

Abstract
Amphipathic peptides with amino acids arranged in alternating patterns of hydrophobic and hydrophilic residues efficiently self‐assemble into β‐sheet bilayer nanoribbons. Hydrophobic side chain functionality is effectively buried in the interior of the putative bilayer of these nanoribbons. This study investigates consequences on self‐assembly of increasing the surface area of aromatic side chain groups that reside in the hydrophobic core of nanoribbons derived from Ac‐(XKXE)2‐NH2 peptides (X = hydrophobic residue). A series of Ac‐(XKXE)2‐NH2 peptides incorporating aromatic amino acids of increasing molecular volume and steric profile (X = phenylalanine [Phe], homophenylalanine [Hph], tryptophan [Trp], 1‐naphthylalanine [1‐Nal], 2‐naphthylalanine [2‐Nal], or biphenylalanine [Bip]) were assessed to determine substitution effects on self‐assembly propensity and on morphology of the resulting nanoribbon structures. Additional studies were conducted to determine the effects of incorporating amino acids of differing steric profile in the hydrophobic core (Ac‐X1KFEFKFE‐NH2 and Ac‐(X1,5KFE)‐NH2 peptides, X = Trp or Bip). Spectroscopic analysis by circular dichroism (CD) and Fourier transform infrared (FT‐IR) spectroscopy indicated β‐sheet formation for all variants. Self‐assembly rate increased with peptide hydrophobicity; increased molecular volume of the hydrophobic side chain groups did not appear to induce kinetic penalties on self‐assembly rates. Transmission electron microscopy (TEM) imaging indicated variation in fibril morphology as a function of amino acid in the X positions. This study confirms that hydrophobicity of amphipathic Ac‐(XKXE)2‐NH2 peptides correlates to self‐assembly propensity and that the hydrophobic core of the resulting nanoribbon bilayers has a significant capacity to accommodate sterically demanding functional groups. These findings provide insight that may be used to guide the exploitation of self‐assembled amphipathic peptides as functional biomaterials.
Keywords: amphipathic, nanoribbons, non‐natural amino acids, self‐assembling peptides, β‐sheet fibrils
Self‐assembling peptide β‐sheet nanoribbons were synthesized with varying side aromatic chain surface areas using natural and non‐natural residues. It was found that the nanoribbon bilayers can accommodate packing of very large side chain groups. The self‐assembly kinetics was accelerated as the hydrophobicity of the amino acids increased.

Abbreviations
- CD
circular dichroism
- FT‐IR
Fourier transform infrared
- HFIP
hexafluoroisopropanol
- HPLC
high‐performance liquid chromatography
- MALDI‐TOF
matrix‐assisted laser desorption ionization‐time of flight
- TEM
transmission electron microscopy
1. INTRODUCTION
Amphipathic peptides composed of alternating hydrophobic and hydrophilic amino acids have a strong propensity to self‐assemble into amyloid‐like β‐sheet nanoribbons.1 This sequence pattern results in peptide side chains to be effectively segregated on opposite faces of the β‐strand (Figure 1). In an aqueous environment, the β‐sheet nanoribbons are formed by face‐to‐face lamination of two β‐sheets. The lamination effectively buries the hydrophobic functionality of the side chains in the interior of the β‐sheet bilayer, leaving the hydrophilic side chain groups exposed to the aqueous environment, thus resulting in highly water‐soluble nanoribbons that can grow to micrometers in length. The nanoribbon architecture and properties allow for inventive applications that include tissue engineering2, 3, 4, 5, 6 and therapeutic drug delivery.7, 8, 9, 10, 11, 12, 13, 14 Although many applications of self‐assembled amphipathic peptide nanoribbons were reported, fundamental questions regarding structure and function of these biomaterials persist, limiting the ability to rationally design next‐generation materials with precisely tuned emergent properties.
FIGURE 1.
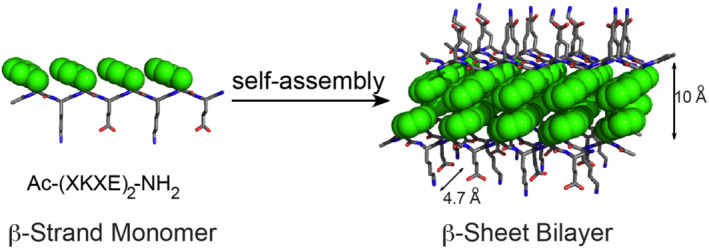
Schematic illustrating self‐assembly of amphipathic Ac‐(XKXE)2‐NH2 peptides into β‐sheet bilayer fibrils. The hydrophobic side chains (X, green) are buried in the interior of the β‐sheet bilayer with the hydrophilic side chain (Lys and Glu) exposed to the aqueous environment. When X = Phe, the distance within the bilayer is 10 Å whereas 4.7 Å between β‐strands. The β‐sheet bilayer axis is perpendicular to the page
The hydrophobic functionality of β‐sheet peptides plays a critical role in driving self‐assembly into β‐sheet bilayer fibrils and also influences the emergent properties of the resulting materials.15, 16, 17 In our previous reports, we characterized the function of hydrophobic and aromatic character in the X position in varying amphipathic sequence patterns for Ac‐(XKAE)2‐NH2, Ac‐(AKXE)2‐NH2, Ac‐(XKXE)2‐NH2, and Ac‐(XK)4‐NH2 peptides.16, 18, 19 We conducted substitution analyses using both aliphatic and aromatic amino acids along with non‐natural residues (Ala, Val, Leu, Ile, phenylalanine [Phe], cyclohexylalanine [Cha], and pentafluorophenylalanine [F5‐Phe]). We reported that self‐assembly propensity increased as the hydrophobicity of X increased. However, the aromatic character at X did not appear to strongly influence self‐assembly propensity. Interestingly, the peptides with aromatic residues did have unique fibril morphologies compared with nonaromatic peptides.
Herein, we seek a more thorough understanding of the relationship between the characteristics of aromaticity of the X residue in Ac‐(XKXE)2‐NH2 and mono‐ and di‐substituted derivatives Ac‐X1KFEFKFE‐NH2 and Ac‐(X1,5KFE)2‐NH2, respectively. We conducted an analysis of peptides that vary in exposed surface area of aromatic X side chain groups using natural and non‐natural amino acids: specifically, phenylalanine (Phe), homophenylalanine (Hph), tryptophan (Trp), 1‐naphthylalanine (1‐Nal), 2‐naphthylalanine (2‐Nal), and biphenylalanine (Bip). It was found that each of these peptides successfully self‐assembled, although some variation in nanoribbon morphology was observed. It was also found that the rate of self‐assembly for these peptides increased as the hydrophobicity of the X residue increased.
Collectively, these studies indicated that amphipathic peptide self‐assembly is tolerant to aromatic changes within the hydrophobic core of the putative β‐sheet bilayer nanoribbons. Insight into the steric packing of the hydrophobic core of amphipathic peptide nanoribbons will facilitate elucidation of design principles for application of these materials in applications, including drug delivery, that require packaging of cargo in the interior of the bilayer. For example, peptide nanoribbons were recently used to harbor hydrophobic drug‐like molecules within the bilayer core for cellular delivery.11, 20, 21 Thus, this work is of significant interest for new biomedical applications.
2. RESULTS AND DISCUSSION
2.1. Experimental rationale and peptide design
The aims were to determine the influences of aromatic side chain functionality with increasing molecular volume and steric profile within the hydrophobic core of amphipathic β‐sheet bilayer nanoribbons. Amphipathic peptides, (XZXZ)n, with nonpolar X residues and polar Z residues have a high propensity to self‐assemble into β‐sheet bilayer nanoribbons.1 In order to gain further insight into the relationship of the X residue and the properties of the resulting self‐assembled materials, we conducted an analysis of peptides that varied in exposed surface area of the X side chain. Specifically, we synthesized Ac‐(XKXE)2‐NH2 peptides in which all X residues were globally and systematically replaced with aromatic amino acids with increasing hydrophobicity and exposed molecular volume. In addition, we report the self‐assembly behavior with replacement of a Phe in only one or two residues, Ac‐X1KFEFKFE‐NH2 and Ac‐(X1,5KFE)2‐NH2, respectively, with either Trp or Bip. Self‐assembly of these peptides was characterized in order to assess the relationship between molecular volume/hydrophobicity of the core amino acids and self‐assembly propensity as well as to examine the tolerance for accommodation of increased steric bulk in the hydrophobic core of the nanoribbon bilayers.
Previous works showed that the aromatic/nonaromatic character of X substituents in Ac‐(XKXE)2‐NH2 and related peptides exerted a dramatic effect on the assembly properties.16, 22 Therefore, in this study, we opted to use only aromatic substituents in order to remove aromaticity as a variable: specifically, two natural, Phe and Trp, and four non‐natural, Hph, 1‐Nal, 2‐Nal, and Bip, residues. The hydrophobicity and molecular volume of the residues are listed in increasing order in Figure 2.
FIGURE 2.

Structures of amino acids incorporated in the X position of Ac‐(XKXE)2‐NH2 variant peptides: phenylalanine (Phe), homophenylalanine (Hph), tryptophan (Trp), 1‐naphthylalanine (1‐Nal), 2‐naphthylalanine (2‐Nal), and biphenylalanine (Bip). Relative hydrophobicity is indicated in the form of partition coefficients (π) between octanol and water relative to glycine (defined as having a partition coefficient of 0 on this scale).23 Higher π values correspond to higher hydrophobicity. The calculated van der Waals volume for each amino acid is also shown in order to indicate the degree of change in steric profile for each amino acid
Of the amino acids utilized herein, Phe is the least hydrophobic (partition coefficient [π] of 1.79) and the smallest exposed volume of the substituents utilized herein (168.03 Å3).23 The additional methylene carbon in Hph results in a moderate increase in both hydrophobicity and volume relative to Phe (π = 2.10 and 186.89 Å3, respectively). The indole side chain of Trp provides further increases in both hydrophobicity and exposed volume (π = 2.25 and 202.39 Å3, respectively). The naphthyl side chains of 1‐Nal and 2‐Nal have similar hydrophobicity (π = 3.08 and 3.15, respectively) and molecular volume (219.60 and 218.82 Å3, respectively) but differ in the spatial orientation of the side chain. Lastly, the Bip amino acid does not have a reported hydrophobicity coefficient. However, we assumed that the 4,4′‐bicyclic ring substitution of the Bip amino acid results in the greatest hydrophobicity (π ≥ 3.15) and exposed volume of the substituents considered herein (250.61 Å3). This collection of amino acid substituents represents a 1.5‐fold increase in molecular volume (from smallest to largest) and a dramatic range of hydrophobic character that allows assessment of these variables on the self‐assembly of this class of amphipathic peptides.
In addition to peptides in which the hydrophobic amino acids are globally replaced, a series of peptides with targeted substitutions were also explored. Structural models based on molecular dynamics simulations indicate that Ac‐(FKFE)2‐NH2 peptides adopt an out‐of‐register antiparallel β‐sheet packing structure (Figure 3).24 Thus, the side chain of the N‐terminal residue of nanoribbons (position 1, X1) is exposed to the aqueous environment and also has unsatisfied hydrogen bond donor/acceptor groups. The remaining hydrophobic residue side chains are paired and buried within the bilayer core of the resulting nanoribbons. We hypothesized that substitution of Phe residues in the Ac‐(FKFE)2‐NH2 peptide with either Trp or Bip would have varying effects on self‐assembly that is dependent on the position of substitution. We chose Trp and Bip as their fully substituted sequences (Ac‐(XKXE)2‐NH2) were the least and most hydrophobic relative to Ac‐(FKFE)2‐NH2 (Table 1). It was expected that substituting Phe 1 with either Trp or Bip (giving Ac‐X1KFEFKFE‐NH2 peptides) would be well tolerated because this residue (X1 in Figure 3) is unpaired in the putative nanoribbon materials. Conversely, it was expected that replacement of both Phe 1 and Phe 5 (giving Ac‐(X1,5KFE)‐NH2 peptides in which X = Trp or Bip) would potentially alter the self‐assembly properties more dramatically because the X5 side chains are proposed to be paired cross‐strand in the nanoribbon bilayer sheets; substitutions at X5 should thus be more sensitive to steric effects when the volume of this side chain is increased.
FIGURE 3.

Proposed packing mode for antiparallel out‐of‐register amphipathic Ac‐X1KFEFKFE‐NH2 and Ac‐ (X1,5KFE)2‐NH2 peptides with tryptophan (Trp) and biphenylalanine (Bip) substitutions at position 1 (X1) or at positions 1 and 5 (X5). As shown schematically in this diagram, the position 1 substituents are unpaired cross‐strand whereas the position 5 substituents are self‐paired
TABLE 1.
Amphipathic peptide sequences and HPLC retention times
| Peptide | Sequence | HPLC retention time (min) |
|---|---|---|
| 1 | Ac‐(FKFE)2‐NH2 | 12.50 |
| 2 | Ac‐(HphKHphE)2‐NH2 | 13.58 |
| 3 | Ac‐(WKWE)2‐NH2 | 12.83 |
| 4 | Ac‐(1‐NalK1‐NalE)2‐NH2 | 14.32 |
| 5 | Ac‐(2‐NalK2‐NalE)2‐NH2 | 14.32 |
| 6 | Ac‐(BipKBipE)2‐NH2 | 15.00 |
| 7 | Ac‐WKFEFKFE‐NH2 | 12.68 |
| 8 | Ac‐(WKFE)2‐NH2 | 12.65 |
| 9 | Ac‐BipKFEFKFE‐NH2 | 13.19 |
| 10 | Ac‐(BipKFE)2‐NH2 | 13.68 |
Abbreviation: HPLC, high‐performance liquid chromatography.
Peptides were synthesized using standard Fmoc solid‐phase peptide synthesis methods as N‐terminal acetyl and C‐terminal amide sequences as previously described.19, 24, 25 Relative peptide hydrophobicity was estimated by high‐performance liquid chromatography (HPLC) analysis. A comparison of HPLC retention times under identical stationary and mobile phase conditions gives reasonable estimates of relative hydrophobicity: longer retention times correspond to higher hydrophobicity with reverse phase stationary phase materials (Table 1 and Figures S1–S10). As expected, the Phe‐containing peptide had the earliest retention time of 12.50 min. However, Ac‐(WKWE)2‐NH2 had an earlier retention time (12.83 min) than the Ac‐(HphKHphE)2‐NH2 peptide (13.58 min), which would not be predicted based on the literature hydrophobicity coefficients for the constituent amino acids of these peptides.23 This earlier retention time for Trp is possibly due to the hydrogen bonding capabilities of the indole side chain that may offset the increased side chain hydrophobicity. HPLC analysis indicated that the Ac‐(1‐NalK1‐NalE)2‐NH2 and Ac‐(2‐NalK2‐NalE)2‐NH2 peptides had identical retention times (14.32 min). As expected, despite the lack of known hydrophobicity coefficient, the Ac‐(BipKBipE)2‐NH2 peptide was the most hydrophobic of the series (retention time of 15.00 min). Thus, the order of hydrophobicity of the Ac‐(XKXE)2‐NH2 peptides studied herein increases in the order: X = Phe < Trp < Hph < 1‐Nal ≈ 2‐Nal < Bip.
The hydrophobicity of Ac‐X1KFEFKFE‐NH2 and Ac‐(X1,5KFE)‐NH2, (X = Trp or Bip), was also assessed by HPLC analysis (peptides 7–10, Table 1). It was found that the Ac‐WKFEFKFE‐NH2 and Ac‐(WKFE)2‐NH2 peptides had similar retention times of 12.68 and 12.65 min, respectively. In comparison, the Ac‐(FKFE)2‐NH2 and Ac‐(WKWE)2‐NH2 peptides had slightly different retention times of 12.50 and 12.83 min, respectively. The Ac‐BipKFEFKFE‐NH2 (13.19 min) and Ac‐(BipKFE)2‐NH2 (13.68 min) peptides were significantly more hydrophobic than the parent Ac‐(FKFE)2‐NH2 peptide, but less hydrophobic than the Ac‐(BipKBipE)2‐NH2 peptide (15.00 min). Thus, this series of mono‐ and di‐substituted variants had a narrower range of hydrophobicity than the global variants (peptides 1–6, Table 1).
2.2. Characterization of Ac‐(XKXE)2‐NH2 variant peptide self‐assembly
Previous studies on the self‐assembly of Ac‐(XKXE)‐NH2 peptides in water indicate that self‐assembly of these peptides occurs very rapidly.16, 19, 24, 25 The increased hydrophobicity of some of the peptide sequences discussed herein, particularly 1‐Nal and 2‐Nal and Bip‐containing peptides, resulted in reduced solubility in water. It was found that the addition of a small amount of hexafluoroisopropanol (HFIP) (5% by volume) to self‐assembly solutions effectively solubilized all peptides.16, 26 Self‐assembly of each peptide was initiated by dissolution of the lyophilized peptide in HFIP followed by dilution in water to 5% HFIP in water at 0.2‐mM peptide concentrations. Self‐assembly was allowed to proceed at room temperature. As described below, the inclusion of HFIP as a co‐solvent (even at 5% v/v) had a decelerating effect on the self‐assembly of these amphipathic peptides.16 Thus, self‐assembly was monitored over a period of 24 h to ensure complete assembly of each peptide studied herein.
Peptide self‐assembly was initially determined as a function of β‐sheet peptide secondary structure using circular dichroism (CD) and Fourier transform infrared (FT‐IR) spectroscopic techniques.19, 24, 25 CD spectra were recorded immediately after sample preparation at 25°C to monitor formation of β‐sheet secondary structure over a span of 24 h at time points of 0, 1, 2, 4, 12, and 24 h (Figures 4 and 7). FT‐IR spectra were recorded at room temperature after the peptides were allowed to assemble for 24 h (Figures 5 and 7).
FIGURE 4.

Circular dichroism spectra in 5% hexafluoroisopropanol/water at time periods of 0, 1, 2, 4, 12, and 24 h for amphipathic Ac‐(XKXE)2‐NH2 peptides in which X is (A) phenylalanine; (B) homophenylalanine; (C) tryptophan; (D) 1‐naphthylalanine; (E) 2‐naphthylalanine; and (F) biphenylalanine
FIGURE 7.

(A) Circular dichroism (CD) spectra of Ac‐WKFEFKFE‐NH2 in 5% hexafluoroisopropanol (HFIP)/water over 24 h; (B) CD spectra for Ac‐(WKFE)2‐NH2 in 5% HFIP/water over 24 h; and (C) Fourier transform infrared spectra for Ac‐BipKFEFKFE‐NH2 and Ac‐(BipKFE)2‐NH2 (24 h, 5% d 2‐HFIP/D2O)
FIGURE 5.

Fourier transform infrared spectra for Ac‐(XKXE)2‐NH2 peptides in which X = phenylalanine, homophenylalanine, tryptophan, 1‐naphthylalanine, 2‐naphthylalanine, or biphenylalanine. Each peptide exhibits a characteristic amide I signal at ~1618 cm−1 indicating β‐sheet secondary structure for all variants
A CD spectrum that is characteristic of β‐sheet nanoribbons was observed for Ac‐(FKFE)2‐NH2 with a strong maximum at 195 nm and minimum at 205 nm (Figure 4A).24, 25, 27 However, the CD spectra for all other Ac‐(XKXE)2‐NH2 derivatives were found to deviate dramatically from canonical β‐sheet nanoribbon spectra. These changes in CD spectra are likely due to handedness, aromaticity, interchromophore distance, and relative orientation of peptide side chains as observed with our previous studies using Nap‐1 and Nap‐2 Ac‐(XKAE)2‐NH2 derivatives.19, 28 Finally, the HFIP co‐solvent slows β‐sheet secondary conformation as evidenced by an intensification of the CD signal over a 24‐h time period for peptides with HPLC retention times from 12.50 to 13.68 except Hph (Figures 4 and 7 and Table 1).
The minute change of an additional β‐alkyl carbon in the Hph residue resulted in a significantly different CD spectrum for the Ac‐(HphKHphE)2‐NH2 variant (Figure 4B). A minimum at 220 nm and a maximum at 195 nm correspond to classical β‐sheet structure. The lack of signal at ~205 nm for the Ac‐(HphKHphE)2‐NH2 peptide implies that π‐stacking or β‐sheet distortion29 is dramatically altered relative to the Ac‐(FKFE)2‐NH2 peptide. The additional degrees of freedom afforded by the β‐alkyl carbon in the Ac‐(HphKHphE)2‐NH2 peptide thus had fairly dramatic effects on β‐sheet packing during self‐assembly. This is also evident because the β‐sheet signal is essentially unchanged over 24 h even in the presence of the HFIP co‐solvent, indicating that the kinetics of self‐assembly is dramatically accelerated for the Ac‐(HphKHphE)2‐NH2 peptide. CD analysis, thus, suggests that increasing the length of the Phe side chain by addition of one alkyl carbon potentially stabilizes β‐sheet formation for the amphipathic Ac‐(HphKHphE)2‐NH2 peptide relative to the parent Ac‐(FKFE)2‐NH2.28
The CD spectrum for Ac‐(WKWE)2‐NH2 diverges dramatically from canonical β‐sheet patterns, although it is similar to data previously reported for a (WKWE)2W‐derived linear peptide that was described as β‐sheet.30 Initially, the CD spectrum had a broad minimum at 210 nm that possibly reflects a mixture of α‐helix and β‐sheet structures due to the inhibitory effect of the HFIP co‐solvent on self‐assembly (Figure 4C).31 Over time, the signal evolved to feature a minimum at 208 nm (possibly an amide n → π* transition) and a maximum at 220 nm (possibly π → π* transition of interacting indole functionality).32 These CD patterns are similar to those previously observed in Trp‐containing β‐hairpin peptides and are indicative of a strong negative–positive exciton‐coupled band between Trp–Trp chromophores within the β‐hairpin.33, 34, 35, 36, 37 Thus, the CD spectrum of the Ac‐(WKWE)2‐NH2 peptide is consistent with an assembled state in which Trp residues interact. Because electronic effects from the Trp residue dominate these spectra, β‐sheet structure requires validation using complementary FT‐IR analysis (discussed below). However, it is interesting that the Ac‐(WKWE)2‐NH2 peptide appears to exhibit overall peptide hydrophobicity and assembly behavior that is kinetically more similar to that of Ac‐(FKFE)2‐NH2 (occurring over 24 h) than to that of Ac‐(HphKHphE)2‐NH2, despite the larger molecular volume and hydrophobicity of Trp relative to Hph side chains. The effect of a change in position of the Hph side chain by a single carbon unit appears to be even more dramatic in this light.
The 1‐Nal‐ and 2‐Nal‐containing peptides also exhibited CD spectra that deviate from classical β‐sheet signatures. The CD spectrum for Ac‐(1‐NalK1‐NalE)2‐NH2 features a single minimum at 230 nm; this minimum is likely indicative of aromatic stacking between 1‐Nal side chains (Figure 4D).38, 39, 40 The CD spectrum for Ac‐(2‐NalK2‐NalE)2‐NH2 displayed a minimum at ~210 nm and a maximum at 230 nm (Figure 4E); as with the 1‐Nal‐containing peptide, this CD spectrum is consistent with aromatic interactions between 2‐naphthyl side chains in 2‐Nal.41, 42 The HFIP co‐solvent did not appear to impair the kinetics of self‐assembly for these peptides, because no change in signal intensity was observed over 24 h. CD spectra of the 1‐Nal and 2‐Nal peptides are dominated by electronic effects from interaction of the naphthyl side chains, making unambiguous assessment of β‐sheet structure difficult without confirmation by other spectroscopic methods (see FT‐IR analysis below).
Lastly, the Ac‐(BipKBipE)2‐NH2 peptide also displayed a unique saddle‐like CD signature (Figure 4F). The Ac‐(BipKBipE)2‐NH2 CD spectrum had minima at 202 and 222 nm, consistent with the characteristic Cotton effect for biphenyl‐containing compounds.43, 44 Electronic effects from the biphenyl side chain dominate the spectrum and suggest self‐assembly via interaction of aromatic groups, although assignment of β‐sheet structure for these assemblies requires additional spectroscopic analysis (as described below). The HFIP co‐solvent had no effect on evolution of signal intensity over time, suggesting that this peptide (as well as the Hph, 1‐Nal, and 2‐Nal peptides) is sufficiently hydrophobic (or possess other unique packing effects) to enable rapid assembly even in the presence of organic co‐solvents with disaggregant properties.
UV spectra were also collected for the molecular assemblies of each peptide derivative (Figure S29). The nanoribbons had absorption signals between 250 and 300 nm consistent with presence of the aromatic amino acids, with Ac‐(HphKHphE)2‐NH2 and Ac‐WKFEFKFE‐NH2 exhibiting the weakest absorbance in this range.45, 46 The mono‐substituted, di‐substituted, and fully substituted Bip derivatives had an overlaying peak at 250 nm. We also observed a long tail over 600 nm that is consistent with elongated π‐stacking or scattering effects from large molecular assemblies.47
FT‐IR analysis of peptides in the amide I region is not sensitive to electronic effects in the same way that CD spectroscopy can be. Therefore, FT‐IR analysis was used to confirm β‐sheet secondary structure for putative assemblies of each variant peptide (Figure 5). β‐Sheet peptides have a characteristic amide I stretch between 1615 and 1635 cm−1.19, 24, 25 As previously reported, the Ac‐(FKFE)2‐NH2 peptide has an amide I stretch at 1618 cm−1, consistent with β‐sheet self‐assembly (Figure 5). Each of the other peptides assessed herein also had FT‐IR spectra consistent with β‐sheet self‐assembly, with amide I stretches from 1616 to 1620 cm−1. Thus, FT‐IR analysis confirms that each Ac‐(XKXE)2‐NH2 variant, regardless of side chain volume or hydrophobicity, effectively forms β‐sheet structures.
2.3. TEM analysis of nanoribbon morphology for Ac‐(XKXE)2‐NH2 peptides
CD and FT‐IR are useful tools for characterizing secondary structure that cannot, however, independently confirm the presence of self‐assembled β‐sheet structures. Imaging methods such as transmission electron microscopy (TEM) can be used to confirm assembly of peptides into higher order nanoribbon/fibril materials and to characterize the morphology of these materials.19, 48 Accordingly, TEM analysis was used to characterize the self‐assembled materials derived from Ac‐(XKXE)2‐NH2 peptides.
As previously reported, the Ac‐(FKFE)2‐NH2 peptide assembled into nanoribbon fibrils.16, 19, 24, 25 Left‐handed helical nanoribbons (pitch of 19.1 ± 1.3 nm) were observed to coexist with flat nanoribbons (Figure 6A); both morphologies had an average ribbon width of 8.8 ± 0.8 nm (Table 2). The helical nanoribbons are present in solution at early time points (minutes to hours) with the flat nanoribbons becoming dominant after extended incubation periods. The evolution of helical fibrils into flat nanoribbons was elucidated with short amphiphilic peptides49; therefore, it can be presumed that the helical structures represent kinetic products whereas the flat nanoribbons are thermodynamic products.50, 51
FIGURE 6.

Transmission electron microscopy images of Ac‐(XKXE)2‐NH2 peptide nanoribbons. (A) X = phenylalanine; (B) X = homophenylalanine; (C) X = tryptophan; (D) X = 1‐naphthylalanine; (E) X = 2‐naphthylalanine; and (F) X = biphenylalanine
TABLE 2.
Nanoribbon morphological differences among variant aromatic amino acids (X)
| Ac‐(XKXE)2‐NH2 | ||||||
|---|---|---|---|---|---|---|
| Phe | Hph | Trp | Bip | 1‐Nal | 2‐Nal | |
| Width (nm) | 8.8 ± 0.8 | 7.6 ± 1.3 | 7.3 ± 1.6 | 5.5 ± 0.9 | 8.2 ± 1.2 | 8.9 ± 1.1 |
| Helical pitch (nm) | 19.1 ± 1.3 | N/A | N/A | N/A | N/A | N/A |
| Ac‐X1KFEFKFE‐NH2 | Ac‐(X1,5KFE)‐NH2 | ||||
|---|---|---|---|---|---|
| Trp | Bip | Trp | Bip | ||
| Width (nm) | 5.3 ± 1.5 | 2.9 ± 0.5 | 7.3 ± 1.5 | 3.2 ± 0.3 to 10.0 ± 1.1 | |
Abbreviations: 1‐Nal, 1‐naphthylalanine; 2‐Nal, 2‐naphthylalanine; Bip, biphenylalanine; Hph, homophenylalanine; N/A, not applicable; Phe, phenylalanine; Trp, tryptophan.
Assemblies of variant Ac‐(XKXE)2‐NH2 peptides display subtle morphological differences relative to assemblies of the parent Ac‐(FKFE)2‐NH2 peptide. Ac‐(HphKHphE)2‐NH2 assemblies (Figure 6B) appeared as 7.6 ± 1.3‐nm width nanoribbon fibrils that lacked the regular helical structure observed at early time points with the Ac‐(FKFE)2‐NH2 peptide. Instead, Ac‐(HphKHphE)2‐NH2 nanoribbons were either flat or gently twisting, without any regularity in the pitch or frequency of the twist. Ac‐(WKWE)2‐NH2 fibrils were predominately flat nanoribbons with a width of 7.3 ± 1.6 nm (Figure 6C). The TEM images for the 1‐Nal variant displayed densely packed fibrils that were primarily flat loosely coiled nanoribbons with widths of 8.2 ± 1.2 nm (Figure 6D). The 2‐Nal peptides assembled into nanoribbons with similar morphology to the 1‐Nal peptides, with widths of 8.9 ± 1.1 nm (Figure 6E). Lastly, Ac‐(BipKBipE)2‐NH2 assembled into densely packed helical nanoribbons that were significantly thinner (5.5 ± 0.9 nm) than the other variants (Figure 6F).
The reason for this subtle variation in fibril morphology as a function of the identity of the hydrophobic residues in Ac‐(XKXE)2‐NH2 peptides is not clearly understood. The fact that morphology does vary, even subtly, implies that the volume and orientation of the side chain groups in the hydrophobic bilayer of these assembled materials do exert an influence on peptide packing within the β‐sheet that is manifested in the overall structure of the assembled material. However, both the 1‐Nal and 2‐Nal Ac‐(XKXE)2‐NH2 peptides had near identical morphologies to those previously observed with Ac‐(XKAE)2‐NH2. These findings indicate that the internal Phe residues may enhance fibril formation as indicated in the CD spectra but not affect fibril morphology (Figure 3). Presumably, out‐of‐register side chain packing of the peptides can directly influence nanoribbon morphology as a function of hydrophobicity in the aqueous environment.
2.4. Self‐assembly of mono‐ and di‐substituted Ac‐X1KFEFKFE‐NH2 and Ac‐(X1,5KFE)‐NH2 variants
The effect of steric factors on the self‐assembly of Ac‐(FKFE)2‐NH2‐derived peptides was also assessed by more subtle mono‐ and di‐substituted Phe 1 and Phe 1/Phe 5 variants. Ac‐X1KFEFKFE‐NH2 and Ac‐(X1,5KFE)‐NH2 peptides in which X is either Trp or Bip were prepared for these studies (Table 1, peptides 7–10). As previously stated, Trp and Bip were chosen due to their relative hydrophobicity to Ac‐(FKFE)2‐NH2. In addition, the positions for these substitutions were chosen due to the differential effects these positions have on core side chain packing based on the putative structure of the β‐sheet materials.19, 50, 51 The Phe 1 side chain is presumably unpaired whereas the Phe 5 side chain self‐pairs cross‐strand within the β‐sheet (Figure 3). Thus, substitutions at position 5 of Ac‐(XKXE)2‐NH2 peptides should exert a more dramatic effect on self‐assembly than substitutions at position 1. In order to maintain consistency with the assessment of peptides 1–6, self‐assembly of peptides 7–10 was also assessed in 5% HFIP/water (v/v). Substitutions with Trp and Bip were explored to assess the effect of both subtle and extreme changes in molecular volume and hydrophobicity.
The mono‐substituted Ac‐WKFEFKFE‐NH2 peptide effectively self‐assembled into β‐sheet materials as evidenced by CD and FT‐IR analyses (Figure 7A,C). The CD spectrum for this peptide was similar to that of Ac‐(FKFE)2‐NH2 β‐sheet nanoribbons, with minima at 205 and 220 nm and a maximum at 195 nm. As with the Ac‐(FKFE)2‐NH2 peptide, the HFIP co‐solvent decelerated the kinetics of self‐assembly, which required ~24 h to reach equilibrium. FT‐IR analysis confirms that Ac‐WKFEFKFE‐NH2 forms β‐sheet structured materials (Figure 7C). Thus, spectral evidence suggests that Phe 1 → Trp substitution has only minimal impact on the self‐assembly of this variant.
The di‐substituted Ac‐(WKFE)2‐NH2 variant exhibited a slightly modified CD pattern likely due to more extensive electronic effects from the cross‐strand interaction of Trp 5 residues within the hydrophobic bilayer (Figure 7B). The CD spectrum of Ac‐(WKFE)2‐NH2 featured prominent minima at 202 and 218 nm that intensified over 24 h. Comparatively, the Ac‐(WKWE)2‐NH2 peptide induced a Cotton effect with a large coupling at 208 nm that appeared after 4 h and intensified over time (Figure 4C).52 The FT‐IR spectrum for Ac‐(WKFE)2‐NH2 was consistent with β‐sheet structure, with an amide I stretch at 1620 cm−1 (Figure 7C). It was observed that the doubly substituted Ac‐(WKFE)2‐NH2 exhibited a more dramatic intensification of CD signal over time relative to the mono‐substituted Ac‐WKFEFKFE‐NH2 peptide, consistent with a more dramatic effect on self‐assembly by substitutions that fall within the core of the bilayer as opposed to the edge of the bilayer.
Bip substitution had a more dramatic effect on self‐assembly of Ac‐(FKFE)2‐NH2‐derived peptides than did Trp substitution. Phe 1 → Bip substitution (Ac‐BipKFEFKFE‐NH2) provided a peptide that, similar to the parent peptide and to Ac‐WKFEFKFE‐NH2, had a CD spectrum with minima at 202 and 218 nm that intensified dramatically over 24 h (Figure 8A). The magnitude of this change was 10‐fold greater for Ac‐BipKFEFKFE‐NH2 than was observed for either Ac‐(FKFE)2‐NH2 or Ac‐WKFEFKFE‐NH2. Thus, dramatic changes in the side chain steric profile at position 1 in these amphipathic sequences can significantly alter the kinetics of self‐assembly even though this side chain is presumably unpaired in the context of assembled nanoribbons (Figure 3).24 The FT‐IR spectrum for Ac‐BipKFEFKFE‐NH2 indicated an amide I stretch at 1618 cm−1, consistent with β‐sheet self‐assembly.
FIGURE 8.

Circular dichroism (CD) and Fourier transform infrared (FT‐IR) spectra for the mono‐ and di‐substituted biphenylalanine (Bip) variants (0.2‐mM peptide). (A) CD spectra of Ac‐BipKFEFKFE‐NH2 in 5% hexafluoroisopropanol (HFIP)/water over 24 h; (B) CD spectra for Ac‐(BipKFE)2‐NH2 in 5% HFIP/water over 24 h; and (C) FT‐IR spectra for Ac‐BipKFEFKFE‐NH2 and Ac‐(BipKFE)2‐NH2 (24 h, 5% d 2‐HFIP/D2O)
Incorporating a second internal Bip substitution at Phe 5 (Ac‐(BipKFE)2‐NH2) again altered the self‐assembly of this peptide relative to the parent peptide and the mono‐substituted Bip 1 variant. The CD spectrum for the Ac‐(BipKFE)2‐NH2 peptide initially featured only a single minimum at 218 nm; over a period of 24 h, a second prominent minimum at 205 nm (attributed to π‐stacking of the Phe side chains) was also observed. This suggests that rearrangement of the packing orientation of the side chain groups can occur within the bilayer core over time. The increased hydrophobicity of the Ac‐(BipKFE)2‐NH2 peptide accelerated the kinetics of self‐assembly, with only minor intensification of the CD spectral signals over time (Figure 8B). Unlike the previously described mono‐ and di‐substituted variants, the Ac‐(BipKFE)2‐NH2 CD signal was seemingly unaffected by the HFIP co‐solvent. The FT‐IR spectrum confirmed β‐sheet structure with an amide I stretch at 1620 cm−1 (Figure 8C).
TEM images of the mono‐ and di‐substituted peptides were obtained to confirm self‐assembly and to assess the fibril morphology of the β‐sheet structures. The fibril morphology for Ac‐WKFEFKFE‐NH2 was very similar to Ac‐(FKFE)2‐NH2 (see Figures 6A and 9A for comparison). In addition to helical nanoribbons that were ~8 nm in width, however, thinner, twisted nanoribbons that were 5.3 ± 1.5 nm wide were also observed (Figure 9A). The Ac‐(WKFE)2‐NH2 peptide had a combination of twisted helical nanoribbons and flat nanotapes with widths averaging 7.3 ± 1.5 nm (Figure 9B). It is evident by these changes in fibril morphology that location of the increased side chain surface area influences the packing mode for β‐sheet fibrils because the N‐terminal substitution had relatively minimal effects compared with the internal substitutions at position 5.
FIGURE 9.

Transmission electron microscopy images for the mono‐ and di‐substituted assembled materials. (A) Ac‐WKFEFKFE‐NH2; (B) Ac‐(WKFE2)‐NH2; (C) Ac‐BipKFEFKFE‐NH2; and (D) Ac‐(BipKFE2)‐NH2
The fibril morphologies for the Bip variants differed more dramatically from the parent peptide. Images of the Ac‐BipKFEFKFE‐NH2 peptide indicated unique bundling of the fibrils that were 2.9 ± 0.5 nm in width, roughly corresponding to the width of a single β‐strand peptide (Figure 9C). These thinner fibril types may result from blockage of edge‐to‐edge alignment of β‐sheets that must account for nanotape widening by the sterically demanding Bip residue that is at the exposed edge of the putative antiparallel β‐sheet. Interestingly, the high degree of bundling of these fibrils may also be due to exposure of the hydrophobic Bip residue; bundling may occur in order to desolvate this functionality.24 Lastly, the Ac‐(BipKFE)2‐NH2 peptide assembled into several morphologies ranging from very thin ribbons averaging 3.2 ± 0.3 nm in width to wider nanoribbons averaging 10.0 ± 1.1 nm in width (Figure 9D). Changes in β‐sheet packing due to incorporation of a single internal Bip residue apparently facilitate self‐assembly into wider nanoribbons. Wang et al. observed a similar phenomenon when aliphatic residues of varying steric bulk were seen to alter the morphology of self‐assembled materials from amyloid‐inspired peptides.29
Changes in fibril morphology as a function of mono‐ or di‐substitution of Phe with more sterically demanding residues are clearly sensitive to the position of substitution. Introducing steric bulk within the center of Ac‐(FKFE)2‐NH2 β‐strands has a more dramatic overall effect on assembled nanoribbon morphology than introduction of steric bulk at the exposed N‐terminal residues. The comparative morphologies for the self‐assembled Ac‐WKFEFKFE‐NH2 and Ac‐BipKFEFKFE‐NH2 peptides were similar. Conversely, the morphologies of self‐assembled materials derived from the Ac‐(WKFE)2‐NH2 and Ac‐(BipKFE)2‐NH2 peptides were dramatically different. This indicates that changes in positions in which cross‐strand pairing within the β‐sheet structure is affected are highly influential on emergent nanoribbon structure. Conversely, the exposed Ν‐terminal residue presumably does not participate in cross‐strand pairing between β‐strands but rather functions to mediate interactions between β‐sheets, which exerts more subtle effects on overall fibril structure. Finally, no association between CD spectra and fibril morphology was observed with the current and previous studies.16, 18, 19, 24, 25 Therefore, future studies will focus on eluding spectroscopic associations with fibril morphology among (XZXZ)n patterned peptides, with X being a hydrophobic residue and Z hydrophilic.
A final set of experiments was conducted to determine the influence of HFIP and temperature on the self‐assembly process.53 In these experiments, the Ac‐(WKFE)2‐NH2 peptide was used as a representative peptide. Solutions of Ac‐(WKFE)2‐NH2 (5% HFIP/water, 0.2‐mM peptide) were assembled 50°C and 70°C for 96 h in both open and closed containers. These higher temperatures were used to assess the effect of HFIP evaporation over the course of the assembly process (the boiling point of HFIP is 58.2°C) and also to determine whether the observed fibrils for this peptide are likely to be thermodynamic products or kinetically trapped assemblies. Assessment of the assemblies formed at both temperatures and in open or sealed containers indicated that assemblies were spectroscopically indistinguishable from those assembled at room temperature in 5% HFIP/water. In addition, TEM images (Figure S30) also show that these assemblies appear to be the same as those observed at room temperature (Figure 9B). Evaporation of HFIP and/or higher temperatures had no impact on the observed structures.
3. CONCLUSION
Amphipathic peptide self‐assembly is sensitive to changes within the hydrophobic core of the putative β‐sheet bilayer nanoribbons. Herein, we reported that Ac‐(XKXE)2‐NH2 peptides with hydrophobic amino acids of increasing surface area and hydrophobicity (X = Phe, Hph, Trp, 1‐Nal, 2‐Nal, or Bip) effectively self‐assemble in all cases, demonstrating that the hydrophobic core of the resulting bilayer nanoribbons has a great capacity to accommodate packing of even very large side chain groups. Interestingly, subtle differences in morphology were observed as a function of hydrophobic X residues with no association to spectroscopic outcomes. This suggests that aromatic/hydrophobic packing effects can also elicit changes in β‐sheet packing (presumably via strand registry or strand twisting) that give rise to the observed variation in nanoribbon appearance. In addition, these studies also confirm that self‐assembly kinetics is accelerated as the hydrophobicity of the core X amino acids increases. These studies clarify the structural tolerances of amphipathic β‐sheet peptide self‐assembly and provide insight into the capacity for appending cargo into the hydrophobic core of these self‐assembled nanoribbon architectures. As such, we aim to investigate the nanoribbon properties with appending cargo under physiological conditions as potential biomaterials and therapeutics. In addition, future studies will focus on evaluation of the molecular basis for the variation in nanoribbon structural morphology as the structure of the core hydrophobic X residues is altered and on understanding how these changes relate to the emergent properties of the self‐assembled materials.
4. EXPERIMENTAL METHODS
4.1. Peptide synthesis, purification, and characterization
Peptides were synthesized on a microwave‐equipped Liberty Peptide Synthesizer (CEM®) using standard Fmoc protection and HBTU/HOBt activation chemistry as previously described.19, 24, 25 Briefly, rink amide resin (Advanced ChemTech, 100–200 mesh, 0.2 mmol g−1) was used as the solid support to provide C‐terminal amide peptides. Peptides were treated with 20% acetic anhydride in dimethylformamide (DMF) to give N‐terminal acetyl sequences. Side chain deprotection and cleavage from the solid support were accomplished by treatment with trifluoroacetic acid (TFA), triisopropylsilane (TIPS), and water (95:2.5:2.5, v/v/v) (room temperature, 1 h). Cleavage solutions were concentrated to 10% of the reaction volume after which peptides were precipitated in ethyl ether and isolated by centrifugation; precipitated peptides were washed by resuspension in ether. The isolated solid peptide was then dissolved in DMSO for purification by HPLC.
Purification of synthetic peptides was conducted using a Shimadzu LC‐AD HPLC instrument over a reverse phase C18 stationary phase (Waters, BEH300, 10 μm, 19 × 250 mm). A binary gradient of acetonitrile and water with 0.1% TFA at 10 ml min−1 was used as the mobile phase, and eluent was monitored by UV absorbance at 215 and 254 nm. Fractions were collected and lyophilized and then analyzed by analytical HPLC (reverse phase C18, Waters, BEH300, 10 μm, 4.6 × 250 mm) to confirm purity (Figures S1–S10). Peptide identity was confirmed by matrix‐assisted laser desorption ionization‐time of flight (MALDI‐TOF) mass spectroscopy (Figures S11–S20). Ac‐(FKFE)2‐NH2 m/z 1162.28 (1162.34 calcd for [MH]+), m/z 1185.25 (1185.32 calcd for [MNa]+), m/z 1201.20 (1201.43 calcd for [MK]+). Ac‐(HphKHphE)2‐NH2 m/z 1219.58 (1219.45 calcd for [MH]+), m/z 1241.57 (1241.43 calcd for [MNa]+), m/z 1257.55 (1257.54 calcd for [MK]+). Ac‐(WKWE)2‐NH2 m/z 1319.51 (1319.49 calcd for [MH]+), m/z 1341.50 (1341.47 calcd for [MNa]+), m/z 1357.47 (1357.58 calcd for [MK]+). Ac‐(1‐NalK1‐Nal‐E)2‐NH2 m/z 1363.47 (1363.58 calcd for [MH]+), m/z 1385.46 (1385.56 calcd for [MNa]+), m/z 1401.43 (1401.67 calcd for [MK]+). Ac‐(2‐NalK2‐Nal‐E)2‐NH2 m/z 1363.54 (1363.58 calcd for [MH]+), m/z 1385.51 (1385.56 calcd for [MNa]+), m/z 1401.47 (1401.67 calcd for [MK]+). Ac‐(BipKBipE)2‐NH2 m/z 1467.17 (1467.73 calcd for [MH]+), m/z 1489.16 (1489.71 calcd for [MNa]+), m/z 1505.13 (1505.82 calcd for [MK]+). Ac‐WKFEFKFE‐NH2 m/z 1202.85 (1202.38 calcd for [MH]+), m/z 1224.85 (1224.36 calcd for [MNa]+), m/z 1240.83 (1240.47 calcd for [MK]+). Ac‐(WKFE)2‐NH2 m/z 1241.78 (1241.42 calcd for [MH]+), m/z 1263.49 (1263.40 calcd for [MNa]+), m/z 1279.47 (1279.51 calcd for [MK]+). Ac‐BipKFEFKFE‐NH2 m/z 1239.59 (1239.44 calcd for [MH]+), m/z 1261.53 (1261.42 calcd for [MNa]+), m/z 1277.51 (1277.53 calcd for [MK]+). Ac‐(BipKFE)2‐NH2 m/z 1314.87 (1315.54 calcd for [MH]+), m/z 1336.85 (1337.52 calcd for [MNa]+), m/z 1352.81 (1353.63 calcd for [MK]+). See Figures S1–S20 and Table S1 for HPLC and MALDI‐MS data.
4.2. Peptide self‐assembly
Peptide self‐assembly was initiated in a solution of water and 1,1,1,3,3,3‐HFIP (95:5, v/v) (pH 3–4 as a consequence of residual TFA from HPLC purification). Self‐assembly was analyzed at a peptide concentration of 0.2 mM quantified according to previously described protocols.16, 24, 25 Lyophilized peptides were dissolved in an acetonitrile:water:TFA mixture (60:39.9:0.1, v/v/v) to maintain an unassembled peptide state. Thus, peptides were analyzed under acidic conditions (pH 3–4) due to residual TFA. As such, all peptides had a net charge of +2 from the lysine side chains. Meanwhile, protonation of the glutamic acid side chains resulted in a neutral charge.
Peptide concentrations were determined from these solutions by analytical HPLC analysis and correlation of HPLC peak area to a standard curve constructed for each peptide (see Figures S21–S28 for standard curves). Standard concentration curves were prepared as described previously, and integrated peak areas were correlated to absolute peptide concentration by amino acid analysis (AIBiotech, Richmond, VA). Aliquots of the desired peptide quantity were frozen, lyophilized, and used immediately in self‐assembly studies. Self‐assembly was initiated by dissolving lyophilized peptides in HFIP followed by addition of water to give a peptide concentration of 0.2 mM (final solvent composition of 5% HFIP by volume); solutions were treated by vortex (1 min) and sonication (5 min) to give optically transparent, homogenous solutions. Self‐assembly was characterized using CD, FT‐IR spectroscopy, and TEM imaging as described in the following sections.
4.3. CD spectroscopy
CD spectra were recorded on an AVIV 202 CD spectrometer. Spectra were obtained using a 0.1‐mm path length quartz cuvette (Hellma) at 25°C from 260 to 190 nm with a 1.0‐nm step, 1.0‐nm bandwidth, and a 3‐s collection time per step. Spectra were collected at 0, 1, 2, 4, 12, and 24 h for each sample. Background subtraction with 5% HFIP/water, conversion to molar ellipticity, and data smoothing with a least squares fit were performed using the AVIV software.
4.4. UV–Vis spectroscopy
UV–Vis spectra were obtained using a Shimadzu UV‐2401PC/2501PC. Prior to UV analysis, peptides were dissolved in HFIP, diluted with water, and assembled as described previously. Samples were allowed to assemble for 24 h (0.2‐mM peptide). Spectra were obtained in 1‐ml cuvettes with a 10‐mm path length from 700 to 190 nm, a sampling interval of 1.0 nm, a slit width of 2.0 nm, and using single scan mode.
4.5. FT‐IR microscopy
FT‐IR spectra were obtained using a Shimadzu 8400 FT‐IR spectrometer. Prior to FT‐IR analysis, TFA counter‐ions were removed from the purified peptides through anion exchange by dissolving the peptides in 40% d 6‐acetonitile/D2O (v/v) with 1% DCl followed by lyophilization. The lyophilized peptides were dissolved in 5% d 2‐HFIP/D2O with 0.1% DCl (0.2‐mM peptide). The peptides were treated by vortex (1 min) and sonication (5 min) to give a homogenous and transparent solution. An aliquot (70 μl) was placed on 25‐mm × 4‐mm CaF2 plates (International Crystal Labs). Spectra were obtained using the Happ–Genzel method from 1550 to 1750 cm−1 with a 2‐cm−1 resolution (512 scans).
4.6. Negative‐stain TEM and electron diffraction
Fibril morphologies of the assembled peptide materials were characterized by TEM analysis. An aliquot of each assembled peptide (10 μl) was applied to 200‐mesh carbon‐coated copper grids. After standing for 30 s, excess fluid was removed by capillary action. Peptide fibrils were then stained by applying 10 μl of 5% uranyl acetate to the grid for 2 min; excess fluid was removed by capillary action. The peptides were washed with water for 1 s (10 μl), then the remaining solvent was removed by capillary action, and the grids were allowed to dry prior to imaging. Electron micrograph images were obtained using a Hitachi 7650 TEM in high‐contrast mode at an accelerating voltage of 80 kV. Fibril dimensions were measured in ImageJ (http://rsbweb.nih.gov/ij/).24, 25 The reported dimensions are the average of at least 100 measurements on distinct fibrils for each self‐assembled peptide.
Supporting information
Figure S1. Analytical HPLC Trace (215 nm) for Ac‐(FKFE)2‐NH2.
Figure S2. Analytical HPLC Trace (215 nm) for Ac‐(HphKHphE)2‐NH2.
Figure S3. Analytical HPLC Trace (215 nm) for Ac‐(WKWE)2‐NH2.
Figure S4. Analytical HPLC Trace (215 nm) for Ac‐(1‐NalK1‐NalE)2‐NH2.
Figure S5. Analytical HPLC Trace (215 nm) for Ac‐(2‐NalK2‐NalE)2‐NH2.
Figure S6. Analytical HPLC Trace (215 nm) for Ac‐(BipKBipE)2‐NH2.
Figure S7. Analytical HPLC Trace (215 nm) for Ac‐WKFEFKFE‐NH2.
Figure S8. Analytical HPLC Trace (215 nm) for Ac‐(WKFE)2‐NH2.
Figure S9. Analytical HPLC Trace (215 nm) for Ac‐BipKFEFKFE‐NH2.
Figure S10. Analytical HPLC Trace (215 nm) for Ac‐(BipKFE)2‐NH2.
Figure S11. MALDI‐TOF‐MS for Ac‐(FKFE)2‐NH2.
Figure S12. MALDI‐TOF‐MS for Ac‐(HphKHphE)2‐NH2.
Figure S13. MALDI‐TOF‐MS data for Ac‐(WKWE)2‐NH2.
Figure S14. MALDI‐TOF‐MS data for Ac‐(1‐NalK1‐NalE)2‐NH2.
Figure S15. MALDI‐TOF‐MS data for Ac‐(2‐NalK2‐NalE)2‐NH2.
Figure S16. MALDI‐TOF‐MS data for Ac‐(BipKBipE)2‐NH2.
Figure S17. MALDI‐TOF‐MS data for Ac‐WKFEFKFE‐NH2.
Figure S18. MALDI‐TOF‐MS data for Ac‐(WKFE)2‐NH2.
Figure S19. MALDI‐TOF‐MS data for Ac‐BipKFEFKFE‐NH2.
Figure S20. MALDI‐TOF‐MS data for Ac‐(BipKFE)2‐NH2.
Table S1. Calculate and Observed m/z for synthetic peptides (MALDI‐TOF‐MS).
Figure S21. HPLC calibration curve (215 nm) for Ac‐(HphKHphE)2‐NH2.
Figure S22. HPLC calibration curve (215 nm) for Ac‐(WKWE)2‐NH2.
Figure S23. HPLC calibration curve (215 nm) for Ac‐(1‐NalK1‐NalE)2‐NH2.
Figure S24. HPLC calibration curve (215 nm) for Ac‐(BipKBipE)2‐NH2.
Figure S25. HPLC calibration curve (215 nm) for Ac‐WKFEFKFE‐NH2.
Figure S26. HPLC calibration curve (215 nm) for Ac‐(WKFE)2‐NH2.
Figure S27. HPLC calibration curve (215 nm) for Ac‐BipKFEFKFE‐NH2.
Figure S28. HPLC calibration curve (215 nm) for Ac‐(BipKFE)2‐NH2.
Figure S29. Normalized absorption spectra of peptide fibrils
Figure S30. TEM images for Ac‐(WKFE)2‐NH2 at 50°C and at 70°C. Nanoribbons were prepared in 5% HFIP/water at 0.2mM peptide for 96 hours. A) Nanoribbons in an open system at 50°C. B) Closed system at 50°C. C) Open system at 70°C. D) Closed system at 70°C. The minor change in morphology in the open system at 70°C is likely a result of evaporation over the 96 hours. Evaporation and temperature show no impact on nanoribbon morphology.
ACKNOWLEDGEMENTS
We gratefully acknowledge Karen Bentley and Dr. Scott Kennedy for assistance with TEM and CD experiments, respectively.
This work was supported by the National Science Foundation (DMR‐1148836 and CHE‐1904528), the National Institutes of Health National Heart, Lung, and Blood Institute (R01 HL138538), and the National Institute of General Medical Sciences (NIGMS) (T32 GM118283). Mass spectroscopy instrumentation was partially supported by grants from the U.S. National Science Foundation (CHE‐0840410 and CHE‐0946653). Manuscript preparation and submission were supported by the NIH through the partnership for Native American Cancer Prevention (NACP), National Cancer Institute (NCI) grant 2U54CA143925‐11, and the Southwest Health Equity Research Collaborative, National Institute on Minority Health and Health Disparities (NIMHD) grant 1U54MD012388‐01, and NIGMS R25GM127199.
Jones CW, Morales CG, Eltiste SL, Yanchik‐Slade FE, Lee NR, Nilsson BL. Capacity for increased surface area in the hydrophobic core of β‐sheet peptide bilayer nanoribbons. J Pep Sci. 2021;27:e3334. 10.1002/psc.3334
REFERENCES
- 1.Bowerman CJ, Nilsson BL. Self‐assembly of amphipathic β‐sheet peptides: insights and applications. Biopolymers. 2012;98(3):169‐184. 10.1002/bip.22058 [DOI] [PubMed] [Google Scholar]
- 2.Galler KM, D'Souza RN, Hartgerink JD. Biomaterials and their potential applications for dental tissue engineering. J Mater Chem. 2010;20(40):8730‐8746. 10.1039/c0jm01207f [DOI] [Google Scholar]
- 3.Yan C, Mackay ME, Czymmek K, Nagarkar RP, Schneider JP, Pochan DJ. Injectable solid peptide hydrogel as a cell carrier: effects of shear flow on hydrogels and cell payload. Langmuir. 2012;28(14):6076‐6087. 10.1021/la2041746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan C, Altunbas A, Yucel T, Nagarkar RP, Schneider JP, Pochan DJ. Injectable solid hydrogel: mechanism of shear‐thinning and immediate recovery of injectable β‐hairpin peptide hydrogels. Soft Matter. 2010;6(20):5143‐5156. 10.1039/C0SM00642D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang S, Gelain F, Zhao X. Designer self‐assembling peptide nanofiber scaffolds for 3D tissue cell cultures. Semin Cancer Biol. 2005;15(5):413‐420. 10.1016/j.semcancer.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 6.Yamada Y, Fichman G, Schneider JP. Serum protein adsorption modulates the toxicity of highly positively charged hydrogel surfaces. ACS Appl Mater Interfaces. 2021;13(7):8006‐8014. 10.1021/acsami.0c21596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakota EL, Wang Y, Danesh FR, Hartgerink JD. Injectable multidomain peptide nanofiber hydrogel as a delivery agent for stem cell secretome. Biomacromolecules. 2011;12(5):1651‐1657. 10.1021/bm200035r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fung SY, Yang H, Bhola PT, et al. Self‐assembling peptide as a potential carrier for hydrophobic anticancer drug ellipticine: complexation, release and in vitro delivery. Adv Funct Mater. 2009;19(1):74‐83. 10.1002/adfm.200800860 [DOI] [Google Scholar]
- 9.Altunbas A, Lee SJ, Rajasekaran SA, Schneider JP, Pochan DJ. Encapsulation of curcumin in self‐assembling peptide hydrogels as injectable drug delivery vehicles. Biomaterials. 2011;32(25):5906‐5914. 10.1016/j.biomaterials.2011.04.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim YB, Lee M. Nanostructures of β‐sheet peptides: steps towards bioactive functional materials. J Mater Chem. 2008;18(7):723‐727. 10.1039/b711188f [DOI] [Google Scholar]
- 11.Lim YB, Kwon OJ, Lee E, Kim PH, Yun CO, Lee M. A cyclic RGD‐coated peptide nanoribbon as a selective intracellular nanocarrier. Org Biomol Chem. 2008;6(11):1944‐1948. 10.1039/b802470g [DOI] [PubMed] [Google Scholar]
- 12.Collier JH, Messersmith PB. Enzymatic modification of self‐assembled peptide structures with tissue transglutaminase. Bioconjug Chem. 2003;14(4):748‐755. 10.1021/bc034017t [DOI] [PubMed] [Google Scholar]
- 13.Eskandari S, Guerin T, Toth I, Stephenson RJ. Recent advances in self‐assembled peptides: implications for targeted drug delivery and vaccine engineering. Adv Drug Deliv Rev. 2017;110‐111:169‐187. 10.1016/j.addr.2016.06.013 [DOI] [PubMed] [Google Scholar]
- 14.Gelain F, Luo Z, Rioult M, Zhang S. Self‐assembling peptide scaffolds in the clinic. NPJ Regen Med. 2021;6(9). 10.1038/s41536-020-00116-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waku T, Tanaka N. Recent advances in nanofibrous assemblies based on β‐sheet‐forming peptides for biomedical applications. Polymer Intern. 2017;66(2):277‐288. 10.1002/pi.5195 [DOI] [Google Scholar]
- 16.Bowerman CJ, Ryan DM, Nissan DA, Nilsson BL. The effect of increasing hydrophobicity on the self‐assembly of amphipathic β‐sheet peptides. Mol Biosyst. 2009;5(9):1058‐1069. 10.1039/b904439f [DOI] [PubMed] [Google Scholar]
- 17.Dehsorkhi A, Castelletto V, Hamley IW. Self‐assembling amphiphilic peptides. J Pept Sci. 2014;20(7):453‐467. 10.1002/psc.2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowerman CJ, Liyanage W, Federation AJ, Nilsson BL. Tuning β‐sheet peptide self‐assembly and hydrogelation behavior by modification of sequence hydrophobicity and aromaticity. Biomacromolecules. 2011;12(7):2735‐2745. 10.1021/bm200510k [DOI] [PubMed] [Google Scholar]
- 19.Betush RJ, Urban JM, Nilsson BL. Balancing hydrophobicity and sequence pattern to influence self‐assembly of amphipathic peptides. Biopolymers [published online ahead of print January 2, 2018]. 2018;110(1). 10.1002/bip.23099. PMID: 29292825. [DOI] [PubMed] [Google Scholar]
- 20.Lim YB, Lee E, Lee M. Cell‐penetrating‐peptide‐coated nanoribbons for intracellular nanocarriers. Angewandte Chem. 2007;46(19):3475‐3478. 10.1002/anie.200604576 [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Wang F, Cui H. Peptide‐based supramolecular hydrogels for delivery of biologics. Bioeng Transl Med. 2016;1(3):306‐322. 10.1002/btm2.10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stankovic IM, Niu S, Hall MB, Zaric SD. Role of aromatic amino acids in amyloid self‐assembly. Int J Biol Macromol. 2020;156:949‐959. 10.1016/j.ijbiomac.2020.03.064 [DOI] [PubMed] [Google Scholar]
- 23.Fauchere JL, Charton M, Kier LB, Verloop A, Pliska V. Amino acid side chain parameters for correlation studies in biology and pharmacology. Int J Pept Protein Res. 1988;32(4):269‐278. 10.1111/j.1399-3011.1988.tb01261.x [DOI] [PubMed] [Google Scholar]
- 24.Lee NR, Bowerman CJ, Nilsson BL. Sequence length determinants for self‐assembly of amphipathic β‐sheet peptides. Biopolymers. 2013;100(6):738‐750. 10.1002/bip.22248 [DOI] [PubMed] [Google Scholar]
- 25.Lee NR, Bowerman CJ, Nilsson BL. Effects of varied sequence pattern on the self‐assembly of amphipathic peptides. Biomacromolecules. 2013;14(9):3267‐3277. 10.1021/bm400876s [DOI] [PubMed] [Google Scholar]
- 26.Yanagi K, Ashizaki M, Yagi H, Sakurai K, Lee YH, Goto Y. Hexafluoroisopropanol induces amyloid fibrils of islet amyloid polypeptide by enhancing both hydrophobic and electrostatic interactions. J Biol Chem. 2011;286(27):23959‐23966. 10.1074/jbc.M111.226688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S. Discovery of the first self‐assembling peptide, study of peptide dynamic behaviors, and G protein‐coupled receptors using an Aviv circular dichroism spectropolarimeter. Biopolymers. 2018;109(8):e23235. 10.1002/bip.23235 [DOI] [PubMed] [Google Scholar]
- 28.De Zotti M, Formaggio F, Crisma M, Peggion C, Moretto A, Toniolo C. Handedness preference and switching of peptide helices. Part I: helices based on protein amino acids. J Pept Sci. 2014;20(5):307‐322. 10.1002/psc.2638 [DOI] [PubMed] [Google Scholar]
- 29.Wang K, Keasling JD, Muller SJ. Effects of the sequence and size of non‐polar residues on self‐assembly of amphiphilic peptides. Int J Biol Macromol. 2005;36(4):232‐240. 10.1016/j.ijbiomac.2005.06.006 [DOI] [PubMed] [Google Scholar]
- 30.Lim YB, Moon KS, Lee M. Stabilization of an alpha helix by β‐sheet‐mediated self‐assembly of a macrocyclic peptide. Angew Chem. 2009;48(9):1601‐1605. 10.1002/anie.200804665 [DOI] [PubMed] [Google Scholar]
- 31.Manavalan P, Johnson WC. Sensitivity of circular dichroism to protein tertiary structure class. Nature. 1983;305(5937):831‐832. 10.1038/305831a0 [DOI] [Google Scholar]
- 32.Kelly SM, Price NC. The use of circular dichroism in the investigation of protein structure and function. Curr Protein Pept Sci. 2000;1(4):349‐384. [DOI] [PubMed] [Google Scholar]
- 33.Roy A, Bour P, Keiderling TA. TD‐DFT modeling of the circular dichroism for a tryptophan zipper peptide with coupled aromatic residues. Chirality. 2009;21(Suppl 1):E163‐E171. 10.1002/chir.20792 [DOI] [PubMed] [Google Scholar]
- 34.Huang R, Wu L, McElheny D, Bour P, Roy A, Keiderling TA. Cross‐strand coupling and site‐specific unfolding thermodynamics of a trpzip β‐hairpin peptide using 13C isotopic labeling and IR spectroscopy. J Phys Chem B. 2009;113(16):5661‐5674. 10.1021/jp9014299 [DOI] [PubMed] [Google Scholar]
- 35.Wu L, McElheny D, Huang R, Keiderling TA. Role of tryptophan–tryptophan interactions in Trpzip β‐hairpin formation, structure, and stability. Biochemistry. 2009;48(43):10362‐10371. 10.1021/bi901249d [DOI] [PubMed] [Google Scholar]
- 36.Wu L, McElheny D, Takekiyo T, Keiderling TA. Geometry and efficacy of cross‐strand Trp/Trp, Trp/Tyr, and Tyr/Tyr aromatic interaction in a β‐hairpin peptide. Biochemistry. 2010;49(22):4705‐4714. 10.1021/bi100491s [DOI] [PubMed] [Google Scholar]
- 37.Wu L, McElheny D, Setnicka V, Hilario J, Keiderling TA. Role of different β‐turns in β‐hairpin conformation and stability studied by optical spectroscopy. Proteins. 2012;80(1):44‐60. 10.1002/prot.23140 [DOI] [PubMed] [Google Scholar]
- 38.Egusa S, Sisido M, Imanishi Y. Side‐chain and main‐chain conformational transitions of poly(l‐1‐ and 2‐naphthylalanine). Polymer J. 1986;18(5):403‐409. 10.1295/polymj.18.403 [DOI] [Google Scholar]
- 39.Dathe M, Nikolenko H, Klose J, Bienert M. Cyclization increases the antimicrobial activity and selectivity of arginine‐ and tryptophan‐containing hexapeptides. Biochemistry. 2004;43(28):9140‐9150. 10.1021/bi035948v [DOI] [PubMed] [Google Scholar]
- 40.Wu JM, Wei SY, Chen HL, Weng KY, Cheng HT, Cheng JW. Solution structure of a novel D‐naphthylalanine substituted peptide with potential antibacterial and antifungal activities. Biopolymers. 2007;88(5):738‐745. 10.1002/bip.20736 [DOI] [PubMed] [Google Scholar]
- 41.Sisido M, Sato Y, Sasaki H, Imanishi Y. Synthesis, structure, and excimer formation of vesicular assemblies carrying 1‐ or 2‐naphthyl chromophores. Langmuir. 1990;6(1):177‐182. 10.1021/la00091a028 [DOI] [Google Scholar]
- 42.Hosoi S, Kamiya M, Ohta T. Novel development of exciton‐coupled circular dichroism based on induced axial chirality. Org Lett. 2001;3(23):3659‐3662. 10.1021/ol010176l [DOI] [PubMed] [Google Scholar]
- 43.Person RV, Monde K, Humpf HU, Berova N, Nakanishi K. A new approach in exciton‐coupled circular dichroism (ECCD)—insertion of an auxiliary stereogenic center. Chirality. 1995;7(3):128‐135. 10.1002/chir.530070304 [DOI] [PubMed] [Google Scholar]
- 44.Formaggio F, Peggion C, Crisma M, et al. Recent contributions of electronic circular dichroism to the investigation of oligopeptide conformations. Chirality. 2004;16(6):388‐397. 10.1002/chir.20051 [DOI] [PubMed] [Google Scholar]
- 45.Singh P, Misra S, Sepay N, et al. Self‐assembling behaviour of a modified aromatic amino acid in competitive medium. Soft Matter. 2020;16(28):6599‐6607. 10.1039/d0sm00584c [DOI] [PubMed] [Google Scholar]
- 46.Pignataro MF, Herrera MG, Dodero VI. Evaluation of peptide/protein self‐assembly and aggregation by spectroscopic methods. Molecules. 2020;25:4854. 10.3390/molecules25204854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uji H, Ogawa J, Itabashi K, Imai T, Kimura S. Compartmentalized host spaces accommodating guest aromatic molecules in a chiral way in a helix‐peptide‐aromatic framework. Chem Commun (Camb). 2018;54(88):12483‐12486. 10.1039/c8cc07380e [DOI] [PubMed] [Google Scholar]
- 48.Kollmer M, Close W, Funk L, et al. Cryo‐EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer's brain tissue. Nat Commun. 2019;10(4760). 10.1038/s41467-019-12683-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang M, Wang J, Zhou P, et al. Nanoribbons self‐assembled from short peptides demonstrate the formation of polar zippers between β‐sheets. Nat Commun. 2018;9(5118). 10.1038/s41467-018-07583-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marini DM, Hwang W, Lauffenburger DA, Zhang S, Kamm RD. Left‐handed helical ribbon intermediates in the self‐assembly of a β‐sheet peptide. Nano Lett. 2002;2(4):295‐299. 10.1021/nl015697g [DOI] [Google Scholar]
- 51.Hwang W, Marini DM, Kamm RD, Zhang S. Supramolecular structure of helical ribbons self‐assembled from a β‐sheet peptide. J Chem Phys. 2003;118(1):389‐397. 10.1063/1.1524618 [DOI] [Google Scholar]
- 52.Telfer SG, McLean TM, Waterland MR. Exciton coupling in coordination compounds. Dalton Trans. 2011;40(13):3097‐3108. 10.1039/c0dt01226b [DOI] [PubMed] [Google Scholar]
- 53.Sinthuvanich C, Nagy‐Smith KJ, Walsh STR, Schneider JP. Triggered formation of anionic hydrogels from self‐assembling acidic peptide amphiphiles. Macromolecules. 2017;50(15):5643‐5651. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analytical HPLC Trace (215 nm) for Ac‐(FKFE)2‐NH2.
Figure S2. Analytical HPLC Trace (215 nm) for Ac‐(HphKHphE)2‐NH2.
Figure S3. Analytical HPLC Trace (215 nm) for Ac‐(WKWE)2‐NH2.
Figure S4. Analytical HPLC Trace (215 nm) for Ac‐(1‐NalK1‐NalE)2‐NH2.
Figure S5. Analytical HPLC Trace (215 nm) for Ac‐(2‐NalK2‐NalE)2‐NH2.
Figure S6. Analytical HPLC Trace (215 nm) for Ac‐(BipKBipE)2‐NH2.
Figure S7. Analytical HPLC Trace (215 nm) for Ac‐WKFEFKFE‐NH2.
Figure S8. Analytical HPLC Trace (215 nm) for Ac‐(WKFE)2‐NH2.
Figure S9. Analytical HPLC Trace (215 nm) for Ac‐BipKFEFKFE‐NH2.
Figure S10. Analytical HPLC Trace (215 nm) for Ac‐(BipKFE)2‐NH2.
Figure S11. MALDI‐TOF‐MS for Ac‐(FKFE)2‐NH2.
Figure S12. MALDI‐TOF‐MS for Ac‐(HphKHphE)2‐NH2.
Figure S13. MALDI‐TOF‐MS data for Ac‐(WKWE)2‐NH2.
Figure S14. MALDI‐TOF‐MS data for Ac‐(1‐NalK1‐NalE)2‐NH2.
Figure S15. MALDI‐TOF‐MS data for Ac‐(2‐NalK2‐NalE)2‐NH2.
Figure S16. MALDI‐TOF‐MS data for Ac‐(BipKBipE)2‐NH2.
Figure S17. MALDI‐TOF‐MS data for Ac‐WKFEFKFE‐NH2.
Figure S18. MALDI‐TOF‐MS data for Ac‐(WKFE)2‐NH2.
Figure S19. MALDI‐TOF‐MS data for Ac‐BipKFEFKFE‐NH2.
Figure S20. MALDI‐TOF‐MS data for Ac‐(BipKFE)2‐NH2.
Table S1. Calculate and Observed m/z for synthetic peptides (MALDI‐TOF‐MS).
Figure S21. HPLC calibration curve (215 nm) for Ac‐(HphKHphE)2‐NH2.
Figure S22. HPLC calibration curve (215 nm) for Ac‐(WKWE)2‐NH2.
Figure S23. HPLC calibration curve (215 nm) for Ac‐(1‐NalK1‐NalE)2‐NH2.
Figure S24. HPLC calibration curve (215 nm) for Ac‐(BipKBipE)2‐NH2.
Figure S25. HPLC calibration curve (215 nm) for Ac‐WKFEFKFE‐NH2.
Figure S26. HPLC calibration curve (215 nm) for Ac‐(WKFE)2‐NH2.
Figure S27. HPLC calibration curve (215 nm) for Ac‐BipKFEFKFE‐NH2.
Figure S28. HPLC calibration curve (215 nm) for Ac‐(BipKFE)2‐NH2.
Figure S29. Normalized absorption spectra of peptide fibrils
Figure S30. TEM images for Ac‐(WKFE)2‐NH2 at 50°C and at 70°C. Nanoribbons were prepared in 5% HFIP/water at 0.2mM peptide for 96 hours. A) Nanoribbons in an open system at 50°C. B) Closed system at 50°C. C) Open system at 70°C. D) Closed system at 70°C. The minor change in morphology in the open system at 70°C is likely a result of evaporation over the 96 hours. Evaporation and temperature show no impact on nanoribbon morphology.