

SUMMARY
The visceral-adipose tissue (VAT) of lean mice hosts a unique population of regulatory-T (Treg) cells that have a distinct transcriptome and T-cell-receptor (TCR) repertoire and regulate local and systemic inflammation and metabolism. Perplexingly, this population disappears in obese mice, limiting the promise of Treg-cell-based therapies for metabolic disorders. We exploited the power of a VAT-Treg TCR-transgenic mouse model to follow the dynamics of, and phenotypic changes in, the VAT-Treg population throughout the development of diet-induced obesity. Our results show that VAT-Treg cells are lost under obesogenic conditions due to down-regulation of their defining transcription factor, PPARγ, coupled with their strikingly enhanced responses to pro-inflammatory cytokines. In particular, the VAT from obese mice (and reportedly humans) was strongly enriched in plasmacytoid dendritic cells that actively express interferon-alpha. These cells were directly toxic to PPARγ+ VAT-Treg cells. Blocking this pathway in obese mice by multiple approaches substantially restored the VAT-Treg population and enhanced insulin sensitivity.
Keywords: Immunometabolism, obesity, adipose tissue, Treg cell, pDC, inflammatory cytokine, interferon, CRISPR-Cas9
Graphical Abstract
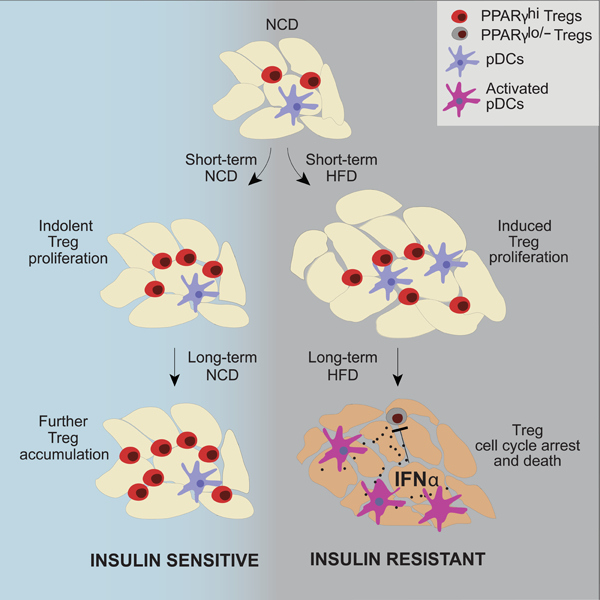
eTOC
Obesity-induced loss of insulin-sensitizing VAT Treg cells hinders the promise of Treg-based therapy for metabolic disorders. Li et al demonstrate that VAT-Treg cells decrease in obese mice in response to elevated levels of IFNα made by local pDCs, which can be targeted to restore VAT-Treg cells and insulin sensitivity.
INTRODUCTION
Individuals with obesity are prone to a cluster of disorders termed the “metabolic syndrome,” which includes type 2 diabetes (T2D), heart disease and stroke. Chronic, low-grade inflammation of the adipose tissue, and eventually systemically, is a major driver of obesity-associated metabolic disorders (Hotamisligil, 2017; Lee et al., 2018). The visceral adipose tissue (VAT) of lean individuals is enriched in classes of immunocytes that keep this inflammation in check in order to maintain tissue homeostasis, notably anti-inflammatory macrophages (MFs), type 2 innate lymphoid cells (ILC2s), and regulatory T (Treg) cells. In states of obesity, however, this anti-inflammatory condition is perturbed, resulting in the accumulation of several types of pro-inflammatory immunocytes; for example pro-inflammatory MFs, natural killer (NK) cells, CD8+ T cells and CD4+ T helper (Th) type 1 cells, resulting in elevated levels of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)α, interleukin (IL)-6, interferons (IFNs) and IL-1β. Such a drastic shift in the inflammatory tenor begs the question of how obesity renders the anti-inflammatory arm of the immune system incapable of properly controlling inflammation and maintaining adipose-tissue homeostasis.
Foxp3+CD4+ Treg cells are critical guardians of the anti-inflammatory state of the VAT in lean individuals (Panduro et al., 2016). Compared with their lymphoid-tissue counterparts, VAT-Treg cells, in particular those in the epididymal fat depot, display a distinct, clonally expanded, repertoire of T cell receptors (TCRs), indicative of specific antigen recognition (Feuerer et al., 2009; Kolodin et al., 2015). In addition, VAT-Treg cells have a unique transcriptome largely driven by the nuclear receptor family member, PPARγ (Cipolletta et al., 2012), the “master-regulator” of adipocyte differentiation. The accumulation of VAT-Treg cells depends critically on the expression of Foxp3, specific TCRs and PPARγ (Li et al., 2018), as well as on interactions with subtypes of VAT mesenchymal stromal cells that produce the cytokine, IL-33 (Vasanthakumar et al., 2015; Kolodin et al., 2015; Spallanzani et al., 2019). VAT-Treg cells are highly represented in 20- to 30-week-old lean mice, reaching as high as 50–80% of the CD4+ T-cell compartment, but are reduced to 10–20% levels with severe obesity (Feuerer et al., 2009; Deiuliis et al., 2011). In addition, the Treg cells that persist in the VAT of obese mice show reduced expression of VAT-Treg signature genes such as Klrg1, Ccr2, Il1rl1 and Il10 (Cipolletta et al., 2015). Depletion of VAT-Treg cells in lean mice leads to increased expression of inflammatory genes and reduced insulin sensitivity, while augmentation of VAT-Treg cells during the course of obesity (e.g. by co-administration of a PPARγ agonist) can improve metabolic indices (Feuerer et al., 2009; Eller et al., 2011; Cipolletta et al., 2012). These findings argue that the dysregulation of VAT-Treg cells is one of the major promoters of obesity-associated inflammation and metabolic abnormalities.
Some progress has been made in identifying the mechanisms underlying the loss of VAT-Treg cells with obesity (Li et al., 2020). However, most of the relevant studies focused on animal models of severe obesity, such as mice fed a long-term (>8–20 weeks) high-fat diet (HFD) or genetically obese mice. Thus, only a late snapshot was taken, and we still lack a detailed view of the dynamics of the VAT-Treg population and its evolution during the onset, progression and late phases of obesity. In addition, such analyses are often complicated by the heterogeneity of Treg cells present in the VAT, potentially consisting of both true antigen-specific VAT-Treg cells that reside in the tissue long-term and polyclonal cells that are circulating through or have been installed temporarily. Consequently, there is often great inter-individual variability in the kinetics and severity of the downstream consequences of obesity. To circumvent these issues, we took advantage of the recently generated vTreg53 TCR-transgenic(tg) mouse line, which hosts a large population of bona fide VAT-Treg cells at a young age (10- to 12-weeks old) and shows more homogeneous inflammatory and metabolic indices (Li et al., 2018). We traced the dynamics of and transcriptional changes in the VAT-Treg population of vTreg53 TCR-tg mice during the development of HFD-feeding-induced obesity. VAT-Treg cells responded in two distinct stages: an early expansion phase characterized by increased activation, proliferation and maturation and then a sharp contraction phase accompanied by a loss of the distinct VAT-Treg transcriptome. The late response was associated with reduced Pparg expression and increased signaling by inflammatory cytokines, notably by IFNα produced by local plasmacytoid dendritic cells (pDCs). These findings highlight new therapeutic options for mitigating the effects of obesity.
RESULTS
The Response of VAT-Treg Cells to HFD Feeding Occurs in Two Distinct Phases
To examine the behavior of VAT-Treg cells during the onset, progression and late phases of obesity, we fed 12-week-old vTreg53 TCR-tg mice either a normal-chow diet (NCD) or an HFD for 2, 4, 8 or 16 weeks; we then examined the frequencies and numbers of clonotype+ Treg cells from the spleen and VAT. As expected, mice switched to an HFD gained significantly more body weight compared with those maintained on an NCD (Fig. 1A). The VAT weight had increased dramatically by as early as 2 weeks of HFD feeding but contracted at later times (8–16 weeks) (Fig. 1A), potentially reflecting increased fibrosis and adipocyte death (Rosen and Spiegelman, 2014). Consistent with previous reports (Feuerer et al., 2009; Cipolletta et al., 2015), while the frequency and number of clonotype+ Treg cells in the spleen of mice fed an NCD remained largely unaltered, those in the VAT increased substantially as the mice aged, starting at around 20% of the CD4+ T cell population and reaching as high as 70–80% by 8–16 weeks (Fig. 1B-D). In contrast, in mice fed an HFD, the VAT-Treg population increased during the early phase (2–4 weeks), attaining more than 60% of the CD4+ T cell compartment, but then declined to only about 20% at 8–16 weeks (Fig. 1B-D).
Fig 1. Evolution of the VAT-Treg compartment of vTreg53 TCR-tg mice with HFD feeding.
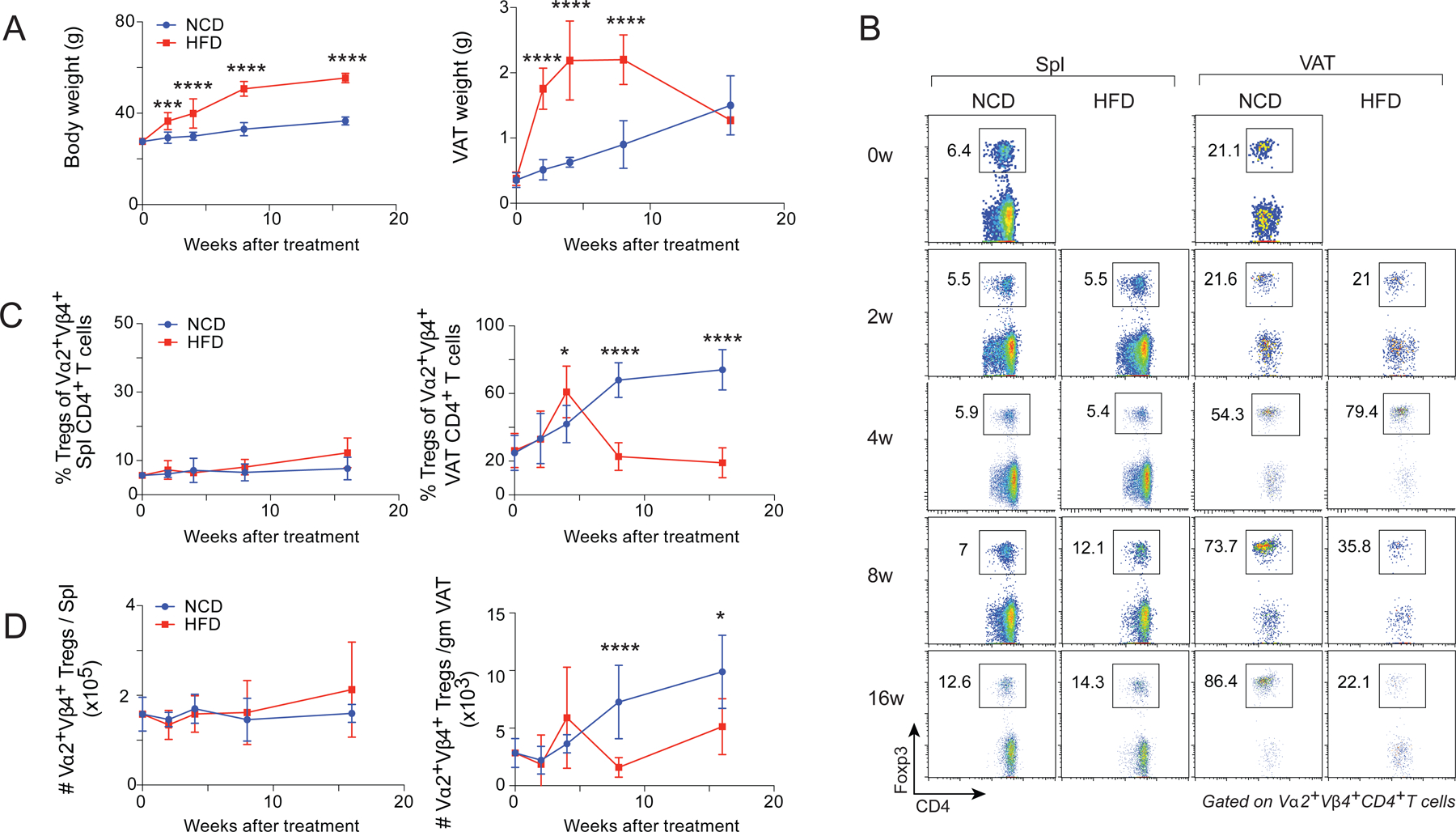
(A-D) 12-week-old male vTreg53 TCR-tg Foxp3-GFP mice were fed either an NCD or an HFD for the indicated durations (n ≥ 4 from 3 or 4 independent experiments, biological replicates). Mean ±SD.
(A) Summary plots of body (left) and VAT (right) weights.
(B) Representative flow cytometry plots (n ≥ 4, biological replicates) of Treg cells. w, weeks of treatment. Numbers in the plots indicate the frequencies of Foxp3+ Treg cells among Vα2+ Vβ4+ CD4+ T cells.
(C) Frequencies of Treg cells among clonotype+ CD4+ T cells from spleen (left) or VAT (right).
(D) Numbers of clonotype+ Treg cells from spleen (left) or VAT (right).
*P < 0.05, ***P < 0.001, ****P < 0.0001 by 2-way ANOVA (A, C, D).
We next compared how the transcriptomes of Treg cells evolved over the course of HFD vs NCD feeding by conducting population-level RNA-seq on double-sorted clonotype+ spleen and VAT-Treg cells. Principal component analysis (PCA) was performed on the four samples (spleen and VAT under NCD and HFD), which were then separated into two plots (NCD and HFD) for easier visualization (Fig 2A). The Treg transcriptomes segregated primarily by their tissue of origin. The transcriptional profile of spleen Treg cells remained quite constant throughout the time-course of both NCD and HFD feeding. In mice fed an NCD, the VAT-Treg transcriptome gradually evolved as the mice aged, becoming more and more distinct from the transcriptional profiles of their splenic counterparts (Fig. 2A, left). On the HFD, the transcriptome of VAT-Treg cells diverged from the transcriptional profiles of splenic Tregs at 2 and 4 weeks, but then gradually lost the distinguishing features and became more like the profiles of splenic Treg cells at 8 and 16 weeks (Fig. 2A, right). In agreement with the PCA, our previously established VAT-Treg up- and down-signatures (Cipolletta et al., 2015) were more prominent in clonotype+ VAT-Treg cells of mice fed an HFD than an NCD at the early time-points, suggesting that either short-term HFD accelerated the maturation of VAT-Treg cells or that there was a higher fraction of bona fide VAT Tregs (as opposed to “interlopers”). In contrast, with prolonged HFD feeding, VAT-Treg cells showed reduced expression of VAT-Treg up-signature genes, indicative of a loss of the distinct VAT-Treg features (Fig. 2B).
Fig 2. Global transcriptional changes induced in VAT-Treg cells by an HFD challenge.
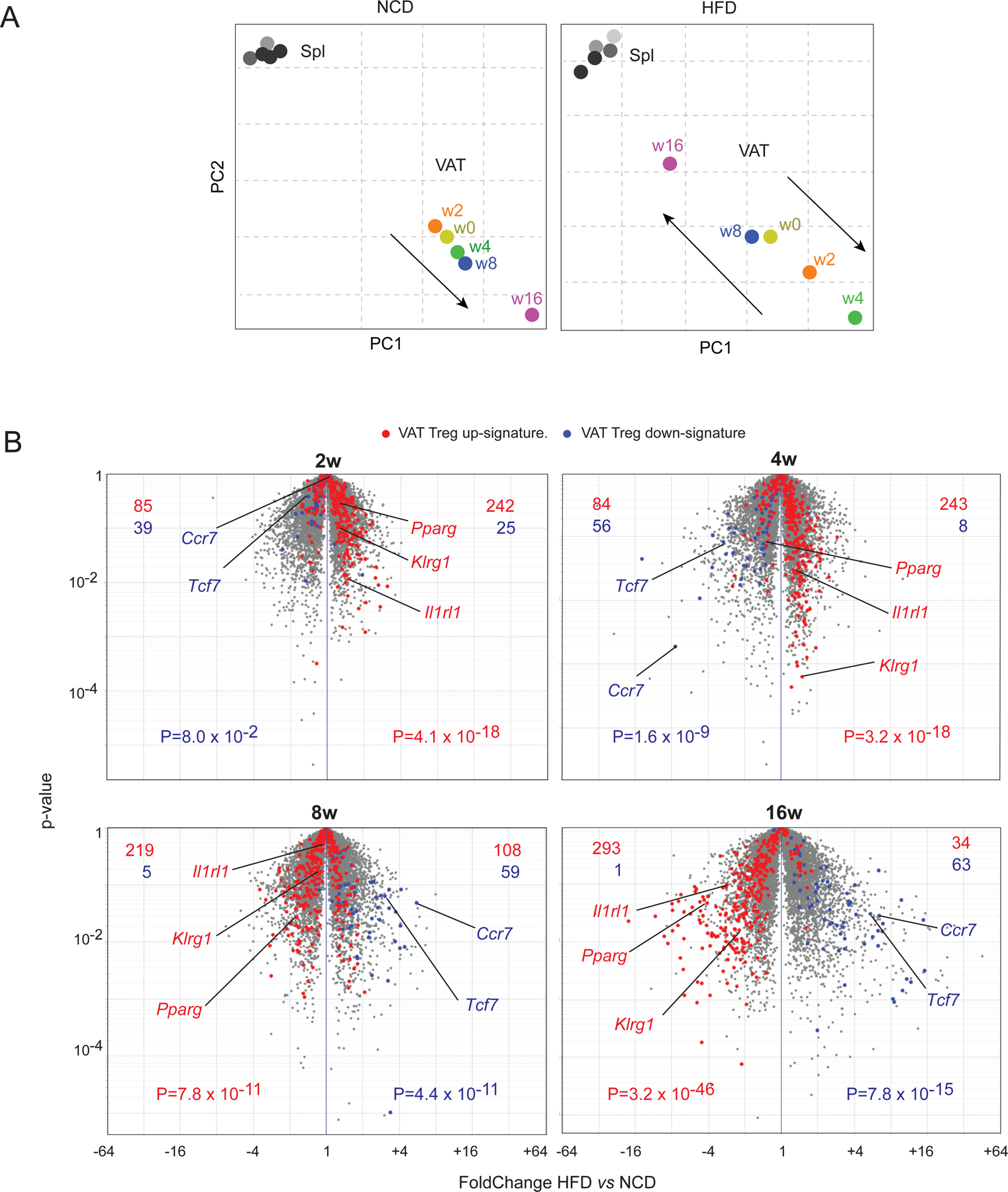
(A, B) Mice were treated as in Fig. 1, and clonotype+ Treg cells were sorted for RNA-seq analysis (n = 2 or 3). Each dot represents the mean of 2–3 biological replicates.
(A) Principal component analyses of transcriptomes of clonotype+ splenic or VAT-Treg cells at various time-points. PCA was done using all samples (Spl and VAT, NCD and HFD) and then separated into two plots for easier visualization. spl, spleen; w, weeks; PC, principal component
(B) Volcano plots comparing transcriptomes of clonotype+ VAT-Treg cells from mice fed a HFD vs NCD for the indicated durations. The VAT Treg up- and down- signatures have been reported (Cipolletta et al., 2015). Representative VAT Treg up- and down- signature genes were highlighted. Figures at the top indicate the number of genes up- (red) or down- (blue) regulated by one or the other population.
P value is determined by a Chi-square test (B).
Collectively, these results indicate that with HFD feeding the size of the VAT depot as well as the representation of VAT-Treg cells evolved in two distinct stages: an early expansion phase and a late contraction phase. The VAT-Treg transcriptome changed in parallel.
The Onset and Progression of Obesity are Associated with Alterations in the Proliferation and Death of VAT-Treg Cells
Gene-Set Enrichment Analysis (GSEA) of the RNA-seq data revealed which pathways were modulated during the onset and progression of obesity. VAT-Treg cells from mice fed an HFD for 2 weeks showed increased expression of genes related to cell activation and proliferation (e.g., Myc targets, E2F targets and the p53 pathway) (Fig. 3A), in agreement with the accelerated accumulation of VAT-Treg cells during the early phases of obesity (Fig. 1B-C). Indeed, a 5-hour in vivo pulse of the nucleoside analog, 5-ethynyl-20-deoxyuridine (EdU), demonstrated a higher proportion of dividing clonotype+ VAT-Treg cells in vTreg53 TCR-tg mice fed an HFD compared with an NCD for 2 weeks (Fig. 3B). Although GSEA also uncovered an increase in transcripts indicative of apoptosis at this time-point, we could not confirm this association by annexin V staining of VAT-Treg cells freshly isolated ex vivo (Fig. 3C). These results suggest that the accelerated accumulation of VAT-Treg cells during the onset of obesity is mainly driven by increased proliferation, rather than by reduced turn-over.
Fig 3. Early changes in VAT-Treg cell dynamics upon HFD feeding.
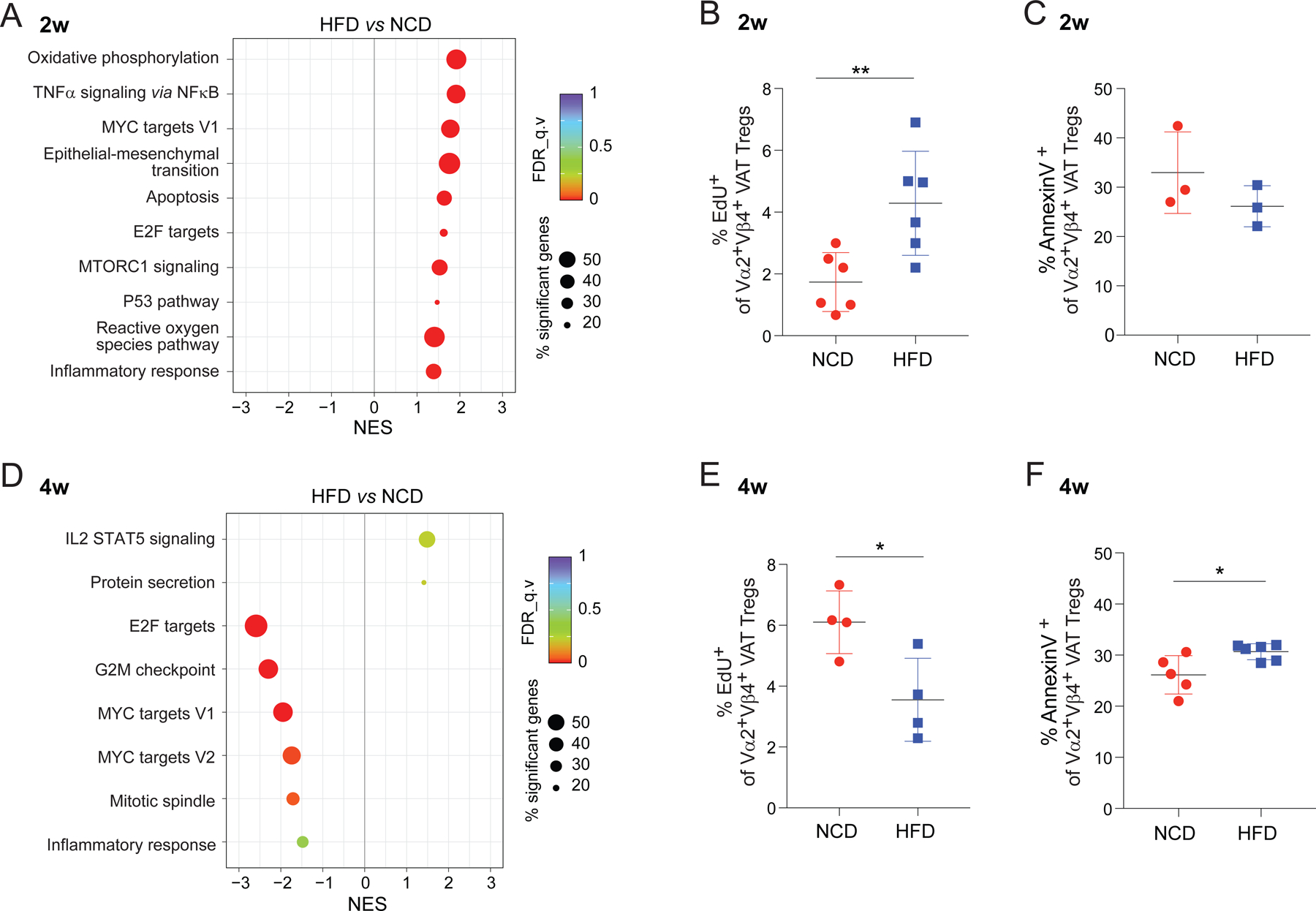
(A) GSEA showing the KEGG pathways most enriched or impoverished in clonotype+ VAT-Treg cells at 2 weeks of HFD, in comparison with NCD, feeding (P < 0.05). NES, normalized enrichment score; FDR, false discovery rate.
(B) Proliferation of clonotype+ VAT-Treg cells at 2 weeks of NCD or HFD feeding. Mice were pulsed for 5 hr with EdU, at which time tissues were flow-cytometrically analyzed for incorporation (n = 6, biological replicates).
(C) Flow-cytometrically determined frequencies of Annexin V+ clonotype+ Treg cells isolated from mice fed NCD or HFD for 2 weeks (n = 3, biological replicates).
(D-F) Same as A-C except mice were fed NCD vs HFD for 4 weeks (n ≥ 4, biological replicates).
Summary plots show data pooled from 2 or 3 independent experiments. Mean ± SD.
*P < 0.05, **P < 0.01 by unpaired Student’s t-test (B, C, E, F).
In striking contrast, VAT-Treg cells from mice fed an HFD for 4 weeks were relatively low in transcripts associated with proliferation and the cell cycle (e.g., E2F targets, G2M checkpoint, Myc targets and mitotic spindle) (Fig. 3D). And flow-cytometric analysis confirmed that the proportion of dividing Treg cells was lower, while that of Annexin V+ cells was greater, in the VAT of mice fed 4 weeks of HFD (compared with NCD) (Fig. 3E, F). Therefore, although still at their peak of accumulation at 4 weeks of HFD feeding (Fig 1B-D), VAT-Treg cells already exhibited clear signs of cell-cycle arrest and greater cell death. Perhaps not surprisingly, by 8 weeks after the introduction of HFD, the residual VAT-Treg survivors no longer showed any differences in cell proliferation or apoptosis (Fig. S1A, B).
Prolonged HFD Feeding is Associated with Loss of the Distinct VAT-Treg Transcriptome and with Increased Responses to Pro-inflammatory Cytokines
At 8 and 16 weeks after the introduction of HFD feeding, GSEA revealed strong inductions of transcripts indicative of responses to the pro-inflammatory cytokines IFNα, TNFα, and IFNγ; while known hallmarks of VAT-Treg cells – including the cholesterol homeostasis, peroxisome, and fatty-acid metabolism pathways – were significantly lower (Fig. 4A, B). k-means clustering of the transcripts differentially expressed by VAT-Treg cells over the course of HFD vs. NCD feeding (P < 0.05; FDR < 0.05; r2 > 0.65) yielded similar findings, but with more detailed kinetics (Fig. 4C). Cluster 1, enriched in transcripts encoding elements of the cholesterol homeostasis pathway (Hmgcr, Hmgcs1, Ldlr, etc.), was maintained at high levels under an NCD but was gradually lost with prolonged HFD feeding. Clusters 2 and 3 -- which included a number of VAT-Treg up-signature transcripts encoding cytokine or chemokine receptors (Ccr2, Cxcr6, Il9r), nuclear receptors (Pparg, Rora), proteins associated with TCR activation (Cd69, Fos, Egr1, Jun), or circadian rhythm components (Nr1d1) -- were increased under NCD feeding and with short durations of HFD feeding but declined with long-term exposure to an HFD. Cluster 4 transcripts, including Ago2, Cd274 and Nfatc2, were maintained at high levels throughout HFD feeding and with short-term NCD feeding, but were reduced with prolonged NCD feeding. Cluster 5 – consisting mainly of transcripts associated with the response to IFNs or TNFα and those encoding TNF family members – was induced under long-term HFD feeding.
Fig 4. The late response of VAT Tregs to an HFD: PPARγ down-regulation and up-regulation of pro-inflammatory cytokine signaling.
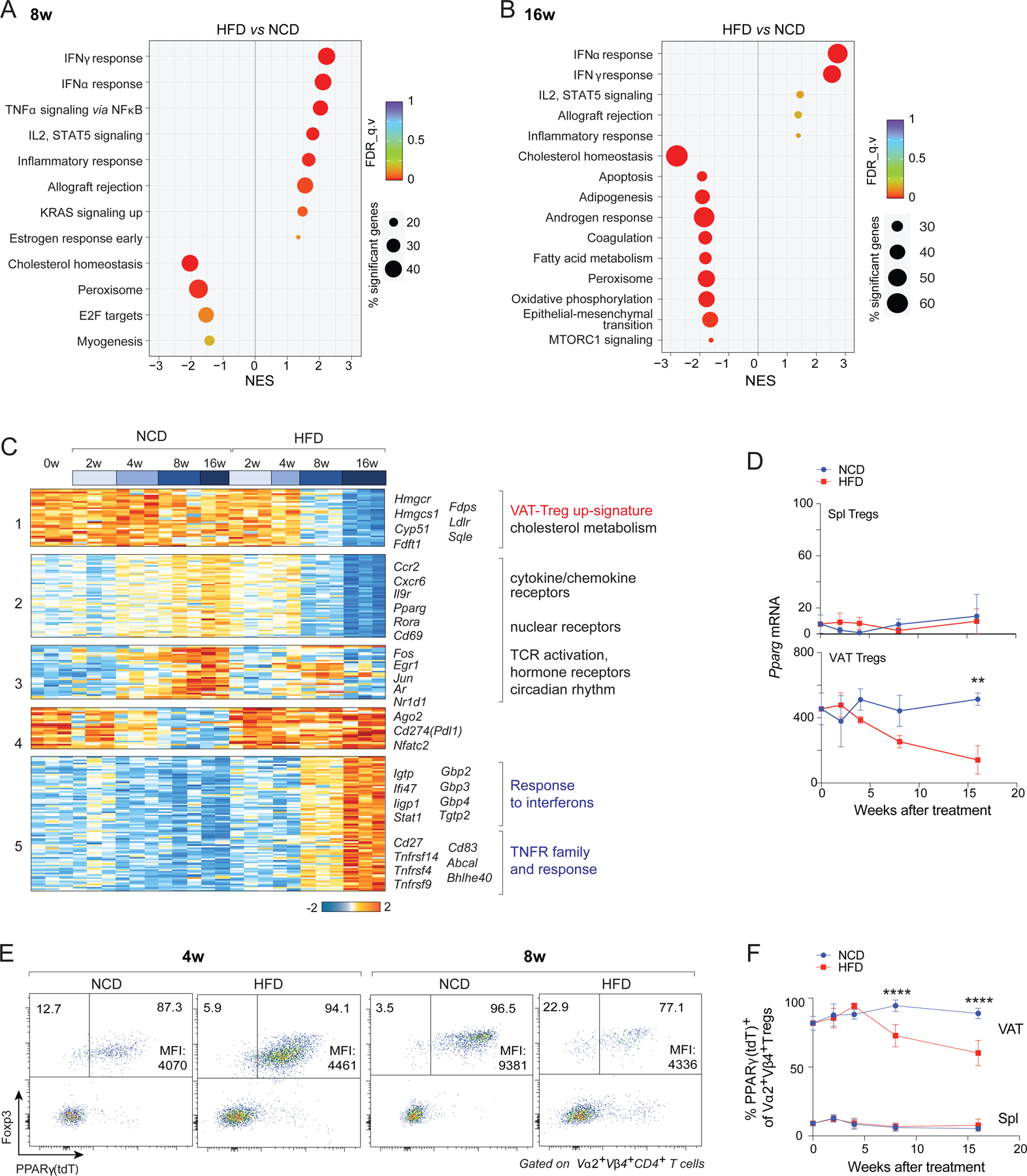
(A, B) GSEA showing top KEGG pathways enriched or impoverished in clonotype+ VAT-Treg cells at 8 (A) or 16 (B) weeks of HFD, compared with NCD, feeding (P < 0.05).
(C) k-means clustering of transcripts expressed in clonotype+ VAT-Treg cells differentially under NCD vs HFD conditions (P < 0.05, FDR < 0.05, r2 ≥ 0.65).
(D) RNA-seq quantification of Pparg transcript levels under the two feeding conditions (n = 2 or 3, biological replicates).
(E) Representative flow-cytometric plots (n ≥ 3, biological replicates) showing frequencies of PPARγ(tdT)+ among clonotype+ VAT-Treg cells. Numbers in the plots indicate the frequencies of PPARγ(tdT)− or PPARγ(tdT)+ cells among clonotype+ Treg cells. Mean fluorescent intensities (MFIs) of PPARγ(tdT) of only the PPARγ(tdT)+ Tregs are indicated.
(F) Summary plot showing frequencies of PPARγ(tdT)+ cells among clonotype+ Treg cells at various times after introduction of NCD or HFD (n ≥ 3, biological replicates). Summary plots show data pooled from two to three independent experiments. Mean ±SD.
**P < 0.01, ****P < 0.0001 by 2-way ANOVA (D, F).
PPARγ is a primary driver of VAT-Treg accumulation and phenotype (Cipolletta et al., 2012). While, as expected, VAT-Treg Pparg transcripts were maintained at high levels in mice on an NCD, they decreased with longer durations of HFD feeding (Fig. 4D). Reduced Pparg expression in VAT-Treg cells of mice on HFD was confirmed by examination of PPARγ-reporter mice, which showed both fewer PPARγ(tdT)+ cells and a lower mean fluorescence intensity (MFI) in those cells that were positive (Fig. 4E, F).
In brief, then, the major transcriptomic hallmarks of long-term HFD feeding were loss of the diagnostic VAT-Treg signature and responses to inflammatory cytokines. These changes did not occur in splenic Tregs from the same mice.
IFNα Directly Inhibits the Proliferation and Survival of PPARγ+ VAT-Treg Cells
The striking induction of transcripts indicative of responses to pro-inflammatory cytokines with prolonged HFD feeding prompted us to investigate whether these cytokines could inhibit the accumulation of VAT-Treg cells. In agreement with previous reports (Ghosh et al., 2016; Hotamisligil et al., 1993), levels of Ifna and Tnfa transcripts were significantly higher in VAT from mice fed 8 weeks on an HFD compared with those on an NCD; the abundance of Ifng transcripts was not higher at this time-point (Fig. 5A). Previous studies have shown that the accumulation of bona fide VAT-Treg cells over time critically depends on the cytokine, IL-33 (Vasanthakumar et al., 2015; Kolodin et al., 2015); however, the amount of IL-33 in VAT is not reduced, but rather increased, under long-term HFD feeding (Spallanzani et al., 2019). Therefore, we hypothesized that pro-inflammatory cytokines might be able to antagonize IL-33-driven accumulation of VAT-Treg cells. We injected two doses of IL-33 (3 days apart) into 8-week-old B6 mice and examined Treg compartments 7 days after the first injection. There was an impressive expansion of the Treg population in VAT (Fig. 5B), and a much weaker one in spleen (Fig. S2A). Injection of IFNα, TNFα, or IFNγ along with IL-33 inhibited IL-33-induced expansion of the VAT-Treg population (Fig. 5B). We did not observe noticeable effects of these cytokines on VAT-Treg cells in the absence of IL-33, likely due to the slow turnover of VAT-Treg cells (Kolodin et al., 2015) and the short duration of treatment.
Fig 5. Roles of IFNα, TNFα and IFNγ in the loss of VAT-Treg cells with HFD feeding.
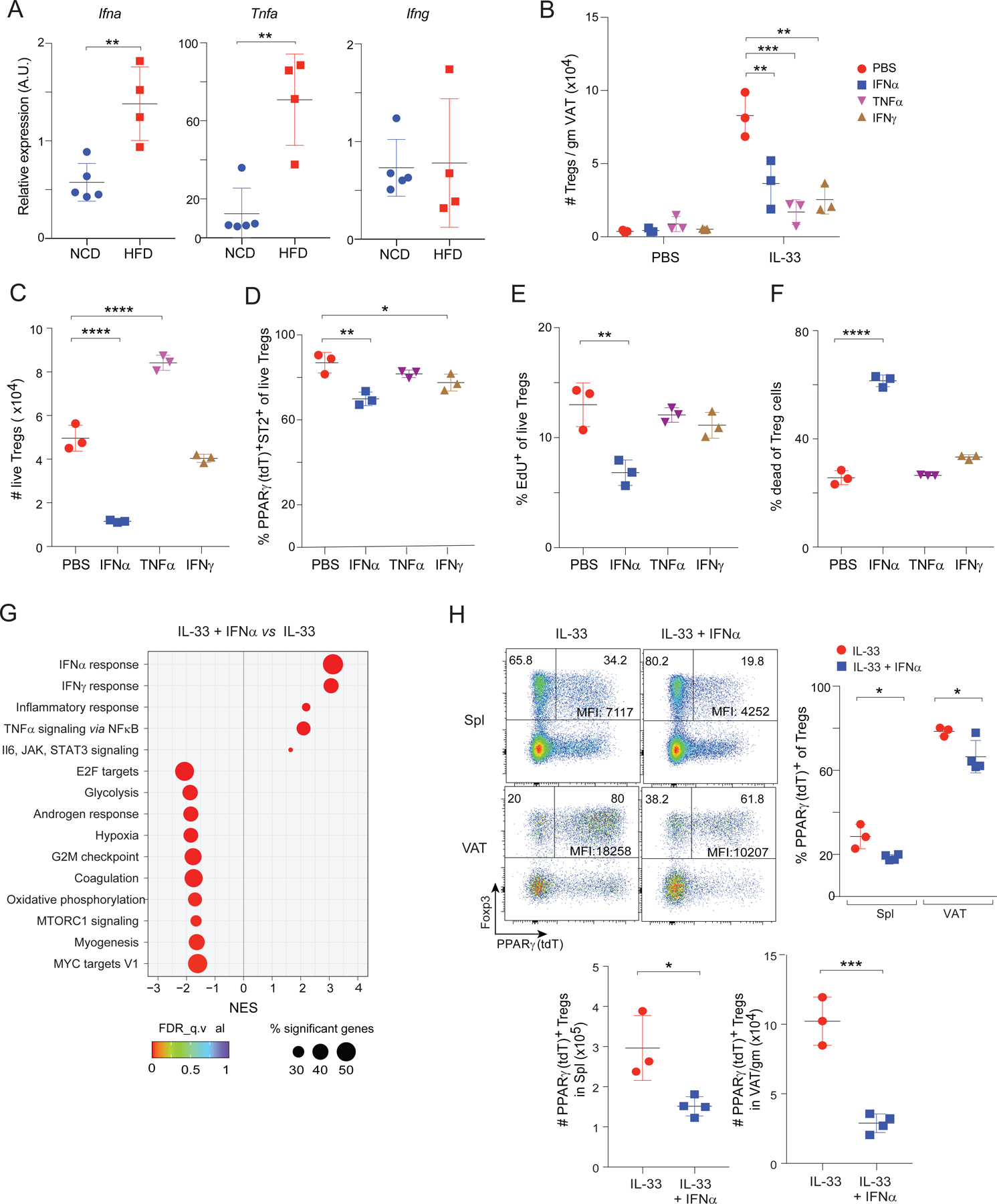
(A) Expression of the indicated cytokine genes in whole VAT tissue from vTreg53 TCR-tg mice fed 8 weeks on NCD or HFD as determined by RT-qPCR. (n ≥ 4, biological replicates).
(B) Number of VAT-Treg cells from 12-week-old B6 mice injected with various cytokines twice in a week (n = 3, biological replicates). PBS, phosphate-buffered saline.
(C-F) VAT-Treg cells were treated with indicated cytokines in vitro for 4 days (n = 3, biological replicates).
(C) Numbers of live VAT-Treg cells.
(D) Frequencies of PPARγ(tdT)+ ST2+ cells among live Treg cells.
(E) Frequencies of live Treg cells that had incorporated EdU in the last 2 hours of culture. (F) Frequencies of dead cells among Treg cells.
(G) Transcriptome analysis of VAT-Treg cells treated in vitro for 3 days (n = 2 or 3, biological replicates). GSEA showing top KEGG pathways enriched or depleted in VAT-Treg cells treated with IL-33+IFNα compared with IL-33 alone.
(H) 8-week-old PPARγ-Tdt Foxp3-GFP mice were injected with two doses of IL-33 alone or IL-33+IFNα in a week (n ≥ 3, biological replicates). Top-left panels: representative flow-cytometric dot-plots of PPARγ(tdT)+ Treg cells. Numbers in the plots indicate the frequencies of PPARγ(tdT)− or PPARγ(tdT)+ cells among Treg cells. MFI of PPARγ(tdT) for only the PPARγ(tdT)+ Treg cells is shown. Top right panels: frequencies of PPARγ(Tdt)+ Treg cells from spleen and VAT. Bottom panels: numbers of PPARγ(tdT)+ Treg cells.
Summary plots show data pooled from two to three independent experiments. Mean ±SD.
*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by unpaired Student’s t-test (A, H bottom), 1-way ANOVA (C), or 2-way ANOVA (B, H, top).
The effect of these pro-inflammatory cytokines could be Treg-cell intrinsic or could reflect indirect influences on other cell-types important for VAT-Treg accumulation, such as ILC2s. Indeed, and in agreement with results from previous studies (Duerr et al., 2016; Moro et al., 2016; Oldenhove et al., 2018; Molofsky et al., 2015), the numbers of ILC2s were reduced by injection of IFNα, TNFα or IFNγ together with IL-33, in comparison with administration of IL-33 alone (Fig. S2B, C). To determine whether any of the pro-inflammatory cytokines could directly antagonize IL-33-driven expansion of bona fide VAT-Tregs, we sorted PPARγ(tdT)+ VAT-Treg cells expanded in vivo with IL-33, and cultured them in vitro with IL-33 plus IFNα, TNFα or IFNγ. By four days of culture, there were much fewer surviving Treg cells with the addition of IFNα, but not with the other two cytokines (Fig. 5C); and the survivors included a lower proportion of PPARγ(tdT)+ST2+ cells (Fig. 5D).
Mechanistically, examination of EdU incorporation and of Live/Dead Violet Dead Cell Stain uptake revealed that IFNα suppressed both the proliferation and survival of VAT-Treg cells, while TNFα and IFNγ did not (Fig. 5E, F). Consistently, GSEA of RNA-seq data on VAT-Tregs treated with IL-33+IFNα vs IL-33 alone showed enrichment of the IFN and inflammatory responses under the former condition, and depletion of pathways associated with cell activation and proliferation (E2F targets, glycolysis, G2M checkpoint, oxidative phosphorylation) (Fig. 5G). We also generated a “16wk-HFD signature” of transcripts that were up- or down-regulated more than 2-fold in VAT Tregs from mice fed 16 weeks on HFD vs NCD (Table S1). Overlay of this signature on a volcano plot comparing gene expression by IFNα+IL-33- vs IL-33-treated VAT Tregs revealed an enrichment of the 16wk-HFD up-regulated gene-set and an impoverishment of the 16wk-HFD down-regulated gene-set in the former group (Fig S2D). Therefore, many of the transcripts changed by long-term HFD feeding were also modulated by IFNα. In addition, expression of Icos, which has been shown to be important for Treg proliferation, was not changed by IFNα treatment in VAT Treg cells (Fig S2E).
We next investigated whether the impact of IFNα was more pronounced in particular subtypes of Treg cells, notably those expressing PPARγ. Injection of IFNα alone into 8-week-old Pparg-tdT Foxp3-GFP mice did not alter the frequencies of PPARγ+ Treg cells in the spleen and VAT, although it did reduce the level of PPARγ in these cells (Fig S2F, G). We have shown previously that IL-33 injection leads to a significant increase in the proportion of PPARγ+ Treg cells (Li et al., 2018). Interestingly, compared with injection of IL-33 alone, co-injection of IFNα and IL-33 into 8-week-old Pparg-tdT Foxp3-GFP mice led to a significant reduction in the proportions and numbers of PPARγ+ Treg cells, as well as PPARγ(tdT) expression levels, in both spleen and VAT (Fig. 5H).
These results indicate that, while multiple pro-inflammatory cytokines were toxic to VAT-Tregs, only IFNα had a direct effect. This cytokine inhibited VAT-Treg accumulation through effects on both proliferation and survival.
Signaling via IFNAR1 on VAT-Treg Cells Drive their Loss with Prolonged HFD Feeding
Type-I IFNs (IFNα and IFNβ) signal through a common heterodimeric receptor consisting of two subunits, IFNAR1 and IFNAR2 (Barrat et al., 2019). To determine whether heightened type-I IFN production was actually responsible for the reduction in VAT-Treg cells under prolonged HFD feeding (and not just capable of causing it), we fed 12-week-old vTreg53 TCR-tg mice an HFD and injected them with either a blocking anti-IFNAR1 monoclonal antibody (mAb) or an IgG isotype-control mAb twice a week for 8 weeks. Blockade of IFNAR1 increased both the proportion and number of clonotype+ Treg cells in VAT but not in the spleen (Fig. 6A, B). In addition, the frequencies and numbers of CD11chi inflammatory macrophages in VAT were lower in mice treated with anti-IFNAR1 compared with those treated with IgG mAb (Fig S3A), likely as a result of higher VAT Treg cells in these mice. Remarkably, while body and VAT weights were comparable for the two groups (Fig. 6C, D), insulin sensitivity was significantly improved by anti-IFNAR1 treatment (Fig. 6E).
Fig 6. Systemic blockade or Treg-specific knock-out of IFNAR1 substantially rescued the VAT-Treg compartment and improved insulin sensitivity in conditions of HFD feeding.
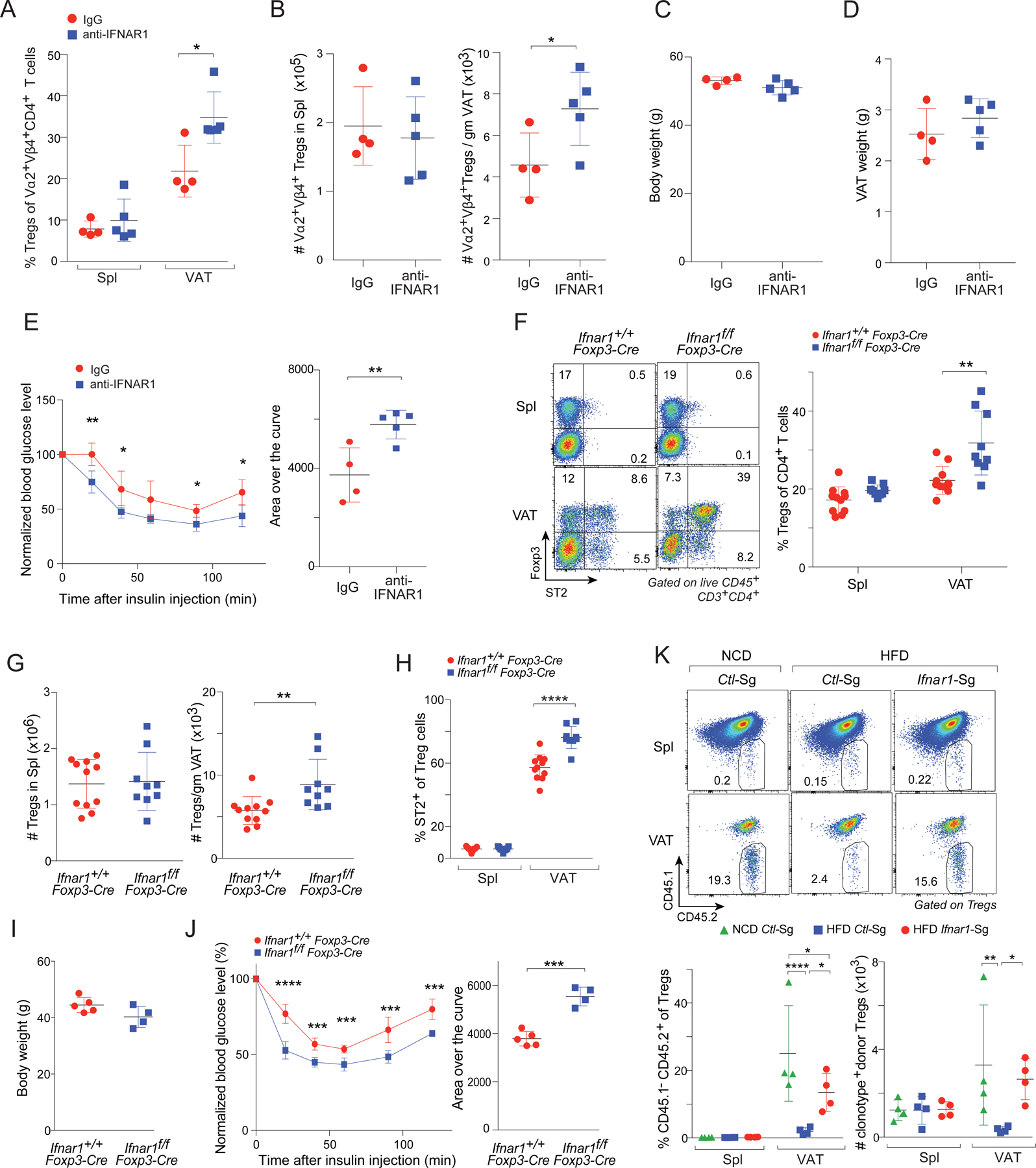
(A-E) 12-week-old vTreg53 TCR-tg mice were fed on HFD and injected with an anti-IFNAR1 or isotype-control Ab (IgG) i.p. twice a week for 8 weeks (n ≥ 4, biological replicates).
(A) Frequencies of clonotype+ Treg cells. Spl, spleen
(B) Numbers of clonotype+ splenic (left) or VAT-(right) Treg cells.
(C) Body weight.
(D) VAT weight.
(E) Insulin-tolerance test (ITT): normalized blood -glucose levels over time (left); area over the curve (AOC) (right).
(F-J) 12-week-old Ifnar1f/f or Ifnar1+/+ Foxp3-cre mice were fed on HFD for 8 weeks.
(F) Frequencies of Treg cells (n ≥ 9, biological replicates). Left: Representative flow-cytometric plots of ST2+ Treg cells. Numbers in the plots indicate the frequencies of cells in each gate. Right: Summary plot.
(G) Numbers of Treg cells in the spleen (left) and VAT (right). (n ≥ 9, biological replicates).
(H) Frequencies of ST2+ cells in Treg cells. (n ≥ 9, biological replicates).
(I) Body weight. (n ≥ 4, biological replicates).
(J) Insulin tolerance test (ITT). normalized blood -glucose levels over time (left); area over the curve (AOC) (right). (n ≥ 4, biological replicates).
(K) Effect of CRISPR/Cas9-mediated ablation of Ifnar1 in vTreg53 TCR-tg Treg cells on their enrichment in VAT following transfer into NCD- or HFD-fed mice (n = 4, biological replicates). Top: representative flow-cytometric dot-plot of donor-derived Treg cells. Numbers in the plots indicate the frequencies of donor-derived cells among total Treg cells. Bottom: frequencies and numbers of donor-derived Treg cells.
Summary plots show data pooled from two to three independent experiments. Mean ±SD.
*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by unpaired Student’s t-test (B, E right, G, J right) or 2-way ANOVA (A, E left, F, H, J left, K bottom).
As many cell types express IFNAR1 and could potentially mediate the benefits observed after anti-IFNAR1 mAb treatment, we determined whether ablation of IFNAR1 on Tregs alone was sufficient to confer these effects. In addition, we wanted to extend our observations to a polyclonal setting. Thus, we generated 12-week-old Ifnar1f/f and Ifnar1+/+ Foxp3-cre mice and fed them an HFD for 8 weeks. Consistent with the anti-IFNAR1 mAb treatment, the frequencies and numbers of VAT Treg cells as well as the fraction of ST2+ Treg cells in VAT were significantly increased in Ifnar1f/f Foxp3-Cre mice compared with Ifnar1+/+ Foxp3-cre littermates (Fig 6F-H). Importantly, Ifnar1f/f Foxp3-Cre mice had body weights that were similar to those of Ifnar1+/+ Foxp3-Cre littermates that had undergone the same treatment but they were significantly more insulin sensitive (Fig 6I, J).
As a second approach to assessing the importance of IFNAR1 specifically on Tregs, we set up an adoptive-transfer system. vTreg53 TCR-tg mice were crossed with mice constitutively expressing CRISPR-associated protein 9 (Cas9) from the Rosa26 locus. Treg cells were purified from pooled spleen and lymph nodes of CD45.1−CD45.2+ TCR-tg+ Cas9KI/WT Foxp3-thy1.1KI/y mice and were transduced with retroviral constructs expressing a single guide RNA (sgRNA) targeting Ifnar1 (Ifnar1-Sg) or a control non-targeting sgRNA (Ctl-Sg). The transduced cells (labeled by GFP) were transferred into 12-week-old CD45.1+CD45.2+ Cas9KI/WT recipients, which were then fed either an NCD or an HFD for 8 weeks (see Fig. S3B for the experimental scheme). As expected, transduction of Ifnar1-Sg resulted in efficient ablation of IFNAR1 on Treg cells (Fig. S3C). In agreement with our previous report (Li et al., 2018), Ctl-Sg-transduced Tg+ Treg cells preferentially accumulated in VAT (vs spleen) after transfer into mice fed an NCD; and this build-up was strongly attenuated in recipient mice fed an HFD (Fig. 6K). Remarkably, ablation of IFNAR1 only on Treg cells largely resurrected the accumulation of transferred Tg+ Treg cells in VAT of mice fed an HFD (Fig. 6K). (Note: In transfer systems like this one and the one below, too few Treg cells accumulate in VAT to be able to impact metabolic indices.)
As an alternative strategy, we transduced Treg cells from CD45.2+ TCR-tg+ Cas9KI/WT mice with retroviral constructs expressing Ifnar1 or Ctl SgRNAs (cells labeled with GFP or mRFP, respectively); mixed them at a 1:1 ratio; and transferred the mixture into recipient mice that were subsequently fed an HFD. After 8 weeks, while the fractions of Ifnar1-Sg-transduced and Ctl-Sg-transduced donor-derived Treg cells were quite similar in the spleen, there were clearly more of the former in the VAT of recipient mice (Fig. S3D).
These findings demonstrate that the suppression of VAT Treg cell accumulation by long-term HFD feeding was largely due to a direct effect of increased type-I IFN levels, and that ablation of IFNAR1 specifically on Tregs was sufficient to attenuate their reduction upon HFD feeding and improve metabolic indices in obese mice. Considered together with the gain-of-function results depicted in Fig. 5, these loss-of-function data constitute a strong argument for the primacy of these pro-inflammatory cytokines.
pDCs are Enriched in VAT with Long-term HFD Feeding, Produces Substantial Levels of IFNα and Suppresses VAT-Treg Accumulation
pDCs are a unique population of immunocytes that secrete large amounts of IFNα in response to viral infections or in certain autoimmune conditions (Liu, 2005). Thus, we wondered whether changes in this cell population might underlie the higher IFNα expression in mice on an HFD. Indeed, the frequency and number of pDCs (identified by the markers PDCA-1 and Ly6c) were strongly elevated in VAT of mice fed an HFD for 8 weeks (Fig. 7A). This increase was evident already 4 weeks after the introduction of HFD (Fig. 7B), correlating well with the time-point when the representation of VAT-Treg cells and their expression of PPARγ began to decline (Figs. 1C and 4D).
Fig 7. IFNα-expressing pDCs increase in the VAT during prolonged HFD feeding and inhibit VAT-Treg accumulation.
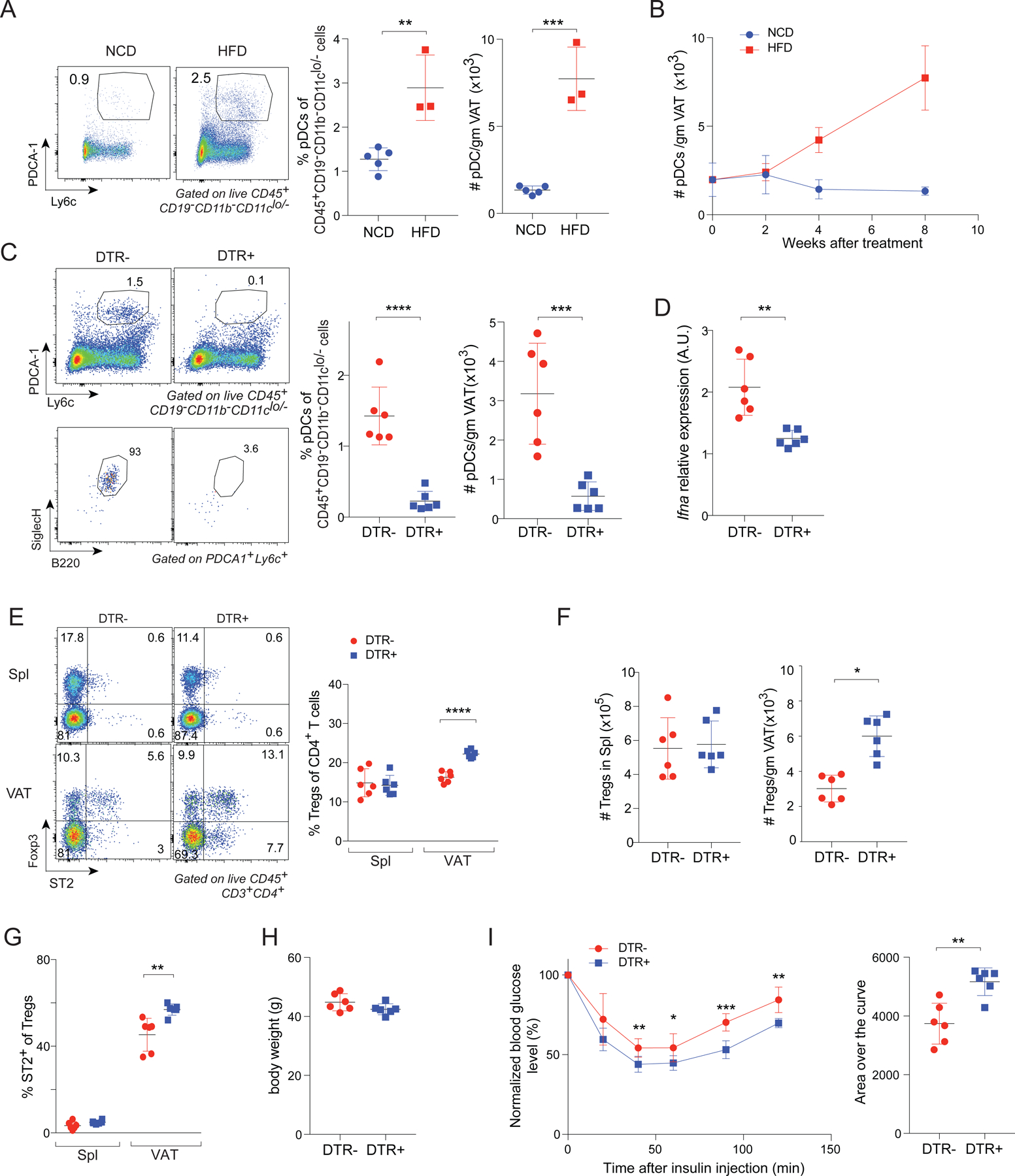
(A) Frequencies and numbers of pDCs from VAT of vTreg53 TCR-tg mice fed on NCD or HFD for 8 weeks (n ≥ 3, biological replicates). Left: representative flow cytometric dot-plots of pDCs. Numbers in the plots indicate the frequencies of pDCs among CD45+ CD19− CD11b− CD11cLo/- cells. Center and right: summary data.
(B) Time-course of pDC numbers from VAT of vTreg53 TCR-tg mice fed a NCD or HFD beginning at 12 weeks of age (n ≥ 3, biological replicates).
(C) pDCs from VAT of HFD-fed, DT-treated Bdca2-Dtr+ or Dtr− mice. (n = 6, biological replicates). Left: representative flow-cytometric dot-plots of pDCs. Numbers in the plots indicate the frequencies of PDCA-1+Ly6c+ cells among CD45+ CD19− CD11b− CD11cLo/- cells (top left) or the frequencies of B220+ SiglecH+ cells among PDCA-1+Ly6c+ cells (bottom left). Center and right: summary data showing frequencies and numbers of pDCs in VAT.
(D) RT-qPCR quantification of Ifna transcript levels from whole VAT tissue of HFD-fed, DT-treated Bdca2-Dtr+ or Dtr− mice (n = 6, biological replicates). A.U., arbitrary units.
(E) Frequencies of Tregs from spleen and VAT of HFD-fed, DT-treated Bdca2-Dtr+ or Dtr− mice (n = 6, biological replicates). Left: representative flow-cytometric dot-plots of ST2+ Treg cells. Numbers in the plots indicate the frequencies of cells in each gate. Right: summary data.
(F) Numbers of Tregs from spleen (left) and VAT (right) of HFD-fed, DT-treated Bdca2-Dtr+ or Dtr− mice (n = 6, biological replicates).
(G) Frequencies ST2+ cells in Spl or VAT Tregs (n = 6, biological replicates).
(H) Body weight (n = 6, biological replicates).
(I) ITT. normalized blood-glucose over time (left); area over the curve (AOC) (right). (n = 6, biological replicates).
Summary plots show data pooled from two to three independent experiments. Mean ±SD.
*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by unpaired Student’s t-test (A, C, D, F, I right) or 2-way ANOVA (E, G, I left).
To evaluate the impact of pDCs on VAT-Treg cells under obesogenic conditions, we obtained Bdca2-Dtr mice, wherein injection of diphtheria toxin (DT) leads to highly specific depletion of pDCs (Swiecki et al., 2010). 10-week-old Bdca2-Dtr+ or Dtr− littermates were fed an HFD for 4 weeks, after which they were continued on an HFD while being injected i.p. with DT twice a week for another 4 weeks. DT was administered only for the latter 4 weeks of HFD feeding to avoid induction of neutralizing Abs upon chronic treatment, and because pDCs started to accumulate only after 4 weeks of HFD feeding (Fig 7B). DT treatment of Bdca2-Dtr+ mice indeed led to a specific depletion of pDCs (identified by markers including PDCA-1, Ly6c, Siglec-H, and B220) and a reduction in Ifna transcript levels compared with those of Dtr− littermates (Fig 7C, D). There were significant increases in the frequencies and numbers of Tregs as well as in their ST2 expression levels on VAT- but not spleen -- Tregs (Fig 7E-G). DT-treated Bdca2-Dtr+ mice had body weights similar to those of Bdca2-Dtr− littermates that received the same treatment, but they were more insulin sensitive (Fig 7H, I).
Because of its translational implications, and although it might not be as specific an approach under inflammatory conditions (Blasius et al., 2006), we also punctually ablated pDCs using a mAb. pDCs in vTreg53 TCR-tg mice were continuously depleted using anti-PDCA-1 and the VAT-Treg population was examined after 8 weeks of HFD feeding. As expected, depletion of VAT pDCs under these conditions resulted in a significant reduction in whole-tissue Ifna transcript levels (Fig. S4A). Strikingly, pDC depletion significantly increased the clonotype+ VAT-Treg population (Fig. S4B), particularly those cells expressing PPARγ (Fig. S4C). Lastly, while the body and VAT weights were comparable between vTreg53 TCR-tg+ mice treated with α-PDCA1 and control-IgG (Fig. S4D, E), pDC-depleted mice exhibited a clear improvement in insulin sensitivity (Fig. S4F).
In short, these results – obtained from both a constitutive and a punctual depletion system – strongly argue that type-I IFN, largely produced by pDCs, was a major driver of the loss of VAT-Treg cells and their PPARγ expression during prolonged HFD feeding.
DISCUSSION
VAT-Treg cells safeguard VAT homeostasis and, thereby, metabolic health. Hence, they have been thought to be a promising therapeutic target for treating obesity-associated metabolic disorders. Unfortunately, the use of Treg-based therapy in this context is hindered by the induction of an adipose-tissue microenvironment that is toxic to VAT-Treg cells in individuals with obesity. The major goals of our study were to explore the dynamics of VAT-Treg cells during the onset, progression and late phases of obesity, and to elucidate mechanisms underlying the eventual dysregulation of VAT-Treg cells. Our data indicate that VAT-Treg cells responded to obesity in two distinct stages. Short-term feeding on an HFD induced expansion of the gonadal VAT depot and accelerated the proliferation and potentially the maturation of VAT-Treg cells. However, prolonged HFD feeding exceeded the expansion capacity of this adipose depot, resulting in regression of the VAT mass. This effect was associated with the accumulation and activation of pDCs that expressed high levels of IFNα, which directly inhibited VAT-Treg accumulation, eventually resulting in a compartment lacking the typical VAT-Treg characteristics.
The impressive increase in VAT-Treg cells shortly after the introduction of an HFD was somewhat unexpected and raises questions as to its causation and biological meaning. Our results argue that this increase was largely due to an augmented proliferation of VAT-Treg cells, likely driven by increased TCR activation, rather than reflecting an enhanced survival of these cells. The precise reason for this elevated TCR activation is currently unknown, but we suspect that it is related to increased maturation of, antigen-presentation by, and/or co-stimulation by antigen presenting cells in VAT upon HFD challenge, as has been described for conventional DCs and MHCII+ MFs (Morris et al., 2013; Macdougall et al., 2018). This initial response of VAT-Treg cells to nutritional perturbations could be a mechanism to dampen the progression of inflammation and thus safeguard organismal metabolism.
Long-term HFD feeding (> 8 weeks) led to a strong reduction in VAT, but not spleen, of Treg cell expression of Pparg transcripts – both the fraction of cells expressing this nuclear receptor and the intensity of their expression. PPARγ is known to be a major and specific orchestrator of the accumulation and phenotype of VAT-Treg cells under lean conditions (Cipolletta et al., 2012). With NCD, mice lacking PPARγ specifically in Treg cells had a greatly reduced VAT (but not spleen) Treg compartment and, perhaps most relevant here, acute administration of a PPARγ antagonist provoked rapid death of bona fide VAT- (but not spleen-) Treg cells. Hence, the reduction of PPARγ expression by VAT-Treg cells with prolonged HFD feeding is almost certainly an important contributor to their dysregulation.
Our data also established that IFNα was a key, direct driver of VAT-Treg loss during the progression of obesity. Increased production of IL-21, IFNγ or TNFα, and elevated activity of STAT3, in VAT have all been suggested to underlie the dysregulation of local Treg cells during obesity (Deng et al., 2017; Fabrizi et al., 2014; Priceman et al., 2013; Cipolletta et al., 2015). Ablation of whole-mouse IL-21 or T-cell-specific loss of STAT3 rescued the VAT-Treg population and improved insulin sensitivity. However, a caveat of these findings is that these mice resisted HFD-induced weight gain, so the restoration of VAT-Treg cells could be secondary to differences in body and VAT weight, rather than from direct effects of these molecules on VAT-Treg cells, themselves. Similarly, although implicated in the in vivo dysregulation of VAT-Treg cells, IFNγ and TNFα did not directly influence VAT-Treg cells in our in vitro culture system, arguing that these two cytokines might inhibit VAT-Treg accumulation in vivo through indirect mechanisms, such as through ILC2s.
In contrast, treatment of purified VAT Treg cells with IFNα directly inhibited their proliferation and induced cell death, while mAb blockade of IFNAR1 signaling or Treg-specific ablation of IFNAR1 by genetic means substantially restored the accumulation of VAT-Treg cells subsequent to prolonged HFD feeding and improved insulin sensitivity without affecting body weight. Previous studies investigating the influence of type-I IFNs on Treg cells were mostly limited to those from lymphoid-organs and the effects could be either positive or negative, depending on the context (Srivastava et al., 2014; Gangaplara et al., 2018; Kawano et al., 2018; Lee et al., 2012; Metidji et al., 2015). VAT Tregs reside in a unique tissue microenvironment and have distinct dependencies on cytokines and growth factors. Therefore, it is important to determine how type-I IFNs affect VAT Tregs specifically and how any effects relate to the dysregulation of VAT Tregs during obesity. Indeed, we found that the PPARγ+ subset of Treg cells was especially sensitive to IFNα-mediated inhibition, which rendered PPARγ+ VAT-Treg cells particularly vulnerable to HFD challenge. Our findings are somewhat similar to a report from Hannibal et al (Hannibal et al., 2017) that deficiencies in type-1 IFN and pDCs prevent diet-induced obesity and accompanying insulin resistance, but they differ in several important regards. For example, Hannibal et al studied mice with a life-long, systemic loss of IFNAR1, which likely explains their observation of resistance to weight gain in the mutant mice, an unfortunate confounder. Also, they looked at effects only on myeloid cell populations.
As major producers of type-I interferons, pDCs are critical for mounting proper anti-viral immune responses, but are also implicated in exacerbating certain autoimmune diseases, like systemic lupus erythematosus (Reizis et al., 2011). pDCs sense single-stranded RNA and microbial DNA via endosomal Toll-like receptor (TLR)-7 and 9, activating a MyD88-dependent signaling cascade that culminates in production of inflammatory mediators, in particular type-1 IFNs. We found that pDCs began to increase in VAT after 4 weeks of HFD feeding, which marked the onset of a regression of VAT mass, reduction in the local Treg compartment, and loss of the bona fide VAT-Treg transcriptome. Depletion of pDCs substantially restored VAT-Treg cell accumulation, increased their PPARγ and ST2 expression, and improved insulin sensitivity in mice fed long-term on HFD. Thus, although VAT-Treg cells initially expanded and matured to counteract HFD-induced inflammation, the progression of obesity resulted in release of extracellular DNA from dying or dead adipocytes, which was sensed by pDCs and induced them to produce large amounts of type-I IFNs, which were toxic to VAT-Treg cells.
Targeting pDCs or the type-I IFN pathway might thus be a promising add-on for Treg-based therapy in the treatment of insulin resistance and metabolic diseases. This approach would allow transfer or induction of VAT-Treg cells to overcome a toxic environment and to effectively exert their beneficial effects. Translation of this strategy to patients is argued by published reports of an enriched population of Tregs in human VAT and its reduction with obesity (Wu et al., 2019); of augmented levels of pDCs, type-I IFNs, and IFN-response transcripts in obese humans (Stefanovic-Racic et al., 2012; Ghosh et al., 2016); and of positive correlations between IFN-response indicators in VAT and levels of inflammatory MFs as well as both local and systemic insulin resistance (Ghosh et al., 2016).
Limitations of Study
One limitation of our study is the absence of experiments to directly translate our findings to humans. Although a reduction in Treg cells and an augmentation in type-I IFN-producing pDCs in VAT from individuals with obesity have been reported, further clinical studies are required to determine whether IFNα producing pDCs indeed directly inhibit the homeostasis of VAT-Treg cells and worsen metabolic indices in subjects with obesity. In addition, the detailed molecular mechanisms by which IFNα produced by pDCs is detrimental to PPARγ+ VAT Treg cells and why these cells are particularly sensitive to IFNα-induced toxicity are still largely unknown and requires further investigation.
STAR METHODS
RESOURCE AVALABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Diane Mathis (Diane_Mathis@hms.harvard.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The accession number for the RNA sequencing data reported in this paper is GEO: GSE174706.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
Male mice were produced in our specific-pathogen-free facilities at Harvard Medical School or Emory University School of Medicine and were fed an NCD (13 kcal% fat, no. 5053, Lab Diet Picolab Mouse Diet 20) until 10–12 weeks of age. They were then either continued on an NCD or switched to an HFD (60 kcal% fat, D12492, Research Diets) for 2, 4, 8, or 16 weeks. Mice were housd in a temperature (22–24°C)-controlled colony room with a 12-h light/dark cycle under specific-pathogen-free (SPF) conditions. Littermates were used for all experiments. B6.CD45.2+ (000664), B6.CD45.1+ (002014), B6.Rosa26-Cas9 (026179) (Platt et al., 2014), B6.Ifnar1fl (028256) (Prigge et al., 2015) and B6.Foxp3-cre (016959) (Rubtsov et al., 2008) mice were obtained from the Jackson Laboratory. vTreg53 TCR-tg and Pparg-tdT mice were generated on the B6 background in our laboratory as previously described (Li et al., 2018). Foxp3-GFP (Bettelli et al., 2006) and Foxp3-Thy1.1 (Liston et al., 2008) mice were obtained from Dr. A. Rudensky and were maintained on the B6 background. B6.Bdca2-Dtr mice were obtained from Dr. M. Colonna and rederived by the Mouse Transgenic and Gene Targeting Core at Emory University. All experiments were performed using littermate controls and repeated at least twice. The animal protocols were approved by the HMS Institutional Animal Use and Care Committee (IACUC) (protocol IS00001257) and Emory University IACUC (PROTO201900197).
Retroviral Transduction and Cell Transfer
Treg cells were sorted from pooled spleen and lymph nodes of 6–8-week-old male CD45.1−CD45.2+ TCR-tg Cas9KI/WT Foxp3-thy1.1KI/y mice, stimulated with Dynabeads™ Mouse T-Activator CD3/CD28 for T-Cell Expansion and Activation (Invitrogen) at 37°C in a 5% CO2 humid atmosphere, transduced with retrovirus expressing sgRNA targeting Ifnar1 or a control non-targeting sgRNA, and were intravenously (i.v.) transferred (0.2×106 per mouse) into 12-week-old male CD45.1+CD45.2+ Cas9KI/WT recipients previously fed with NCD. In one set of experiment, both the Ifnar1- and Ctl-sgRNA expressing constructs also encoded GFP, and transduced cells were transferred into separate groups of recipient mice subsequently fed with either NCD or HFD for 8 weeks. In another set of experiments, the Ifnar1-sgRNA construct co-expressed GFP, while the Ctl-sgRNA construct co-expressed mRFP. The transduced cells were mixed at a 1:1 ratio, and the mixture was transferred into recipient mice subsequently fed on HFD for 8 weeks.
Analysis of Cell Proliferation and Death
For monitoring of cell division in vivo, 40 µg/g body weight EdU (Invitrogen) was intraperitoneally (i.p.) injected into mice fed with NCD or HFD, and 5 hrs later, cells were processed for detection by the Click-iT Plus EdU Cell Proliferation Kit following the manufacturer’s instructions (Invitrogen). For detection of Treg-cell death directly ex vivo, freshly isolated cells were stained with fluorochome-labeled Annexin V according to the manufacturer’s instructions (BioLegend). For monitoring of VAT-Treg cell division in vitro, PPARγ+ Treg cells were sorted from VAT of 8–10-week-old Pparg-tdTKI/KI Foxp3-GFPKI/y male mice that were injected with 2ug recombinant mouse IL-33 (BioLegend) twice in a week, and were cultured in the presence of 2000 U/ml recombinant human IL-2 (Peprotech) and 10 ng/ml IL-33 with or without 50ng/ml TNFα, IFNγ, or IFNα (BioLegend) for 4 days at 37°C in a 5% CO2 humid atmosphere. In the last 2 hrs of culture, cells were pulsed with 10 µM EdU, and cell proliferation and death were detected by the Click-iT Plus EdU Cell Proliferation Kit and LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit, respectively.
In Vivo Cytokine Treatment
For in vivo cytokine treatment, 8-week-old B6 or Pparg-tdT Foxp3-GFP male mice were i.p.-injected with 2 µg TNFα, IFNγ, or IFNα with or without 2 µg IL-33 twice in a week. The frequencies and numbers of spleen- and VAT-Treg cells as well as ILC2s were analyzed on day 7 after the first injection.
METHOD DETAILS
Male mice were asphyxiated with CO2 and perfused with 10–20ml of phosphate-buffered saline (PBS). Epididymal VAT was excised, minced and digested for 20 min with 1.5 mg/ml collagenase type II (Sigma) in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 2% fetal calf serum (FCS) in a 37°C water bath with shaking. The digested materials were filtered through a sieve and then a 40 µm nylon cell strainer, and the stromal vascular fraction (SVF) was collected after centrifugation at 450 x g for 10 min. For T lymphocyte analysis, cells were stained with anti-CD45.1 (A20), -CD45.2 (104), -CD3 (17A2), -CD4 (GK1.5), -CD25 (PC61), -KLRG1 (2F1/KLRG1), -IFNAR1 (MAR1–5A3), and/or -TCR Vα2 (B20.1) mAbs (all from BioLegend); anti-ST2 (RMST2–2) (eBioscience); anti-TCR Vβ4 (KT4), -Thy1.1 (OX-7), and/or -Thy1.2 (53–2.1) mAbs (all from BD Biosciences), and/or LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Invitrogen). Cells were either fixed, permeabilized, and intracellularly stained for Foxp3 (FJK-16 s) according to the manufacturer’s instructions (eBioscience) or were fixed by 2% paraformaldehyde (PFA) in PBS for 20 min before analysis. For myeloid cell analysis, cells were stained with anti-CD45 (30-F11), -CD11b (M1/70), -CD11c (N418), -F4/80 (BM8), -CD19 (1D3/CD19), -PDCA1 (927), -B220 (RA3–6B2), -SiglecH (551), and/or - Ly6c (HK1.4) mAbs (all from BioLegend). All Abs were used at a 1 to 100 dilution for staining. Cells were acquired with LSRII or FACSymphony A3 flow cytometers (BD Biosciences) and sorted with MoFlo Astrios EQ cell sorter or MoFlo Legacy (Beckman Coulter), Data were analyzed using FlowJo software.
In vivo Antibody Treatment
For the IFNAR1-blockade experiments, 12-week-old male vTreg53 TCR-tg mice were fed with HFD, and were injected i.p. with 250 µg anti-mouse IFNAR1 mAb (BioXCell, clone MAR1–5A3) or mouse IgG1 isotype control (BioXCell, clone MOPC-21) twice a week for 8 weeks. Any effects of the Ab treatments on metabolic parameters were determined 7 weeks after the introduction of HFD, and the mice were euthanized for flow cytometric analysis 8 weeks after HFD introduction.
Depletion of Plasmacytoid Dendritic Cells
For the pDC-depletion experiments using anti-PDCA-1 Ab, 12-week-old male vTreg53 TCR-tg mice were fed with HFD, and were injected i.p. with 250ug anti-mouse PDCA-1 (BioXcell, clone 927) or Rat IgG2b isotype control (BioXCell, clone LTF-2) twice a week for 8 weeks. For the pDC-depletion experiments using Bdca2-Dtr mice, 10-week-old Bdca2-Dtr+ or Dtr− male mice were fed on HFD for 4 weeks and were then continued on HFD and injected i.p. with 5ng/g body weight DT (Sigma) twice a week for another 4 weeks. Any effects of pDC depletion on metabolic parameters were determined 7 weeks after the introduction of HFD, and the mice were euthanized for flow cytometric analysis 8 weeks after HFD introduction.
Metabolic Studies
10–12-week-old male mice with specific genotypes were fed with HFD, and were injected i.p. with mAbs twice a week for 8 weeks, or injected i.p. with DT twice a week for 4 weeks (between 5 and 8 weeks after HFD treatment). At 7 weeks after the introduction of HFD, mice were subjected to an insulin tolerance test (ITT). Briefly, they were fasted for 4 hours before being injected i.p. with insulin (1 unit/kg body weight, Humulin R, Lilly). Blood-glucose levels were measured before and 20, 40, 60, 90, and 120 minutes after insulin injection, and were normalized to the blood-glucose levels before insulin injection. Area over the curve (AOC) was calculated using GraphPad Prism.
Quantification of Gene Expression by RT-qPCR
RNA from adipose tissue was extracted and purified using RNeasy Lipid Tissue Mini Kit (QIAGEN) according to the manufacturer’s instructions. cDNA was synthesized using SuperScript II Reverse Transcriptase (Thermo Fischer Scientific). Real-time quantitative PCR was performed using SYBR Green-based assays (Applied Biosystems). Transcript values were normalized to those from the mouse Tbp gene (FWD: ACCCTTCACCAATGACTCCTATG; REV: TGACTGCAGCAAATCGCTTGG). A primer set that detects most Ifna isoforms was employed for PCR titration of transcript levels (FWD: CCTGCTGGCTGTGAGGA; REV: GGAAGACAGGGCTCTCCAG).
RNA-Seq Library Preparation and Data Analysis
For the HFD time-course experiment, 12-week-old male vTreg53 TCR-tg Foxp3-GFPKI/y mice were fed with either NCD or HFD for 2, 4, 8, or 16 weeks, and biological replicates of 1,000 clonotype+ spleen- or VAT-Treg cells were double-sorted by Moflo into 5ul Buffer TCL (QIAGEN) with 1% 2-Mercaptoethanol (Sigma). For the IFNα treatment experiments, PPARγ+ Treg cells were sorted from VAT of 8–10-week-old Pparg-tdTKI/KI Foxp3-GFPKI/y male mice injected with 2 µg IL-33 twice in a week, and were cultured in the presence of 2000 U/ml IL-2 and 10 ng/ml IL-33 with or without 50ng/ml IFNα. Three days after culture, biological replicates of 1,000 Treg cells were double-sorted by Moflo into 5ul Buffer TCL (QIAGEN) with 1% 2-Mercaptoethanol (Sigma). Smart-Seq2 libraries were prepared by the Broad Technology Labs, and were sequenced using the Broad Genomics Platform (Picelli et al., 2014). Briefly, total RNA was captured and purified on RNAClean XP beads (Beckman Coulter). Polyadenylated mRNA was then selected using an anchored oligo(dT) primer and converted to cDNA via reverse transcription. First-strand cDNA was subjected to limited PCR amplification, followed by transposon-based fragmentation using the Nextera XT DNA Library Preparation Kit (Illumina). Samples were then PCR-amplified using bar-coded primers such that each sample carried a specific combination of Illumina P5 and P7 bar-codes and were pooled prior to sequencing. Sequencing was performed on an Illumina NextSeq500 instrument using 2×25bp or 2×38bp reads. Transcripts were quantified by the Broad Technology Labs computational pipeline with Cuffquant version 2.2.1 (Trapnell et al., 2012). Normalized reads were further filtered by minimal expression and coefficient of variation. Data were analyzed by Population PCA and Multiplot Studio in the GenePattern software package and GSEA (Subramanian et al., 2005). To identify gene expression dynamics across HFD and NCD time-series data, a two-step regression modeling strategy using maSigPro was applied (Nueda et al., 2014). An alpha of 0.05 was selected to account for multiple hypothesis testing and a false discovery rate of 5% were applied to first identify differential gene expression. To identify co-expression of genes during the time-series, silhouette analysis partitioned significant genes into 5 clusters (cluster correlation, r2 > 0.65), and visualized as heatmaps (Barter and Yu, 2018). Compilation of the VAT-Treg signature gene-sets was described previously (Cipolletta et al., 2015).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were routinely presented as mean ± SD. Normal distribution of populations at the level was calculated. Unless stated otherwise, significance was assessed by Student’s t-test for two-groups comparisons or ANOVA for multiple-group comparisons using GraphPad Prism 7.0. To determine the enrichment of certain gene signatures in RNA-seq data-sets, we used a Chi-square test. P = *, < 0.05; **, < 0.01; ***, < 0.001; ****, < 0.0001. n represents the number of mice and is reported in the figure legends. Mice with the same genotypes were assigned randomly to groups. No samples were excluded and blinding methods were not used. No statistical method was used to predetermine sample size. Sample sizes were chosen based on similar previous publications (Kolodin et al., 2015). Data showed a continuous normal distribution.
Supplementary Material
Table S1 (Related to Fig. 5): List of transcripts up- or down-regulated by two-fold or more in VAT Treg cells from vTreg53 TCR-tg mice fed 16 weeks on HFD vs. NCD.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD45.1 (clone A20) | Biolegend | Cat# 110724 |
| anti-CD45.2 (clone 104) | Biolegend | Cat# 109824 |
| anti-CD3 (clone 17A2) | Biolegend | Cat# 100214 |
| anti-CD4 (clone GK1.5) | Biolegend | Cat# 100422 |
| anti-CD25 (clone PC61) | Biolegend | Cat# 102026 |
| anti-KLRG1 (clone 2F1/KLRG1) | Biolegend | Cat# 138416 |
| anti-IFNAR1 (clone MAR1–5A3) | Biolegend | Cat# 127312 |
| anti-TCR Vα2 (clone B20.1) | Biolegend | Cat# 127806 |
| anti-CD45 (clone 30-F11) | Biolegend | Cat# 103126 |
| anti-CD11b (clone M1/70) | Biolegend | Cat# 101230 |
| anti-CD11c (clone N418) | Biolegend | Cat# 117310 |
| anti-F4/80 (clone BM8) | Biolegend | Cat# 123114 |
| anti-CD19 (clone 1D3/CD19) | Biolegend | Cat# 152408 |
| anti-PDCA1 (clone 927) | Biolegend | Cat# 127014 |
| anti-B220 (clone RA3–6B2) | Biolegend | Cat# 103232 |
| anti-SiglecH (clone 551) | Biolegend | Cat# 129606 |
| anti-Ly6c (clone HK1.4) | Biolegend | Cat# 128005 |
| anti-ST2 (clone RMST2–2) | eBioscience | Cat# 46–9335-80 |
| anti-Foxp3 (clone FJK-16s) | eBioscience | Cat# 17–5773-82 |
| anti-TCR Vβ4 (clone KT4) | BD Biosciences | Cat# 553366 |
| anti-Thy1.1 (clone OX-7) | BD Biosciences | Cat# 561409 |
| anti-Thy1.2 (clone 53–2.1) | BD Biosciences | Cat#561641 |
| InVivoMAb anti-IFNAR1 (clone MAR1–5A3) | BioXCell | Cat# BE0241 |
| InVivoMAb IgG1 isotype control (clone MOPC-21) | BioXCell | Cat# BE0083 |
| InVivoMAb anti-PDCA1 (clone 927) | BioXCell | Cat# BE0311 |
| InVivoMAb IgG2b isotype control (clone LTF-2) | BioXCell | Cat# BE0090 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human IL-2 | Peprotech | Cat# 200–02 |
| Recombinant mouse IL-33 (carrier-free) | Biolegend | Cat# 580506 |
| Recombinant Mouse IFN-α (carrier-free) | Biolegend | Cat# 752806 |
| Recombinant Mouse TNF-α (carrier-free) | Biolegend | Cat# 575206 |
| Recombinant Mouse IFN-γ (carrier-free) | Biolegend | Cat# 575306 |
| Insulin | Eli Lilly | Humulin R U-100 |
| D-Glucose | Thermo Fisher | Cat# D16–500 |
| Collagenase type II | Sigma | Cat# C6885 |
| 2-Mercaptoethanol | Sigma | Cat# M7522 |
| Diphtheria Toxin | Sigma | Cat# D0564 |
| Critical commercial assays | ||
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00–5523-00 |
| TransIT®−293 Transfection Reagent | Mirus | Cat# MIR 2705 |
| Click-iT™ Plus EdU Pacific Blue™ Flow Cytometry Assay Kit | Invitrogen | Cat# C10636 |
| LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Cat# L10119 |
| Annexin V | Biolegend | Cat# 640920 |
| Annexin V Binding Buffer | Biolegend | Cat# 422201 |
| SuperScript™ III Reverse Transcriptase | Invitrogen | Cat# 18080093 |
| Dynabeads™ Untouched™ Mouse CD4 Cells Kit | Thermo Fisher | Cat# 11415D |
| Dynabeads™ Mouse T-Activator CD3/CD28 for T-Cell Expansion and Activation | Thermo Fisher | Cat# 11453D |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat# 74804 |
| RNAClean XP beads | Beckman Coulter | Cat# A63987 |
| Nextera DNA Sample Prep Kit | Illumina | Cat# FC-121–1030 |
| Deposited data | ||
| RNA-Seq data | This paper | GSE174706 |
| Experimental models: Cell lines | ||
| Platinum-E Retroviral Packaging Cell Line | CELL BIOLABS | RV-101 |
| Experimental models: Organisms/strains | ||
| Mouse: vTreg53 TCR Tg / B6 | Li et al., 2018 | N/A |
| Mouse: Pparg-Tdt / B6 | Li et al., 2018 | N/A |
| Mouse: B6.CD45.1 | Jackson Laboratory | 002014 |
| Mouse: B6.CD45.2 | Jackson Laboratory | 000664 |
| B6.Foxp3-cre | Jackson Laboratory | 016959 |
| B6.Rosa26-Cas9 | Jackson Laboratory | 026179 |
| B6.Ifnar1fl | Jackson Laboratory | 028256 |
| Mouse: B6. Foxp3-GFP | Bettelli et al., 2006 | N/A |
| Mouse: B6. Foxp3-Thy1.1 | Liston et al., 2008 | N/A |
| Mouse: B6.Bdca2-Dtr | Swiecki et al., 2010 | N/A |
| Oligonucleotides | ||
| sgRNA targeting Ifnar1 sequence: 5’ CTTCTAAACGTACTTCTGGG 3’ | This paper | N/A |
| qPCR primer for Tbp FWD: 5’ ACCCTTCACCAATGACTCCTATG | This paper | N/A |
| qPCR primer for Tbp REV: 5’ TGACTGCAGCAAATCGCTTGG | This paper | N/A |
| qPCR primer for Ifna FWD: 5’ CCTGCTGGCTGTGAGGA | This paper | N/A |
| qPCR primer for Ifna REV: 5’ GGAAGACAGGGCTCTCCAG | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: MSCV-Ctl-sg-GFP | This paper | N/A |
| Plasmid: MSCV-Ctl-sg-RFP | This paper | N/A |
| Plasmid: MSCV-Ifnar1-sg-GFP | This paper | N/A |
| Software and algorithms | ||
| Cuffquant version 2.2.1 | Trapnell et al., 2012; | http://cole-trapnell-lab.github.io/cufflinks/install/ |
| GenePattern software package | Broad Institute | http://software.broadinstitute.org/cancer/software/genepattern/ |
| R | The R Foundation | https://www.r-project.org |
| GSEA | Broad Institute | http://www.gsea-msigdb.org/gsea/index.jsp |
| PRISM | GraphPad | https://www.graphpad.com |
| FlowJo | FlowJo, LLC | https://www.flowjo.com |
| Other | ||
| NCD: Picolab Mouse Diet 20 with 13 kcal% Fat | Lab Diet | No. 5053 |
| HFD: Rodent Diet With 60 kcal% Fat | Research Diets | Cat#: D12492 |
Highlights.
Long-term, but not short-term, HFD feeding induces the loss of VAT-Treg cells
Obesity reduces VAT-Treg Pparg transcripts while inducing IFN-response transcripts
Obesity-induced IFNα, made by local pDCs, is directly toxic to VAT-Treg cells
Blocking the pDC-IFNα axis restores VAT-Treg cells and insulin sensitivity
ACKNOWLEDGEMENTS
We thank A. Ortiz-Lopez, K. Hattori, L. Yang, K. Seddu, A. Mann, G. Spallanzani, T. Xiao, A. Munoz-Rojas, K. Langston, C. Laplace, C. Elkins, B. Arbasi, L. Cervantes, C. Arane and A. Wood for experimental help, and Drs. V. Kuchroo, M. Colonna, S. Gilfillan, and A. Rudensky for mouse lines. Derivation of certain mouse lines was done by the Emory Mouse Transgenic and Gene Targeting Core (supported by NIH award No. UL1TR000454)._Cell sorting and flow cytometry were performed at the HSCI/DRC Flow Core (Supported by NIH Grant No. P30 DK036836) and Emory Flow Cytometry Core (subsidized by the Emory University School of Medicine and supported by NIH award No. UL1TR002378). This work was supported by grants from the NIH (2R01 DK092541, 5RC2DK116691) and the JPB Foundation to D.M, and the Emory University School of Medicine Startup Fund to C.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Barrat FJ, Crow MK, and Ivashkiv LB (2019). Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. 20, 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter RL and Yu B. (2018). Superheat: An R package for creating beautiful and extendable heatmaps for visualizing complex data. J. Comput. Graph. Stat. 27, 910–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, and Kuchroo VK (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 441, 235–238. [DOI] [PubMed] [Google Scholar]
- Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, and Colonna M. (2006). Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. 177, 3260–3265. [DOI] [PubMed] [Google Scholar]
- Cipolletta D, Cohen P, Spiegelman BM, Benoist C, and Mathis D. (2015). Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARγ effects. Proc Natl Acad Sci U S A. 112, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, and Mathis D. (2012). PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 486, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, Perry K, Hazey J, Kampfrath T, Kollengode M et al. (2011). Visceral adipose inflammation in obesity is associated with critical alterations in T regulatory cell numbers. PLoS. ONE. 6, e16376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng T, Liu J, Deng Y, Minze L, Xiao X, Wright V, Yu R, Li XC, Blaszczak A, Bergin S et al. (2017). Adipocyte adaptive immunity mediates diet-induced adipose inflammation and insulin resistance by decreasing adipose Treg cells. Nature Comm. 8, 15725. [Google Scholar]
- Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, Eva MM, Gauchat JF, Qureshi ST, Mazer BD et al. (2016). Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. 17, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, Wolf D, Patsch W, Rosenkranz AR, and Eller P. (2011). Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. 60, 2954–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizi M, Marchetti V, Mavilio M, Marino A, Casagrande V, Cavalera M, Moreno-Navarrete JM, Mezza T, Sorice GP, Fiorentino L et al. (2014). IL-21 is a major negative regulator of IRF4-dependent lipolysis affecting Tregs in adipose tissue and systemic insulin sensitivity. Diabetes. 63, 2086–2096. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S et al. (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale AS, Glass DD, Lopez-Ocasio M, Baur R, Kanakabandi K, Porcella SF et al. (2018). Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 14, e1006985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AR, Bhattacharya R, Bhattacharya S, Nargis T, Rahaman O, Duttagupta P, Raychaudhuri D, Liu CS, Roy S, Ghosh P et al. (2016). Adipose recruitment and activation of plasmacytoid dendritic cells fuel metaflammation. Diabetes. 65, 3440–3452. [DOI] [PubMed] [Google Scholar]
- Hannibal TD, Schmidt-Christensen A, Nilsson J, Fransen-Pettersson N, Hansen L, and Holmberg D. (2017). Deficiency in plasmacytoid dendritic cells and type I interferon signalling prevents diet-induced obesity and insulin resistance in mice. Diabetologia. 60, 2033–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2017). Foundations of immunometabolism and Implications for metabolic health and disease. Immunity. 47, 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, and Spiegelman BM (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Zavidij O, Park J, Moschetta M, Kokubun K, Mouhieddine TH, Manier S, Mishima Y, Murakami N, Bustoros M et al. (2018). Blocking IFNAR1 inhibits multiple myeloma-driven Treg expansion and immunosuppression. J. Clin. Invest. 128, 2487–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodin D, van PN, Li C, Magnuson AM, Cipolletta D, Miller CM, Wagers A, Germain RN, Benoist C, and Mathis D. (2015). Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab. 21, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Li X, Kim JC, Lee J, Gonzalez-Navajas JM, Hong SH, Park IK, Rhee JH, and Raz E. (2012). Type I interferons maintain Foxp3 expression and T-regulatory cell functions under inflammatory conditions in mice. Gastroenterology. 143, 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Wollam J, and Olefsky JM (2018). An integrated view of immunometabolism. Cell. 172, 22–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Dispirito JR, Zemmour D, Spallanzani RG, Kuswanto W, Benoist C, and Mathis D. (2018). TCR transgenic mice reveal stepwise, multi-site acquisition of the distinctive fat-Treg phenotype. Cell. 174, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Spallanzani RG, and Mathis D. (2020). Visceral adipose tissue Tregs and the cells that nurture them. Immunol. Rev. [DOI] [PubMed]
- Liston A, Nutsch KM, Farr AG, Lund JM, Rasmussen JP, Koni PA, and Rudensky AY (2008). Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc Natl. Acad Sci U S. A. 105, 11903–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ (2005). IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev Immunol. 23, 275–306. [DOI] [PubMed] [Google Scholar]
- Macdougall CE, Wood EG, Loschko J, Scagliotti V, Cassidy FC, Robinson ME, Feldhahn N, Castellano L, Voisin MB, Marelli-Berg F et al. (2018). Visceral adipose tissue immune homeostasis is regulated by the crosstalk between adipocytes and dendritic cell subsets. Cell Metab. 27, 588–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metidji A, Rieder SA, Glass DD, Cremer I, Punkosdy GA, and Shevach EM (2015). IFN-α/β receptor signaling promotes regulatory T cell development and function under stress conditions. J Immunol. 194, 4265–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AB, Van GF, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, Bluestone JA, and Locksley RM (2015). Interleukin-33 and interferon-γ counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity. 43, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, Fukunaga K, Asano K, Betsuyaku T, and Koyasu S. (2016). Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. 17, 76–86. [DOI] [PubMed] [Google Scholar]
- Morris DL, Cho KW, Delproposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G, Singer K, and Lumeng CN (2013). Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes. 62, 2762–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nueda MJ, Tarazona S, and Conesa A. (2014). Next maSigPro: updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics. 30, 2598–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenhove G, Boucquey E, Taquin A, Acolty V, Bonetti L, Ryffel B, Le BM, Englebert K, Boon L, and Moser M. (2018). PD-1 Is involved in the dysregulation of type 2 innate lymphoid cells in a murine model of obesity. Cell Rep. 25, 2053–2060. [DOI] [PubMed] [Google Scholar]
- Panduro M, Benoist C, and Mathis D. (2016). Tissue Tregs. Annu. Rev Immunol. 34, 609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, and Sandberg R. (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priceman SJ, Kujawski M, Shen S, Cherryholmes GA, Lee H, Zhang C, Kruper L, Mortimer J, Jove R, Riggs AD et al. (2013). Regulation of adipose tissue T cell subsets by Stat3 is crucial for diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A. 110, 13079–13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge JR, Hoyt TR, Dobrinen E, Capecchi MR, Schmidt EE, and Meissner N. (2015). Type I IFNs act upon hematopoietic progenitors to protect and maintain hematopoiesis during pneumocystis lung infection in mice. J. Immunol. 195, 5347–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizis B, Bunin A, Ghosh HS, Lewis KL, and Sisirak V. (2011). Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev Immunol. 29, 163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED and Spiegelman BM (2014). What we talk about when we talk about fat. Cell. 156, 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr. et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 28, 546–558. [DOI] [PubMed] [Google Scholar]
- Spallanzani RG, Zemmour D, Xiao T, Jayewickreme T, Li C, Bryce PJ, Benoist C, and Mathis D. (2019). Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol. 4, eaaw3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Koch MA, Pepper M, and Campbell DJ (2014). Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J Exp. Med. 211, 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, Sipula IJ, Dedousis N, Scott DK, Morel PA et al. (2012). Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes. 61, 2330–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl. Acad Sci U S. A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Gilfillan S, Vermi W, Wang Y, and Colonna M. (2010). Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8+ T cell accrual. Immunity. 33, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL et al. (2015). The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 16, 276–285. [DOI] [PubMed] [Google Scholar]
- Wu D, Han JM, Yu X, Lam AJ, Hoeppli RE, Pesenacker AM, Huang Q, Chen V, Speake C, Yorke E et al. (2019). Characterization of regulatory T cells in obese omental adipose tissue in humans. Eur. J. Immunol. 49, 336–347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Related to Fig. 5): List of transcripts up- or down-regulated by two-fold or more in VAT Treg cells from vTreg53 TCR-tg mice fed 16 weeks on HFD vs. NCD.
Data Availability Statement
The accession number for the RNA sequencing data reported in this paper is GEO: GSE174706.