

Summary
Dysfunction in the parvalbumin (PV) subclass of GABAergic interneurons is implicated in several neurodevelopmental disorders that evolve in severity with postnatal developmental stages. Understanding the molecular underpinnings of the postnatal changes in the function of PV interneurons has been limited by the difficulty in the isolation of pure adult PV interneurons and high-quality RNA. Here, we describe our protocol for the isolation of pure young adult PV interneurons and preparation of high-quality RNA from these cells.
For complete details on the use and execution of this protocol, please refer to Joseph et al. (2021).
Subject areas: Cell isolation, Flow cytometry/mass cytometry, Molecular biology, Neuroscience
Graphical abstract
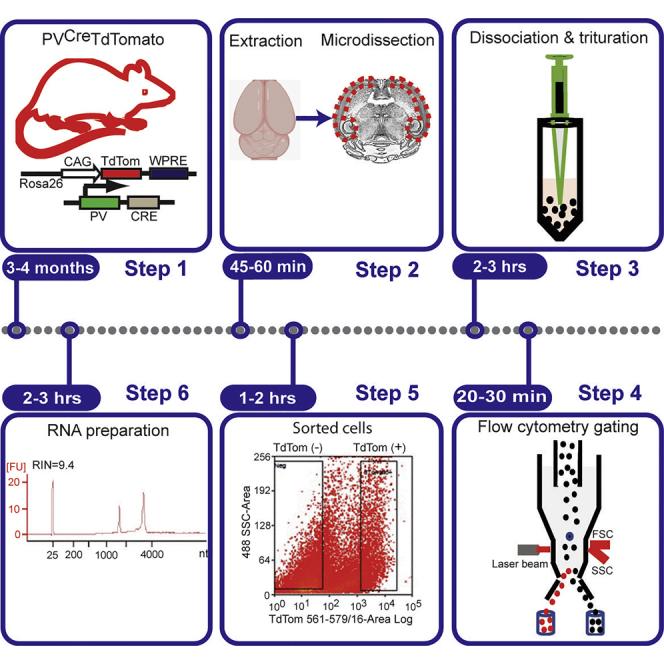
Highlights
-
•
A protocol for isolation of fluorescently labeled young adult parvalbumin interneurons
-
•
FACS sorting of cortical/hippocampal parvalbumin interneurons
-
•
Generation of high-quality RNA for genome-wide sequencing
Dysfunction in the parvalbumin (PV) subclass of GABAergic interneurons is implicated in several neurodevelopmental disorders that evolve in severity with postnatal developmental stages. Understanding the molecular underpinnings of the postnatal changes in the function of PV interneurons has been limited by the difficulty in the isolation of pure adult PV interneurons and high-quality RNA. Here, we describe our protocol for the isolation of pure young adult PV interneurons and preparation of high-quality RNA from these cells.
Before you begin
We have found that various protocols used for the isolation of embryonic and early postnatal interneurons result in low survival of young adult PV interneurons and low-quality RNA. This is likely due to the sensitivity of these young adult interneurons to damage by enzymatic dissociation and mechanical trituration, leading to cell death and low-quality RNA. Therefore, the main improvements in this protocol to increase yield and quality of RNA are the following: preparing acute slices with focus on ensuring viability; using a mild enzymatic and mechanical dissociation; supplementing the dissociating solutions with trehalose, a disaccharide that has been shown to improve cell viability in dissociated neuronal preparations (Saxena et al., 2012).
Experimental considerations
-
1.
This protocol can also be used for isolation of young adult pyramidal cells and other GABAergic interneuron subtypes. Ensure that your core facility has the instruments compatible for fluorescence-activated cell sorting (FACS) and for RNA integrity assessment before starting.
-
2.
To ensure success, perform pilot experiments to improve on speed and to set appropriate conditions for FACS.
-
3.
Wear protective gear (gloves, goggles, masks, lab coat).
Generation of transgenic mice
Timing: 3–4 months
The PVcreTdTomato mouse is now commercially available from the Jackson lab (027395 - C57BL/6-Tg(Pvalb-TdTomato)15Gfng/J). However, we generated our PVcreTdTomato by crossing heterozygous PVCre mice [B6;129P2-Pvalbtm1(cre)Arbr/J; RRID:IMSR_JAX:008069] to homozygous Ai14 mice. If the goal is to compare conditional knock-out (CKO) of a particular gene of interest in PV interneurons to controls, then female PVCreTdTomato double heterozygotes mice can be crossed to male mice with the gene of interest flanked by LoxP sites mice to generate CKO PVCreTdTomato mice and PVCreTdTomato littermate controls. Mice should be genotyped for Cre and TdTomato using end-PCR. We typically use the primers listed in key resources table for Cre and TdTomato (Fulp et al., 2008; Guo et al., 2000). Cre primers should result in gene products of ∼350 bp and the TdTomato primers in gene products of ∼350 bp in PVCreTdTomato mice.
Note: Please make sure to follow all relevant guidelines and regulations set by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by your Institutional Animal Care and Use Committee.
Preparation on the night before the experiment
-
4.
Set up bubbling station near the vibratome: We use refillable compressed gas cylinders attached to an in-wall line served by our building facilities (Figure 1A, left panel). However, if such infrastructure is not available, attach the line from the regulator of the gas cylinder to a Masterflex Manifold Fittings with Female Luer Locks (Cole-Farmer, SK-30600-43; Figure 1A, red arrow to right panel). Each fitting of the Masterflex Manifold can be connected to gas diffusers and inlet solvent filters using proper size tubing. Note that an additional line with small tubing and no bubbler attached is needed to equilibrate small volumes. Make sure the gas cylinder is secured and the regulator is properly fitted.
-
5.
Set an anesthesia induction area near the vibratome: We use a portable isoflurane induction system supplied the Veterinary Department of the Children’s Hospital of Philadelphia. This system is connected to an induction chamber and isoflurane is delivered through a vaporizer with 100% O2 as a carrier gas. However, if such mobile anesthesia induction system is not available, attach a line from the regulator of a 100% O2 gas cylinder to a Vaporizer. Connect the vaporizer to an induction chamber and a line from the chamber to a fume hood to allow for scavenging of excess anesthetic.
-
6.
Make two separate 500 mg/mL trehalose stock solutions (One in EBSS and the other in NB) and store at −20°C. This solution should be used as fresh as possible and not be stored for future preparations.
-
7.
Make sure gas cylinders (O2 and mixed O2/CO2) have enough volume to complete the whole procedure.
-
8.
Prepare ice dissection trays (2) by filling the chamber outside the 35 mm Sylgard dish with water and freeze at −20°C. See materials and equipment section below for detailed instructions on how to construct dissection trays.
-
9.
Prepare cutting solution and store overnight at 4°C.
Cutting solution (pH 7.35, 310–315 mOsm), 500 mL
| Reagent | Final concentration (mM) | Amount |
|---|---|---|
| Sucrose | 180 | 30.81 g |
| NaH2PO4 | 1.25 | 0.1 g |
| KCl | 2.5 | 0.1 g |
| Glucose | 11 | 1g |
| NaHCO3 | 26 | 1.1 g |
| NaAscorbate | 5 | 0.5g |
| NaPyruvate | 3 | 0.17g |
| Thiourea | 2 | 0.08g |
| MgSO4 | 10 | 5 mL of 1M stock |
| CaCl2 | 0.5 | 250μL of 1M stock |
| ddH2O | n/a | Add to 500 mL |
| Total | n/a | 500 mL |
CRITICAL: Use the volumetric flask when adding water to final volume. Do not add MgSO4 and CaCl2 to solution until after bubbling with carbogen (95% O2 /5% CO2) for 30 min on the morning of the experiment to saturate cutting solution.
Figure 1.

Bubbling station and initial setup for brain slicing
(A) A secured refillable compressed 95 O2/5 CO2 mixed gas tank attached to an in-wall line (Left panel). Right panel shows the various ports at the end of the in-wall line and the connected glass diffusers used to bubble microdissection and cutting solutions. Red arrow in the right panel points to a Masterflex Manifold Fittings setup that can be used as an alternative to an in-wall line.
(B) Cutting solution with ice crystals on the walls of the bottle bubbling on ice.
(C) Photograph of the vibratome setup for the maintenance of a cold cutting environment. Ice is tightly packed in the accessible spaces between the buffer chamber (Dashed arrow) and the vibratome ice tray (Solid arrow). Scale bars: 20 mm.
Preparation on the morning of the experiment:
-
10.
Get the mice from the animal facility and hold them in a cage with bedding, food, and water until anesthesia induction.
-
11.
Saturate cutting solution with oxygen for ∼ 25–30 min by bubbling with carbogen (95% O2 /5% CO2) then add drugs as suggested above in the critical section (Figure 1A, right panel). Place the saturated solution in a −80°C or −20°C freezer until ice crystals start to form on the surface of the solution and walls of the bottle. This should take about ∼30 min at −80°C and ∼50 min at −20°C.
-
12.
Fill a bucket with ice and place it next to the bubbling station. Take out the cutting solution out of the freezer and shake it or break ice chunks into a slushy and place the bottle on the ice with constant bubbling using the glass diffuser (See Figure 1B). Place ice around the vibratome slicing chamber to maintain a cold cutting environment (Figure 1C).
-
13.
Prepare microdissection solution.
Microdissection solution (pH 7.35, 310–315 mOsm), 500 mL
| Reagent | Final concentration (mM) | Amount |
|---|---|---|
| NaCl | 115 | 3.36 g |
| KCl | 3.5 | 0.26 g |
| NaH2PO4 | 1.25 | 0.1 g |
| NaHCO3 | 26 | 1.1 g |
| Glucose | 11 | 1.98 g |
| NaAscorbate | 5 | 0.5g |
| NaPyruvate | 3 | 0.17g |
| Thiourea | 2 | 0.08g |
| CaCl2 | 2.5 | 1.25 mL of 1M stock |
| MgSO4 | 1.0 | 0.5 mL of 1M stock |
| ddH2O | n/a | Add to 500 mL |
| Total | n/a | 500mL |
Note: All brain extraction solutions should be made as fresh as possible and not be stored for future use. We also recommend using water of high purity to avoid trace metals and other contaminants that can compromise slice health. Dedicate a set of glassware for fresh tissue preparations and make sure that glassware used, at least the very time, are thoroughly cleaned by rinsing with dilute (0.1N) nitric acid followed by copious amounts of pure water.
-
14.
Turn on the hybridization oven/incubator and set it at 39°C. When our oven/incubator is set at that temperature, the actual chamber is 37°C according to thermometer reading.
-
15.Prepare solutions from papain kit with few modifications to manufacturer’s instructions (Worthington Biochemical Corporation, see Figure 2 for reagents included in the kit).
-
a.EBSS with bicarbonate and phenol red (vial 1), equilibrate with sterile O2: CO2
-
b.Ovomucoid protease inhibitor (OPI) with BSA (Ovomucoid inhibitor 10 mg with albumin 10 mg, vial 4)Note: Add 32mL EBSS (vial 1) to OPI with BSA (vial 4). Allow contents to dissolve. Aliquot OPI into 5mL and keep at 4°C for up to 6 months.
-
i.Prepare EBSS#1 solution in a 15 mL Falcon tube: 9 mL EBSS from papain kit, and 1 mL of 500 mg/mL trehalose in EBSS
-
ii.Prepare EBSS#1-papain solution: Add 5 mL of EBSS#1 solution to one vial of papain and incubate the vial for 10 min at 37°C or until solution appears clear and then transfer it to 15 mL falcon tube to equilibrate with 95/5 O2CO2 until color turns orangeish (see pH scale included in kit). Bubble above the liquid using a tubing line without inlet solvent filter to avoid spillage.
-
iii.Prepare DNase 1 solution: Add 500μL of regular EBSS to vial 3 (1000 units of DNAse1) and mix gently (AVOID VIGOROUS MIXING). Keep on ice until use.
-
iv.EBSS#2-Papain-DNAse1 solution (20units/mL papain and 0.005% DNAse): Add 250μL of DNAse solution to Papain-EBSS#1 solution for a total of 5.25 mL volume just prior to adding dissected tissue.
-
i.
-
a.
-
16.
Prepare FACS solution and keep it on ice.
FACS sorting solution, 20mL
| Reagent | Final concentration (%) | Amount (mL) |
|---|---|---|
| Neural basal (NB) | n/a | 15 |
| Fetal bovine serum (FBS) | 10 | 2 |
| Bovine serum albumin (BSA; 30%) | 1.5 | 1 |
| Trehalose | 5 | 2 mL of 500mg/mL stock |
| Total | n/a | 20 |
Note: Do not store this solution for future use.
Figure 2.

Components of the papain dissociation system
(A) Photograph of the reagents used in the preparation of enzymatic and dissociation solutions including deoxyribonuclease 1 (5 small glass bottles), papain (5 medium size glass bottles), ovomucoid protease inhibitor (1 large glass bottle), and Earl’s balanced salt solution (1 large plastic media bottle).
(B) Closeup view of these reagents with an already prepared ovomucoid inhibitor-Albumin mixture (Large glass bottle). Scale bars (A) 10 mm and (B) 20 mm.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Papain dissociation system | Worthington | Cat#: LK003150 |
| Chemicals, peptides, and recombinant proteins | ||
| Neurobasal A medium | Thermo Fisher Scientific | Cat#: 12348017 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat#: A3840301 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#: A9576 |
| Trehalose | Sigma-Aldrich | Cat#: 90208 |
| KCl | Sigma Millipore | Cat#: 7447407 |
| NaCl | Sigma Millipore | Cat#: 7647145 |
| Sodium ascorbate | Sigma Millipore | Cat#: A4034 |
| Sodium Pyruvate | Sigma Millipore | Cat#: P8574 |
| Thiourea | Sigma Millipore | Cat#: T8656 |
| Sucrose | Sigma Millipore | Cat#: S7903 |
| Sodium bicarbonate | Sigma Millipore | Cat#: S5761 |
| Sodium Phosphate Monobasic | Sigma Millipore | Cat#: S381 |
| Glucose | Sigma Millipore | Cat#: G7021 |
| Magnesium Sulfate | Sigma Millipore | Cat#: M7506 |
| Calcium Chloride | Sigma Millipore | Cat#: C1016 |
| b Agar | Invitrogen | Cat # 30391-023 |
| b Isoflurane | VetOne | Cat#: V1-502017 |
| b Acetone | Fisher Scientific | Cat#: A18-500 |
| d DEPC-Treated water | Thermo Fisher Scientific | Cat#: AM-9915 |
| d RNA ZAP | Thermo Fisher Scientific | Cat#: AM9780 |
| d TRIzol LS | Thermo Fisher Scientific | Cat#: 10296010 |
| d Isopropanol | VWR | Cat#: 76204-296 |
| d Chloroform | VWR | Cat#: IC0219400290 |
| d Sodium acetate (3M) | Sigma Millipore | Cat#: 71196 |
| d Linear Acrylamide | Thermo Fisher Scientific | Cat#: AM9520 |
| d Ethanol absolute | VWR | Cat#: 71001-754 |
| Oligonucleotides | ||
| Cre (Forw: GGC GCG CGG TCT GGC AGT AAA AAC | Thermo Fisher Scientific | N/A |
| Rev: CCC TGA TCC TGG CAA TTT CGG CTA TA | Thermo Fisher Scientific | N/A |
| TdTom (Forw: CTC TGC TGC CTC CTG GCT TCT | Thermo Fisher Scientific | N/A |
| Rev: CGA GGC GGA TCA CAA GCA ATA | Thermo Fisher Scientific | N/A |
| Experimental models: organisms/strains | ||
| Mouse: Rosa-CAG-LSL-tdTomato-WPRE | The Jackson Laboratory | RRID:IMSR_JAX:007914 |
| Mouse: B6; 129P2-Pvalbtm1 (cre)Arbr/J | The Jackson Laboratory | RRID:IMSR_JAX:008069 |
| Other | ||
| a Vapor pressure osmometer 5520 | Wescor | Cat#: SS-140 |
| a Mettler Toledo SevenEasy pH meter | Sigma Millipore | Cat#: Z654272 |
| a Milli-Q® Water Purification System | Sigma Millipore | Cat#: Z00QSV0US |
| a Solvent inlet filters | Thermo Fisher Scientific | Cat#: A-302 |
| a Gas dispersion tube | Sigma Millipore | Cat#: Z408700 |
| a Storage media bottle (500 mL) | VWR | Cat#: 10754-818 |
| a Storage media bottle (100 mL) | VWR | Cat#: 89000-242 |
| a Volumetric flask (500 mL) | VWR | Cat#: 10545-998 |
| a Beaker (800 mL) | VWR | Cat#: 10754-958 |
| a Magnetic Stirring Bars | VWR | Cat#: 58948-025 |
| a Magnetic Stirrer | VWR | Cat#: 10153-314 |
| a Analytical balance | Sartorius | Cat#: BSA124S |
| a Tubing (1/2” ID) | VWR | Cat#: 89412-478 |
| a Tubing (1/4” ID) | VWR | Cat#: 89412-468 |
| a Tubing (1/8” ID) | VWR | Cat#: 89412-464 |
| a Tubing (1/16” ID) | VWR | Cat#: 89412-462 |
| a Carbogen: 95% O2/5% CO2 | Air gas | Cat#: Z02OX952000033 |
| b Leica VT1200S Vibratome | VWR | Cat#: 76001-018 |
| b Oxygen (100%) | Air gas | Cat#: OX 200 |
| b Isoflurane Vaporizer | VWR | Cat#: 89012-506 |
| b Induction chamber | VetEquip | Cat#: 89012-702 |
| b Micro Dissecting scissors | Roboz Surgical Store | Cat#: RS-5914SC |
| b Forceps | Roboz Surgical Store | Cat#: RS-4902 |
| b Standard Scissors | Fine Science Tools | Cat#: 14014-17 |
| b Scalpel Handle | Roboz Surgical Store | Cat#: RS-9843 |
| b Scalpel Blades | Roboz Surgical Store | Cat#: RS-9801-10 |
| b Bone Rongeurs | Roboz Surgical Store | Cat#: RS-8300 |
| b Tissue Forceps | Roboz Surgical Store | Cat#: RS-8102 |
| b Feather Double Edge Blade | Electron Microscopy Sciences | Cat#: 72002-01 |
| b Flat Squared Tip Spatula | Electron Microscopy Sciences | Cat#: 78326-13 |
| b Long Curved Tip Spatula | Electron Microscopy Sciences | Cat#: 78326-12 |
| b Sable Brushes | Ted Pella | Cat#: 11812 |
| b Filter Paper (42.5 mm) | VWR | Cat#: 28460-019 |
| b Filter Paper (9 cm) | VWR | Cat#: 28313-057 |
| b Loctite Super Glue | Office Depot | Cat#: A11364076 |
| b Crystallizing Dishes | VWR | Cat#: 10754-772 |
| b Transfer pipette | VWR | Cat#: 16001-174 |
| b Nylon Mesh | Warner Instruments | Cat#: 13890-046 |
| b Disposable Beakers (100 mL) | VWR | Cat#: 13890-046 |
| b Sylgard | World Precision Instruments | Cat#: SYLG184 |
| b Norit A Charcoal | Sigma Millipore | Cat#: 53663 |
| b Petri Dish (35 mm) | VWR | Cat#: 10799-192 |
| b Petri Dish (150 mm) | VWR | Cat#: 25382-470 |
| b Petri Dish (60 mm) | VWR | Cat#: 25384-328 |
| b Glass petri dish (20 mm) | VWR | Cat#: 75845-524 |
| c Benchtop centrifuge | Thermo Fisher Scientific | Cat#: 75009912 |
| c Falcon Tube (15 mL) | VWR | Cat#: 62406-200 |
| c Falcon Tube (50 mL) | VWR | Cat#: 21008-940 |
| c Serological Pipettes (10 mL) | Thermo Fisher Scientific | Cat#: 07-000-376 |
| c Hybridization Oven/Incubator with Rocker | VWR | Cat#: 97005-252 |
| c Trypan Blue solution | Sigma Millipore | Cat#: T8154 |
| c Hemocytometer | Sigma Millipore | Cat#: Z359629 |
| d Micro-centrifuge | Thermo Fisher Scientific | Cat#: UX-17703-05 |
| d Bioanalyzer 2100 | Agilent | Cat#: G2939BA |
| d MoFlo Astrios Cell Sorter | Beckman Coulter | Cat#: B52102 |
| d Nanodrop 8000 | Thermo Fisher Scientific | Cat#: ND-8000-GL |
| d Eppendorf tube (1.5 mL) | VWR | Cat#: 89166-280 |
For Cutting & Recovery solutions
For Brain Slicing
For Micro-dissection and Dissociation
For FACS and RNA Prep
Materials and equipment
Animal and dissociation reagents
Young adult male PVcreTdTomato mice postnatal days of age 35–40 (P35-40).
Papain kit with:
-
•
Earle's Balanced Salt Solution (EBSS) with bicarbonate and phenol red.
-
•
Papain containing L-cysteine and EDTA.
-
•
Deoxyribonuclease I (DNase).
-
•
Ovomucoid protease inhibitor.
Neurobasal (NB) A medium without phenol red.
Trehalose (500 mg/mL stock in EBBS and NB).
Bovine Serum Albumin (BSA) solution (30%).
Fetal bovine serum (FBS), heat inactivated.
Note: We used male mice because of our general interest in the role of the X linked Aristaless related homeobox gene in the functional maintenance of maturing GABAergic interneurons, but female mice can also be used for the isolation of PV interneurons using this protocol. While the TdTomato reporter line used in our protocol offers superior brightness, reporters such as mCherry and enhanced GFP amongst others can also be used. In addition, we used NB as sorting and collection buffer since it is the most commonly used medium in neuronal cell culture. Although other types of media have been used successfully in the isolation of embryonic and early postnatal neurons, we strongly recommend prior testing of their efficacy in supporting healthy young adult PV interneurons before a complete run through this protocol. Regardless of the chosen medium, we strongly recommend that it is free of phenol red in order to avoid autofluorescence during FACS sorting.
Materials and Equipment for preparing Cutting and Microdissection solutions
Glassware and tools for preparing solutions: Storage media bottle (2×, 500 and 100 mL) volumetric flasks (500 mL), large beakers (800 mL), glass dispersion tubes, and magnetic stirrer and bars, solvent inlet filter 1/16” OD, carbogen (95 O2/5 CO2 mixed gas), tubing (½” ¼, 1/8, and 1/16 inner diameters).
Vapor Pressure Osmometer. It must be calibrated frequently, and the thermocouple must be cleaned regularly in order to obtain accurate measurements.
Analytical balance
pH meter: Must be strictly dedicated for fixatives-free usage.
Ultrapure Water: We use the Milli-Q high purity water system from Sigma-Millipore, but other commercially available water systems provide water of comparable purity.
Materials and Equipment for brain slicing
Surgical tools: Standard scissors, micro-dissecting scissors, fine forceps, tissue forceps, long curved tip spatula, flat squared tip spatula, scalpel handle and #10 blades, plastic transfer pipette, Loctite super glue, filter paper (9 cm and 42.5 mm diameter), glass petri dish (20 mm), double-edge razor blade, and acetone.
Agar block: Prepare 4% agar block to be used as support in the back of the mounted brain during slicing. We typically prepare it by adding 8 g agar to 200 mL Milli-Q-water and microwave until agar is completely dissolved. Pour it in a 150 mm petri dish and allow it to cure into a block at RT (21°C–23°C) or at 4°C. The agar block can be stored at 4°C for up two months.
Ice tray for brain extraction and microdissection: We use a custom-made ice tray with an inner dish with darkened Sylgard to aid with the microdissection (See Figure 3A).
Note: Sylgard dishes are commercially available at Living Systems Instrumentations (Cat#: DD-50-S-BLK), but we prepare our own by thoroughly mixing 300g of liquid A and 30g of liquid B of the Sylgard 184 kit. To darken sylgard, we add to 2g of Norit A Charcoal Powder to the mixture and thoroughly mix. The darkened mixture is then poured into a 35mm Petri dishes to about a 1/3 to ½ full and allowed to cure in a dust-free area (e.g., Fume hood) for at least 24 h prior to using. During the pouring process, bubbles will inevitably form, and it is important to get rid of them. This can be done by using a small butane torch above the dish but take care not to overheat Sylgard.
Figure 3.

Custom-made ice tray and multi-slice holder
(A) The ice tray consists of a 35 mm dish filled up to ½ with Sylgard glued to the center of a 60 mm dish. The resultant outer zone of the 60 mm dish is then filled with water and the whole contraption is kept in the -20C until use.
(B) The multi-slice holder was constructed first by cutting the middle third of the wall of two 100 mL Polypropylene disposable beakers, which were then inserted into one another with a Nylon mesh in between. Three screws were attached to the side of the chamber for height and weight in order to prevent slice holder from floating. Scale bars: 20 mm.
Anesthesia induction: Vaporizer, pure oxygen gas, induction chamber, isoflurane.
Note: For anesthesia induction, we use isoflurane (1–2%) delivered via 100% Oxygen, but other anesthesia cocktails (e.g., mixture of 100 mg/kg ketamine and 10 mg/kg xylazine) can be used. Please make sure that the anesthetic cocktail of choice is approved by your institutional care committee.
Vibratome and multi-slice incubation chamber.
Note: We use the Leica VT1200S, but healthy slices can be obtained by using other models when optimally calibrated. Also, we use a custom-made multi-slice holder placed in a crystallization dish as a slice incubation chamber (See Figure 3B). However, the commercially available Brain Slice Keeper-4 quad (BSK- 4, AutoMate Scientific) and the Brain/Tissue slice system (BSC-PC, cat#: 65–0076; Warner Instruments) incubation chambers are reliable alternatives.
Materials and Equipment for microdissection and tissue dissociation
Tools & equipment: Scalpel with blade, sable brush, ice tray, tabletop centrifuge, and hybridization oven/incubator with a rocker platform.
Trituration: Falcon tubes (15 and 50 mL) and individually wrapped serological pipettes (10 mL).
Materials and Equipment for FACS and RNA Preparation
Reagents: RNAse ZAP, DEPC-treated water, Ethanol (100% and 75% in DEPC-treated water), isopropanol, chloroform, and TRIzol LS, low bind Eppendorf tubes (1.5 mL), linear acrylamide.
Equipment: Micro-centrifuge, Nanodrop 8000 and Bioanalyzer 2100, FACS machine.
Note: Please use personal protective equipment when handling the reagents as they may be quite hazardous. Also, we use the MoFlo Astrios (Beckman Coulter), but similar results can be obtained with other cell sorters using proper settings.
Step-by-step method details
Slicing and microdissection
-
1.
Fill another bucket with ice and pour ∼80 mL of the slushy cutting solution in a 100 mL storage bottle and place it in the ice bucket along with a tissue forceps and a pair of standard and micro-dissecting scissors (See Figure 4A for setup). Fill the custom-made or commercial incubation chamber with micro-dissecting solution and place it on ice next to the bubbling cutting solution. Keep this solution saturated using a solvent inlet bubbler line.
Note: We generally do not perform transcardial perfusion prior to brain extraction, but it is a common practice that may prove to be beneficial especially if this protocol is applied to aged mice. In such scenario, perfusion can be done manually using a 30mL syringe filled with approximately 25mL of slushy solution. This perfusion syringe should be prepared in advance and kept on ice until processing (See Figure 4A).
-
2.
Place a double-edge razor blade in a 20 mm glass petri dish with acetone for 5 min. Scrape residual glue off the blade and rinse with copious amount of water. Mount the blade on the vibratome (See Figure 1C) and set up the optimal slicing settings for your machine. We cut 350μm-thick slices using an advancing speed of 0.1–0.15 mm/s and 1 mm amplitude to obtain healthy slices with the Leica VT-1200, but slices of similar quality can be obtained using other models, providing that proper settings are used.
-
3.
Move the ice bucket with the cutting solution containing syringe and 100 mL storage bottle as well as tools to the dissection area. Remove the ice tray from the freezer and rinse the inner dish containing the Sylgard in order to dispose ice from accidental water spillage. Now add the slushy cold cutting solution from the 100 mL storage bottle to the Sylgard dish and bubble it with the inlet solvent diffuser in the dissection area.
-
4.
Place the mouse in the induction chamber and turn on oxygen tank and vaporizer filled with isoflurane (1–2%) and wait until limb and eye reflexes have disappeared. Note that approved anesthetic cocktails can be used instead of isoflurane (i.e., intraperitoneal injection of the ketamine–xylazine mixture). When preparing slices from two animals (e.g., a control and an experimental) at the same time, we recommend processing each animal separately from anesthesia to brain extraction (See step 7). We do not recommend the processing of more than two animals in the same preparation. Also, a second ice tray is required to keep the brains from the animals apart in order to avoid mix up.
-
5.
Decapitate the animal using the standard scissors. Place the head in the slushy cutting solution in the ice tray and cut the skin longitudinally along the skull median line and laterally as well using the micro-dissecting scissors in order to fully expose the skull.
-
6.
Cut the skull bone longitudinally along the median line, advancing from back to front with extreme care in order to prevent damage to the brain with the scissors tip. Make one last transverse cut along the nasal bone, then use the bone rongeurs to pull the skull top from the brain.
-
7.
Gently scoop the brain using a long-curved tip spatula and cut out the cerebellum and olfactory bulbs while in the slushy bubbling solution (Figures 4B and 4C). Allow the brain to recover for 2–3 min in the ice tray while bubbling using the solvent inlet filter line. At this point, it is possible to begin the brain extraction process for another animal beginning with the anesthesia induction in step 4.
-
8.
Place a 42.5 mm round filter on the back of the glass petri dish and dampen it with some cold cutting solution. Gently place the brain on the soaked filter paper using a flat squared tip spatula. Remove excess cutting solution using a transfer pipette or 2–3 pieces of filter paper sequentially.
-
9.
Apply a dab of glue on the surface of the specimen holder in front of the agar block (Figure 4D) and use a strip of filter paper to lift the brain from the back of the petri dish and place it on the glue. Make sure the back of the brain is in contact with the agar block. Note that lifting the brain with the filter paper strip can be done by first folding ¼ inch of the filter to create an L shape strip and touch the surface of the brain with the smaller folded portion to lift it and place the ventral side of the brain on the glue. This approach allows for the slicing of both brain hemispheres in the transverse plane. If you are processing two brains, we recommend mounting the brains in a line to mitigate the risk of mixing the slices as they come off the blade. This arrangement, however, prolongs the slicing time as opposed to mounting side-by-side.
Note: we generally place a 4% agar block on the specimen holder prior to mounting the brain in order to prevent advancing force of the blade from dislodging it during slicing (See Figure 4D). The brain is mounted between the block and the advancing blade holder. We strongly recommend using such agar block as it serves an important role in generating healthy slices.
-
10.
Place the specimen holder with brain attached to the stage chamber and quickly fill it with ice-slushy cutting solution (Figure 5A). Set up a cutting window if your vibratome allows for it as it significantly reduces the cutting time. Remove the inlet solvent bubbler from the ice tray and place it in the stage chamber as far away as possible from the cutting blade (Figure 5A) and cut the tip of a transfer pipette to create an opening large enough to capture the slices as they come off the blade. Cut 350μm-thick transverse slices. If you are slicing two brains, take the necessary care to prevent mixing them (See steps 4 and 9 for tips).
-
11.
Carefully pick up each slice as they come off the blade using the wide mouth transfer pipette and place the slices inside the bubbling incubation chamber sitting on ice (Figure 5B, pictured off ice for clarity). If you are slicing two brains, make sure to have a dedicated commercial (See note in materials and equipment/brain slicing for suggestions) or custom (Figure 3B) incubation chamber for each brain.
-
12.
Move the slices to an ice tray with bubbling recovery solution for microdissection under a microscope immediately after collecting the last slice from the vibratome (Figure 5C).
-
13.
Adjust the optical settings on the dissection microscope in order to have clear view of the cortex and hippocampus (Figure 5D). Using a sable brush to gently hold the slice in place, cut the cortex and hippocampus from the subcortical brain structures of both hemispheres with a scalpel blade (Figure 5E). Mince tissue into chunks as small as possible and collect the hippocampal/cortical tissue chunks along with some microdissection solution in a 15 mL Falcon tube and proceed to incubation.
Figure 4.

Brain extraction and mounting setup
(A) Ice bucket containing the dissection tools, perfusion syringe with slushy cutting solution, and a 100 mL storage bottle with slushy cutting solution as well.
(B) An extracted brain in an ice tray containing bubbling cutting solution and the arrow points to the inlet solvent filter used for bubbling. This brain was perfused with slushy cutting solution prior to extraction in order to obtain a high-quality image.
(C) Image of the brain following removal of the cerebellum and olfactory bulbs.
(D) Specimen holder with a glued agar block used a bumper support to prevent dislodging of the brain during slicing. Scale bars: (Main panels) 20 mm and (Insets) 5 mm.
Figure 5.

Slicing and microdissection setup
(A) Final view of the slicing setup with the mounted on the specimen holder and submerged in bubbling slushy cutting solution.
(B) Slices in bubbling recovering or microdissection solution. The slices are generally kept on ice, but they were briefly taken off the ice for the picture.
(C) Final set up for microdissection with the slices in an ice tray with bubbling microdissection solution, microscope, and an additional light source on the left.
(D) A close up view of the transverse or horizontal slices prior to microdissection of the cortex and hippocampus.
(E) A close up view of the slices following microdissection of the cortex (Dashed arrow) and hippocampus (Solid arrow). Scale bars: (Main panels) 20 mm and (Insets) 5 mm.
Incubation
-
14.
Remove microdissection solution from 15 mL falcon tube with minced cortical and hippocampal tissue chunks using a transfer pipette and quickly add ∼2.6 mL of EBSS#1-papain-DNase mixture to the Falcon tube.
-
15.
Displace the air in vial with a carbogen tubing line with no inlet solvent filter attached at room temperature (RT; 21°C–23°C) for 2–3 min and immediately cap the vial. Do not bubble gas through the solution.
-
16.
Seal the cap of the Falcon tube with parafilm and wrap it in foil. Tape the sealed and wrapped Falcon tube to a rocking platform in the hybridization oven/incubator and incubate for ∼90 min at 37⁰C.
-
17.In the meantime, prepare the following:
-
a.Set a centrifuge for 15 mL tubes to RT (21°C–23°C) for short spins.
-
b.Precoat Eppendorf LoBind Microcentrifuge or 15 mL Falcon tubes for collecting FACS cells by filling them with 30% BSA incubate at RT (21°C–23°C) until after final resuspension. We generally collect our FACS cells in a 15 mL falcon tube.
-
c.EBSS#2 (RESUSPENSION) solution in a 50 mL Falcon tube: 9.6 mL EBSS + 700μL OPI + 500μL DNAse 1, and 1.2cc 50% Trehalose in EBSS for a total volume of 12 mL for each brain and place on ice.
-
d.OPI discontinuous density gradient medium: 5 mL OPI in EBSS + 0.6 mL of 50% Trehalose in EBSS for a total volume 5.6 mL per brain and place on ice.
-
a.
Trituration
-
18.
Triturate the mixture 8–10 times using a 10 mL serological pipette. Let the non-dissociated chunks settle for a couple minutes and collect the cloudy suspension for centrifugation in a 15 mL Falcon tube. This is important for FACS efficiency.
-
19.
Centrifuge the suspension at 300g(∼1800 RPM) for 5 min at RT (21–23°).
-
20.
Discard the supernatant and gently re-suspend the pellet in 3 mL of mixed EBSS#2 solution by trituration with a 1 mL micropipette while avoiding air bubbles.
-
21.
Slowly and gently layer the cell suspension to the OPI discontinuous density gradient medium previously prepared and do not agitate. The cell suspension can be transferred to the OPI medium by slowly expelling it from the transfer pipette along the wall of the Falcon tube. This step is critical for a cell suspension with minimal cellular debris.
-
22.
Centrifuge at 70 g (∼800 RPM) for 6 min at RT (21–23°).
-
23.
Discard the supernatant and re-suspend the pellet 2 mL of FACS solution prepared in the morning using a 1 mL micropipette.
-
24.
Count cells: Take 50 μL of cell suspension and add it to 250μL of 0.4% trypan blue. Mix well and mount on hemocytometer. Count the live, unstained cells (Live cells do not take up trypan blue) in 4 corner squares on the hemocytometer. Take the average cell count and multiply by 10,000 (1×104). Multiply by five to correct for the 1:5 dilution from the trypan blue addition. The final value is the number of viable cells/mL in the original cell suspension.
FACS sorting
-
25.
Prepare FACS collection tubes: Aspirate BSA from 15 mL Falcon tubes or the Eppendorf LoBind Microcentrifuge tubes and add the desired solution for collecting FACS cells (e.g., 0.1–2 mL FACS solution or 750μL TRIzol LS/250μL sorted volume). Note that we generally collect our cells in a 15 mL Falcon tube with a starting volume of 2 mL FACS solution.
-
26.
Confirm that the flow cytometer tubing was rinsed with one run of RNase free dH2O, then bleach followed by RNase free dH2O.
-
27.
Confirm that the nozzle size is set up to ensure smooth passage of neurons. We normally use a 100 μm nozzle with a sheath pressure of 45 psi. We generally excite TdTomato with an argon-ion laser (488 nm) and detect using emission spectra between 564 nm and 606 nm. Finally, limit sorting to 60–75 min per sample.
-
28.
Confirm the sorting will be conducted at slow speed to prevent damage to neurons.
-
29.Take a few cells at the beginning of sorting and observe under a fluorescent microscope to ensure cells are:
-
a.Well dissociated and viable.
-
b.Tomato red positive.
-
c.Use proper forward (FSC) and side scatter (SSC) properties to exclude debris and doublets (Figure 6).
-
d.check the intensity of the fluorescence and report it.
-
e.Gating can be adjusted at this stage to ensure that fluorescence positive cells alone are collected.
-
a.
-
30.
Set up a timer at the start of sorting and collect TdTomato+ cells in either Eppendorf micro-centrifuge or 15 mL Falcon tubes with FACS solution or TRIzol LS. If you choose to collect directly in TRIzol LS, limit your collection volume to 250μL as noted above in step 25 because TRIzol LS allows at most a 30% sample volume for optimal RNA isolation as per manufacturer’s instructions (ThermoFisher Scientific). More importantly, restrict total sorting time to 1–2 h to prevent RNA degradation.
-
31.
After sorting, spin the cells at 140 g (∼1300 RPM) for 10 min at 4°C and discard the supernatant. Cells pellet can be kept at −80°C for until RNA preparation at a later time point. While freezing cells in TRIzol LS at −80°C for up to a month is generally tolerable, we recommend immediate RNA extraction from cells collected in such solution in order to maximize recovery.
Figure 6.

Fluorescence activated cell sorting of PV interneurons with high TdTomato expression
FACS plots from two representative preparations of dissociated neuronal suspension showing modest (A) and high (B) yield of TdTom+ cells. (I) Total neuronal population in both preparations were selected by forward and side scatter profiles. (II) Fluorescence gates were established by comparing TdTomato positive and negative cells. (III) TdTomato positive cells were selected for RNA extraction. (IV) Histograms of fluorescence emission of sorted TdTomato positive cells versus event number.
RNA extraction and purification
We use the TRIzol LS method for RNA extraction and purification with few modifications from the manufacturer’s instructions (ThermoFisher Scientific), but other methods such as the Invitrogen purelink kit can be used with modifications to obtain high quality RNA (Saxena et al., 2012).
-
32.Preparation:
-
a.Reagents: Isopropanol, Chloroform, TRIzol LS, RNase Zap, linear acrylamide, DEPC-treated water, sodium acetate (3M), 200 proof ethanol.
-
b.75% ethanol in DEPC-treated water on ice.
-
c.Turn on centrifuge and set temperature to 4°C
-
d.Take out linear acrylamide out of the freezer and keep at RT.
-
e.Set up water bath to 37°C.
-
f.Clean and wipe tools, working area, and tubes with RNase Zap.
-
a.
-
33.
Thaw your FACS sample on ice. If you have a frozen pellet of FACS cells, add 1 mL of TRIzol LS while thawing. Homogenize sample by intermittent vortexing for 2 min and then by pipetting up and down (10×).
-
34.
Incubate the homogenized sample at room temp for 5 min to permit complete dissociation of the nucleoprotein complex.
-
35.
Add 267μL of chloroform per 1 mL of TRIzol LS reagent used for homogenization. Cap the tube securely and shake vigorously by hand for 15 s.
-
36.
Incubate for 15 min at RT (21°–23°) and then spin a centrifuge at 12,000 g for 15 min at 4°C.
-
37.
Remove the aqueous phase by decanting into a clean new tube. Avoid the organic phase as it contains mostly DNA and, thus, will affect the quality of the extracted RNA.
-
38.
Add 2μL of linear acrylamide to the aqueous phase and gently mix by inverting the tube.
-
39.
Add 666μL of 100% isopropanol per 1 mL TRIzol LS used for homogenization and gently mix by inverting the tube for 15 s.
-
40.
Incubate at RT (21°–23°) for 10 min and then spin at 12,000 g for 15 min at 4°C.
-
41.
Remove the supernatant without disturbing the RNA pellet and resuspend the pellet in 100μL of DEPC-treated water.
-
42.
Add 200μL of 100% ethanol and 10μL of 3M sodium acetate to the resuspended RNA. Briefly Vortex to mix the solution and store in a −20°C freezer for at least 1 h.
-
43.
Spin at 14000 g for 30 min at 4°C and discard the supernatant.
-
44.
Wash the RNA pellet with 1 mL of 75% cold ethanol (Made with DEPC-treated water) by gently inverting the tube.
-
45.
Spin at 14000 g for 10 min at 4°C and discard the supernatant. Air dry the pellet for about 5 min to allow residual ethanol to evaporate. Do not allow pellet to over dry as that tends to interfere with solubility.
-
46.
Resuspend pellet in ∼25μL DEPC-treated water and obtain a rough estimate of RNA yield using a nanodrop or fluorescent based methods (Qubit for example). Accurate yield and quality of RNA can be readily obtained using a bioanalyzer 2100 with a Pico/Nano chip. At this point RNA sample can be stored at −80°C until use for RNAseq or other experimental plans.
Expected outcomes
PV interneurons/TdTomato+ cells density and FACS
Recent advances in Cre recombination technologies have led to the development of fluorescently labeled GABAergic interneuron subtypes, paving the way for the identification and collection of individual subtype of GABAergic interneurons for molecular profiling (Taniguchi et al., 2011). Significant progress has been made in the molecular profiling of embryonic and early postnatal GABAergic interneurons, partly due to the relative ease in which these immature neurons can be efficiently isolated. Evidently, the complex nature of the extracellular matrix and sensitivity to enzymatic treatment have hampered the isolation of fluorescently labeled adult GABAergic. While emergent studies have described various approaches to overcome the technical barriers limiting the isolation of sufficient healthy GABAergic neurons from mature adult and aging animals (Hrvatin et al., 2018; Mardinly et al., 2016; Paul et al., 2017; Tasic et al., 2016; Zeisel et al., 2015), we ran into difficulties in applying any particular published method when isolating sufficient young adult PV interneurons for the extraction of high integrity RNA. Here, we describe a protocol that leads to highly purified young adult PV interneurons from the PVcreTdTomato mouse. Using the papain dissociation enzyme, protective slice preparation, and gentle mechanical dissociation, we typically sort on average more than 670,000 TdTomato+ PV interneurons per brain (n=36 mice) by using proper FACS gating procedures (Figure 6).
RNA quality
High quality RNA is a limiting factor for genome-wide sequencing and other sequencing method. From our experience, the amount and quality of RNA appears to be dependent on yield, suggesting that RNAse accumulation as a result of excessive cell death affects the quality of RNA extracted from the surviving FACS of young adult PV interneurons. We typically recover on average 2.5 μg/μL of RNA from sorted PV interneurons in each brain. While the integrity of RNA varies from preparation to preparation, we routinely measure RIN scores in the range of 7–9 (Figure 7).
Figure 7.

Total RNA samples from Sorted TdTomato positive interneurons show high integrity scores
(A) RNA samples from eleven different preparations were ran on an Agilent 2100 Bioanalyzer: L, Ladder/marker; A-K denote samples 1–11.
(B–L) The fluorescence plots or electropherograms of samples 1–11, showing lower marker, 18S, and 28S peaks in sequential order.
(M) RIN scores and RNA concentration for the 11 samples. Note that sample 11 or K was our very first preparation and the low RIN was likely due very low TdTomato+ cell yield and unhealthy sorted cells.
Limitations
This protocol is designed for isolation of PV interneurons from freshly dissociated brains from young adult mice (P35-40). While it represents a good starting point for aged mice, modifications may be needed in order to obtain enough viable PV interneurons for high quality RNA. N-Methyl-D-glucamine-based (NMDG) cutting/recovery solution has been described as an improved approach to prepare healthy slices from aged animals (Ting et al., 2014) and should be considered for animals older than P35-40. In addition, alternatives to enzymatic dissociation may need to be considered in addition to modifications to density gradient filtration as the older tissue may lead to higher amount of cellular debris, which can interfere with the flow sorting and RNA quality.
Another limitation is the amount of time required to perform this protocol. As noted in the step-by-step instructions, FACS of healthy young adult PV interneurons requires meticulous processing with careful trituration and density gradient filtration to remove cellular debris and clumps that might otherwise clog the nozzle or impede sorting. Although manual sorting (Paul et al., 2017) circumvents many of these issues, it is limited by the number of cells that can be collected and by the inherent subconscious bias of the selector. However, it must be noted that FACS represents a more non-bias approach for isolating cells for the comprehensive cataloging of their cellular profile. The time-consuming nature of this process and the many centrifugation steps along with sorting itself could contribute to the resultant transcriptomic or proteomic profile of these cells. For that purpose, we highly recommend that control and experimental animals are processed simultaneously.
Troubleshooting
Problem 1
Incomplete dissociation of tissue or dissociation takes too long (steps 16 and 18).
Potential solution
If this problem occurs, it is likely that the enzyme-tissue ratio is off or enzymatic digestion is not effective enough.
In the case of tissue-enzyme ratio, it is important to follow manufacturer’s instruction in regard to the amount of enzyme to use per mg of tissue. In addition, we advise using fresh batch of enzymes or adjust to higher concentrations should incomplete dissociation persists. Finally, consider limiting the time enzymes are kept at RT (21°–23°) or 37°C before the dissociation to maximize activity.
Problem 2
Excessive amounts of debris in cell suspension (steps 21 and 24).
Potential solution
This problem could be due to incomplete enzymatic and/or mechanical disruption or technical issue with OPI gradient.
Due to sensitivity of young adult PV interneurons, we recommend minor adjustment to the trituration steps by adding slightly more mechanical disruption. This should be done by pipetting up and down a bit more with a 10 mL serological pipette rather than using pipettes with smaller openings. We did not use a cell strainer, but the use of proper cell strainer size (∼70 μm or larger) might be a viable option to reduce debris. Note that, in this case, cell suspension should be added gently to the strainer with a 10 mL pipette to reduce the amount of debris seeping through. Smaller mesh size is likely to produce cleaner suspension, but the yield is likely to be lower. Finally, we recommend that cell suspension be layered gently to the OPI discontinuous density gradient solution to prevent complete mixture and excessive dense cellular debris in the cellular fraction upon centrifugation.
Problem 3
Low cell yield in suspension (step 24).
Potential solution
PV interneurons represent a small percentage of cells in the cortical/hippocampal suspension. Therefore, it is important to have a high cell yield in the suspension in order for enough PV interneurons to be sorted in a timely manner. Some potential reasons for low cell yield in suspension includes issues with slicing/microdissection solutions, slow preparation of slices, incomplete digestion, damaged cells due to prolonged enzymatic dissociation, and aggressive trituration leading to excessive cell death.
We recommend several practice sessions in order to perform all the steps outlined in the protocol in a precise and timely manner. Ideally, brain extraction should take 45 s or less. Mounting of the brain on the vibratome and slicing should be completed in about 20 min. Also, it is critical that the cutting and recovery solutions are properly made (pH measures at 7.3–7.4 and osmolarity at 310–315mOm). Optimize the enzymatic dissociation by using higher enzyme concentrations if the digestion takes too long or lower concentrations if cells are too sensitive. To reduce the impact of prolonged exposure of cells to papain, equal amount of EBSS#2 or FACS solution can be added to the enzyme solution to dilute its concentration just prior to trituration. We elected to triturate without diluting the enzymatic solution because of the mild nature of papain, but we cannot rule out the possibility that it could have improved our preparations even more. Mechanical disruption is important for the dissociation of cells from tissue chunks, but it will also damage cells, so find the proper balance between mechanical disruption force and cell yield. We recommend slightly prolonged trituration with the low mechanical force using a 10 mL serological pipette to achieve that balance. Work to minimize the time spent between steps in the protocol from anesthesia induction to the preparation of RNA from isolated PV interneurons. If slicing continues to be a point of delay, consider transcardial perfusion of the anesthetized mouse with cold cutting solution prior to brain extraction. This approach can decrease body temperature, thereby reducing hypoxic damage to the brain prior to extraction. Finally, we recommend regular confirmation of cell viability at every step between digestion and homogenization.
Problem 4
Low RNA yield (step 46).
Potential solution
This problem could be due to low cell yield, excessive cell death, and RNase contamination.
See potential solutions for problems 1–3 to increase cell yield. Excessive cell death could lead to the release of RNAse in cell suspension, leading to the isolation of degraded RNA. We routinely generate cell suspension with less than 2% of dead or dying cells. Finally, ensure that your RNA workstation and pipettes are cleaned with RNAse ZAP and dedicated to RNA use only. Given that RNAse ZAP contains a fair amount of detergent, we do not recommend its use in the dissection area nor on tools use for the preparation of acute slices for electrophysiological recordings.
Problem 5
Low RIN Values (<7) (step 46).
Potential solution
While a RIN value of 7 is reasonable for a lot of applications, we recommend RIN values in the range of 8–9 for RNAseq. The problem with low RIN is likely due to degradation of RNA as a result of RNase contamination.
As mentioned in the solution to problem 4, ensure RNase-free techniques are employed throughout the protocol by using a bench and equipment dedicated to RNA use only. It is important that bacterial DNA preps are not carried out in the RNA area as RNAse is commonly used at high concentrations in such preps. In addition, change gloves frequently, use filter pipette tips, and make all solutions using RNase-free water. Finally, always keep samples and solutions on ice unless otherwise stated.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Eric D. Marsh (marshe@chop.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code.
Acknowledgments
The authors thank Dr. Florin Tuluc from the Flow Cytometry Core at the Children’s Hospital of Philadelphia. We are also grateful for advice and suggestions from members of the Marsh laboratory during the conduct of this research. This work was supported by the National Institute of Neurological Disorders and Stroke grant (Grant #: RO1 NS082761 to E.D.M., 2018); The Stiftung zur Förderung der Medizinischen Forschung der Christian-Albrechts-Universität zu Kiel (Fellowship grant to M.V.D., 2017); the National Institute of Child Health and Human Development (Grant #: 5U54HD086984 to the Institutional Intellectual Developmental Disabilities Research Center Cellular Neuroscience Core at the Children’s Hospital of Philadelphia, 2019); and the Epilepsy Foundation (Postdoctoral fellowship #: 367394 to D.J.J., 2015).
Author contributions
Conceptualization, E.D.M., M.V.D., and D.J.J.; methodology, E.D.M., D.J.J, Y.H., A.G.C., and M.V.D.; investigation, D.J.J., Y.H., M.V.D., A.J.M., and R.R.; writing – original draft, D.J.J. and E.D.M.; writing – review & editing, A.G.C., A.J.M., and M.V.D.; visualization, D.J.J., A.G.C., and A.J.M.; supervision, E.D.M.; funding acquisition, E.D.M., M.V.D., and D.J.J.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Donald J. Joseph, Email: josephd1@chop.edu.
Eric D. Marsh, Email: marshe@chop.edu.
References
- Fulp C.T., Cho G., Marsh E.D., Nasrallah I.M., Labosky P.A., Golden J.A. Identification of Arx transcriptional targets in the developing basal forebrain. Hum. Mol. Genet. 2008;17:3740–3760. doi: 10.1093/hmg/ddn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H., Mao C., Jin X.L., Wang H., Tu Y.T., Avasthi P.P., Li Y. Cre-mediated cerebellum- and hippocampus-restricted gene mutation in mouse brain. Biochem. Biophys. Res. Commun. 2000;269:149–154. doi: 10.1006/bbrc.2000.2263. [DOI] [PubMed] [Google Scholar]
- Hrvatin S., Hochbaum D.R., Nagy M.A., Cicconet M., Robertson K., Cheadle L., Zilionis R., Ratner A., Borges-Monroy R., Klein A.M. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat. Neurosci. 2018;21:120–129. doi: 10.1038/s41593-017-0029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph D.J., Von Deimling M., Hasegawa Y., Cristancho A.G., Ahrens-Nicklas R.C., Rogers S.L., Risbud R., McCoy A.J., Marsh E.D. Postnatal Arx transcriptional activity regulates functional properties of PV interneurons. iScience. 2021;24:101999. doi: 10.1016/j.isci.2020.101999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly A.R., Spiegel I., Patrizi A., Centofante E., Bazinet J.E., Tzeng C.P., Mandel-Brehm C., Harmin D.A., Adesnik H., Fagiolini M., Greenberg M.E. Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature. 2016;531:371–375. doi: 10.1038/nature17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul A., Crow M., Raudales R., He M., Gillis J., Huang Z.J. transcriptional architecture of synaptic communication delineates GABAergic neuron identity. Cell. 2017;171:522–539 e520. doi: 10.1016/j.cell.2017.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A., Wagatsuma A., Noro Y., Kuji T., Asaka-Oba A., Watahiki A., Gurnot C., Fagiolini M., Hensch T.K., Carninci P. Trehalose-enhanced isolation of neuronal sub-types from adult mouse brain. Biotechniques. 2012;52:381–385. doi: 10.2144/0000113878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H., He M., Wu P., Kim S., Paik R., Sugino K., Kvitsiani D., Fu Y., Lu J., Lin Y. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B., Menon V., Nguyen T.N., Kim T.K., Jarsky T., Yao Z., Levi B., Gray L.T., Sorensen S.A., Dolbeare T. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19:335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting J.T., Daigle T.L., Chen Q., Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol. 2014;1183:221–242. doi: 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A., Munoz-Manchado A.B., Codeluppi S., Lonnerberg P., La Manno G., Jureus A., Marques S., Munguba H., He L., Betsholtz C. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code.