

SUMMARY
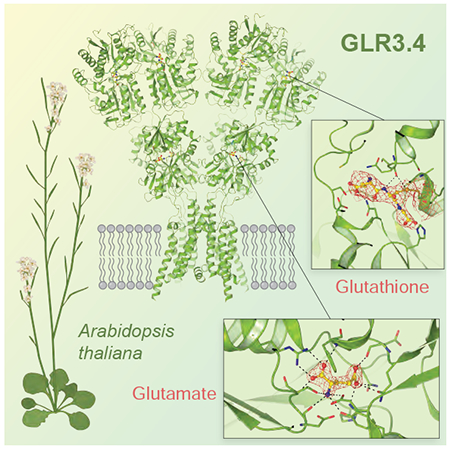
Glutamate receptor-like channels (GLRs) play vital roles in various physiological processes in plants, such as wound response, stomatal aperture control, seed germination, root development, innate immune response, pollen tube growth, and morphogenesis. Despite the importance of GLRs, knowledge about their molecular organization is limited. Here we employ X-ray crystallography and single-particle cryo-EM to solve structures of the Arabidopsis thaliana GLR3.4. Our structures reveal the tetrameric assembly of GLR3.4 subunits into a three-layer domain architecture, reminiscent of animal ionotropic glutamate receptors (iGluRs). However, the non-swapped arrangement between layers of GLR3.4 domains, binding of glutathione through S-glutathionylation of cysteine C205 inside the amino-terminal domain clamshell, unique symmetry, inter-domain interfaces, and ligand specificity distinguish GLR3.4 from representatives of the iGluR family and suggest distinct features of the GLR gating mechanism. Our work elaborates on the principles of GLR architecture and symmetry and provides a molecular template for deciphering GLR-dependent signaling mechanisms in plants.
Graphical Abstract
eTOC Blurb
Green et al. solve structures of Arabidopsis thaliana glutamate receptor-like channel GLR3.4 that shows tetrameric subunit assembly with three-layer architecture, similar to its mammalian homologs, ionotropic glutamate receptors, but with distinct symmetry, inter-domain interfaces, ligand specificity and non-swapped domain arrangement between layers of ligand-binding and glutathione-bound amino-terminal domains.
INTRODUCTION
Glutamate receptor-like channels (GLRs) have been identified as homologs of mammalian ionotropic glutamate receptors (iGluRs), which mediate the majority of the excitatory neurotransmission in the central nervous system (Traynelis et al., 2010). GLR homologs have been identified in both vascular and non-vascular plants (Lam et al., 1998; Wudick et al., 2018a) and in genomes across the entire plant evolutionary tree, including Chlamydomonas, chlorophytes, mosses, ferns, gymnosperms, and angiosperms (De Bortoli et al., 2016; Wudick et al., 2018a). Based on phylogenetic analysis, the 20 GLRs identified in the model flowering plant Arabidopsis thaliana have been organized into 3 different clades: GLR1, GLR2, and GLR3 (Lam et al., 1998). In contrast to iGluRs, which are mainly targeted to the plasma membrane, GLRs have not only been located in the plasma membrane but also located in the plant mitochondria, chloroplasts, tonoplast, endoplasmic reticulum, and sperm cell (endo)membranes (Teardo et al., 2015; Vincill et al., 2013; Wudick et al., 2018a). GLRs play vital roles in various physiological processes in plants, such as cell signaling, metabolism, wound response, stomatal aperture, seed germination, root development, innate immune response, pollen tube growth, and water loss (Wudick et al., 2018a). Given the diverse range of crucial physiological roles, targeting GLRs can have practical applications, especially related to biotic and abiotic environmental stressors, which constitute a major problem for sustainability (Jones et al., 2008). Genetic efforts to explore GLR potential have been hindered by the apparent high functional redundancy and a large number of genes coding for GLRs in the majority of angiosperms, including crops. An alternative to traditional strategies for regulating GLR functions in plants could utilize approaches that target their specific molecular features (De Bortoli et al., 2016; Michard et al., 2017; Wudick et al., 2018a; Wudick et al., 2018b).
The molecularly oriented approaches to harness GLR function rely on the detailed knowledge of GLR structural organization. Recent studies of the GLR3.2 and GLR3.3 ligand-binding domains (LBDs), from the model organism Arabidopsis thaliana, provided the first glimpse to the fragment of GLR structure responsible for ligand binding (Alfieri et al., 2020; Gangwar et al., 2021). GLR3.2 and GLR3.3 LBD structures identified similar overall folds to iGluRs’ LBDs, with the distinct features in the ligand-binding pocket that are responsible for the promiscuous GLR activation by various amino acids. Despite the important insights from the recent structural studies of GLR LBDs, structures of other GLR domains, principles of their assembly into the full-length receptor, its architecture, and structural basis of gating have remained an enigma. To fill this gap in knowledge, we embarked on the endeavor to determine the structure of a full-length GLR.
RESULTS
Functional characterization and structure determination
We screened several full-length GLRs from the three different clades using fluorescence-detection size-exclusion chromatography (FSEC) (Kawate and Gouaux, 2006) and identified GLR3.4 from Arabidopsis thaliana as a promising candidate for structural studies. GLR3.4 expresses at the plasma membrane of root phloem cells, vascular bundles, mesophyll cells, and guard cells (Meyerhoff et al., 2005; Vincill et al., 2013). We show that GLR3.4 localizes specifically to the aperture of the pollen grain (Figure 1A) and appears to be associated with cell polarity, from the formation of the early bulge, which will develop into the pollen tube, to the retention in the apical tips after the formation of the callose plugs, or in root phloem cells (Vincill et al., 2013) (Figure S1A–C). Cell polarity in Arabidopsis pollen tubes has also been characterized by the tip-focused influx of calcium (Ca2+) (Michard et al., 2011; Michard et al., 2017). Supporting the role of GLR3.4 in this cell polarity process, two mutant alleles of Atglr3.4 showed reduced pollen tube Ca2+ fluxes compared to wild type (Figure S1D).
Figure 1. Functional, biochemical, and structural characterization of GLR3.4.

(A) Voltage-dependence of whole-cell patch-clamp currents recorded from COS-7 cells expressing GLR3.4, CNIH1 and CNIH4 before (black circles) and after (red circles) application of 1 mM Glu (n = 3, mean ± SEM). The control (white circles) are endogenous currents recorded from COS-7 cells transformed with the empty vector (n = 5, mean ± SEM). Inset shows specific GFP labeling of GLR3.4 at the plasma membrane of the pollen grain aperture, an outgrowth spot where the pollen tube will emerge.
(B) Changes in cytosolic Ca2+ measured in COS-7 cells expressing the Ca2+ sensor Yellow CaMeleon 3.6 (YC3.6) and expressing either CNIH1+4 alone (control, white circles) or GLR3.4 and CNIH1+4, in response to application of 14.5 mM Ca2+ in the absence (black circles; n = 27, mean ± SEM) or presence (red circles; n = 33, mean ± SEM) of 1 mM Glu.
(C) FSEC trace and SDS-PAGE gel for purified GLR3.4.
(D) Cryo-EM density for 3.57-Å 3D reconstruction of GLR3.4, with four subunits colored in green, pink, blue, and orange; and non-protein densities representing carbohydrates and ligands colored in purple.
See also Figures S1–S3 and Table S1.
GLR3.4 was reported to mediate currents activated by a broad range of amino acids (Meyerhoff et al., 2005; Stephens et al., 2008; Vincill et al., 2012). GLR3.4 expressing COS-7 cells conducted robust ionic currents in response to application of Glu or Asn only when co-expressed with CORNICHON HOMOLOG (CNIH) proteins, CNIH1 and/or CNIH4 (Figure 1A, Figure S2A–D), consistent with the recently discovered role of CNIHs in GLR sorting and activation (Wudick et al., 2018b). GLR3.4 activation in the presence of Glu or Asn resulted in an increase in cytosolic calcium (Figure 1B, Figure S2E–F), which may work as a signaling mechanism in plants (Michard et al., 2011; Mousavi et al., 2013; Ortiz-Ramírez et al., 2017).
We expressed full-length GLR3.4 in HEK 293 cells, purified the protein to sufficient purity and homogeneity (Figure 1C), and subjected it to single-particle cryo-EM. We reconstructed a 3D map to an overall 3.57 Å resolution (Figure 1D), with the core of the molecule resolved at ∼3 Å resolution (Figure S3, Table S1, Supplementary Movie 1). The map quality was adequate for building each subunit of the GLR3.4 tetramer, excluding the S2-M4 linkers and loops connecting M1 to M2 and M2 to M3, which were not represented clearly by resolved density.
Architecture and symmetry
The Y-shape of the 175 Å-tall GLR3.4 structure resembles the shape of non-NMDA subtype iGluR structures (Burada et al., 2020b; Meyerson et al., 2016; Sobolevsky et al., 2009) (Figure 2A). Similar to all iGluRs, the structure of GLR3.4 has a three-layer architecture, which includes the amino-terminal domain (ATD) layer at the top, the ligand-binding domain (LBD) layer in the middle, and the transmembrane domain (TMD) layer at the base of the “Y”. The ATDs and LBDs of subunits A/B and C/D form dimers with monomers related by two-fold rotational symmetry. The GLR3.4 tetramer has an overall two-fold rotational symmetry that relates one ATD dimer to another, one LBD dimer to the second, and half of the TMD to another half. The axes of local two-fold rotational symmetry for the LBD and ATD dimers coincide and make a 20° angle with the axis of the overall 2-fold rotational symmetry of the tetramer. This is very different from the domain organization of AMPA, kainate, and NMDA subtypes of iGluRs where the ATD and LBD dimers have distinct axes of local two-fold rotational symmetry. An even more striking feature of GLR3.4 architecture is the lack of domain swapping between the ATD and LBD dimers, which is an inherent property of all major iGluR subtypes. The only exception among iGluRs are orphan delta receptors, which have been recently proposed to follow non-swapped domain architecture similar to GLR3.4 based on low-resolution cryo-EM reconstructions (Burada et al., 2020a, b).
Figure 2. Overall architecture and symmetry.

(A) GLR3.4 structure viewed parallel to the membrane (grey bars), with four subunits colored differently, molecules of GSH and Glu shown as space-filling models and carbohydrates as sticks. The axes of the overall and local two-fold symmetries are shown as dotted and solid lines, respectively.
(B-D) ATD (B), LBD (C) and TMD (D) layers viewed extracellularly, with the overall (large ovals) and local (small ovals) two-fold and four-fold (square) symmetries indicated.
(E) ATD and LBD dimers formed by subunits A and B, viewed perpendicular to the axis of the local two-fold symmetry and showing their broad and narrow faces.
See also Figures S4–S5.
We confirmed the unorthodox subunit and domain arrangement of GLR3.4 by introducing cysteine substitutions at intersubunit interfaces and observing redox-dependent dimer formation as a result of spontaneous subunit crosslinking (Figure S4). There are five intersubunit interfaces that keep the four GLR3.4 subunits together by means of hydrogen bonds, hydrophobic and ionic interactions (Figure S4). In the ATD layer, the intradimer interfaces between subunits A and B as well as C and D are large (3,180 Å2) and involve both the upper (L1) and lower (L2) lobes. In contrast, the interdimer interface between subunits B and D, which is mediated primarily by ionic interactions between R436 and E431, is small (209 Å2) but keeps the A/B and C/D dimers from falling apart. Similarly, in the LBD layer, the intradimer interfaces between subunits A and B as well as C and D are large (800 Å2), while the interdimer interface between subunits A and C is small (218 Å2) and mediated by hydrogen bonds between T717 and N719. The four GLR3.4 subunits interact most extensively at the TMD, where each of the four pairs of the neighboring subunits (A/B, B/C, C/D and D/A) contribute a large (1,547 Å2) intersubunit interface. Apart from numerous hydrophobic interactions, which are typical for transmembrane regions, these interfaces are also stabilized by hydrogen bonds between residues T613, E615, L686, N689, S690, T693, T697, S698, L700, K856, and S857 that belong to the adjacent subunits.
The symmetrical dimer of dimers organization of GLR3.4 is maintained over the ATD and LBD layers (Figure 2B–C) but changes to pseudo four-fold symmetrical organization in the TMD layer (Figure 2D). Correspondingly, interfaces between four GLR3.4 subunits are equivalent at the level of TMD but distinct between the ATDs and LBDs, with only the proximal pairs contributing to the interdimer interfaces. Interestingly, the pairs of proximal and distal subunits in GLR3.4 are exactly the same as in iGluRs’ swapped-topology, despite the lack of domain swapping between the ATD and LBD layers. Indeed, the A/C pair is distal in the ATD layer and proximal in the LBD layer, while the B/D pair is proximal in the ATD layer and distal in the LBD layer (Figure 2B–C). Switching of the proximal and distal subunit pairs becomes possible due to the perpendicular orientation of the ATD and LBD dimers with respect to each other (Figure 2E).
The overall two-fold rotational symmetry of the GLR3.4 tetramer with pseudo four-fold symmetrical TMD (Figure 2D) is a result of two diagonal pairs, A/C and B/D, of chemically identical subunits adapting two different conformations (Figure S5A). While their extracellular domains related by the local two-fold rotational symmetry have the same conformations, their TMDs are displaced in a 93° rotation (Figure S5B). As a result of such dramatic conformational changes, the A/C and B/D subunit pairs play different structural roles in the extracellular portion of GLR3.4, while contributing equally to the ion channel. As a reflection of the different structural roles of the A/C and B/D subunit pairs, the subunits B and D ATDs are in direct contact, while the subunits A and C ATDs are separated by more than 70 Å (Figure S5C–D).
Amino terminal domains
The ATDs of GLR3.4 adapt a clamshell architecture that was previously observed in ATDs of iGluRs (Hansen et al., 2010), bacterial periplasmic amino-acid binding proteins (Trakhanov et al., 2005), extracellular domain of the natriuretic peptide receptor (He et al., 2001) and ligand-binding domains of family C G-protein-coupled receptors, including metabotropic glutamate (Koehl et al., 2019) and GABAB receptors (Papasergi-Scott et al., 2020; Park et al., 2020; Shaye et al., 2020). The dimer-of-dimers assembly of ATDs in iGluRs is important for their tetrameric stability but functionally these domains are orphan in AMPA and kainate subtypes due to the open clamshells that have not been reported to bind any ligand and pronounced interfaces between the upper L1 and lower L2 lobes that restrain clamshell opening/closing transformations (Jin et al., 2009). NMDA receptors, which bind the inhibitor ion Zn2+ inside the GluN2B subunit ATD clamshell and conduct the conformation changes in the ATDs to the LBDs and the ion channel (Jalali-Yazdi et al., 2018; Karakas et al., 2009), are an exception. Since the overall domain organization and shape of GLR3.4 resembles more AMPA and kainate than NMDA receptors, we expected the ATD clamshells to be similar to non-NMDA receptors. Against these expectations, we found that the GLR3.4 ATDs are all bound to glutathione (GSH) through S-glutathionylation of cysteine C205 (Figure 3A).
Figure 3. Amino-terminal domain.

(A) Structure of the GLR3.4 ATD with secondary structure elements labeled. The ATD-LBD linker domain is colored red. The inset shows a closeup view of the GSH binding pocket. GSH is covalently bound to C205 and shown in ball-and-stick representation. Binding pocket residues are shown in sticks and their interactions with GSH are indicated by dashed lines. Density for GSH is shown as a blue mesh.
(B) Spectrum annotation of the peptide containing C205 modified by GSH.
(C) Representative whole-cell patch-clamp recordings from COS-7 cells expressing GLR3.4:CNIH1:CNIH4 prior to treatment (background; without exogenous GSH or Glu), after application of 1 mM Glu (+Glu) or after application of 1 mM Glu and 1 mM GSH together (+Glu+GSH).
D) Voltage-dependencies of currents recorded from COS-7 cells expressing GLR3.4:CNIH1:CNIH4 (like those shown in C) in comparison with the ones recorded from COS-7 cells expressing CNIH1 and CNIH4 only, before and after application of 1 mM Glu and 1 mM GSH (n=8 for GLR3.4 background condition, n=5 for GLR3.4 +Glu, n=5 for GLR3.4+Glu+GSH, n=5 for CNIH1:CNIH4 background, and n=5 for CNIH1:CNIH4+Glu+GSH).
(E) Voltage-dependencies of currents recorded from COS-7 cells expressing wild-type GLR3.4 (n=6), GLR3.4(C205A) (n=4), and CNIH1:CNIH4 (n=5) before and after application of 1 mM GSH. Both wild type and mutant GLR constructs were co-transfected with CNIH1 and CNIH4. All data shown in D and E are mean ± SEM.
(F-G) Superposition of the GLR3.4 ATD with GluA2 ATD (blue, PDB ID: 3H5V) (F) and mGluR1 ligand-binding domain (yellow, PDB ID: 1EWK) (G). Closure by 16° of the GLR3.4 ATD clamshell compared to the GluA2 ATD clamshell is indicated by the green arrow.
(H) Closeup view of the GLR3.4 interface between ATD and LBD, with the contributing residues shown as sticks.
(I) Amino acid sequence alignment for the ATD-LBD linker region of AtGLR3.4 (NP_001030971.1), AtGLR3.1 (NP_028351.2), AtGLR3.2 (NP_567981.1), AtGLR3.3 (NP_174978.1), AtGLR3.6 (NP_190716.3), AtGLR3.7 (NP_565744.1), AtGLR2.1 (NP_198062.2), AtGLR1.4 (NP_187408.2), AMPA subtype rat GluA2 (NP_058957), kainate subtype rat GluK2 (P42260.2), delta subtype rat GluD1 (NP_077354.1) and GluD2 (NP_077355.1), NMDA subtype rat GluN1 (EDL93606.1), GluN3A (NP_612555.1) and GluN2A (NP_036705.3) and AvGluR1 (ADW94593.1). Numbering is for the mature protein. Secondary structure elements are shown as cylinders (α-helices), arrows (β-strands), and lines (loops) and colored according to domains, ATD (purple) and LBD (orange). Conserved residues are highlighted in blue.
The binding of GSH in the middle of the ATD clamshell explains why GSH has been previously described as an activator of GLRs (Qi et al., 2006). The reversible covalent linking to cysteine C205 endows GSH with the potential to act as a biological switch and be integral in several critical oxidative signaling events (Xiong et al., 2011), including stress response in plants (Dixon et al., 2005). Apart from its disulfide bond to C205, GSH is held in its binding pocket by hydrogen bonds to the backbone carbonyl of Q133 and the amide nitrogen of N259 as well as several non-bonded contacts. As a result of all these interactions, GSH binding is strong, signified by the clearly resolved density that unambiguously identifies the position and pose of this molecule in its binding pocket (Figure 3A, inset). Covalent modification of C205 by GSH was also confirmed by the standard bottom-up proteomics approach (Figure 3B). Wild-type GLR3.4 and C205A mutant protein samples digested in reducing and non-reducing conditions were subjected to mass spectrometry analysis. Samples digested in reducing conditions showed no GSH modification. However, when the digest was carried out in non-reducing conditions, GSH was detected at the residue 205 in the wild-type protein but not in the C205A mutant.
We tested a potential role of GSH in GLR3.4 function by heterologously expressing the channel in COS-7 cells. When GLR3.4 was co-expressed with CNIH1 and CNIH4, Glu with GSH elicited larger currents than Glu alone (Figure 3C–D). This activating effect of GSH was impaired by C205A mutation (Figure 3E), suggesting that cysteine C205 and the GSH binding pocket identified in the ATD play an important role in GLR3.4 function. On the other hand, significant GSH-induced potentiation of GLR3.4(C205A) currents indicates that there is a C205A-independent component of activation by GSH. For example, this component can be mediated by GSH binding to the same site in the ATD but without covalent attachment to C205.
To infer whether GSH binding has the potential to produce conformational changes in the GLR3.4 protein, we compared its ATD to the ATD of the AMPA subtype iGluR GluA2, which has the same overall fold but does not bind to any ligands. Strikingly, GSH-bound clamshell of the GLR3.4 ATD is 16° more closed than the ATD clamshell of GluA2 (Figure 3F). This conformation is reminiscent of the conformation of mGluR ligand-binding domain after it closes in response to the binding of agonist glutamate (Figure 3G). Thus, a comparison of the GLR3.4 ATD to the domains of a similar fold suggests that GSH binding can cause conformational changes in the GLR protein, which might contribute to gating of its ion channel.
For conformational changes in the ATD to be effectively transmitted to the LBD and TMD, the ATD must have a strong structural connection to LBD, such as the one observed in NMDA receptor (Karakas and Furukawa, 2014; Lee et al., 2014) and which is largely missing in AMPA, kainate or delta receptors (Burada et al., 2020b; Meyerson et al., 2016; Sobolevsky et al., 2009). In GLR3.4, a unique ATD-LBD linker plays the role of an inter-domain connector, creating a strong interface between the ATD and LBD through a network of hydrophobic (residues W485, F487, L494, Y534, P535, V536, P809, L810) and electrostatic (residues L248, R483, N489, N490, Y534, P535, P809, D813) interactions (Figure 3H). Such a strong connection between the ATD and LBD ensures that conformational changes in the ATD are allosterically transmitted to the LBD. This interaction appears highly specific to GLRs, as the ATD-LBD linker amino acid sequence alignment shows low similarity between GLRs and iGluRs (Figure 3I). On the other hand, the majority of residues in the ATD-LBD linker are conserved among GLRs and highly conserved among clade 3 GLRs (Figure 3I).
Ligand-binding domains
LBDs are typically the drivers of conformational changes in iGluRs that start with agonist binding to the LBD and end up with the opening of the ion channel for current conductance. To get a high-resolution view of GLR3.4 LBD and ligand binding, we purified this domain separately and solved crystal structures of the isolated LBD in complex with agonists glutamate, serine, and methionine (Table S2). The structure of GLR3.4 LBD has a clamshell architecture (Figure 4A; Figure S6A–C) that is typical for iGluRs and similar to the recently published structures of isolated GLR3.2 and GLR3.3 LBDs (Figure S6D). The agonist binds in the middle of the LBD clamshell, to the binding site between its upper D1 and lower D2 lobes, resulting in closed clamshell conformation (Figure 4B), and agonist binding can be monitored using microscale thermophoresis (Figure S6E). The serine-bound LBD structure was solved without adding the ligand to the protein, indicating serine’s endogenous origin (see Methods). Accordingly, in thermophoresis experiments, serine binding was not detected, likely because it was already bound to the LBD (Figure S6E). As a consequence, the apparent dissociation constants for glutamate and methionine unlikely report their true affinities but rather effective concentrations to outcompete endogenous serine.
Figure 4. Ligand-binding domain.

(A) Structure of the Glu-bound isolated GLR3.4 LBD with secondary structure elements labeled. The inset shows a closeup view of the Glu binding pocket, with Glu shown in ball-and-stick representation and the omit map density for Glu at 5σ shown as purple mesh. Binding pocket residues are shown in sticks and their interactions with Glu are indicated by dashed lines.
(B) Superposition of the Glu-bound isolated GLR3.4 LBD (blue) with Glu-bound isolated GluA2 LBD (green, PDB ID: 1FTJ) and isolated apo-state GluA2 LBD (pink, PDB ID: 1FTO). Opening of the apo-state GluA2 LBD clamshell by 26° compared to the Glu-bound LBD clamshells is indicated by the pink arrow.
C-E) Glu-bound dimers of isolated GLR3.4 LBD (C), GluA2 LBD (PDB ID: 1FTJ) (D) and GluK2 LBD (PDB ID: 3G3F) (E), with the distances between upper and lower lobes indicated. The two-fold symmetry axes are shown as vertical black lines.
(F-H) LBD dimers from the full-length structures of GLR3.4 (F), GluA2 in complex with GSG1L (PDB ID: 5VHZ) (G) and GluK2 (PDB ID: 5KUF) (H), in complex with Glu, L-quisqualate and 2S,4R-4-methylglutamate, respectively, and superposed with the corresponding isolated LBD dimers (C-E) shown in grey. Rearrangements of LBDs in the full-length structures compared to isolated LBDs are indicated by purple arrows.
The key interactions with agonists and binding residues are conserved among iGluRs and GLRs (Alfieri et al., 2020; Gangwar et al., 2021). For all three agonists, the guanidinium group of R92 (R577 in the full-length GLR3.4) and the backbone amines of T87 and F139 are hydrogen bonded to the carboxyl group of the ligand, while the backbone carbonyl oxygen of D85, the hydroxyl groups of T87 and Y186 and the carboxyl group of E183 coordinate the amino group of the ligand. The ligand side chains are stabilized differently. The side-chain carboxyl group of glutamate and thioether group of methionine are additionally coordinated by the guanidinium group of R16, amide groups of N64, and Q135, phenol group of Y67, and the backbone carbonyl oxygen of D136 (Figure 4A, inset; Figure S6C). These interactions are missing in the case of serine, which lacks the bulky side chain. Instead, a water occupies the void, where it is stabilized by hydrogen bonds with the hydroxymethyl side chain of serine, the guanidinium group of R16, amide group of Q135, and the backbone carbonyl oxygen of D136 (Figure S6B). Water molecules played a similar role in the binding of glycine to the LBDs of GLR3.2 and GLR3.3 (Alfieri et al., 2020; Gangwar et al., 2021). Altogether, these data illustrate how the ligand-binding pocket of GLRs evolved to bind differently sized amino acids by exploiting the same interactions for binding the conserved amino acid core and adjusting the fit of the side chains with water. This explains the ligand-binding promiscuity of GLRs, which are activated by at least 12 of the 20 proteinogenic amino acids and the non-proteinogenic amino-acid ACC (1-aminocyclopropane-1-carboxylic acid) (Forde and Roberts, 2014; Kong et al., 2016; Michard et al., 2011; Mou et al., 2020; Tapken et al., 2013; Vincill et al., 2012; Vincill et al., 2013; Wudick et al., 2018a; Wudick et al., 2018b), in contrast to vertebrate iGluRs that are only activated by specific amino acids (Traynelis et al., 2010). Interestingly, ACC was shown to be a partial agonist for the NMDA receptor GluN1 subunit (Inanobe et al., 2005), while invertebrate iGluRs can also be activated by different amino acids (Lomash et al., 2013).
In our crystal structures, the isolated LBDs form dimers that show back-to-back arrangement of monomers (Figure 4C), typical for the open state LBD dimers in iGluRs. Indeed, the dimers are held together through D1-D1 interfaces, reminiscent of GluA2 (Figure 4D), and GluK2 (Figure 4E) LBD dimers. Interestingly, the LBD dimers in the context of full-length GLR3.4 show very different assembly of monomers, with about the same distance between the upper lobes as in the isolated LBD structures but a much shorter distance between their bottom lobes (Figure 4F). The reduced separation of the D2 lobes in dimers of agonist-bound LBDs is the characteristic feature of iGluR desensitized state (Figure 4G–H). In contrast to the open state, which also has LBD clamshells closed with agonists, the desensitized state is characterized by a non-conducting ion channel.
Ion channel
Similar to iGluRs, the ion channel of GLR3.4 is assembled of the TMDs of four subunits in a four-fold symmetrical manner, each TMD contributing the transmembrane helices M1, M3, and M4 and a re-entrant intracellular loop M2 (Figures 2D, 5A). The extracellular part of the channel’s ion conduction pathway is lined by the M3 helices that form the gate. Its intracellular part, which is typically responsible for ion selectivity, is lined by the non-helical portions of M2, apparently disordered in our cryo-EM maps. M1 and M4 are on the periphery of the ion channel and contact the surrounding TMD membrane lipid. M1 helices are preceded by short pre-M1 helices that form an extracellular collar around the ion permeation pathway, which in AMPA receptors harbors binding sites for non-competitive inhibitors, including the antiepileptic drug Perampanel (Yelshanskaya et al., 2016).
Figure 5. Ion channel.

(A) Ion conduction pathway (grey) in GLR3.4, with residues lining the pore shown as sticks. Only two of four GLR3.4 subunits are shown, with the front and back subunits removed for clarity.
(B) Pore radius calculated using HOLE. The dashed line corresponds to 1.4 Å (radius of a water molecule).
(C) Amino acid sequence alignment for the M3 region of GLRs and iGluRs. The gate-forming residues are indicated by asterisks.
Measurements of the pore radius (Figure 5A–B) suggest that the ion channel is in the closed non-conducting conformation. The pore’s narrow constriction is formed by the extracellular portions of the M3 helices and contributed by four rings of residues, including T693, T697, T701, and L705. The homologous residues in iGluRs that also form the pore’s narrow constriction are either part of the highly conserved SYTANLAAF motif or just C-terminal to it (Figure 5C). The distinct character of gate-forming residues in GLR3.4 compared to iGluRs suggest that gating and permeation properties of GLRs might differ from iGluRs.
DISCUSSION
In the GLR3.4 structure, the ATDs (Figure 3) and LBDs (Figure 4) are bound to agonist molecules. The ATDs are bound to GSH and adapt closed clamshell conformations. The LBDs also adapt closed clamshell conformations and are modelled bound to Glu, which we added to the protein before subjecting it to cryo-EM. There is a possibility, however, that the LBDs in the full-length GLR3.4 structure are bound to Ser, similar to the isolated LBD, which was found bound to Ser of endogenous origin (Figure S6, Methods). In fact, it is practically impossible to distinguish Glu from Ser in cryo-EM maps at 3.57 Å resolution (Figures 1 and S3, Supplementary Movie 1). Independent of whether the LBDs are bound to Glu or Ser, their closed clamshell conformations are identical (Figure S6A).
Closed clamshell conformations observed for the ATDs and LBDs are active conformations of the clamshell domains, which can drive the opening of the GLR3.4 ion channel. However, the ion channel in the GLR3.4 structure is in a closed non-conducting state (Figure 5), suggesting that the structure represents a desensitized or inactivated state. Supporting this idea is the arrangement of monomers in the GLR3.4 LBD dimers, where the lower lobes D2 are separated by a much smaller distance (Figure 4F) than expected from dimers representing the active state (Figure 4C–E), recently confirmed by the open-state structures of the intact AMPA receptor (Chen et al., 2017; Twomey et al., 2017). Consistently, the smaller separation of the D2 lobes was also observed in the desensitized states of AMPA (Figure 4G) and kainate (Figure 4H) receptors. The conformational state of the GLR3.4 structure, therefore, suggests common features of the gating mechanisms of GLRs and iGluRs. At the level of LBD, gating is triggered by the binding of agonist that presumably leads to individual clamshell closures and separation of the D2 lobes (Figure 6). Applied to the LBD-TMD linkers, the separation of the D2 lobes would cause ion channel opening. During desensitization, the monomers of the LBD dimers could rearrange to decrease the distance between the D2 lobes, relieve the strain on the LBD-TMD linkers and convert the channel to the closed non-conducting state.
Figure 6. GLR3.4 gating.

(A) In the absence of AtCNIHs (HEK cells), GLR3.4 adapts desensitized or inactivated conformation, where GSH is bound to ATD (purple), Glu is bound to LBD (orange) and the channel (green) is in closed, non-conducting conformation.
(B) In the presence of AtCNIHs (grey, COS cells), ligand-induced conformational changes in the extracellular domain (red arrows) can lead to opening of the ion channel and ion conductance (blue arrow).
Despite the apparent similarities in the general principles of gating between GLRs and iGluRs, there are substantial differences. First, in COS-7 cells, GLR3.4 requires CNIHs to conduct currents (Figure 1, Figure S2). The majority of iGluRs conduct currents in the absence of CNIHs, although surface expression and gating of AMPA-subtype iGluRs are strongly regulated by CNIHs (Nakagawa, 2019; Schwenk et al., 2009). The only iGluR exception is GluD-subtype channels, which alone conduct currents only if they acquire mutations in the pore region (Kohda et al., 2000; Yadav et al., 2011). Interestingly, these silent iGluRs are the only iGluRs that share GLR3.4’s non-swapped arrangement of ATD and LBD domains (Burada et al., 2020a, b). Whether GluDs can mediate currents by binding to CNIHs or other auxiliary subunits, remains undiscovered. Similarly, the possible roles of CNIHs in GLR trafficking to the cell surface and acting as chaperones to alter subunit conformations and assembly will require further investigation.
Secondly, agonist molecules can bind not only to the LBD but also to the ATD (Figure 3), suggesting that GLR3.4’s channel opening can be triggered from both extracellular domain layers (Figure 6). The unique architecture of GLR3.4 with non-swapped pairs of the ATD and LBD dimers and tight ATD-LBD linkage through the connecting domain are likely important for communicating conformational changes in the ATD and LBD layers to the ion channel opening. Similarly, the unique GLR3.4 architecture allows the arrangement of monomers within the LBD dimers, which is clearly different from AMPA and kainate receptors (Figure 4F–H). Accordingly, the distinct conformational ensemble may be an indication that GLRs and iGluRs have principally different desensitization mechanisms. Additional structural and functional studies are necessary to decipher the GLR gating mechanisms, its regulation by CNIHs, and the way it shapes GLRs’ functions in plants.
Limitations of the study
According to our experiments, function of GLR3.4 requires CNIH auxiliary subunits. In contrast, our full-length structure of GLR3.4 is determined in the absence of CNIH subunits. Solving structures of GLR3.4-CNIH complexes will be necessary to confirm the physiological relevance of the GLR3.4 structure and to better understand the molecular bases of GLR3.4 function. Our full-length structure of GLR3.4 reports only a single conformation, which we interpret as a desensitized state. We propose that GLR3.4 can undergo conformational changes and open its ion channel to carry out GLR3.4 physiological functions. To verify this hypothesis and to characterize gating and conformational ensemble of GLR3.4 more completely, it will be necessary to solve structures of GLR3.4 and GLR3.4-CNIH complexes in the closed and open states. The unique modulation of GLR3.4 function by GSH binding to the ATD is particularly interesting as it has not been observed in iGluRs. To understand the molecular mechanism of this modulation, it will be necessary to solve structures of GLR3.4 and GLR3.4-CNIH complexes in the absence of bound GSH.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for the resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alexander Sobolevsky (as4005@cumc.columbia.edu).
Materials availability
New materials listed in key resources are available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ampicillin | Sigma | Cat# A8351 |
| Kanamycin | Fisher scientific | Cat# BP906-5 |
| Tetracycline | Fisher scientific | Cat# BP912 |
| IPTG | Zymo Research | Cat# I1001-5 |
| Tris | Fisher scientific | Cat# BP152-1 |
| NaCl | Fisher scientific | Cat# BP358–212 |
| L-Glutamate | Sigma | Cat# 49621 |
| L-Methionine | Sigma | Cat# M9625 |
| MgSO4 | Fluka | Cat# 13143 |
| DNAse | Sigma | Cat# DN25-1 |
| PMSF | Acros Organics | Cat# 215740500 |
| 2-Mercaptoethanol (BME) | Acros Organics | Cat# 125470100 |
| Ni-Affinity Resin | Takara | Cat# 635660 |
| Imidazole | Acros Organics | Cat# 301870025 |
| Thrombin | Haematologic Technologies | Cat# HCT-0020 |
| Glycerol | Fisher scientific | Cat# BP229-4 |
| MES buffer | Sigma | Cat# M2933 |
| PEG 2000 MME | Fluka | Cat# 81321 |
| Ammonium Sulfate | Fisher scientific | Cat# A702-500 |
| Glycine | Jena Biosciences | Cat# CS-507L |
| Ammonium acetate | Fisher scientific | Cat# BP326-500 |
| Sodium Acetate | Fisher scientific | Cat# S209-500 |
| CAA | Sigma-Aldrich | Cat# C0267 |
| TCEP | Sigma-Aldrich | Cat# C4706 |
| Urea | Fischer Scientific | Cat# BP169-500 |
| LysC | Wako Chemicals | Cat# 129-02541 |
| Trypsin | Promega | Cat# V5280 |
| Formic Acid | Sigma-Aldrich | Cat# F0507 |
| CDS Empore™ SDB-RPS | Fischer Scientific | Cat# 13-110-023 |
| Dulbecco’s Modified Eagle’s Medium | Gibco | Cat# 10566024 |
| Fetal bovine serum | Gibco | Cat# 16140071 |
| Sf-900 III SFM | Gibco | Cat# 12658027 |
| Freestyle 293 expression medium | Gibco | Cat# 12338018 |
| DMEM media | Corning | Cat# 10-013-CV |
| Sodium butyrate | ACROS Organics | Cat# 263191000 |
| Lipofectamine 2000 | Invitrogen | Cat#11668027 |
| Penicillin Streptomycin Fungizone | Cytiva HyClone | Cat# SV3007901 |
| FugeneHD | Promega | Cat# E2311 |
| EGTA | Sigma | Cat# E4378 |
| Bis-Tris Propane | RPI | Cat# B78000100.0 |
| HEPES | Sigma | Cat# H3375-250G |
| D-mannitol | Fisher | Cat# M120-500 |
| Ca-Gluconate | Sigma | Cat# C8231-100G |
| Deposited Data | ||
| Coordinates of GLR3.4-S1S2-Glu | This paper | PDB: 7LZ0 |
| Coordinates of GLR3.4-S1S2-Ser | This paper | PDB: 7LZ1 |
| Coordinates of GLR3.4-S1S2-Met | This paper | PDB: 7LZ2 |
| Coordinates of full-length GLR3.4 | This paper | PDB: 7LZH |
| Cryo-EM map of full-length GLR3.4 | This paper | EMD-23606 |
| Coordinates of ATD and micelle subtracted GLR3.4 | This paper | PDB: 7LZI |
| Cryo-EM map of ATD and micelle subtracted GLR3.4 | This paper | EMD-23607 |
| MS raw files and output tables | This paper | PXD024563 |
| S1S2 of AtGLR3.2 | (Gangwar et al., 2021) | PDB: 6VE8 |
| S1S2 of AtGLR3.3 | (Alfieri et al., 2020) | PDB: 6R8A |
| Experimental Models: Cell Lines | ||
| HEK293S GnTI− | ATCC | Cat#CRL-3022 |
| Sf9 | Gibco | Cat#12659017 |
| COS-7 | ATTC | CRL-1651 |
| E. coli Origami B (DE3) | Novagen | Cat# 70837 |
| Experimental Models: Organisms/Strains | ||
| Agrobacterium tumefaciens strain GV3101 | GoldBio | CC-207-A |
| Arabidopsis thaliana col0 ecotype | ABRC | CS70000 |
| Arabidopsis thaliana tDNA mutant atglr3.4-1 | ABRC | Salk_201768C |
| Arabidopsis thaliana tDNA mutant atglr3.4-2 | ABRC | Salk_079842 |
| Recombinant DNA | ||
| pEF1-YC3.6 | Dr. Jörg Kudla lab, Univ. Muenster, Germany | N/A |
| pcDNA3 | Invitrogen | N/A |
| pcDNA3-atglr3.4 | This paper | N/A |
| pCDNA3-atglr3.4(C205A) | This paper | N/A |
| pCI | Promega | N/A |
| pCI-atCNIH1 | (Wudick et al., 2018) | N/A |
| pCI-atCNIH4 | (Wudick et al., 2018) | N/A |
| pDONR201 | Invitrogen | N/A |
| pDONR201-atglr3.4(gDNA) | This paper | N/A |
| pDS_EF1-XB-glr3.4-CFP | Dr. Edgar Spalding, University of Wisconsin, Madison | N/A |
| pIRES-CD8 | Clontech | N/A |
| pK7FWG2-Lat52 | (Wudick et al., 2018) | N/A |
| pSoup | Hellens, R.P. | GenBank: EU048870.1 |
| Oligonucleotides | ||
| AtGLR3.4 amplification primer: 5’-gtcgactccgccaccatgggatttttggtgatgataagag -3’ | This paper | N/A |
| AtGLR3.4 amplification primer: 5’- cggcaccagagtaatttcgccatgttgtgattgtga -3’ | This paper | N/A |
| B193 5’ – CACTATAGGGCGAATTGGGTACCGTCGACATAC TCGACTCAGAAGG – 3’ | This paper | N/A |
| B204 5’ – AGGGAACAAAAGCTGGGGAATTCAGGTCACTG GATTTTGGTTTTAGG – 3’ | This paper | N/A |
| B278 5’ – CGTAATACGACTCACTATAGGGCGAATTGGGT ACC GTCGACGAGTCAGTAATAAACG – 3’ | This paper | N/A |
| B279 5’ – TCGTTCAGCTTTTTTGTACAAACTTGTGATATCA CTAGTCTGTTAATCAGAAAAACTCAG – 3’ | This paper | N/A |
| B394 5’ – GGGGACAAGTTTGTACAAAAAAGCAGGCTTCA TGGGATTTTTGGTGATGATAA – 3’ | This paper | N/A |
| B395 5’ – GGGGACCACTTTGTACAAGAAAGCTGGGTCAG TAATTTCGCCATGTTGTGAT – 3’ | This paper | N/A |
| B579 5’ – TGCAGATATCCATCACACTGGCGGCCGCATGG GATTTTTGGTGATGATAAGAG – 3’ | This paper | N/A |
| B580 5’ – ATAGGGCCCTCTAGATGCATGCTCGAGTTAAGT AATTTCGCCATGTTGTGATTGT – 3’ | This paper | N/A |
| C328 5’ – CCCGTTCCTGCCAGCCTCATCATCAACGAATA – 3’ | This paper | N/A |
| C329 5’ – CGTTGATGATGAGGCTGGCAGGAACGGGAT ATC – 3’ | This paper | N/A |
| Software and Algorithms | ||
| Leginon | (Suloway et al., 2005) | http://emg.nysbc.org/redmine/projects/leginon/wiki/Leginon_Homepage |
| gCTF | (Zhang, 2016) | http://www.mrc-lmb.cam.ac.uk/kzhang/Gctf/ |
| Motioncor2 | (Zheng et al., 2017b) | http://msg.ucsf.edu/em/software/motioncor2.html |
| RELION 2.0 | (Kimanius et al., 2016) | http://www2.mrc-lmb.cam.ac.uk/relion/ |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Pymol (Schrödinger) | (DeLano, 2002) | http://www.pymol.org |
| PHENIX | (Adams et al., 2010) | https://www.phenix-online.org/ |
| CCP4 | (Winn et al., 2011) | http://www.ccp4.ac.uk/ |
| COOT | (Emsley and Cowtan, 2004) | http://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot |
| XDS | (Kabsch, 2010) | http://xds.mpimf-heidelberg.mpg.de/ |
| Swiss-Model | (Waterhouse et al., 2018) | https://swissmodel.expasy.org/ |
| PDBsum | (Laskowski et al., 2018) |
https://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html |
| SigmaPlot 11.0 | Systat Software Inc. | Systatsoftware.com |
| Origin 2015 Sr2 | OriginLab Corporation | https://www.originlab.com/doc/ |
| pFind | (Chi et al., 2018) | http://pfind.ict.ac.cn/ |
| PDBePISA | (Krissinel and Henrick, 2007) | http://www.ebi.ac.uk/pdbe/prot_int/pistart.html |
| CheckMyMetal | (Zheng et al., 2017a) | https://cmm.minorlab.org/ |
| Other | ||
| DeltaVision Elite Deconvolution/TIRF microscope system | GE Healthcare | Part # 53-851206-001 |
| CF-1.2/1.3-2Au 200 mesh holey carbon grids | Protochips | Cat#CF-1.2/1.3-2Au |
| Gold wire | Ted Pella, Inc. | Cat#21-10 |
| Ion Exchange Hi-Trap Q HP column | GE Healthcare | Cat# 17-1154-01 |
| Size Exclusion Superose 10/300 column | GE Healthcare | Cat# 17-5172-01 |
Data and code availability
Cryo-EM map for full-length GLR3.4 and after ATD and micelle subtraction have been deposited to the EMDB with the accession codes EMD-23606 and EMD-23607, respectively. Coordinates for full-length GLR3.4 and after ATD and micelle subtraction have been deposited to the PDB with the accession codes 7LZH and 7LZI, respectively. Coordinates and structure factors for GLR3.4-S1S2Glu, GLR3.4-S1S2Ser and GLR3.4-S1S2Met structures have been deposited to the PDB with the accession codes 7LZ0, 7LZ1 and 7LZ2, respectively. Raw files and full output tables for mass spectrometry results are available through PRIDE repository with identifier PXD024563 (reviewer access username: reviewer_pxd024563@ebi.ac.uk, password: UDehuNMe). This study did not generate new code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Expression of the full-length GLR3.4 protein was performed in HEK 293S GnTI− cells (ATCC) that were cultured in the Freestyle 293 expression medium (GIBCO) at 37°C and 5% CO2. Baculovirus for infecting HEK 293S GnTI− cells was produced in Sf9 cells (GIBCO) that were cultured in the Sf-900 III SFM media (GIBCO) at 27°C. S1S2 protein expression was performed in Escherichia coli Origami B (DE3) cells (Novagen). Cells were cultured in LB media at 37°C until OD600 reached the value of 1.0-1.2, then cooled down to 20°C, induced with 250 μM IPTG and incubated for another 20 hours at 20°C. COS-7 cells for calcium imaging experiments were maintained at 37°C and 5% CO2 in Dulbecco’s Modified Eagle’s Medium, supplemented with 5 % fetal bovine serum and 1 % penicillin/streptomycin.
METHOD DETAILS
Constructs
The full-length Arabidopsis thaliana GLR3.4 (residues 1-959; Uniprot Q8GXJ4, NCBI NP_001030971.1) was introduced into a pEG BacMam vector for baculovirus-based protein expression in mammalian cells (Goehring et al., 2014), with the C-terminal thrombin cleavage site (LVPRG), followed by eGFP and streptavidin affinity tag (WSHPQFEK). The GLR3.4-S1S2 construct for expression of the isolated LBD was made by introducing S1 (residues 492-601) and S2 (residues 709-842) fragments of GLR3.4 connected by the glycine-threonine (GT) linker into a pET22b vector (Novagen), with the N-terminal 8xHis affinity tag followed by the thrombin cleavage site. The boundaries of S1 and S2 were determined based on sequence alignment of GLR3.4 with GluA2 and GLR3.2 LBDs (Armstrong and Gouaux, 2000; Armstrong et al., 1998; Sobolevsky et al., 2009).
For expression in COS-7 cells, full-length GLR3.4 cDNA was amplified by PCR from vector pDS_EF1-XB-glr3.4-CFP (from Spalding’s lab) with the primer pair B579/B580. The resulting PCR product was cloned into NotI/XhoI digested pcDNA3 via GIA (Gibson Isothermal Assembly) to yield pcDNA3-atglr3.4. In order to generate the mutant pCDNA3-atglr3.4(C205A), two PCRs were performed using pcDNA3-atglr3.4 as template with the primer pairs B579/C328 and C329/B580. The resulting PCR products were cloned into NotI/XhoI digested pCDNA3 via GIA.
For expression in Arabidopsis pollen, a custom gateway destination vector, pGreenII-OLE1p::OLE1::eGFP atUBQ10p::Gateway::eGFP, was generated. The pLat52 Gateway cassette with C-terminal eGFP was PCR amplified with the primer pair B193/B204 using pK7FWG2-Lat52 (Wudick et al., 2018b) as template and the resulting product was digested with Spel and purified. PCR using Arabidopsis col0 gDNA was performed with the primer pair B278/B279 to amplify the atUBQ10 (At4g05320) promoter. Both PCR products were then cloned into KpnI/EcoRI digested pGreenII-OLE1p::OLE1::eGFP Lat52p::Gateway (Wudick et al., 2018b) via GIA. The genomic coding sequence of atglr3.4 was amplified with primer pair B394/B395 via PCR using Arabidiopsis col0 gDNA as template. The resulting PCR product was then used in a Gateway BP reaction with pDONR201 to create the donor clone, pDONR201-atglr3.4. A Gateway LR reaction was performed with donor clone pDONR201-atglr3.4(gDNA) and destination clone pGreenII-OLE1p::OLE1::eGFP atUBQ10p::Gateway::eGFP to generate the expression clone pGreenII-OLE1p::OLE1::eGFP atUBQ10p::atglr3.4(gDNA)::eGFP.
Plant Transformation
Expression construct pGreenII-OLE1p::OLE1::eGFP atUBQ10p::atglr3.4(gDNA)::eGFP was transformed into Agrobacteria strain GV3101 harboring the pSoup helper plasmid via electroporation. Arabidopsis thaliana col0 plants were then transformed via the floral dip method according to standard protocols (Wudick et al., 2018b).
GLR3.4-S1S2 expression and purification
GLR3.4-S1S2 was transformed into Escherichia coli Origami B (DE3) cells and grown in LB media supplemented with 100 μg/ml ampicillin, 15 μg/ml kanamycin and 12.5 μg/ml tetracycline. The freshly inoculated culture was grown at 37°C until OD600 reached the value of 0.8-1.2. Then cells were cooled down to 20°C, induced with 250 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) and incubated in the orbital shaker for 20 hours at 20°C. The cells were harvested by centrifugation at 5,000 rpm for 15 min at 4°C and washed with the buffer containing 150 mM NaCl and 20 mM Tris pH 8.0.
For GLR3.4-S1S2Glu, all buffers in the following purification steps were supplemented with 1 mM L-glutamate. First, the cells were resuspended in the lysis buffer containing 200 mM NaCl, 20 mM Tris pH 7.5, 1 mM β-mercaptoethanol (βME), 1 mM phenylmethysulfonyl fluoride (PMSF), 200 μg/ml lysozyme, 5% glycerol and DNAse and disrupted by sonication. After the lysate was centrifuged at 40,000 rpm using the Ti45 rotor for 1 hour at 4°C, the supernatant was mixed with His60 Ni superflow resin (Takara) and rotated for 2 hours at 4°C. The protein-bound resin was washed with the buffer containing 200 mM NaCl, 20 mM Tris pH 7.5, 1 mM βME and 20 mM imidazole and the protein was eluted using the same buffer but containing 200 mM instead of 20 mM imidazole. The protein was 3x diluted in the buffer containing 150 mM NaCl, 20 mM Tris pH 7.5, 1 mM βME and 5% glycerol and subjected to thrombin digestion (1:500 w/w) for 1.5 hour at 22°C. The protein was further purified using ion-exchange Hi-Trap Sepharose-SP (GE Healthcare).
For GLR3.4-S1S2Met and GLR3.4-S1S2Ser, protein purification was similar, except 1 mM L-glutamate was omitted from all purification buffers. In addition, instead of ion exchange chromatography, we used size-exclusion chromatography with the Superdex 16/60 column (GE Healthcare) and the buffer containing 150 mM NaCl, 20 mM Tris pH 7.5, 1 mM βME, and 5% glycerol. The purified protein was evaluated using SDS-PAGE and FSEC (Superpose 10/30 column, GE Healthcare) and supplemented with the corresponding ligand (Met) before subjecting it to crystallization.
GLR3.4-S1S2 crystallization and structure determination
Purified GLR3.4-S1S2Glu protein concentrated to ∼8-10 mg/ml was first subjected to crystallization screening using Mosquito robot (TTP Labtech) in sitting drop vapor diffusion 96-well crystallization plates at 4°C. After two weeks, small octagonal crystals appeared in the condition consisting of 2 M ammonium sulfate and 0.1 M Sodium Acetate pH 4.6. For GLR3.4-S1S2Met, 1 mM methionine was added to the purified protein and the ligand-protein mixture was incubated for 30-40 min on ice before crystallization screening with Mosquito. By not adding any ligands to the protein we intended to solve the S1S2 structure in the apo state, but instead ended up solving the GLR3.4-S1S2Ser structure with serine of an apparent endogenous origin. Small octagon-shaped crystals of GLR3.4-S1S2Met and GLR3.4-S1S2Ser grew in 2 M ammonium sulfate at 4°C. All crystals were cryoprotected using crystal growth buffers supplemented with 25% glycerol and flash-frozen in liquid nitrogen. Crystal diffraction data were collected at the beamline 24-ID-C of the Advanced Photon Source (APS) and processed using XDS(Kabsch, 2010) and Aimless as a part of the CCP4 suite (Winn et al., 2011).
The structure of GLR3.4-S1S2Glu was solved by molecular replacement using Phaser (McCoy, 2007) and GLR3.2-S1S2Gly (PDB 6VEA) as a search probe. The model was refined by alternating cycles of building in COOT (Emsley and Cowtan, 2004) and automatic refinement in Phenix or Refmac (Adams et al., 2010). The structures of GLR3.4-S1S2Met and GLR3.4-S1S2Ser were solved by molecular replacement using the GLR3.4-S1S2Glu structure as a search probe. Water molecules were added manually in Coot. Analysis of possible inclusion of metal ions in the S1S2 crystal structures was performed using the CheckMyMetal validation tool (https://cmm.minorlab.org/) (Zheng et al., 2017a). All structural figures were prepared in Pymol (DeLano, 2002).
Measurements of ligand binding to GLR3.4-S1S2 using microscale thermophoresis
GLR3.4-S1S2 was purified as described above, except the N-terminal 8xHis tag was not cleaved by thrombin and remained with the protein. The unlabeled GLR3.4-S1S2 protein was subjected to microscale thermophoresis (MST) in the Monolith NT Label free (Nano Temper Technologies), which detected its intrinsic tryptophan fluorescence. The 1.5-μM purified protein stock solution was made using the interaction buffer consisted of 150 mM NaCl and 20 mM Tris pH 7.5. The stock solutions of the ligands L-glutamate, L-methionine and L-serine were prepared at the concentration of 16 mM and serially diluted using the same buffer. For interaction measurements, the protein and ligand solutions were mixed at the 1:1 (v/v) ratio, with the protein concentration kept constant and the ligand concentration varied. The interaction mix was incubated at room temperature for 5-10 min and subjected to thermophoresis measurements in 16 capillaries with medium MST and 20% LED power at 24°C. For each ligand, the experiment was repeated 3-4 times and the data was averaged, normalized and fitted with the logistic equation using the Origin 2015 software (OriginLab Corporation).
Full length GLR3.4 expression and purification
The GLR3.4 bacmid and baculovirus were made by using the standard methods (Goehring et al., 2014). The P2 virus was produced in Sf9 cells (GIBCO, 12659017) and added to HEK 293S GnTI− cells (ATCC, CRL-3022) incubated at 37°C and 5% CO2. Cells were supplemented with 10 mM sodium butyrate 12 hours post infection and the temperature was changed to 30°C. Cell were harvested 72 hours post infection using low-speed centrifugation (5,500 g, 10 min), washed using 1X PBS pH 8.0 and pelleted again (5,500 g, 15 min). The pellet was resuspended in 50 ml per 1 L of the initial cell culture lysis buffer consisting of 150 mM NaCl, 20 mM Tris pH 8.0, 1 mM βME, 0.8 μM aprotinin, 2 μg/ml leupeptin, 2 μM pepstatin A and 1 mM PMSF and lysed by sonication. The lysate was centrifuged (9,900 g, 15 min) to remove cell debris and the supernatant was subjected to ultracentrifugation (186,000 g, 40 min) to pellet cell membranes. The membranes were mechanically homogenized and solubilized for 2 hours in the buffer containing 150 mM NaCl, 20 mM Tris-HCl pH 8.0, 1 mM βME and 1% digitonin (Cayman Chemical Company, 14952). Insoluble material was removed by ultracentrifugation (186,000 g, 40 min), while the supernatant was added to the pre-equilibrated Streptavidin-linked resin (2 ml resin per 1L of the initial cell culture) and the mixture was rotated for 10-14 hours at 4°C. The protein-bound resin was washed with 25 ml of the buffer containing 150 mM NaCl, 20 mM Tris-HCl pH 8.0 and 0.05% digitonin and the protein was eluted using the same buffer supplemented with 2.5 mM D-desthiobiotin. The eluted protein was subjected to thrombin digestion (1:300 w/w) at 22°C for 1 hour. The digest reaction was injected into Superose 6 10/30 size-exclusion column (GE Healthcare) equilibrated with the buffer containing 150 mM NaCl, 20 mM Tris-HCl pH 8.0, and 0.05% digitonin. The tetrameric GLR3.4 peak fractions were pooled, concentrated to ∼2.5 mg/ml and used for cryo-EM sample preparation. All the steps, unless otherwise noted, were performed at 4°C.
Cryo-EM sample preparation and data collection
Au/Au grids were prepared as described in the literature (Russo and Passmore, 2014). Briefly, grids were prepared by first coating C-flat (Protochips, Inc., Morrisville, NC) CF-1.2/1.3-2Au 200 mesh holey carbon grids with ∼50 nm of gold using an Edwards Auto 306 evaporator. Subsequently, an Ar/O2 plasma treatment (6 minutes, 50 watts, 35.0 sccm Ar, 11.5 sccm O2) was used to remove the carbon with a Gatan Solarus (model 950) Advanced Plasma Cleaning System. The grids were again plasma treated (H2/O2, 20 s, 10 watts, 6.4 sccm H2, 27.5 sccm O2) prior to sample application in order to make their surfaces hydrophilic. Purified GLR3.4 protein was mixed with 1 mM L-Glutamate and incubated on ice for ∼30 min before grid preparation. A Vitrobot Mark IV (FEI) was used to plunge-freeze the grids after the application of 3 μl protein solution with 100% humidity at 4°C, a blot time of 3 seconds, blot force set to 3, and a wait time of 20 seconds. The grids were clipped and loaded into a 300 kV Titan Krios microscope equipped with Gatan K3 direct electron detection camera. 11,854 micrographs were collected in counting mode with a pixel size of 0.83 Å (∼105,000x magnification) across a defocus range of −1.0 μm to −2 μm. The total dose of 58 e−Å−2 was attained by using a dose rate of ∼16.0 e−pixel−1s−1 across 50 frames for 2.5 seconds total exposure time.
Image processing
The total of 11,854 micrographs were collected as two datasets, dataset 1 (6,119 micrographs) and dataset 2 (5,735 micrographs) and initially processed in Relion 3.1 (Zivanov et al., 2018). Frame alignment was done using MotionCor2 (Zheng et al., 2017b). CTF estimation was performed using Gctf (Zhang, 2016) on non-dose-weighted micrographs while subsequent data processing was done on dose-weighted micrographs. For each data set, ∼3,000 particles were manually selected to generate 2D classes that were used as templates to autopick 1,044,465 particles from dataset 1 and 1,116,729 particles from dataset 2. The particle images were 4×4 binned to a pixel size of 3.32 Å/pixel and subjected to 3D classification, each dataset into ten classes. A density map was generated in Chimera from the crystal structure of GluA2 (PDB ID: 3KG2), low-pass filtered to 40 Å, and used as an initial reference. The best 3D classes contained 190,076 particles for dataset 1 and 145,948 particles for dataset 2, which were further unbinned to a pixel size of 0.83 Å/pixel and re-extracted. These particles were cleaned up by 3D classification and subsequently subjected to Bayesian polishing and CTF refinement. After refinement and postprocessing, the resulting 109,759 particles for dataset 1 and 117,856 particles for dataset 2 produced 3D maps at the resolution of 5.0 and 4.2 Å, respectively. Then the particles for datasets 1 and 2 were joined and subjected to micelle subtraction and multiple rounds of 3D classification to reduce particle heterogeneity. After additional beam tilt CTF refinement, followed by regular refinement and postprocessing, the remaining 118,592 cleaned up particles produced a map at the resolution of 3.73 Å. These particles were imported into cryoSPARC (Punjani et al., 2017) and subjected to several rounds of 2D clean up, homogeneous and non-uniform refinement with C2 symmetry, resulting in a final map at 3.57 Å resolution (110,630 particles). Focused classification and focused refinement were performed to improve density in the TMD region (Figure S3D). The total of 227,615 combined particles from the two datasets were particle subtracted in Relion 3.1 by providing a soft mask around the LBD-TMD region. The subtracted particles were refined, cleaned by several rounds of 2D classification in Relion 3.1 and refined again with a soft mask to yield a final map at 4.39 Å resolution (174,044 particles). The overall resolution was estimated using the Fourier shell correlation (FSC=0.143) criterion on masking-effect-corrected FSC curves calculated between two independent half-maps (Chen et al., 2013; Scheres, 2012). The local resolutions were estimated with unfiltered half-maps using ResMap (Kucukelbir et al., 2014) and EM density maps were visualized using UCSF Chimera (Pettersen et al., 2004).
Model building
For model building of the LBD, the GLR3.4-S1S2Glu crystal structure was used as a guide. To guide model building of the ATD and TMD, homology models of these domains were created using Swiss-Model (Waterhouse et al., 2018) and the GluA2 structure (PDB ID: 3KG2) as a template. The remaining parts of GLR3.4 were built de novo using cryo-EM density as a guide. The resulting model was real space refined in Phenix (Afonine et al., 2012) and visualized using Chimera (Pettersen et al., 2004) or Pymol (DeLano, 2002). The analysis of intersubunit interfaces as well as surface area calculations were performed using the PDBePISA web service (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) (Krissinel and Henrick, 2007).
Cysteine Crosslinking
Cysteine substitutions were introduced using conventional PCR-based methods. Constructs were verified by sequencing over the entire length of the GLR3.4 coding region. Wild type and cysteine-substituted GLR3.4 proteins were expressed and purified as described above, except 1% lauryl maltose neopentyl glycol (LMNG) detergent (Anatrace) was used instead of digitonin for protein extraction. Binding of protein to streptavidin-linked resin lasted for 1 hour. Instead of 0.05% of digitonin, 0.1 mM LMNG was in the buffers that were used to wash the protein-bound resin, elute the protein, and for size-exclusion chromatography. For crosslinking, ∼2.5 μg of protein was subjected to denaturing conditions by addition of SDS sample buffer in the absence (non-reducing condition) or presence (reducing condition) of 600 mM dithiothreitol (DTT). The protein samples were then run on a 4-15% gradient SDS-PAGE gel and protein bands were visualized by AquaStain Protein Gel Stain (Bulldog Bio). The oligomeric state of wild type and cysteine-substituted GLR3.4 was assessed by subjecting ∼2 μg of protein to FSEC.
COS-7 cells transfection and Ca2+ imaging
Protocols for COS-7 cells transfection and Ca2+ imaging were adapted from the previous studies (Ortiz-Ramírez et al., 2017). COS-7 cells (Sigma-Aldrich) were maintained at 37°C and 5% CO2 in Dulbecco’s Modified Eagle’s Medium, supplemented with 5% fetal bovine serum and 1% penicillin/streptomycin (Gibco), and transfected at low passage (P < 7). Cells were plated at a density at 50% confluence in 35-mm diameter dishes and transfected using FugeneHD (Promega) as specified by the supplier. Cells were co-transfected with pCI-AtCNIH4 (0.2 μg), pCI-AtCNIH1 (0.2 μg), pCI-AtGLR3.4 (0.8 μg) and pEF1-YC3.6 (0.5 μg). The co-transfection with pCI-AtCNIH4 and pCI-AtCNIH1 was previously shown to enhance functional expression and activity of GLRs (Wudick et al., 2018b). In experiments illustrated in Figure S2E, pCI-AtCNIH4 was increased to 0.4 μg (red circles) and replaced by empty pCI plasmid in cells expressing AtGLR3.4 alone (black circles). Cells were used for imaging 38 to 41 hours after transfection. They were washed in a Ca2+-free solution (1 mM EGTA, 10 mM Bis-Tris propane buffered to pH 7.3 with HEPES and set to 350 mOsmol with D-mannitol). Cells were imaged in the Ca2+-free solution for 1.5 min before the addition of Ca2+ to a final concentration of 14.5 mM (using Ca-Gluconate). The ligands (Glu, Asn, or Gly; 0.5 or 1.0 mM) were added at the beginning, before Ca2+ was introduced. L-glutamate, Glycine and L-Asparagine were prepared as 100 mM stock solutions (Sigma-Aldrich, St. Louis, MO, USA). L-Glu stock solution was adjusted to pH 7.0 with Bis-tris propane. Time-lapse acquisition was performed with a sampling interval of 30 secs. 8 to 12 cells were imaged in each dish using the stage position recording tool of the microscope system. Imaging was performed at room temperature using a DeltaVision Elite Deconvolution/TIRF microscope system (Olympus inverted IX-71) under a 60X lens (1.2NA UPLSAPO water /WD 0.28 mm). A xenon lamp from the DeltaVision system was used with a CFP excitation filter (438-424 nm). Two simultaneous emission records were captured: YFP emission (548-522 nm) and CFP emission (475-424 nm). To minimize bleaching, the laser was set to 2%. YFP and CFP imaging were recorded with 0.6 sec exposure time. Images were processed using ImageJ. Ratios were obtained after background subtraction and signal clipping using the “Ratio-plus” plug-in for ImageJ. The signal of each channel was averaged in a circle in the middle of the cell (with 100-200 pixel diameter, depending on the size of the cell). The YFP/CFP ratio was obtained by dividing the emission recorded for YFP (548-522 nm) by the one recorded for CFP (475-424 nm). No significant bleaching or ratio drift was observed in our experimental conditions.
Patch-clamp recordings
For patch-clamp recordings, COS-7 cells were maintained and transfected according to the protocol described in the COS-7 cells transfection and Ca2+ imaging section. Cells were cotransfected with pCI-AtCNIH4 (0.2 μg), pCI-AtCNIH1 (0.2 μg), pCDNA3-AtGLR3.4 (0.4 μg) and pIRES-CD8 (0.05 μg), the latter to select transfected cells with the help of anti-CD8 antibody-coated beads (Dynabeads, Thermo Fisher Scientific, USA). In experiments described in Figure S2B, the quantity of pCDNA3-AtGLR3.4 was increased to 0.8 μg. Patch-clamp was performed in the whole-cell configuration, 39 to 45 hours after transfection. Pipettes were pulled using a P97 puller (Sutter Instrument, Novato, CA, USA). Their resistance was 3-5 MΩ in the bath solution. Whole-cell currents were recorded at the sampling frequency of 2-4 kHz and filtered at 1-2 kHz using an Axopatch 200A amplifier with an Axon 1200 DigiData analog-to-digital converter and pCLAMP software (Molecular Devices, Sunnyvale, CA, USA). The pipette solution contained 150 mM Na-Gluconate, 3 mM MgCl2, 4 mM HCl, 5 mM EGTA, and 10 mM Bis-tris-propane pH 7.2 (adjusted using HEPES). The bath solution contained 20 mM Ca-Gluconate, 10 mM Na-Gluconate and 10 mM Bis-tris propane pH 6.5 (adjusted using MES). Solutions were adjusted to 350 mOsmol with D-mannitol. L-glutamate, Glycine and L-Asparagine were prepared as 100 mM stock solutions (Sigma-Aldrich, St. Louis, MO, USA). L-Glu stock solution was adjusted to pH 7.0 with Bis-tris propane. In experiments illustrated in Figure S2C, washout was performed by gravity perfusion at the rate of 1 ml per minute. For the experiments with GSH, the pipette solution contained: 150 mM NaCl, 3 mM MgCl2, 10 mM BTP, 5 mM EGTA, and 100 nM free Ca2+. pH was adjusted to 7.3 with HEPES and osmolarity to 350 mOsmol with D-mannitol. Bathing solution contained: 15 mM CaCl2 and 10 mM BTP. pH was adjusted to 7.3 with HEPES and osmolarity to 350 mOsmol with D-mannitol. The voltage protocol during current acquisition followed the sequence: (1) cells were clamped at a holding potential of 0 mV, and (2) 1.5-second voltage pulses were applied in the range of −100 to +60 mV with the 20-mV steps. The holding potential was returned to 0 mV for 1.1 seconds between the pulses.
Extracellular calcium influx measurements
Extracellular Ca2+ influx was measured at the tip of the pollen tubes using the ion-selective vibrating probe with simultaneous growth rate monitoring, as described previously (Wudick et al., 2018b). Measurements were performed in two Arabidopsis thaliana independent T-DNA insertion lines, Atglr3.4-1 (SALK_201768) and Atglr3.4-2 (SALK_079842), while Col-0 was used as wild-type. Plants were grown under short-day conditions (12 hours of light at 22°C and 12 hours of dark at 18°C, with an irradiance of ca. 100 μmol m−2sec−1). Arabidopsis pollen grains were collected from fresh flowers and then germinated in liquid medium containing 500 μM KCl, 500 μM CaCl2, 125 μM MgSO4, 0.005% H3BO3, 125 μM HEPES pH 7.5 and 16% sucrose. Pollen grains were incubated at 21.5°C for at least 3 hours. Growing pollen tubes longer than 150 μm were selected for flux and growth rate measurements.
Confocal microscopy of Arabidopsis pollen
Wild-type Col-0 pollen stably expressing UBQ10:GLR3.4-GFP was mounted in germination medium (500 μM KCl, 500 μM CaCl2, 125 μM MgSO4, 0.005% H3BO3, 125 μM HEPES pH 7.5 and 16% sucrose) and visualized using the 63×/1.4 plan-apochromat objective of a Zeiss LSM700 laser scanning confocal microscope using the 488 nm laser for excitation (emission recorded at 500-530 nm). Image analysis was performed using ImageJ (http://rsbweb.nih.gov/ij/).
Liquid chromatography and mass spectrometry
Purified full length GLR3.4 was alkylated with 40 mM 2-chloroacetamide (CAA, Sigma-Aldrich) followed by the addition of Urea (Sigma-Aldrich) to a final concentration of 8 M. For reducing conditions, tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) was added to a final concentration of 10 mM. The resulting solution was heated to 37°C for 1 h. Next, the samples were diluted with 50 mM ammonium bicarbonate (Sigma-Aldrich) and digested by a combination of LysC (Wako) and Trypsin (Promega). The final peptide mixtures were acidified with formic acid (Sigma-Aldrich) to a final concentration of 1% and then desalted with CDS Empore™ SDB-RPS (Fischer Scientific) in house-packed stage-tips (Rappsilber et al., 2007).
Desalted peptides were injected into an EASY-Spray PepMap RSLC C18 50 cm × 75 μm column (Thermo Scientific), which was coupled to the Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific). Peptides were eluted at the flow rate of 250 nL/min with a non-linear 120-min gradient of 5-30% buffer B consisted of 0.1% (v/v) formic acid and 100% acetonitrile. The column temperature was maintained at 50°C throughout the entire experiment. Thermo Scientific™ Orbitrap Fusion™ Tribrid™ mass spectrometer was used for peptide MS/MS analysis. Survey scans of peptide precursors were performed from 350 to 1500 m/z at 120K FWHM resolution (at 200 m/z) with a 2 × 105 ion count target and a maximum injection time of 60 ms. The instrument was set to run in top speed mode with 3 s cycles for the survey and the MS/MS scans. After the survey scan, tandem MS was performed on the most abundant precursors exhibiting a charge state from 2 to 6 of greater than 2 × 105 intensity by isolating them in the quadrupole at 1.6 Th. HCD fragmentation was applied with 30% collision energy and the resulting fragments were detected using the auto: m/z Normal in the Orbitrap. The AGC target for MS/MS was set to 5 × 104 and the maximum injection time limited to 30 ms. The dynamic exclusion was set to 30 s with a 10 ppm mass tolerance around the precursor and its isotopes. Monoisotopic precursor selection was enabled.
The acquired raw data were analyzed with pFind software platform (Chi et al., 2018) against reduced database based on the reviewed Arabidopsis thaliana proteins (16,268 entries, version 2021_02, downloaded from UniProt) from which 1,000 random entries plus GLR3.4 were retained. Full-specific mode with TrypsinKR_C and up to two missed cleavages were used. Precursor tolerance and Fragment tolerance were set to 20 ppm. Modification search was performed in an open mode with Carbamidomethyl (C), Glutathione (C), and Oxidation (M) set as dynamic modifications. In case of Glutathione, a neutral loss was defined as 129.1148 Da. The FDR was set as 1% with the peptides mass in the range of 600-10,000 Da and the length between 6 and 100 residues. The raw files and full output tables are available through PRIDE repository with identifier PXD024563 (reviewer access username: reviewer_pxd024563@ebi.ac.uk, password: UDehuNMe).
QUANTIFICATION AND STATISTICAL ANALYSIS
Calcium imaging and confocal microscopy data (Figure 1B, S1 and S2E–F) were analyzed using ImageJ. Mass spectrometry data (Figure 3B) was analyzed using pFind. Statistical analysis was performed using SigmaPlot 11.0 (Figures 1A–B, 3C-E, S1D and S2) and Origin 2015 Sr2 (Figure S6E). In all figure legends, n represents the number of independent biological replicates. All quantitative data were presented as mean ± SEM. Statistical analyses were conducted using one-way ANOVA followed by the post-hoc Dunnett test (Figures S1D and S2D). Values of p < 0.05 were considered statistically significant.
Supplementary Material
Supplementary Movie 1, Related to Figure 1
The movie shows rotating cryo-EM density for the entire GLR3.4 tetramer or a single subunit, with or without the molecular model, before or after ATD and micelle subtraction.
Highlights.
Cryo-EM structure of Arabidopsis thaliana glutamate receptor-like channel GLR3.4
Glutathione regulates channel activity by binding to C205 in amino-terminal domain
Crystal structures of GLR3.4 ligand-binding domain illustrate ligand promiscuity
Amino-terminal and ligand-binding domain layers show non-swapped domain arrangement
ACKNOWLEDGMENTS
We thank R. Grassucci, Z. Zhang, Y.-C. Chi, and L. Zheng (Columbia University Cryo-Electron Microscopy Center) for help with microscope operation and cryo-EM data collection, Surajit Banerjee (beamline 24-ID-C of the Advanced Photon Source) for assistance with crystallographic data collection, Rajesh Kumar Soni (Proteomics and Macromolecular Crystallography Shared Resource, Herbert Irving Comprehensive Cancer Center, Columbia University) for Mass Spectrometry data acquisition, and Renato Bruni (New York Structural Biology Center) for help with MST. The crystallographic data were collected at the beamline 24-ID-C of the Advanced Photon Source, one of the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on the 24-ID-C beamline is funded by an NIH-ORIP HEI grant (S10 RR029205). A.I.S. was supported by the NIH (R01 CA206573, R01 NS083660, R01 NS107253) and the NSF (1818086). J.A.F. was supported by the NIH (R01 GM131043) and the NSF (MCB1714993 and MCB1930165). M.M.W. was supported by the Deutsche Forschungsgemeinschaft (DFG, 267205415-SFB 1208).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfieri A, Doccula FG, Pederzoli R, Grenzi M, Bonza MC, Luoni L, Candeo A, Romano Armada N, Barbiroli A, Valentini G, et al. (2020). The structural bases for agonist diversity in an Arabidopsis thaliana glutamate receptor-like channel. Proc Natl Acad Sci U S A 117, 752–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N, and Gouaux E (2000). Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ, and Gouaux E (1998). Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 395, 913–917. [DOI] [PubMed] [Google Scholar]
- Burada AP, Vinnakota R, and Kumar J (2020a). The architecture of GluD2 ionotropic delta glutamate receptor elucidated by cryo-EM. J Struct Biol 211, 107546. [DOI] [PubMed] [Google Scholar]
- Burada AP, Vinnakota R, and Kumar J (2020b). Cryo-EM structures of the ionotropic glutamate receptor GluD1 reveal a non-swapped architecture. Nat Struct Mol Biol 27, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, and Henderson R (2013). High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhao Y, Wang Y, Shekhar M, Tajkhorshid E, and Gouaux E (2017). Activation and Desensitization Mechanism of AMPA Receptor-TARP Complex by Cryo-EM. Cell 170, 1234–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Liu C, Yang H, Zeng WF, Wu L, Zhou WJ, Wang RM, Niu XN, Ding YH, Zhang Y, et al. (2018). Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat Biotechnol. 36, 1059–1061. [DOI] [PubMed] [Google Scholar]
- De Bortoli S, Teardo E, Szabò I, Morosinotto T, and Alboresi A (2016). Evolutionary insight into the ionotropic glutamate receptor superfamily of photosynthetic organisms. Biophys Chem 218, 14–26. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002). The PyMOL Molecular Graphics System (San Carlos, CA, USA: DeLano Scientific; ). [Google Scholar]
- Dixon DP, Skipsey M, Grundy NM, and Edwards R (2005). Stress-induced protein S-glutathionylation in Arabidopsis. Plant Physiol 138, 2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Forde BG, and Roberts MR (2014). Glutamate receptor-like channels in plants: a role as amino acid sensors in plant defence? F1000Prime Rep 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangwar SP, Green MN, Michard E, Simon AA, Feijo JA, and Sobolevsky AI (2021). Structure of the Arabidopsis Glutamate Receptor-like Channel GLR3.2 Ligand-Binding Domain. Structure 29, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring A, Lee CH, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, and Gouaux E (2014). Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc 9, 2574–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H, and Traynelis SF (2010). Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol 78, 535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Chow D, Martick MM, and Garcia KC (2001). Allosteric activation of a spring-loaded natriuretic peptide dimer by hormone. Science 293, 1657–1662. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Furukawa H, and Gouaux E (2005). Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron 47, 71–84. [DOI] [PubMed] [Google Scholar]
- Jalali-Yazdi F, Chowdhury S, Yoshioka C, and Gouaux E (2018). Mechanisms for Zinc and Proton Inhibition of the GluN1/GluN2A NMDA Receptor. Cell 175, 1520–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Singh SK, Gu S, Furukawa H, Sobolevsky AI, Zhou J, Jin Y, and Gouaux E (2009). Crystal structure and association behaviour of the GluR2 amino-terminal domain. Embo J 28, 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AM, Chory J, Dangl JL, Estelle M, Jacobsen SE, Meyerowitz EM, Nordborg M, and Weigel D (2008). The impact of Arabidopsis on human health: diversifying our portfolio. Cell 133, 939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, and Furukawa H (2014). Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E, Simorowski N, and Furukawa H (2009). Structure of the zinc-bound amino-terminal domain of the NMDA receptor NR2B subunit. Embo J 28, 3910–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawate T, and Gouaux E (2006). Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681. [DOI] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5, e18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A, Hu H, Feng D, Sun B, Zhang Y, Robertson MJ, Chu M, Kobilka TS, Laeremans T, Steyaert J, et al. (2019). Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohda K, Wang Y, and Yuzaki M (2000). Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci 3, 315–322. [DOI] [PubMed] [Google Scholar]
- Kong D, Hu HC, Okuma E, Lee Y, Lee HS, Munemasa S, Cho D, Ju C, Pedoeim L, Rodriguez B, et al. (2016). L-Met Activates Arabidopsis GLR Ca. Cell Rep 17, 2553–2561. [DOI] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, and Tagare HD (2014). Quantifying the local resolution of cryo-EM density maps. Nat Methods 11, 63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HM, Chiu J, Hsieh MH, Meisel L, Oliveira IC, Shin M, and Coruzzi G (1998). Glutamate-receptor genes in plants. Nature 396, 125–126. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Jablonska J, Pravda L, Varekova RS, and Thornton JM (2018). PDBsum: Structural summaries of PDB entries. Protein Sci 27, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X, and Gouaux E (2014). NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomash S, Chittori S, Brown P, and Mayer ML (2013). Anions mediate ligand binding in Adineta vaga glutamate receptor ion channels. Structure 21, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ (2007). Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhoff O, Müller K, Roelfsema MR, Latz A, Lacombe B, Hedrich R, Dietrich P, and Becker D (2005). AtGLR3.4, a glutamate receptor channel-like gene is sensitive to touch and cold. Planta 222, 418–427. [DOI] [PubMed] [Google Scholar]
- Meyerson JR, Chittori S, Merk A, Rao P, Han TH, Serpe M, Mayer ML, and Subramaniam S (2016). Structural basis of kainate subtype glutamate receptor desensitization. Nature 537, 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michard E, Lima PT, Borges F, Silva AC, Portes MT, Carvalho JE, Gilliham M, Liu LH, Obermeyer G, and Feijó JA (2011). Glutamate receptor-like genes form Ca2+ channels in pollen tubes and are regulated by pistil D-serine. Science 332, 434–437. [DOI] [PubMed] [Google Scholar]
- Michard E, Simon AA, Tavares B, Wudick MM, and Feijó JA (2017). Signaling with Ions: The Keystone for Apical Cell Growth and Morphogenesis in Pollen Tubes. Plant Physiol 173, 91–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou W, Kao YT, Michard E, Simon AA, Li D, Wudick MM, Lizzio MA, Feijó JA, and Chang C (2020). Ethylene-independent signaling by the ethylene precursor ACC in Arabidopsis ovular pollen tube attraction. Nat Commun 11, 4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi SA, Chauvin A, Pascaud F, Kellenberger S, and Farmer EE (2013). GLUTAMATE RECEPTOR-LIKE genes mediate leaf-to-leaf wound signalling. Nature 500, 422–426. [DOI] [PubMed] [Google Scholar]
- Nakagawa T (2019). Structures of the AMPA receptor in complex with its auxiliary subunit cornichon. Science 366, 1259–1263. [DOI] [PubMed] [Google Scholar]
- Ortiz-Ramírez C, Michard E, Simon AA, Damineli DSC, Hernández-Coronado M, Becker JD, and Feijó JA (2017). GLUTAMATE RECEPTOR-LIKE channels are essential for chemotaxis and reproduction in mosses. Nature 549, 91–95. [DOI] [PubMed] [Google Scholar]
- Papasergi-Scott MM, Robertson MJ, Seven AB, Panova O, Mathiesen JM, and Skiniotis G (2020). Structures of metabotropic GABA. Nature 584, 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Fu Z, Frangaj A, Liu J, Mosyak L, Shen T, Slavkovich VN, Ray KM, Taura J, Cao B, et al. (2020). Structure of human GABA. Nature 584, 304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Qi Z, Stephens NR, and Spalding EP (2006). Calcium entry mediated by GLR3.3, an Arabidopsis glutamate receptor with a broad agonist profile. Plant Physiol 142, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Russo CJ, and Passmore LA (2014). Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, et al. (2009). Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science 323, 1313–1319. [DOI] [PubMed] [Google Scholar]
- Shaye H, Ishchenko A, Lam JH, Han GW, Xue L, Rondard P, Pin JP, Katritch V, Gati C, and Cherezov V (2020). Structural basis of the activation of a metabotropic GABA receptor. Nature 584, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, and Gouaux E (2009). X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens NR, Qi Z, and Spalding EP (2008). Glutamate receptor subtypes evidenced by differences in desensitization and dependence on the GLR3.3 and GLR3.4 genes. Plant Physiol 146, 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Tapken D, Anschütz U, Liu LH, Huelsken T, Seebohm G, Becker D, and Hollmann M (2013). A plant homolog of animal glutamate receptors is an ion channel gated by multiple hydrophobic amino acids. Sci Signal 6, ra47. [DOI] [PubMed] [Google Scholar]
- Teardo E, Carraretto L, De Bortoli S, Costa A, Behera S, Wagner R, Lo Schiavo F, Formentin E, and Szabo I (2015). Alternative splicing-mediated targeting of the Arabidopsis GLUTAMATE RECEPTOR3.5 to mitochondria affects organelle morphology. Plant Physiol 167, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakhanov S, Vyas NK, Luecke H, Kristensen DM, Ma J, and Quiocho FA (2005). Ligand-free and -bound structures of the binding protein (LivJ) of the Escherichia coli ABC leucine/isoleucine/valine transport system: Trajectory and dynamics of the interdomain rotation and ligand specificity. Biochemistry 44, 6597–6608. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J, and Sobolevsky AI (2017). Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature 549, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincill ED, Bieck AM, and Spalding EP (2012). Ca(2+) conduction by an amino acid-gated ion channel related to glutamate receptors. Plant Physiol 159, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincill ED, Clarin AE, Molenda JN, and Spalding EP (2013). Interacting glutamate receptor-like proteins in Phloem regulate lateral root initiation in Arabidopsis. Plant Cell 25, 1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wudick MM, Michard E, Oliveira Nunes C, and Feijo JA (2018a). Comparing Plant and Animal Glutamate Receptors: Common Traits but Different Fates? J Exp Bot 69, 4151–4163. [DOI] [PubMed] [Google Scholar]
- Wudick MM, Portes MT, Michard E, Rosas-Santiago P, Lizzio MA, Nunes CO, Campos C, Santa Cruz Damineli D, Carvalho JC, Lima PT, et al. (2018b). CORNICHON sorting and regulation of GLR channels underlie pollen tube Ca. Science 360, 533–536. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Uys JD, Tew KD, and Townsend DM (2011). S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signal 15, 233–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav R, Rimerman R, Scofield MA, and Dravid SM (2011). Mutations in the transmembrane domain M3 generate spontaneously open orphan glutamate δ1 receptor. Brain Res 1382, 1–8. [DOI] [PubMed] [Google Scholar]
- Yelshanskaya MV, Singh AK, Sampson JM, Narangoda C, Kurnikova M, and Sobolevsky AI (2016). Structural bases of noncompetitive inhibition of AMPA subtype ionotropic glutamate receptors by antiepileptic drugs. Neuron 91, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, and Minor W (2017a). CheckMyMetal: a macromolecular metal-binding validation tool. Acta Crystallogr D Struct Biol 73, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017b). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1, Related to Figure 1
The movie shows rotating cryo-EM density for the entire GLR3.4 tetramer or a single subunit, with or without the molecular model, before or after ATD and micelle subtraction.
Data Availability Statement
Cryo-EM map for full-length GLR3.4 and after ATD and micelle subtraction have been deposited to the EMDB with the accession codes EMD-23606 and EMD-23607, respectively. Coordinates for full-length GLR3.4 and after ATD and micelle subtraction have been deposited to the PDB with the accession codes 7LZH and 7LZI, respectively. Coordinates and structure factors for GLR3.4-S1S2Glu, GLR3.4-S1S2Ser and GLR3.4-S1S2Met structures have been deposited to the PDB with the accession codes 7LZ0, 7LZ1 and 7LZ2, respectively. Raw files and full output tables for mass spectrometry results are available through PRIDE repository with identifier PXD024563 (reviewer access username: reviewer_pxd024563@ebi.ac.uk, password: UDehuNMe). This study did not generate new code.