilitating engagement of antiviral T cells with cancer cells[67]. This pMHC-IgG recombinant protein can redirect bystander T cells without the need for conjugation of peptides to tumor-targeting antibodies.
Collectively, viral peptide-based biomaterials represent promising approaches to redirect antiviral T cells against cancer in the TME. This strategy presents several advantages for T cell-based immunotherapies by stimulating antiviral T cells in tumors. First, local re-stimulation of known antiviral immunity not only recruits endogenous antiviral T cells to fight cancer, but also broadly activates innate and adaptive immunity in the TME. The latter could potentially enhance the antitumor activity of adoptively transferred T cells against immunosuppressive solid tumors. Second, localized activation of virus-specific T cells in the tumor reduces the risk of systemic toxicity associated with BiTEs and cytokine therapies (e.g., IL-2), which would otherwise stimulate a broad diversity of T cells[176–178]. However, cellular presentation of viral peptides is restricted to individual patients’ specific human leukocyte antigen (HLA) alleles, but certain alleles are found at high prevalence (e.g., >30% of the US populations exhibit HLA A2.1 allele[179]). Using peptides specific to these alleles would allow broad application of viral peptide-based approaches. Lastly, redirecting endogenous virus-specific T cells against cancer mitigates the need for adoptive transfer of engineered T cells and could therefore reduce manufacturing costs compared to current T cell therapies.
4.2. Delivering immunomodulators to T cells
Administering stimulatory cytokines (e.g., IL-2, IL-15) or TME-modulating factors (e.g., transforming growth factor beta (TGFβ) inhibitor, adenosine receptor inhibitor) are two approaches for augmenting anti-tumor T cell activity that have been explored in preclinical and clinical studies to increase response rates and extend ACT to solid tumors[180–183]. However, systemic delivery of these immunomodulators can cause dose-limiting toxicity[184–187]; therefore, delivery of immunomodulators to the TME or to sites in proximity to T cells is crucial. To mitigate toxicity associated with systemic administration, nanomaterials have been explored for targeted delivery of a broad range of cargo, including cytokines, small molecules, and nucleic acids, to augment T cell functions. One approach is to leverage T cells themselves as delivery vehicles to infiltrate tumors and delivery cargo. For example, cytokine-loaded nanoparticle backpacks conjugated to the surface of adoptively transferred T cells have been demonstrated to preferentially release cargo in the TME (Figure 8b). This has been shown with cytokines that were crosslinked into nanogels using reduction-sensitive disulfide bonds, which release the immunostimulatory cytokines in response to elevated reducing conditions on the surface of T cells during TCR activation by tumor cells[69]. In preclinical models, T cells carrying cytokine backpacks amplified T cell expansion by 16-fold in tumors compared to systemic cytokine injections while limiting systemic stimulation. This backpacking approach allowed at least eight-fold higher doses of IL-2 and IL-15 to be administered without toxicity, substantially widening the therapeutic window of cytokine treatments to support T cell therapies. Supported by their therapeutic potency against murine cancer models (B16-F10 melanoma, U87-MG glioblastoma), cytokine-backpacked T cells have recently entered clinical trials for a variety of solid tumor types (NCT03815682). This cell-conjugated nanomaterial approach can also be used to tether small molecule-supporting drugs to T cells (e.g., TGFβ inhibitor)[188–190] and is applicable to other immune cells, such as macrophages[191], for cancer immunotherapy.
Drug-loaded nanomaterials that directly target lymphocytes in vivo through chemically conjugated targeting moieties that bind to T cell surface receptors have also been recently reported[70–72]. The vast majority of nanomaterials delivered by IV injection accumulate in the liver and spleen, with less than 5% of injected dose accumulating in tumors[192, 193]. Although this biodistribution profile is known to severely limit tumor targeting of nanomaterials, it favors delivery to T cells as they are present in high number in the circulating blood and spleen[194]. Therefore, in contrast to poor tumor targeting of nanomaterials, functionalizing PLGA nanoparticles with antibodies that bound to CD8+ T cells by surface CD8a markers targeted >80% CD8+ T cells in the blood, spleen, and tumor at 1 hour after injection[70]. In the context of adoptive cell therapies, liposomes with antibodies targeting antigens on transferred T cells reached >95% of transferred T cells and allowed repeated doses of IL-2 to be delivered. The repeated IL-2 doses expanded the transferred T cells three-fold in vivo compared to a control group treated with T cells alone, while avoiding systemic toxicity of IL-2 treatment[71]. Moreover, T cells can be used to carry immunomodulatory molecules to the TME. Huang and colleagues have shown that T cells actively concentrated their payloads in mouse tumors by two orders of magnitudes higher than nanoparticles[195]. Consistent with this result, PLGA nanoparticles functionalized with anti-PD1 antibody targeted endogenous exhausted T cells and delivered TLR7/8 agonist to the TME. This strategy led to pronounced therapeutic activity against solid tumors that was absent from particle formulations lacking the targeting moiety or equivalent doses of the free drug[70].
Collectively, these studies highlight the use of nanomaterials to target, modulate, or enhance engineered T cells. Moving forward, nanomaterials that enable genetic modulation of endogenous or adoptively transferred T cells in situ would offer new opportunities to modulate genes that currently cannot be targeted pharmacologically by small molecules or biologics. For example, a recent study using CRISPR for genome-wide screening discovered that knockout of three gene targets (SOCS1, TCEB2, and RASA2) in human T cells enhanced both proliferation and in vitro anti-cancer function[196]. While promising, those gene targets are currently undruggable by small molecules or biologics. Therefore, nanomaterials that can deliver gene modulators (e.g., siRNA, mRNA, CRISPR-Cas9) to T cells have the potential to augment the anti-cancer efficacy of T cell therapies. These advances would require new formulations of lipid nanoparticles to be developed for delivery of nucleic acids, as has been demonstrated recently for delivery of siRNA to splenic T cells[147].
4.3. Remote control of engineered T cells
The push to reduce toxicity and improve response rates against solid tumors has motivated strategies to increase the precision of engineered T cells. An emerging approach is remote control of T cell responses using externally applied triggers such as small molecules, light, or heat[197]. These strategies rely on the unique properties of biomaterials to amplify or transduce such cues (e.g., the use of plasmonic nanomaterials to convert incident light into heat) and increasingly sophisticated genetic circuitry to allow T cells to sense-and-respond to these inputs. Such remotely controlled systems have the potential to tune the duration and strength of T cell responses, as well as localize signals to tumors or secondary lymphoid tissues such as draining lymph nodes. Here we highlight work that interfaces biomaterials with synthetic biology to achieve remote control of engineered T cells.
Recombinant proteins have been designed as pharmacodynamic inducers to allow remote- and user-control of CAR T cell activity toward specific tumor antigens[73–78, 198–200]. A shared feature of these designs is that the intracellular signaling components are separated from the extracellular antigen binding domain. The intracellular components and extracellular domain can only assemble into a functional CAR complex in the presence of the pharmacodynamic inducer[198]. Therefore, these CAR constructs remain inactive until they sense both the inducer and the target tumor antigen. Furthermore, T cell activity can be tuned and reversibly controlled by titrating the dose of pharmacodynamic inducers. Pharmacodynamic inducers have been developed with small molecules and antibody-based adaptors. For example, rapamycin and its derivatives, which are FDA-approved small-molecule drugs, have been used to control T cells with ON-switch CARs[199, 200]. These CARs consist of an intracellular signaling component and an extracellular binding region, each as a separate polypeptide, and a rapamycin-inducible heterodimerization domain[199, 200]. Administration of rapamycin turns on CAR T cell activity against cancer cells. This approach resulted in a significant reduction of tumor burden in a humanized mouse model of leukemia[201].
Another approach is to use antibody-based adaptors, which can control both T cell activity and antigen specificity[76, 78, 202–204]. These antibody-based adaptors comprise a tumor-targeting antibody (e.g., IgG, Fab, scFv) and a second moiety (e.g., exogenous peptides, FITC, biotin) that selectively binds the CAR molecules[76, 78, 202–204]. CAR T cell activity is thus strictly dependent on the formation of the ternary complex between the CAR-T cell, adaptor, and tumor antigen. This approach not only affords controllable T cell activity but also enables engineered T cells to target a variety of tumor antigens by changing the antibody specificity of the adaptors[198]. One example is a split, universal, and programmable (SUPRA) CAR system that was composed of various pairs of universal orthogonal CARs expressed by engineered T cells and corresponding tumor-specific (e.g., HER2, Mes, Axl) scFv adaptors that engaged the CARs through leucine zipper interactions (Figure 9a and b)[78]. Notably, the addition of a competitive adaptor that blocked activation of CAR T cells reduced the cytokine production by the SUPRA CAR T cells in vivo, indicating the potential of the SUPRA system to mitigate CRS. Another approach is to use CD19-antibody fusion proteins as pharmacodynamic adaptors to redirect FDA-approved anti-CD19 CAR T cells against other tumor antigens[205]. This approach could leverage FDA-approved CAR T cell therapies to accelerate clinical translation.
Figure 9. Remote control of engineered T cells through biomaterials.
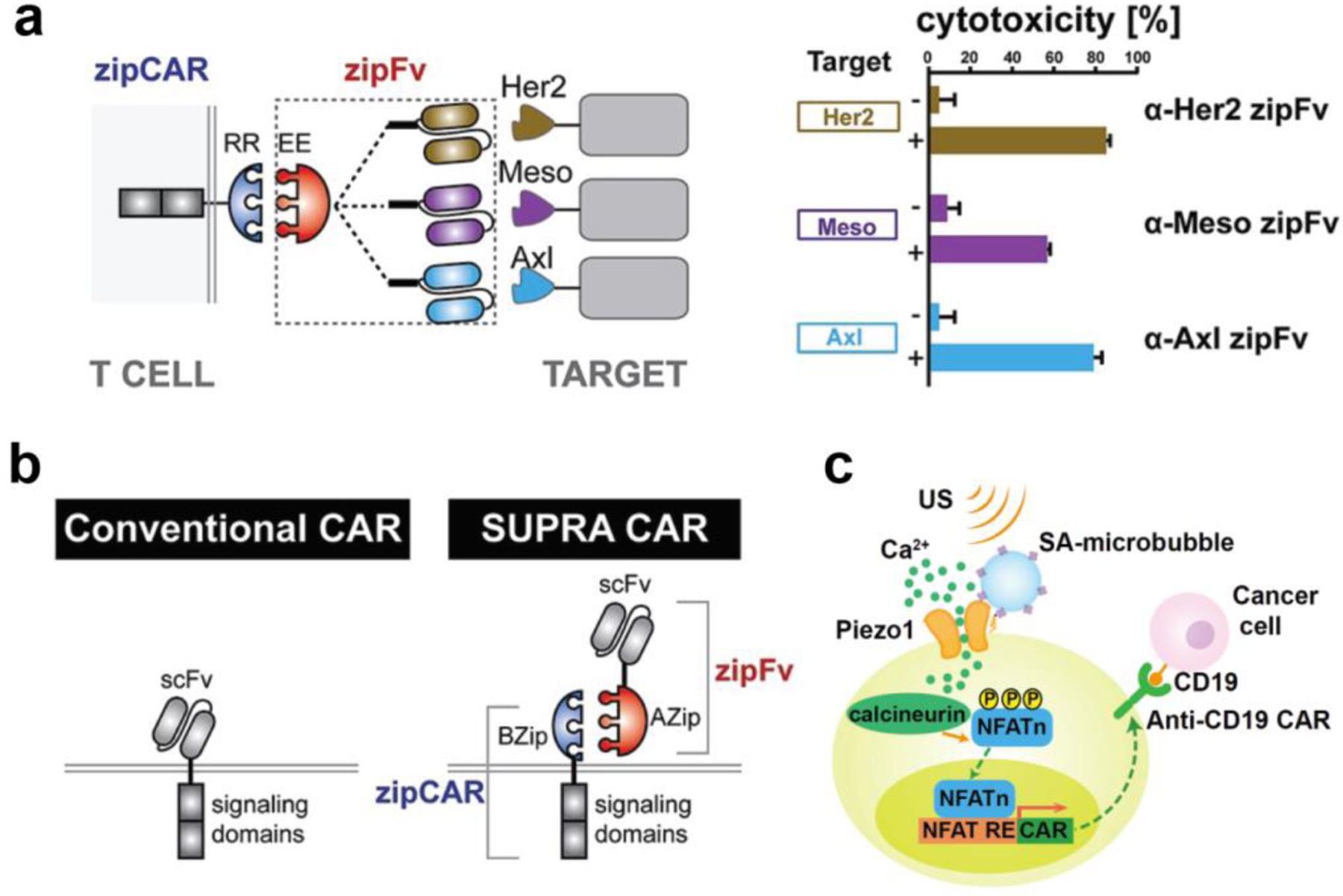
(a) A SUPRA CAR system targets multiple tumor antigens using zipFv designed with different antigen-targeting specificities. Engineered T cells with zipCAR demonstrate cytotoxicity against K562 cells expressing Her2, Mesothelin (Meso), or Axl. Reproduced with permission.[78] Copyright 2018, Elsevier. (b) Comparison of conventional and SUPRA CARs. T cells engineered with the SUPRA CAR system offer controllable activity and antigen-specificity through binding of signaling zipCARs to antibody-based adaptor zipFv. Reproduced with permission.[78] Copyright 2018, Elsevier. (c) Ultrasound-induced cell activation and CAR expression. Microbubbles functionalized with RGD peptides are coupled to the surface of T cells. Upon exposure to ultrasound waves, microbubbles amplify the ultrasound signals to activate mechanosensitive Piezo1 ion channels that trigger calcium influx, activating calcium-induced CAR expression on engineered T cells. Reproduced with permission.[79] Copyright 2018, National Academy of Sciences. SUPRA CAR, split, universal, and programmable chimeric antigen receptor; scFv, single-chain variable fragment; US, ultrasound; NFAT, nuclear factor of activated T cells; NFAT RE, NFAT response element.
Biomaterials can also enable remote control of CAR T cells with high precision[79–81, 206], which is important to both avoid systemic toxicities and enhance T-cell activity against solid tumors. Unlike pharmacodynamic adaptors that lack spatial resolution, other biomaterials have been developed to confine remote triggers, such as ultrasound and heat, within local tumor tissues. For example, a recent study used microbubbles, consisting of a gas surrounded by a lipid shell, to amplify low-frequency ultrasound for the activation of mechanically sensitive Piezo1 ion channels on the surface of T cells (Figure 9c)[79]. The activated Piezo1 ion channels could then trigger calcium influx in response to an ultrasound signal to activate calcium-induced CAR expression on engineered T cells[79]. An important feature of this approach is the use of microbubbles modified with RGD peptides for the coupling of microbubbles to Piezo1 channels on T cells. This proximity facilitated the transduction of ultrasound triggers and the spatial control of CAR T cell activation.
Another approach for remote control of T cells is the use of localized hyperthermia to tune the activities of T cells genetically engineered with the capacity to respond to heat (Figure 8c)[80, 81]. Hyperthermia[207] has a longstanding history in thermal medicine and is used for clinical applications such as thermal ablation of tumors[208], increasing transport of therapeutic molecules[209] and sensitization of cancer cells to chemotherapy[210]. Transient exposure to mild hyperthermia (40–42°C) is well-tolerated due to the induction of heat shock response (HSR), a highly conserved protective mechanism to cellular stress that triggers transient expression of cytoprotective genes[211]. Leveraging this endogenous pathway, Miller and colleagues constructed and screened panels of synthetic thermal gene switches containing combinations of endogenous promoters and DNA motifs that drive transcriptional responses following mild hyperthermia. The optimized thermal gene switches triggered transgene expression of T cells in response to small elevations in temperature (3–5 °C), but not to orthogonal cellular stresses like hypoxia. Gamboa et al. further demonstrated the use of thermal gene switches to control the expression of a catalytically dead CRISPR-associated protein (dCas9)[212] to suppress or activate endogenous genes, including a key T cell effector molecule GzmB[206]. For in vivo control, Miller and team showed that photothermal heating of plasmonic gold nanorods could be used to activate T cells engineered with thermal gene switches (Figure 8c) to produce broad classes of immunostimulatory agents (e.g., CARs, BiTEs, and IL-15 superagonist) to enhance key T cell functions like proliferation and antigen-specific cytotoxicity. Notably, the heat-induced release of IL-15 superagonist in the TME significantly enhanced anti-tumor activity of the CAR T cells and overall survival of mice bearing solid tumors in both syngeneic and humanized mouse models[80].
Taken together, these studies highlight how different classes of biomaterials are being harnessed to interface with engineered T cell therapies for remote control. Further advances in protein engineering and nanomaterials will continue to expand the immunoengineering toolbox to allow for combinations of orthogonal switches to independently control T cell ON/OFF states and targeting specificity. To ensure clinical success, the biodistribution and half-life of antibody-based inducers and transducers (e.g., microbubbles, gold nanorods) are important factors to be considered for the design of dosing regimens. Moreover, tissue localization and penetration depths of remote triggers (e.g., ultrasound and heat source) will also need to be considered to ensure the accessibility and precision of remote triggers to engineered T cells in tumor tissues. In this regard, the existing suite of medical platforms, such as MRI-guided focused ultrasound and intracranial laser heating, could be leveraged as remote triggers for engineered T cell therapies.
5. Conclusion
Research in biomaterials is making inroads into synthetic T cell therapies by providing new strategies to increase the affordability of these treatments, anticipate clinical outcomes, and improve therapeutic efficacy. In this review, we examined emerging strategies in three frontier arenas comprising manufacturing, monitoring, and modulation (Table 1). In manufacturing, one central opportunity is the development of nonviral platforms for gene delivery to T cells as viral vectors remain the gold standard despite challenges associated with safety and transfection efficiency. Nanomaterials such as lipid- or polymer-based formulations have the potential to solve these issues and may even lead to the ability to deliver transgenes directly to circulating T cells without the need for a complex ex vivo pipeline. An improved manufacturing process will result in dramatically reduced overall costs, minimized time from diagnosis to treatment, and broaden patient access to these therapies. We further discussed the need to develop or identify biomarkers for predictive monitoring of patient response to therapy. Technologies ranging from microfluidic immunoassays to in vivo activity-based sensors are providing new avenues for densely multiplexed and multiparametric analysis of immune cells. These diagnostics have the potential to unveil immunological features of response and resistance earlier on treatment to improve clinical decisions. Finally, we reviewed biomaterials that respond to endogenous or exogenous cues to localize and enhance anti-tumor T cell activity. These strategies could lead to new ways to redirect pre-existing antiviral immunity against tumors or remotely control the activity of engineered T cells to enhance therapy while limiting systemic toxicity.
As the intersection between biomaterials and synthetic immunity continues to rapidly expand, new biomaterials should be devised with a view towards translation to cGMP-grade production and extensive evaluation in preclinical models recapitulating human cancers[213, 214]. Reproducible and scalable chemistry, manufacturing, and controls also require careful consideration with an emphasis on simplicity in material designs in light of the challenges of regulatory approval. For these emerging technologies to be realized in the clinical practice of engineered T cell therapy, rigorous evaluation in carefully selected patient populations is required to establish safety and efficacy profiles. Solving these challenges could significantly improve outcomes for patients with intractable disease and contribute to the goal of democratizing T cell therapies.
Acknowledgements
The authors apologize to those investigators whose work could not be referenced because of space limitation. The authors thank Dr. Shreyas N. Dahotre, Lena Gamboa, and Ali H. Zamat for helpful feedback on the content of this progress report. The authors also thanks Qingyang Zhao for his help on the literature search. This work was funded by the NIH Director’s New Innovator Award (DP2HD091793), NIH R01 Research Project Grant (R01CA237210–01), the Shurl and Kay Curci Foundation, and the NSF (ECCS-1542174). Q.M. is supported by the NSF GRFP (DGE-1650044). A.S. is supported by the Georgia Tech President’s Fellowship, the Emory Laney Graduate Fellowship, and the NSF GRFP (DGE-1650044). This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (ECCS-1542174). G.A.K. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. F.Y.S. and Q.M. contributed equally to this work.
Biography

Dr. Gabe Kwong is an Associate Professor and the Wallace H. Coulter Distinguished Faculty Fellow in the Department of Biomedical Engineering at Georgia Tech and Emory School of Medicine. The research in his laboratory integrates engineering approaches with discoveries in immunology to advance human health. His transdisciplinary team pioneers new technologies for early disease detection and engineered cell therapies to treat complex diseases like cancer. His work has been published in leading journals and broadly featured by the media. Dr. Kwong holds over 25 issued or pending patents in biomedical engineering.

Fang-Yi “Ida” Su is a postdoctoral fellow in the Coulter Department of Biomedical Engineering at the Georgia Institute of Technology and Emory University. Ida earned her B.S. and M.S. from National Tsing Hua University in Taiwan. Prior to her postdoctoral training with Dr. Gabe Kwong, Ida received her Ph.D. in Bioengineering from University of Washington under the supervision of Dr. Patrick Stayton. Her research interests include nanotechnology, immunoengineering, and cell programming.

Quoc Mac is an NSF GRFP fellow and obtained his Ph.D. in biomedical engineering from the Coulter Department of Biomedical Engineering at the Georgia Institute of Technology and Emory University in 2021 under the guidance of Dr. Gabe Kwong. He received his B.S. in chemistry with highest honors and highest distinction from the University of North Carolina at Chapel Hill in 2015. His research centers on engineering noninvasive and predictive diagnostic technologies with a focus on T cell sensing for transplant rejection, cancer immunotherapies, and other immunological conditions.

Anirudh Sivakumar is an NSF GRFP fellow and Ph.D. candidate in the Coulter Department of Biomedical Engineering at the Georgia Institute of Technology and Emory University. He graduated with B.S. degrees in chemical engineering and mathematics with highest honors from the University of Texas at Austin, then joined Dr. Gabe Kwong’s group in 2018. His research currently focuses on engineering programmable biosensors for precision diagnostics, especially in the context of immunological disorders and immunotherapies.
Footnotes
Competing interests
G.A.K. is co-founder of and serves as consultant to Glympse Bio. This review paper includes studies that could affect his personal financial status. The terms of this arrangement have been reviewed and approved by Georgia Tech in accordance with its conflict-of-interest policies.
Contributor Information
Fang-Yi Su, The Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology & Emory University, Atlanta, GA 30332, USA..
Quoc D. Mac, The Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology & Emory University, Atlanta, GA 30332, USA.
Anirudh Sivakumar, The Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology & Emory University, Atlanta, GA 30332, USA..
Gabriel A. Kwong, The Wallace H. Coulter Department of Biomedical Engineering, Institute for Electronics and Nanotechnology, Parker H. Petit Institute of Bioengineering and Bioscience, Integrated Cancer Research Center, Georgia Immunoengineering Consortium, Winship Cancer Institute, Emory University, Georgia Institute of Technology & Emory University, Atlanta, GA 30332, USA.
References
- [1].Fenton OS, Olafson KN, Pillai PS, Mitchell MJ, Langer R, Adv Mater 2018, e1705328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Coburn JM, Kaplan DL, Bioconjug Chem 2015, 26, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Perry J, Chambers A, Spithoff K, Laperriere N, Curr Oncol 2007, 14, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen DS, Mellman I, Immunity 2013, 39, 1. [DOI] [PubMed] [Google Scholar]
- [5].Rosenberg SA, J Immunol 2014, 192, 5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, Ali CF, Chang TK, Konakova M, Pena RL, Kanhere RS, Kirksey YM, Ji C, Wang Y, Huang J, Sweeney TD, Kantak SS, Doberstein SK, Clin Cancer Res 2016, 22, 680. [DOI] [PubMed] [Google Scholar]
- [7].Turecek PL, Bossard MJ, Schoetens F, Ivens IA, J Pharm Sci 2016, 105, 460. [DOI] [PubMed] [Google Scholar]
- [8].Waldman AD, Fritz JM, Lenardo MJ, Nat Rev Immunol 2020, 20, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, Wasik M, Levine BL, Lacey SF, Melenhorst JJ, Porter DL, June CH, N Engl J Med 2017, 377, 2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vairy S, Garcia JL, Teira P, Bittencourt H, Drug Des Devel Ther 2018, 12, 3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vormittag P, Gunn R, Ghorashian S, Veraitch FS, Current opinion in biotechnology 2018, 53, 164. [DOI] [PubMed] [Google Scholar]
- [12].Levine BL, Miskin J, Wonnacott K, Keir C, Molecular therapy. Methods & clinical development 2017, 4, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M, Cell Death & Disease 2018, 9, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shah NN, Fry TJ, Nature reviews. Clinical oncology 2019, 16, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, Westin J, Gulbis AM, Loghin ME, de Groot JF, Adkins S, Davis SE, Rezvani K, Hwu P, Shpall EJ, Nature reviews. Clinical oncology 2018, 15, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Maude SL, Barrett D, Teachey DT, Grupp SA, Cancer journal (Sudbury, Mass.) 2014, 20, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Olden BR, Cheng Y, Yu JL, Pun SH, Journal of Controlled Release 2018, 282, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ, Nano Letters 2020, 20, 1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS, Stephan MT, Nature Nanotechnology 2017, 12, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT, Nature Communications 2020, 11, 6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, Jena B, Dawson MJ, Kumaresan PR, Su S, Maiti S, Dai J, Moriarity B, Forget M-A, Senyukov V, Orozco A, Liu T, McCarty J, Jackson RN, Moyes JS, Rondon G, Qazilbash M, Ciurea S, Alousi A, Nieto Y, Rezvani K, Marin D, Popat U, Hosing C, Shpall EJ, Kantarjian H, Keating M, Wierda W, Do KA, Largaespada DA, Lee DA, Hackett PB, Champlin RE, Cooper LJN, The Journal of Clinical Investigation 2016, 126, 3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chicaybam L, Abdo L, Viegas M, Marques LVC, de Sousa P, Batista-Silva LR, Alves-Monteiro V, Bonecker S, Monte-Mór B, Bonamino MH, Gene Therapy 2020, 27, 85. [DOI] [PubMed] [Google Scholar]
- [23].Morita D, Nishio N, Saito S, Tanaka M, Kawashima N, Okuno Y, Suzuki S, Matsuda K, Maeda Y, Wilson MH, Dotti G, Rooney CM, Takahashi Y, Nakazawa Y, Molecular Therapy - Methods & Clinical Development 2018, 8, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, Ivics Z, Hudecek M, Leukemia 2017, 31, 186. [DOI] [PubMed] [Google Scholar]
- [25].Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M, Nature 2017, 543, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, Castermans E, thor Straten P, Blank C, Haanen JB, Heemskerk MH, Schumacher TN, Nature Methods 2009, 6, 520. [DOI] [PubMed] [Google Scholar]
- [27].Newell EW, Klein LO, Yu W, Davis MM, Nature Methods 2009, 6, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O’Brien S, Gherardini PF, Herati RS, Huang AC, Chang K-M, Newell EW, Bovenschen N, Pe’er D, Albelda SM, Wherry EJ, Immunity 2018, 48, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Winkler F, Bengsch B, Front. Immunol 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, Ward JP, Wong SC, Schreiber RD, Newell EW, Nature Communications 2017, 8, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fehlings M, Jhunjhunwala S, Kowanetz M, O’Gorman WE, Hegde PS, Sumatoh H, Lee BH, Nardin A, Becht E, Flynn S, Ballinger M, Newell EW, Yadav M, Journal for ImmunoTherapy of Cancer 2019, 7, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nowicki TS, Berent-Maoz B, Cheung-Lau G, Huang RR, Wang X, Tsoi J, Kaplan-Lefko P, Cabrera P, Tran J, Pang J, Macabali M, Garcilazo IP, Carretero IB, Kalbasi A, Cochran AJ, Grasso CS, Hu-Lieskovan S, Chmielowski B, Comin-Anduix B, Singh A, Ribas A, Clinical Cancer Research 2019, 25, 2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Parisi G, Saco JD, Salazar FB, Tsoi J, Krystofinski P, Puig-Saus C, Zhang R, Zhou J, Cheung-Lau GC, Garcia AJ, Grasso CS, Tavaré R, Hu-Lieskovan S, Mackay S, Zalevsky J, Bernatchez C, Diab A, Wu AM, Comin-Anduix B, Charych D, Ribas A, Nature Communications 2020, 11, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mair F, Erickson JR, Voillet V, Simoni Y, Bi T, Tyznik AJ, Martin J, Gottardo R, Newell EW, Prlic M, Cell Reports 2020, 31, 107499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nakamoto M, Corselli M, Taylor I, Saksena S, The Journal of Immunology 2019, 202, 189.18. [Google Scholar]
- [36].Varadarajan N, Kwon DS, Law KM, Ogunniyi AO, Anahtar MN, Richter JM, Walker BD, Love JC, Proceedings of the National Academy of Sciences 2012, 109, 3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Han Q, Bradshaw EM, Nilsson B, Hafler DA, Love JC, Lab Chip 2010, 10, 1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Han Q, Bagheri N, Bradshaw EM, Hafler DA, Lauffenburger DA, Love JC, Proceedings of the National Academy of Sciences 2012, 109, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Torres AJ, Contento RL, Gordo S, Wucherpfennig KW, Love JC, Lab Chip 2012, 13, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kwong GA, Radu CG, Hwang K, Shu CJ, Ma C, Koya RC, Comin-Anduix B, Hadrup SR, Bailey RC, Witte ON, Schumacher TN, Ribas A, Heath JR, J. Am. Chem. Soc 2009, 131, 9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, Koya RC, Liu C-C, Kwong GA, Radu CG, Ribas A, Heath JR, Nature Medicine 2011, 17, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shi Q, Qin L, Wei W, Geng F, Fan R, Shin YS, Guo D, Hood L, Mischel PS, Heath JR, Proceedings of the National Academy of Sciences 2012, 109, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lu Y, Chen JJ, Mu L, Xue Q, Wu Y, Wu P-H, Li J, Vortmeyer AO, Miller-Jensen K, Wirtz D, Fan R, Anal. Chem 2013, 85, 2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ma C, Cheung AF, Chodon T, Koya RC, Wu Z, Ng C, Avramis E, Cochran AJ, Witte ON, Baltimore D, Chmielowski B, Economou JS, Comin-Anduix B, Ribas A, Heath JR, Cancer Discov 2013, 3, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lu Y, Xue Q, Eisele MR, Sulistijo ES, Brower K, Han L, Amir E-AD, Pe’er D, Miller-Jensen K, Fan R, Proceedings of the National Academy of Sciences of the United States of America 2015, 112, E607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dura B, Liu Y, Voldman J, Lab Chip 2014, 14, 2783. [DOI] [PubMed] [Google Scholar]
- [47].Dura B, Dougan SK, Barisa M, Hoehl MM, Lo CT, Ploegh HL, Voldman J, Nature Communications 2015, 6, 5940. [DOI] [PubMed] [Google Scholar]
- [48].Dura B, Servos MM, Barry RM, Ploegh HL, Dougan SK, Voldman J, Proceedings of the National Academy of Sciences 2016, 113, E3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Larimer BM, Wehrenberg-Klee E, Caraballo A, Mahmood U, J Nucl Med 2016, 57, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tavaré R, Escuin-Ordinas H, Mok S, McCracken MN, Zettlitz KA, Salazar FB, Witte ON, Ribas A, Wu AM, Cancer Research 2016, 76, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C, Cragnolini JJ, Bierie B, Gostissa M, Gorman J, Grotenbreg GM, Bhan A, Weinberg RA, Ploegh HL, The Journal of Experimental Medicine 2017, 214, 2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Seo JW, Tavaré R, Mahakian LM, Silvestrini MT, Tam S, Ingham ES, Salazar FB, Borowsky AD, Wu AM, Ferrara KW, Clinical Cancer Research 2018, 24, 4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kristensen LK, Christensen C, Alfsen MZ, Cold S, Nielsen CH, Kjaer A, Molecular Imaging and Biology 2020, 22, 1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hao L, Zhao RT, Ngambenjawong C, Fleming HE, Bhatia SN, bioRxiv 2020, 2020.06.17.157180. [Google Scholar]
- [55].Mac Q, Xu C, Bowen JR, Sivakumar A, Phuengkham H, Su F-Y, Stentz SZ, Sim H, Harris AM, Li TT, Qiu P, Kwong GA, bioRxiv 2020, 2020.12.10.420265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kwon EJ, Dudani JS, Bhatia SN, Nat Biomed Eng 2017, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dudani JS, Buss CG, Akana RTK, Kwong GA, Bhatia SN, Adv Funct Mater 2016, 26, 2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kwong GA, Dudani JS, Carrodeguas E, Mazumdar EV, Zekavat SM, Bhatia SN, Proc Natl Acad Sci U S A 2015, 112, 12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lin KY, Kwong GA, Warren AD, Wood DK, Bhatia SN, ACS Nano 2013, 7, 9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Warren AD, Gaylord ST, Ngan KC, Dumont Milutinovic M, Kwong GA, Bhatia SN, Walt DR, J Am Chem Soc 2014, 136, 13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kwong GA, von Maltzahn G, Murugappan G, Abudayyeh O, Mo S, Papayannopoulos IA, Sverdlov DY, Liu SB, Warren AD, Popov Y, Schuppan D, Bhatia SN, Nat Biotechnol 2013, 31, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mac QD, Mathews DV, Kahla JA, Stoffers CM, Delmas OM, Holt BA, Adams AB, Kwong GA, Nature Biomedical Engineering 2019, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kirkpatrick JD, Warren AD, Soleimany AP, Westcott PMK, Voog JC, Martin-Alonso C, Fleming HE, Tammela T, Jacks T, Bhatia SN, Sci Transl Med 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, Pennell CA, Blazar BR, Chen CC, Geller MA, Vezys V, Masopust D, Nature Communications 2019, 10, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gerber H-P, Senter PD, Grewal IS, MAbs 2009, 1, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sefrin JP, Hillringhaus L, Mundigl O, Mann K, Ziegler-Landesberger D, Seul H, Tabares G, Knoblauch D, Leinenbach A, Friligou I, Dziadek S, Offringa R, Lifke V, Lifke A, Front. Immunol 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Schmittnaegel M, Levitsky V, Hoffmann E, Georges G, Mundigl O, Klein C, Knoetgen H, Cancer Immunology Research 2015, 3, 764. [DOI] [PubMed] [Google Scholar]
- [68].Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ, Nature Medicine 2010, 16, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tang L, Zheng Y, Melo MB, Mabardi L, Castaño AP, Xie Y-Q, Li N, Kudchodkar SB, Wong HC, Jeng EK, Maus MV, Irvine DJ, Nature Biotechnology 2018, 36, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schmid D, Park CG, Hartl CA, Subedi N, Cartwright AN, Puerto RB, Zheng Y, Maiarana J, Freeman GJ, Wucherpfennig KW, Irvine DJ, Goldberg MS, Nature Communications 2017, 8, 1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zheng Y, Stephan MT, Gai SA, Abraham W, Shearer A, Irvine DJ, Journal of Controlled Release 2013, 172, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Yang Y-SS, Moynihan KD, Bekdemir A, Dichwalkar TM, Noh MM, Watson N, Melo M, Ingram J, Suh H, Ploegh H, Stellacci FR, Irvine DJ, Biomaterials science 2018, 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP, Blood cancer journal 2016, 6, e458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, Campana D, Cancer Res 2014, 74, 93. [DOI] [PubMed] [Google Scholar]
- [75].Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, Fonslow BR, Kochenderfer JN, Wright TM, Schultz PG, Young TS, Kim CH, Cao Y, Proc Natl Acad Sci U S A 2016, 113, E450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, Ma J, Nunez V, Shen J, Woods AK, Wright TM, Schultz PG, Kim CH, Young TS, Proceedings of the National Academy of Sciences 2016, 113, E459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Viaud S, Ma JSY, Hardy IR, Hampton EN, Benish B, Sherwood L, Nunez V, Ackerman CJ, Khialeeva E, Weglarz M, Lee SC, Woods AK, Young TS, Proceedings of the National Academy of Sciences 2018, 115, E10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Cho JH, Collins JJ, Wong WW, Cell 2018, 173, 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Pan Y, Yoon S, Sun J, Huang Z, Lee C, Allen M, Wu Y, Chang Y-J, Sadelain M, Shung KK, Chien S, Wang Y, Proceedings of the National Academy of Sciences 2018, 115, 992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Miller IC, Sun L-K, Harris AM, Gamboa L, Zamat A, Kwong GA, bioRxiv 2020, 2020.04.26.062703. [Google Scholar]
- [81].Miller IC, Gamboa Castro M, Maenza J, Weis JP, Kwong GA, ACS Synthetic Biology 2018, 7, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Spink K, Steinsapir A, Cell and Gene Therapy Insights 2018, 4, 1105. [Google Scholar]
- [83].Piscopo NJ, Mueller KP, Das A, Hematti P, Murphy WL, Palecek SP, Capitini CM, Saha K, Biotechnology journal 2018, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zhao Y, Stepto H, Schneider CK, Human gene therapy methods 2017, 28, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Marcucci KT, Jadlowsky JK, Hwang W-T, Suhoski-Davis M, Gonzalez VE, Kulikovskaya I, Gupta M, Lacey SF, Plesa G, Chew A, Melenhorst JJ, Levine BL, June CH, Molecular Therapy 2018, 26, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ, EMBO molecular medicine 2017, 9, 1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].DiTommaso T, Cole JM, Cassereau L, Buggé JA, Hanson JLS, Bridgen DT, Stokes BD, Loughhead SM, Beutel BA, Gilbert JB, Nussbaum K, Sorrentino A, Toggweiler J, Schmidt T, Gyuelveszi G, Bernstein H, Sharei A, Proceedings of the National Academy of Sciences 2018, 115, E10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hudecek M, Ivics Z, Current Opinion in Genetics & Development 2018, 52, 100. [DOI] [PubMed] [Google Scholar]
- [89].Sharei A, Zoldan J, Adamo A, Sim WY, Cho N, Jackson E, Mao S, Schneider S, Han M-J, Lytton-Jean A, Basto PA, Jhunjhunwala S, Lee J, Heller DA, Kang JW, Hartoularos GC, Kim K-S, Anderson DG, Langer R, Jensen KF, Proceedings of the National Academy of Sciences 2013, 110, 2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ding X, Stewart MP, Sharei A, Weaver JC, Langer RS, Jensen KF, Nature Biomedical Engineering 2017, 1, 0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, Lambert A, Stoddard BL, Kiem HP, Stephan MT, Nature Communications 2017, 8, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Mislick KA, Baldeschwieler JD, Proc Natl Acad Sci U S A 1996, 93, 12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG, Nature Reviews Genetics 2014, 15, 541. [DOI] [PubMed] [Google Scholar]
- [94].Nature Reviews Materials 2021, 6, 99. [Google Scholar]
- [95].Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, Tobin V, Nguyen DN, Lee MR, Putnam AL, Ferris AL, Chen JW, Schickel J-N, Pellerin L, Carmody D, Alkorta-Aranburu G, del Gaudio D, Matsumoto H, Morell M, Mao Y, Cho M, Quadros RM, Gurumurthy CB, Smith B, Haugwitz M, Hughes SH, Weissman JS, Schumann K, Esensten JH, May AP, Ashworth A, Kupfer GM, Greeley SAW, Bacchetta R, Meffre E, Roncarolo MG, Romberg N, Herold KC, Ribas A, Leonetti MD, Marson A, Nature 2018, 559, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Olden BR, Cheng E, Cheng Y, Pun SH, Biomater Sci 2019, 7, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Harris E, Elmer JJ, Biotechnology Progress, n/a, e3066. [DOI] [PubMed] [Google Scholar]
- [98].Bobardt MD, Armand-Ugón M, Clotet I, Zhang Z, David G, Este JA, Gallay PA, Virology 2004, 325, 389. [DOI] [PubMed] [Google Scholar]
- [99].Kedmi R, Veiga N, Ramishetti S, Goldsmith M, Rosenblum D, Dammes N, Hazan-Halevy I, Nahary L, Leviatan-Ben-Arye S, Harlev M, Behlke M, Benhar I, Lieberman J, Peer D, Nature Nanotechnology 2018, 13, 214. [DOI] [PubMed] [Google Scholar]
- [100].Ramishetti S, Kedmi R, Goldsmith M, Leonard F, Sprague AG, Godin B, Gozin M, Cullis PR, Dykxhoorn DM, Peer D, ACS Nano 2015, 9, 6706. [DOI] [PubMed] [Google Scholar]
- [101].Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, Costa C, Wels WS, Modlich U, Stripecke R, Verhoeyen E, Buchholz CJ, EMBO molecular medicine 2018, 10, e9158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Agarwal S, Weidner T, Thalheimer FB, Buchholz CJ, OncoImmunology 2019, 8, e1671761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Agarwal S, Hanauer JDS, Frank AM, Riechert V, Thalheimer FB, Buchholz CJ, Molecular Therapy 2020, 28, 1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Shirley JL, de Jong YP, Terhorst C, Herzog RW, Molecular Therapy 2020, 28, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Shirley JL, de Jong YP, Terhorst C, Herzog RW, Molecular therapy : the journal of the American Society of Gene Therapy 2020, 28, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Izsvák Z, Ivics Z, Molecular Therapy 2004, 9, 147. [DOI] [PubMed] [Google Scholar]
- [107].Han J, Gao F, Geng S, Ye X, Wang T, Du P, Cai Z, Fu Z, Zhao Z, Shi L, Li Q, Cai J, Molecular Cancer Therapeutics 2020, 19, 178. [DOI] [PubMed] [Google Scholar]
- [108].Gracey Maniar LE, Maniar JM, Chen Z-Y, Lu J, Fire AZ, Kay MA, Molecular therapy : the journal of the American Society of Gene Therapy 2013, 21, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Dietz WM, Skinner NE, Hamilton SE, Jund MD, Heitfeld SM, Litterman AJ, Hwu P, Chen ZY, Salazar AM, Ohlfest JR, Blazar BR, Pennell CA, Osborn MJ, Molecular therapy : the journal of the American Society of Gene Therapy 2013, 21, 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hudecek M, Izsvák Z, Johnen S, Renner M, Thumann G, Ivics Z, Critical Reviews in Biochemistry and Molecular Biology 2017, 52, 355. [DOI] [PubMed] [Google Scholar]
- [111].Crudele JM, Chamberlain JS, Nature Communications 2018, 9, 3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rahimi H, Salehiabar M, Charmi J, Barsbay M, Ghaffarlou M, Roohi Razlighi M, Davaran S, Khalilov R, Sugiyama M, Nosrati H, Kaboli S, Danafar H, Webster TJ, Nano Today 2020, 34, 100895. [Google Scholar]
- [113].Eoh J, Gu L, Biomaterials Science 2019, 7, 1240. [DOI] [PubMed] [Google Scholar]
- [114].Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, Klichinsky M, Shestova O, Patel PR, Kulikovskaya I, Nazimuddin F, Bhoj VG, Orlando EJ, Fry TJ, Bitter H, Maude SL, Levine BL, Nobles CL, Bushman FD, Young RM, Scholler J, Gill SI, June CH, Grupp SA, Lacey SF, Melenhorst JJ, Nature Medicine 2018, 24, 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ, Nature Nanotechnology 2020, 15, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Cevaal PM, Ali A, Czuba-Wojnilowicz E, Symons J, Lewin SR, Cortez-Jugo C, Caruso F, ACS Nano 2021. [DOI] [PubMed] [Google Scholar]
- [117].Nishino M, Ramaiya NH, Hatabu H, Hodi FS, Nature reviews. Clinical oncology 2017, 14, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Krebs S, Ponomarev V, Slovin S, Schoder H, J Nucl Med 2019, 60, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Krekorian M, Fruhwirth GO, Srinivas M, Figdor CG, Heskamp S, Witney TH, Aarntzen E, Theranostics 2019, 9, 7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Dahotre SN, Chang YM, Wieland A, Stammen SR, Kwong GA, Proceedings of the National Academy of Sciences 2018, 115, 4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhang SQ, Ma KY, Schonnesen AA, Zhang M, He C, Sun E, Williams CM, Jia W, Jiang N, Nat Biotechnol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJS, McGranahan N, Rosenthal R, Straten t. P., Szallasi Z, Svane IM, Swanton C, Quezada SA, Jakobsen SN, Eklund AC, Hadrup SR, Nature Biotechnology 2016, 34, 1037. [DOI] [PubMed] [Google Scholar]
- [123].Woodham AW, Zeigler SH, Zeyang EL, Kolifrath SC, Cheloha RW, Rashidian M, Chaparro RJ, Seidel RD, Garforth SJ, Dearling JL, Mesyngier M, Duddempudi PK, Packard AB, Almo SC, Ploegh HL, Nature Methods 2020, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Larimer BM, Wehrenberg-Klee E, Dubois F, Mehta A, Kalomeris T, Flaherty K, Boland G, Mahmood U, Cancer Res 2017, 77, 2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Larimer BM, Bloch E, Nesti S, Austin EE, Wehrenberg-Klee E, Boland G, Mahmood U, Clinical Cancer Research 2019, 25, 1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Gibson HM, McKnight BN, Malysa A, Dyson G, Wiesend WN, McCarthy CE, Reyes J, Wei W-Z, Viola-Villegas NT, Cancer Research 2018, 78, 5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].McKinnon KM, Curr Protoc Immunol 2018, 120, 5 1 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Seder RA, Darrah PA, Roederer M, Nat Rev Immunol 2008, 8, 247. [DOI] [PubMed] [Google Scholar]
- [129].Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, Pumphrey NJ, Jakobsen BK, Price DA, Ladell K, Sewell AK, Clin Exp Immunol 2015, 180, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Nikolich-Zugich J, Slifka MK, Messaoudi I, Nat Rev Immunol 2004, 4, 123. [DOI] [PubMed] [Google Scholar]
- [131].Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebaek E, Svane IM, Schumacher TN, Thor Straten P, Hadrup SR, Cancer Res 2012, 72, 1642. [DOI] [PubMed] [Google Scholar]
- [132].Chattopadhyay PK, Gierahn TM, Roederer M, Love JC, Nat Immunol 2014, 15, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Davis MM, Altman JD, Newell EW, Nat Rev Immunol 2011, 11, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Spitzer Matthew H., Nolan Garry P., Cell 2016, 165, 780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Hartmann FJ, Bendall SC, Nature Reviews Rheumatology 2020, 16, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJ, McGranahan N, Rosenthal R, Straten PT, Szallasi Z, Svane IM, Swanton C, Quezada SA, Jakobsen SN, Eklund AC, Hadrup SR, Nat Biotechnol 2016, 34, 1037. [DOI] [PubMed] [Google Scholar]
- [137].Zhang S-Q, Ma K-Y, Schonnesen AA, Zhang M, He C, Sun E, Williams CM, Jia W, Jiang N, Nature Biotechnology 2018, 36, 1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Dahotre SN, Chang YM, Romanov AM, Kwong GA, Anal. Chem. 2019, 91, 2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Kacherovsky N, Cardle II, Cheng EL, Yu JL, Baldwin ML, Salipante SJ, Jensen MC, Pun SH, Nat Biomed Eng 2019, 3, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Heath JR, Ribas A, Mischel PS, Nat Rev Drug Discov 2016, 15, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Labib M, Kelley SO, Nature Reviews Chemistry 2020, 4, 143. [DOI] [PubMed] [Google Scholar]
- [142].Corrie SR, Plebanski M, Molecular Immunology 2018, 98, 28. [DOI] [PubMed] [Google Scholar]
- [143].Di Modugno F, Colosi C, Trono P, Antonacci G, Ruocco G, Nistico P, J Exp Clin Cancer Res 2019, 38, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Gu L, Mooney DJ, Nat Rev Cancer 2016, 16, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, Miccolis C, Ceccarello E, Adriani G, Raimondi MT, Kamm RD, Bertoletti A, JCI Insight 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Wallstabe L, Göttlich C, Nelke LC, Kühnemundt J, Schwarz T, Nerreter T, Einsele H, Walles H, Dandekar G, Nietzer SL, Hudecek M, JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Lokugamage MP, Sago CD, Gan Z, Krupczak BR, Dahlman JE, Advanced Materials 2019, 31, 1902251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].McCracken MN, Tavaré R, Witte ON, Wu AM, in Advances in Immunology, Vol. 131 (Eds: F. W. Alt, F. W. Alt), 2016, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Griessinger CM, Olafsen T, Mascioni A, Jiang ZK, Zamilpa C, Jia F, Torgov M, Romero JM, Marchioni F, Satpayev D, Lee C, Zhang G, Nayak TK, Pincha M, Amann M, Mohan PLB, Richard M, Nicolini VG, Sam J, Claus C, Ferrara C, Brünker P, Bacac M, Umana P, Rüttinger D, Wilson IA, Gudas J, Klein C, Tessier JJL, Cancer Research 2020, 80, 2903. [DOI] [PubMed] [Google Scholar]
- [150].Dudani JS, Warren AD, Bhatia SN, Annual Review of Cancer Biology 2018, 2, 353. [Google Scholar]
- [151].Barr DB, Wilder LC, Caudill SP, Gonzalez AJ, Needham LL, Pirkle JL, Environ Health Perspect 2005, 113, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Jones CA, McQuillan GM, Kusek JW, Eberhardt MS, Herman WH, Coresh J, Salive M, Jones CP, Agodoa LY, Am J Kidney Dis 1998, 32, 992. [DOI] [PubMed] [Google Scholar]
- [153].Cullen SP, Brunet M, Martin SJ, Cell Death Differ 2010, 17, 616. [DOI] [PubMed] [Google Scholar]
- [154].Zhuang Q, Holt BA, Kwong GA, Qiu P, PLoS Comput Biol 2019, 15, e1006909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Widen JC, Tholen M, Yim JJ, Antaris A, Casey KM, Rogalla S, Klaassen A, Sorger J, Bogyo M, Nat Biomed Eng 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Badeau BA, Comerford MP, Arakawa CK, Shadish JA, DeForest CA, Nat Chem 2018, 10, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Holt BA, Kwong GA, Nat Commun 2020, 11, 5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Holt BA, Curro I, Kwong GA, bioRxiv 2019, 556951. [Google Scholar]
- [159].Brown CE, Mackall CL, Nat Rev Immunol 2019, 19, 73. [DOI] [PubMed] [Google Scholar]
- [160].Brudno JN, Kochenderfer JN, Blood 2016, 127, 3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Fang L, Zhao Z, Wang J, Zhang P, Ding Y, Jiang Y, Wang D, Li Y, Science Advances 2020, 6, eaba4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Liu X, Feng Z, Wang C, Su Q, Song H, Zhang C, Huang P, Liang XJ, Dong A, Kong D, Wang W, Biomaterials 2020, 230, 119649. [DOI] [PubMed] [Google Scholar]
- [163].Chao Y, Liang C, Tao H, Du Y, Wu D, Dong Z, Jin Q, Chen G, Xu J, Xiao Z, Chen Q, Wang C, Chen J, Liu Z, Science Advances 2020, 6, eaaz4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Yang P, Song H, Qin Y, Huang P, Zhang C, Kong D, Wang W, Nano Lett 2018, 18, 4377. [DOI] [PubMed] [Google Scholar]
- [165].Park CG, Hartl CA, Schmid D, Carmona EM, Kim H-J, Goldberg MS, Science Translational Medicine 2018, 10, eaar1916. [DOI] [PubMed] [Google Scholar]
- [166].Dahotre SN, Romanov AM, Su F-Y, Kwong GA, Advanced Therapeutics 2021, n/a, 2100034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Millar DG, Ramjiawan RR, Kawaguchi K, Gupta N, Chen J, Zhang S, Nojiri T, Ho WW, Aoki S, Jung K, Chen I, Shi F, Heather JM, Shigeta K, Morton LT, Sepulveda S, Wan L, Joseph R, Minogue E, Khatri A, Bardia A, Ellisen LW, Corcoran RB, Hata AN, Pai SI, Jain RK, Fukumura D, Duda DG, Cobbold M, Nature Biotechnology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, Duan K, Ang N, Poidinger M, Lee YY, Larbi A, Khng AJ, Tan E, Fu C, Mathew R, Teo M, Lim WT, Toh CK, Ong B-H, Koh T, Hillmer AM, Takano A, Lim TKH, Tan EH, Zhai W, Tan DSW, Tan IB, Newell EW, Nature 2018, 557, 575. [DOI] [PubMed] [Google Scholar]
- [169].Zuhair M, Smit GSA, Wallis G, Jabbar F, Smith C, Devleesschauwer B, Griffiths P, Reviews in medical virology 2019, 29, e2034. [DOI] [PubMed] [Google Scholar]
- [170].Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, Klenerman P, J Immunol 2003, 170, 2022. [DOI] [PubMed] [Google Scholar]
- [171].Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, Moss PA, J Immunol 2002, 169, 1984. [DOI] [PubMed] [Google Scholar]
- [172].Ahmed R, Gray D, Science 1996, 272, 54. [DOI] [PubMed] [Google Scholar]
- [173].Wherry EJ, Ahmed R, J Virol 2004, 78, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Anderson KG, Stromnes IM, Greenberg PD, Cancer Cell 2017, 31, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Bumbaca B, Li Z, Shah DK, Drug Metab Pharmacokinet 2019, 34, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Goebeler ME, Bargou RC, Nature reviews. Clinical oncology 2020, 17, 418. [DOI] [PubMed] [Google Scholar]
- [177].Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL, Blood 1997, 90, 2541. [PubMed] [Google Scholar]
- [178].Skrombolas D, Frelinger JG, Expert Rev Clin Immunol 2014, 10, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Ellis JM, Henson V, Slack R, Ng J, Hartzman RJ, Katovich Hurley C, Human Immunology 2000, 61, 334. [DOI] [PubMed] [Google Scholar]
- [180].Stüber T, Monjezi R, Wallstabe L, Kühnemundt J, Nietzer SL, Dandekar G, Wöckel A, Einsele H, Wischhusen J, Hudecek M, Journal for ImmunoTherapy of Cancer 2020, 8, e000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, Abedi M, Davies RA, Cabral HJ, Al-Homsi AS, Cohen SI, The Prostate 2016, 76, 1257. [DOI] [PubMed] [Google Scholar]
- [182].Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, Pise-Masison CA, Radonovich MF, Brady JN, Restifo NP, Berzofsky JA, Leonard WJ, J Exp Med 2005, 201, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S, Slaney CY, Johnstone RW, Trapani JA, Stagg J, Loi S, Kats L, Gyorki D, Kershaw MH, Darcy PK, The Journal of clinical investigation 2017, 127, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1999, 17, 2105. [DOI] [PubMed] [Google Scholar]
- [185].Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1995, 13, 688. [DOI] [PubMed] [Google Scholar]
- [186].Sportès C, Babb RR, Krumlauf MC, Hakim FT, Steinberg SM, Chow CK, Brown MR, Fleisher TA, Noel P, Maric I, Stetler-Stevenson M, Engel J, Buffet R, Morre M, Amato RJ, Pecora A, Mackall CL, Gress RE, Clinical Cancer Research 2010, 16, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Schmidt J, Ferk P, The Journal of pharmacy and pharmacology 2017, 69, 790. [DOI] [PubMed] [Google Scholar]
- [188].Siriwon N, Kim YJ, Siegler E, Chen X, Rohrs JA, Liu Y, Wang P, Cancer immunology research 2018, 6, 812. [DOI] [PubMed] [Google Scholar]
- [189].Zheng Y, Tang L, Mabardi L, Kumari S, Irvine DJ, ACS Nano 2017, 11, 3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ, Biomaterials 2012, 33, 5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Shields CW, Evans MA, Wang LL-W, Baugh N, Iyer S, Wu D, Zhao Z, Pusuluri A, Ukidve A, Pan DC, Mitragotri S, Science Advances 2020, 6, eaaz6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Taurin S, Nehoff H, Greish K, Journal of controlled release : official journal of the Controlled Release Society 2012, 164, 265. [DOI] [PubMed] [Google Scholar]
- [193].Khawar IA, Kim JH, Kuh HJ, Journal of controlled release : official journal of the Controlled Release Society 2015, 201, 78. [DOI] [PubMed] [Google Scholar]
- [194].J. A. Alberts B, Lewis J, et al. , Molecular Biology of the Cell 4th edition., New York: Garland Science, 2002. [Google Scholar]
- [195].Huang B, Abraham WD, Zheng Y, Bustamante López SC, Luo SS, Irvine DJ, Science Translational Medicine 2015, 7, 291ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, Li PJ, Diolaiti ME, Ashworth A, Marson A, Cell 2018, 175, 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Gamboa L, Zamat AH, Kwong GA, Theranostics 2020, 10, 3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Arndt C, Fasslrinner F, Loureiro LR, Koristka S, Feldmann A, Bachmann M, Cancers (Basel) 2020, 12, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA, Science (New York, N.Y.) 2015, 350, aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Leung W-H, Gay J, Martin U, Garrett TE, Horton HM, Certo MT, Blazar BR, Morgan RA, Gregory PD, Jarjour J, Astrakhan A, JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA, Science 2015, 350, aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ Jr., Cancer Res 2012, 72, 1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Cartellieri M, Feldmann A, Koristka S, Arndt C, Loff S, Ehninger A, von Bonin M, Bejestani EP, Ehninger G, Bachmann MP, Blood cancer journal 2016, 6, e458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Ma JSY, Kim JY, Kazane SA, Choi S-H, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB, Fonslow BR, Kochenderfer JN, Wright TM, Schultz PG, Young TS, Kim CH, Cao Y, Proceedings of the National Academy of Sciences of the United States of America 2016, 113, E450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Klesmith JR, Su L, Wu L, Schrack IA, Dufort FJ, Birt A, Ambrose C, Hackel BJ, Lobb RR, Rennert PD, Molecular pharmaceutics 2019, 16, 3544. [DOI] [PubMed] [Google Scholar]
- [206].Gamboa L, Phung EV, Li H, Meyers JP, Hart AC, Miller IC, Kwong GA, ACS Chemical Biology 2020, 15, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Brace CL, Curr Probl Diagn Radiol 2009, 38, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Chu KF, Dupuy DE, Nature Reviews Cancer 2014, 14, 199. [DOI] [PubMed] [Google Scholar]
- [209].van Driel WJ, Koole SN, Sikorska K, Schagen van Leeuwen JH, Schreuder HWR, Hermans RHM, de Hingh IHJT, van der Velden J, Arts HJ, Massuger LFAG, Aalbers AGJ, Verwaal VJ, Kieffer JM, Van de Vijver KK, van Tinteren H, Aaronson NK, Sonke GS, New England Journal of Medicine 2018, 378, 230. [DOI] [PubMed] [Google Scholar]
- [210].Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, Felix R, Schlag PM, The Lancet. Oncology 2002, 3, 487. [DOI] [PubMed] [Google Scholar]
- [211].Nikfarjam M, Muralidharan V, Christophi C, The Journal of surgical research 2005, 127, 208. [DOI] [PubMed] [Google Scholar]
- [212].Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA, Cell 2013, 152, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Das R, Strowig T, Verma R, Koduru S, Hafemann A, Hopf S, Kocoglu MH, Borsotti C, Zhang L, Branagan A, Eynon E, Manz MG, Flavell RA, Dhodapkar MV, Nat Med 2016, 22, 1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].DuPage M, Dooley AL, Jacks T, Nat Protoc 2009, 4, 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]