

Abstract
Background:
Aberrant angiogenesis may play a role in the development of Alzheimer’s disease and related dementia.
Objective:
To explore the relationship between angiogenesis activity and evidence of neurodegeneration among older adults.
Methods:
Cross-sectional study of 49 older adults clinically characterized as cognitively normal, mild cognitive impairment, or early Alzheimer’s disease. In addition to neuroimaging, we completed assays on peripheral blood, including: vascular endothelial growth factor, tumor necrosis factor, fibroblast growth factor, and amyloid-β peptide 40. We used advanced polychromatic flow cytometry to phenotype circulating mononuclear cells to assess angiogenesis activity.
Results:
Although we documented differences in cognitive performance, structural changes on neuroimaging, and burden of amyloid and tau on positron emission tomography, angiogenesis activity did not vary by group. Interestingly, VEGF levels were shown to be increased among subjects with mild cognitive impairment. In ANCOVA models controlling for age, sex, intracranial volume, and monocyte subpopulations, angiogenesis activity was correlated with increased white matter hyperintensities.
Conclusion:
We demonstrate a significant association between angiogenesis activity and cerebrovascular disease. To better understand the potential of angiogenesis as an intervention target, longitudinal studies are needed.
Keywords: Alzheimer’s disease, biomarkers, pathologic angiogenesis, vascular dementia
INTRODUCTION
Chronic cerebral hypoperfusion may play a larger initial role in the etiology of Alzheimer’s disease (AD) dementia than previously understood [1, 2]. Cerebral hypoperfusion generally results in the upregulation of angiogenesis [3]. The body controls angiogenesis through a complex interplay of humoral and cellular factors, and angiogenesis takes place in the adult brain just as it takes place in other tissues [4]. In the context of AD, prior research describes dysregulation of humoral factors involved in angiogenesis, such as vascular endothelial growth factor (VEGF) and the interaction of inflammation and angiogenesis in response to hypoxia [3, 4]. Whether these changes are protective or ultimately neurotoxic is uncertain, and both could be true at different stages of the pathologic process of AD [5]. In other words, initial upregulation could be adaptive but continued upregulation could ultimately be toxic, just as may be true for the role of neuroinflammation [6]. Angiogenesis is also notable as an important component of the neurogenesis pathway which itself is dysregulated in AD [7].
Aberrant angiogenesis is defined as inappropriately under-active or inappropriately over-active angiogenesis. Over-active angiogenesis may be present in conditions such as cancer and macular degeneration, for example [8–10]. Normal aging has been associated with a gradual decline in angiogenesis capacity, although there is substantial inter-individual variation [11, 12]. In some individuals, however, an inappropriately under-active angiogenesis response to chronic cerebral hypoperfusion may increase the risk of AD while the capacity to upregulate angiogenesis may offer protection [4, 5, 13]. Thus, the potential to modulate angiogenesis may offer a therapeutic target in the prevention and treatment of AD [5].
In addition to humoral factors such as VEGF, cellular factors also play a fundamental role in angiogenesis. Recent studies have demonstrated increased levels of peripherally circulating endothelial cells in response to local areas of hypoperfusion such as myocardial infarction and stroke [14, 15]. In contrast, Lee et al. reported that circulating endothelial cells were decreased in subjects newly diagnosed with AD compared with controls matched on age, sex, and Framingham risk score [16]. A detailed understanding of how endothelial and hematopoietic progenitor cells function during angiogenesis has been hampered by the heterogeneity of the cell populations studied [17–19]. Advances in data analysis techniques and instrumentation have revolutionized the field of flow cytometry, allowing for a more comprehensive understanding of the cellular biology of rare blood cell subsets [20–23]. We have previously demonstrated that relative levels of angiogenesis activity can be identified through the measurement of two phenotypically and functionally distinct subpopulations of circulating hematopoietic stem and progenitor cells (CHSPCs) in the peripheral blood [17, 18, 24]. While these circulating subsets do not directly integrate as replacement endothelial cells, they play key proangiogenic roles in promoting vascular repair or angiogenesis and have been studied as biomarkers for the angiogenic state [25, 26].
In the current study, we hypothesized that aberrant angiogenesis would be correlated with clinical and neuroimaging evidence of cognitive impairment, cerebral hypoperfusion, and neurodegeneration. We designed our subject sampling approach to simulate changes in angiogenesis over time that may be occurring over the course of a dementing illness and in the context of aging and comorbid conditions. A central premise of the current study is that peripheral markers of angiogenesis activity (cellular and humoral) reflect angiogenesis activity in the central nervous system. This premise is consistent with recent literature documenting the impact of chronic systemic inflammation on neurodegeneration [6, 27, 28]. We performed a cross-sectional study to describe the association of angiogenesis activity in peripheral blood samples with cognitive function and dementia biomarkers among 49 older adults meeting consensus diagnosis criteria for: 1) normal cognition; 2) amnestic mild cognitive impairment; and 3) late-onset AD. This preliminary study is a first step in ultimately testing the hypothesis that age-related decline in angiogenesis capacity is an initiating event in the cascade of events leading to neuronal cell death and dementia.
METHODS
This study was approved by the Indiana University Purdue University – Indianapolis Institutional Review Board and conducted from May 2016-April 2019. All study participants or their legally authorized representatives signed informed consent for participation in the Indiana Alzheimer Disease Center. All clinical assessments, including collection of blood samples and completion of neuroimaging took place in the Indiana Alzheimer Disease Center. Clinical and cognitive data were curated by the National Alzheimer’s Coordinating Center and the National Centralized Repository for Alzheimer’s Disease and Related Dementias [29]. A specific time of day was not targeted for blood collection; however, all samples utilized for this study were collected between 9 am and 2 pm. This was true simply due to practical constraints in that blood was drawn at times to accommodate the operating hours for the clinic and laboratory.
Recruitment
Participants aged 60 years and older were recruited through the Indiana Alzheimer Disease Center and local Memory Care Practices. We targeted recruitment of three groups of subjects:
Group 1: cognitively normal (CN) older adults;
Group 2: patients with amnestic mild cognitive impairment (MCI); and
Group 3: patients with early stage clinically diagnosed AD dementia.
Patients with dementia due to subtypes other than AD were excluded from this study. Patients with a history of cancer, schizophrenia, bipolar disease, traumatic brain injury, trauma or surgery in the prior 12 months, myocardial infarction or stroke in the prior 12 months, and other neurodegenerative diseases such as Parkinson’s disease were excluded. We also excluded any patient treated with anti-angiogenic medications for any indication.
Clinical assessment
Through the resources of the Indiana Alzheimer Disease Center, all subjects completed a Uniform Data Set (UDS 3.0) Neuropsychological Battery assessment consisting of a comprehensive clinical and cognitive assessment, which includes tests of executive function, memory and language, visuospatial skills, and attention at baseline in order to determine eligibility, current symptoms, and cognitive status [29]. Diagnosis was determined by clinical consensus using the following guidelines: 1) CN: normal performance on all cognitive tests including the short and long delay portions of the Rey Auditory Verbal Learning Test (RAVLT) and Craft Stories (UDS 3.0); 2) MCI: 1.5 standard deviation (SD) or more deficit on the memory tests; 3) Clinical AD dementia defined using standardized criteria [30]. The consensus diagnosis was assigned after all data collection, including neuroimaging, but blinded to data on participants’ markers of angiogenesis activity. Additional data collection included education level, comorbid medical conditions, and self-reported and informant reported perceptions of cognitive complaints using the 20-item Cognitive Change Index [31].
Measurement of circulating hematopoietic stem and progenitor cells
Our group previously reported a novel polychromatic flow cytometry (PFC) assay which defines multiple cell subpopulations including circulating endothelial cells (CEC; CD31brightCD34 +CD45−AC133−), endothelial colony forming cells (ECFC; CD31brightCD34 +CD45−AC133+CD146+CD105+ CD14−CD41a−CD235a−LIVE/DEAD−), and CHS PCs [18, 32, 33]. Further, the assay actually detects two phenotypically and functionally distinct CHSPC subsets, one being pro-angiogenic (CD31+CD 34brightCD45dimAC133+CD14−CD41a−CD235a−LIVE/DEAD−) and the other non-angiogenic (CD31+CD34brightCD45dimAC133−CD14−CD41a−CD235a−LIVE/DEAD−). The conjugated monoclonal antibodies used in this study included anti-human: CD34 (BD Biosciences Cat# 550761, RRID:AB_393871),]; CD45 (Thermo Fisher Scientific Cat# MHCD4527, RRID:AB_10372213); CD31 (BD Biosciences Cat# 555445, RRID:AB_395838); AC133 (Miltenyi Biotec Cat# 130-113-106, RRID: AB_2725935); CD14 (Thermo Fisher Scientific Cat# MHCD1418, RRID:AB_10371748); CD16 (BD Biosciences Cat# 557744, RRID:AB_396850) LIVE/DEAD (Life Technologies, Cat#L34964), anti-human CD235a (glyA, R&D Systems) conjugated to Pacific Blue (PacB, Thermo Fisher Scientific) and the amine reactive viability dye, LiveDead (Thermo Fisher Scientific). Prior to use, each lot of antibody was individually titered to determine the optimal staining concentration.
In summary, mononuclear cells isolated with Ficoll density gradient centrifugation were stained with antibodies against the above-mentioned cell surface antigens (i.e., CD34, AC133, CD31, CD45, CD14, and CD16 as well as a viability marker (LIVE/DEAD) and glycophorin A, for the exclusion of dead cells and red blood cells, respectively) and incubated for 30 min at 4°C. Cells were then washed twice in PBS supplemented with 2% fetal bovine serum, fixed in 300 μl 1% paraformaldehyde (Sigma Aldrich, St. Louis, MO) and acquired on a BD LSRII flow cytometer (BD, Franklin Lakes, NJ, USA) equipped with a 405 nm violet laser, 488 nm blue laser, and 633 nm red laser. Fluorescence minus one (FMO) controls were used as positive gating controls. Acquisition files were exported as FCS 3.0 files and analyzed using FlowJo software, version 8.7.3 (Tree Star, Inc).
Figure 1 demonstrates the gating strategy utilized for identifying pro-angiogenic and non-angiogenic CHSPCs. The analyses ultimately result in an individual estimate of the CHSPC (Pro-CHSPCs:NonCHSPC) ratio in the peripheral blood. In the remainder of this manuscript, we refer to this value as the “angiogenesis ratio”. Using an analogous gating strategy, and in addition to total monocytes, we also identified the following four subpopulations of monocytes based on their surface antigen characteristics: a) activated inflammatory (LIVECD 45+CD16++CD14dim); b) inflammatory (LIVE CD45+CD16+CD14+); c) intermediate (LIVECD 45+CD16+CD14+); and d) classical (LIVECD 45+CD16−CD14++).
Fig. 1.
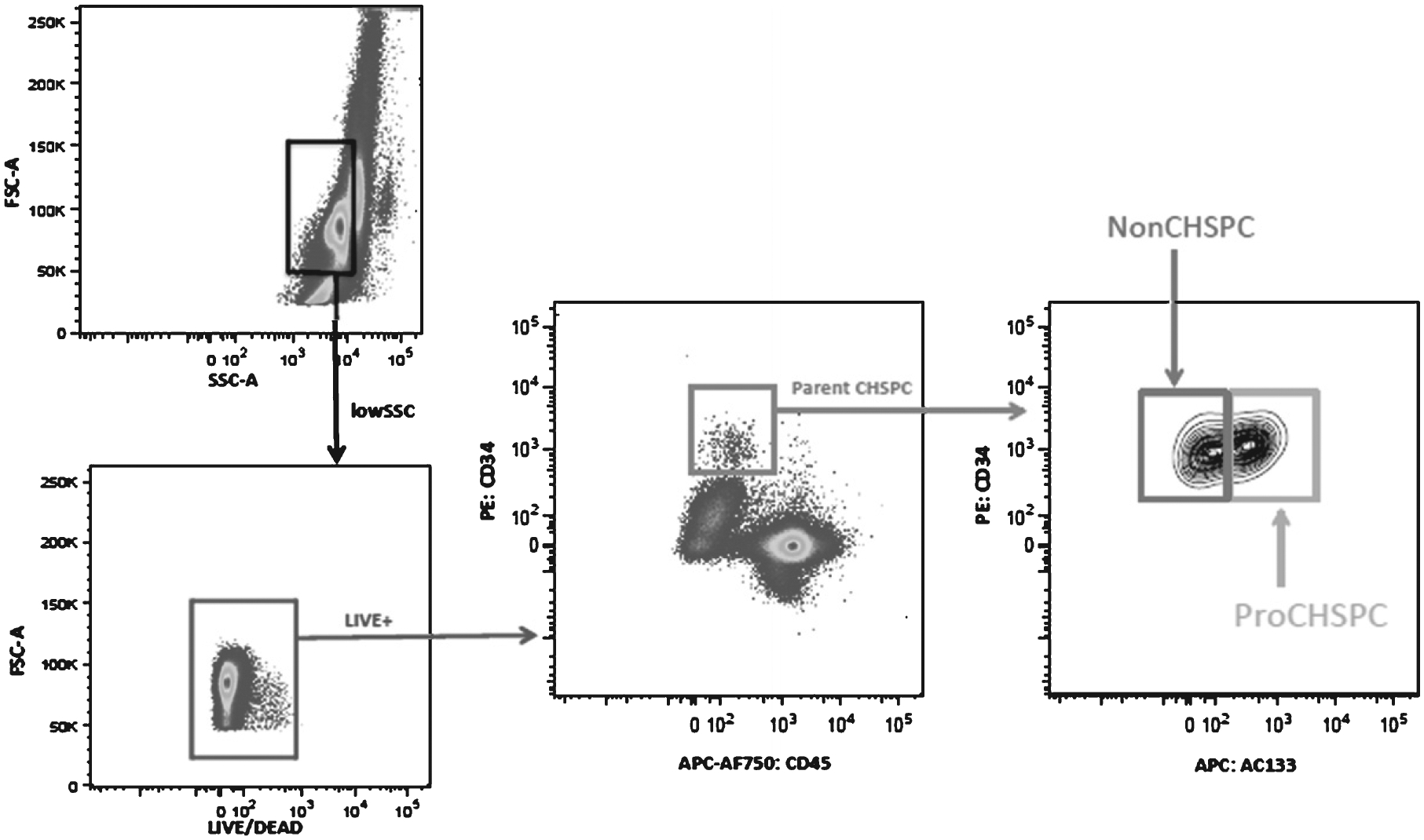
Representative plots showing the gating strategy to identify pro-angiogenic circulating hematopoietic stem and progenitor cells (Pro-CHSPC) and non-angiogenic circulating hematopoietic stem and progenitor cells (Non-CHSPC) by polychromatic flow cytometry. In step 1, low SSC monocytes were selected (black gate) and in step 2 dead cells were removed (blue gate). In step 3, parent CHSPCs were identified (red gate) and in step 4 these CHSPCs are divided into Non-CHSPC and Pro-CHSPC subsets.
Measurement of serum biomarkers
We completed enzyme-linked immunosorbent assays (ELISA) on blood samples to obtain concurrent values for vascular endothelial growth factor (VEGF), tumor necrosis factor (TNFα), fibroblast growth factor (FGF2), and amyloid-β peptide 40. Plasma was isolated by centrifugation at 2200 rpm for 15 min at 4°C. It was aliquoted and stored at −80°C until thawed once and analyzed. Levels of Human TNFα, Human VEGF, and Human FGF2 were measured using Quantikine® enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN). Levels of human amyloid-β peptide 40 were measured using the solid phase sandwich enzyme-linked immunosorbent assay kit (IBL America, Minneapolis, MN).
Neuroimaging
Forty-two participants completed a magnetic resonance imaging (MRI) scan sequence. The MRI scans were performed on a research-dedicated Siemens Prisma 3T magnet and included a magnetization-prepared rapid acquisition with gradient echo (MPRAGE), following the Alzheimer’s Disease Neuroimaging Initiative (ADNI)-2 sequence (http://adni.loni.usc.edu), a 1 mm isotropic fluidattenuated inversion recovery (FLAIR), and a 3D pseudo-continuous arterial spin labeling (pCASL) scan. The pCASL scan parameters are as follows: 2.5 × 2.5 × 2.5 mm3 voxel size, 3790 ms TR, 40.7 ms TE, 10 Turbo Factor, 7 Labeled-Control image pairs, 1800 ms Post Label Delay.
The MPRAGE was analyzed using Freesurfer version 6 to extract total intracranial volume and hippocampal volume. The FLAIR was analyzed using the lesion segmentation toolbox (LST) in Statistical Parametric Mapping 8 (SPM8) to calculate global white matter hyperintensities (WMHI). Using SPM12, the pCASL scans were motion corrected and aligned to the MPRAGE. Then, mean cerebral blood flow (CBF) images were calculated, masked for grey matter, and abnormally high and low CBF values (CBF<0 or CBF>200) were removed. In addition, visual quality control was conducted and 7 participants were removed for the CBF analyses only. Finally, CBF images were normalized to Montreal Neurologic Institute (MNI) space and smoothed with a full-width half-maximum Gaussian kernel. Global grey matter CBF and hippocampal CBF were extracted from subject-specific regions of interest (ROIs) generated using Freesurfer version 6.
Thirty-eight participants underwent amyloid PET scans, collected according to the ADNI-3 [18F]florbetapir or [18F]florbetaben PET protocols [32] (http://adni.loni.usc.edu) on a research-dedicated Siemens Biograph mCT. Briefly, approximately 10 mCi of [18F]florbetapir or 8 mCi of [18F]florbetaben was injected intravenously, and after a 50-min or 90-min uptake period, respectively, continuous listmode PET data were acquired for 20 min. A computerized tomography scan was also collected for scatter and attenuation corrected for both tracers. For both tracers, the listmode data was rebinned into four 5-min frames using scanner software (Siemens; Knoxville, TN) and ordered subset expectation maximization (OSEM) was applied using ADNI parameters (http://adni.loni.usc.edu). Using SPM8, the PET data was aligned to the MPRAGE, motion corrected, normalized to MNI space, and averaged to create a 50–70 min [18F]florbetapir or 90–110 min [18F]florbetaben static image. Then, standardized uptake value ratio images (SUVR) were created by intensity normalizing the images using a whole cerebellar ROI taken from the Centiloid project (http://www.gaain.orig/centiloid-project/) [33]. The resulting SUVR images were converted to Centiloid units as previously described [34]. Finally, the [18F]florbetapir and [18F]florbetaben Centiloid scans were smoothed using an 8 mm FWHM Gaussian kernel. Regional [18F]florbetapir and [18F]florbetaben Centiloid data was extracted from an average bilateral global cortical, lateral parietal, and precuneus ROI created from the average parcellation from 30 independent CN older adults from ADNI-2.
Twenty-three individuals also underwent a [18F]flortaucipir PET scan on the same research-dedicated Siemens mCT. Approximately 10 mCi of [18F]flortaucipir was injected IV, and after a 75-min uptake, participants underwent a 30-min scan using continuous listmode data acquisition. The listmode PET data was rebinned into six 5-min frames and reconstructed using standard scanner software (Siemens; Knoxville, TN) using OSEM, with correction for scatter and random coincident events, attenuation, and radionuclide decay. The middle four 5-min frames (80–100 min) were motion corrected, normalized to MNI space, averaged to create an 80–100 min static image, intensity normalized to the cerebellar crus to create SUVR images, and smoothed with an 8 mm FWHM Gaussian kernel using SPM8. [18F]Flortaucipir SUVR was extracted using subject-specific ROIs generated from Freesurfer version 6 for target regions known to show tau binding in AD, namely the bilateral mean medial temporal lobe (MTL, average of entorhinal cortex, fusiform, and parahippocampal gyri), bilateral mean inferior parietal gyri, and the bilateral mean lateral temporal lobe (LTL, average of inferior temporal gyri, middle temporal gyri, superior temporal gyri).
Data analysis
Descriptive statistics, including means and standard deviations for continuous variables and counts and percentages for categorical variables, were calculated for the three groups of subjects (Normal, MCI, and AD dementia). Additionally, Spearman correlation coefficients were calculated between the angiogenesis ratio, selected serum biomarkers and cognitive test scores, and t-tests were used for testing whether the correlation coefficients were significantly different from 0. Analysis of variance (ANOVA) models and Fisher’s exact tests were used to compare the groups on demographic, clinical, and neuroimaging variables and serum biomarkers.Following a significant overall test for continuous variables, Dunnett’s test or Sidak adjustment for multiple comparisons were used to compare each group against the normal subjects. Analysis of covariance (ANCOVA) models were used to evaluate the association of the angiogenesis ratio with neuroimaging measures adjusting for age, sex, and intracranial volume. All analysis was performed using SAS version 9.4.
RESULTS
We enrolled a total of 49 subjects; seven subjects did not complete any neuroimaging; 39 subjects completed all study procedures. Among the enrolled subjects, 20 (41%) were CN, 17 (34%) were diagnosed with MCI, and 12 (25%) were diagnosed with AD. Table 1 compares the three study groups on demographic, clinical, and neuroimaging variables. Subjects with MCI were older and less likely to be African-American than the other two groups, and subjects with AD had less education, but these differences were not statistically significant. As expected, patients with AD or MCI performed significantly lower on cognitive testing and informants reported greater cognitive symptoms. Cardiovascular risk factors were prevalent but similar between groups. Neuroimaging demonstrated significant differences between study groups in hippocampal volume, as well as amyloid and tau burden. There were no differences in measures of cerebral blood flow or total WMHI. Figure 2 displays a wide variation in angiogenesis ratios between subjects, but no clear relation to age, sex, or diagnosis.
Table 1.
Comparison of diagnosis groups on demographic, clinical, and neuroimaging variables¥
| Variable name | Alzheimer’s (n = 12) | Mild cognitive impairment (n = 17) | Normal (n = 20) | p |
|---|---|---|---|---|
| Age at Visit (mean years) | 71.37 ± 8.01 | 76.35 ± 5.82 | 70.73 ± 8.04 | 0.0589 |
| Sex | 0.5762 | |||
| Female | 8 (66.7%) | 8 (47.1%) | 12 (60.0%) | |
| Male | 4 (33.3%) | 9 (52.9%) | 8 (40.0%) | |
| Subject Race | 0.1972 | |||
| African American | 4 (33.3%) | 3 (17.6%) | 9 (45.0%) | |
| White | 8 (66.7%) | 14 (82.4%) | 11 (55.0%) | |
| Education (mean years) | 14.50 ± 3.34 | 15.63 ± 2.22 | 16.35 ± 2.62 | 0.1828 |
| Clinical Characteristics | ||||
| Cognitive Change Index (CCI) | 34.50 ± 9.26 | 31.69 ± 12.75 | 25.05 ± 9.30 | 0.1125 |
| Self CCI Total Score | 53.25 ± 14.66 | 49.50 ± 20.16 | 37.60 ± 14.91 | 0.0753 |
| Informant CCI Total Score | 77.45 ± 20.20 | 52.27 ± 14.30 | 28.28 ± 13.96 | <0.0001*,† |
| Montreal Cognitive Assessment | 10.00 ± 6.22 | 21.12 ± 3.39 | 24.75 ± 3.74 | <0.0001*,† |
| Global Clinical Dementia Rating | 1.42 ± 0.76 | 0.50 ± 0.00 | 0.10 ± 0.21 | <0.0001*,† |
| Body Mass Index (kg/m2) | 29.60 ± 5.68 | 27.23 ± 3.17 | 27.57 ± 5.80 | 0.4457 |
| Hypertension (UDS Health History) | 6 (50.0%) | 12 (70.6%) | 11 (55.0%) | 0.5694 |
| Diabetes (UDS Health History) | 0 (0.0%) | 3 (17.6%) | 4 (20.0%) | 0.3390 |
| Hypercholesterolemia (UDS Health History) | 5 (41.7%) | 14 (82.4%) | 11 (55.0%) | 0.0596 |
| Cardiovascular disease | 1 (8.3%) | 1 (5.9%) | 5 (25.0%) | 0.3006 |
| Congestive Heart Failure (UDS Health History) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Neuroimaging Variables | ||||
| MRI data (n = 42) | (n = 9) | (n = 14) | (n = 19) | |
| Total Intracranial volume | 1,365,890 ± 94,169 | 1,480,401 ± 136,342 | 1,482,096 ± 215,244 | 0.2140 |
| Bilateral mean hippocampal volume | 2,855.6 ± 356.36 | 3,120.0 ± 464.00 | 3,653.8 ± 445.40 | <0.0001*,† |
| Cerebral blood flow in the global cerebral cortex | 25.83 ± 5.07 | 32.34 ± 7.71 | 30.40 ± 8.08 | 0.1653 |
| Cerebral blood flow in the bilateral hippocampus | 18.79 ± 6.83 | 23.13 ± 10.26 | 22.65 ± 7.72 | 0.4537 |
| Total white matter hyperintensities | 1.78 ± 1.73 | 3.33 ± 5.15 | 1.37 ± 2.38 | 0.2750 |
| Amyloid PET data (n = 38) | (n = 8) | (n = 12) | (n = 18) | |
| Amyloid burden‡ bilateral global cortical grey matter | 48.70 ± 15.50 | 15.67 ± 27.95 | −4.97 ± 13.03 | <0.0001*,† |
| Amyloid burden‡ bilateral lateral parietal lobe | 51.03 ± 18.53 | 19.37 ± 29.91 | −3.08 ± 15.34 | <0.0001*,† |
| Amyloid burden‡ bilateral precuneus | 73.35 ± 21.06 | 36.84 ± 38.53 | 5.41 ± 19.76 | <0.0001*,† |
| Tau PET data (n = 23) | (n = 3) | (n = 8) | (n = 12) | |
| Tau burden¶ in the medial temporal lobe | 2.34 ± 0.20 | 1.24 ± 0.21 | 1.15 ± 0.08 | <0.0001* |
| Tau burden¶ in the inferior parietal lobule | 2.75 ± 1.12 | 1.22 ± 0.26 | 1.11 ± 0.08 | <0.0001* |
| Tau burden¶ in the lateral temporal lobe | 2.38 ± 0.62 | 1.20 ± 0.23 | 1.12 ± 0.08 | <0.0001* |
Results are from univariate analysis of variance models for continuous variables and Fisher’s exact tests for categorical variables;
Pairwise Dunnett or Sidak adjusted comparison between Dementia and Normal is significant (p < 0.05);
Pairwise Dunnett or Sidak adjusted comparison between MCI and Normal is significant (p < 0.05);
Mean Centiloid value;
Mean [18F] flortaucipir SUVR.
Fig. 2.
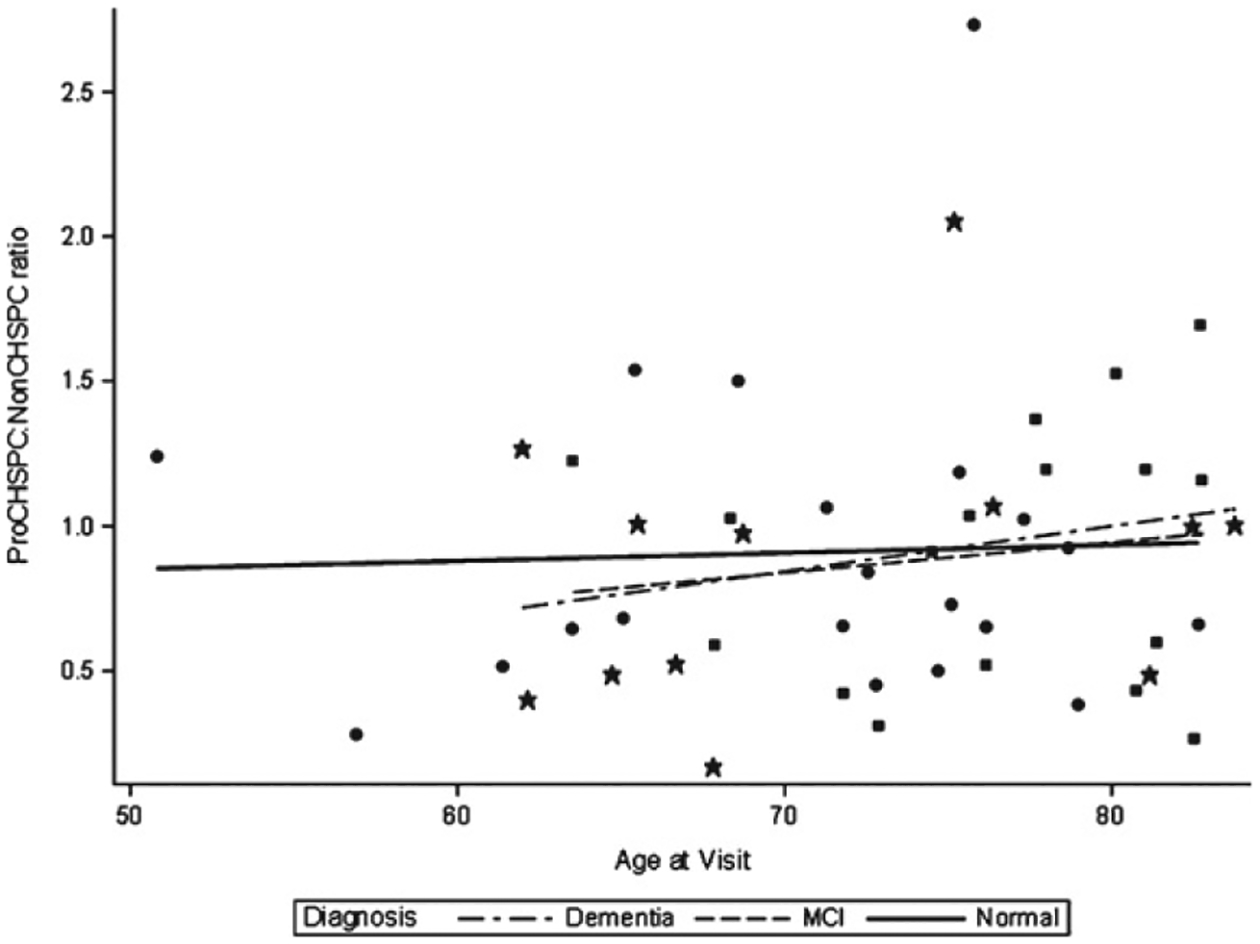
Distribution of Angiogenesis Ratio by Age and Diagnosis Group. Star, dementia; Square, mild cognitive impairment (MCI); Circle, normal.
Table 2 compares the serum biomarkers, including the angiogenesis ratio, among the diagnostic groups. Subjects with MCI had significantly higher levels of VEGF but the angiogenesis ratio and the other biomarkers did not vary between groups. In data not shown in the table, the angiogenesis ratio was not significantly correlated with VEGF, TNF-α, FGF, or amyloid-β peptide 40, nor were these four biomarkers significantly correlated with each other. Furthermore, total monocyte population and subpopulations did not vary significantly between the study groups. The angiogenesis ratio was not significantly correlated with the Montreal Cognitive Assessment score or the self-reported or informant-reported Cognitive Change Index.
Table 2.
Comparison of diagnosis groups on serum biomarkers¥
| Variable name | Alzheimer’s (n = 12) | Mild cognitive impairment (n = 17) | Normal (n = 20) | p |
|---|---|---|---|---|
| Angiogenesis ratio* | 0.87 ± 0.50 | 0.91 ± 0.44 | 0.91 ± 0.55 | 0.9681 |
| Tumor Necrosis Factor alpha† | 7.07 ± 2.95 | 7.74 ± 3.25 | 6.86 ± 2.76 | 0.6755 |
| Fibroblast Growth Factor† | 15.12 ± 13.74 | 15.79 ± 10.03 | 16.30 ± 14.15 | 0.9718 |
| Vascular Endothelial Growth Factor† | 53.76 ± 47.57 | 84.38 ± 52.35 | 43.38 ± 25.24 | 0.0167‡ |
| APOE ε4 allele carrier | 7 (63.6%) | 6 (40.0%) | 8 (40.0%) | 0.4513 |
| Serum B amyloid† | 277.46 ± 40.75 | 279.28 ± 77.40 | 297.43 ± 63.81 | 0.6421 |
| Total Monocytes** | 26.52 ± 11.87 | 27.53 ± 16.09 | 26.95 ± 11.74 | 0.9794 |
| Activated Inflammatory Monocytes** | 0.21 ± 0.27 | 0.52 ± 1.11 | 0.40 ± 0.70 | 0.6079 |
| Intermediate Monocytes** | 2.47 ± 2.23 | 2.55 ± 1.46 | 2.98 ± 2.50 | 0.7522 |
| Inflammatory Monocytes** | 7.64 ± 4.81 | 11.10 ± 6.00 | 8.41 ± 4.66 | 0.1593 |
| Classical Monocytes** | 68.47 ± 13.21 | 67.70 ± 15.28 | 61.66 ± 18.19 | 0.4015 |
Results are from univariate analysis of variance models for continuous variables and Fisher’s exact tests for categorical variables.
Pro-angiogenic to non-angiogenic circulating hematopoietic stem and progenitor cells.
mean and standard deviation values in pg/mL.
Pairwise Dunnett or Sidak adjusted comparison between MCI and Normal is significant (p < 0.05).
See text for monocyte cell surface antigen characterization.
Table 3 shows the results of the pre-planned analysis of covariance models exploring the association of the angiogenesis ratio with neuroimaging findings including white matter hyperintensities, mean bilateral hippocampal volume, hippocampal blood flow, and cerebral blood flow. These neuroimaging findings were chosen because of the a priori hypothesis that the angiogenesis ratio would be elevated among those subjects with evidence of cerebral hypoperfusion. Controlling for age, sex, and intracranial volume, total white matter hyperintensities were significantly correlated with the angiogenesis ratio. The association between the angiogenesis ratio and white matter hyperintensities remain significant when we repeated these models controlling for each of the monocyte populations. We found no significant association between angiogenesis ratio and amyloid load (n = 41) or tau load (n = 26).
Table 3.
Results from analysis Of covariance (ANCOVA) of models of angiogenesis ratio* on neuroimaging measures† adjusted for age, sex, and intracranial volume‡
| Model | Dependent variable | Covariates | ParameterEstimate | StandardError | p |
|---|---|---|---|---|---|
| 1 | Total white matter hyperintensities | Age at Visit | 0.19 | 0.07 | 0.0059 |
| Sex: Female | 2.48 | 1.37 | 0.0792 | ||
| Total Intracranial volume*,‡ | 0.74 | 0.040 | 0.0704 | ||
| Angiogenesis ratio*,‡ | 3.47 | 1.00 | 0.0013 | ||
| 2 | Hippocampal Volume | Age at Visit | −25.67 | 9.66 | 0.0115 |
| Sex: Female | 248.46 | 201.90 | 0.2262 | ||
| Total Intracranial volume*,‡ | 160 | 60 | 0.0110 | ||
| Angiogenesis ratio*,‡ | −83.98 | 146.51 | 0.5700 | ||
| 3 | Hippocampal Blood Flow | Age at Visit | −0.22 | 0.19 | 0.2446 |
| Sex: Female | 5.78 | 3.83 | 0.1424 | ||
| Total Intracranial volume*,‡ | 0.055 | 1.2 | 0.9615 | ||
| Angiogenesis ratio*,‡ | −0.49 | 2.87 | 0.8644 | ||
| 4 | Cerebral Blood Flow | Age at Visit | −0.26 | 0.16 | 0.1219 |
| Sex: Female | 6.52 | 3.37 | 0.0625 | ||
| Total Intracranial volume*,‡ | 1.6 | 0.99 | 0.1195 | ||
| Angiogenesis ratio*,‡ | 0.48 | 2.53 | 0.8495 |
Pro-angiogenic to non-angiogenic circulating hematopoietic stem and progenitor cells;
N = 46;
Total intracranial volume in 100,000 mm3.
DISCUSSION
To our knowledge, this study is the first to report on cellular markers of angiogenesis in a well-characterized sample of older adults with cognitive impairment and dementia as well as cognitively normal older adults. This cross-sectional sample, all of whom completed standardized assessments, represents three distinct diagnostic groups with a range of ages, cognitive performance, and neuropathology. In designing the study in this fashion, we sought to simulate changes in angiogenesis over time that may be occurring over the course of a dementing illness and in the context of aging and comorbid conditions. We demonstrated a positive association between the angiogenesis ratio and the burden of total white matter hyperintensities. We did not identify an association between the angiogenesis ratio and global cerebral blood flow, hippocampal volume, or burden of amyloid or tau pathology. These preliminary results demonstrate the potential to explore the role of angiogenesis in the development of AD dementia as well as related dementia. To better characterize changes in angiogenesis activity over time, a next step project would be to collect repeated samples over a period of many years in well-characterized clinical populations.
Angiogenesis, either peripherally or in the central nervous system, is a complex process involving numerous local and distal humoral and cellular factors. Angiogenesis can be upregulated or down-regulated by multiple stimuli including appropriate responses to hypoxia, tissue injury, inflammation, or disease. Numerous circulating cell subsets participate in repairing damaged vessels and care must be taken to use detection strategies that reflect distinct changes in those heterogeneous cell populations related to a specific disorder or process that may be influenced by many biologic variables over time [17, 18, 35]. Like most fundamental processes, angiogenesis is controlled by both stimuli to activate endothelium and stimuli to serve as a negative feedback loop. Age-related changes, in general, are believed to result in a less vigorous angiogenesis response while age-related disease, such as cancer or macular degeneration, may represent inappropriate upregulation of angiogenesis. Many observational data over at least a century point to a role for hypoperfusion in the development of vascular dementia and also AD dementia. Angiogenesis and neovascularization in the central nervous system may also be counterproductive and result in the over-production of amyloid-β and other neurotoxins [4].
Given the role of angiogenesis in the development and maintenance of the neurovascular unit in the central nervous system, many prior researchers have posited a role for aberrant angiogenesis in neurodegenerative diseases [36]. Improvement in angiogenesis, for example, has been hypothesized as the mechanism of action by which exercise might delay cognitive impairment. Furthermore, modulation of angiogenesis by medications could offer a new therapeutic target in the prevention of AD. To date, however, there are no in vitro models to simulate neurovascular dysfunction over a period of decades [36]. Thus, most of the prior literature in humans focuses on cross-sectional observational data [4, 5].
VEGF is perhaps the most widely studied angiogenesis factor in the context of AD and in the related field of study in neuroinflammation. While VEGF is a critical factor in the cascade of events controlling angiogenesis, VEGF is also implicated in aberrant angiogenesis, including in the setting of cancer and neovascularization in the retina. Furthermore, VEGF has also been reported to increase vascular permeability which might promote appropriate or inappropriate neuroinflammation, and our group has previously reported that pro-angiogenic CHSPCs secrete VEGF [24]. In the setting of AD, the role of VEGF remains uncertain. Previous studies have reported both increased and decreased VEGF activity in subjects with AD dementia, including studies of the cerebrospinal fluid in well-characterized subjects [37, 38]. As noted earlier, much of the literature exploring the relationship between VEGF and AD is cross-sectional and/or focuses on post-mortem studies [39]. In the related field of inflammation and cognition, in contrast, longitudinal studies have recently shown that baseline peripheral blood biomarkers such a C-reactive protein are associated with cognitive decline 20 years later [6, 27]. Another important contribution of angiogenesis is its role in neurogenesis which is thought to be critical for maintaining hippocampal function and hence episodic memory processes [7].
Our study has three important limitations. First, the sample size is relatively small. Even with a small sample size, richly phenotyped studies such as this are expensive, and the present study is only possible by leveraging the resources of an NIA-designated ADRC. The angiogenesis assay itself must be completed within 2 h of sample collection and therefore presents an additional challenge. Second, we report cross-sectional data. Over the course of middle and later life, in addition to changes associated with aging and chronic conditions, any given individual might experience multiple episodes of acute illness or trauma that might require an appropriate angiogenesis response. Thus, angiogenesis more likely represents a current “state” rather than an enduring trait. However, as in the case of neuroinflammation, it is possible that some individuals develop chronic states that results in pathology [6]. Third, the study design assumes that peripherally circulating hematopoietic stem and progenitor cells represent the angiogenesis “state” in the central nervous system. While the angiogenesis ratio has been reported to reflect states of low [17, 18] or high angiogenesis [24] in patients with cardiovascular disease or cancer, respectively, further studies will be required to prospectively assess the use of the ratio as a biomarker in patients with neurocognitive disorders.
In conclusion, lower levels of angiogenesis activity are associated with a greater burden of small vessel disease as reflected by white matter hyperintensities on neuroimaging. However, in this small study, angiogenesis activity was not significantly associated with clinical measures of cognitive function or neuroimaging measures of amyloid or tau burden. Future research would benefit from a longitudinal design with repeated measures of angiogenesis activity in larger clinical samples to capture the dynamic nature of the angiogenesis state of older adults over time.
ACKNOWLEDGMENTS
We are grateful to the patients and their family members who made this investigation possible.
We thank Emily Sims and Mathew Repass of the Angio BioCore at the Indiana University Simon Cancer Center (IUSCC) for processing the peripheral blood samples for this study and for running the flow cytometry instrument. We also acknowledge the assistance and state-of the art facilities of the Flow Cytometry Resource Facility at the IUSCC. We thank Eileen Tallman, John D. West, Rachael Deardorff, and Cecily Swinford and Drs. James Fletcher, Gary Hutchins and Karmen Yoder of the Department of Radiology and Imaging Sciences and the Indiana Alzheimer Disease Center for their assistance with neuroimaging acquisition and/or analysis.
This work was supported, in part, by grants from the National Institute on Aging [R21AG051932 (CMC), P30 AG010133 (AJS), R01 AG019771 (AJS) and K01 AG049050 (SLR)] and the Clinical Research Center within the Indiana CTSI [UL1TR001108]. In addition, IU received PET tracer precursor support from Avid Radiopharmaceuticals.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-1293r2).
REFERENCES
- [1].de la Torre JC (2004) Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol 3, 184–190. [DOI] [PubMed] [Google Scholar]
- [2].Purnell C, Gao S, Callahan CM, Hendrie HC (2009) Cardiovascular risk factors and incident Alzheimer disease: A systematic review of the literature. Alzheimer Dis Assoc Disord 23, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vagnucci AH Jr., Li WW (2003) Alzheimer’s disease and angiogenesis. Lancet 361, 605–608. [DOI] [PubMed] [Google Scholar]
- [5].Govindpani K, McNamara LG, Smith NR, Vinnakota C, Waldvogel HJ, Faull RL, Kwakowsky A (2019) Vascular dysfunction in Alzheimer’s disease: A prelude to the pathological process or a consequence of it? J Clin Med 8, E651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Walker KA (2018) Inflammation and neurodegeneration: Chronicity matters. Aging (Albany NY) 11, 3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Horgusluoglu E, Nudelman K, Nho K, Saykin AJ (2017) Adult neurogenesis and neurodegenerative diseases: A systems biology perspective. Am J Med Genet B Neuropsychiatr Genet 174, 93–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim LA, D’Amore PA (2012) A brief history of anti-VEGF for the treatment of ocular angiogenesis. Am J Pathol 181, 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438, 932–936. [DOI] [PubMed] [Google Scholar]
- [10].Felmeden DC, Blann AD, Lip GY (2003) Angiogenesis: Basic pathophysiology and implications for disease. Eur Heart J 24, 586–603. [DOI] [PubMed] [Google Scholar]
- [11].Ambrose CT (2015) A therapeutic approach for senile dementias: Neuroangiogenesis. J Alzheimers Dis 43, 1–17. [DOI] [PubMed] [Google Scholar]
- [12].Lahteenvuo J, Rosenzweig A (2012) Effects of aging on angiogenesis. Circ Res 110, 1252–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pimentel-Coelho PM, Rivest S (2012) The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer’s disease. Eur J Neurosci 35, 1917–1937. [DOI] [PubMed] [Google Scholar]
- [14].Guven H, Shepherd RM, Bach RG, Capoccia BJ, Link DC (2006) The number of endothelial progenitor cell colonies in the blood is increased in patients with angiographically significant coronary artery disease. J Am Coll Cardiol 48, 1579–1587. [DOI] [PubMed] [Google Scholar]
- [15].Greenberg DA, Jin K (2013) Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci 70, 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee ST, Chu K, Jung KH, Jeon D, Bahn JJ, Kim JH, Kun Lee S, Kim M, Roh JK (2010) Dysfunctional characteristics of circulating angiogenic cells in Alzheimer’s disease. J Alzheimers Dis 19, 1231–1240. [DOI] [PubMed] [Google Scholar]
- [17].Mund JA, Estes ML, Yoder MC, Ingram DA Jr., Case J (2012) Flow cytometric identification and functional characterization of immature and mature circulating endothelial cells. Arterioscler Thromb Vasc Biol 32, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mund JA, Case J (2011) The ontogeny of endothelial progenitor cells through flow cytometry. Curr Opin Hematol 18, 166–170. [DOI] [PubMed] [Google Scholar]
- [19].Salter AB, Meadows SK, Muramoto GG, Himburg H, Doan P, Daher P, Russell L, Chen B, Chao NJ, Chute JP (2009) Endothelial progenitor cell infusion induces hematopoietic stem cell reconstitution in vivo. Blood 113, 2104–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Herzenberg LA, Tung J, Moore WA, Herzenberg LA, Parks DR (2006) Interpreting flow cytometry data: A guide for the perplexed. Nat Immunol 7, 681–685. [DOI] [PubMed] [Google Scholar]
- [21].Parks DR, Roederer M, Moore WA (2006) A new “Logicle” display method avoids deceptive effects of logarithmic scaling for low signals and compensated data. Cytometry A 69, 541–551. [DOI] [PubMed] [Google Scholar]
- [22].Baumgarth N, Roederer M (2000) A practical approach to multicolor flow cytometry for immunophenotyping. J Immunol Methods 243, 77–97. [DOI] [PubMed] [Google Scholar]
- [23].Kaleem Z (2006) Flow cytometric analysis of lymphomas: Current status and usefulness. Arch Pathol Lab Med 130, 1850–1858. [DOI] [PubMed] [Google Scholar]
- [24].Mund JA, Shannon H, Sinn AL, Cai S, Wang H, Pradhan KR, Pollok KE, Case J (2013) Human proangiogenic circulating hematopoietic stem and progenitor cells promote tumor growth in an orthotopic melanoma xenograft model. Angiogenesis 16, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fadini GP, Avogaro A (2010) Cell-based methods for ex vivo evaluation of human endothelial biology. Cardiovasc Res 87, 12–21. [DOI] [PubMed] [Google Scholar]
- [26].Medina RJ, Barber CL, Sabatier F, Dignat-George F, Melero-Martin JM, Khosrotehrani K, Ohneda O, Randi AM, Chan JKY, Yamaguchi T, Van Hinsbergh VWM, Yoder MC, Stitt AW (2017) Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Transl Med 6, 1316–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Walker KA, Gottesman RF, Wu A, Knopman DS, Gross AL, Mosley TH Jr., Selvin E, Windham BG (2019) Systemic inflammation during midlife and cognitive change over 20 years: The ARIC Study. Neurology 92, e1256–e1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Walker KA, Walston J, Gottesman RF, Kucharska-Newton A, Palta P, Windham BG (2019) Midlife systemic inflammation is associated with frailty in later life: The ARIC Study. J Gerontol A Biol Sci Med Sci 74, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weintraub S, Besser L, Dodge HH, Teylan M, Ferris S, Goldstein FC, Giordani B, Kramer J, Loewenstein D, Marson D, Mungas D, Salmon D, Welsh-Bohmer K, Zhou XH, Shirk SD, Atri A, Kukull WA, Phelps C, Morris JC (2018) Version 3 of the Alzheimer Disease Centers’ Neuropsychological Test Battery in the Uniform Data Set (UDS). Alzheimer Dis Assoc Disord 32, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carillo MC, Thies B, Weintraub S, Phelps CH (2011) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging and the Alzheimer’s Association workgroup. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rattanabannakit C, Risacher SL, Gao S, Lane KA, Brown SA, McDonald BC, Unverzagt FW, Apostolova LG, Saykin AJ, Farlow MR (2016) The Cognitive Change Index as a measure of self and informant perception of cognitive decline: Relation to neuropsychological tests. J Alzheimers Dis 51, 1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, Price JC, Reiman EM, Skovronsky D, Koeppe RA, Alzheimer’s Disease Neuroimaging Initiative (2010) The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement 6, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD Sr., Jagust WJ, Johnson KA, Mathis CA, Minhas D, Pontecorvo MJ, Rowe CC, Skovronsky DM, Mintun MA (2015) The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 11, 1–15 e11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Risacher SL, Tallman EF, West JD, Yoder KK, Hutchins GD, Fletcher JW, Gao S, Kareken DA, Farlow MR, Apostolova LG, Saykin AJ (2017) Olfactory identification in subjective cognitive decline and mild cognitive impairment: Association with tau but not amyloid positron emission tomography. Alzheimers Dement (Amst) 9, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Huizer K, Mustafa DAM, Spelt JC, Kros JM, Sacchetti A (2017) Improving the characterization of endothelial progenitor cell subsets by an optimized FACS protocol. PLoS One 12, e0184895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Parkes I, Chintawar S, Cader MZ (2018) Neurovascular dysfunction in dementia - human cellular models and molecular mechanisms. Clin Sci (Lond) 132, 399–418. [DOI] [PubMed] [Google Scholar]
- [37].Hohman TJ, Bell SP, Jefferson AL, for the Alzheimer’s Disease Neuroimaging Initiative (2015) The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: Exploring interactions with biomarkers of Alzheimer disease. JAMA Neurol 72, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chakraborty A, Chatterjee M, Twaalfhoven H, Del Campo Milan M, Teunissen CE, Scheltens P, Fontijn RD, van Der Flier WM, de Vries HE (2018) Vascular endothelial growth factor remains unchanged in cerebrospinal fluid of patients with Alzheimer’s disease and vascular dementia. Alzheimers Res Ther 10, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Harris R, Miners JS, Allen S, Love S (2018) VEGFR1 and VEGFR2 in Alzheimer’s disease. J Alzheimers Dis 61, 741–752. [DOI] [PubMed] [Google Scholar]