

Abstract
Calcium-dependent cardiac muscle contraction is regulated by the protein complex troponin (cTn), and specifically by the regulatory N-terminal domain (N-cTnC) which contains one active Ca2+ binding site (site II). It has been previously shown that cardiac muscle contractility and functionality is affected by mutations in N-cTnC which alter calcium binding affinity. Here, we describe the application of adaptive steered molecular dynamics to characterize the influence of N-cTnC mutations on site II calcium binding affinity. We observed the correct trends for all of the studied calcium sensitizing and desensitizing mutants, in conjunction with loop II perturbations. Additionally, the potential of mean force accuracy was shown to increase substantially with increasingly slower speeds and using fewer trajectories. This study presents a novel approach to computationally estimate the Ca2+ binding affinity of N-cTnC structures and is a valuable potential tool to support the design and characterization of novel mutations with potential therapeutic benefits.
Graphical Abstract
Introduction
Calcium-dependent cardiac muscle contraction occurs through a multitude of proteins and filaments operating cohesively, and is regulated by the protein complex troponin (cTn). Troponin’s functionality is modulated through its three subunits: troponin C (cTnC), troponin I (cTnI), and troponin T (cTnT). cTnT anchors the protein complex to tropomyosin (Tm). cTnI inhibits the actomyosin interaction1. cTnC, the calcium binding subunit, when bound to Ca2+ induces a conformational change in cTnI, dissociating it from actin. Tm is then able to slide along the actin filament away from the myosin binding site, thus allowing myosin to bind to actin, forming a cross bridge, the first step in the cross-bridge cycle. cTnC is a two-domain subunit, with each domain containing two EF-hand (helix-loop-helix) structural motifs. The C-domain, referred to as the structural domain, contains site III and site IV which under physiological conditions are consistently occupied by either Ca2+ or Mg2+ 2. The regulatory, N-terminal domain (N-cTnC), contains one active binding site (site II) 3. Site II has been characterized as having low affinity and high selectivity to Ca2+ 4, 5, with a N-cTnC Ca2+ KD=7μM, measured using steady state fluorescence at 15 °C 6. The inactivity of site I (residues 28–41) is attributed to non-conserved residue mutations in the Ca2+ coordinating sites of the loop, compared to the skeletal isoform which contains two active binding sites in the regulatory domain 7.
Cardiac muscle contractility and functionality has been shown to be affected by mutations in the regulatory domain of cTnC. The mutation either increases or decreases Ca2+ sensitivity based on the substituted residue identity and its location in the tertiary structure relative to site II 6. Recent work has shown success in designing mutations (F20Q, V44Q, L48Q, M81Q) that increase Ca2+ sensitivity 6. Additionally, designed mutations that decrease Ca2+ sensitivity have been reported: I61Q 8 and D73N 9. It has been shown in vivo that expression of mutant cTn complexes has the potential to restore cardiac muscle contractility and functionality after a myocardial infarction event, thus supporting the claim that mutated cTn structures combined with gene therapy offer a therapeutic strategy for treatment of heart disease 10–12.
In addition to experimental characterization of mutational effects on N-cTnC Ca2+ sensitivity, computational methods to study protein dynamics such as conventional and accelerated Molecular Dynamics (MD)8, 13–24, as well as free energy methods such as Umbrella Sampling (US)13, 25–27 and Thermodynamic Integration (TI) 28 have been used to support experimental findings. The free energy studies were partly motivated by the desire to accurately calculate the Ca2+ binding affinity of known sensitizing and desensitizing mutations. This could potentially support and accelerate the design and characterization of novel mutations with therapeutic benefits for further experimental validation. However, limitations of free energy methods such as inaccuracies in forcefield descriptions of divalent metal cations29–31, non-classical electronic effects32, potential insufficient sampling of the thermodynamic ensemble33, and high computational cost34 have hampered these calculations.
Steered Molecular Dynamics (SMD) has the potential to overcome some of the limitations discussed above. It serves as a biased integration methodology to obtain the potential of mean force (PMF) along a chosen pulling path. In this method a pseudo particle applies a steering force in order to move along the reaction coordinate at a particular velocity. The non-equilibrium work conducted on the system during the SMD simulation can be related to the free energy difference between the initial and final states of the simulation 35. However, the main limitation of this method comes from requiring thousands of individual nonequilibrium trajectories to achieve convergence of the PMF, especially for long distance paths. An improvement upon this method was introduced by Hernandez and coworkers 36 termed Adaptive Steered Molecular Dynamics (ASMD). The strength of ASMD arises from the ability to split the reaction coordinate into a number of smaller stages, where the Jarzynski average (JA) 37 is calculated for each stage over a number of individual trajectories. Then the JA serves as the starting reference point for the next sequential stage of the reaction coordinate. This method has been shown to greatly reduce the number of individual nonequilibrium trajectories needed to converge the PMF from thousands to only a few hundred 38, 39. Thus, ASMD offers a potential solution to the previously described limitations of insufficient sampling and cost efficiency that have been obstacles for other free energy methods.
In this work, we utilized the ASMD method to obtain the PMF of Ca2+ binding to wildtype N-cTnC, several designed Ca2+ sensitizing and desensitizing mutants, and perturbations in loop II of the regulatory domain of cTnC. We show that based on the ASMD theory with increasingly slower pull speeds, the number of individual nonequilibrium trajectories needed to converge the PMF is greatly reduced. The obtained PMFs of all mutants exhibited the correct trend of either increased or decreased binding affinity, making this a viable computational tool for characterizing cTnC sensitizing and desensitizing mutants.
Methods
Preparation of Initial Structure for Molecular Dynamics
The structure of the Ca2+-bound N-terminal domain of cardiac troponin was obtained from PDB 1AP4 40. The representative model was solvated using the tLeap module of AMBER 18 41 in a 40 Å TIP3P 42 water box and neutralized with Na+ ions. The forcefield used for the simulations was ff14SB 43. Energy minimizations were performed on the entire system. The first minimization minimized the solvent with the protein and cations held fixed for a maximum of 10000 cycles, where the first 10 iteration were performed using the steepest descent algorithm and the rest used the conjugate gradient algorithm. The second energy minimization minimized the entire system for a maximum of 10000 cycles, where the first 10 iterations were performed using the steepest descent algorithm and the remaining cycles utilized the conjugate gradient algorithm. The system was subsequently heated to 300 K over a span of 500 ps using the Langevin thermostat 44, followed by 500 ps of density equilibration using the Berendsen barostat 45. The entire system was then set to equilibrate over a span of 800 ps (with production settings) at constant pressure and temperature (300 K).
Adaptative Steered Molecular Dynamics (ASMD)
To study the unbinding process of Ca2+ from N-cTnC site II, we used adaptive steered molecular dynamics (ASMD). The simulations started with the equilibrated system as detailed above. ASMD was performed by restraining the distance between the center of mass of the α carbons in the residues Ser69, Gly70, and Thr71 for a 20Å pull from the original starting distance between the Ca2+ ion and the calculated center of mass point. The initial distance between the center of mass of the α carbons and Ca2+ of the pulling coordinate was determined using the CPPTRAJ 46 distance function. The pulling was performed using a force constant of 7.2 kcal mol−1 Å−2 and pulling speeds of 1 Å/ns and 0.5 Å/ns. All ASMD simulations were split into ten stages. For a pulling speed of 1 Å/ns, each stage was stimulated for 2 ns with either 100 or 150 trajectories per stage. For a pulling speed of 0.5 Å/ns, each stage was simulated for 4 ns and 50 trajectories per stage. Upon completion of the SMD pulls for any stage (N) of the simulation, the JA was calculated via the “ASMD.py” script provided by Hernandez and colleagues in the AMBER Advanced Tutorial 26. The final coordinates of the trajectory whose work most closely matched the JA were used to initialize the subsequent stage (N+1) of the simulation. All ASMD simulations were performed with the GPU implementation of PMEMD47 from the AMBER 18 package.
Mutant N-cTnC Structures
All Ca2+ sensitizing mutants (F20Q, V44Q, L48Q, M81Q), Ca2+ desensitizing mutants (I61Q, D73N), and loop II mutants (D65A, D67A, E76A, D65A/D67A/E76A, D65A,D67A,S69A,T71A,E76A) were constructed using the protein mutagenesis tool in PyMOL48. The 1AP4 representative N-cTnC structure served as the base model to which the mutations were applied. Mutant structures were solvated, minimized, and equilibrated as detailed for the wildtype structure. ASMD simulations were performed with a pull speed of 0.5 Å/ns and 50 trajectories per stage and using the GPU implementation of PMEMD.
Results and Discussion
Validating PMF Accuracy with Slower Pull Speeds and Fewer Trajectories for N-cTnC
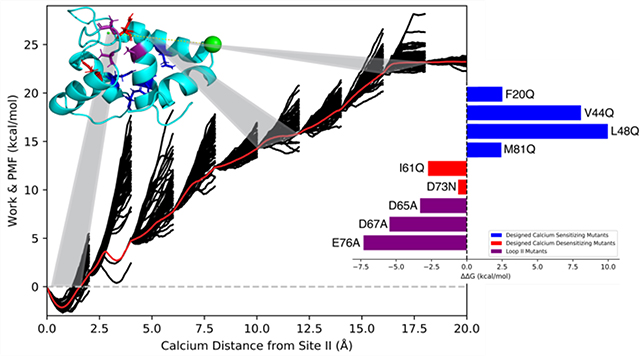
It has been previously shown that ASMD accuracy can be improved via two ways: an infinitely slow pull rate (such that all intermediate states are in thermodynamic equilibrium) and increasing the number of trajectories per stage 38, 39. For Ca2+ binding to cTnC, our results support the established notion that an increasingly slow pull rate has a larger impact on increasing the PMF accuracy than increasing the number of trajectories per stage (Figure 1). We conducted ASMD simulations on the regulatory domain of cTnC (N-cTnC) using two different pull speeds (1Å /ns and 0.5 Å/ns) with varying numbers of trajectories per stage (tps) to create the PMF of Ca2+ binding to site II. Figure 1 supports the claim that at a constant pull rate, increasing the number of trajectories improves PMF accuracy especially at long distances. The PMF containing 150 tps showed an improvement (−3.5 kcal/mol) over the PMF containing 100 tps. However, with an even slower pull rate we were able to achieve better results with fewer trajectories. The PMF obtained using a pull speed of 0.5 Å/ns and only 50 tps exhibited an improved accuracy with a lower absolute binding energy compared to the PMF obtained from a 1Å /ns pull rate and 150 tps. ASMD utilizes nonequilibrium trajectories to provide an unbiased sampling of the reaction coordinate 49. The benefit of slower sampling speeds is that statistically more favorable trajectories are explored more frequently compared to the high energy alternatives (Figure 2A), thus accounting for the improved accuracy of the PMF while using fewer trajectories. The Cα RMSD was calculated for all protein residues (Figure 2B) and for loop II residues (Figure 2C) over the duration of the simulation for the resulting Jarzynski averaged PMF trajectory. These values show that the protein structure remained stable during the course of the simulation, thus ensuring a physiologically relevant structure. GPU acceleration through the PMEMD.cuda program allowed for these slow rate ASMD simulations to be performed at a relatively low computational cost. Based on this data we proceeded with a 0.5 Å/ns pull speed and 50 tps for all mutant ASMD simulations.
Figure 1.

Comparison ofthe PMF of Ca2+ binding to site II of N-cTnC at múltiple pulí speeds and number of trajectòries per stage (tps).The solid black Une shows the pulí speed of 0.5 Á/ns using 50 tps, while the red dashed curve shows the pulí speed of 1 Á/ns using 100 tps, and the red dotted curve shows the pulí speed of 1 Á/ns using 150 tps.
Figure 2.

Analysis of Ca2+-bound N-cTnC ASMD. (A) The PMF ofwildtype N-cTnC obtained at constant NPT using ASMD in 10 stages with 50tps, and a pulling speed of 0.5 Á/ns.The work traces W(£t’ )forthe 50 individual trajectòries in each stage are shown in thin black curves, and the PMF W obtained from the Jarzynski Equality is shown asa thin red curve. A representative structure ofWT N-cTnC [PDB 1AP4] is shown with the pulí path of calcium (dashed yellow line) and highlighted sections corresponding to specific ASMD stages. (B)The RMSDof Ca atoms of N-cTnC (residues 1–89) throughout the entirety of the Jarzynski Averaged trajectory. (C)The RMSD of Ca atoms of loop II N-cTnC (residues 65–76) throughout the entirety of the Jarzynski Averaged trajectory.
Adaptive Steered Molecular Dynamics of Ca2+ Sensitivity Altering Mutants Successfully Identify Calcium Sensitization and Desensitization
The PMF was obtained for wildtype N-cTnC, calcium sensitizing cTnC mutants (F20Q, V44Q, L48Q, M81Q) [Figure 3A], calcium desensitizing cTnC mutants (I61Q, D73N) [Figure 3B], and loop II cTnC mutants (D65A, D67A, E76A, D65A/D67A/E76A, D65A/D67A/S69A/T71A/E76A) [Figure 3C] using a 0.5 Å/ns pull speed and 50 tps. We successfully predicted the correct trends for mutagenesis effects on N-cTnC Ca2+ binding affinity. The ΔG of Ca2+ binding was estimated to be the difference between the free energies along the obtained PMF when Ca2+ was 15 Å from its initial starting position in site II (x = 15Å) and the initial starting position (x = 0Å). This reference position was chosen instead of the end of the pull length (x = 20Å) due to observed secondary interactions between Ca2+ and helix A for pull lengths larger than 15Å. These observed secondary interactions were attributed to be an artifact of the ASMD simulations due to the vector along which Ca2+ was pulled. Therefore, these interactions are physiologically unlikely to occur and were ignored. The reference point of 15 Å from the initial starting position was chosen based on having negligible electrostatic interactions between Ca2+ and residues in loop II, in addition to being far enough away from helix A such that secondary interactions were minimal.
Figure 3.

PMFs of Ca2+ binding to wild type N-cTnC and mutants: (A) designed calcium sensitizing mutations, (B) designed calcium desensitizing mutations, (C) loop II mutations.
The calculated ΔΔG values for the calcium sensitizing and desensitizing mutants followed the anticipated trends. However, the ΔΔG values of the sensitizing mutations (F20Q, V44Q, L48Q, and M81Q) were consistently overestimated compared to the derived experimental values6, with predicted ΔΔG values of 2.5, 8.0, 10.0, and 2.5 kcal/mol respectively. L48Q showed the greatest increase in binding affinity in agreement with experimental findings. The calcium desensitizing mutations (I61Q and D73N) exhibited negative ΔΔG values, correctly reflecting the trend observed in experimental pCa50 measurements 8, 9. The predicted ΔΔG values calculated for I61Q and D73N were −2.7 and −0.6 kcal/mol, respectively. Additionally, three individual mutations of chelating residues in loop II were simulated to characterize the effects of mutating specific Ca2+ coordinating residues. Residues Asp65, Asp67, and Glu76 were selected since these residues were expected to have the greatest impact on Ca2+ binding affinity. This was supported by the significant difference in predicted ΔΔG values (−3.3, −5.5, and −7.3 kcal/mol respectively). The ΔΔG values between WT N-cTnC and all single mutant N-cTnC structures are summarized in Figure 4. The PMFs of the triple mutant (D65A/D67A/E76A) and quintuple mutant (D65A/D67A/S69A/T71A/E76A) showed that calcium binding was effectively completely removed from loop II. The ΔG of Ca2+ binding obtained from the PMFs were 1.13 and 2.25 kcal/mol, respectively. The PMFs also indicated that at the initial pulling distances (distances less than 10Å) the ΔG of binding was essentially 0 kcal/mol (Figure 3C), suggesting that nonbonded ligand-protein interactions within site II were effectively removed. It was not until further distances that work was done on the system, thus supporting the claim that at long distances any work done on the system was an attribute of secondary protein-ligand interactions.
Figure 4.

Relative AAG valúes between mutant and wíld type N-cTnC structures. AG valúes of calcium binding wereobtained from the respective PMFs at a distance of Ca2+ 15Áfrom its ¡nitially bound position ¡n site II.
Limitations
ASMD has proven to be a dependable method for characterizing the relative Ca2+ binding affinities of mutant N-cTnC structures. The sign of the ΔΔG values was correctly identified for all investigated mutations. However, this method was not without limitations. The discrepancies between in vivo and in silico ΔG values can be attributed to several factors. We speculate that the use of a non-polarizable forcefield contributed to overestimation of the mutational effects on Ca2+ binding. Non-polarizable forcefields add nonbonded interactions in a pairwise manner, greatly simplifying the interactions of ligand with protein and solvent. Additionally, the exact reaction coordinate of calcium binding remains unknown, making it exceedingly difficult to accurately and efficiently sample all possible potential binding paths. The explored path has been shown to introduce work onto the system via secondary protein-ligand interactions which presumably do not occur in vivo. Therefore, it is possible that a more biologically relevant pulling path could be explored.
Conclusions
We utilized an adaptive steered molecular dynamics protocol in order to characterize the binding affinity of Ca2+ to wildtype and several mutant regulatory domain cTnC structures. It was shown that more accurate PMFs were generated faster and with fewer trajectories per stage with slower pull rates. The pull rate of 0.5 Å/ns with 50 tps proved to have the highest PMF accuracy while still maintaining relatively low computational cost. ASMD of WT N-cTnC and Ca2+ sensitivity modulating mutations supported the experimentally confirmed ΔΔG value trends. We also successfully characterized the effects of several loop II perturbations on Ca2+ binding affinity. The inclusion of multisite alanine mutations at coordinating residues in loop II proved to effectively minimize any nonbonded ligand-protein interactions, thus resulting in negligible work to be done on the system. In conclusion, we have shown that ASMD is a valuable method for the characterization of ligand binding affinity in cTnC. The sign of the ΔΔG values was correctly identified for all investigated mutations. This positions ASMD as a prime tool to computationally study novel designed calcium sensitivity altering mutants for potential therapeutic application. It is our hope that this method can support and supplement experimental efforts for the design and characterization of novel mutations with therapeutic benefits.
Data and Software Availability
The wildtype cTnC protein structure (1AP4) was available in the Protein Data Bank (PDB) at https://www.rcsb.org/. All mutations were created using the wildtype protein structure as the base model within the PyMOL software using the protein mutagenesis tool. All ASMD simulations were performed within the Amber18 software framework. Initial simulation file preparation scripts were obtained from the AMBER Advanced Tutorial 26 at http://ambermd.org/tutorials/advanced/tutorial26/. Scripts from this tutorial were modified based on the system specifications and for usage on the Ohio Supercomputer Center.
Acknowledgements
The authors would like to thank Joseph Fernandez and Dr. Christopher Hadad for helpful discussions regarding ASMD methodology. We would also like to thank members of the Lindert Lab for helpful discussions relating to this work. Additionally, we would like to thank the Ohio Supercomputer Center50 for valuable computational resources. This work was supported by the NIH (R01 HL137015 to S.L.).
References
- 1.Ohtsuki I; Morimoto S, Troponin. In Encyclopedia of Biological Chemistry (Second Edition), Lennarz WJ; Lane MD, Eds. Academic Press: Waltham, 2013; pp 445–449. [Google Scholar]
- 2.Li MX; Hwang PM, Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holroyde MJ; Robertson SP; Johnson JD; Solaro RJ; Potter JD, The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J Biol Chem 1980, 255, 11688–11693. [PubMed] [Google Scholar]
- 4.Marston S; Zamora JE, Troponin structure and function: a view of recent progress. J Muscle Res Cell Motil 2020, 41, 71–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sia SK; Li MX; Spyracopoulos L; Gagné SM; Liu W; Putkey JA; Sykes BD, Structure of cardiac muscle troponin C unexpectedly reveals a closed regulatory domain. J Biol Chem 1997, 272, 18216–18221. [DOI] [PubMed] [Google Scholar]
- 6.Tikunova SB; Davis JP, Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J Biol Chem 2004, 279, 35341–35352. [DOI] [PubMed] [Google Scholar]
- 7.Herzberg O; Moult J; James MNG, CONFORMATIONAL FLEXIBILITY OF TROPONIN C11Supported by the Medical Research Council of Canada, an Alberta Heritage Foundation of Medical Research Fellowship (to O.H.) and an Alberta Heritage Foundation for Medical Research Visiting Scientist Fellowship (to J.M.). In Calcium-Binding Proteins in Health and Disease, Norman AW; Vanaman TC; Means AR, Eds. Academic Press: 1987; pp 312–322. [Google Scholar]
- 8.Wang D; McCully ME; Luo Z; McMichael J; Tu AY; Daggett V; Regnier M, Structural and functional consequences of cardiac troponin C L57Q and I61Q Ca(2+)-desensitizing variants. Arch Biochem Biophys 2013, 535, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McConnell BK; Singh S; Fan Q; Hernandez A; Portillo JP; Reiser PJ; Tikunova SB, Knock-in mice harboring a Ca(2+) desensitizing mutation in cardiac troponin C develop early onset dilated cardiomyopathy. Front Physiol 2015, 6, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shettigar V; Zhang B; Little SC; Salhi HE; Hansen BJ; Li N; Zhang J; Roof SR; Ho H-T; Brunello L; Lerch JK; Weisleder N; Fedorov VV; Accornero F; Rafael-Fortney JA; Gyorke S; Janssen PML; Biesiadecki BJ; Ziolo MT; Davis JP, Rationally engineered Troponin C modulates in vivo cardiac function and performance in health and disease. Nat. Commun 2016, 7, 10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feest ER; Steven Korte F; Tu AY; Dai J; Razumova MV; Murry CE; Regnier M, Thin filament incorporation of an engineered cardiac troponin C variant (L48Q) enhances contractility in intact cardiomyocytes from healthy and infarcted hearts. J Mol Cell Cardiol 2014, 72, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis J; Davis LC; Correll RN; Makarewich CA; Schwanekamp JA; Moussavi-Harami F; Wang D; York AJ; Wu H; Houser SR; Seidman CE; Seidman JG; Regnier M; Metzger JM; Wu JC; Molkentin JD, A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens CM; Rayani K; Singh G; Lotfalisalmasi B; Tieleman DP; Tibbits GF, Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations. J Biol Chem 2017, 292, 11915–11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim CC; Yang H; Yang M; Wang CK; Shi J; Berg EA; Pimentel DR; Gwathmey JK; Hajjar RJ; Helmes M; Costello CE; Huo S; Liao R, A novel mutant cardiac troponin C disrupts molecular motions critical for calcium binding affinity and cardiomyocyte contractility. Biophys J 2008, 94, 3577–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindert S; Kekenes-Huskey PM; Huber G; Pierce L; McCammon JA, Dynamics and calcium association to the N-terminal regulatory domain of human cardiac troponin C: a multiscale computational study. J Phys Chem B 2012, 116, 8449–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindert S; Kekenes-Huskey PM; McCammon JA, Long-timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in troponin C. Biophys J 2012, 103, 1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kekenes-Huskey PM; Lindert S; McCammon JA, Molecular basis of calcium-sensitizing and desensitizing mutations of the human cardiac troponin C regulatory domain: a multi-scale simulation study. PLoS Comput Biol 2012, 8, e1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y; Lindert S; Kekenes-Huskey P; Rao VS; Solaro RJ; Rosevear PR; Amaro R; McCulloch AD; McCammon JA; Regnier M, Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys J 2014, 107, 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Y; Rao V; Tu AY; Lindert S; Wang D; Oxenford L; McCulloch AD; McCammon JA; Regnier M, Troponin I Mutations R146G and R21C Alter Cardiac Troponin Function, Contractile Properties, and Modulation by Protein Kinase A (PKA)-mediated Phosphorylation. J Biol Chem 2015, 290, 27749–27766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindert S; Cheng Y; Kekenes-Huskey P; Regnier M; McCammon JA, Effects of HCM cTnI mutation R145G on troponin structure and modulation by PKA phosphorylation elucidated by molecular dynamics simulations. Biophys J 2015, 108, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowman JD; Lindert S, Computational Studies of Cardiac and Skeletal Troponin. Front Mol Biosci 2019, 6, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewan S; McCabe KJ; Regnier M; McCulloch AD; Lindert S, Molecular Effects of cTnC DCM Mutations on Calcium Sensitivity and Myofilament Activation-An Integrated Multiscale Modeling Study. J Phys Chem B 2016, 120, 8264–8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dvornikov AV; Smolin N; Zhang M; Martin JL; Robia SL; de Tombe PP, Restrictive Cardiomyopathy Troponin I R145W Mutation Does Not Perturb Myofilament Length-dependent Activation in Human Cardiac Sarcomeres. J Biol Chem 2016, 291, 21817–21828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zamora JE; Papadaki M; Messer AE; Marston SB; Gould IR, Troponin structure: its modulation by Ca2+ and phosphorylation studied by molecular dynamics simulations. Physical Chemistry Chemical Physics 2016, 18, 20691–20707. [DOI] [PubMed] [Google Scholar]
- 25.Bowman JD; Lindert S, Molecular Dynamics and Umbrella Sampling Simulations Elucidate Differences in Troponin C Isoform and Mutant Hydrophobic Patch Exposure. J Phys Chem B 2018, 122, 7874–7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowman JD; Coldren WH; Lindert S, Mechanism of Cardiac Troponin C Calcium Sensitivity Modulation by Small Molecules Illuminated by Umbrella Sampling Simulations. J Chem Inf Model 2019, 59, 2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens CM; Rayani K; Genge CE; Singh G; Liang B; Roller JM; Li C; Li AY; Tieleman DP; van Petegem F; Tibbits GF, Characterization of Zebrafish Cardiac and Slow Skeletal Troponin C Paralogs by MD Simulation and ITC. Biophys J 2016, 111, 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rayani K; Seffernick J; Li AY; Davis JP; Spuches AM; Van Petegem F; Solaro RJ; Lindert S; Tibbits GF, Binding of calcium and magnesium to human cardiac Troponin C. J Biol Chem 2021, 100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo J; Aksimentiev A, Improved Parametrization of Li+, Na+, K+, and Mg2+ Ions for All-Atom Molecular Dynamics Simulations of Nucleic Acid Systems. J. Phys. Chem. Lett 2012, 3, 45–50. [Google Scholar]
- 30.Saxena A; Sept D, Multisite Ion Models That Improve Coordination and Free Energy Calculations in Molecular Dynamics Simulations. J Chem Theory Comput 2013, 9, 3538–3542. [DOI] [PubMed] [Google Scholar]
- 31.Saxena A; García AE, Multisite ion model in concentrated solutions of divalent cations (MgCl2 and CaCl2): osmotic pressure calculations. J Phys Chem B 2015, 119, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller WH; Cotton SJ, Classical molecular dynamics simulation of electronically non-adiabatic processes. Faraday Discuss. 2016, 195, 9–30. [DOI] [PubMed] [Google Scholar]
- 33.Mobley DL; Gilson MK, Predicting Binding Free Energies: Frontiers and Benchmarks. Annu Rev Biophys 2017, 46, 531–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adcock SA; McCammon JA, Molecular dynamics: survey of methods for simulating the activity of proteins. Chem Rev 2006, 106, 1589–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Izrailev S; Stepaniants S; Isralewitz B; Kosztin D; Lu H; Molnar F; Wriggers W; Schulten K In Steered Molecular Dynamics, Computational Molecular Dynamics: Challenges, Methods, Ideas, Berlin, Heidelberg, 1999//; Deuflhard P; Hermans J; Leimkuhler B; Mark AE; Reich S; Skeel RD, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 1999; pp 39–65. [Google Scholar]
- 36.Ozer G; Quirk S; Hernandez R, Adaptive steered molecular dynamics: Validation of the selection criterion and benchmarking energetics in vacuum. J. Chem. Phys 2012, 136, 215104. [DOI] [PubMed] [Google Scholar]
- 37.Jarzynski C, Nonequilibrium Equality for Free Energy Differences. Phys. Rev. Lett 1997, 78, 2690–2693. [Google Scholar]
- 38.Ozer G; Quirk S; Hernandez R, Thermodynamics of Decaalanine Stretching in Water Obtained by Adaptive Steered Molecular Dynamics Simulations. Journal of Chemical Theory and Computation 2012, 8, 4837–4844. [DOI] [PubMed] [Google Scholar]
- 39.Ozer G; Valeev EF; Quirk S; Hernandez R, Adaptive Steered Molecular Dynamics of the Long-Distance Unfolding of Neuropeptide Y. J. Chem. Theory Comput 2010, 6, 3026–3038. [DOI] [PubMed] [Google Scholar]
- 40.Spyracopoulos L; Li MX; Sia SK; Gagné SM; Chandra M; Solaro RJ; Sykes BD, Calcium-Induced Structural Transition in the Regulatory Domain of Human Cardiac Troponin C. Biochemistry 1997, 36, 12138–12146. [DOI] [PubMed] [Google Scholar]
- 41.Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TEI; Cruzeiro VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK; Gohlke H; Goetz AW; Greene D; Harris R; Homeyer N; Huang Y; Izadi S; Kovalenko A; Kurtzman T; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein DJ; Merz KM; Miao Y; Monard G; Nguyen C; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Schott-Verdugo S; Shen J; Simmerling CL; Smith J; SalomonFerrer R; Swails J; Walker RC; Wang J; Wei H; Wolf RM; Wu X; Xiao L; York DM; Kollman PA AMBER 2018 University of California, San Francisco., 2018. [Google Scholar]
- 42.Jorgensen WL; Madura JD, Quantum and statistical mechanical studies of liquids. 25. Solvation and conformation of methanol in water. J. Am. Chem. Soc 1983, 105, 1407–1413. [Google Scholar]
- 43.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loncharich RJ; Brooks BR; Pastor RW, Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers 1992, 32, 523–535. [DOI] [PubMed] [Google Scholar]
- 45.Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR, Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- 46.Roe DR; Cheatham TE, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- 47.Götz AW; Williamson MJ; Xu D; Poole D; Le Grand S; Walker RC, Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput 2012, 8, 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC. [Google Scholar]
- 49.Zhuang Y; Bureau HR; Quirk S; Hernandez R, Adaptive steered molecular dynamics of biomolecules. Mol. Simul 2020, 1–12. [Google Scholar]
- 50.Ohio Supercomputer Center. Ohio Technology Consortium of the Ohio Board of Regents, 1987. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The wildtype cTnC protein structure (1AP4) was available in the Protein Data Bank (PDB) at https://www.rcsb.org/. All mutations were created using the wildtype protein structure as the base model within the PyMOL software using the protein mutagenesis tool. All ASMD simulations were performed within the Amber18 software framework. Initial simulation file preparation scripts were obtained from the AMBER Advanced Tutorial 26 at http://ambermd.org/tutorials/advanced/tutorial26/. Scripts from this tutorial were modified based on the system specifications and for usage on the Ohio Supercomputer Center.