

SUMMARY
Metastasis is a complex and poorly understood process. In pancreatic cancer, loss of the transforming growth factor (TGF)-β/BMP effector SMAD4 is correlated with changes in altered histopathological transitions, metastatic disease, and poor prognosis. In this study, we use isogenic cancer cell lines to identify SMAD4 regulated genes that contribute to the development of metastatic colonization. We perform an in vivo screen identifying FOSL1 as both a SMAD4 target and sufficient to drive colonization to the lung. The targeting of these genes early in treatment may provide a therapeutic benefit.
Graphical abstract
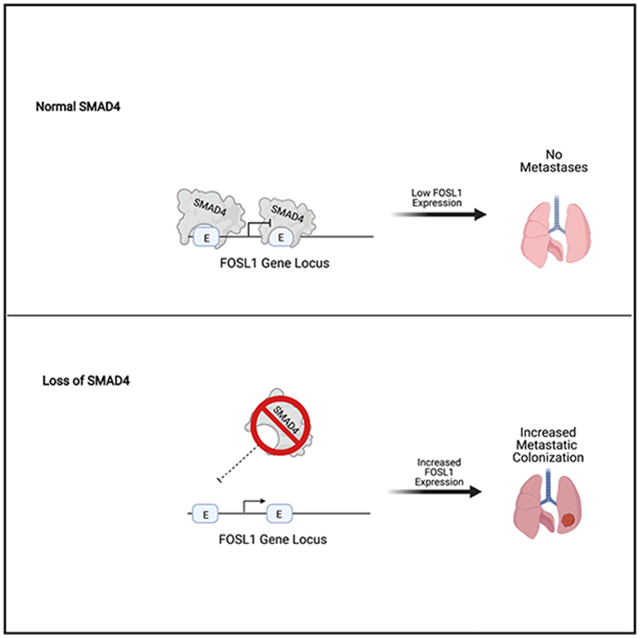
In brief
Loss of SMAD4 is associated with poor outcome in pancreatic cancer. Using an in vivo, isogenic metastatic colonization assay, Dai et al. identified SMAD4-regulated genes that affect metastasis but not primary tumor growth. FOSL1 is a SMAD4-regulated gene that is necessary and sufficient to drive metastatic colonization.
INTRODUCTION
Survival following a pancreas cancer diagnosis is poor (Gordon-Dseagu et al., 2018; Maisonneuve, 2019; McGuigan et al., 2018; Siegel et al., 2019), in part due to the late stage at which this malignancy is diagnosed and its profound resistance to current therapies. Indeed, fewer than 20% of pancreatic ductal adenocarcinoma (PDAC) cases are confined to the pancreas at the time of diagnosis (Siegel et al., 2019), and the molecular mechanisms that drive dissemination remain incompletely understood. Genome characterization studies of primary tumors have defined the mutational landscape of PDAC: activating mutations of the KRAS oncogene occur in most PDAC cases and are accompanied by loss of tumor suppressor genes, including CDKN2A, TP53, and SMAD4 (Raphael et al., 2017; Waddell et al., 2015; Witkiewicz et al., 2015).
Clinical studies have linked SMAD4 loss, which occurs in 55% of patients (Blackford et al., 2009; Hahn et al., 1996), with increased metastatic burden and worse prognosis in PDAC (Blackford et al., 2009; Iacobuzio-Donahue et al., 2009; Oshima et al., 2013) and other cancers (Ding et al., 2011; Miyaki et al., 1999; Xue et al., 2014). Within pancreatic cancer patients at 5 years, the rate of synchronous local and distant failure rate was 14.9% in the SMAD4-deficient cohort compared to 5.3% within the functional SMAD4 cohort (Herman et al., 2018). In genetically engineered mouse models, KRASG12D initiates tumorigenesis, and the additional loss of SMAD4 induces distinct histopathological progression (Bardeesy et al., 2006; Izeradjene et al., 2007; Kojima et al., 2007). Furthermore, (Bardeesy et al., 2006) showed that 6 of 16 mice showed evidence of liver metastases in the Pdx1-Cre LSL-KrasG12D Smad4lox/lox Ink4a/Arflox/+ mouse model of pancreatic cancer. In addition, SMAD4 loss is associated with changes in epithelial-to-mesenchymal transition (EMT) status (Ioannou et al., 2018; Wang et al., 2019) and extracellular matrix remodeling (Bardeesy et al., 2006; Klein-Scory et al., 2007); however, the mechanisms by which SMAD4 loss fosters aggressive disease have not been defined.
Mechanistically, SMAD family proteins act downstream of transforming growth factor (TGF)-β and BMP signaling (Massagué and Chen, 2000). The binding of ligands to TGF-β and BMP receptors triggers the phosphorylation of receptor SMADs (SMAD2/3) that, in turn, induce recruitment of SMAD4 to SMAD2/3 (Moustakas and Heldin, 2009). This complex then translocates to the nucleus and regulates gene transcription (Chen et al., 2005; Feng and Derynck, 2005). SMAD4 loss thus disrupts TGF-β and BMP signaling (Grau et al., 1997). The importance of TGF-β signaling in metastasis is underscored by the decrease of liver metastases with the loss of TGF-β in the KPC mouse model (Zhong et al., 2017). However, it remains unclear either in what stage of metastasis that loss of SMAD4 participates or the gene program that facilitates metastatic disease. To shed light on details of SMAD4 function in this context, we identified SMAD4-regulated genes that drive metastatic colonization to the lung by performing an in vivo screen in a panel of SMAD4 isogenic PDAC cell lines. In this study, we identify the increased expression of FOSL1 as a direct driver of SMAD4 loss-mediated metastatic colonization.
RESULTS
SMAD4 loss does not affect cell proliferation rate in vitro or in vivo
To interrogate the role of SMAD4 in pancreatic cancer progression, we created SMAD4 isogenic cell line pairs in the SMAD4 wild-type (WT) PDAC cell lines, PANC1 and PATU8902, by disrupting SMAD4 using CRISPR-Cas9 genome editing. In SMAD4 null HPAC and PANC0327 cells, we reconstituted SMAD4 expression by lentiviral-mediated stable integration of a V5 epitope-tagged SMAD4 cDNA. We confirmed the deletion and expression of SMAD4 in each of these cell lines by immunoblotting (Figure 1A) and verified ectopic expression of SMAD4 in HPAC and PANC0327 cells (Data S1A).
Figure 1. Generation and characterization of SMAD4 isogenic PDA cell lines.

(A) Immunoblot of SMAD4 gene deletion in SMAD4-WT PDAC cell lines (PANC1 and PATU8902) and SMAD4 re-expression in SMAD4 null PDAC cell lines (HPAC and PANC0327).
(B) qRT-PCR of SERPINE1 mRNA expression in SMAD4 isogenic PDAC cell lines treated for 24 h with 10 ng/mL TGF-β. Mean ± SD of three biological replicates is shown.
(C) Longitudinal quantification of tumor volume in mice orthotopically implanted with LacZ- or SMAD4-expressing HPAC cells with bioluminescence imaging (BLI) (red indicates HPAC SMAD4; blue indicates HPAC LacZ). Total flux was measured weekly during 42 days (n = 8 for each genotype; standard deviation is reported in error bars).
(D) Ultrasound reported tumor volume in mice orthotopically implanted with LacZ- or SMAD4-expressing HPAC cells at 42 days after injection (n = 8 for each cohort; standard deviation is reported in error bars)
(E) Progression-free survival of SMAD4-WT (blue) and SMAD4 mutant or SMAD4 null (red) TCGA pancreatic cancer primary resected tumors (p = 0.0179); see Table S1.
(F) Quantification of tumor area (percentage of tumor area per lung lobe) in lungs of immunodeficient mice i.v. inoculated with SMAD4 isogenic pancreatic cancer cells; each lobe is presented as an independent data point. n = 3 or 4 mice (t test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001); see Table S2.
(G) Longitudinal quantification of BLI of mice tail vein inoculated with LacZ- or SMAD4-expressing HPAC cells with BLI (red indicates HPAC SMAD4; blue indicates HPAC LacZ; t test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n = 7 for each cohort).
(H) Representative immunofluorescent images of mice i.v. inoculated with LacZ- or SMAD4-expressing HPAC cells at 12 weeks following inoculation. SMAD4 cells are stained in yellow (scale bars, 100 μm).
To confirm the functional status of SMAD4 in these isogenic cells, we measured expression of a benchmark target gene SERPINE1 (Samarakoon et al., 2009) using qRT-PCR. As expected, we found that SMAD4 deletion significantly (p < 0.0001) diminished SERPINE1 activation in response to TGF-β treatment in PANC1 and PATU8902 cells. Conversely, SMAD4 re-expression significantly (p < 0.0001) increased TGF-β-induced SERPINE1 activation in HPAC and PANC0327 cells (Figure 1B). Furthermore, we failed to observe an effect on the phosphorylation of other SMAD family members with the loss of SMAD4 in the PATU8902 cell line and a slight increase in expression and phosphorylation of SMAD1 with the overexpression of SMAD4 in the HPAC cell line as determined by immunoblotting (Data S1B).
To test for effects of SMAD4 levels on cell proliferation, we measured population doublings of these PDAC isogenic cell lines (Data S1C) as well as tumor growth kinetics following subcutaneous implantation of cells in immunodeficient mice (Data S1D). We also implanted HPAC SMAD4 overexpression lines orthotopically into murine pancreas and found that the resulting tumors grew at a similar rate during the course of 42 days (Figures 1C and 1D). Notably, these tumors also showed no obvious histological differences. We found that manipulation of SMAD4 levels did not alter cell proliferation rates in cell culture or affect the growth of primary tumors grown subcutaneously or in the pancreas. Taken together, these observations demonstrate that despite modifying the expression of a known SMAD4 regulated gene, SERPINE1, the expression of SMAD4 in the selected PDAC cell lines did not affect either in vitro proliferation or the in vivo growth of primary tumors.
SMAD4 plays a role in promoting metastatic disease
To better understand the clinical impact of SMAD4 loss in PDAC, we analyzed The Cancer Genome Atlas (TCGA) PDAC (Raphael et al., 2017) dataset of primary resected cancers and compared survival rates of patients whose tumors expressed or lacked SMAD4. Specifically, we integrated data from copy number alteration, mutation status, and mRNA expression to group the patients into SMAD4-WT (n = 114) and SMAD4-altered (n = 71) (Table S1). We confirmed that patients whose tumors harbored WT SMAD4 in the primary tumor experienced longer progression-free survival (p = 0.0179) (Figure 1E).
We then assessed whether SMAD4 modulates PDAC progression in an experimental model of metastatic colonization. Specifically, we tested the ability of SMAD4 isogenic cells to colonize the lung after intravenous (i.v.) injection in immunodeficient mice. Lungs were collected 12 weeks post-injection or when mice displayed respiratory difficulty. Dissected lungs were stained with hematoxylin and eosin, and the percentage of tumor area per lung lobe was quantified. Deletion of SMAD4 in PANC1 cells showed a significant increase with guide RNA #1 (p < 0.05) and a substantial increase in tumor area in the lungs with guide RNA #2 (Figure 1F) compared to a guide targeting luciferase, and deletion of SMAD4 by both guides in PATU8902 cells led to significantly increased metastatic colonization (p < 0.01). Conversely, restoring SMAD4 expression in the SMAD4 null PDAC lines potently abrogated metastatic colonization and outgrowth of tumor cells in the mouse lung. In this model, HPAC cells reconstituted with V5-luciferase efficiently metastasized to the lung, occupying >30% of the lung at the endpoint, whereas SMAD4 overexpression led to a striking suppression of lung metastatic colonization (Figure 1F). This reduction in the ability to metastasize was also seen when SMAD4 was overexpressed in the PANC0327 line. Taken together, these studies showed that loss of SMAD4 is associated with both an increase in number of lesions in the lung as well as an increase in the median size of these lesions. In contrast, we found that increased expression of SMAD4 led to lung lesions that are smaller and less abundant (Table S2).
Given the complex nature of colonization, we sought to understand whether SMAD4 overexpression blocked the initial colonization of the lung or blocked the outgrowth of the lesion once present in the lung. Due to the fact that through bioluminescence imaging (BLI) of the lung we identified an initial signal from SMAD4 overexpression cells versus LacZ controls followed by a stable signal, we hypothesized that there was a defect in metastatic outgrowth (Figure 1G). We used immunofluorescence to identify SMAD4-expressing cells in the lung from HPAC cells expressing SMAD4 or LacZ. We found SMAD4-expressing cells in the lung lesions, indicating that SMAD4 expression did not affect initial colonization but instead repressed the ability of these cells to grow in the lung (Figure 1H).
In addition, we performed a competition assay by creating a 1:1 mixture of LacZ- and SMAD4-expressing HPAC cells and then (1) co-culturing this mixture in vitro, (2) implanting the mixture subcutaneously, or (3) injecting the mixture i.v. in vivo. Consistent with our previous observation that SMAD4 re-expression did not alter in vitro proliferation and primary tumor growth kinetics, we found that the relative ratio of LacZ- and SMAD4-expressing HPAC cells remained stable after 4 weeks of in vitro co-culture or after subcutaneous co-implantation. In stark contrast, we found that 4 weeks after i.v. co-injection, the metastatic tumors in the lung consist of almost exclusively LacZ-expressing cells (Data S2A). These observations demonstrate that SMAD4 expression does not affect cell proliferation but is a potent suppressor of PDAC metastatic colonization.
Identification of SMAD4-regulated genes that regulate metastatic colonization
SMAD4 is known to regulate transcription downstream of TGF-β- and BMP-related signaling pathways. To better understand the role of SMAD-mediated gene transcription in metastasis, we used a multi-component integrative RNA sequencing (RNA-seq) strategy. To identify genes regulated by SMAD4, we interrogated in vitro and in vivo transcription profiles of the SMAD4 isogenic lines. Specifically, we performed RNA-seq on samples from two categories: (1) SMAD4 isogenic PDAC cell pairs treated with or without TGF-β in vitro, and (2) SMAD4-WT and SMAD4 null tumors (Figure 2A).
Figure 2. Gene overexpression metastatic colonization screen.

(A) Schematic of RNA sequencing (RNA-seq) conditions. RNA-seq was performed on SMAD4 isogenic pancreatic cell lines treated with or without TGF-β and tumors formed by SMAD4 isogenic pancreatic cells. The heatmap summarizes significantly differentially regulated gene expression through multi-component RNA-seq analysis; see Table S3.
(B) A volcano plot depicting the GO groups enriched in the up (positive) and down (negative) SMAD4-related genes. Red dots signify metastasis-related gene ontologies.
(C) Schematic of ORF library in vivo metastasis screen. ORF library transduced SMAD4-expressing HPAC cells are split into one of three arms, i.e., (1) pre-injection T0 cell pellet, (2) in vitro proliferation for six population doublings, and (3) i.v. inoculation into immunodeficient mice, to screen for genes that promote metastatic colonization to the lung, which is completely suppressed in the SMAD4-expressing HPAC cell line.
(D) Longitudinal BLI in the chest of mice following i.v. inoculation of ORF library-transduced SMAD4-expressing HPAC cells (n = 6, standard deviation is reported in error bars).
(E) Summary of in vivo pooled screen. The x axis shows penetrance, which was calculated as (times each ORF was more than 5% of total tumor reads)/(number of mice in which the ORF was assessed). They axis shows maximum enrichment, which was calculated as (maximum percentage of ORF in any metastatic tumor) – (percentage of the ORF in pre-injection cell pellet). The same five negative control ORFs were included in each sub-pool and were not enriched in the metastatic tumors.
For tumors, we performed RNA-seq on (1) subcutaneous tumors formed by isogenic control LacZ- or SMAD4-expressing HPAC cells, (2) matched subcutaneous tumors formed from SMAD4-WT PATU8902 cells expressing a doxycycline-inducible shSMAD4 short hairpin RNA (shRNA) or a corresponding seed control shRNA (Buehler et al., 2012), and (3) micro-dissected tumors from lung tissue harboring metastatic tumor formed from PATU8902 cells that were either SMAD4-WT or SMAD4-deleted. From these in vitro and in vivo conditions, we used a threshold of p < 0.05 and a fold change greater than 1.5 to classify genes as SMAD4 target genes.
From this analysis, we identified 135 genes that were upregulated and 254 genes that were downregulated by SMAD4 expression (Table S3). This gene list includes benchmark SMAD4 transcription targets, such as SERPINE1 (Samarakoon et al., 2009), TGFBI (Chiao et al., 1999), ADAM19 (Chan et al., 2008), ID1-3 (Kang et al., 2003), as well as many other putative SMAD4 gene targets. Gene Ontology (GO) analysis revealed an enrichment for processes and pathways with known roles in metastasis (noted in red), including cell migration/motility and extracellular matrix remodeling (Figure 2B).
SMAD4-regulated drivers of metastatic colonization
We hypothesized that SMAD4 suppresses metastasis through transcriptional modulation of the identified target genes. In principle, SMAD4 may either upregulate a set of metastasis-suppressive genes that normally inhibit cellular processes involved in tumor seeding and inhibit growth at secondary sites or downregulate genes that normally promote such processes. To evaluate this hypothesis, we designed a lentiviral cDNA (open reading frame [ORF]) expression library composed of the SMAD4-downregulated genes. We introduced these ORFs into the SMAD4-expressing HPAC cell line, a model in which the metastatic phenotype is completely suppressed by SMAD4 re-expression, to identify genes promoting the metastatic phenotype.
We divided the candidate gene list into three barcoded ORF sub-pools. Each sub-pool contained the same five negative control ORFs (Table S4; sub-pools 1–3 contained 90, 85, and 65 candidate ORFs). We transduced the SMAD4-expressing HPAC cells with each sub-pool in six independent infection replicate experiments. As outlined in Figure 2C, we then performed an in vivo ORF screen to identify individual genes that promoted the formation of metastatic foci in the lung. Specifically, following puromycin selection and in vitro culture, we divided the transduced cells into three groups: (1) a pre-injection sample, (2) an in vitro proliferation group of 1 × 106 cells from each infection replicate further cultured for six population doublings, and (3) an in vivo group of 1 × 106 cells from each pool injected (i.v.) into immunodeficient mice.
To gauge the rate of metastatic tumor growth of the barcoded ORF library-transduced cells, we included LacZ- and SMAD4-expressing HPAC cells as positive and negative controls. All mice were imaged weekly to monitor tumor development in the lung. As expected, unmanipulated SMAD4-expressing HPAC cells failed to form metastatic tumors in the lung (Figure 2D). We found that SMAD4-expressing cells transduced with the sub-libraries exhibited increased signal in lungs over time, albeit at a much slower rate compared to LacZ-expressing HPAC cells, which readily colonize and continue to form metastatic tumors in the lung. This observation suggested that a subset of target genes in each pool promoted a metastatic phenotype. To identify these genes, at 11 weeks post-injection, the pooled in vivo screening cohorts were euthanized, and genomic DNA was extracted from entire lungs. Barcodes associated with each ORF were quantified by massively parallel sequencing after PCR amplification (STAR Methods). In parallel, we sequenced the pre-injection cell sample and the cells remaining at the endpoint of the in vitro proliferation.
In the in vitro proliferation cell samples, the ratio of ORFs remained tightly correlated among biological replicates (average N×N Pearson correlation = 0.997, n = 6 for each sub-group; Data S2B). Similarly, samples derived from the pre-injection cells also showed an equal representation of ORFs. Taken together, these results confirm that there was no bias in the representation of genes due to proliferation effects. Most metastatic tumors contained a small number of highly represented ORFs, as is evident in the strong shifts in the cumulative distribution of ORFs between the initial (T0) time point and lung metastatic lesions (Data S2C). In the lung metastasis samples, the representation of ORFs showed an average N×N Pearson correlation of 0.607 (n = 5 for pool 1 and pool 3, and n = 6 for pool 2), indicating potential drivers of metastasis.
From these experiments, we identified 37 ORFs representing 36 genes that exhibited representation of more than 5% in mice from which the lung metastatic tumors were collected. Certain ORFs, such as LCN2, were found enriched in all mice assessed; other ORFs were less penetrant but significantly enriched in some but not all mice. We considered 11 ORFs that exhibited more than 40% penetrance and more than 10% maximum enrichment to be strong drivers of metastatic colonization (Figure 2E). The metastatic phenotype induced by these candidate genes was in stark contrast to the negative control ORFs that were never detected at more than 0.3% or enriched more than Δ% = −0.1% (enrichment was calculated by taking the % in metastatic tumor minus the % in pre-injection cell pellet for each barcode].
Among these 11 candidate genes we focused on MAGEA3, NTRK2, LCN2, and FOSL1 for downstream validation based on prior data that suggested involvement in metastasis in other cancers. We first determined whether the expression of these candidates correlated with SMAD4 expression. In the isogenic LacZ- and SMAD4-expressing HPAC cell lines, we found that MAGEA3, NTRK2, LCN2, and FOSL1 were, as expected, transcriptionally downregulated by SMAD4 after 8 h of TGF-β treatment. MAGEA3 and FOSL1 were also significantly (p < 0.001) downregulated in xenograft tumors formed from SMAD4-expressing HPAC cells compared to tumors formed from LacZ-expressing control HPAC cells (Figure 3A).
Figure 3. Arrayed validation of candidate genes from the pooled metastasis screen.

(A) (Left) qRT-PCR of LacZ- and SMAD4-expressing HPAC cells treated 8 h with TGF-β showing downregulation of MAGEA3, NTRK2, and LCN2 mRNA by SMAD4, and TGF-β-induced SMAD4-dependent downregulation of FOSL1 mRNA. (Right) qRT-PCR of LacZ- and SMAD4-expressing HPAC xenograft tumors showing downregulation of MAGEA3 and FOSL1 expression. Mean of three biological replicates is shown (t test, **p < 0.01, ***p < 0.001; standard deviation is reported in error bars)
(B) Overexpression of “hit” genes in HPAC-SMAD4 cells. All ORFs are V5-tagged except for NTRK2, which is immunoblotted with TRKB antibody.
(C) Waterfall plot and statistical summary of chest BLI at 10 weeks following i.v. inoculation (t test, *p < 0.05, **p < 0.01, ***p < 0.001).
(D) 2D cell culture growth rate of HPAC SMAD4 overexpression cells with overexpressed NTRK2, LCN2, FOSL1, MAGEA3, or GFP control. Time points were taken every 24 h during the course of 4 days. Cell count is reported (n = 3, error bars reflect standard deviation).
(E) Gene correlation graphs correlation of the hit genes NTRK2 (Pearson’s r = 0.37, p = 7.45e—7), MAGEA3 (Pearson’s r = 0.02, p = 0.762), LCN2 (Pearson’s r = −0.44, p = 2.12e—9), and FOSL1 (Pearson’sr= −0.48, p = 4.15e—11) in TCGA pan-cancer pancreatic cancer cohort with SMAD4. SMAD4 copy number status is indicated by point color (blue indicates homozygous deletion, light blue indicates heterozygous deletion, gray indicates diploid, and light red indicates gain).
(F) Kaplan-Meier overall survival analysis of TCGA Pancreatic Cancer cohort split by top third (red) and bottom third (blue) of FOSL1 expression (log-rank p = 0.0052).
To validate the metastasis-promoting potential of these four candidates, we generated individual stable luciferase-expressing HPAC-SMAD4 lines that also overexpress each of the ORFs (Figure 3B) and injected each i.v. into immunodeficient mice and performed weekly BLI. Cells expressing FOSL1, LCN2, and MAGEA3 developed metastatic tumors in the lung with high penetrance and at a rate comparable to the control LacZ-expressing HPAC line (Figure 3C). NTRK2 promoted metastatic colonization in a subset of mice but failed to reach statistical significance. In contrast, negative control ORFs including GFP or hcRed did not promote metastasis of the SMAD4-expressing HPAC cells. Importantly we next identified if the overexpression of each gene led to increased proliferation. With the expression of each gene individually we identified either no difference in in vitro growth rate (FOSL1 and MAGEA3) compared to GFP control or significantly reduced growth rate (NTRK2 and LCN2) (Figure 3D). In a pooled population containing all overexpressed genes we identified either slight (MAGEA3) or non-significant (FOSL1, NTRK2, and LCN2) increases in growth rate compared to GFP control (Extended Data 2D).
To determine whether LCN2, MAGEA4, FOSL1, and NTRK2 were involved in a shared pathway, we performed a STRING-db analysis and found a network consisting of LCN2, FOSL1, and NTRK2 (Extended Data 2E). This finding suggests these three proteins work in concert to lead to lung colonization.
We further analyzed the pancreatic cancer TCGA cohort (Raphael et al., 2017) for correlations between the expression of each target gene and SMAD4 expression/copy number. We found a significant inverse correlation between SMAD4 (expression and copy number) and FOSL1 as well as LCN2 expression (Figure 3E). In addition, high FOSL1 expression levels were associated with worse overall patient survival (p = 0.0052) (Figure 3F). We next analyzed publicly available SMAD4 chromatin immunoprecipitation sequencing (ChIP-seq) data and interrogated SMAD4 binding at the gene locus of each of the target genes (Cistrome Data Browser, cistrome.org/db). We failed to identify significant SMAD4 binding at the MAGEA3 or NTRK2 gene loci. However, we confirmed that SMAD4 binds to the enhancer (as defined by the consensus ENCODE H3K27ac signal) and promoter (5 kb upstream of the transcriptional start site) regions surrounding FOSL1 and LCN2 (Data S3). These analyses support direct regulation of FOSL1 and LCN2 by SMAD4.
SMAD4 directly regulates FOSL1 expression
Since FOSL1 was the strongest candidate that was downregulated in both the in vitro and in vivo expression experiments, was validated in the metastasis-promoting assay, and was negatively correlated with SMAD4 in patient samples, we focused mechanistic studies on FOSL1.
To confirm the functional relevance of FOSL1, we suppressed FOSL1 in the luciferase-labeled SMAD4-low line, HPAC, and identified changes in lung colonization after tail vein injection. Using two distinct shRNAs targeting FOSL1, we observed a reduction in the number of mice that expressed metastatic lesions compared to mice harboring tumors expressing the scrambled control. Specifically, we found luciferase signals in 6 of 10 mice in the control group and 1 of 10 mice (p = 0.057) and 0 of 10 (p = 0.0108) mice expressing tumors harboring FOSL1-specific shRNAs (Figure 4A).
Figure 4. SMAD4 suppresses FOSL1 through recruitment to and modulation of a FOSL1 enhancer.

(A) Representative bioluminescent signal images of mice (n = 10 for each cohort) 12 weeks after tail vein inoculation with HPAC cell lines with sh-scramble, sh-FOSL1 hairpin 1, or sh-FOSL1 hairpin 2.
(B) Immunoblot showing reduced FOSL1 expression in xenograft tumors formed from SMAD4-expressing HPAC cells compared to those formed from LacZ-expressing HPAC cells.
(C) qRT-PCR of FOSL1 in PANC1 cells with SMAD4 gene deletion treated 24 h with TGF-β. Mean of three biological replicates is shown (t test, **p < 0.01; standard deviation is reported in error bars)
(D) ChIP qPCR on FOSL1 promoter, enhancer, and gene body in LacZ- and SMAD4-expressing HPAC cells treated with TGF-β. Mean of four (for ChIP/SMAD4) or five (for ChIP/H3K27Ac) biological replicates is shown (standard deviation is reported in error bars).
(E) ChIP qPCR on FOSL1 promoter, enhancer, and gene body in PANC1 cells with SMAD4 gene deletion treated with TGF-β. Mean of three (for ChIP/SMAD4) or six (for ChIP/H3K27Ac) biological replicates is shown (standard deviation is reported in error bars).
Supporting the hypothesis that SMAD4 directly binds the gene locus and regulates FOSL1 levels, we observed reduced FOSL1 protein expression in xenograft tumors formed from SMAD4-re-constituted HPAC cells compared to LacZ control tumor (Figure 4B). Moreover, FOSL1 mRNA expression is downregulated by TGF-β treatment in SMAD4-WT PANC1 cells and is restored following CRISPR-Cas9-mediated SMAD4 gene deletion (Figure 4C).
To better understand SMAD4 recruitment at the FOSL1 genomic locus, we ectopically expressed LacZ or SMAD4 in the SMAD4 null HPAC cells and performed SMAD4 ChIP-qPCR following TGF-β treatment. Specifically, we examined binding to the FOSL1 promoter, enhancer, and gene body, as well as an alpha satellite repeat (ASR) region at centromeric heterochromatin. We found that in SMAD4 reconstituted HPAC cells, SMAD4 is primarily bound to the FOSL1 enhancer region, and this binding is augmented by TGF-β treatment (Figure 4D, upper panel). In contrast, qPCR with primers specific to the FOSL1 promoter and gene body, as well as the negative control ASR region, showed minimal SMAD4 enrichment at these loci (Figure 4D, upper panel). To confirm an active epigenetic signature at the FOSL1 enhancer in these conditions, we used ChIP-qPCR to measure chromatin binding of acetylated histone H3 Lys27 (H3K27ac) at the gene regions described above. We found that the FOSL1 enhancer activity is reduced with TGF-β treatment in the SMAD4-expressing HPAC cells (Figure 4D, lower panel).
In a reciprocal experiment using SMAD4-WT PANC1 cells, we found that SMAD4 bound to the FOSL1 enhancer at a low basal level. TGF-β treatment greatly increased SMAD4 binding at the FOSL1 enhancer, accompanied by reduced binding of AcH3K27 (Figure 4E). The reduction of FOSL1 enhancer activity by TGF-β was dependent on functional SMAD4, as SMAD4 gene deletion in PANC1 cells reversed this phenotype. Collectively, these observations indicate that SMAD4 acts at a FOSL1 enhancer region to mediate TGF-β-dependent FOSL1 transcription repression by modulating FOSL1 enhancer activity.
To identify potential downstream targets of FOSL1 that are disrupted by SMAD4 suppression, we utilized TCGA PDAC dataset to identify genes that were significantly correlated with expression of FOSL1 and negatively correlated with SMAD4 expression (Data S4A). We then placed these genes into a String-db network to identify convergent pathways and found an extensive protein network focused on Rho/Rac signaling proteins (Data S4B). Furthermore, the network contained genes that scored in the lung colonization screen (boxed in blue), indicating that SMAD4 loss may mediate colonization of the lung through direct activation of FOSL1, in turn regulating a number of Rho/Rac-associated proteins.
DISCUSSION
Metastasis is a complex, multi-step process. TGF-β has been shown to play a critical role in a number of steps in the metastatic cascade, including evasion of immune surveillance (Gorelik and Flavell, 2001), EMT (Miettinen et al., 1994; Oft et al., 1996), and colonization of the lung (Padua et al., 2008). SMAD4 has been shown to be the effector protein in many of these TGF-β-driven processes, but the mechanisms by which TGF-β and SMAD4 regulate the metastatic program remain incompletely defined.
In this study, we provide evidence of a SMAD4-mediated transcriptional mechanism that regulates metastatic colonization in PDAC. Specifically, we identified SMAD4 target genes that drive metastatic colonization through preferential binding of SMAD4 to enhancer regions. We further showed that a direct SMAD4 transcriptional target is FOSL1, which, when expressed in PDAC cells expressing SMAD4, drives metastatic colonization and subsequent outgrowth. FOSL1 is a member of the FOS family of transcription factors. This AP1-related transcription factor is transcriptionally dysregulated in a subset of T cell leukemias and CD30-positive lymphomas (Chen et al., 2012; Iwai et al., 2001). In addition, FOSL1 expression is upregulated in metastatic lung (Román et al., 2019) and prostate (Luo et al., 2018) cancers and is associated with EMT. Taken together, these observations suggest that dysregulation of FOSL1 contributes to cancer progression in several cell lineages and that in PDAC, loss of SMAD4 leads directly to increased FOSL1 expression.
Prior studies have reported an association between SMAD4 loss, poor prognosis, and a higher likelihood of metastatic disease (Ijichi et al., 2006; Kojima et al., 2007; Shugang et al., 2016). In a genetically engineered mouse model of pancreatic cancer, heterozygous loss of SMAD4 was shown to contribute to metastasis through RUNX3-dependent mechanisms (Whittle et al., 2015). RUNX3 did not score in the screen described herein, indicating that RUNX3 could be important for phenotypes not measured in our assay, which focuses on extravasation and colonization into metastatic sites. Indeed, given the many steps required to fully program metastatic progression, it is likely that SMAD4 loss alters the expression of genes acting at different points in the development of a fully metastatic lesion. Moreover, since we identified 254 genes downregulated by SMAD4, we speculate that these other genes cooperate with RUNX3 and FOSL1 to program the full metastatic phenotype. Another important area of metastasis to explore is the temporal regulation of genes and identification of those genes that are dysregulated early in the metastatic cascade versus later time points. This study is taken at a single endpoint during advanced colonization of the lung. We think that this is an important area of understanding and ultimately treating metastatic disease, but more studies should identify those genes that are dysregulated earlier in the process.
More generally, these findings reinforce the emerging concept that metastatic potential is determined largely by the combination of genetic alterations that initiate the primary tumor and subsequent transcriptional reprogramming through epigenetic mechanisms. Sequencing studies of matched primary tumors and metastatic lesions have shown that primary and metastatic lesions share a common precursor but continue to evolve from each other (Brastianos et al., 2015; Campbell et al., 2010; Makohon-Moore et al., 2017; Yachida et al., 2010) and exhibit clear transcriptional changes.
To improve patient outcomes, both the genomic programs driving tumor initiation and transcriptional modifications driving the progression of the tumor need to be targeted. Specifically, we have shown that the metastatic colonization is driven in part by enhancer reprogramming. Taken together, these findings show a potential therapeutic avenue in treating late-stage pancreatic cancer patients that could drastically improve outcomes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William Hahn (William_Hahn@dfci.harvard.edu).
Materials availability
Plasmids and other reagents generated in this study are available by request to lead contact, William Hahn (William_Hahn@dfci.harvard.edu).
Data and code availability
- The RNA-seq generated during this study will be available on GEO (GSE148248) available as of the date of publication.
- No novel code was written for the analysis of the dataset.
- Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Studies
This research project has been approved by the IACUC at Dana-Farber Cancer Institute (protocol 04-101) and is in compliance with the Animal Welfare Act and the Office of Laboratory Welfare of the NIH. All studies were completed in female CrTac:NCr-Foxn1nu mice, ages 6-8 weeks.
Cell lines
Pancreatic cancer cell lines were obtained from the Cancer Dependency Map (Broad Institute), and Lenti-X 293T cells were obtained from Clontech. Cell lines were routinely screened for mycoplasma throughout the study. PANC1, PATU8902, HPAC, MiaPaCa2, and L3.3 were maintained in DMEM (GIBCO) supplemented with 10% fetal bovine serum (Sigma Aldrich), and PANC0327 was maintained in RPMI-1640 supplemented with 15% fetal bovine serum. All media was supplemented with 2mM glutamine, 100U/mL penicillin, and 100μg/mL streptomycin (GIBCO).
METHOD DETAILS
Generation of isogenic cell lines
Lentivirus was generated in 293T cells by transfecting with VSVG, delta8.9 and ORF, sgRNA, or shRNA plasmids using TransIT (Mirus Bio). Virus was harvested 72 hr. after transfection. Cas9-expressing PANC1 and PATU8902 cells were generated by lentiviral transduction with pLX311-Cas9 and selection in blasticidin. sgCTRL and sgSMAD4 lines were further derived by lentiviral transduction with pXPR003-sgCTRL or −sgSMAD4 and subsequent selection with puromycin. Expression of luciferase or SMAD4 in HPAC and PANC0327 cells were achieved by lentiviral transduction with pLX307-luciferase or pLX307-SMAD4. For bioluminescent imaging, LacZ or SMAD4 were cloned into the FUW-Luc-mCherry-Hygro vector by Gibson Assembly, and parental HPAC cells were transduced with the lentiviral plasmids and selected with hygromycin. Overexpression of experimental ORFs for arrayed validation experiments were achieved by further introducing the pLX317-ORF vectors and subsequent selection with puromycin.
ORF library
Pool1 and Pool2 of the ORF library were arrayed from the pLX317 barcoded human ORFeome V8.1 library (Yang et al., 2011) by the Broad Institute Genetic Perturbation Platform. Pool3 contains the ORFs that were not available from the ORFeome and were cloned in-house. Specifically, we first generated a small array of pLX317 vectors with barcodes that were distinct from the ones in ORFeome V8.1, and gateway-cloned the ORFs into this vector with Phusion high-fidelity DNA polymerase or Q5 high-fidelity DNA Polymerase (New England Biolabs), from either the Harvard PlasmID Repository (Zuo et al., 2007) or a cDNA library generated from HPAC cells.
Pooled in vivo metastasis screen
To titer the virus for each ORF library subpool, 3×106 cells were seeded per well in a 12-well plate and were infected with different amounts of virus (0, 50, 100,150, 200, 400 μL), with a final concentration of 8 μg/mL polybrene. Cells were centrifuged for 2 hours at 2000 rpm at 30°C. Approximately 8 hr. after infection, cells from each infection were seeded into duplicate wells in a 6-well plate. 24 hr. after infection, one well was treated with 2μg/mL of puromycin and one with media alone. After 3 days of selection, cells were counted to determine the amount of virus that resulted in ~25%–30% infection efficiency, and this amount of virus was used in the screen. For the in vivo metastasis screen, 3×106 SMAD4-expressing HPAC cells were infected with each ORF virus pool in 6 replicates with ~25%–30% infection efficiency. 3×106 cells per well were seeded in 12-well plates and were infected with the amount of virus determined during optimization, with a final polybrene concentration of 8 μg/mL. Plates were spun for 2 hr. at 2000 rpm at 30°C. Approximately 8 hr. after infection, each replicate was split into a 10cm dish. 24 hours after infection, cells were selected in 2 μg/mL puromycin for 3 days and expanded in puromycin-free media for 4 days prior to mouse injection. On the day of injection, 1×106 cells from each replicate was saved as a pre-injection cell pellet (termed T0), 1×106 cells used for tail vein injection into 1 female CrTac:NCr-Foxn1nu mouse (termed MET), and 1×106 cells cultured in vitro for another 6 population doublings (termed 6PD). LacZ- and SMAD4-expressing cells were also injected on the same day as positive and negative controls. Injected mice were imaged 4 days post inoculation, and then once weekly to monitor BLI change. Cohorts of ORF-library injected mice were euthanized at week 11 and whole lungs were snap frozen. One mouse each from pool1 and pool3 died prematurely from anesthesia and imaging and were excluded from analysis. Genomic DNA was extracted from the entire lung using the QiaAmp DNA Blood Maxi Kit (QIAGEN). The barcodes corresponding to each ORF were amplified using PCR and analyzed by next-generation sequencing. Enriched barcodes were analyzed as follows: (1) the percentage of each barcode in each T0, 6PD, and MET sample was calculated, (2) enrichment was calculated by taking (% in metastasis sample - % in T0 sample) for each barcode. Barcodes that were significantly presented in a MET sample (>5%) was considered to have scored in that mouse. For each ORF, we consider both the penetrance and the maximum enrichment (Δ%).
Cell proliferation assay
5×104 cells were seeded in 12-well plates in technical triplicates and passaged subconfluent every 3–4 days. Cells were counted at each passage, and number of cell doublings was calculated. Two biological replicates were performed. For the daily assay 5×104 cells were plated into a 12-well plate and counted every 24 hr.
In vivo tumor injections and experimental metastasis
For xenograft tumor growth, 2 million cells resuspended in 200 μL PBS (Clontech) were injected subcutaneously into the bilateral flanks of female CrTac:NCr-Foxn1nu mice (ages 6-8 weeks) (Taconic Laboratories). Tumor growth was monitored by digital caliper measurement and tumor volume calculated using V = (W(2) × L)/2. Mice were euthanized when tumor volume exceeded 2000mm3. For all metastasis experiments, 1×106 cells resuspended in 200μL PBS were injected into the lateral tail vein of female CrTac:NCr-Foxn1nu mice (ages 6-8 weeks) (Taconic Laboratories). Bioluminescent imaging was performed 4 days post injection, and weekly thereafter; BLI signal in Region of Interest (ROI) gated to the chest area is reported.
Competition assay
LacZ- and SMAD4-expressing HPAC cells were each transduced with a barcode vector (pXPR003-sgGFP #3 (BC1), and pXPR003-sgGFP #4(BC2)) at low MOI. Cells were mixed at 1:1 and co-cultured in vitro, or the mixture was implanted subcutaneously or injected IV in CrTac:NCr-Foxn1nu nude mice. 4 weeks following in vitro or in vivo competition, genomic DNA was extracted using QiaAmp DNA Blood Maxi kit (QIAGEN) or DNeasy Blood and Tissue kit, and the relative abundance of barcodes were quantified by Taqman qPCR using TaqMan Universal PCR Master Mix (Life Technologies).
Preparation of samples for Integrative RNA-seq of in vitro and in vivo conditions
RNA-seq was performed on in vitro and in vivo cell and tumor samples. Specifically, we performed RNA-seq on the following in vitro samples from parental and derivative cell lines: (1) PANC1 and PATU8902 cells with SMAD4 gene-deletion via CRISPR-Cas9 using 2 SMAD4-targeting sgRNA or a control sgRNA, and (2) HPAC and PANC0327 cells with restored SMAD4 expression or expression of Luciferase as negative control. Each parental or derivative cell line was treated either with vehicle (1mg/mL BSA, 4mM HCl) or 10ng/mL TGFβ for 24 hr. For in vivo conditions, we performed RNA-seq on the following tumors: (1) matched subcutaneous xenograft tumors from isogenic Lac-Z or SMAD4-expressing HPAC cells harvested at ~300mm3, (2) matched xenograft tumors formed from PATU8902 cells expressing a doxycycline-inducible shSMAD4 shRNA, or its corresponding seed control shRNA (shSMAD4 c7-9), and (3) micro-dissected metastatic tumor from isogenic PATU8902 cells with SMAD4-WT or SMAD4 null genetic status. To obtain matched xenograft tumors formed from PATU8902 cells with dox-inducible shSMAD4, we generated 4 PATU8902 derivative lines, expressing dox-inducible shSMAD4#1, shSMAD4#2, or their corresponding seed control hairpins. Xenograft tumors were first formed in the absence of shRNA knockdown, and when tumors reached ~100mm3, shRNA was induced for 6 days with doxycycline diet (625ppm for 0.6-2 mg/kg in diet) prior to tumor harvest. Snap frozen xenograft tumors were transitioned in RNAlater-Ice (Life Technologies) at −20°C overnight prior to RNA extraction using RNeasy Plus Mini kit (QIAGEN). Metastatic tumors from isogenic PATU8902 cell pairs with SMAD4-WT or SMAD4 null genetic status were micro-dissected from mouse lungs embedded in OCT compound (Tissue Tek), and total RNA was extracted using RNeasy Micro kit (QIAGEN). RNA-seq was performed on 2 biological replicates for each in vitro sample, and 3 biological replicates for each in vivo sample.
RNA sequencing and analysis
First strand cDNA was generated using Oligo(dT)12-18 Primer (Invitrogen) and AffinityScript Multiple Temperature Reverse Transcriptase (Agilent) from 80ng of total RNA prepared from microdissected metastatic tumors, or 1.5 μg of total RNA from all other samples. Second strand cDNA was synthesized using an mRNA Second Strand Synthesis Module (NEB) and washed with Agencourt AMPure XP beads (Beckman Coulter). Libraries were prepared by tagmentation (Nextera XT DNA Sample Preparation Kit, Illumina) using index primers (Nextera XT Index kit, Illumina) to facilitate multiplexing. Libraries were pooled and sequenced on the Illumina NextSeq 500 sequencer (paired-end, 150 bp). Image analysis and base calling were done using the standard Illumina pipeline, and then demultiplexed into FASTQ files. Reads were aligned with Tophat 2.0.2 (Ghosh and Chan, 2016) using the human hg19 transcriptome and genome annotation from the UCSC genome browser. Transcripts were assembled and tested for abundance and differential expression using HTseq (Anders et al., 2015) and DEseq2 (Love et al., 2014). Genes that scored as differentially regulated by SMAD4 in any of the following categories were considered for the screen: (1) at least 4 of 17 total in vitro and in vivo conditions, (2) at least 2 of 3 arms (in vitro without TGFβ, in vitro with TGFβ, and in vivo), and (3) at least 2 tumor sample conditions. Data has been deposited under GSE148248.
Immunoblots and antibodies
Cells were lysed with RIPA Buffer (Sigma) containing protease inhibitors (cOmplete, Roche). 20-40 μg of cell lysate per sample was separated on a 4%–12% Bis-Tris gel (Invitrogen) and transferred to nitrocellulose membrane using the iBlot system (Life Technologies).
Quantitative PCR for gene expression
RNA was isolated using RNeasy Plus Mini Kit (QIAGEN). cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), and analyzed by quantitative PCR (qPCR) using Power SYBR Green PCR Master Mix (Invitrogen) on a Light Cycler 480 II PCR system (Roche) or a BioRad CFX384 Real-Time System (BioRad) according to the manufacturer’s recommendations. Relative expression was calculated using the ΔΔCt method with ACTB for normalization across samples.
ChIP
ChIP assays were carried out on parental and derivative HPAC and PANC1 cultures of approximately 4-8 million cells per sample and per epitope. Cells were cross-linked for 10 min in 1% formaldehyde at room temperature. This reaction was subsequently quenched in 125 mM glycine for 5 min. Chromatin from formaldehyde-fixed cells was solubilized in Myers ChIP RIPA buffer (1xPBS/1% NP-40/0.5% sodium deoxycholate/0.1% SDS, freshly supplemented with protease inhibitor), and fragmented to a size range of 200–700 bases with a Covaris E220 focused-ultrasonicator (Covaris). Chromatin was then immunoprecipitated overnight at 4°C with the indicated antibodies coupled to equal part mixtures of protein A and protein-G Dynabeads (Life Technologies). The beads were washed 5 times with Myers ChIP RIPA buffer, once with LiCl wash buffer (10mM Tris-HCl pH7.5/500mM LiCl/1% NP-40/1% sodium deoxycholate), once with TE buffer (10mM Tris-HCl pH7.5/0.1mM Na2EDTA), and then eluted with IP elution buffer (1% SDS/0.1M NaHCO3). After crosslink reversal, RNase A, and proteinase K treatment, immunoprecipitated DNA was extracted with QIAquick PCR Purification kit (QIAGEN) and analyzed with qPCR using the %input method.
Gene Ontology
Gene ontology enrichment analysis was performed on biological processes using http://geneontology.org.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of tumor area in experimental metastasis
Whole slide images were acquired from H&E stained slides using a Vectra 3.0 Automated Quantitative Pathology Imaging System (Perkin Elmer, Inc). Classification and quantification of tumor area and normal lung tissue was performed by a blinded research pathologist utilizing the Halo Image Analysis Platform (Indica Labs).
Statistical analysis
Graphs in the manuscript were created using Graphpad software and statistical analysis was run through the same program. All data represent the average of at least three independent experiments, unless otherwise indicated. Significance was calculated by two-tail Student’s t test. Differences were considered significant when p value was < 0.05. Specific statistical considerations are found in each legend including number of samples (n), standard error (SEM), and p value.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| V5 | Invitrogen | R96025 |
| V5/HRP | Invitrogen | R961-25 |
| SMAD4 | Santa Cruz Biotechnology | B-8; sc-7966 |
| beta-actin/HRP | Santa Cruz Biotechnology | sc-47778 HRP |
| TRK | Cell Signaling Technology | A7H6R; #92991 |
| FOSL1 | Cell Signaling Technology | D80B4, #5281 |
| SMAD4 | Cell Signaling Technology | D3M6U, #38454 |
| Histone H3 Ac-K27 | AbCam | ab4729 |
| SMAD1 | Santa Cruz | sc-7965 |
| Phospho-Smad1/5/8 | Cell Signaling Technology | 41D10 |
| Phospho-SMAD2 | Cell Signaling Technology | 3101S |
| GAPDH | Cell Signaling Technology | 14C10 |
| Bacterial and virus strains | ||
| Metastasis Candidate ORF - Pool1 | Broad Institute | This manuscript |
| Metastasis Candidate ORF - Pool2 | Broad Institute | This manuscript |
| Metastasis Candidate ORF - Pool3 | Broad Institute | This manuscript |
| Chemicals, peptides, and recombinant proteins | ||
| Blasticidin | ThermoFisher Scientific | A1113903 |
| Puromycin | ThermoFisher Scientific | A1113803 |
| Hygromycin | Life Technologies | 10687010 |
| Doxycycline | Clontech | 3P 631311 |
| Recombinant TGFβ | Peprotech | AF-100-21C |
| Critical commercial assays | ||
| QiaAmp DNA Blood Maxi Kit | QIAGEN | 51194 |
| DNeasy Blood and Tissue kit | QIAGEN | 69506 |
| RNAlater-ICE Frozen Tissue Transition Solution | ThermoFisher | AM7030 |
| Phusion high-fidelity DNA polymerase | New England Biolabs | M0530 |
| TaqMan Universal PCR Master Mix | ThermoFisher | 4364338 |
| RNeasy Plus Mini kit | QIAGEN | 74134 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | 4368813 |
| Power SYBR Green PCR Master Mix | Invitrogen | 4367659 |
| Deposited data | ||
| SMAD4 RNA-seq | This manuscript | GSE148248 |
| Experimental models: Cell lines | ||
| Lenti-X 293T | Takara | 632180 |
| PANC1 | Broad Institute | Project Achilles |
| PATU8902 | Broad Institute | Project Achilles |
| HPAC | Broad Institute | Project Achilles |
| MiaPaCa2 | Broad Institute | Project Achilles |
| L3.3 | Broad Institute | Project Achilles |
| PANC0327 | Broad Institute | Project Achilles |
| Experimental models: Organisms/strains | ||
| CrTac:NCr-Foxn1nu | Taconic | NCRNU-F |
| Recombinant DNA | ||
| pXPR003-sgCTRL | Broad Institute | This study |
| pXPR003-sgSMAD4 | Broad Institute | This study |
| pLX307-luciferase | Broad Institute | This study |
| pLX307-SMAD4 | Broad Institute | This study |
| pXPR003-sgGFP | Broad Institute | This study |
| PLKO shFOSL1-1 | Broad Institute | This study |
| PLKO shFOSL1-2 | Broad Institute | This study |
| FUW-LacZ-Luciferase-mCherry | This manuscript | This study |
| FUW-SMAD4-Luciferase-mCherry | This manuscript | This study |
| Software and algorithms | ||
| Tophat 2.0.2 | NA | Ghosh and Chan, 2016 |
| HTseq | NA | Anders et al., 2015 |
| DEseq | NA | Love et al., 2014 |
Highlights.
SMAD4 loss does not alter primary tumor growth rate
SMAD4 represses FOSL1 expression
FOSL1 is necessary and sufficient to drive metastatic colonization
Loss of SMAD4 has a direct role in facilitating metastasis
ACKNOWLEDGMENTS
This work was supported by the National Cancer Institute’s Office of Cancer Genomics Cancer Target Discovery and Development (CTD2) initiative (U01 CA176058 to W.C.H.) as well as AACR Basic Cancer Research postdoctoral fellowship 15-40-01-DAIC, an American Cancer Society postdoctoral fellowship PF-17-207-01-CSM (to C.D.), NCI grant K00 CA212221 (to J.P.R), American Cancer Society Mentored Research Scholar Grant MRSG-18-202-01, and by Department of Defense CDMRP W81XWH-19-1-0281 (to A.L.H.). We thank Quang-De Nguyen from the Lurie Family Imaging Center and Catherine Sypher from the DFCI Animal Research Facility for assistance with imaging. The authors would like to thank members of the Hahn and Cichowski labs for their helpful comments.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109443.
DECLARATION OF INTERESTS
W.C.H. is a consultant for Thermo Fisher Scientific, Solasta Ventures, MPM Capital, KSQ Therapeutics, iTeos, Tyra Biosciences, iTeos, Frontier Medicines, Jubliant Therapeutics, RAPPTA Therapeutics, and Paraxel. A.J.A. has consulted for Oncorus, Inc., Arrakis Therapeutics, and Merck & Co., Inc., and has research funding from Mirati Therapeutics and Deerfield, Inc. that is unrelated to this project. A.O.G. is a share and option holder of 10X Genomics. The remaining authors declare no competing interests.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, and DePinho RA (2006). Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 20, 3130–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JCH, Leary RJ, Eshleman JR, et al. (2009). SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin. Cancer Res 15, 4674–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, et al. (2015). Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 5, 1164–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler E, Chen YC, and Martin S (2012). C911: A bench-level control for sequence specific siRNA off-target effects. PLoS ONE 7, e51942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. (2010). The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MWY, Huang YW, Hartman-Frey C, Kuo CT, Deatherage D, Qin H, Cheng ASL, Yan PS, Davuluri RV, Huang THM, et al. (2008). Aberrant transforming growth factor β1 signaling and SMAD4 nuclear translocation confer epigenetic repression of ADAM19 in ovarian cancer. Neoplasia 10, 908–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HB, Rud JG, Lin K, and Xu L (2005). Nuclear targeting of transforming growth factor-β-activated Smad complexes. J. Biol. Chem 280, 21329–21336. [DOI] [PubMed] [Google Scholar]
- Chen HT, Tsou HK, Chang CH, and Tang CH (2012). Hepatocyte growth factor increases osteopontin expression in human osteoblasts through PI3K, Akt, c-Src, and AP-1 signaling pathway. PLoS ONE 7, e38378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao PJ, Hunt KK, Grau AM, Abramian A, Fleming J, Zhang W, Breslin T, Abbruzzese JL, and Evans DB (1999). Tumor suppressor gene Smad4/DPC4, its downstream target genes, and regulation of cell cycle. Ann. NY Acad. Sci 880, 31–37. [DOI] [PubMed] [Google Scholar]
- Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, et al. (2011). SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X-H, and Derynck R (2005). Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol 21, 659–693. [DOI] [PubMed] [Google Scholar]
- Ghosh S, and Chan CKK (2016). Analysis of RNA-seq data using TopHat and cufflinks. Methods Mol. Biol 1374, 339–361. [DOI] [PubMed] [Google Scholar]
- Gordon-Dseagu VL, Devesa SS, Goggins M, and Stolzenberg-Solomon R (2018). Pancreatic cancer incidence trends: Evidence from the Surveillance, Epidemiology and End Results (SEER) population-based data. Int. J. Epidemiol 47, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, and Flavell RA (2001). Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat. Med 7, 1118–1122. [DOI] [PubMed] [Google Scholar]
- Grau AM, Zhang L, Wang W, Ruan S, Evans DB, Abbruzzese JL, Zhang W, and Chiao PJ (1997). Induction of p21waf1 expression and growth inhibition by transforming growth factor β involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res. 57, 3929–3934. [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, and Kern SE (1996). DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271, 350–353. [DOI] [PubMed] [Google Scholar]
- Herman JM, Jabbour SK, Lin SH, Deek MP, Hsu CC, Fishman EK, Kim S, Cameron JL, Chekmareva M, Laheru DA, et al. (2018). Smad4 loss correlates with higher rates of local and distant failure in pancreatic adenocarcinoma patients receiving adjuvant chemoradiation. Pancreas 47, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, Vilardell F, Wang Z, Keller JW, Banerjee P, et al. (2009). DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol 27, 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, Wright CVE, and Moses HL (2006). Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-β signaling in cooperation with active Kras expression. Genes Dev. 20, 3147–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou M, Kouvaras E, Papamichali R, Samara M, Chiotoglou I, and Koukoulis G (2018). Smad4 and epithelial-mesenchymal transition proteins in colorectal carcinoma: An immunohistochemical study. J. Mol. Histol 49, 235–244. [DOI] [PubMed] [Google Scholar]
- Iwai K, Mori N, Oie M, Yamamoto N, and Fujii M (2001). Human T-cell leukemia virus type 1 tax protein activates transcription through AP-1 site by inducing DNA binding activity in T cells. Virology 279, 38–46. [DOI] [PubMed] [Google Scholar]
- Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, and Hingorani SR (2007). KrasG12D and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 11,229–243. [DOI] [PubMed] [Google Scholar]
- Kang Y, Chen C-R, and Massagué J (2003). A self-enabling TGFβ response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell 11, 915–926. [DOI] [PubMed] [Google Scholar]
- Klein-Scory S, Zapatka M, Eilert-Micus C, Hoppe S, Schwarz E, Schmiegel W, Hahn SA, and Schwarte-Waldhoff I (2007). High-level inducible Smad4-reexpression in the cervical cancer cell line C4-II is associated with a gene expression profile that predicts a preferential role of Smad4 in extracellular matrix composition. BMC Cancer 7, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, Grizzle WE, and Klug CA (2007). Inactivation of Smad4 accelerates KrasG12D-mediated pancreatic neoplasia. Cancer Res. 67, 8121–8130. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo YZ, He P, and Qiu MX (2018). FOSL1 enhances growth and metastasis of human prostate cancer cells through epithelial mesenchymal transition pathway. Eur. Rev. Med. Pharmacol. Sci 22, 8609–8615. [DOI] [PubMed] [Google Scholar]
- Maisonneuve P (2019). Epidemiology and burden of pancreatic cancer. Presse Med. 48, e113–e123. [DOI] [PubMed] [Google Scholar]
- Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, Chatterjee K, Wong F, Jiao Y, Kohutek ZA, et al. (2017). Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet 49, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, and Chen YG (2000). Controlling TGF-β signaling. Genes Dev. 14, 627–644. [PubMed] [Google Scholar]
- McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, and McCain RS (2018). Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol 24, 4846–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen PJ, Ebner R, Lopez AR, and Derynck R (1994). TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J. Cell Biol 127, 2021–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, Hishima T, Koike M, Shitara N, Iwama T, et al. (1999). Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 18, 3098–3103. [DOI] [PubMed] [Google Scholar]
- Moustakas A, and Heldin CH (2009). The regulation of TGFβ signal transduction. Development 136, 3699–3714. [DOI] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwarz H, Beug H, and Reichmann E (1996). TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 10, 2462–2477. [DOI] [PubMed] [Google Scholar]
- Oshima M, Okano K, Muraki S, Haba R, Maeba T, Suzuki Y, and Yachida S (2013). Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann. Surg 258, 336–346. [DOI] [PubMed] [Google Scholar]
- Padua D, Zhang XHF, Wang Q, Nadal C, Gerald WL, Gomis RR, and Massagué J (2008). TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, Robertson AG, Cherniack AD, Gupta M, Getz G, et al. ; Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu; Cancer Genome Atlas Research Network (2017). Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Román M, López I, Guruceaga E, Baraibar I, Ecay M, Collantes M, Nadal E, Vallejo A, Cadenas S, Miguel ME, et al. (2019). Inhibitor of differentiation-1 sustains mutant KRAS-driven progression, maintenance, and metastasis of lung adenocarcinoma via regulation of a FOSL1 network. Cancer Res. 79, 625–638. [DOI] [PubMed] [Google Scholar]
- Samarakoon R, Higgins CE, Higgins SP, and Higgins PJ (2009). TGF-β1-induced expression of the poor prognosis SERPINE1/PAI-1 gene requires EGFR signaling: A new target for anti-EGFR therapy. J. Oncol 2009, 342391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugang X, Hongfa Y, Jianpeng L, Xu Z, Jingqi F, Xiangxiang L, and Wei L (2016). Prognostic value of SMAD4 in pancreatic cancer: A meta-analysis. Transl. Oncol 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2019). Cancer statistics, 2019. CA Cancer J. Clin 69, 7–34. [DOI] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. ; Australian Pancreatic Cancer Genome Initiative (2015). Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li Y, Zhan S, Zhang L, Zhang S, Tang Q, Li M, Tan Z, Liu S, and Xing X (2019). SMAD4 Y353C promotes the progression of PDAC. BMC Cancer 19, 1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, Wood LD, Goggins M, Hruban RH, Chang AE, et al. (2015). RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell 161, 1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. (2015). Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Hung M, Huang S, Xue J, Lin X, Chiu W, Chen Y, Yu G, and Liu M (2014). Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer metastasis. J. Clin. Invest 124, 564–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. (2010). Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Macgregor-Das A, Saunders T, Whittle MC, Makohon-Moore A, Kohutek ZA, Poling J, Herbst BT, Javier BM, Cope L, et al. (2017). Mutant p53 together with TGFβ signaling influence organ-specific hematogenous colonization patterns of pancreatic cancer. Clin. Cancer Res 23, 1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo D, Mohr SE, Hu Y, Taycher E, Rolfs A, Kramer J, Williamson J, and LaBaer J (2007). PlasmID: A centralized repository for plasmid clone information and distribution. Nucleic Acids Res. 35, D680–D684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
- The RNA-seq generated during this study will be available on GEO (GSE148248) available as of the date of publication.
- No novel code was written for the analysis of the dataset.
- Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request