

Abstract
In an earlier study of pi-expansive ruthenium complexes for photodynamic and photochemo therapies it was shown that a pair of structural isomers differing only in the connection point of a naphthalene residue exhibited vastly different biological activity. These isomers are further explored in this paper through the activity of their functionalized derivatives. In normoxia, the inactive 2-NIP isomer (5) can be made as photocytotoxic as the active 1-NIP isomer (1) by functionalizing with methyl or methoxy groups, while methoxy variants of the 1-NIP isomer became inactive. In all cases singlet oxygen sensitization quantum yield was below 1%. Hypoxic photocytotoxicity was attenuated, with only three of the series showing any activity, notwithstanding the photodissociative ligands. The results here are consistent with the earlier findings in that seemingly minor structural modifications on the non-strained ligand can dramatically modulate the normoxic and hypoxic activity of these strained compounds, and that these changes appear to exert a greater influence on photocytotoxicity than singlet oxygen sensitization or rates of photosubstitution in cell-free conditions.
1. INTRODUCTION
Globally, cancer remains a leading cause of morbidity, with nearly 10 million deaths worldwide in 2020 (1). New therapeutic strategies continue to be developed, but there remains an urgent need for treatment options with greater selectivity for tumors than healthy tissue. One approach for enhancing specificity could make use of prodrugs that become active only in the cancerous environment. In this way, light-activated compounds can be used to direct treatment with spatial and temporal control, avoiding the deleterious side-effects often found in more conventional systemic approaches.
Photodynamic therapy (PDT) is an established example of such a strategy, with a number of clinically approved examples (2–7). The PDT mechanism consists of the interaction of photons with an otherwise non-toxic photosensitizer (PS) to produce cytotoxic singlet oxygen (1O2) and other reactive oxygen species (ROS). In contrast, the related therapeutic approach of photochemotherapy (PCT), which has not been clinically investigated, involves an irreversible photochemical reaction that leads to photocytotoxicity (Scheme 1) (8–11).
Scheme 1.

General photophysical and photochemical pathways for anticancer activity in a Ru(II) complex.
The advantage of the PDT strategy is that singlet oxygen sensitization is in principle catalytic, with a single PS molecule producing many equivalents of cytotoxic singlet oxygen. By contrast, PCT is stoichiometric, likely necessitating higher concentrations of the light-responsive compound to invoke useful cytotoxicity. Photoinduced ligand ejection, for example, offers localized modalities such as targeted drug release (if the ligand is pharmacologically active)(12–22) or cisplatin-like interactions with DNA at newly opened coordination sites (8, 23–25). Importantly, PCT mechanisms do not necessarily rely on oxygen, a feature that could be exploited in hypoxic tumors. While few compounds to date have been shown to present good hypoxic activity (26–32), efforts are actively underway to design improved PCT agents with high photocytotoxicity in low-oxygen conditions.
Polypyridyl-type metal complexes have been used as a synthetically convenient, photoreactive framework by several groups (12, 13, 20, 21, 26, 33–40), in addition to our own (8, 19, 22, 28, 41, 42), for the design of PSs for PDT and PCT. This coordination complex motif is attractive owing to its modular design: individual ligands can be chosen to manipulate important properties such as the absorption wavelength window, solubility, biological activity, etc. Unlike the traditional tetrapyrrole macrocycles that have been used for PDT, metal complexes present a number of excited states that differ in character, including the type of the excited state and its multiplicity, and the photophysics and energetics of each of these excited states can be tuned systematically by molecular design (6, 43–49). Long-lived 3IL (intraligand) and 3ILCT (intraligand charge transfer) excited states can be accessed with π-expansive ligands, and these systems produce extremely high singlet oxygen quantum yields and the most potent PDT activity observed to this date (6). Alternatively, photodissociative 3MC (metal centered) states are ideal for PCT applications, and ligands that cause steric crowding at the metal center, e.g., 6,6′-dimethyl-2,2′-bipyridine (6,6′-dmb), lower the energy of the 3MC state relative to the 3MLCT state, making its population favorable in the excited state relaxation pathway (50, 51). By manipulating the energetics of the 3IL and 3MC states in complexes having both of these features, there is the possibility of controlling which mechanism dominates: PDT-active long-lived triplets, PCT-active dissociative states, or some environmentally sensitive combination of the two for dual-action (22, 42, 52, 53).
It was within this conceptual framework that we recently examined a series of ruthenium (Ru) complexes incorporating two 6,6′-dmb combined with an imidazo[4,5-f][1,10]phenathroline (IP) ligand appended to a variety of π-systems with high to low 3IL energies (30). This design was to provide 3IL states to sensitize 1O2 for potent PDT cytotoxicity in normoxia, combined with an alternate photodissociative PCT pathway in hypoxia. In our previous study, we found that photodissociation in cell-free environments did not necessarily lead to photocytotoxicity in hypoxia. There was no clear correlation between normoxic or hypoxic photocytotoxicity (which varied significantly) and the half-life (t1/2) for ligand photosubstitution in water (which were all similar) or the estimated 3IL state energy (which varied substantially). Of the compounds investigated (π groups=benzene, fluorene, phenanthrene, two naphthalene isomers, pyrene, and anthracene), only one exhibited substantial photocytotoxicity in hypoxia. Compound 1 (Chart 1), containing the 1-NIP ligand, had a phototherapeutic index (PI, ratio of dark to light photocytotoxicity) of 22 in normoxia and 15 in hypoxia. This was surprising, in that the other structural isomer of naphthalene, compound 5 (containing the 2-NIP ligand), which differed only in the link position between naphthalene and IP, was completely inactive in both conditions. Notably, the hypoxic PI of 1 was among the largest reported at the time (30), but its singlet oxygen quantum yield and photodissociation rates did not stand out in any way from the rest of the series. Clearly, some other features play an important role.
Chart 1.
Structures of compounds 1‒7.
This result is consistent with other recent findings by our group that demonstrate that seemingly innocuous structural changes can have a very significant influence on a compound’s photocytotoxicity in normoxia and in hypoxia (28, 29). The present study not only validates the findings from our previous report regarding the activity of 1 and the inactivity of 5, but it explores in greater depth the apparent dichotomy of these two NIP isomers. We now show that functionalization of the naphthalene rings can modulate the properties that led to photocytotoxic performance in one but not the other. Compounds 2–4 are 1-NIP derivatives of parent 1 with different groups substituted at different positions on the naphthalene rings, whereas 6 and 7 are 2-NIP variants of parent 5. The choice of simple substituents highlights the fact that very subtle structural modifications can have a large impact on the properties of Ru(II) complexes used for PDT or PCT applications.
2. MATERIALS AND METHODS
Unless otherwise specified, all reagents and solvents were purchased from commercial sources and used without further purification. Water used for all biological experiments was deionized to a resistivity ≥ 18.2 MΩ·cm (type 1) using either a Barnstead or Milli-Q® filtration system.
2.1. Instrumentation
Microwave reactions were performed in a CEM Discover microwave reactor. Flash chromatography relied on the Teledyne Isco CombiFlash® EZ Prep system with Silicycle SiliaSep silica flash cartridges (FLH-R10030B-ISO25). Size-exclusion chromatography was performed on a gravity column packed with Sephadex® LH-20. NMR spectra were collected using a JEOL ECA 500 NMR spectrometer (1H, 1H-1H COSY) and JEOL 400 NMR spectrometer (1H) at the NMR facility at the University of North Carolina at Greensboro (UNCG) and Agilent 700 MHz NMR spectrometer (1H-1H COSY) at the Joint School of Nanoscience and Nanoengineering at Greensboro (JSNN). The chemical shifts are reported in parts per million (ppm) and were referenced to the residual solvent peaks. ESI mass spectra were obtained using a Thermo Fisher LTQ Orbitrap XL coupled to a Water’s Acquity Ultra Performance Liquid Chromatography (UPLC) stack using a BEH C18 column at UNCG’s Triad Mass Spectrometry facility. HPLC analyses were carried out on an Agilent/Hewlett Packard 1100 series instrument (ChemStation Rev. A. 10.02 software) using a Hypersil GOLD C18 column (Thermo 25005–254630, guard 25003–014001) with an A–B gradient (40 min run; 98% → 5% A; A=0.1% formic acid in water, B=0.1% formic acid in acetonitrile (MeCN); see Table S1). Reported retention times are accurate to within ± 0.1 min.
2.2. Synthesis
The Ru(6,6′-dmb)2Cl2·2H2O intermediate was prepared following an adapted literature procedure (42, 54). The syntheses of IP-based ligands followed a revised literature procedure (30, 55) and are described below. The synthesis and isolation of Ru(II) complexes 1–7 followed a slightly modified procedure to the previously reported Ru(6,6′-dmb)2(IP-Ar) family (30, 42). Compounds 1 and 5 were synthesized previously by our group (30). Complexes 1–7 were isolated as PF6− salts due to their increased solubility in organic solvents then converted to their Cl− salts for structural characterization via 1H NMR, 1H-1H COSY NMR, and ESI+ mass spectrometry. Acceptable purity (≥95%) was confirmed by 1H NMR and HPLC. The water-soluble Cl− salts were used for photobiological, spectroscopic, and photochemical experiments.
4MEO1N. 1,10-phenanthroline-5,6-dione (168 mg, 0.80 mmol), 4-methoxy-1-napthaldehyde (149 mg, 0.80 mmol), and ammonium acetate (616 mg, 8.00 mmol) were added to a microwave vessel followed by the addition of glacial acetic acid (4 mL). The vial was subjected to microwave irradiation at 180°C for 15 minutes. The reaction mixture was transferred to a beaker and diluted with deionized H2O (5 mL), then neutralized (pH 7) via the dropwise addition of aqueous NH4OH. The obtained precipitate was washed with cold deionized water (50 mL) and diethyl ether (70 mL). The final product was a brown solid (211 mg, 70 %). Rf = 0.83 (2% H2O, 43% CHCl3, 29% MeOH, 1% NH4OH, 25% acetone). 1H NMR (400 MHz, DMSO-d6, ppm): δ 9.17 (d, J = 8.0 Hz, 1H; i), 9.06 (dd, J = 4.0, 1.0 Hz, 2H; a), 8.97 (m, 2H; c), 8.33 (d, J = 8.3 Hz, 1H; f), 8.10 (d, J = 8.1 Hz, 1H; d), 7.85 (m, 2H; b), 7.72 (t, J = 8.0 Hz, 1H; g), 7.63 (t, J = 8.0 Hz, 1H; h), 7.24 (d, J = 8.4 Hz, 1H; e), 4.10 (s, 3H; 4-OMe). HPLC retention time: 24.19 min (95% purity by peak area).
6MEO2N. 1,10-phenanthroline-5,6-dione (168 mg, 0.80 mmol), 6-methoxy-2-napthaldehyde (149 mg, 0.80 mmol), and ammonium acetate (616 mg, 8.00 mmol) were combined and treated according to the procedure described for the synthesis of 4MEO1N to yield a red-brown solid (294 mg, 97%). Rf = 0.83 (2% H2O, 43% CHCl3, 29% MeOH, 1% NH4OH, 25% acetone). 1H NMR (400 MHz, DMSO-d6, ppm): δ 9.05 (dd, J = 4.2, 1.6 Hz, 2H; a), 8.97 (dd, J = 8.2, 1.6 Hz, 2H; c), 8.75 (s, 1H; i), 8.39 (dd, J = 8.6, 1.6 Hz, 1H; d), 8.07–8.01 (m, 2H; e, h), 7.86 (dd, J = 8.0, 4.4 Hz, 2H; b), 7.44 (d, J = 2.4 Hz, 1H; f), 7.28 (dd, J = 8.8, 2.4 Hz, 1H; g), 3.93 (s, 3H; 6-OMe). HPLC retention time: 17.30 min (89% purity by peak area).
DMEO1N. 1,10-phenanthroline-5,6-dione (168 mg, 0.80 mmol), 4,7-methoxy-1-napthaldehyde (173 mg, 0.80 mmol), and ammonium acetate (616 mg, 8.00 mmol) were combined and treated according to the procedure described for the synthesis of 4MEO1N to yield a light-brown solid (339 mg, quantitative yield). Rf = 0.80 (2% H2O, 43% CHCl3, 29% MeOH, 1% NH4OH, 25% acetone). 1H NMR (400 MHz, DMSO-d6, ppm): δ 9.06 (dd, J = 4.4, 1.2 Hz, 2H; a), 9.02–8.96 (m, 3H; c,h), 8.23 (d, J = 9.2 Hz, 1H; f), 8.15 (d, J = 8.4 Hz, 1H; d), 7.86 (m, 2H; b), 7.28 (dd, J = 9.2, 2.8 Hz, 1H; g), 7.11 (d, 8.4 Hz, 1H; e), 4.08 (s, 3H; 4-OMe), 3.95 (s, 3H; 7-OMe). HPLC retention time: 18.79 min (82% purity by peak area).
4F1N. 1,10-phenanthroline-5,6-dione (168 mg, 0.80 mmol), 4-fluoro-1-napthaldehyde (139 mg, 0.80 mmol), and ammonium acetate (616 mg, 8.0 mmol) were combined and treated according to the procedure described for the synthesis of 4MEO1N to yield a dark-brown solid (293 mg, quantitative yield). Rf = 0.74 (2% H2O, 43% CHCl3, 29% MeOH, 1% NH4OH, 25% acetone). 1H NMR (400 MHz, DMSO-d6, ppm): δ 9.22 (d, J = 8.2 Hz, 1H; i), 9.07 (dd, J = 4.4, 1.6 Hz, 2H; a), 8.97 (dd, J = 7.6, 1.0 Hz, 2H; c), 8.23 (d, J = 8.0 Hz, 1H; g), 8.16 (dd, J = 8.0, 5.6 Hz, 1H; d), 7.87 (dd, J = 8.1, 4.4 Hz, 2H; b), 7.80 (m, 2H; f,h), 7.64 (dd, J = 10.4, 8.4 Hz, 1H; e). HPLC retention time: 12.53 min (83% purity by peak area).
7M2N. 1,10-phenanthroline-5,6-dione (117 mg, 0.56 mmol), 7-methyl-2-napthaldehyde (95 mg, 0.56 mmol), and ammonium acetate (308 mg, 4.0 mmol) were combined and treated according to the procedure described for the synthesis of 4MEO1N to yield a tan solid (180 mg, 89%). Rf = 0.75 (2% H2O, 43% CHCl3, 29% MeOH, 1% NH4OH, 25% acetone). 1H NMR (400 MHz, DMSO-d6, ppm): δ 9.06 (dd, J = 4.4, 2.0 Hz, 2H; a), 8.99 (dd, J = 8.2, 1.6 Hz, 2H; c), 8.73 (d, J = 1.6 Hz, 1H; i), 8.37 (dd, J = 8.8, 2.0 Hz, 1H; d), 8.10 (d, J = 8.8 Hz, 1H; e), 7.92 (d, J = 8.4 Hz, 1H; f), 7.87 (m, 3H; b,h), 7.46 (dd, J = 8.4, 1.5 Hz, 1H; g), 2.54 (s, 3H, 7-Me). HPLC retention time: 21.02 min (83% purity by peak area).
[Ru(6,6′-dmb)2(4MEO1N)]Cl2 (2). Ru(6,6′-dmb)2Cl2·2H2O (92 mg, 0.16 mmol) and 4MEO1N (60 mg, 0.16 mmol) were added to a microwave vessel containing argon-purged ethylene glycol (3 mL) and subjected to microwave irradiation at 180°C for 15 minutes. The resulting dark-red mixture was transferred to a separatory funnel with deionized water (20 mL) and CH2Cl2 (30 mL). After gentle mixing, the CH2Cl2 layer was drained and the remaining aqueous layer was washed with CH2Cl2 (30 mL portions) until the CH2Cl2 layer appeared colorless. At that point, CH2Cl2 (30 mL) and saturated aqueous KPF6 (5 mL) was added and the mixture was shaken gently and allowed to settle overnight to facilitate transfer of the product from the aqueous layer to the CH2Cl2 layer, at which point the CH2Cl2 layer was collected. Additional CH2Cl2 (20mL × 2) was used to further extract the product from the aqueous layer to maximize yield. Then, the CH2Cl2 layers were combined and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography with gradient of MeCN, 10% water in MeCN, then 7.5% water with 0.5% aqueous saturated KNO3 in MeCN. The product eluted last, and product-containing fractions (red) were concentrated under reduced pressure to obtain red solid. The solid, which is the desired complex [Ru(6,6′-dmb)2(4MEO1N)]2+ as a NO3−/PF6− salt mixtures, was dissolved in deionized H2O (20 mL) and transferred to a separatory funnel. CH2Cl2 (30 mL) and saturated aqueous KPF6 (3 mL) was added in order to convert the product to a PF6− salt. This mixture was gently shaken and allowed to settle over time. The CH2Cl2 layer was drained off into a round bottom flask, and more CH2Cl2 (30 mL) was added. This was repeated once. The combined CH2Cl2 layers were concentrated under reduced pressure until dry to give [Ru(6,6′-dmb)2(4MEO1N)](PF6)2 (42 mg, 23%). The PF6− salt [Ru(6,6′-dmb)2(4MEO1N)](PF6)2 (42 mg) was dissolved in 1–2 mL of MeCN to which an equal volume of MeOH was added with sonication, then loaded onto a 20×150 mm column of HCl-treated Amberlite IRA-410 (30 g) suspended in MeOH. The sample was eluted with MeOH over 2 h and then dried under reduced pressure to yield the Cl− salt [Ru(6,6′-dmb)2(4MEO1N)]Cl2 in quantitative yield (33 mg). The Cl− salt [Ru(6,6′-dmb)2(4MEO1N)]Cl2 was further purified using size-exclusion chromatography on Sephadex LH-20 with MeOH as the eluent (31 mg, 21%). Rf = 0.22 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (500 MHz, MeOD-d3, ppm): δ 9.01 (d, J = 7.5 Hz, 2H; c), 8.76 (dd, J = 8.0, 1.0 Hz, 2H; 3), 8.63 (d, J = 8.2 Hz, 1H; i), 8.56 (dd, J = 8.5, 1.0 Hz, 2H; 3’), 8.39 (d, J = 8.1 Hz, 1H; f), 8.33 (dd, J = 5.5, 1.0 Hz, 2H; a), 8.28 (t, J = 8.0 Hz, 2H; 4), 7.97 (d, J = 8.0 Hz, 1H; d), 7.85 (dd, J = 8.0, 5.5 Hz, 2H; b), 7.74 (t, J = 8.0 Hz, 2H; 4’), 7.67 (dd, J = 7.5, 1.0 Hz, 2H; 5), 7.65–7.57 (m, 2H; g,h), 7.15 (d, J = 8.0 Hz, 1H; e), 7.00 (dd, J = 7.5, 1.0 Hz, 2H; 5’), 4.13 (s, 3H; 4-OMe), 1.98 (s, 6H; 6-Me), 1.64 (s, 6H; 6’-Me). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C48H40N8ORu 423.1179; Found 423.1176. [M-2Cl-H]+ Calcd for C48H39N8ORu 845.2285; Found 845.2288. HPLC retention time: 20.35 min (99% purity by peak area).
[Ru(6,6′-dmb)2(DMEO1N)]Cl2 (3). Ru(6,6′-dmb)2Cl2·2H2O (92 mg,0.16 mmol) and DMEO1N (65 mg, 0.16 mmol) were combined and treated according to the procedure described for 2 to yield an orange solid (29 mg, 15%). The PF6− salt was converted to its corresponding Cl− salt in quantitative yield using the procedure described for 2. The Cl− salt was further purified using size-exclusion chromatography on Sephadex LH-20 with MeOH as the eluent (18 mg, 12%). Rf = 0.14 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (500MHz, MeOD-d3, ppm): δ 9.00 (dd, J = 8.3, 1.0 Hz, 2H; c), 8.74 (dd, J = 8.2, 1.0 Hz, 2H; 3), 8.54 (dd, J = 8.0, 1.0 Hz, 2H; 3’), 8.31–8.24 (m, 6H; a,f,4,h), 7.95 (d, J = 8.2 Hz, 1H; d), 7.82 (dd, J = 8.2, 5.5 Hz, 2H; b), 7.72 (t, J = 8.0 Hz, 2H; 4’), 7.65 (dd, J = 7.8, 1.0 Hz, 2H; 5), 7.20 (dd, J = 9.3, 2.7 Hz, 1H; g), 6.99 (d, J = 8.0 Hz, 1H; e), 6.98 (dd, J = 7.5, 1.0 Hz, 2H; 5’), 4.10 (s, 3H; 4-OMe), 3.88 (s, 3H; 7-OMe), 1.98 (s, 6H; 6-Me), 1.64 (s, 6H; 6’-Me). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C49H42N8O2Ru 438.1232; Found: 438.1233. [M-2Cl-H]+ Calcd for C49H41N8O2Ru 875.2390; Found 875.2406. HPLC retention time: 11.91 min (98% purity by peak area).
[Ru(6,6′-dmb)2(4F1N)]Cl2 (4). Ru(6,6′-dmb)2Cl2·2H2O (92 mg, 0.16 mmol) and 4F1N (58 mg, 0.16 mmol) were combined and treated according to the procedure described for 2 to yield a red solid (19 mg, 11%). The PF6− salt was converted to its corresponding Cl− salt in quantitative yield using the procedure described for 2. The Cl− salt was further purified using size-exclusion chromatography on Sephadex LH-20 with MeOH as the eluent (15 mg, 10%). Rf = 0.13 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (700 MHz, MeOD-d3, ppm): δ 9.01 (d, J = 8.2 Hz, 2H; c), 8.76 (d, J = 8.2 Hz, 2H; 3), 8.72 (d, J = 7.9 Hz, 1H; i), 8.56 (d, J = 8.1 Hz, 2H; 3’), 8.35 (dd, J = 5.5, 1.0 Hz, 2H; a), 8.28 (t, J = 7.7 Hz, 2H; 4), 8.26 (m, 1H; g), 8.02 (dd, J = 8.0, 5.5 Hz, 1H; d), 7.86 (dd, J = 8.2, 5.5 Hz, 2H; b), 7.77–7.71 (m, 4H; 4’,f,h), 7.67 (d, J = 7.7 Hz, 2H; 5), 7.44 (dd, J = 9.8, 7.7 Hz, 1H; e), 7.01 (d, J = 7.7 Hz, 2H; 5’), 1.98 (s, 6H, 6-Me), 1.64 (s, 6H, 6’-Me). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C47H37FN8Ru 417.1079; Found: 417.1081. [M-2Cl-H]+ Calcd for C47H36FN8Ru 833.2085; Found 833.2093. HPLC retention time: 20.24 min (95% purity by peak area).
[Ru(6,6′-dmb)2(6MEO2N)]Cl2 (6). Ru(6,6′-dmb)2Cl2·2H2O (92 mg, 0.16 mmol) and 6MEO2N (60 mg, 0.16 mmol) were combined and treated according to the procedure described for 2 to yield a red solid (28 mg, 15%). The PF6− salt was converted to its corresponding Cl− salt in quantitative yield using the procedure described for 2. The Cl− salt was further purified using size-exclusion chromatography on Sephadex LH-20 with MeOH as the eluent (23 mg, 16%). Rf = 0.20 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (500 MHz, MeOD-d3, ppm): δ 9.10 (dd, J = 8.2, 1.0 Hz, 2H; c), 8.76 (dd, J = 7.8, 1.0 Hz, 2H; 3), 8.70 (d, 1.5 Hz, 1H; i), 8.56 (dd, J = 7.9, 1.0 Hz, 2H; 3’) 8.34–8.26 (m, 5H; a,d,4), 8.01 (d, J = 9.0 Hz, 1H; e), 7.95 (d, J = 9.0 Hz, 1H; h), 7.86 (dd, J = 8.3, 5.5 Hz, 2H; b), 7.73 (t, J = 8.0 Hz, 2H; 4’), 7.67 (dd, J = 8.0, 1.0 Hz, 2H; 5), 7.36 (d, J = 2.5 Hz, 1H; f), 7.25 (dd, J = 9.0, 2.5 Hz, 1H; g), 6.99 (dd, J = 7.5, 1.0 Hz, 2H; 5’), 3.96 (s, 3H; 6-OMe), 1.97 (s, 6H; 6-Me), 1.63 (s, 6H; 6’-Me). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C48H40N8ORu 423.1179; Found 423.1178. [M-2Cl-H]+ Calcd for C48H39N8ORu 845.2285; Found 845.2291. HPLC retention time: 14.77 min (96% purity by peak area).
[Ru(6,6′-dmb)2(7M2N)]Cl2 (7). Ru(6,6′-dmb)2Cl2·2H2O (92 mg, 0.16 mmol) and 7M2N (57mg, 0.16 mmol) were combined and treated according to the procedure described in 2 to yield a red solid (55 mg, 31%). The PF6− salt was converted to its corresponding Cl− salt in quantitative yield using the procedure described for 2.. The Cl− salt was further purified using size-exclusion chromatography on Sephadex LH-20 with MeOH as the eluent (43 mg, 30%). Rf = 0.14 (0.5% KNO3, 7.5% H2O, 92% MeCN). 1H NMR (500MHz, MeOD-d3, ppm): δ 9.18–9.00 (m, 2H; c), 8.76 (dd, J = 8.5, 1.0 Hz, 2H; 3), 8.68 (d, J = 1.0 Hz, 1H; i), 8.54 (dd, J = 8.0, 1.0 Hz, 2H; 3’), 8.31 (dd, J = 5.5, 1.0 Hz, 2H; a), 8.29–8.24 (m, 3H; d,4), 8.02 (d, J = 8.5 Hz, 1H; e), 7.87–7.83 (m, 3H; b,f), 7.81 (d, J = 1.0 Hz, 1H; h), 7.72 (t, J = 8.0 Hz, 2H; 4’), 7.65 (dd, J = 7.5, 1.0 Hz, 2H; 5), 7.45 (dd, J = 8.5, 1.5 Hz, 1H; g), 6.98 (dd, J = 7.5, 1.0 Hz, 2H; 5’), 2.56 (s, 3H, 7-Me), 1.97 (s, 6H; 6-Me), 1.63 (s, 6H; 6’-Me). HRMS (ESI+) m/z: [M-2Cl]2+ Calcd for C50H40N8Ru 415.1204; Found 415.1201. [M-2Cl-H]+ Calcd for C50H39N8Ru 829.2336; Found 823.2336. HPLC retention time: 9.76 min. (95% purity by peak area).
2.3. Spectroscopy
2.3.1. UV-visible spectroscopy
Ultraviolet-visible absorption spectra were measured on a Jasco V730 spectrometer at room temperature in acetonitrile using 5 mm pathlength quartz cuvettes. Compounds 1 and 5 were reported previously in water (30).
2.3.2. Singlet oxygen sensitization
Quantum yields for singlet oxygen sensitization (ΦΔ) were measured using the intensity of the 1O2 emission band, centered around 1276 nm, as measured on a PTI Quantamaster emission spectrometer equipped with a Hamamatsu R5509–42 NIR PMT. The instrument was internally corrected for wavelength-dependent nonlinearities in lamp output and detector sensitivities.
[Ru(bpy)3]2+ was used as the actinometry standard (ΦΔ = 0.56 in aerated MeCN(56)) in Equation 1, where I denotes the emission integration, A is the UV-Vis absorption of the solution at the excitation wavelength, and η is the solvent’s refractive index (η2/ηS2=1 in this case, since MeCN was used for both). The standard is indicated by the subscript S.
| Equation 1 |
These measurements were performed as 20 μM solutions of complexes in acetonitrile prepared from a 1 mM stock in CD3OD (to facilitate dissolution of the chloride salts; final 2% v/v CD3OD). The local MLCT absorption maximum in the UV-vis spectrum (around 463 nm) was chosen for the excitation wavelength. The emission was collected between 1200–1350 nm using a 1000 nm long-pass filter, and baseline corrected.
2.4. Photosubstitution
Percent conversion of the intact metal complex to all photoproducts was estimated by comparing integrated peak areas using light treatments that were identical to those used in the cell-based experiments. The assumption is that the photoproducts formed are photosubstitution products resulting from the loss of one or two 6,6’-dmb ligands, or partial dechelation. Complexes 1–7 were diluted from 5 mM stocks (10% v/v DMSO:H2O) to 50 μM solutions in a deep-well plate using type 1 water (≥18.2 MΩ·cm) with a final 0.1% v/v DMSO composition. Solutions were then dispensed to 96-well plates in duplicate at 100 μL well−1. Dark treated plates were covered in aluminum foil and placed in a drawer while light treated plates were treated with 100 J cm−2 delivered at 18–22 mW cm−2 using cool white visible (400–700 nm), green 523 nm, and red 633 nm light sources (Figure S26). Plates were covered with low evaporation lids during the irradiation period (~1.5 h). Samples were then transferred to glass vials, the solvent removed in vacuo, compounds dissolved in HPLC grade MeOH to approximately 100 μM, and syringe filtered with 0.2 μm PTFE-membrane (PP-housing) filters prior to HPLC injection (30 μL). Aqueous samples were prepared in this manner due to insolubility of photoproducts and their removal by filtration if kept in aqueous solvents. Samples were otherwise eluted using the same column conditions and solvent gradient as reported in the synthetic methodology (Table S1).
Retention times and peak areas were partially analyzed in R (57) using the dplyr (58) and tidyr (59) packages. Unreacted complexes were identified using the dark-treated samples and referenced to the original chromatograms ran when first synthesized. Free 6,6′-dmb ligand was identified with retention time of 8.40 min, whereas metal complex species (dark and light-treated) eluted between 12–26 min. The peak area of unreacted PS was normalized with the sum peak area of metal species for any given sample at the 285 nm detection wavelength. Percent impurity values were calculated using Equation 2. Percent photosubstitution values were calculated using Equation 3 with correction where A is area and f denotes the fraction of impurities from the original sample.
| Equation 2 |
| Equation 3 |
2.5. Cell Culture and Photobiology
Compounds 1–7 were screened on 96-well plates for dose-response ranging from 1×10−3 to 300 μM in accordance with our standard assay (6, 30).
2.5.1. Cell culture
In general, cells were cultured using standard aseptic technique and no antibiotics.
SKMEL28. Male human melanoma cell line SK-MEL-28 [SKMEL28] (ATCC HTB-72) was subcultured as previously described (30) at 5% USP-grade CO2, ≥ 90% relative humidity, and 37°C in a water-jacketed incubator (ThermoFisher, Thermo Scientific 4110). Complete media was 10% FB essence (VWR, 10803–034) in EMEM with 1% L-glutamine (Corning, 15–010-CV). Split ratios were commonly performed between 1:2–1:5 (150,000–400,000 cells mL−2). Cells were used at passages 14 and 16 from receipt. For cytotoxicity and photocytotoxicity screens, cells were seeded at 10,000 cells well−1.
2.5.2. Cellular assays
General dose-response screens were performed on 96-well plates in hypoxia and normoxia following our standard assay (6, 30). Plates were stacked up to 2-plates high in an incubator to facilitate rapid temperature equilibrium. The drug-to-light interval (DLI) was between 17 and 22 h for both normoxic and hypoxic experiments. Additionally, final viability measurements were conducted using the resazurin viability assay, read fluorometrically on a Molecular Devices M2e (15 s shake, bottom-read, λexc 530 nm, long-pass 570 nm, λem 620 nm).
2.5.3. Hypoxia cytotoxicity and photocytotoxicity
Following our recent example (30), we screened the activity of our PS family in hypoxia (1% O2) to evaluate oxygen independence relative to a compound known to be highly oxygen-dependent for its activity [Ru(bpy)2(dppn)]Cl2 (60, 61). This was performed 4 days after the normoxic screen. Hypoxic cell culture was conducted inside a Biospherix Xvivo X3 at 1% O2 (5% CO2, 37 °C). Following cell adhesion post-seeding (2–3 h) in normoxia, plates were incubated at 1% O2 for 2–3 h before dispensing compound dilutions. After an overnight incubation in hypoxia, plates to be irradiated were covered with film (VWR, 89134–428) to maintain the low oxygen environment during light treatment. Following light treatment, films were removed and all low oxygen plates were replaced to the normoxic incubator and incubated overnight before final viability measurements.
2.5.4. Light devices and protocols.
For photobiological evaluation, we used three visible light sources. For standard screens cells were treated at a fluence of 100 J cm−2 and irradiance of 18–22 mW cm−2. Light sources included a cool white LED panel (SOLLA-CREE, 400–700 nm, maxima ~450 nm) and two UHP-LEDs (Prizmatix, 523 and 633 nm). Their spectral output is included in Figure S26.
2.5.5. Data processing and statistics
All results from the endpoint-based resazurin assay were background subtracted with negative controls (media and DPBS) and normalized relative to positive cell controls. Any negative values were assumed to be a mismatch of background (i.e., fluorescence quenching) and assigned as zero values. Likewise, at high compound concentration, background fluorescence and/or quenching is often observed for this class of compounds. Zero values were assigned for these cases when indicated by several consecutive concentrations for a given treatment (dark or light). Additional verification was conducted via light microscopy before finalizing data corrections. Further discussion of assay limits for these compound types is provided in a recent review.(6)
Resazurin data over a wide concentration range was fit to both a three-parameter log-logistic and logistic models using GraphPad Prism 8.4.0 according to Equation 4 and Equation 5 (four-parameter shown) where bottom is constrained to equal zero and X is equal to concentration.
| Equation 4 |
| Equation 5 |
Experiments were done in triplicate with replicates plotted ± standard deviation (SD) on a given plot. Reported EC50 values are ± SEM for a given experiment; these denote the effective concentration to reduce relative cell viability by 50% of the fitted curve (EC50) ± standard error of the mean (SEM). Phototherapeutic indices (PI) are reported as the ratio of dark to light EC50 values and used as a measure of light-induced potency. Summary activity plots were used for quickly comparing potency of the compounds (Log EC50 and PI) and include SEM from log-logistic fits where applicable (Log EC50).
3. RESULTS
3.1. Synthesis
Compounds 1–7 (Chart 1) were prepared by reacting the Ru(6,6′-dmb)2Cl2·2H2O intermediate with the corresponding naphthalene-substituted imidazo[4,5-f][1,10]phenanthroline (IP-Ar) ligand in ethylene glycol using microwave irradiation for 15 min at 180°C. The desired products were precipitated as their PF6− salts and purified on silica gel using 5–10% H2O containing 0.5% KNO3 in acetonitrile (MeCN) as the eluent. The resulting NO3−/PF6− salt mixtures were converted to their pure PF6− salts. The Cl− salts were obtained from corresponding PF6− salts in quantitative yield via anion metathesis on HCl-treated Amberlite IRA-410 resin (Alfa-Aesar, A1773436) with methanol as the eluent. The Cl− salt was then further purified using size-exclusion chromatography on Sephadex LH-20 with methanol as the eluent, affording final products in 10–30% yields. Final complexes were a racemic mixture of Δ/Λ isomers. The ligands 4MEO1N, DMEO1N, 4F1N, 6MEO2N, and 7M2N were characterized by TLC, 1H NMR and HPLC (Figures S1–S5, S16–S20). Complexes 2–4, 6, and 7 were characterized by TLC, 1H NMR and 1H-1H COSY NMR (Figures S6–S10), high-resolution ESI+-MS (Figures S11–S15), HPLC (Figures S21–S25), and UV-Vis spectroscopy (Figure 2).
Figure 2:

UV-visible spectra of the series at room temperature in acetonitrile.
The signals in the 1D 1H NMR spectra of 2–4, 6, and 7 were assigned using 2D 1H-1H COSY NMR spectra (Figures S6–S10). The 1H NMR signals corresponding to the coligand 6,6′-dmb (3—5, 3’—5’, 6-Me, 6’-Me) and the imidazophenanthroline core (a—c) were readily identified using assignments from the previously reported [Ru(bpy)3]Cl2 complex (62) as well as the [Ru(6,6′-dmb)2(IP-Ar)]Cl2 and oligothienyl [Ru(6,6′-dmb)2(IP-nT)]Cl2 families (30, 42). These signals appeared in the following order: c, 3, 3’, 4, b, 4’, 5, 5’ (Figure 1). The 1H NMR signals corresponding to the substituted naphthalenes were assigned with the help of assignments from previously reported imidazo[4,5-f][1,10]phenanthroline naphthalene complexes (30), and were confirmed by studying their 1H-1H COSY correlations (Figures S6–S10) and J values (see experimental section). The assignments for complexes 2–4, 6, and 7 as well as the J values extracted from their 1H NMR spectra, were used to assign the 1H NMR signals for ligands 4MEO1N, DMEO1N, 4F1N, 6MEO2N, and 7M2N. The location of the signals corresponding to the naphthalene unit in the ligands agreed well with that of the complexes.
Figure 1.

Summary of aromatic 1H NMR assignments for final complexes. Top: Structures of 1–5 with labels for each distinct hydrogen. Bottom: Overlay of 1H NMR spectra (500 MHz, MeOD-d3) of compounds 1–5 with assignments.
3.2. Spectroscopy
The UV-vis spectra of the substituted compounds are shown in Figure 2 and tabulated in Table 1. The spectra of parent compounds 1 and 5 are published elsewhere (30). The spectra are dominated by a strong absorption near 300 nm that we ascribe to ππ* transitions on the 6,6′-dmb ligands. Weak shoulders are due to substituted naphthalene transition (εmax=6.0×103 at 275 nm for naphthalene (63)). The Ru2+(dπ) ⟶LL(π*) MLCT transition is located at 463±1 nm across the series, indicating the same π* acceptor orbital is involved in each compound, with no contribution by the pendent naphthalene substituents.
Table 1:
Spectroscopic data. Data for 1 and 5 from previous work.
| Cmpd | ΦΔ | Selected absorptions (nm and log10 ε) |
|---|---|---|
| 1 | <0.01 | 219 (4.85), 256 (4.62), 297 (4.83), 434 (4.02), 464 (4.18) |
| 2 | <0.01 | 301 (4.85), 435 (4.13), 463 (4.25) |
| 3 | <0.01 | 255 (4.87), 300 (4.96), 352 (4.58), 439 (4.27), 464 (4.35) |
| 4 | <0.01 | 262 (4.67), 299 (4.91), 435 (4.14), 462 (4.27) |
| 5 | <0.01 | 210 (4.72), 293 (4.82), 433 (3.99), 465 (4.15) |
| 6 | <0.01 | 298 (4.92), 333 (4.70), 434 (4.14), 464 (4.28) |
| 7 | <0.01 | 296 (4.83), 433 (4.01), 463 (4.14) |
The compounds produced very weak singlet oxygen sensitization quantum yields, with ΦΔ≤1% in each case, consistent with an excited state pathway that does not involve a long-lived 3IL state.
3.3. Photosubstitution
To test the degree of photosubstitution under cell-free conditions, aqueous solutions of 1–7 were irradiated using the same light parameters employed in our standard photocytotoxicity assays (6). A fluence of 100 J cm−2 (18–22 mW cm−2) was delivered from a broadband visible (400–700 nm) light source or monochromatic green (523 nm) or red (633 nm) LEDs. Following irradiation, the photolyzed samples were transferred to methanol for quantification by HPLC, where the percent conversion to photoproducts was estimated as the ratio of intact PS to total Ru species based on MLCT absorption >400 nm. The major photoconversion pathway was assumed to be photosubstitution involving dissociation of a 6,6′-dmb ligand (Rt = 8.40 min) and solvation of the residual Ru center with an aqua ligand,(8, 16, 42, 64) as was previously observed for 1 and 5 (30). Water was chosen as the solvent to estimate the degree of photosubstitution in the simplest case with identical light treatments (100 J cm−2) to parallel the results of cell-based experiments (Table 2).
Table 2.
HPLC analysis of conversion to photoproducts under cell-free conditions for 1–7.
| Conversion to photoproducts (%) | |||
|---|---|---|---|
| Cmpd | Visiblea | Greenb | Redc |
| 1 | 100 | 100 | 6.7 |
| 2 | 94.0 | 94.2 | 1.6 |
| 3 | 90.4 | 92.3 | 0.6 |
| 4 | 96.7 | 86.6 | 4.2 |
| 5 | 91.4 | 93.4 | 4.5 |
| 6 | 85.7 | 89.0 | 9.0 |
| 7 | 93.7 | 98.5 | 2.3 |
Light treatments were 100 J cm−2 delivered at 18–22 mW cm−2 with avisible (400–700 nm), bgreen 523 nm, and cred 633 nm. A value of 100% was assigned if no peak detected from intact PS.
All complexes were stable in the absence of light over the course of the cell-free experiment (12–24 h, 20°C), including heating to ~50°C to remove water and storage in MeOH for HPLC analysis (12–24 h). Treatments with visible or green light produced near complete photoconversion (85.7–100%) for all compounds. Only 4 exhibited an appreciable wavelength dependence, with 96.7% photoconversion using visible light and 86.6% with green light. The complexes were relatively photoinert using longer wavelengths of light. Only two complexes had photosubstitution values >5% with red light (6.7% for 6 and 9.0% for 1). This inactivity is consistent with compounds 1–7 having minimal absorption beyond 600 nm (Figure 2). Accordingly, only visible and green light were expected to lead to photocytotoxicity in the in vitro experiments.
3.3. Cellular Assays
3.4.1. Normoxia (18.5–21% O2)
Compounds 1–7 were evaluated for their cytotoxicities (dark plates) and photocytotoxicites (light plates) against SKMEL28 melanoma cells using the resazurin viability assay according to our standard assay (6, 30). Cells were first seeded to well plates and dosed with compound between 1 nM and 300 μM. After overnight incubation, the dark plates were left in the incubator while the light plates were irradiated (DLI=17–21.5 h) using a fluence of 100 J cm−2 delivered at an irradiance of 18–22 mW cm−2. Following irradiation, the plates were returned to the incubator for two days prior to cell viability determination of both the dark- and light-treated samples with the resazurin dye. The dark and light EC50 values, or the effective concentrations to reduce relative viability by 50%, were obtained from sigmoidal fits of the dose-response curves (Table 3, Figure 3). The phototherapeutic indices (PIs) were calculated as the ratio of dark to light EC50 values and represent the light amplification of cytotoxicity (Table 3, Figure 4).
Table 3.
Cytotoxicity and photocytotoxicity of 1–7 in normoxic (~18.5% O2, top) or hypoxic (1% O2, bottom) treated SK-MEL-28 melanoma cells.
| Resazurin-based Cell Viability | ||||||||
|---|---|---|---|---|---|---|---|---|
| EC50 ± SEM (μM) | PId | |||||||
| Complex | Oxygen% | Dark | Visiblea | Greenb | Redc | Visiblea | Greenb | Redc |
| 1 | ~18.5 | 45.0 ± 7.3 | 2.81 ± 0.09 | 23.3 ± 0.8 | 31.9 ± 1.9 | 16 | 2 | 1 |
| 2 | ~18.5 | 42.3 ± 6.8 | 33.5 ± 1.1 | 34.3 ± 2.4 | 45.1 ± 7.2 | 1 | 1 | 1 |
| 3 | ~18.5 | 37.7 ± 6.3 | 27.2 ± 1.0 | 32.3 ± 3.0 | 43.2 ± 7.4 | 1 | 1 | 1 |
| 4 | ~18.5 | 36.1 ± 3.5 | 3.68 ± 0.20 | 23.1 ± 0.7 | 47.2 ± 6.0 | 10 | 2 | 1 |
| 5 | ~18.5 | 31.3 ± 1.5 | 23.5 ± 1.0 | 29.1 ± 1.2 | 34.0 ± 1.8 | 1 | 1 | 1 |
| 6 | ~18.5 | 35.9 ± 3.6 | 4.64 ± 0.20 | 21.4 ± 0.7 | 25.9 ± 0.8 | 8 | 2 | 1 |
| 7 | ~18.5 | 24.8 ± 0.9 | 2.54 ± 0.09 | 7.07 ± 0.1 | 25.8 ± 0.8 | 10 | 4 | 1 |
| 1 | 1 | 97.5 ± 6.7 | 8.66 ± 0.27 | 44.0 ± 2.7 | 56.8 ± 5.6 | 11 | 2 | 2 |
| 2 | 1 | 45.1 ± 6.8 | 44.5 ± 2.6 | 26.2 ± 1.7 | 48.5 ± 7.0 | 1 | 2 | 1 |
| 3 | 1 | 46.6 ± 5.6 | 33.6 ± 1.5 | 20.6 ± 0.6 | 49.3 ± 6.8 | 1 | 2 | 1 |
| 4 | 1 | 61.4 ± 4.7 | 21.2 ± 1.7 | 30.1 ± 1.6 | 54.8 ± 5.6 | 3 | 2 | 1 |
| 5 | 1 | 74.3 ± 2.7 | 57.6 ± 2.3 | 46.8 ± 1.1 | 65.2 ± 3.0 | 1 | 2 | 1 |
| 6 | 1 | 57.8 ± 6.7 | 32.6 ± 4.5 | 51.7 ± 4.2 | 54.8 ± 4.2 | 2 | 1 | 1 |
| 7 | 1 | 32.0 ± 0.7 | 6.95 ± 0.29 | 20.6 ± 1.0 | 28.4 ± 2.8 | 5 | 2 | 1 |
Light treatments were approximately 100 J cm−2 delivered at 18–22 mW cm−2 with acool white visible (400–700 nm), bgreen 523 nm, cred 633 nm, and dPI = phototherapeutic index. Hypoxic and normoxic experiments were ran within 4 days of each other.
Figure 3.
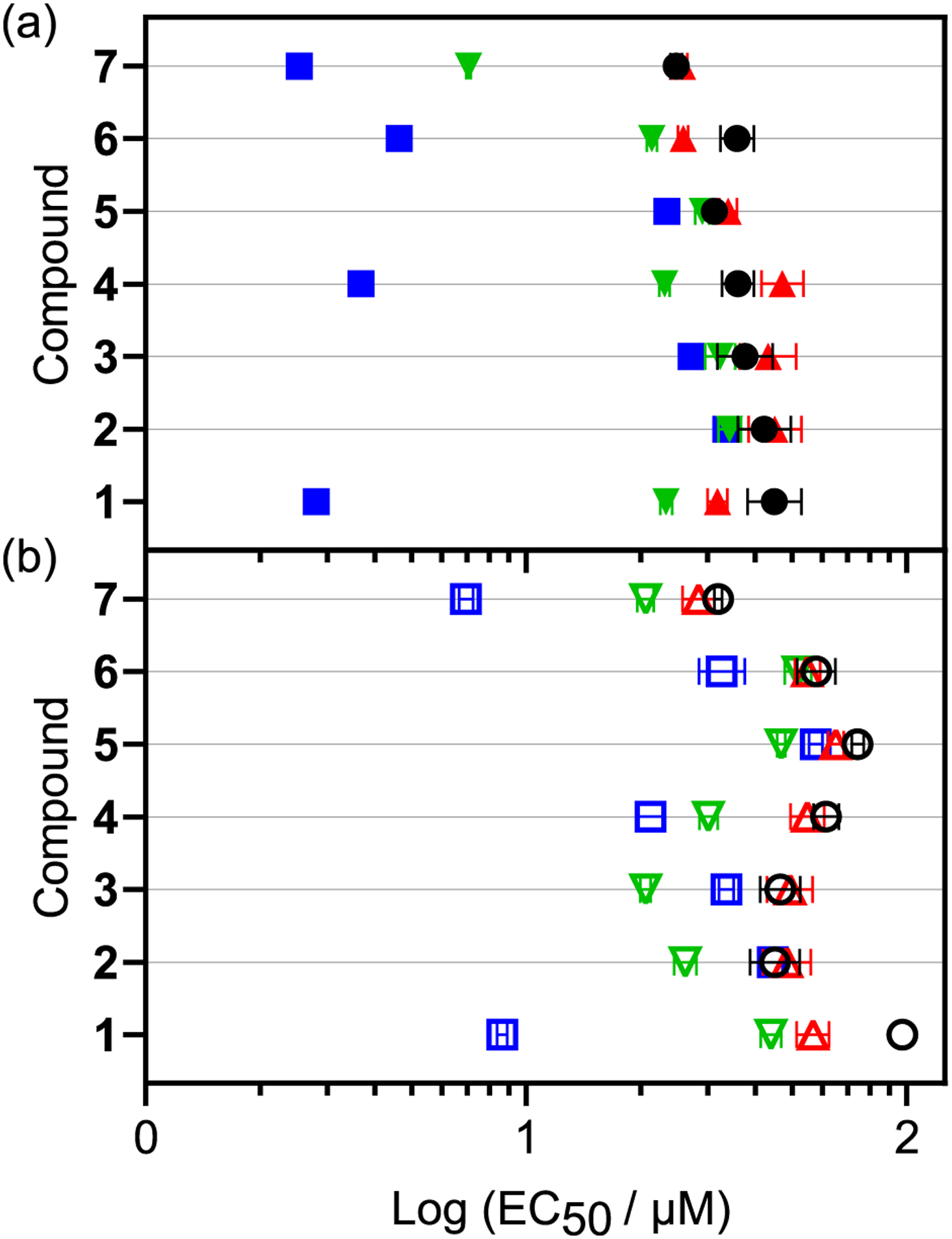
Biological and photobiological log (EC50 ± SEM) activities of 1–7 under (a) normoxic and (b) hypoxic 1% O2 conditions. Treatments include dark or sham treatments (black circle) and 100 J cm−2 at 18–22 mW cm−2 using cool white vis (400–700 nm; blue square), green 523 nm (inverted triangle), or red 633 nm (triangle) light sources.
Figure 4.

Phototherapeutic indices (PIs) of 1–7 under normoxic (filled symbols) and (b) hypoxic 1% O2 conditions (open symbols). PIs are taken as the ratio of dark to light EC50 values.
All seven compounds were slightly cytotoxic in the dark, with EC50 values ranging from 24.8 to 45.0 μM. The unsubstituted reference compounds 1 and 5 had dark EC50 values of 45.0 and 31.3 μM, respectively, in agreement with our previous report (30). Functionalization of compound 1 at C4 led to a slight increase in this cytotoxicity, where methoxy substitution at this position gave an EC50 value of 42.3 μM (2) and fluorination gave an EC50 value of 36.1 μM (4). Addition of a second methoxy group at C8 further increased the cytotoxicity and led to an EC50 value of 37.7 μM (3). Substitution of 5 did not produce a consistent trend. Methoxy substitution at C6 resulted in decreased cytotoxicity (EC50=35.9 μM for 6) whereas C7 methylation increased cytotoxicity (EC50=24.8 μM for 7).
Red light had no effect on the cytotoxicities of the compounds in normoxia, with the dark and light EC50 values being almost equal and producing PIs near 1. This was expected given that red light absorption was minimal and did not produce any appreciable photosubstitution. Because all of the compounds undergo ligand photosubstitution with visible and green light (Table 2), it was expected that these wavelengths would produce an increase in cytotoxicity upon irradiation. However, we previously found that the position of the naphthalene unit appended to the IP ligand plays a critical role (30), where 1 yielded a PIblue of 22 (EC50=2.43 μM) with monochromatic blue light, while 5 was completely inactive (PIblue=1). We wondered whether this extreme difference between parent compounds 1 and 5 could be modulated even further with substitution on the naphthalene rings.
Similar to what we observed previously, 1 was photocytotoxic with broadband visible light (EC50=2.81 μM, PIvis=16) and less active with green light (EC50=23.3 μM, PIgreen=2), while 5 was inactive. The EC50 value was slightly larger and PI thus lower for 1 in this study due to the fact that our previous study used monochromatic blue light, and the present study employed broadband visible light.
Three of the five new compounds were considered active. Of these active compounds (4, 6, and 7), visible light was much more effective (EC50=2.54–3.68 μM, PIvis=8–16) than green (EC50=7.07–23.1 μM, PIgreen=2–4). Interestingly, appending methoxy substituents to the naphthalene system of 1 completely suppressed its activity. Compounds 2 and 3, the methoxy analogs of 1, had EC50 values in both the dark and light >20 μM and PIs near 1. Fluorination of 1 at C4, on the other hand, retained the single-digit micromolar activity of 1, with 4 having an EC50 of 3.68 μM and PI of 10 for visible light.
Another interesting observation was that substitution of the inactive compound 5 with a methyl group at C7 led to activity for 7 that rivaled 1 as most potent compound, with an EC50 value of 2.54 μM and PIvis of 10 (versus 2.81 μM and 16, respectively, for 1). Unlike the methoxy derivatives of 1, the methoxy analog of 5 at the C6 position activated the compound (PIvis= 8, EC50=4.64 μM), albeit to a lesser extent than methylation at C7.
3.4.2. Hypoxia (1% O2)
Since they were designed as PCT agents for low-oxygen conditions, the compounds were evaluated in hypoxia (1% O2) using a protocol similar to the normoxic screen but with hypoxia maintained up to and during the light treatment. Our standard hypoxia assay protocol (28–30) involves preincubating the cells under hypoxic conditions prior to compound addition, applying a film over the well plates before removal from the hypoxia chamber for light treatment outside the hypoxic chamber, and incubating the cells in normoxia after irradiation (since the resazurin cell viability assay used for quantifying live versus dead cells is based on metabolic respiration). We have confirmed that this is a robust method for testing photocytotoxicity in hypoxia (28–30), and provides low oxygen tension at the time of illumination. This was verified by the inactivity of [Ru(bpy)2(dppn)]Cl2, a PS known to be highly oxygen-dependent (60, 61).
Compounds 2, 3, and 7 were similar in their cytotoxicities under both normoxia and hypoxia. However, the rest of the compounds exhibited differences in their normoxic and hypoxic cytotoxicities (Figure 3b). The increased resistance of hypoxic cells to cytotoxic compounds is well-known, especially in solid tumors (65–67), and has been generally attributed to HIF-1 expression, alternate metabolic pathways, and decreased pH (68). The dark cytotoxicities of 1, 4, 5, and 6 were reduced by up to 2.5-fold in hypoxia. The cells were most resistant to parent scaffolds 1 and 5, which had EC50 values in hypoxia of 74.3 and 97.5 μM, respectively. The reason for the difference in normoxic and hypoxic sensitivity for only some of the compounds within the relatively small compound series is not clear, underscoring that minor structural changes can have major impact even on the oxygen-dependence of the baseline dark cytotoxicity.
Compared to dark cytotoxicity, photocytotoxicity tends to be much more sensitive to hypoxia. Most photosensitizers that exhibit excellent activity in normoxia are not photocytotoxic in hypoxia and have PIs near 1 (69). Despite having photochemical reactivity that is presumably oxygen independent, many PCT agents are also inactive in hypoxia such as compounds 2, 3, 5, and other examples (17, 30). However, we previously reported single-digit micromolar photocytotoxicity for 1 in hypoxia with blue light (PIblue=15) (30). Notably, its hypoxic PI was almost as large as its normoxic PIblue of 22. These values were slightly attenuated in the present study, owing to the use of broadband visible light rather than monochromatic blue (453 nm). However, 1 still gave single-digit micromolar photocytotoxicity (light EC50=8.66 μM) with a hypoxic PI of 11 (relative to PInorm=16).
As expected, the compounds that were not photocytotoxic in normoxia (2, 3, and 5) were also inactive in hypoxia. However, two of the three new compounds that were photocytotoxic in normoxia maintained some of their activity in hypoxia, with PIvis values in the range of 3–5. Interestingly, 4 and 7 both had PIvis values of 10 in normoxia but were affected differently by hypoxia. Compound 7 gave a single-digit micromolar EC50 value of 6.95 μM and PIvis= 5, while 4 was less active (EC50=21.2 μM, PIvis =3). Despite being similar in potency to 4 and 7 in normoxia, compound 6 was relatively inactive in hypoxia with a PIvis value of only 2.
Since the singlet oxygen quantum yields for all compounds in this family are less than 1%, any observed photocytotoxicity is assumed to stem from photoreactions that take place from accessible 3MC states, namely photosubstitution reactions. However, the relative potencies of the compounds in terms of photocytotoxicity does not appear to be related to their extinction coefficients or their percent photoconversion under cell free conditions. Nevertheless, there are factors that we did not probe as part of this study that could be responsible. For example, we observed previously that the photodissociation rates change dramatically with wavelength. Shorter-wavelength blue light led to faster photosubstitution rates and were related to larger PIs (30). This is evidence that relaxation pathways to the dissociative state are complex and in competition with other deactivation pathways. It must be considered that the cellular environment under hypoxia is sufficiently different that it impacts the photosubstitution efficiency, as well as the fact that the products may be less active under these conditions.
Collectively, these photocytotoxicity studies in normoxia and in hypoxia corroborate the previously reported importance of the isomeric connectivity of the IP ligand, as the unsubstituted 1-NIP scaffold (1) is superior to the unsubstituted 2-NIP scaffold (5). However, electronic tuning of the expansive ligand through the addition of substituents to these naphthalene groups can completely abolish the activities of these complexes, or enhance them, depending on the identities of the substituents. The nature of the substituent also influences whether a compound that is active in normoxia will be active in hypoxia. Consequently, both the point of connectivity to IP and the substituents on the naphthalene unit can drastically modify the activity, providing two independent handles for tuning the activity. Once we understand the relationship between the photocytotoxicity and the light parameters under biologically relevant conditions, it should be possible to rationally tune these systems for even better potency in hypoxia.
4. CONCLUSION
In our previous study (30), we observed that the identity and point of connection of aryl groups appended to the IP ligand greatly influenced the biological activities of the resulting Ru(II) complexes. Compounds 1 and 5, containing naphthalene appended to IP but differing in the attachment position, exhibited drastically different photocytotoxicities despite similar photochemical reactivities. Compound 1 was the best among the series, and isomer 5 was completely inactive. In the present study, we demonstrate that the activity of a small family of strained Ru(II) complexes derived from 1-NIP or 2-NIP ligands is strongly affected by simple functionalization of the non-strain-inducing IP-based ligand that does not photodissociate.
In normoxia, we show that the inactive 5 can be activated by adding either methoxy (6) or methyl groups (7) to the 2-NIP ligand, leading to photocytotoxicity rivaling that of compound 1 in the case of 7. In contrast, fluorine substitution (4) of 1 led to performance loss and ranking third most active, while methoxy (2 and 3) substitutions on 1 completely inactivated the 1-NIP scaffold. In hypoxia, the activities of both compound groups were attenuated with or without light treatment. Drug resistance (dark) varied widely in hypoxia based on the NIP scaffold and its substituents, with dark EC50 values increasing up to 2.5-fold relative to their normoxic values. Photocytotoxicities were likewise attenuated to where only three compounds were sufficiently active in hypoxia compared to four in normoxia, the parent scaffold 1 (PIvis= 11) followed by a derivative of the 2-NIP ligand (7, PIvis= 5) then 1-NIP ligand (4, PIvis=3). Despite similar activity as either 4 or 7 in normoxia, compound 6 lost most of its potency in hypoxia (PI≤2).
This study further corroborates that the NIP-containing ligand, while not photodissociative, is essential to photobiological activity as a nondissociative ligand. The NIP group has the capacity to drastically modulate the photocytotoxicities of these compounds. This has been demonstrated in terms of its connectivity to the chelating IP ligand as well as its substituents. In a study of limited structural scope, we have shown that substituents can both activate the compound’s photocytotoxic behavior and completely suppress it. Additionally, these modifications affect the oxygen dependence of the photocytotoxicity. In all conditions, compound 1 remained the top performer in terms of its PI values in normoxia and in hypoxia. However, 5, which is inactive, was improved dramatically with methyl substitution, whereby compound 7 was the second most active compound in both normoxia and in hypoxia.
The reason for these differences is not yet clear. The compounds would not be expected to differ substantially in their lowest excited singlet state energies, based on the similarity of their ground state absorption spectra in regions where they were excited in the photochemical and photobiological experiments. Additionally, each of the compounds were poor singlet oxygen generators, with singlet oxygen quantum yields less than 1%, suggesting that the photochemical (and oxygen-independent) pathway is the channel that leads to any observed photocytotoxicity. The compounds had similar photochemical conversions under cell-free conditions under the employed light dose, with visible light being the most effective and red light not producing any observable photoproducts. However, the rate of photosubstitution was not probed as part of this study, and may differ among the compounds. It has already been shown to differ between the wavelengths for a given compound in our previous study, whereby visible light led to much faster photosubstitution compared to green light delivered at the same irradiance. These distinctions may reflect differences in the singlet excited states initially populated and access to photodissociative 3MC states as well as differences in the 3MCLT and 3MC energies. It is also possible that the singlet oxygen quantum yields and photosubstitution efficiencies may differ in the cellular environment and that cellular accumulation and subcellular distribution are further influencers. In addition, pathways other than singlet oxygen sensitization or photosubstitution may play a role. For example, ROS such as superoxide or hydroxyl radicals can be formed via electron transfer reactions. Finally, any photosubstitution products formed could participate in one or more of these processes. Efforts are underway to understand these aspects, which will be critical to rationally designing optimal PCT agents of this type.
While the full picture is evolving, our structure-activity investigation from this relatively small set of related compounds sets the stage for the rational design of PCT agents that are photocytotoxic in hypoxia. By demonstrating the importance of distal groups on the non-strain inducing ligands to the activity of the overall complex, we highlight that the identity of the distal group, and its connectivity to the chelating ligand, as well as its substituents, have a defining role to play in further optimization of these systems. These additional features provide new mechanisms to control the activity of PCT agents.
Supplementary Material
5.2. Acknowledgments
S.A.M. and C.G.C. thank the National Cancer Institute (NCI) of the National Institutes of Health (NIH) (Award R01CA222227) and E.C.G. thanks the National Institute of General Medical Sciences (NIGMS) of the NIH (Award R01GM107586) for support. The content in this review is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. S.A.M. also acknowledges the Canadian Institutes of Health Research (CIHR), the Natural Sciences and Engineering Council of Canada (NSERC), the Canadian Foundation for Innovation (CFI), and the Nova Scotia Research and Innovation Trust (NSRIT) for support. S.A.M. also thanks Dr. Daniel Todd as UNCG’s Triad Mass Spectrometry Facility manager and his assistants Jennifer Simpson and Diane Wallace. We thank Dr. Franklin Moy for his experimental support and instrument maintenance as UNCG’s NMR facility manager.
Footnotes
Synthetic characterization (1D and 2D NMR, HRMS, HPLC) and (photo)biological data are included in the Supporting Information. This material is available free of charge via the Internet at https://chemistry-europe.onlinelibrary.wiley.com/journal/18607187.
S.A.M. has a potential research conflict of interest due to a financial interest with Theralase Technologies, Inc. and PhotoDynamic, Inc. A management plan has been created to preserve objectivity in research in accordance with UTA policy.
6. REFERENCES
- 1.Ferlay J, Ervik M, Lam F, Colombet M, Mery L, Pineros M, Znaor A, Soerjomataram I and Bray F (2020) Global Cancer Observatory: Cancer Today All Cancers Fact Sheet. WHO International Agency for Research on Cancer. [Google Scholar]
- 2.Usuda J, Kato H, Okunaka T, Furukawa K, Tsutsui H, Yamada K, Suga Y, Honda H, Nagatsuka Y, Ohira T, Tsuboi M and Hirano T (2006) Photodynamic therapy (PDT) for lung cancers. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 1, 489–493. [PubMed] [Google Scholar]
- 3.Plaetzer K, Krammer B, Berlanda J, Berr F and Kiesslich T (2009) Photophysics and photochemistry of photodynamic therapy: fundamental aspects. Lasers Med. Sci 24, 259–268. 10.1007/s10103-008-0539-1. [DOI] [PubMed] [Google Scholar]
- 4.Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S, Pogue BW and Hasan T (2010) Imaging and Photodynamic Therapy: Mechanisms, Monitoring, and Optimization. Chem. Rev 110, 2795–2838. 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Straten D, Mashayekhi V, de Bruijn H, Oliveira S and Robinson D (2017) Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 9, 19. 10.3390/cancers9020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monro S, Colón KL, Yin H, Roque J, Konda P, Gujar S, Thummel RP, Lilge L, Cameron CG and McFarland SA (2019) Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev 119, 797–828. 10.1021/acs.chemrev.8b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McFarland SA, Mandel A, Dumoulin-White R and Gasser G (2020) Metal-based photosensitizers for photodynamic therapy: the future of multimodal oncology? Curr. Opin. Chem. Biol 56, 23–27. 10.1016/j.cbpa.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howerton BS, Heidary DK and Glazer EC (2012) Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents. J. Am. Chem. Soc 134, 8324–8327. 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]
- 9.Huisman M, White JK, Lewalski VG, Podgorski I, Turro C and Kodanko JJ (2016) Caging the uncageable: using metal complex release for photochemical control over irreversible inhibition. Chem. Commun 52, 12590–12593. 10.1039/C6CC07083C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White JK, Schmehl RH and Turro C (2017) An overview of photosubstitution reactions of Ru(II) imine complexes and their application in photobiology and photodynamic therapy. Inorg Chim Acta 454, 7–20. 10.1016/j.ica.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnet S (2018) Why develop photoactivated chemotherapy? Dalton Trans. 47, 10330–10343. 10.1039/C8DT01585F. [DOI] [PubMed] [Google Scholar]
- 12.Respondek T, Garner RN, Herroon MK, Podgorski I, Turro C and Kodanko JJ (2011) Light Activation of a Cysteine Protease Inhibitor: Caging of a Peptidomimetic Nitrile with Ru II (bpy) 2. J. Am. Chem. Soc 133, 17164–17167. 10.1021/ja208084s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karaoun N and Renfrew AK (2015) A luminescent ruthenium(ii) complex for light-triggered drug release and live cell imaging. Chem. Commun 51, 14038–14041. 10.1039/C5CC05172J. [DOI] [PubMed] [Google Scholar]
- 14.Zamora A, Denning CA, Heidary DK, Wachter E, Nease LA, Ruiz J and Glazer EC (2017) Ruthenium-containing P450 inhibitors for dual enzyme inhibition and DNA damage. Dalton Trans. 46, 2165–2173. 10.1039/C6DT04405K. [DOI] [PubMed] [Google Scholar]
- 15.Li A, Yadav R, White JK, Herroon MK, Callahan BP, Podgorski I, Turro C, Scott EE and Kodanko JJ (2017) Illuminating cytochrome P450 binding: Ru(II)-caged inhibitors of CYP17A1. Chem. Commun 53, 3673–3676. 10.1039/C7CC01459G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuello-Garibo JA, Meijer MS and Bonnet S (2017) To cage or to be caged? The cytotoxic species in ruthenium-based photoactivated chemotherapy is not always the metal. Chem. Commun 53, 6768–6771. 10.1039/C7CC03469E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lameijer LN, Ernst D, Hopkins SL, Meijer MS, Askes SHC, Le Dévédec SE and Bonnet S (2017) A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. Int. Ed 56, 11549–11553. 10.1002/anie.201703890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohrabaugh TN, Rohrabaugh AM, Kodanko JJ, White JK and Turro C (2018) Photoactivation of imatinib–antibody conjugate using low-energy visible light from Ru( ii )-polypyridyl cages. Chem. Commun 54, 5193–5196. 10.1039/C8CC01348A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Havrylyuk D, Deshpande M, Parkin S and Glazer EC (2018) Ru(II) complexes with diazine ligands: electronic modulation of the coordinating group is key to the design of “dual action” photoactivated agents. Chem. Commun 54, 12487–12490. 10.1039/C8CC05809A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arora K, Herroon M, Al-Afyouni MH, Toupin NP, Rohrabaugh TN, Loftus LM, Podgorski I, Turro C and Kodanko JJ (2018) Catch and Release Photosensitizers: Combining Dual-Action Ruthenium Complexes with Protease Inactivation for Targeting Invasive Cancers. J. Am. Chem. Soc 140, 14367–14380. 10.1021/jacs.8b08853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li A, Turro C and Kodanko JJ (2018) Ru(II) polypyridyl complexes as photocages for bioactive compounds containing nitriles and aromatic heterocycles. Chem. Commun 54, 1280–1290. 10.1039/C7CC09000E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Havrylyuk D, Stevens K, Parkin S and Glazer EC (2020) Toward Optimal Ru(II) Photocages: Balancing Photochemistry, Stability, and Biocompatibility Through Fine Tuning of Steric, Electronic, and Physiochemical Features. Inorg. Chem 59, 1006–1013. 10.1021/acs.inorgchem.9b02065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wachter E, Heidary DK, Howerton BS, Parkin S and Glazer EC (2012) Light-activated ruthenium complexes photobind DNA and are cytotoxic in the photodynamic therapy window. Chem. Commun 48, 9649. 10.1039/c2cc33359g. [DOI] [PubMed] [Google Scholar]
- 24.Glazer EC (2013) Light-Activated Metal Complexes that Covalently Modify DNA. Isr. J Chem 53, 391–400. 10.1002/ijch.201300019. [DOI] [Google Scholar]
- 25.Wachter E, Zamora A, Heidary DK, Ruiz J and Glazer EC (2016) Geometry matters: inverse cytotoxic relationship for cis/trans-Ru(II) polypyridyl complexes from cis/trans-[PtCl 2$ (NH 32$ ]. Chem Commun 52, 10121–10124. 10.1039/C6CC04813G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Rixel VHS, Ramu V, Auyeung AB, Beztsinna N, Leger DY, Lameijer LN, Hilt ST, Le Dévédec SE, Yildiz T, Betancourt T, Gildner MB, Hudnall TW, Sol V, Liagre B, Kornienko A and Bonnet S (2019) Photo-Uncaging of a Microtubule-Targeted Rigidin Analogue in Hypoxic Cancer Cells and in a Xenograft Mouse Model. J. Am. Chem. Soc 141, 18444–18454. 10.1021/jacs.9b07225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bevernaegie R, Doix B, Bastien E, Diman A, Decottignies A, Feron O and Elias B (2019) Exploring the Phototoxicity of Hypoxic Active Iridium(III)-Based Sensitizers in 3D Tumor Spheroids. J. Am. Chem. Soc 141, 18486–18491. 10.1021/jacs.9b07723. [DOI] [PubMed] [Google Scholar]
- 28.Roque JA III, Barrett PC, Cole HD, Lifshits LM, Shi G, Monro S, von Dohlen D, Kim S, Russo N, Deep G, Cameron CG, Alberto ME and McFarland SA (2020) Breaking the barrier: an osmium photosensitizer with unprecedented hypoxic phototoxicity for real world photodynamic therapy. Chem. Sci 11, 9784–9806. 10.1039/D0SC03008B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roque JA, Barrett PC, Cole HD, Lifshits LM, Bradner E, Shi G, von Dohlen D, Kim S, Russo N, Deep G, Cameron CG, Alberto ME and McFarland SA (2020) Os(II) Oligothienyl Complexes as a Hypoxia-Active Photosensitizer Class for Photodynamic Therapy. Inorg. Chem 59, 16341–16360. 10.1021/acs.inorgchem.0c02137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roque J, Havrylyuk D, Barrett PC, Sainuddin T, McCain J, Colón K, Sparks WT, Bradner E, Monro S, Heidary D, Cameron CG, Glazer EC and McFarland SA (2020) Strained, Photoejecting Ru(II) Complexes that are Cytotoxic Under Hypoxic Conditions. Photochem. Photobiol 96, 327–339. 10.1111/php.13174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou X-Q, Xiao M, Ramu V, Hilgendorf J, Li X, Papadopoulou P, Siegler MA, Kros A, Sun W and Bonnet S (2020) The Self-Assembly of a Cyclometalated Palladium Photosensitizer into Protein-Stabilized Nanorods Triggers Drug Uptake In Vitro and In Vivo. J. Am. Chem. Soc 142, 10383–10399. 10.1021/jacs.0c01369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuang S, Sun L, Zhang X, Liao X, Rees TW, Zeng L, Chen Y, Zhang X, Ji L and Chao H (2020) A Mitochondrion‐Localized Two‐Photon Photosensitizer Generating Carbon Radicals Against Hypoxic Tumors. Angew. Chem. Int. Ed 59, 20697–20703. 10.1002/anie.202009888. [DOI] [PubMed] [Google Scholar]
- 33.Zayat L, Calero C, Alborés P, Baraldo L and Etchenique R (2003) A New Strategy for Neurochemical Photodelivery: Metal–Ligand Heterolytic Cleavage. J. Am. Chem. Soc 125, 882–883. 10.1021/ja0278943. [DOI] [PubMed] [Google Scholar]
- 34.Nikolenko V, Yuste R, Zayat L, Baraldo LM and Etchenique R (2005) Two-photon uncaging of neurochemicals using inorganic metal complexes. Chem. Commun 1752. 10.1039/b418572b. [DOI] [PubMed] [Google Scholar]
- 35.Zayat L, Salierno M and Etchenique R (2006) Ruthenium(II) Bipyridyl Complexes as Photolabile Caging Groups for Amines. Inorg. Chem 45, 1728–1731. 10.1021/ic0512983. [DOI] [PubMed] [Google Scholar]
- 36.Goldbach RE, Rodriguez-Garcia I, van Lenthe JH, Siegler MA and Bonnet S (2011) N-Acetylmethionine and Biotin as Photocleavable Protective Groups for Ruthenium Polypyridyl Complexes. Chem. - Eur. J 17, 9924–9929. 10.1002/chem.201101541. [DOI] [PubMed] [Google Scholar]
- 37.San Miguel V, Álvarez M, Filevich O, Etchenique R and del Campo A (2012) Multiphoton Reactive Surfaces Using Ruthenium(II) Photocleavable Cages. Langmuir 28, 1217–1221. 10.1021/la2033687. [DOI] [PubMed] [Google Scholar]
- 38.Zayat L, Filevich O, Baraldo LM and Etchenique R (2013) Ruthenium polypyridyl phototriggers: from beginnings to perspectives. Philos. Trans. R. Soc. Math. Phys. Eng. Sci 371, 20120330. 10.1098/rsta.2012.0330. [DOI] [PubMed] [Google Scholar]
- 39.Chan H, Ghrayche JB, Wei J and Renfrew AK (2017) Photolabile Ruthenium(II)-Purine Complexes: Phototoxicity, DNA Binding, and Light-Triggered Drug Release: Photolabile Ruthenium(II)-Purine Complexes: Phototoxicity, DNA Binding, and Light-Triggered Drug Release. Eur. J. Inorg. Chem 2017, 1679–1686. 10.1002/ejic.201601137. [DOI] [Google Scholar]
- 40.Wei J and Renfrew AK (2018) Photolabile ruthenium complexes to cage and release a highly cytotoxic anticancer agent. J. Inorg. Biochem 179, 146–153. 10.1016/j.jinorgbio.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 41.Hidayatullah AN, Wachter E, Heidary DK, Parkin S and Glazer EC (2014) Photoactive Ru(II) Complexes With Dioxinophenanthroline Ligands Are Potent Cytotoxic Agents. Inorg. Chem 53, 10030–10032. 10.1021/ic5017164. [DOI] [PubMed] [Google Scholar]
- 42.Sainuddin T, Pinto M, Yin H, Hetu M, Colpitts J and McFarland SA (2016) Strained ruthenium metal–organic dyads as photocisplatin agents with dual action. J. Inorg. Biochem 158, 45–54. 10.1016/j.jinorgbio.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P and von Zelewsky A (1988) Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev 84, 85–277. 10.1016/0010-8545(88)80032-8. [DOI] [Google Scholar]
- 44.Kalyanasundaram K (1992) Photochemistry of polypyridine and pophyrin complexes. Acad. Pr, London. [Google Scholar]
- 45.Balzani V, Juris A, Venturi M, Campagna S and Serroni S (1996) Luminescent and Redox-Active Polynuclear Transition Metal Complexes. Chem. Rev 96, 759–834. 10.1021/cr941154y. [DOI] [PubMed] [Google Scholar]
- 46.Balzani V, Credi A and Venturi M (1998) Photochemistry and photophysics of coordination compounds: An extended view. Coord. Chem. Rev 171, 3–16. 10.1016/S0010-8545(98)90005-4. [DOI] [Google Scholar]
- 47.Balzani V and Juris A (2001) Photochemistry and photophysics of Ru(II) polypyridine complexes in the Bologna group. From early studies to recent developments. Coord. Chem. Rev 211, 97–115. 10.1016/S0010-8545(00)00274-5. [DOI] [Google Scholar]
- 48.McClenaghan ND, Leydet Y, Maubert B, Indelli MT and Campagna S (2005) Excited-state equilibration: a process leading to long-lived metal-to-ligand charge transfer luminescence in supramolecular systems. Coord. Chem. Rev 249, 1336–1350. 10.1016/j.ccr.2004.12.017. [DOI] [Google Scholar]
- 49.Campagna S, Puntoriero F, Nastasi F, Bergamini G and Balzani V (2007) Photochemistry and Photophysics of Coordination Compounds: Ruthenium. In Photochemistry and Photophysics of Coordination Compounds I Vol. 280 (Edited by Balzani V and Campagna S), pp. 117–214. Springer Berlin Heidelberg, Berlin, Heidelberg. 10.1007/128_2007_133. [DOI] [Google Scholar]
- 50.Sun Q, Mosquera-Vazquez S, Suffren Y, Hankache J, Amstutz N, Lawson Daku LM, Vauthey E and Hauser A (2015) On the role of ligand-field states for the photophysical properties of ruthenium(II) polypyridyl complexes. Coord. Chem. Rev 282–283, 87–99. 10.1016/j.ccr.2014.07.004. [DOI] [Google Scholar]
- 51.Sun Q, Dereka B, Vauthey E, Lawson Daku LM and Hauser A (2017) Ultrafast transient IR spectroscopy and DFT calculations of ruthenium(II) polypyridyl complexes. Chem. Sci 8, 223–230. 10.1039/C6SC01220E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loftus LM, White JK, Albani BA, Kohler L, Kodanko JJ, Thummel RP, Dunbar KR and Turro C (2016) New Ru(II) Complex for Dual Activity: Photoinduced Ligand Release and 1O2 Production. Chem. - Eur. J 22, 3704–3708. 10.1002/chem.201504800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toupin NP, Nadella S, Steinke SJ, Turro C and Kodanko JJ (2020) Dual-Action Ru(II) Complexes with Bulky π-Expansive Ligands: Phototoxicity without DNA Intercalation. Inorg. Chem 59, 3919–3933. 10.1021/acs.inorgchem.9b03585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collin JP and Sauvage JP (1986) Synthesis and study of mononuclear ruthenium(II) complexes of sterically hindering diimine chelates. Implications for the catalytic oxidation of water to molecular oxygen. Inorg. Chem 25, 135–141. 10.1021/ic00222a008. [DOI] [Google Scholar]
- 55.Wang Z (2010) Radziszewski Reaction. In Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons, Inc. 10.1002/9780470638859.conrr518. [DOI] [Google Scholar]
- 56.DeRosa MC and Crutchley RJ (2002) Photosensitized singlet oxygen and its applications. Coord. Chem. Rev 233–234, 351–371. 10.1016/S0010-8545(02)00034-6. [DOI] [Google Scholar]
- 57.R Core Team (2020) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 58.Wickham H, François R, Henry L and Müller K (2020) dplyr: A Grammar of Data Manipulation. [Google Scholar]
- 59.Wickham H and Henry L (2020) tidyr: Tidy Messy Data. [Google Scholar]
- 60.Sun Y, Joyce LE, Dickson NM and Turro C (2010) Efficient DNA photocleavage by [Ru(bpy)2(dppn)]2+ with visible light. Chem. Commun 46, 2426–2428. 10.1039/B925574E. [DOI] [PubMed] [Google Scholar]
- 61.Reichardt C, Monro S, Sobotta FH, Colón KL, Sainuddin T, Stephenson M, Sampson E, Roque J III, Yin H, Brendel JC, Cameron CG, McFarland S and Dietzek B (2019) Predictive Strength of Photophysical Measurements for in Vitro Photobiological Activity in a Series of Ru(II) Polypyridyl Complexes Derived from π-Extended Ligands. Inorg. Chem 58, 3156–3166. 10.1021/acs.inorgchem.8b03223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pazderski L, Pawlak T, Sitkowski J, Kozerski L and Szłyk E (2010) 1$ H NMR assignment corrections and 1$ H, 13$ C, 15$ N NMR coordination shifts structural correlations in Fe(II), Ru(II) and Os(II) cationic complexes with 2,2′-bipyridine and 1,10-phenanthroline. Magn. Reson. Chem 48, 450–457. 10.1002/mrc.2600. [DOI] [PubMed] [Google Scholar]
- 63.Berlman I (1971) Handbook of florescence spectra of Aromatic Molecules. Elsevier Science, Oxford. [Google Scholar]
- 64.Dickerson M, Howerton B, Bae Y and Glazer CE, (2016) Light-sensitive ruthenium complex-loaded cross-linked polymeric nanoassemblies for the treatment of cancer. J. Mater. Chem. B 4, 394–408. 10.1039/C5TB01613D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lindsay D, Garvey CM, Mumenthaler SM and Foo J (2016) Leveraging Hypoxia-Activated Prodrugs to Prevent Drug Resistance in Solid Tumors. PLOS Comput. Biol 12, e1005077. 10.1371/journal.pcbi.1005077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fer ND and Rapisarda A (2014) Exploiting “Hif Addiction” For Cancer Therapy. In Hypoxia and Cancer (Edited by Melillo G) Cancer Drug Discovery and Development, pp. 329–341. Springer New York, New York, NY. 10.1007/978-1-4614-9167-5_14. [DOI] [Google Scholar]
- 67.Pérez-Velázquez J, Gevertz JL, Karolak A and Rejniak KA (2016) Microenvironmental Niches and Sanctuaries: A Route to Acquired Resistance. In Systems Biology of Tumor Microenvironment Vol. Vol. 936 (Edited by Rejniak KA) Advances in Experimental Medicine and Biology, pp. 149–164. Springer International Publishing, Cham. 10.1007/978-3-319-42023-3_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jing X, Yang F, Shao C, Wei K, Xie M, Shen H and Shu Y (2019) Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 18, 157. 10.1186/s12943-019-1089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Casas A, Perotti C, Di Venosa G and Batlle A (2015) Mechanisms of Resistance to Photodynamic Therapy: An Update. In Resistance to Photodynamic Therapy in Cancer Vol. Vol. 5 (Edited by Rapozzi V and Jori G) Resistance to Targeted Anti-Cancer Therapeutics, pp. 29–63. Springer International Publishing, Cham. 10.1007/978-3-319-12730-9_2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.