

Abstract
The first general method for the rapid, chemoselective, and modular functionalization of serine residues in native polypeptides is reported using a reagent platform based on P(V) oxidation state. This redox-economic approach can be used to append nearly any kind of cargo onto serine, generating a stable, benign, and hydrophilic phosphorothioate linkage. The method tolerates all other known nucleophilic functional groups of naturally occurring proteinogenic amino acids. A variety of applications can be envisaged by this expansion of the toolbox of site-selective bioconjugation methods.
Graphical Abstract

It is no surprise that of the twenty naturally-occurring amino acid residues bearing reactive groups, serine is rarely chosen as a handle for selective derivatization (Figure 1A).1–4 This lack of serine functionalization clearly stems not from a lack of desire, but rather the unmet challenge of differentiating the nucleophilicity of serine residues from other nucleophilic sidechains and water (chemoselectivity) and single residue among its multiple copies (site-selectivity).1–7 A process that could achieve this differentiation would be inherently valuable, as it would add a new dimension to the toolbox of options available for chemoselective modification of peptides and the site-selective labeling of proteins. Whereas nature has pointed to a possible solution to this problem using enzymatic machinery and phosphorus(V) reactivity,8–16 there is currently no general method (chemoenzymatic or otherwise) to site-selectively attach a precise cargo to a serine residue in a native biomolecule.1–3,17 Kinases are typically responsible for the phosphorylation of various amino acid residues such as serine (Figure 1B).8–16 This process suggests that P(V)-based electrophiles could have an innate preference for alcohol-based nucleophiles.18 In parallel with this biological precedent, chemical inspiration for P(V)-based bioconjugation stems from the new class of phosphorous reagents (Ψ and Π) which react with both oxygen- and carbon-based nucleophiles in a rapid and predictable way.19,20 In this Communication, Ψ-loaded reagents (Ψ-modules) are shown to exhibit strikingly selective reactivity towards serine. This method enables both selective post-translational labeling and bioconjugation of serine residues.
FIGURE 1.
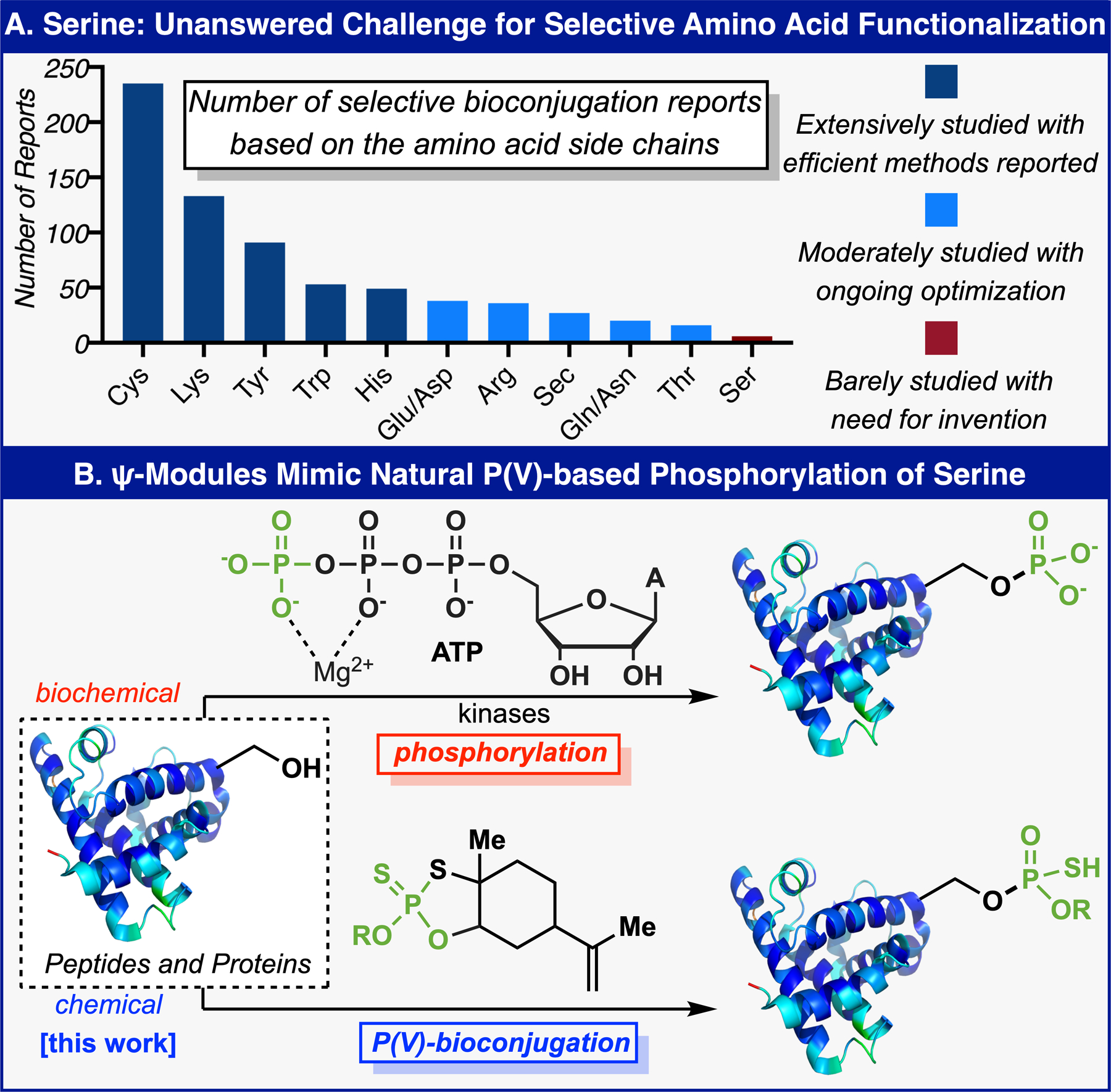
(A) Serine: Unanswered Challenge for Selective Amino Acid Functionalization. (B) PSI Reagents Mimic Natural P(V)-based Phosphorylation of Serine.
In 2018, a set of reagents based upon a limonene-scaffold fused to an oxathiophospholane heterocycle were introduced for the purpose of simplifying stereocontrolled access to phosphorothioate linkages (Phosphorous-Sulfur Incorporation, abbreviated PSI or Ψ).19 This redox-economic approach avoided the extraneous steps associated with a P(III)-based manifold, yet retained the high reactivity associated with phosphoramidites and related systems.19 Shortly thereafter, related limonene-based reagents were introduced to access both phosphines and methylphosphonates through stereocontrolled P–C bond formation (Phosphorous Incorporation, abbreviated PI or Π).20
During the studies using PSI reagent, it was sporadically observed that oxygen-based nucleophiles would react in preference to other heteroatoms such as sulfur and nitrogen.19,20 After optimization of the coupling step (see supporting information, Figure S23), the generality of these observations was explored systematically in the context of amino-acid functionalization as depicted in Figure 2A. Thus, competitive coupling experiments between amino acids revealed a striking selectivity for serine functionalization using Ψ-module P(V)-1 in the presence of cysteine, lysine, tyrosine and selenocysteine, even at high concentration (0.1 M). These preliminary studies established the viability of such an approach, and represent selectivities on par with traditional protein labeling methods (e.g. NHS esters for lysine, iodoacetamide for cysteine, and MTAD for tyrosine/tryptophan).1–4 Interestingly, reagent P(V)-1 under similar conditions displayed a respectable selectivity of 7:1 favoring serine over threonine. Transitioning to more realistic models for chemoselective labeling, competitive coupling experiments were then pursued using small peptides with Ψ-modules P(V)-1, P(V)-2a, and P(V)-3 (Figure 2B). Good-to-excellent conversions were observed within 15 minutes, and unless otherwise noted, only one product was formed with no epimerization. Exquisite selectivity was obtained for serine at both 50 mM and 100 mM reagent concentration (10 mM peptide concentration) when competing with cysteine, lysine, histidine, glutamine, methionine, tryptophan, and tyrosine. When employing cysteine containing peptides, excess EtSH was added to avoid the formation of adducts between cysteine and the thiirane by-product of the Ψ-module. In addition, a reductive quench (DTT) is used to reduce any S–S bond formed during the reaction.21,22 When competing with threonine, a useful level of selectivity (determined by H-coupled 31P NMR) was observed at 50 mM of reagents P(V)-1 (8:1), P(V)-2a (6:1), or reagent P(V)-3 (7:1) delivering compounds 15, 16 and 17 in 95%, 72% and 94%, respectively.1–4
FIGURE 2.

(A) Competitive Experiments Between Amino Acids. (B) Competitive Experiments Using Small Tetrapeptides (10 mM). a Using EtSH (5 equiv).
More realistic applications of this method were then explored in the context of longer peptides containing a variety of nucleophilic amino acids (Figure 3A). Excellent selectivity for serine and satisfactory conversions were always observed using a variety of Ψ-modules (Figure 3A). As an example, peptides SI-14 was selectively labelled at the serine position to afford peptide 31, 32, and 33 in 23%, 36% and 17% conversions using reagents P(V)-1, P(V)-2b, and P(V)-3, respectively. In addition to these linear examples, the functionalization of cyclic peptides was explored due to their importance and relevance in the context of medicinal chemistry (Figure 3B).23–25 The developed conditions were first applied to the diversification of a cyclic peptide containing several nucleophilic functionalities, affording 61% yield of the desired product 34 within 10 minutes. An oxytocin-derivative, bearing a sensitive disulfide bridge and a tyrosine, successfully underwent coupling in 42% yield, with complete selectivity for the serine residue.26–28 Finally, vancomycin was selected as a key example with an impressive functional group array: one carboxylic acid, one primary amine, one primary amide, three phenolic alcohols, five secondary alcohols, and a single primary alcohol.29–33 Nevertheless, both reagents P(V)-1 and P(V)-3 demonstrated remarkable selectivity for the primary alcohol site affording compounds 36 and 37 in 19% and 21% yields, respectively. The labeled position was confirmed by H-coupled 31P NMR; assignment was simple as the molecule bears only one primary alcohol.
FIGURE 3.
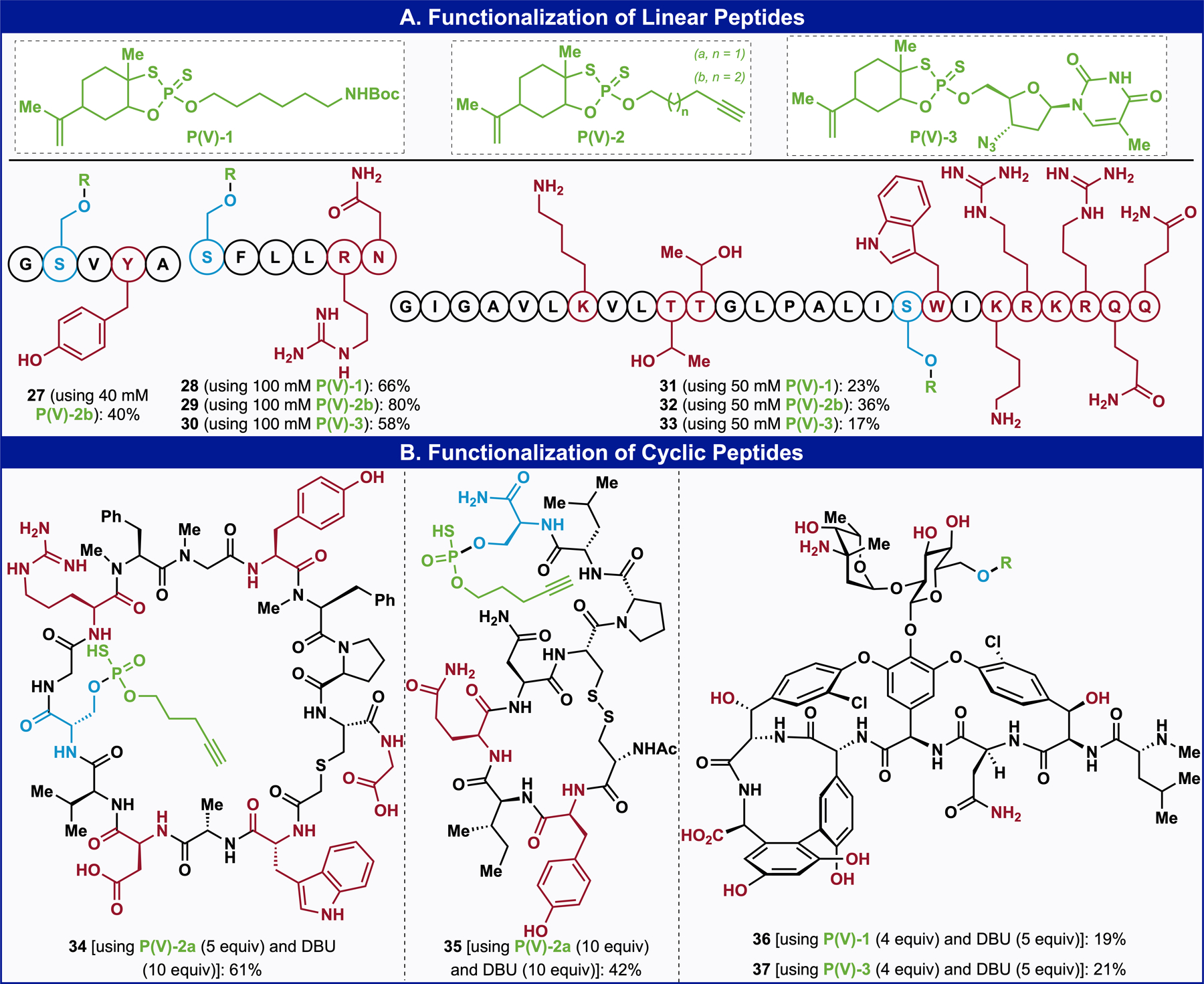
(A) Peptide Scope. (B) Functionalization of Cyclic Peptide.
Next, we challenged this chemical method for the installation of various Ψ-loaded reagents on proteins. The protein ubiquitin was primarily chosen for its relevance in proteasome targeting.34–36 When ubiquitin 38 was vortexed with P(V)-2b reagent and DBU, it resulted in mono-labeled ubiquitin 39 in 37% conversion (Figure 4A). Labeled ubiquitin 39 was digested with trypsin and analyzed by LC-MS/MS (see SI for details). The resulting MS/MS data were searched using the Mascot search algorithm using a variable modification of 176 Da and a database containing the ubiquitin sequence. MS/MS of P(V) tagged peptide (ESTLHLVLR, E64-R72, m/z 622.31 [M+2H]2+) confirmed the site-selective labeling of Ser residue (S65; kinetic labeling). Subsequently, other Ψ-modules were screened for the labeling of ubiquitin. P(V)-1, P(V)-3, and P(V)-4 reagents afforded the mono-labeling of ubiquitin in 32% (40), 40% (41), and 20% (42) conversions respectively. In addition, the method was efficiently translated to the labeling of the DNA binding protein 434 repressor37–39 43 using P(V)-2b; reaction of these components delivered 44 in 35% conversion (Figure 4B). Further advances in the design of Ψ-reagents that are more reactive, yet more stable in aqueous conditions, and cysteine compatible will facilitate further use of this new approach toward the chemo- and regioselective modification of serine residues on proteins.
FIGURE 4.

Functionalization of Proteins (A) Functionalization of Ubiquitin. (B) Functionalization of 434 Repressor.
DFT studies were undertaken to rationalize the selectivity observed for serine in the coupling step (Figure 5).40 These studies support a stepwise AN+DN phosphorylation mechanism (Figure 5A) with limiting barriers for the formation (TS1) or collapse (TS2) of a trigonal bipyramidal pentacoordinated (TBP, INT1) intermediate (Figure 5B). The remaining steps of the reaction coordinate involve a ring flip of the limonene skeleton (TS3) followed by SN2 displacement of the phosphorylated amino acid and simultaneous release of the cyclohexene sulfide (TS4) with an exothermic balance (ΔG ≈ −37 kcal/mol).41 Transition structures TS3 represent the highest barrier within the sequence of conformational distorsions42 required to transform chair INT2 into chair INT3 (See SI for details).43 The transition structures leading to the TBP, TS1, display the backside attack of the nucleophile to the P-O ring bond in a late, intermediate-like arrangement. TS1 shows a large extent of RX–P (where X = O, or N) bond formation and concomitant proton transfer to the nitrogen atom of DBU acting as a general base. Amino acids with alcohol and amine side chains display a rate-limiting formation of the trigonal bipyramidal intermediate with barriers that increase in the order Ser < Thr << Tyr << Lys (TS1, Figure 5B). A ΔΔG‡ of 0.6 kcal/mol between TS1-Ser and TS1-Thr is in qualitative agreement with the lower but competitive reactivity of threonine, whereas a ΔΔG‡ of 4.9 kcal/mol between TS1-Ser and TS1-Tyr is consistent with the limited reactivity of tyrosine. Similarly, TS1-Ser is favored relative to TS1-Lys by ≈ 9 kcal/mol in agreement with the lower acidity of the primary amine and greater steric congestion during the partial deprotonation by DBU. A search for the transition structures corresponding to the collapse of the TBP intermediates led to structures TS2, which depict the dissociation of the ring S–P bond accompanied by pseudorotation44 and reduction of the O–P–O angle as the tetrahedral phosphorylated P(V) product forms (Figure 5A and SI for details). The formation of a strong O–P bond and the axial arrangement of the methoxy group facilitate the dissociation of the weaker S–P bond (See SI for details).45 For amino acids with alcohol side chains, transition structures TS2 leading to the collapse of the TBP intermediates are ≈ 8 kcal/mol more stable than the transition structures TS1 forming the TBP intermediates. In contrast with the reaction coordinate for the alcohol, the collapse of the TBP-Cys intermediate is disfavored relative to its formation (cf. TS1-Cys and TS2-Cys, Figure 5B). The existence of a low barrier for decomposition of the TBP intermediate back to reactants has been used to explain the slow thiolysis of phosphate triesters.46 In agreement with experiment, the limiting barrier for the phosphorylation of cysteine (TS2-Cys, ΔG‡ = +22.2 kcal/mol) is estimated to be 2.7 kcal/mol higher than the limiting barrier for the phosphorylation of serine (TS1-Ser, ΔG‡ = +19.5 kcal/mol). A comparison of TS2-Cys and TS2-Ser geometries shows the reluctance of cysteine to pseudorotate (S–P–Oring 147.0° in TS2-Cys versus 111.2° in TS2-Ser), become equatorial, and stabilize ring P–S dissociation via the axial-axial arrangement of the OMe group and the ring P–S bond (See SI). The higher pseudorotation barrier of cysteine relative to serine is consistent with the preference of good leaving groups (e.g. cysteine) to occupy an axial position.47 It worth noting that the selectivity for serine versus cysteine and tyrosine may simply be associated with a higher degree of reversibility. Taken all together, these results rationalize the serine selectivity observed for the coupling step.
FIGURE 5.
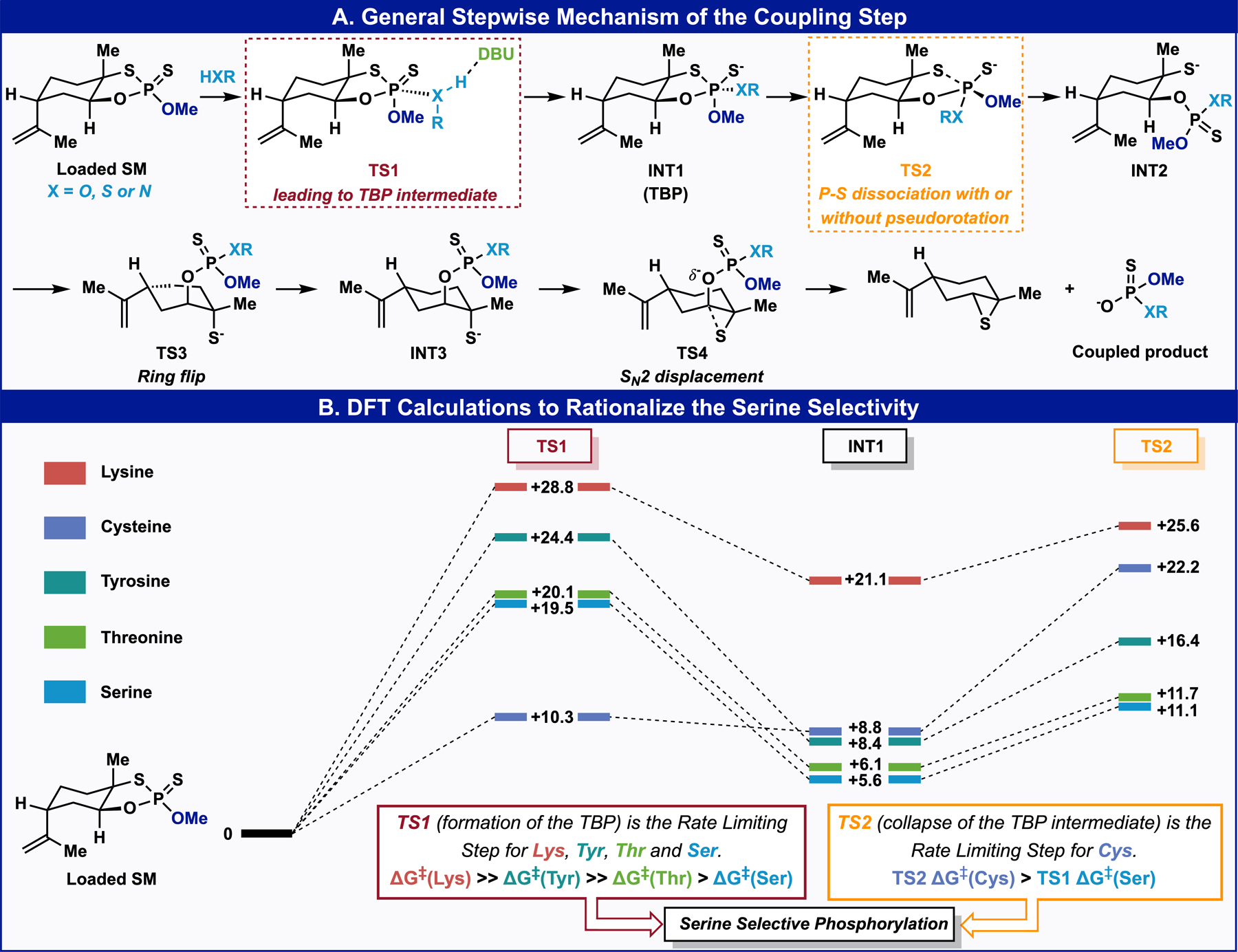
(A) General Stepwise Mechanism of the Coupling Step. (B) DFT Calculations to Rationalize the Serine Selectivity.
Expanding on our previous work on the chemistry of the Ψ-reagent system, a chemoselective, rapid, and robust method for the direct functionalization of serine residues in the context of both peptides and proteins has been developed. The presented methodology displays excellent chemoselectivity for serine residues under the optimized solvent conditions and will enable practitioners in this field to append virtually any kind of cargo desired in an orthogonal fashion. Applications of these findings to bioconjugation, chemical biology, as well as the pursuit of novel materials can all be envisaged.48 Development of new PSI-reagents that are compatible to aqueous conditions and cysteine residues will also facilitate further use of this new approach toward the chemo- and regioselective modification of serine residues on proteins.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided by Bristol-Myers Squibb, NIH (GM-118176), the Marie Skłodowska-Curie Global Fellowships (749359-EnanSET, N.M.P) within the European Union research and innovation framework programme (2014-2020), FCT Portugal (IF/00624/2015) and the Royal Society (URF\R\180019). DTF was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1 TR002551 and linked award TL1 TR002551. Authors are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (Scripps Research) for assistance with nuclear magnetic resonance (NMR) spectroscopy, to Dr. Jason Chen, Brittany Sanchez and Emily Sturgell (Scripps Automated Synthesis Facility) for assistance with HPLC, HRMS and LCMS. We finally thank Dr Victor L. Ayora and Lavinia Dunsmore for 434 repressor protein sample.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. The Supporting Information contains all experimental procedures, analysis, the detail of DFT calculations and compound characterization data.
REFERENCES
- (1).Algar W. Russ, D. P, Medintz Igor L., Chemoselective and Bioorthogonal Ligation Reactions: Concepts and Applications. John Wiley & Sons: 2017. [Google Scholar]
- (2).deGruyter JN; Malins LR; Baran PS Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Hoyt EA; Cal PMSD; Oliveira BL; Bernardes GJL Contemporary approaches to site-selective protein modification. Nat. Rev. Chem 2019, 3, 147–171. [Google Scholar]; (b) Rawale DG; Thakur K; Adusumalli SR; Rai V Chemical methods for selective labeling of proteins. Eur. J. Org. Chem 2019, 6749–6763. [Google Scholar]
- (4).Boutureira O; Bernardes GJL Advances in chemical protein modification. Chem. Rev 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- (5).Bruice TC; Fife TH; Bruno JJ; Brandon NE Hydroxyl group catalysis. II. The reactivity of the hydroxyl group of serine. The nucleophilicity of alcohols and the ease of hydrolysis of their acetyl esters as related to their pKa. Biochemistry 1962, 1, 7–12. [DOI] [PubMed] [Google Scholar]
- (6).Neet KE; Koshland DE The conversion of serine at the active site of subtilisin to cysteine: a “chemical mutation”. Proc. Natl. Acad. Sci. USA 1966, 56, 1606–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Polgar L; Bender ML A new enzyme containing a synthetically formed active site. thiol-subtilisin. J. Am. Chem. Soc 1966, 88, 3153–3154. [Google Scholar]
- (8).Jackson MD; Denu JM Molecular reactions of protein phosphatases-insights from structure and chemistry. Chem. Rev 2001, 101, 2313–2340. [DOI] [PubMed] [Google Scholar]
- (9).Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- (10).Kennelly PJ Protein phosphatases-a phylogenetic perspective. Chem. Rev 2001, 101, 2291–2312. [DOI] [PubMed] [Google Scholar]
- (11).Cohen P The regulation of protein function by multisite phosphorylation – a 25-year update. Trends Biochem. Sci 2000, 25, 596–601. [DOI] [PubMed] [Google Scholar]
- (12).Johnson LN; Lewis RJ Structural basis for control by phosphorylation. Chem. Rev 2001, 101, 2209–2242. [DOI] [PubMed] [Google Scholar]
- (13).Chen C; Ha BH; Thevenin AG; Lou HJ; Zhang R; Yip KY; Peterson JR; Gerstein M; Kim PM; Filippakopoulos P; Knapp S; Boggon TJ; Turk BE Identification of a major determinant for serine-threonine kinase phosphoacceptor specificity. Mol. Cell 2014, 53, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tagliabracci VS; Wiley SE; Guo X; Kinch LN; Durrant E; Wen J; Xiao J; Cui J; Nguyen KB; Engel JL; Coon JJ; Grishin N; Pinna LA; Pagliarini DJ; Dixon JE A single kinase generates the majority of the secreted phosphoproteome. Cell 2015, 161, 1619–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Miller CJ; Turk BE Homing in: mechanisms of substrate targeting by protein kinases. Trends Biochem. Sci 2018, 43, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Westheimer FH Why nature chose phosphates. Science 1987, 235, 1173–1178. [DOI] [PubMed] [Google Scholar]
- (17).Liu H; Li X Serine/Threonine Ligation: Origin, Mechanistic Aspects, and Applications. Acc. Chem. Res 2018, 51, 1643–1655. [DOI] [PubMed] [Google Scholar]
- (18).For a recent report on selective phosphorylation of histidine residue, see:; Jia S; He D; Chang CJ Bioinspired Thiophosphorodichloridate Reagents for Chemoselective Histidine Bioconjugation. J. Am. Chem. Soc 2019, 141, 7294–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Knouse KW; deGruyter JN; Schmidt MA; Zheng B; Vantourout JC; Kingston C; Mercer SE; McDonald IM; Olson RE; Zhu Y; Hang C; Zhu J; Yuan C; Wang Q; Park P; Eastgate MD; Baran PS Unlocking P(V): Reagents for chiral phosphorothioate synthesis. Science 2018, 361, 1234–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Xu D; Rivas-Bascón N; Padial, N. M; Knouse KW; Zheng B; Vantourout JC; Schmidt MA; Eastgate MD; Baran PS Enantiodivergent Formation of C-P Bonds: Synthesis of P-Chiral Phosphines and Methylphosphonate Oligonucleotides. J. Am. Chem. Soc 2020, 142, 5785–5792. [DOI] [PubMed] [Google Scholar]
- (21).Lu BY; Chang JY Rapid and irreversible reduction of protein disulfide bonds. Anal. Biochem 2010, 405, 67–72. [DOI] [PubMed] [Google Scholar]
- (22).Singh R; Whitesides GM Reagents for Rapid Reduction of Native Disulfide Bonds in Proteins. Bioorg. Chem 1994, 22, 109–115. [Google Scholar]
- (23).Zorzi A; Deyle K; Heinis C Cyclic peptide therapeutics: past, present and future. Curr. Opin. Chem. Biol 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]
- (24).Jing X; Jin K A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev 2020, 40, 753–810. [DOI] [PubMed] [Google Scholar]
- (25).Lee AC; Harris JL; Khanna KK; Hong JH A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci 2019, 20, 2383–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ichinose W; Cherepanov SM; Shabalova AA; Yokoyama S; Yuhi T; Yamaguchi H; Watanabe A; Yamamoto Y; Okamoto H; Horike S; Terakawa J; Daikoku T; Watanabe M; Mano N; Higashida H; Shuto S Development of a Highly Potent Analogue and a Long-Acting Analogue of Oxytocin for the Treatment of Social Impairment-Like Behaviors. J. Med. Chem 2019, 62, 3297–3310. [DOI] [PubMed] [Google Scholar]
- (27).Gimpl G; Fahrenholz F The oxytocin receptor system: structure, function, and regulation. Physiol. Rev 2001, 81, 629–683. [DOI] [PubMed] [Google Scholar]
- (28).Bakermans-Kranenburg MJ; van IJMH Sniffing around oxytocin: review and meta-analyses of trials in healthy and clinical groups with implications for pharmacotherapy. Transl. Psychiatry 2013, 3, e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Okano A; Isley NA; Boger DL Peripheral modifications of [Ψ[CH2NH]Tpg4]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc. Natl. Acad. Sci. U.S.A 2017, 114, E5052–E5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Boger DL; Miyazaki S; Kim SH; Wu JH; Castle SL; Loiseleur O; Jin Q Total Synthesis of the Vancomycin Aglycon. J. Am. Chem. Soc 1999, 121, 10004–10011. [Google Scholar]
- (31).Nicolaou KC; Mitchell HJ; Jain NF; Winssinger N; Hughes R; Bando T Total Synthesis of Vancomycin. Angew. Chem. Int. Ed 1999, 38, 240–244. [Google Scholar]
- (32).Monteiro JF; Hahn SR; Goncalves J; Fresco P Vancomycin therapeutic drug monitoring and population pharmacokinetic models in special patient subpopulations. Pharmacol Res. Perspect 2018, 6, e00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Boger DL Vancomycin, teicoplanin, and ramoplanin: synthetic and mechanistic studies. Med. Res. Rev 2001, 21, 356–381. [DOI] [PubMed] [Google Scholar]
- (34).Pickart CM Ubiquitin in chains. Trends in Biochem. Sci 2000, 25, 544–548. [DOI] [PubMed] [Google Scholar]
- (35).Swatek KN; Komander D Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Glickman MH; Ciechanover A The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev 2002, 82, 373–428. [DOI] [PubMed] [Google Scholar]
- (37).Bushman FD; Ptashne M Activation of transcription by the bacteriophage 434 repressor. Proc. Natl. Acad. Sci. U.S.A 1986, 83, 9353–9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rodgers DW; Harrison SC The complex between phage 434 repressor DNA-binding domain and operator site OR3: structural differences between consensus and non-consensus half-sites. Structure 1993, 1, 227–240. [DOI] [PubMed] [Google Scholar]
- (39).Koudelka GB Recognition of DNA structure by 434 repressor. Nucleic Acids Res. 1998, 26, 669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40). See SI for references regarding the DFT calculations process. Based on reported pKa values of the side chain functional groups, Ser, Thr, and Tyr were modeled following a general base catalysis path, whereas Cys was modeled as a fully deprotonated nucleophile.
- (41).(a) Uchimaru T; Stec WJ; Taira K Mechanism of the Chemoselective and Stereoselective Ring Opening of Oxathiaphospholanes: An Ab Initio Study. J. Org. Chem 1997, 62, 5793–5800. [Google Scholar]; (b) Uchimaru T; Stec WJ; Tsuzuki S; Hirose T; Tanabe K; Taira K Ab Initio Investigation on Nucleophilic Ring opening of 1, 3, 2-Oxathiaphospholane: Nucleophilic Substitution at Phosphorus Coupled with Pseudorotation. Chem. Phys. Lett 1996, 263, 691–696. [Google Scholar]
- (42).Dixon DA; Komornicki A Ab Initio Conformational Analysis of Cyclohexane. J. Phys. Chem 1990, 94, 5630–5636. [Google Scholar]
- (43).The S–C–C–O dihedral angle was scanned at fixed intervals and the highest energy geometry of the resulting potential energy was fully optimized to afford a twisted half-chair transition structure.
- (44).(a) Westheimer FH Pseudo-Rotation in the Hydrolysis of Phosphate Esters. Acc. Chem. Res 1968, 1, 70–78. [Google Scholar]; (b) Boyd DB Mechanism of Hydrolysis of Cyclic Phosphate Esters. J. Am. Chem. Soc 1969, 91, 1200–1205. [Google Scholar]
- (45).Park K-H; Kim M-H; Um I-H Kinetic Study on Nucleophilic Displacement Reactions of Phenyl Y-Substituted Phenyl Carbonates with 1,8-Diazabicyclo [5.4.0] undec-7-ene: Effects of Amine Nature on Reaction Mechanism. Bull. Korean Chem. Soc 2016, 37, 77–81. [Google Scholar]
- (46).Arantes GM; Chaimovich H Thiolysis and Alcoholysis of Phosphate Tri- and Monoesters with Alkyl and Aryl Leaving Groups. An ab Initio Study in the Gas Phase. J. Phys. Chem. A 2005, 109, 5625–5635. [DOI] [PubMed] [Google Scholar]
- (47).DeBruin KE; Petersen JR Steric and Electronic Effects on the Stereochemistry of the Alkaline Hydrolysis of Acyclic Dialkoxyphosphonium Salts. Pseudorotation of Intermediates in Phosphorus Ester Reactions. J. Org. Chem 1972, 37, 2272–2278. [Google Scholar]
- (48).Flood DT; Knouse KW; Vantourout JC; Sanchez BB; Sturgell EJ; Chen JS; Baran PS; Dawson PE Synthetic Elaboration of Native DNA by RASS (SENDR) ChemRxiv Preprint 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.