

Abstract
Rationale:
Despite contemporary therapy, coronary artery disease (CAD) remains a leading cause of mortality. Genetic variants at ADAMTS7 have been associated with CAD and the loss of ADAMTS7 is protective for atherosclerosis. ADAMTS7 (a disintegrin and metalloproteinase with thrombospondin motifs 7) is a secreted metalloproteinase and complex proteoglycan, yet the mechanism linking ADAMTS7 to CAD risk remains unresolved.
Objective:
To investigate the role of ADAMTS7 catalytic function in vascular smooth muscle cellular migration and during atherosclerosis.
Methods and Results:
We established a new purification strategy for full-length mouse ADAMTS7 and demonstrated the loss of activity in the catalytic mutant form of ADAMTS7. To test if the enzymatic activity of ADAMTS7 mediates atherosclerosis, we generated a catalytically inactive mutant mouse allele and compared it to the Adamts7 knockout. Using two models of atherosclerosis, we found that reducing either ADAMTS7 dosage or catalytic function decreased the burden of atherosclerosis. We demonstrate impaired vascular smooth muscle migration in both Adamts7 catalytic mutant and null cells using a lateral migration wound healing assay. Expression of the wild-type allele rescued the migration phenotype in Adamts7 null cells while expression of the catalytic mutant protein did not. We then characterized a human ADAMTS7 coding variant rs3825807 (Ser214Pro) associated with reduced CAD risk. This variant had a hypomorphic effect on ADAMTS7 secretion and migration of vascular smooth muscle cells (VSMC), findings consistent with our mouse studies.
Conclusions:
We demonstrated that loss of ADAMTS7 catalytic function protects against atherosclerosis via phenotype switch of VSMCs and that the atherosclerosis protective effects could be mediated by a loss-of-function coding variant associated with CAD risk. In aggregate, we provide compelling evidence that dosage of ADAMTS7 and catalytic function are responsible for the atherosclerotic phenotype, suggesting that the catalytic domain would be an attractive therapeutic target for CAD.
Keywords: ADAMTS7, single nucleotide polymorphisms, atherosclerosis, vascular smooth muscle cell, coronary artery disease, protease, Genetically Altered and Transgenic Models
Graphical Abstract
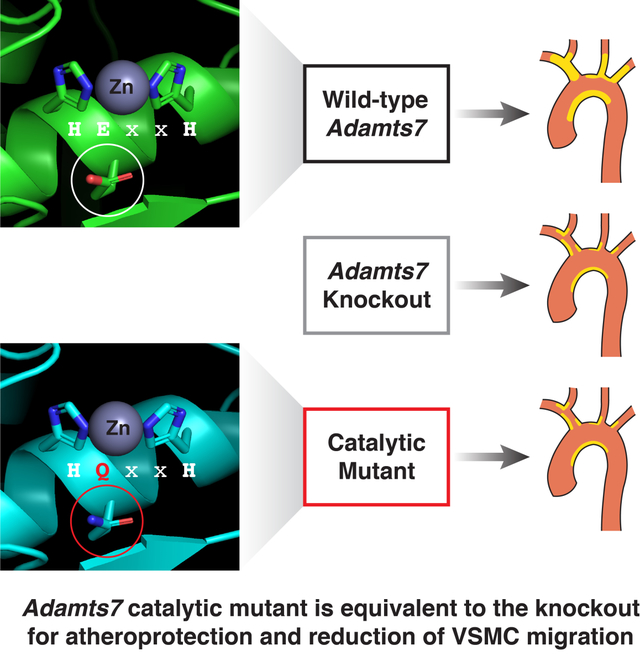
The secreted metalloproteinase ADAMTS7 has been associated with CAD and loss of ADAMTS7 is protective for atherosclerosis, yet a connection between enzymatic function and CAD risk remains unresolved. We therefore generated a catalytic mutant mouse allele and compared it to the Adamts7 knockout. Using two models of atherosclerosis, we found that a reduction of ADAMTS7 dosage or catalytic function reduced the burden of atherosclerosis. We then characterized a coding variant, rs3825807 (p.Ser214Pro), associated with reduced CAD risk. This variant had a hypomorphic effect on ADAMTS7 secretion and migration of vascular smooth muscle cells, findings consistent with our mouse studies. In aggregate, we provide compelling evidence that loss of ADAMTS7 enzymatic function protects against atherosclerosis.
INTRODUCTION
Coronary artery disease is a major cause of mortality worldwide and newly identified genetic risk factors are currently being evaluated as targets for therapeutic development1. Multiple genome wide association studies (GWAS) have identified variants within ADAMTS72, 3 and in the flanking intergenic regions as associated with CAD4, 5. ADAMTS7 risk variants are also associated with a high-risk plaque phenotype and an increase in secondary cardiovascular events6–9. Initial characterization of a coding variant rs3825807 (p.Ser214Pro) has demonstrated a beneficial effect from less ADAMTS7 function10. A recent study demonstrated that Adamts7 deficiency in hyperlipidemic ApoE (apolipoprotein E) knockout (KO) or Ldlr (low density lipoprotein receptor) KO background mice results in reduced atherosclerotic lesion formation, suggesting that ADAMTS7 is pro-atherogenic11. Further studies performed in rodent vascular injury models support a role for ADAMTS7 loss of function as protective against vascular smooth muscle cell migration and proliferation in neointima formation11–14. Despite all of this genetic evidence, it is presently unclear if the catalytic function of ADAMTS7 directly contributes to the atherosclerosis phenotype.
ADAMTS7 belongs to a family of secreted zinc metalloproteinases containing elaborate carboxyl-terminal ancillary domains15. The catalytic domain of all 19 ADAMTS enzymes is preceded by a signal peptide and prodomain that serves as a chaperone and contains a “cysteine switch” to engage with the catalytic zinc to maintain enzyme latency16. The ADAMTS catalytic domain is followed by a set of disintegrin, thrombospondin, cysteine-rich and spacer domains thought to confer substrate specificity and localization17. ADAMTS7 contains a total of eight thrombospondin repeats and a highly glycosylated mucin domain with a chondroitin sulfate GAG attachment site rendering ADAMTS7 as both an enzyme and a proteoglycan18. It is notable that there are seven ADAMTS-like (ADAMTSL) proteins which lack the catalytic domain; however, ADAMTSL loss-of-function of mutations are associated with diseases, indicating a role for the ancillary domains in matricellular biology separate from catalytic function15. ADAMTS7, and its close paralog ADAMTS12, are defined as cartilage oligomeric protein (COMP) proteases, capable of associating with COMP via their ancillary carboxyl-terminal domains19, 20. Regulated cleavage of COMP or other extracellular matrix (ECM) proteins modulates VSMC migration10, 21. However, a role for the auxiliary domains of ADAMTS7 on VSMC migration and atherogenesis remains a distinct possibility independent from the enzymatic function.
In the current work, we demonstrate that the catalytic activity of ADAMTS7 is essential for its pro-atherogenic effects and that the atherosclerosis protective effects could be mediated by a loss of function ADAMTS7 coding variant associated with CAD risk.
METHODS
Data Availability.
The authors declare that all supporting data are available within the article and its online supplementary files.
Expanded methods sections for the expression vectors, mammalian cell culture, western blot, RT-PCR expression analysis, vascular smooth muscle cell migration assay, generation of Adamts7 mouse alleles, full-length ADAMTS7 purification and Thrombospondin 1 (TSP1) cleavage assay, atherogenic mouse models, atherosclerotic lesion measurement, immunohistochemistry and β-galactosidase (β-gal) staining are available in the online-only Data Supplement. Representative images were selected to represent quantification results shown with the images. Assay procedures and quantification analysis for in vivo experiments (Oil-Red O staining, α-SMA staining and β-gal staining) were performed by scientists (T.M., B.B), who are not aware of mouse genotype. Different scientists (B.T.M., N.R.P.) performed genotyping for all the mice.
Adamts7 mouse alleles used in the atherosclerosis studies.
Publicly available Adamts7 tm1a (KOMP) Wtsi “knock-out first/conditional ready” mice were obtained from UC Davis and resuscitated on the C57BL/6J strain background. Adamts7 tm1a mice were bred with EIIa-Cre mice (JAX 003724, B6 background) to remove the PGK-Neo and loxP flanked exons, generating the tm1b “LacZ tagged null allele”. The Adamts7 E373Q catalytic mutant allele was generated using CRISPR (clustered regularly interspaced short palindromic repeats) HDR (Homology-Directed Repair) at the Harvard Genome Modification Facility on the C57BL/6J strain background. For experiments on the ApoE KO atherogenic background, both mouse strains were independently backcrossed to the ApoE KO line (JAX 002052, B6 background). All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committees of the Broad Institute of MIT and Harvard.
Statistical analysis.
To detect significant differences between 2 groups, two-tailed Student t test or one-way ANOVA (analysis of variance) with Tukey’s test was applied to data, which show normality, and Mann-Whitney test or Kruskal-Wallis test with Dunn’s multiple comparison test was used for data without evidence for normality. In repeated measures data, we applied two-way RM (repeated measurements) ANOVA with Sidak’s multiple comparisons test for comparison of 2 groups or Tukey’s multiple comparisons test for comparison of 3 or more groups. In data where two factors (genotype and sex) were considered, and assumptions of normality were met, we applied two-way ANOVA with Sidak’s multiple comparisons test for comparison of 2 groups or Tukey’s multiple comparisons test for comparison of 3 or more groups. Kolmogorov-Smirnov test was applied to test normality of the data. A value of P<0.05 was considered statistically significant. No experiment -wide / across-test or multiple test correction was applied. For all data, error bars indicate SEM. For statistical analysis, Graphpad Prism 8 (GraphPad Software) was used. Statistical details were shown in Online Table I.
RESULTS
Purification of full-length mouse ADAMTS7 active enzyme and E373Q catalytic mutant.
Prior reports have illustrated the importance of the carboxyl terminal thrombospondin repeats of ADAMTS7 for substrate interaction and specificity, yet it remains unknown whether ADAMTS7 catalytic activity mediates atherosclerosis. Furthermore, it has proven challenging to purify full-length ADAMTS7 protein13, 19, 22. We therefore sought to express and purify full-length ADAMTS7 to establish an in vitro cleavage assay. We first evaluated the expression of mouse and human full-length ADAMTS7 with a C-terminal 3x-Flag tag in adherent HEK293T cells. For each construct we generated a catalytic mutant form using a glutamate to glutamine substitution (EQ) in the conserved HExxH active site, generating mouse Glu373Gln (E373Q) or human Glu389Gln (E389Q), respectively (Fig. 1A). In the total cell lysate, ADAMTS7 migrates as two prominent bands: an immature ~180 kDa full-length form and a highly glycosylated ~250 kDa mature form. Although we detected similar levels of expression in the total cell lysate for mouse and human ADAMTS7, mouse ADAMTS7 was present at much higher levels in the media consistent with previous reports18, 22 (Fig. 1B). We also observed a trend of higher amounts of the catalytic mutant form in the media, possibly due to a lack of cis or trans auto-cleavage within the full-length ADAMTS7 protein. Initial characterization of full-length ADAMTS7 in the media described a mature highly glycosylated form migrating at around 250 kDa, with an additional population migrating in a smear >300 kDa associated with a chondroitin sulfate glycosaminoglycan (CS-GAG) attachment within the mucin domain18 (Fig. 1B). CS-GAG attachment typically occurs at Ser-Gly dipeptides and in the paralog ADAMTS12, CS-GAG attachment was prevented by mutating the serine residues within a conserved “SGSG” dipeptide region in the mucin domain to alanine23 (Fig. 1A). Within ADAMTS7 orthologs, a similar “SGSGS” region within the mucin domain is conserved in mammals, with conservation of the central serine extending to all vertebrates. Similar to the strategy used in ADAMTS12, we selectively mutated the “SGSGS” serine residues to alanine (referred to as S3A) in the presumptive mucin domain CS-GAG attachment site in both mouse and human ADAMTS7. The S3A variants eliminated the >300 kDa smear in the media associated with CS-GAG proteoglycan attachment for the Wild-type (WT) and EQ forms in mouse and human ADAMTS7 (Fig. 1B). Our results are consistent with the reported treatment of full-length mouse ADAMTS7 with chondroitinase ABC to remove the attached CS-GAG from the mucin domain18. Based on our construct design and test expression results, the mouse S3A versions represented full-length, non-proteoglycan forms of ADAMTS7 most amenable to purification.
Figure 1: Full-length mouse ADAMTS7 purification and in vitro TSP1 cleavage.
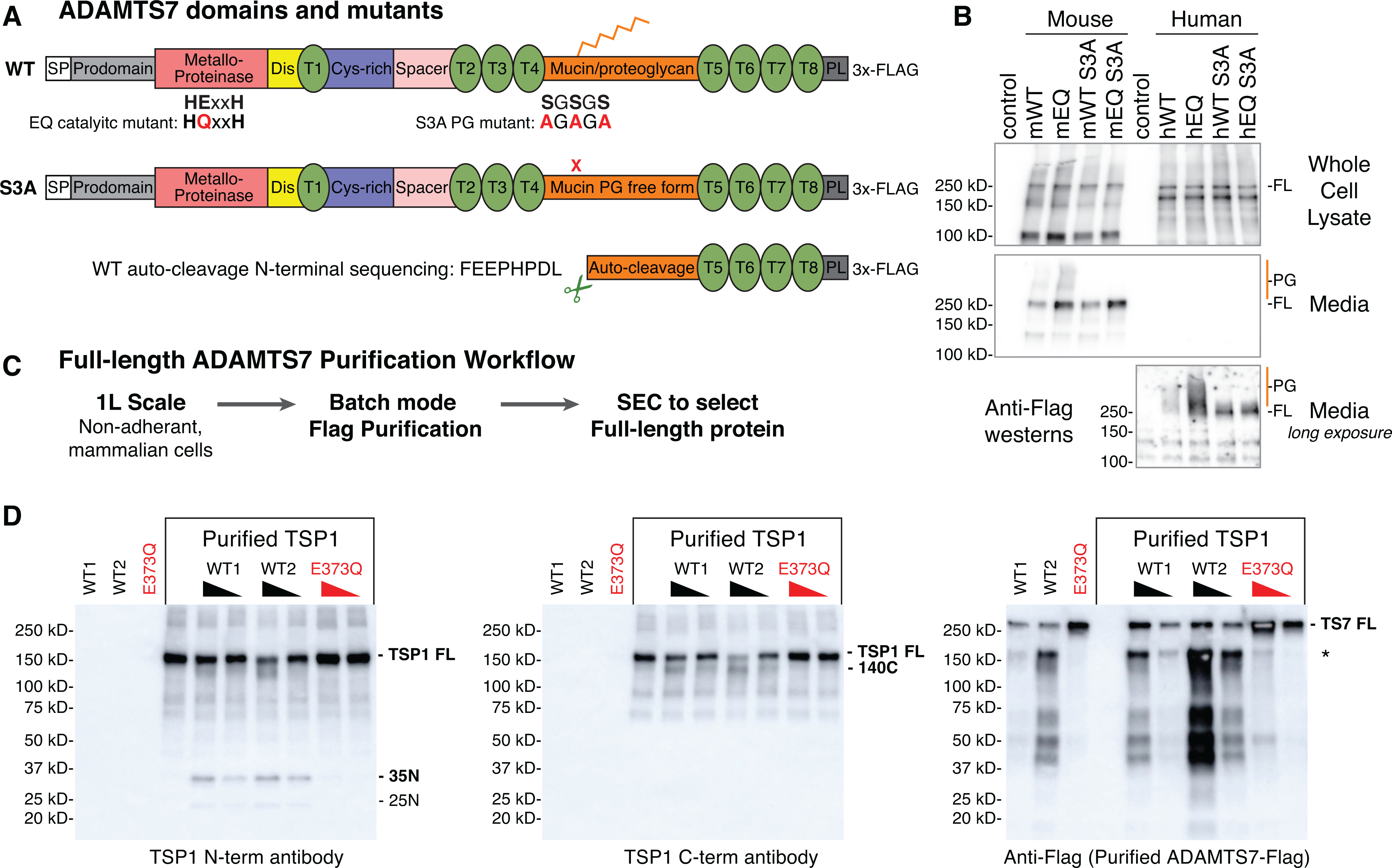
A, ADAMTS7 protein domains and locations of the glutamate to glutamine (EQ) catalytic mutant and serine to alanine (S3A) substitutions to prevent proteoglycan (PG) attachment. Abbreviated ADAMTS7 domains: signal peptide (SP), disintegrin (Dis), thrombospondin repeats (T), cysteine-rich (Cys-rich), protease and lacunin (PL). B, expression of full-length (FL) mouse and human ADAMTS7 3xFlag proteins in the whole cell lysate and secreted in the media under reducing conditions, detected by western blot using the M2-HRP antibody. High molecular weight proteoglycan (PG) species are marked in orange. A longer exposure of the conditioned media was required to visualize the secreted human ADAMTS7 proteins. C, full-length ADAMTS7 two step purification workflow used to purify WT S3A active enzyme and EQ S3A negative control protein. D, Thrombospondin1 in vitro cleavage by purified full-length mouse ADAMTS7 WT protein (* indicates co-purified auto-cleavage band). Western blots under reducing conditions to resolve proteolytic TSP1 bands generated by active ADAMTS7 WT purified enzyme. Presence of the E373Q catalytic mutation in the purified full-length mouse ADAMTS7 EQ protein ablated the catalytic activity.
Next, we purified full-length mouse ADAMTS7 WT and E373Q in the context of the S3A variant from non-adherent Expi293F cells using a two-step purification strategy (Fig. 1C). Following the Flag capture and elution step, both the WT S3A and EQ S3A displayed similar SEC profiles with peak B containing mostly high molecular weight aggregates, primary peak C containing the full-length 250 kDa mature form of ADAMTS7 and peak D containing the Flag peptide (Online Fig. IA–D). Within the WT S3A Flag and SEC fractions we noted the presence of band at ~150 kDa that contained the carboxyl terminal 3x-Flag tag and was purified along with the full-length WT S3A protein. Peptide sequencing of the lower band or the total purified WT S3A protein identified a potential auto-cleavage site at 1061.1062 (SYGS|FEEP) within the mucin domain that was absent in the EQ purification. This Flag tagged proteolytic fragment lacks the catalytic domain, therefore we collected SEC fractions containing the full-length ADAMTS7 along with differing amounts of mucin domain auto-cleavage product. ADAMTS7 was previously shown to cleave TSP1 by western blot, generating a 140 kDa cleavage product from the full-length 170 kDa monomeric TSP113. To test the catalytic activity of purified mouse ADAMTS7 WT, we independently tested WT1 (mostly full-length) and WT2 (full-length and mucin domain auto-cleavage) along with the purified E373Q catalytic mutant control protein (EQ) (Online Fig. IB,D). TSP1 cleavage products at 140 kDa were detected using an antibody to the C-terminal region in a dosage dependent manner for both WT1 and WT2 ADAMTS7 similar to previous results13 (Fig. 1D). Using an antibody to the N-terminal region of TSP1, we detected cleavage products migrating at 35 kDa and 25 kDa, suggesting ADAMTS7 cleavage at the proteolytically sensitive oligomerization domain of TSP124, 25. N-terminal TSP1 cleavage was dose dependent for the WT1 and WT2 purified proteins and absent in the EQ catalytic mutant control. Furthermore, this demonstrates that the E373Q substitution results in loss of catalytic activity.
Genetic ablation of ADAMTS7 catalytic function in the full-length protein.
To determine if ADAMTS7 catalytic activity mediates atherosclerosis, we generated a catalytic mutant mouse and compared it to the loss of function allele. To eliminate catalytic function, we employed the same glutamate to glutamine substitution to preserve the helical structure of the HExxH motif while preventing the generation of a reactive nucleophile at the zinc metalloprotease active site16 (Fig. 2A). The conserved metalloproteinase HExxH motif is present in mouse ADAMTS7 and the corresponding mouse ADAMTS7 E373Q mutation (CAG to CAC) was generated using CRISPR-HDR (Fig. 2B). From this approach, the Cas9 PAM (protospacer adjacent motif) site located in Ala371 (GCC) was also altered synonymously to prevent additional editing (GCT) resulting in two nucleotide positions changed in the E373Q catalytic mutant allele. We examined Adamts7 +/E373Q heterozygotes and observed no evidence of allelic expression imbalance from these modified positions (Fig. 2C). To generate a KO allele, we obtained the publicly available mouse Adamts7 tm1a “KO-first/conditional ready” allele which carries gene-trap LacZ reporter inserted into intron 4 ahead of loxP flanked exons 5 and 6. This allele was bred to EIIa-Cre mice for germline deletion of the loxP flanked exons encoding part of the catalytic domain to generate the Adamts7 tm1b KO allele used in our study (Online Fig. IIA). To test for Adamts7 expression in the heart using our homozygous mouse alleles, we used two probe sets located at the exon 4–5 boundary (absent in the tm1b allele) and at the exon 23–24 boundary of the last intron. As expected, no detectable signal was present from the exon 4–5 probe in the Adamts7 KO (tm1b homozygotes). Residual signal in the KO was present with the exon 23–24 probe, similar to what was observed in Adamts7 tm1a homozygous KO11. No difference in Adamts7 mRNA expression levels was observed between WT and E373Q/E373Q mice for either probe set (Fig. 2D).
Figure 2: Generation and characterization of the mouse Adamts7 E373Q catalytic mutant allele.
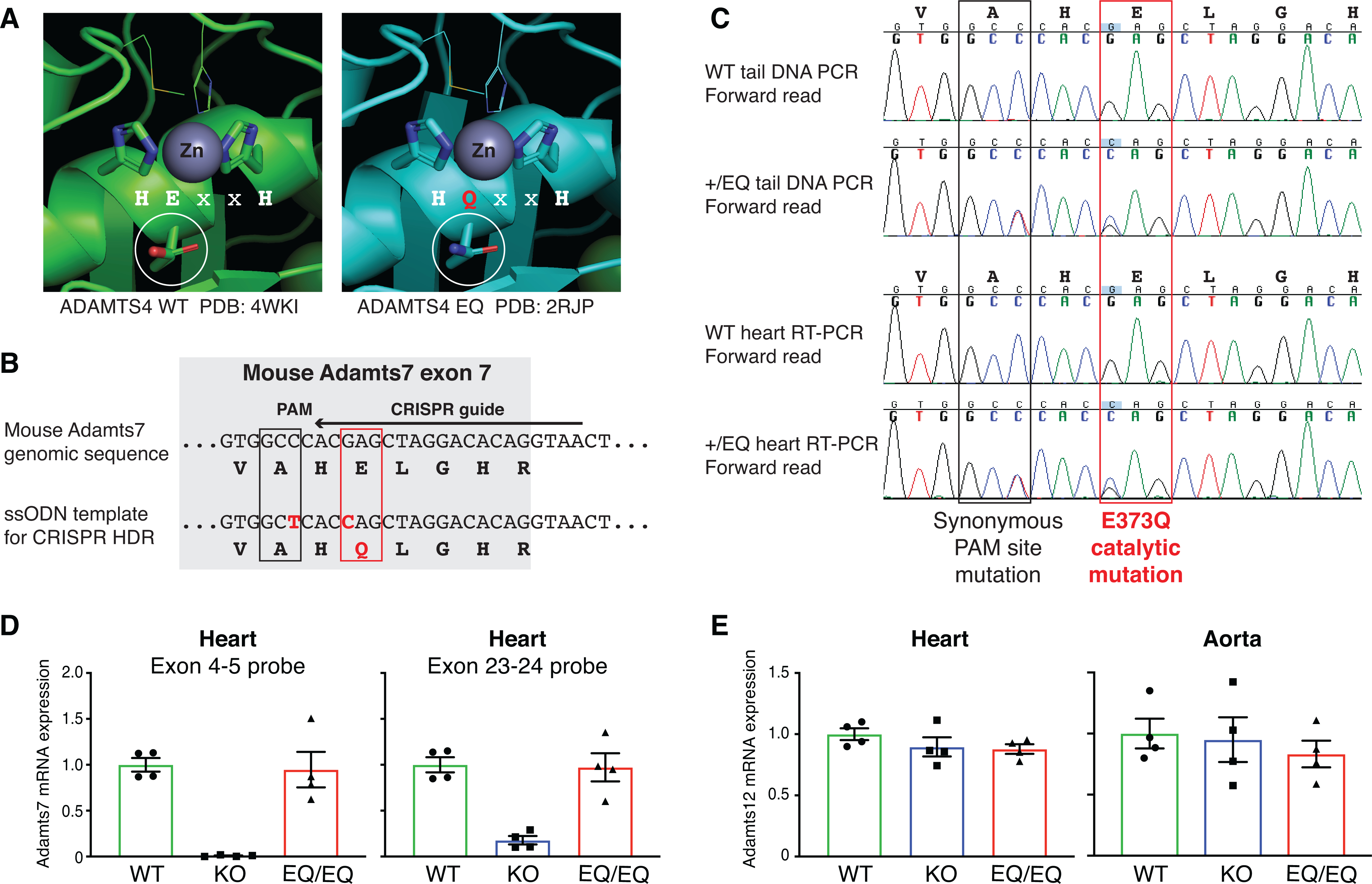
A, Structural context of the ADAMTS7 HExxH to HQxxH catalytic mutation. Crystal structures from ADAMTS4 WT (PDB:4WKI)33 and ADAMTS4 EQ (PDB: 2RJP)34 were aligned and annotated in PyMOL to highlight the catalytic residues and visualize the Zinc metal in the active site. The E to Q substitution preserves the tertiary structure of ADAMTS4 and the residues “VAHELGH” in the active site are conserved in ADAMTS7. B, Schematic of the Adamts7 E373Q catalytic mutant allele within exon 7. To generate the mutant allele, c. 1117G->C (p. E373Q) mutation at the Adamts7 catalytic domain and c. 1113C->T (p. A371A) to disrupt the PAM site were induced by CRISPR Homology-Directed Repair (HDR). C, Sanger sequencing of genomic tail DNA PCR and heart mRNA RT-PCR from WT and heterozygous +/E373Q mice. Representative forward reads show no evidence of allelic expression imbalance at the two nucleotide substitutions. D, Adamts7 mRNA expression level in the heart from WT, Adamts7 −/− (KO) and homozygous E373Q/E373Q (EQ/EQ) mice were measured by real time quantitative polymerase chain reaction (qPCR) using 2 TaqMan probe sets (exon 4–5 boundary and exon 23–24 boundary). n=4 per group. E, Adamts12 mRNA expression levels were analyzed by real time qPCR in the heart and aorta harvested from WT, KO and EQ/EQ mice. n=4 per group. A Kruskal-Wallis test with Dunn’s multiple comparison test was applied to D and E.
ADAMTS12 is known to share the same domain organization as ADAMTS7 and the two paralogs likely evolved from a gene duplication event18, 26. A recent paper reported compensatory regulation between Adamts7 and Adamts12 transcription levels, notably with upregulation of Adamts12 in the tendon from Adamts7 KO mice23. Therefore, we evaluated Adamts12 mRNA expression in the heart and aorta from WT, Adamts7 KO and E373Q/E373Q mice. We found no evidence of compensatory regulation of Adamts12 in these tissues (Fig. 2E).
Genetic inhibition of ADAMTS7 catalytic function prevents atherosclerosis
Using our characterized Adamts7 KO and catalytic mutant alleles, we examined the development of atherosclerosis in two different atherogenic backgrounds. In the first model, adult mice were injected with an AAV8 (adeno-associated virus) gain of function mutant form of PCSK9 (proprotein convertase subtilisin/kexin type 9) (AAV8-mPCSK9 D377Y) and placed on a high fat Western diet for 16 weeks to induce atherosclerosis27 (Online Fig. IIB). Following a single injection, plasma PCSK9 levels reached peak expression within one week and persisted for 16 weeks during the atherogenesis study (Online Fig. IIIA,D). Plasma total cholesterol levels were >15mM within one week and were persistently high, consistent with the significant reduction of LDLR in the liver at week 16 (Online Fig. IIIB,C,E,F). To examine the expression of ADAMTS7 throughout development of atherosclerosis, we utilized the LacZ reporter from heterozygous Adamts7 +/tm1b mice. We harvested aortic roots from heterozygous Adamts7 +/tm1b mice, which were injected with AAV8-mPCSK9 D377Y, after 6, 8, 12 and 16 weeks on the high fat diet (Fig. 3A). β-gal staining was evident 6 weeks post injection before plaque formation and present in early plaques at 8 weeks post injection (Fig. 3B–D). Fewer β-gal positive cells were observed at 12 and 16 weeks post injection, consistent with previous reports of transient ADAMTS7-LacZ expression during atherogenesis11 (Fig. 3B,E-F). Alterations in ADAMTS7 expression were not evident after 4 weeks on the high fat diet (Online Fig. IIC–D) and no β-gal staining was detected in littermate WT control atherosclerotic plaques or in the heart (Online Fig. IIE).
Figure 3. Transient expression of ADAMTS7 during atherogenesis.
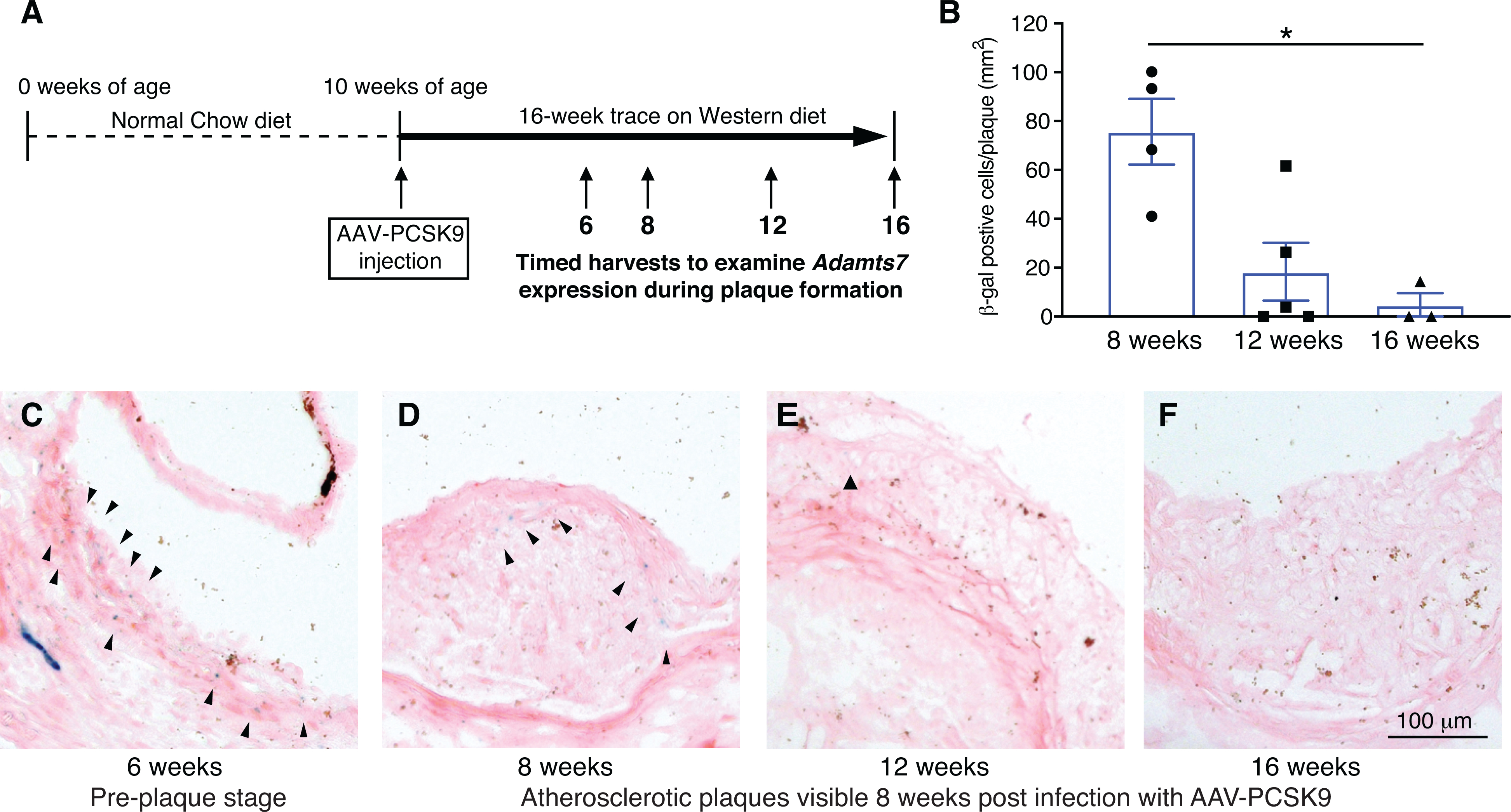
ADAMTS7 expression reported by the LacZ gene trap tm1b allele in heterozygotes during atherogenesis at 6, 8, 12 and 16 weeks post infection with AAV8-PCSK9. A, Stages examined in the atherogenic model post infection with AAV8-PCSK9. B, Quantitative analysis of β-gal positive cells in the plaque (n=3 to 5 per group, data points represent individual animals). A Kruskal-Wallis test with Dunn’s multiple comparison test was applied, *P<0.05. C-F, Representative photomicrographs of β-gal staining in the aortic sinus at the indicated weeks after AAV8-PCSK9 injection.
To confirm the effect of Adamts7 deficiency on the development of atherosclerosis, we examined male and female Adamts7 KO mice compared to littermate controls. Adamts7 KO mice showed significantly decreased lesion formation in en face analysis of the aortic arch region (mean ±SEM for all en face results: control littermates 23.9 ±1.2% versus Adamts7 KO mice 19.0 ±1.1%; P=0.031 [male] and control littermates 25.0 ±1.2% versus Adamts7 KO mice 21.1 ±1.1%; P=0.024 [female]; Fig. 4A,D). From the same mice, we harvested and sectioned aortic roots to quantitatively assess plaque progression and composition. There was no significant difference in plaque burden in the aortic root between 2 groups (Fig. 4B,E). By the immunohistochemical analysis of aortic root plaques composition, we observed a significant reduction in SMC (smooth muscle cell) content in Adamts7 KO mice compared to control littermates (Fig. 4C,F). Macrophage accumulation and collagen content in the aortic plaque were not affected by Adamts7 deficiency (Online Fig. IIIG,H).
Figure 4. Decreased plaque formation in Adamts7 knockouts and in homozygous catalytic mutants in the AAV-PCSK9 atherogenic mouse model.
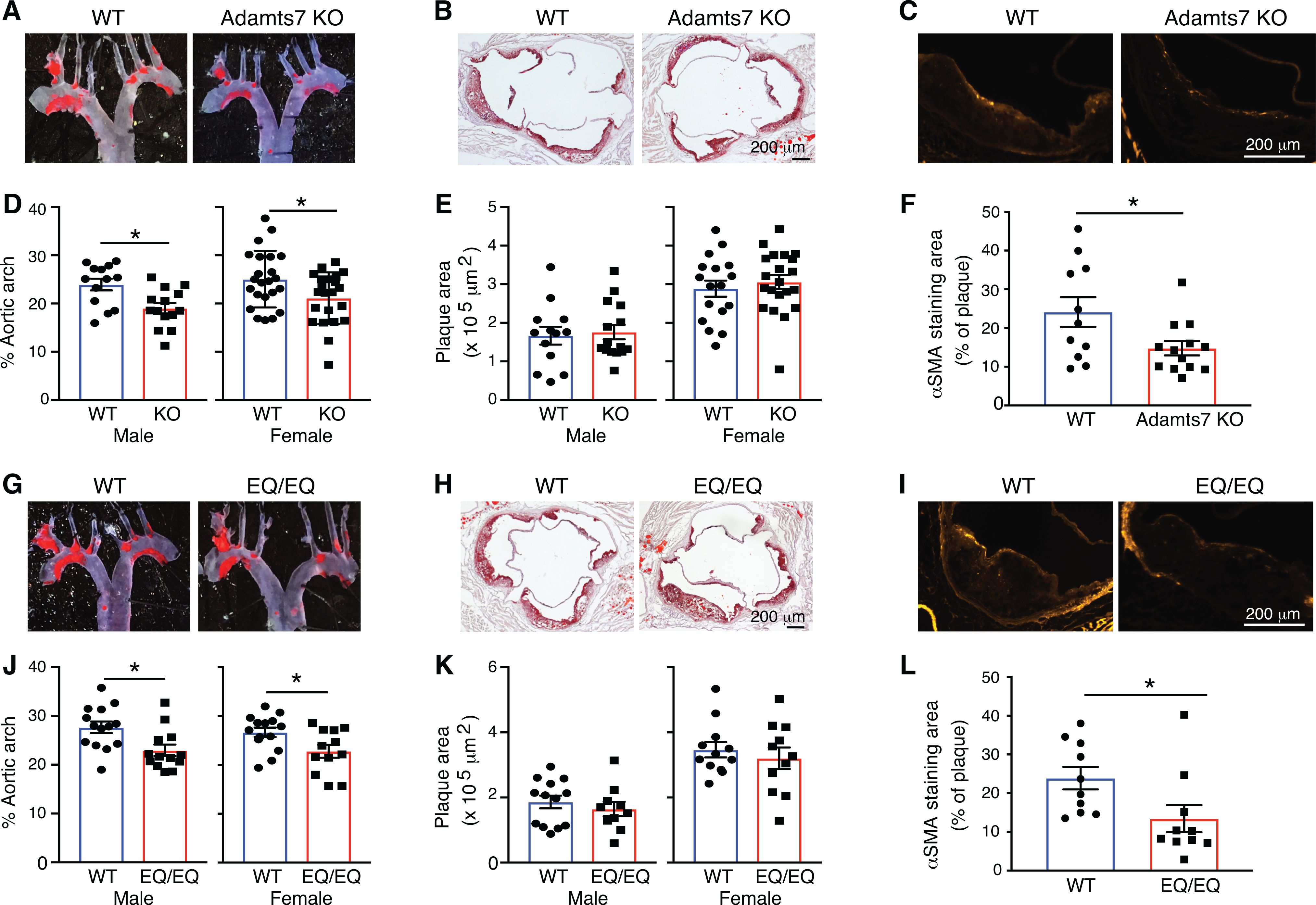
At the age of 10 weeks, mice were injected with rAAV8-D377Y-mPCSK9 and challenged a western diet for an additional 16 weeks to stimulate atherosclerosis. The mice were euthanized at 26 weeks of age for evaluation of atherosclerosis in the aortic arch and aortic root. A, B, D and E, Representative photomicrographs of Oil-Red O staining and quantitative analysis of atherosclerotic lesion area in the aortas from littermate controls and Adamts7 KO (D-E, males, n=13 to 15 per group; females, n=17 to 23 per group). G, H, J and K, From a separate +/EQ x +/EQ cross, littermate controls and EQ/EQ homozygotes were evaluated for atherosclerosis (J-K, males, n=10 to 14 per group; females, n=11 to 14 per group). C,F,I and L, Representative sections and quantitative analyses of α-SMA positive smooth muscle cells in the aortic sinus (n=10 to 12 per group). Data points represent individual animals, error bars indicate means ±SEM. Two-way ANOVA with Sidak’s multiple comparison test was applied to D, E, J and K. A two-tailed Student’s test was applied to F, which showed normality and Mann-Whitney test was applied to L, which did not pass a normality test. Normality was tested by Kolmogorov-Smirnov test. *P<0.05.
Next, we examined male and female Adamts7 E373Q/E373Q homozygotes compared to littermate controls. Adamts7 E373Q/E373Q mice showed significantly decreased lesion formation in en face analysis of aortic arch (control littermates 27.7 ±1.2% versus E373Q/E373Q mice 22.9 ±1.2%; P=0.010 [male] and control littermates 26.7 ±1.0% versus E373Q/E373Q mice 22.8 ±1.3%; P=0.045 [female]; Fig. 4G,J). In parallel with the en face analysis, cross-sectional analysis of aortic roots was performed. There was no significant difference in plaque burden in the aortic root between 2 groups (Fig. 4H,K). Consistent with the reduced SMC content in the aortic plaque from Adamts7 KO mice, a significant reduction in SMC content was observed in Adamts7 E373Q/E373Q mice (Fig. 4I,L). Collectively, these results show that atherosclerosis-related phenotype of the catalytically inactive ADAMTS7 mice mirrors the atherosclerosis phenotype of Adamts7 KO mice.
ADAMTS7 dosage and catalytic activity controls atherogenesis.
In the second mouse model, we backcrossed the Adamts7 alleles to the atherogenic ApoE KO background. To assess the dosage effects of Adamts7 loss of function, we examined WT, heterozygous and homozygous KO mice exposed to a high-fat Western diet for 10 weeks. On this background, we observed a decreased lesion formation in the aortic arch of Adamts7 KO mice compared to WT mice, which is consistent with previous reports11 (Fig. 5A). Quantitation of the en face results showed a significant difference between WT and KO (control littermates 21.6 ±1.4% versus Adamts7 KO mice 15.8 ±1.5%; P=0.027 [male] and control littermates 29.5 ±1.9% versus Adamts7 KO mice 22.7 ±2.0%; P=0.014 [female]; Fig. 5B–C). A comparison between WT and heterozygotes missing one functional Adamts7 allele displayed a similar trend suggesting benefits from 50% loss of function (control littermates 21.6 ±1.4% versus Adamts7 +/tm1b mice 18.4 ±1.2%; P=0.27 [male] and control littermates 29.5 ±1.9% versus Adamts7 +/tm1b mice 24.4 ±1.0%; P=0.033 [female]; Fig. 5A–C). Next, we analyzed the plaque content from aortic root sections on the ApoE KO background. Similar to our results on the AAV-PCSK9 model, we did not observe a significant difference in aortic root for any of the genotypes on the ApoE KO background (Online Fig. IV A–C).
Figure 5: Adamts7 dosage and catalytic dependent effects on the ApoE KO atherogenic background.
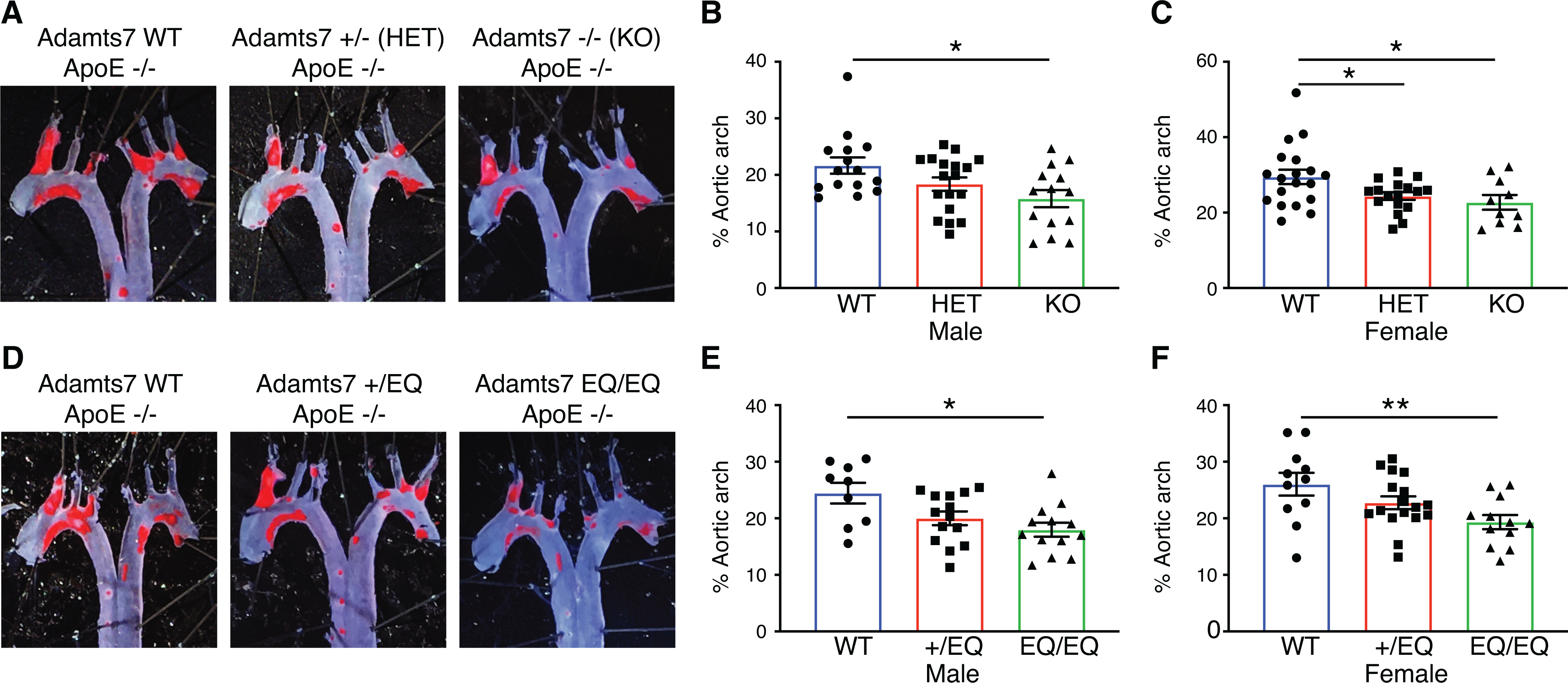
Mice from heterozygous intercrosses for the tm1b loss of function allele or the E373Q catalytic mutant allele, were challenged with 10 weeks of high fat diet to stimulate atherosclerosis on the ApoE KO background. The mice were euthanized at 20 weeks of age for evaluation of atherosclerosis. A and D, Representative photomicrographs of Oil-Red O staining and quantitative analysis of atherosclerotic lesion area in the aortic arch. B, male WT vs Adamts7 +/− (HET) vs Adamts7 −/− (KO), n=14 to 18 per group; C, female WT vs HET vs KO, n=10 to 19 per group; E, male WT vs +/EQ vs EQ/EQ, n=9 to 14 per group; F, female WT vs +/EQ vs EQ/EQ, n=11 to 17 per each group. Data points represent individual animals, error bars indicate means ±SEM, and two-way ANOVA with Tukey’s test was applied. Normality was tested by Kolmogorov-Smirnov test. *P<0.05, **P<0.01.
In a parallel cross, we examined WT, heterozygous Adamts7 +/E373Q and homozygous Adamts7 E373Q/E373Q mice after administration of a high-fat Western diet for 10 weeks. From this cross, we observed a significant decrease in lesion formation in the aortic arch region between WT and Adamts7 E373Q/E373Q mice both in male and female mice (control littermates 24.5 ±1.8% versus Adamts7 E373Q/E373Q mice 18.0 ±1.2%; P=0.011 [male] and control littermates 26.0 ±2.0% versus Adamts7 E373Q/E373Q mice 19.3 ±1.2%; P=0.0057 [female], Fig. 5D–F). We also observed an intermediate phenotype in Adamts7 +/E373Q heterozygous animals approaching significance for both sexes (control littermates 24.5 ±1.8% versus Adamts7 +/E373Q mice 20.0 ±1.2%; P=0.10 [male] and control littermates 26.0 ±2.0% versus Adamts7 +/E373Q mice 22.8 ±1.1%; P=0.21 [female], Fig. 5D–F). No significant difference in lesion formation in the aortic root was observed (Online Fig. IVD–F). Collectively our results on the ApoE KO atherogenic background corroborate our data from the AAV-PCSK9 atherogenic model and support dosage-dependent protective effects from ADAMTS7 catalytic inhibition.
Adamts7 catalytic function is responsible for VSMC migration.
ADAMTS7 has previously been implicated in the regulation of VSMC migration10–13. To reveal the effect of ADAMTS7 catalytic inactivation on VSMC migration, we performed wound healing assays on the primary VSMCs harvested from WT, Adamts7 KO and E373Q/E373Q mice. We verified that ADAMTS7 catalytic inactivation and Adamts7 gene KO did not affect Adamts7 or Adamt12 mRNA expression in primary VSMCs (Online Fig. VA–C). Our results from the wound healing assay validated reduced migration of Adamts7 KO VSMCs compared with WT VSMCs consistent with the previous literature11 (Fig. 6A–B). Subsequently VSMCs from E373Q/E373Q mice were tested for migration and showed less migratory activity compared to littermate WT VSMCs (Fig. 6C–D), supporting a role for ADAMTS7 catalytic function in promoting VSMC migration.
Figure 6. ADAMTS7 catalytic function is responsible for ADAMTS7-mediated VSMC migration.
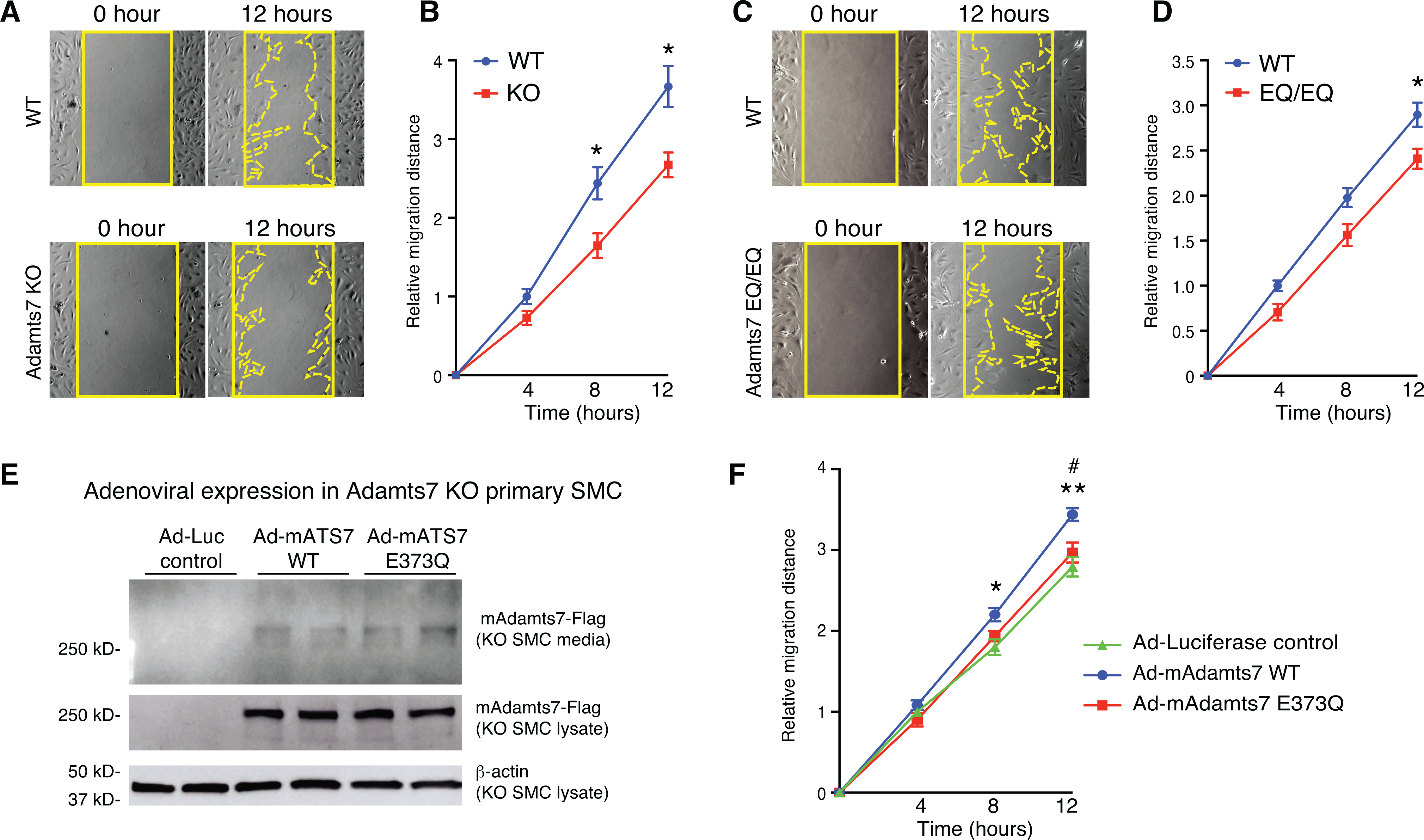
A-D, Migration of primary VSMCs from Adamts7 −/− (KO) and homozygous EQ/EQ catalytic mutant mice was assessed by wound healing assay. A and C, Representative photomicrographs of the wound at 0 and 12 hours in indicated genotypes of VSMCs are shown. Solid line indicates wound edge. Dotted line indicates migration edge. B and D, The distance of migration is quantified at 4, 8 and 12 hours. n=8 to 9 per group. E, Representative images of western blot in conditioned media and cell lysates from adenovirus-infected primary Adamts7 KO VSMCs. Cell lysates and conditioned media collected from Ad-Luciferase-, Ad-mAdamts7 (mAts7)-WT- and Ad-mAts7-E373Q-infected primary Adamts7 KO VSMCs were subjected to western blot analysis with anti-Flag antibody. F, Migration of primary Adamts7 KO VSMCs infected with Ad-Luciferase, Ad-mAdamts7 WT and Ad-E373Q was assessed by wound healing assay. The mean distance of migration is quantified at 4, 8 and 12 hours. n= 8 to 9 per group. Error bars indicate ±SEM. Two-way RM ANOVA with Sidak’s multiple comparisons test was applied to B, D. *P<0.05. Two-way RM ANOVA with Tukey’s test was applied to F. Normality was tested by Kolmogorov-Smirnov test. **P<0.01, (Ad-Luciferase vs Ad-mAts7 WT), #P<0.05 (Ad-mAts7-WT vs Ad-mAts7–373Q).
To test whether catalytic inactivation of ADAMTS7 counteracts pro-migratory effects of ADAMTS7, VSMCs from Adamts7 KO mice were infected with Ad-Luciferase, Ad-mouse Adamts7 WT (mAts7 WT) or Ad-mouse Adamts7-E373Q (mE373Q) and were subject to wound healing assays. Western blot analyses confirmed no differences in protein levels in cultured media and cell lysates between ADAMTS7-WT and ADAMTS7-E373Q (Fig. 6E). As expected, overexpression of ADAMTS7-WT, but not E373Q, promoted VSMC migration, indicating that pro-migratory effects were abolished by the E373Q substitution without affecting the protein levels (Fig. 6F). In summary our in vitro studies indicate that ADAMTS7 catalytic activity promotes VSMC migration.
ADAMTS7 coding variant associated with CAD risk affects secretion and VSMC migration.
Having established that inhibition of ADAMTS7 catalytic activity prevents atherosclerosis and VSMC migration, we then sought to determine the impact of a coding variant in ADAMTS7 that is associated with CAD risk. A nonsynonymous variant in ADAMTS7, rs3825807, results in serine to proline substitution at amino acid 214. The proline allele, with an allele frequency of 0.43 in individuals of European ancestry, has been associated with reduced risk for CAD3. To further examine the importance of this coding variant, we generated full-length human expression vectors (1–1686 a.a. with a C-terminal 3xFlag tag) carrying either the common risk allele Ser214 or the protective allele Pro214. Transfection into HEK293T cells shows that Ser214 is secreted into the media in a dose dependent manner while Pro214 is hindered at all doses tested (Fig. 7A–B). Analysis of the lysate shows similar levels for Ser214 and Pro214 expressed proteins at both the highly glycosylated 270kDa mature and immature 180kDa forms (Fig. 7C). Next we transferred the full-length ADAMTS7 alleles into an adenovirus expression system and tested the secretion phenotype in human coronary artery smooth muscle cells (HCA-SMC). Once again we observed a reduction in Pro214 secretion into the media compared to the Ser214 allele (Fig. 7D–E; Online Fig. VD). In contrast, we detected an increased level of mature Pro214 ADAMTS7 in the HCASMC lysates (Fig. 7F).
Figure 7. ADAMTS7 Ser214 CAD risk variant rs3825807 displays increased secretion and function compared to the Pro214 protective variant or catalytic mutant.
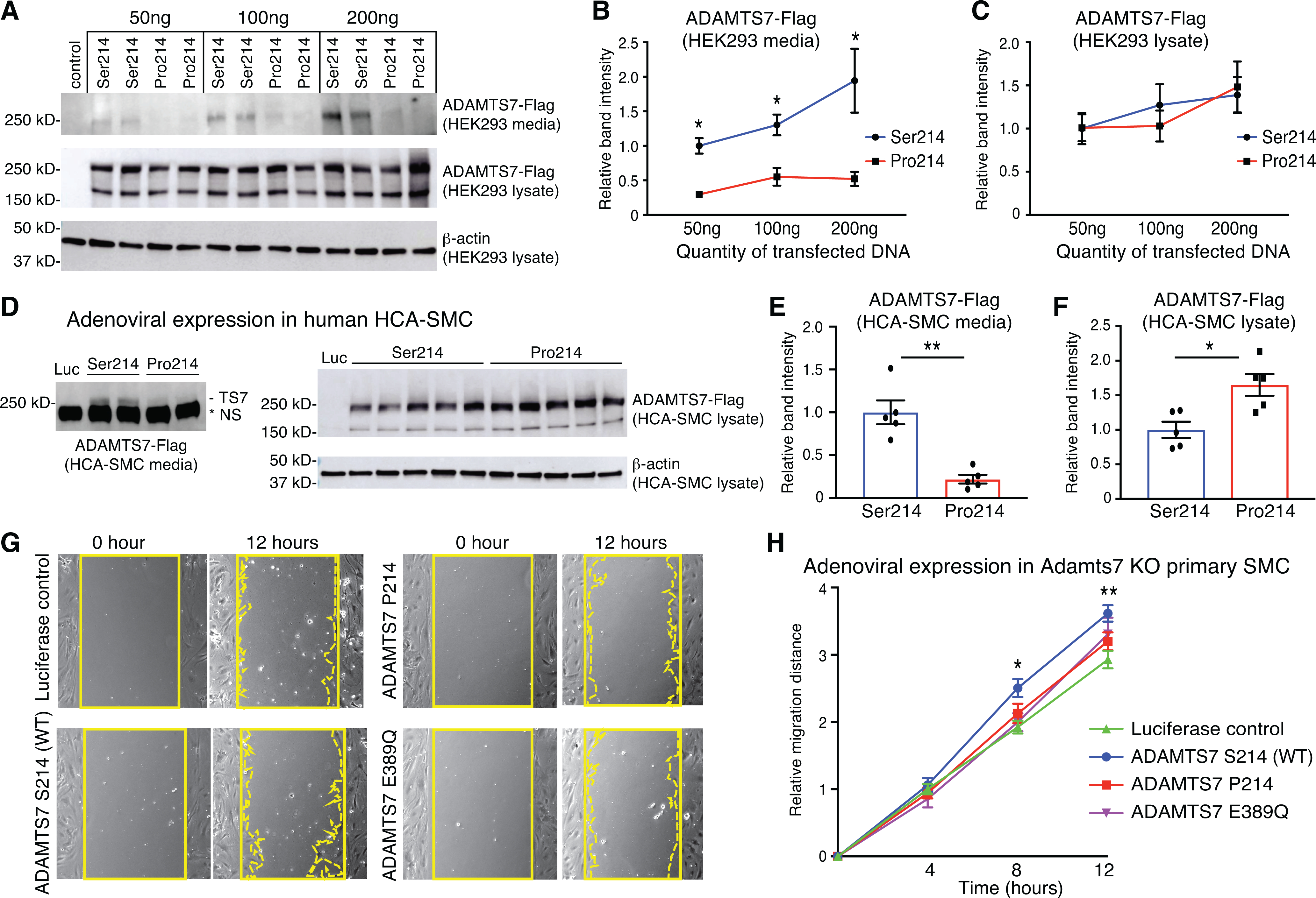
A-C, Representative images and quantifications of western blots from HEK 293 cells transfected with empty vector control, full-length ADAMTS7 risk variant Ser214 or Pro214. A, Full-length ADAMTS7 3xFlag protein was detected by anti-Flag antibodies from conditioned media and cell lysates. B-C, Relative band intensities were quantified from n=4 per group. D-F, Representative images and quantifications of western blot in conditioned media and cell lysates from adenovirus-infected HCA-SMCs infected with Ad-Luciferase (Luc), Ad-ADAMTS7-Ser214 or Ad-ADAMTS7-Pro214. D, Full-length ADAMTS7 3xFlag protein was detected by western blot in conditioned media and cell lysates. NS (non-specific) band migrating faster than ADAMTS7-Flag is present in Luc control. E-F, Relative band intensities were quantified (n=5 per group). Error bars indicate means ±SEM, Mann-Whitney test was applied to B, C, E and F, *P<0.05, **P<0.01. G, Migration of primary Adamts7 KO VSMCs infected with Ad-Luciferase, Ad-ADAMTS7-Ser214 (WT), Ad-ADAMTS7-Pro214 or the Ad-ADAMTS7 catalytic mutant E389Q was assessed by wound healing assay. Representative photomicrographs of the wound at 0 and 12 hours in VSMCs infected with indicated adenovirus are shown. Solid line indicates wound edge. Dotted line indicates migration edge. H, Migration was quantified at 4, 8 and 12 hours (n= 8 to 9 per group, two-way RM ANOVA with Tukey’s test. Normality was tested by Kolmogorov-Smirnov test. *P<0.05, **P<0.01 for LUC vs Ser214).
Finally, to test the effects of the human risk and protective alleles, we harvested primary VSMC from the Adamts7 KO mice and analyzed the lateral migration induced by adenoviral expression of human ADAMTS7 (Fig. 7G). Expression of the human Ser214 risk allele resulted in the highest amount of VSMC migration which was significant compared to a Luciferase control (Fig. 7H). There was a reduction in migration for the Pro214 allele, consistent with the observed reduction of ADAMTS7 levels detected in the media. Because ADAMTS7 catalytic function has been implicated in regulation of VSMC migration (Fig. 6), the ADAMTS7 E389Q (EQ) catalytic mutation made in the Ser214 allele construct was transferred to an adenovirus expression system to test if the reduction in migration for the Pro214 allele is identical with that for the ablation of ADAMTS7 catalytic function. Expression of the Pro214 and EQ catalytic mutant were comparable to Ser214 allele (Online Fig. VE). However, the amount of induced migration from the ADAMTS7 EQ catalytic mutant was reduced compared to the Ser214 allele, and was equivalent to the hypomorphic Pro214 allele (Fig. 7H). Our results suggest that loss of catalytic activity may be functionally equivalent to lower levels of ADAMTS7 expression in VSMC.
DISCUSSION
Population based genetic studies have identified more than a hundred disease associated genetic loci for CAD. However, it remains challenging to leverage these associations into causative genes, biological mechanisms and ultimately into new therapies 28. In most cases GWAS provides a location with candidate gene(s) but does not intrinsically define directionality or mechanism for disease. This understanding is essential for translating a GWAS hit into a druggable target and requires validation in cell-based assays and model organisms. From the integrative analysis of multiple variants, there is support for ADAMTS7 as the causal gene at the human chromosome 15q25 locus2–4. ADAMTS7 loss of function correlates with atheroprotection in the mouse and provides support for a therapeutic antagonist approach11. As a secreted enzyme in a gene family with several examples of small molecule29 and antibody-based inhibitors30, ADAMTS7 is a potential therapeutic target, yet a mechanism for disease independent of catalytic function remained a distinct possibility.
Here, we have demonstrated for the first time that protease activity is essential for proatherogenic effects of ADAMTS7. We purified full-length ADAMTS7 through modification of the mucin domain chondroitin sulfate attachment site and demonstrated the of loss of protease activity for the catalytic mutant form in an in vitro TSP1 cleavage assay. Next, we generated a catalytically inactive ADAMTS7 mouse line and compared this to an Adamts7 KO mouse line in two different atherogenic backgrounds. In each case, we observed a critical role for both ADAMTS7 catalytic activity and dosage in atherosclerosis. In aggregate, our in vivo studies provide compelling evidence that inhibiting ADAMTS7 catalytic inhibition can be a promising therapeutic approach for atherosclerosis, an effect equivalent to genetic depletion of ADAMTS7.
In both of our atherogenic mouse models, we observed that a reduction in atherosclerosis development was most prominent in the aortic arch and brachiocephalic region. We did observe a reduction in atherosclerosis in the lower sections of the aorta but the timeframes of atherogenesis we choose for our study did not result in a robust accumulation in plaques for these regions in our animal facility. Therefore, a limitation in our study is that most of the beneficial effects were based from measurements in the thoracic aortic region. Assessment of the aortic root plaques at a different point during atherosclerosis progression in our mice may have revealed a significant difference in lesion volume similar to previously reported results11. Although we did not detect a significant difference in the amount of plaque in the aortic root, we did observe a reduction in α-smooth muscle actin (SMA) staining at this location consistent with a decrease in SMC content in both Adamts7 KO and catalytic mutant homozygotes. Careful investigation of ADAMTS7 expression by β-gal staining in Adamts7 +/tm1b heterozygotes revealed a transient window of expression in developing and early plaques in the aortic root. Remarkably this expression did not persist in later stages of plaque development. Our observations are consistent with a transient ADAMTS7 expression response given an atherogenic or vascular injury challenge11, 12. Future studies applying single cell RNA sequencing technologies may help to identify the VSMC subtypes that transiently express ADAMTS7 during these processes. Coupled with our in vitro results showing a reduction in SMC migration in both loss of function and catalytic mutants, and rescue of migration with only the active form of ADAMTS7, our data supports a model in which ADAMTS7 facilitates atherosclerosis through VSMC phenotypic switching mediated through catalytic function.
In the active form of full-length mouse ADAMTS7, we consistently observed a lower band at 150 kDa in the media. Purification using a carboxyl terminal Flag tag retained both the full-length and 150 kDa product from the WT construct, in contrast to only the full-length EQ protein from the catalytic mutant. Amino terminal sequencing identified the WT 150 kDa band beginning at phenylalanine 1062 to support a predominant ADAMTS7 auto-cleavage event at 1061.1062 (SYGS|FEEP) nearby the CS-GAG attachment site in the mucin domain. Removal of the amino acids 1062–1657 would preserve a CS-GAG tethered enzyme that lacks a carboxyl terminus normally thought to be required for substrate recognition. The mouse ADAMTS7 auto-cleavage site is adjacent to one of the few highly conserved regions within the mucin domain and is partially conserved in human ADAMTS7 1080.1081 (SYGP|SEEP), although we were unable to confirm auto-cleavage for WT human ADAMTS7. Whether or not this is a regulated event, the consequence could be a shift in exosite substrate specificity to the remaining catalytic domain.
We have also examined the biological effect of a CAD associated coding variant, rs3825807 (Ser214Pro). Although this position is not conserved in rodents, it is located in the prodomain within a predicted alpha helix upstream from the Furin cleavage site at the start of the metalloproteinase domain. The hypomorphic secretion of the full-length Pro214 protective allele is similar to those published with a truncated ADAMTS7 (1–460 a.a.) containing only the prodomain and catalytic domain10. We found that the Pro214 allele resulted in hypomorphic function due to both decreased ADAMTS7 secretion and reduced vascular smooth muscle cell migration. Expression of the full-length Ser214 risk variant stimulated VSMC migration compared to control, Pro214 or EQ catalytic mutant variants in cells devoid of ADAMTS7 function. Thus, our results are in keeping with prior reports that this variant is likely to explain at least a portion of the CAD associated risk at this locus6–10.
Before ADAMTS7 was reported as CAD GWAS locus, it was demonstrated that ADAMTS7 knockdown prevented, while ADAMTS7 overexpression increased, neointima formation in a rat carotid artery vascular injury model12. Mouse knockout studies confirmed that total loss of function reduced neointima formation in the carotid13 and femoral arteries11. We have yet to examine our catalytic mutant mice in a vascular injury model to determine if this phenotype is mediated purely through catalytic function or if the additional domains of ADAMTS7 contribute to non-catalytic protein-protein based interactions in the extracellular matrix. For example, it was shown that catalytic mutations in the paralog ADAMTS12 were able to partially rescue the Adamts12 loss of function effects in an aortic ring endothelial cell sprouting assay31. Additionally, there has been a lack of characterized ADAMTS7 substrate cleavage sites for the reported substrates COMP and TSP1 that may contribute to the observed vascular phenotype. Although we were able to show in vitro cleavage of TSP1, we were unable to identify endogenous TSP1 cleavage fragments generated from ADAMTS7 activity. Thus, a limitation to our study would be a direct link to an ADAMTS7 substrate with biological effects that is largely unchanged in both our loss of function and catalytic mutant homozygotes. Recently an unbiased ADAMTS7 substrate cleavage site identification method using terminal amine isotopic labeling of substrates (TAILS) was employed to search for candidate sites from a human fibroblast secretome32. Further work will be needed to link these candidate cleavage sites, or others identified by similar TAILS experiments, to hone in an endogenous substrate or substrates responsible for the vascular phenotypes observed in ADAMTS7 catalytic mutant mice.
In conclusion, our current study provides compelling evidence that ADAMTS7 dosage and catalytic activity contribute to the atherosclerosis phenotype. These findings support the hypothesis that therapeutic inhibition of ADAMTS7 catalytic activity can reduce atherosclerosis.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
ADAMTS7 (a distintegrin and metalloproteinase with thrombospondin motifs 7) has been identified as a locus for coronary artery disease (CAD) from human population studies.
Complete Adamts7 loss of function in mice results in protection from atherosclerosis suggesting that ADAMTS7 is pro-atherogenic.
ADAMTS7 is a large multidomain secreted enzyme considered to be a therapeutic target for CAD, however it is unclear if ADAMTS7 enzymatic function controls the progression of atherosclerosis.
What New Information Does This Article Contribute?
We generated a catalytic mutant form of ADAMTS7 and studied the effects in mouse models of atherosclerosis to demonstrate that loss of ADAMTS7 protease function is equivalent to the knockout phenotype.
Catalytic mutant vascular smooth muscle cells show reduced migration similar to knockout cells; and these effects correlate with the protective human coding variant which we show to have reduced secretion compared to the ADAMTS7 risk variant.
The current study supports a role for ADAMTS7 protease function during atherosclerosis and suggest that therapies targeting ADAMTS7 catalytic activity will have beneficial effects.
ACKNOWLEDGEMENTS
This work was supported by a research grant from Bayer AG within the Cardiovascular Disease Initiative at the Broad Institute. T.M. was supported by a Uehara Memorial Foundation Research Fellowship. P.T.E. is supported by the Fondation Leducq (14CVD01), the NIH (1RO1HL092577, R01HL128914, and K24HL105780) and the American Heart Association (18SFRN34110082).
COMPETING INTERESTS
This work was supported by a research grant from Bayer AG within the Cardiovascular Disease Initiative at the Broad Institute. T.M. performed this work at the Broad Institute and is now an employee of Verve Therapeutics. S. K. is an employee of Verve Therapeutics, and holds equity in Verve Therapeutics, Maze Therapeutics, Catabasis, and San Therapeutics. He is a member of the scientific advisory boards for Regeneron Genetics Center and Corvidia Therapeutics; he has served as a consultant for Acceleron, Eli Lilly, Novartis, Merck, Novo Nordisk, Novo Ventures, Ionis, Alnylam, Aegerion, Haug Partners, Noble Insights, Leerink Partners, Bayer Healthcare, Illumina, Color Genomics, MedGenome, Quest, and Medscape; he reports patents related to a method of identifying and treating a person having a predisposition to or afflicted with cardiometabolic disease (20180010185) and a genetics risk predictor (20190017119). P.T.E. is supported by a grant from Bayer AG to the Broad Institute focused on the genetics and therapeutics of cardiovascular diseases, and he has served on advisory boards or consulted for Bayer AG, Quest Diagnostics, MyoKardia and Novartis. The remaining authors declare no competing interests.
Nonstandard Abbreviation and Acronyms:
- CAD
coronary artery disease
- GWAS
genome wide association study
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs
- ADAMTSL
ADAMTS-like
- CS-GAG
chondroitin sulfate glycosaminoglycan
- COMP
cartilage oligomeric protein
- TSP1
thrombospondin 1
- VSMC
vascular smooth muscle cell
- HCA-SMC
human coronary artery smooth muscle cell
- SMC
smooth muscle cell
- SMA
α-smooth muscle actin
- WT
wild-type
- EQ
glutamate to glutamine substitution
- KO
knockout
- APOE
apolipoprotein E
- LDLR
low density lipoprotein receptor
- E373Q
Glu373Gln
- E389Q
Glu389Gln
- β-gal
β-galactosidase
- PCSK9
proprotein convertase subtilisin/kexin type 9
- AAV
adeno-associated virus
- TAILS
terminal amine isotopic labeling
Footnotes
SUPPLEMENTARY MATERIALS
Expanded Materials and Methods
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Khera AV and Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, Burnett MS, Devaney JM, Knouff CW, Thompson JR, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377:383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coronary Artery Disease Genetics C. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–44. [DOI] [PubMed] [Google Scholar]
- 5.Dichgans M, Malik R, Konig IR, Rosand J, Clarke R, Gretarsdottir S, Thorleifsson G, Mitchell BD, Assimes TL, Levi C, et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: a genome-wide analysis of common variants. Stroke. 2014;45:24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pereira A, Palma Dos Reis R, Rodrigues R, Sousa AC, Gomes S, Borges S, Ornelas I, Freitas AI, Guerra G, Henriques E, et al. Association of ADAMTS7 gene polymorphism with cardiovascular survival in coronary artery disease. Physiol Genomics. 2016;48:810–815. [DOI] [PubMed] [Google Scholar]
- 7.Chan K, Pu X, Sandesara P, Poston RN, Simpson IA, Quyyumi AA, Ye S and Patel RS. Genetic Variation at the ADAMTS7 Locus is Associated With Reduced Severity of Coronary Artery Disease. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bengtsson E, Hultman K, Duner P, Asciutto G, Almgren P, Orho-Melander M, Melander O, Nilsson J, Hultgardh-Nilsson A and Goncalves I. ADAMTS-7 is associated with a high-risk plaque phenotype in human atherosclerosis. Sci Rep. 2017;7:3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li HW, Shen M, Gao PY, Li ZR, Cao JL, Zhang WL, Sui BB, Wang YX and Wang YJ. Association between ADAMTS7 polymorphism and carotid artery plaque vulnerability. Medicine (Baltimore). 2019;98:e17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pu X, Xiao Q, Kiechl S, Chan K, Ng FL, Gor S, Poston RN, Fang C, Patel A, Senver EC, et al. ADAMTS7 cleavage and vascular smooth muscle cell migration is affected by a coronary-artery-disease-associated variant. Am J Hum Genet. 2013;92:366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bauer RC, Tohyama J, Cui J, Cheng L, Yang J, Zhang X, Ou K, Paschos GK, Zheng XL, Parmacek MS, et al. Knockout of Adamts7, a novel coronary artery disease locus in humans, reduces atherosclerosis in mice. Circulation. 2015;131:1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Zheng J, Bai X, Liu B, Liu CJ, Xu Q, Zhu Y, Wang N, Kong W and Wang X. ADAMTS-7 mediates vascular smooth muscle cell migration and neointima formation in balloon-injured rat arteries. Circ Res. 2009;104:688–98. [DOI] [PubMed] [Google Scholar]
- 13.Kessler T, Zhang L, Liu Z, Yin X, Huang Y, Wang Y, Fu Y, Mayr M, Ge Q, Xu Q, et al. ADAMTS-7 inhibits re-endothelialization of injured arteries and promotes vascular remodeling through cleavage of thrombospondin-1. Circulation. 2015;131:1191–201. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Yu F, Wang L, Zheng J, Du Y, Huang Y, Liu B, Wang X and Kong W. ADAMTS-7 promotes vascular smooth muscle cells proliferation in vitro and in vivo. Sci China Life Sci. 2015;58:674–81. [DOI] [PubMed] [Google Scholar]
- 15.Mead TJ and Apte SS. ADAMTS proteins in human disorders. Matrix Biol. 2018;71–72:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerda-Costa N and Gomis-Ruth FX. Architecture and function of metallopeptidase catalytic domains. Protein Sci. 2014;23:123–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelwick R, Desanlis I, Wheeler GN and Edwards DR. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015;16:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somerville RP, Longpre JM, Apel ED, Lewis RM, Wang LW, Sanes JR, Leduc R and Apte SS. ADAMTS7B, the full-length product of the ADAMTS7 gene, is a chondroitin sulfate proteoglycan containing a mucin domain. J Biol Chem. 2004;279:35159–75. [DOI] [PubMed] [Google Scholar]
- 19.Liu CJ, Kong W, Ilalov K, Yu S, Xu K, Prazak L, Fajardo M, Sehgal B and Di Cesare PE. ADAMTS-7: a metalloproteinase that directly binds to and degrades cartilage oligomeric matrix protein. FASEB J. 2006;20:988–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu CJ, Kong W, Xu K, Luan Y, Ilalov K, Sehgal B, Yu S, Howell RD and Di Cesare PE. ADAMTS-12 associates with and degrades cartilage oligomeric matrix protein. J Biol Chem. 2006;281:15800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riessen R, Fenchel M, Chen H, Axel DI, Karsch KR and Lawler J. Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:47–54. [DOI] [PubMed] [Google Scholar]
- 22.de Groot R ADAMTS7: Recombinant Protein Expression and Purification. Methods Mol Biol. 2020;2043:63–73. [DOI] [PubMed] [Google Scholar]
- 23.Mead TJ, McCulloch DR, Ho JC, Du Y, Adams SM, Birk DE and Apte SS. The metalloproteinase-proteoglycans ADAMTS7 and ADAMTS12 provide an innate, tendon-specific protective mechanism against heterotopic ossification. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlson CB, Lawler J and Mosher DF. Structures of thrombospondins. Cell Mol Life Sci. 2008;65:672–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee NV, Sato M, Annis DS, Loo JA, Wu L, Mosher DF and Iruela-Arispe ML. ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J. 2006;25:5270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cal S, Arguelles JM, Fernandez PL and Lopez-Otin C. Identification, characterization, and intracellular processing of ADAM-TS12, a novel human disintegrin with a complex structural organization involving multiple thrombospondin-1 repeats. J Biol Chem. 2001;276:17932–40. [DOI] [PubMed] [Google Scholar]
- 27.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG and Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–9. [DOI] [PubMed] [Google Scholar]
- 28.Erdmann J, Kessler T, Munoz Venegas L and Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114:1241–1257. [DOI] [PubMed] [Google Scholar]
- 29.Muller M, Kessler T, Schunkert H, Erdmann J and Tennstedt S. Classification of ADAMTS binding sites: The first step toward selective ADAMTS7 inhibitors. Biochem Biophys Res Commun. 2016;471:380–5. [DOI] [PubMed] [Google Scholar]
- 30.Santamaria S and de Groot R. Monoclonal antibodies against metzincin targets. Br J Pharmacol. 2019;176:52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El Hour M, Moncada-Pazos A, Blacher S, Masset A, Cal S, Berndt S, Detilleux J, Host L, Obaya AJ, Maillard C, et al. Higher sensitivity of Adamts12-deficient mice to tumor growth and angiogenesis. Oncogene. 2010;29:3025–32. [DOI] [PubMed] [Google Scholar]
- 32.Colige A, Monseur C, Crawley JTB, Santamaria S and de Groot R. Proteomic discovery of substrates of the cardiovascular protease ADAMTS7. J Biol Chem. 2019;294:8037–8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durham TB, Klimkowski VJ, Rito CJ, Marimuthu J, Toth JL, Liu C, Durbin JD, Stout SL, Adams L, Swearingen C, et al. Identification of potent and selective hydantoin inhibitors of aggrecanase-1 and aggrecanase-2 that are efficacious in both chemical and surgical models of osteoarthritis. J Med Chem. 2014;57:10476–85. [DOI] [PubMed] [Google Scholar]
- 34.Mosyak L, Georgiadis K, Shane T, Svenson K, Hebert T, McDonagh T, Mackie S, Olland S, Lin L, Zhong X, et al. Crystal structures of the two major aggrecan degrading enzymes, ADAMTS4 and ADAMTS5. Protein Sci. 2008;17:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metz RP, Patterson JL and Wilson E. Vascular smooth muscle cells: isolation, culture, and characterization. Methods Mol Biol. 2012;843:169–76. [DOI] [PubMed] [Google Scholar]
- 36.Sasaki N, Yamashita T, Takeda M, Shinohara M, Nakajima K, Tawa H, Usui T and Hirata K. Oral anti-CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation. 2009;120:1996–2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article and its online supplementary files.
Expanded methods sections for the expression vectors, mammalian cell culture, western blot, RT-PCR expression analysis, vascular smooth muscle cell migration assay, generation of Adamts7 mouse alleles, full-length ADAMTS7 purification and Thrombospondin 1 (TSP1) cleavage assay, atherogenic mouse models, atherosclerotic lesion measurement, immunohistochemistry and β-galactosidase (β-gal) staining are available in the online-only Data Supplement. Representative images were selected to represent quantification results shown with the images. Assay procedures and quantification analysis for in vivo experiments (Oil-Red O staining, α-SMA staining and β-gal staining) were performed by scientists (T.M., B.B), who are not aware of mouse genotype. Different scientists (B.T.M., N.R.P.) performed genotyping for all the mice.