

Abstract
We propose a novel approach for building a classification/identification framework based on the full complement of RNA post-transcriptional modifications (rPTMs) expressed by an organism at basal conditions. The approach relies on advanced mass spectrometric techniques to characterize the products of exonuclease digestion of total RNA extracts. Sample profiles comprising identities and relative abundances of all detected rPTM were used to train and test the capabilities of different of machine learning (ML) algorithms. Each algorithm proved capable of identifying rigorous decision rules for differentiating closely related classes and correctly assigning unlabeled samples. The ML classifiers resolved different members of the Enterobacteriaceae family, alternative E. coli serotypes, a series of S. cerevisiae knockout mutants, and primary cells of H. sapiens central nervous system, which shared very similar genetic backgrounds. The excellent levels of accuracy and resolving power achieved by training on a limited number of classes were successfully replicated when the number of classes was significantly increased to escalate complexity. A dendrogram generated from ML-curated data exhibited a hierarchical organization that closely resembled those afforded by established taxonomic systems. Finer clustering patterns revealed the extensive effects induced by the deletion of a single pivotal gene. This information provided a putative roadmap for exploring the roles of rPTMs in their respective regulatory networks, which will be essential to decipher the epitranscriptomics code. The ubiquitous presence of RNA in virtually all living organisms promises to enable the broadest possible range of applications, with significant implications in the diagnosis of RNA-related diseases.
Graphical Abstract
Since the dawn of time, humans have felt the instinctive need to classify animals and plants according to common features, origin, and behavior. In the 18th century, Carl Linnæus ushered in modern taxonomy by publishing Systema Naturæ, in which he proposed a hierarchical classification system that divided the natural world in animal, plant, and mineral kingdoms, and used a binomial nomenclature to uniquely identify the different species.1 Over the years, the system has been the object of frequent upgrades in response to our steadily expanding knowledge and the introduction of ever more stringent classification criteria. For the past three decades, the small subunit of ribosomal RNA (SSU rRNA) has been embraced as the gold standard for species classification and identification.2 The fact that ribosomal operon size, nucleotide sequence, and secondary structure are highly conserved across species allows SSU rRNA to differentiate organisms at the genus and family levels. This characteristic has been successfully leveraged not only to create a robust classification framework, but also to guide the assignment of unknown samples to the correct species, as demonstrated by the utilization of SSU rRNA to identify organisms present in the gut microbiome, which are responsible for a series of intestinal and extra-intestinal diseases.3 However, a broader diffusion of these types of applications has been thus far hampered by the incidence of horizontal gene transfer, intra-genomic heterogeneity, and formation of mosaic-like structures, which limit the ability of SSU rRNA to discriminate closely related organisms.4 For this reason, searching for putative features capable of supporting unambiguous classification/identification operations remains a common theme in “omics” research.
The ubiquitous presence of RNA post-transcriptional modifications (rPTMs) across all kingdoms of life is substantiated by pervasive biological functions that are gradually emerging. New technologies for rPTM analysis have helped gain valuable insights into the significance of the ever growing number of known variants of canonical ribonucleotides.5,6 The identity and abundance of these modifications is governed by the activity of specific enzymes, which reflects the development, health, and metabolic state of a cell, as well as the environment to which the cell was exposed.7 The fact that such enzymes and corresponding regulatory pathways are firmly encoded in the cell’s genome, and are thus the result of a well-defined evolutionary trajectory, suggests that the end products could represent unique differentiating features. Consistent with this hypothesis, the study of tRNA distribution revealed the presence of organism- and organ-specific rPTMs.8,9 In earlier work, we found that this observation was not limited to an individual class of RNA, but extended instead to the entire assortment of RNA in the cell. Indeed, we observed that rPTM profiles obtained from S. cerevisiae’s total RNA were markedly different from those of HeLa and other human cell lines.10–12 Therefore, establishing unequivocal links between rPTM identity/incidence and type of organism could promote RNA modifications as an ideal platform for creating a robust classification/identification framework with a broad range of possible applications.
The majority of current detection technologies utilizes Next Generation Sequencing (NGS) to analyze samples that were either enriched by immunoprecipitation with rPTM-specific anti-bodies,13 contained abortive strands generated by halting reverse-transcription,14 or exhibited site-specific mutations induced by targeted chemical reactions.15 Although such technologies have been developed pre-eminently to obtain sequence location, they are routinely applied also to assess the incidence of individual rPTMs at full transcriptome levels. In contrast, mass spectrometric (MS) analysis can identify virtually all rPTMs present in a sample according to unique mass, fragmentation, and ion mobility characteristics.16,17 In earlier work, we have demonstrated that the analysis of a mono-ribonucleotide mixture generated by exonuclease digestion of a total RNA extract can provide a very accurate representation of the landscape of rPTMs contained in a biological sample.10–12 To evaluate the merits of entire panels of RNA modifications as possible differentiating features, we now compared the global rPTM profiles of a wide range of organisms at basal conditions to avoid any ambiguities introduced by abnormal metabolic, health, and environmental circumstances. Prompted by the complexity of such profiles, which may comprise tens of different types of rPTMs, we explored the application of machine learning (ML) to help rationalize the experimental data and identify rigorous classification rules. We then assessed the ability of such rules to differentiate organisms with very close evolutionary trajectories. We finally compared the class organization obtained from ML-curated data with those provided by established phylogenetic systems to evaluate possible dissimilarities induced by the utilization of a different set of differentiating criteria and the implementation of strictly agnostic, data-driven classification rules.
EXPERIMENTAL SECTION
Biological samples.
The study examined the following biological samples: E. coli MG1655 (serotype OR:H48:K) and CDC EDL 933 (serotype O157:H7); S. typhimurium; L. monocytogenes; S. pneumoniae; H. salinarum; S. cerevisiae strain BY4741 (defined here as wildtype, WT) and knockout mutants trf4Δ::kanMX, rit1Δ::kanMX, and set1Δ::kanMX derived from the S288C strain; A. thaliana; A. superba; A. aegypti; HeLa and HEK 293T cell lines; human glioblastoma astrocytoma (U251-MG); human primary neurons, astrocytes and microglia. All samples were obtained either from commercial sources or collaborators, and authenticated according to established procedures. Each organism or cell line was grown under established conditions considered as basal by the respective field (see Supporting Information, S.I.).
Sample preparation.
The analytical workflow has been described in great detail in ref.10–12 and S.I. Briefly, each sample was lysed and extracted by using TRIzol (ThermoFisher Scientific) according to the vendor recommendations. For S. cerevisiae samples, this treatment was performed in the presence of 0.5 mm glass beads to aid lysis. For A. thaliana and A. superba, the material was frozen in liquid N2 and homogenized in a mortar before extraction. The aqueous layer containing primarily RNA and low-level DNA contamination was digested with DNase I (ThermoFisher) and ethanol-precipitated overnight to eliminate the latter. The total-RNA was treated with nuclease P1 and phosphodiesterase from snake venom (Sigma Aldrich) to obtain the desired mononucleotide mixture.10–12
Mass spectrometry.
Immediately before analysis, each total-RNA digest was diluted to 4 ng/μL in 10 mM MS-grade ammonium acetate (Sigma Aldrich) and 10% isopropanol. Each sample was analyzed either on a Waters Synapt G2 HDMS ion mobility spectrometry mass spectrometer (IMS-MS) or a Thermo Scientific LTQ-Orbitrap Velos, as previously described.10–12 The former provided heat maps that offered comprehensive representations of the entire complement of rPTMs present in the sample.10 The latter provided accurate mass and fragmentation data that were used to confirm the IMS-MS assignments. Conditions and figures of merit are detailed in S. I.
Data interpretation and machine learning.
The rPTMs in each sample were recognized by searching experimental masses against a non-redundant database containing the masses of all known RNA modifications.10 The relative abundance of each rPTM was expressed relative to the cumulative abundance of the canonical (i.e., unmodified) nucleotides present in the total-RNA digest.10 Replicate analyses (indicated with N in the text) were carried out to enable a proper assessment of statistical significance. The data were then used to train/test machine learning (ML) classifiers based on the K-nearest neighbors (KNN), naïve Bayes (NB), and gradient boosting (GB) algorithms included in Scikit-learn v.0.23 (https://scikit-learn.org/stable/). Accuracy, precision, and recall were calculated directly by the software. Euclidean distances between relevant features were used as input for an agglomerative clustering procedure supported by the sklearn.metrics.pairwise.euclidean_distances program of the Scikit-learn v.0.23 package (see S.I.).
RESULTS AND DISCUSSION
Profiling RNA post-transcriptional modifications.
The full complement of RNA rPTMs expressed by a cell is determined by a complex combination of genetic, regulatory, and metabolic factors. On one hand, the writer and eraser enzymes responsible for establishing or removing each modification are encoded in the cell’s genome. On the other, the expression and activity of such enzymes are subjected to the control of regulatory mechanisms that reflect the actual developmental, metabolic, environmental, and health state of the cell. Therefore, global rPTM analysis can in principle afford a comprehensive view of the effects of any of these variables, if all the others are kept constant.
These observations keenly underscored the need to base our classification/identification framework on the most representative possible rPTM profiles, capable of identifying unambiguously the respective classes. For this reason, we only examined organisms grown/secured under conditions that are generally considered as basal or “standard” by the respective fields (see S.I.). We purposely refrained from pursuing variations induced by biological/environmental factors, which were instead the object of prior reports and are currently being explored for possible diagnostic applications.10–12 The decision to rely on comprehensive analysis of total RNA, rather than pursuing the isolation of any specific type of RNA, was made to expedite the analysis and increase sample throughput, as well as to eliminate possible sources of variability introduced by the differential effects of biological/environmental factors on the expression and complexion of different RNAs. At the same time, no particular effort was made to control for putative differential effects of TRIzol extraction on the yield of specific RNA types, to finely tune exonuclease digestion to account for the different susceptibility of 2’-O-methylated or phosphorothioate modifications, or to assess the chemical stability of individual rPTMs, such as mnm5s2U,18 during the entire sample preparation workflow. Instead, the same exact protocol was applied to all samples in the study, from harvest to analysis. This way, all samples of a given class produced self-consistent profiles that were accurately representative of such class under the selected experimental conditions. The always possible presence of analytical bias between classes did not frustrate the ability of their respective profiles to be uniquely representative under the established conditions.
The need to streamline the analytical workflow and minimize any sample-to-sample bias was also addressed by analyzing the digestion mixtures by nanoflow electrospray (nanospray) with no front-end chromatographic separation. All analyses were carried out on either ion mobility spectrometry (IMS) or high-resolution mass spectrometers, a described earlier.10 Although these approaches could readily support the use of isolated standards or stable isotope labels to determine absolute rPTM levels, it is usually preferable to calculate relative abundances from their signal intensities in relation to the cumulative signal of all mixture components, or just the unmodified canonical nucleotides in the sample (see S.I.). The latter is particularly effective when comparing the relative effects of selected variables, which does not require knowing actual cellular concentrations.10
Representative data obtained from E. coli OR:H48:K, H. salinarum, S. cerevisiae BY4741, A. thaliana, and H. sapiens HeLa cells, which represent respectively the kingdoms of Bacteria, Archaea, Fungi, Plantae and Animalia, are summarized in Table S1 of S.I. Each column constitutes the average of five different samples (i.e., biological replicates) that were individually analyzed five times each (i.e., technical replicates) for a total N = 25 replicates per organism. The color gradient represents relative abundances expressed as percentage of the sum of the abundances of the canonical bases (i.e., AvP units, see Experimental of S.I.). The relative intensities exhibited by the E. coli OR:H48:K samples (first column) were used as references to assess the statistical significance of corresponding values obtained from the other samples, which enabled the assignment of different colors to relative intensities producing p values no greater than 0.05. The treatment afforded an immediate appreciation of the astonishing diversity of the rPTMs expressed by the various organisms. No sample exhibited fewer than 24 rPTMs, whereas their number increased steadily as a function of organism complexity. A close examination revealed that some rPTMs were present in all organisms, whereas others were observed only in some of them. As a whole, the modification landscape of each individual organism looked significantly different from those of all the others in the table, thus suggesting that rPTM profiles may constitute valid differentiating features in possible classification/identification applications.
Integrating rPTM analysis with machine learning.
The wealth of information afforded by rPTM profiles poses numerous challenges to effective data interpretation, evaluation, and organization. In previous work, we resorted to Venn diagrams to dissect rPTM profiles, visualize the presence of features across samples, and recognize differences and communalities in unambiguous fashion.12 However, the complexity of these diagrams tends to increase geometrically with the number of classes under consideration, which hobbles the ability to create robust classification systems and limits the types of correlations that could be drawn. Furthermore, Venn diagrams fail to account for the relative abundances of the various features, thus overlooking precious information. In contrast, classifiers based on machine learning (ML) algorithms exhibit the intrinsic ability to handle multidimensional problems, like the creation of a putative classification/identification platform based on rPTM analysis. The multidimensional nature of this problem is substantiated not only by the type of pairwise information (i.e., identity/abundance) for each rPTM, but also by the large number of rPTMs (i.e., features) in a typical profile, that of organisms (i.e., classes) to be included in the sought-after framework, and that of the replicate analyses (N) necessary to ensure statistical significance. For this reason, we explored the ability of supervised ML algorithms to account for all these variables and to turn the challenge of an ever-expanding dataset into a positive force driving the learning process. In particular, we evaluated the merits of the k nearest neighbor (KNN),19 naïve Bayes (NB),20 and gradient boosting (GB)21 algorithms. All of them utilize labeled input data to carry out supervised training operations that are aimed at learning the best possible function, or set of rules, capable of predicting the correct output for new unlabeled data. Briefly, the KNN algorithm utilizes vectors to represent the identities and relative abundances of the rPTMs in each profile, considered here as an individual input datapoint. Such vectors are plotted in n-dimensional space (where n is the number of features) and used to calculate the distance between the datapoints in the training set. Subsets of k datapoints with the lowest mutual distances identify decision boundaries between classes, which can be subsequently utilized to assign any unlabeled datapoint submitted to the classifier.19 NB classifiers are based on the assumption that the value assumed by any given feature is independent from those of all the others in the class. This assumption supports a probability model in which the decision rule can simply consist of picking the most probable assignment estimated from the training set).20 In contrast, GB classifiers involve sequential decision trees that seek to correctly assign each sample to a given class by minimizing the residual error incurred in the previous round of predictions.21 The final outcome is therefore achieved through a stepwise process in which an ensemble of weak prediction models leads to the correct assignment by successive approximations.
The initial dataset comprising the rPTM profiles of the above representative organisms was randomly split into training and testing subsets of 70:30 proportions to eliminate the possibility that testing putative classification rules on the exact same data used to distill them might lead to self-fulfilling predictions (see Experimental). Supervised training followed an iterative leave-one-out (LOO)21 approach according to which each datapoint was in turn excluded from the input, while the remaining ones were used to identify putative classification rules. The excluded input was then submitted to the algorithm as unlabeled data to assess the predictive power of such rules. The process was repeated N-1 times (where N is the number of datapoints/replicates) to allow for the exclusion of each input at least once, thus eliminating possible bias and providing cross-validation. Therefore, each predictive model was produced through an extensive series of iterations between rule-formulation and cross-validation steps, which constitutes a distinctive characteristic of these ML approaches. As exemplified by the plot obtained from the GB classifier (Figure 1a), the results of the LOO process are typically expressed in a graphic format in which predicted and actual labels are plotted on the x- and y-axis, respectively. A color gradient is used to indicate the frequency by which a given assignment was made during the entire process. Analogous plots obtained for the KNN and NB classifiers are shown in Figure S1a and S2a of S. I.
Figure 1.
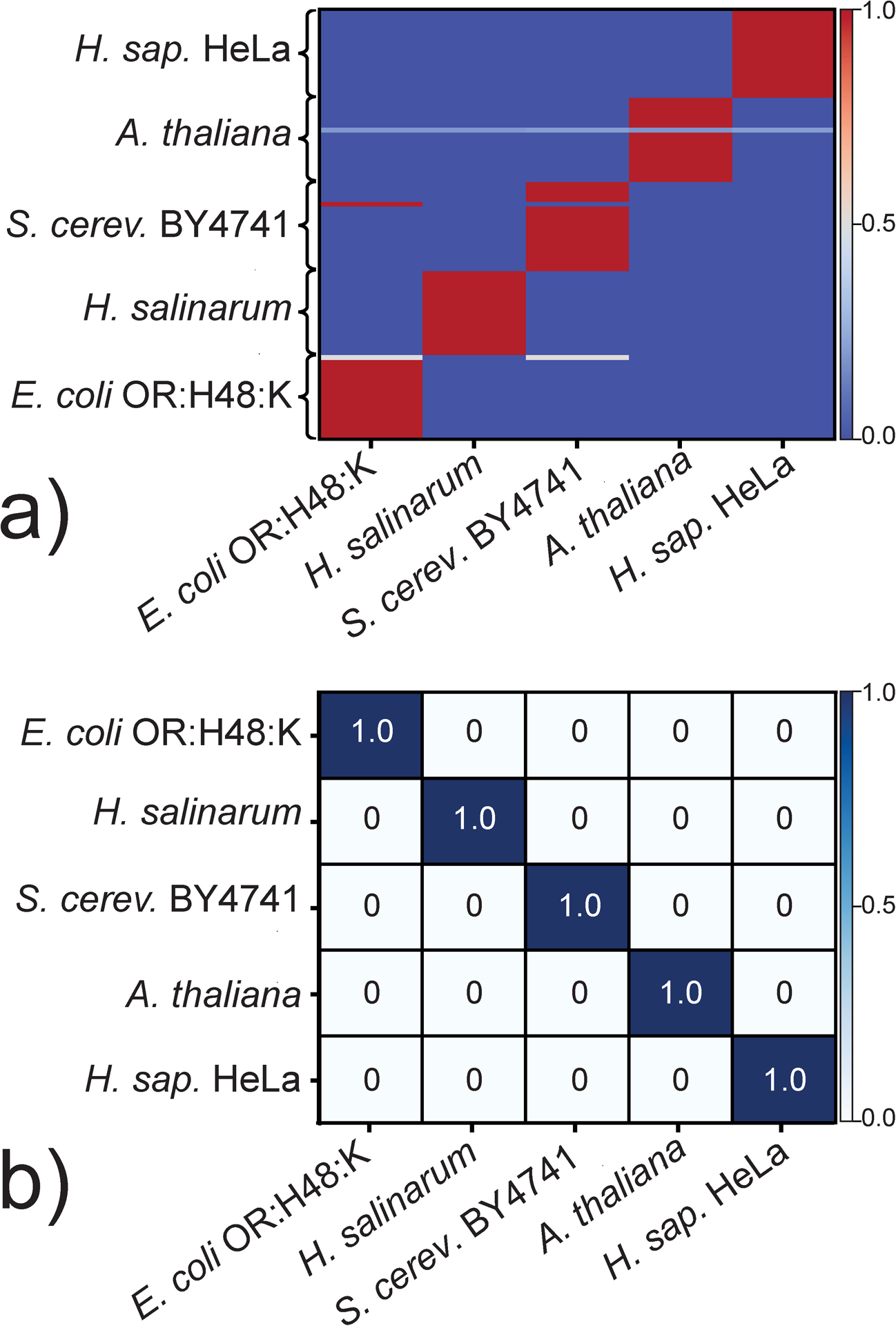
Results obtained by training a) and testing b) the gradient boosting (GB) algorithm on the representative data summarized in Table S1 of S.I. (see Experimental). The initial dataset, which comprised rPTM profiles acquired in 25 replicate analyses per organism (i.e., N = 25), was randomly split into training and testing sets of 70:30 proportions. Panel a) Frequency by which a training datapoint on the y-axis was assigned to a class on the x-axis during leave-one-out (LOO) iterations between rule-formulation and cross-validation steps. Panel b) Frequency by which testing datapoints of a given class plotted on the y-axis were assigned to one of the classes on the x-axis. Color gradients convey frequencies of assignment.
A close examination of these types of plots can provide valuable information on the robustness of the training process, as well as the possible quality of the underlying experimental data. For example, the fact that all datapoints of H. salinarum and H. sapiens HeLa were consistently assigned to the correct classes afforded a measure of the excellent potential afforded by this classification approach (Figure 1a). At the same time, the fact that one of the profiles of A. thaliana was assigned with similar frequencies to all classes under consideration suggested possible problems with sample integrity or analytical performance. Indeed, visual inspection of the original nanospray-MS data revealed undesirable deterioration of the signal-to-noise ratio across the board, which could be ascribed either to significant losses of analytes during sample preparation, or possible spray instabilities that might have reduced the overall sensitivity of analysis. In contrast, the observation that one of the S. cerevisiae BY4741 profiles was erroneously assigned as E. coli OR:H48:K 90% of the times, or that one of the E. coli OR:H48:K was mistaken for S. cerevisiae BY4741 50% of the times could be explained by possible contamination during sample manipulation. It should be noted that the supervised nature of the learning process precluded the ability to recognize the exact source of the contamination. Indeed, the classifiers learn the identity of a sample from its given label. If mixed samples are employed, the classifiers will place them in a separate “mixed” class, if they were labeled as “mixed”, or strive to group them with one of the mixture components, if they were labeled as such. The important outcome of this experiment, however, was the absence of cases in which multiple profiles of a certain organism were consistently mis-assigned to a specific class, and vice versa, which would signal systematic failure of the classifier to properly differentiate between classes.
After training was completed, the performance of the predictive models was evaluated by submitting the testing subset (30% of the total) as unlabeled datapoints, and then determining the frequency by which they were assigned to the correct classes. The results were displayed in the form of confusion matrices in which predicted and actual labels were plotted on the xand y-axis, respectively. The frequency of assignment was provided in both color-gradient and numerical format. The confusion matrix obtained from the GB classifier indicated that the algorithm had properly assigned all the unlabeled datapoints to the correct classes (Figure 1b). This very desirable outcome was shared with the KNN but not the NB classifier, as shown by the respective matrices in Figure S1b and S2b of S.I. The testing results were also expressed in terms of accuracy, precision, and recall, which provide a very effective way for comparing performance. Indeed, accuracy measures the overall ability of a classifier to provide correct assignments, whereas precision is the percentage of true positive assignments over the total of true and false positives (see Experimental of S.I.). Recall is instead the percentage of true positives over all positives, true and false. As shown in Table S2 of S.I., KNN and GB achieved the highest possible marks across the board, whereas NB afforded somewhat inferior values. A possible cause for this outcome could be traced to the corresponding training process, during which at least one E. coli OR:H48:K datapoint was consistently mis-assigned a S. cerevisiae BY4741, and vice-versa one S. cerevisiae BY4741 as E. coli OR:H48:K (Figure S2a). This fact likely led to a weaker predictive model that proved to be error-prone during the testing phase (Figure S2b).
A closer look at the predictive models can readily reveal the weight of individual rPTMs in differentiating the various organisms. This type of information can offer valuable insights not only into the inner workings of the classifier itself, but also into the possible biological significance of specific features. For example, the importance plot obtained from the GB classifier identified t6A, monomethyl-adenosines, and mo5U as exerting the greater influence on class differentiation (Figure 2a). Their weights, however, were not found to be proportional to the respective relative abundances in the rPTM profiles, but depended instead on their mutual distributions across the classes. In fact, the weighty features were not among the most abundant in any particular class (Table 1S of S.I.), but displayed excellent clustering between profiles of the same class (Figure 2b). At the same time, rPTMs with comparable expression levels in all organisms contributed little to class differentiation, whereas those with widely different abundances possessed much greater differentiating power. It should be noted that the very principles at the basis of the KNN and NB classifiers are not conducive to the identification of features wielding any particular influence on the formulation of classification rules. Therefore, no analogous importance plots were obtained from these classifiers.
Figure 2.
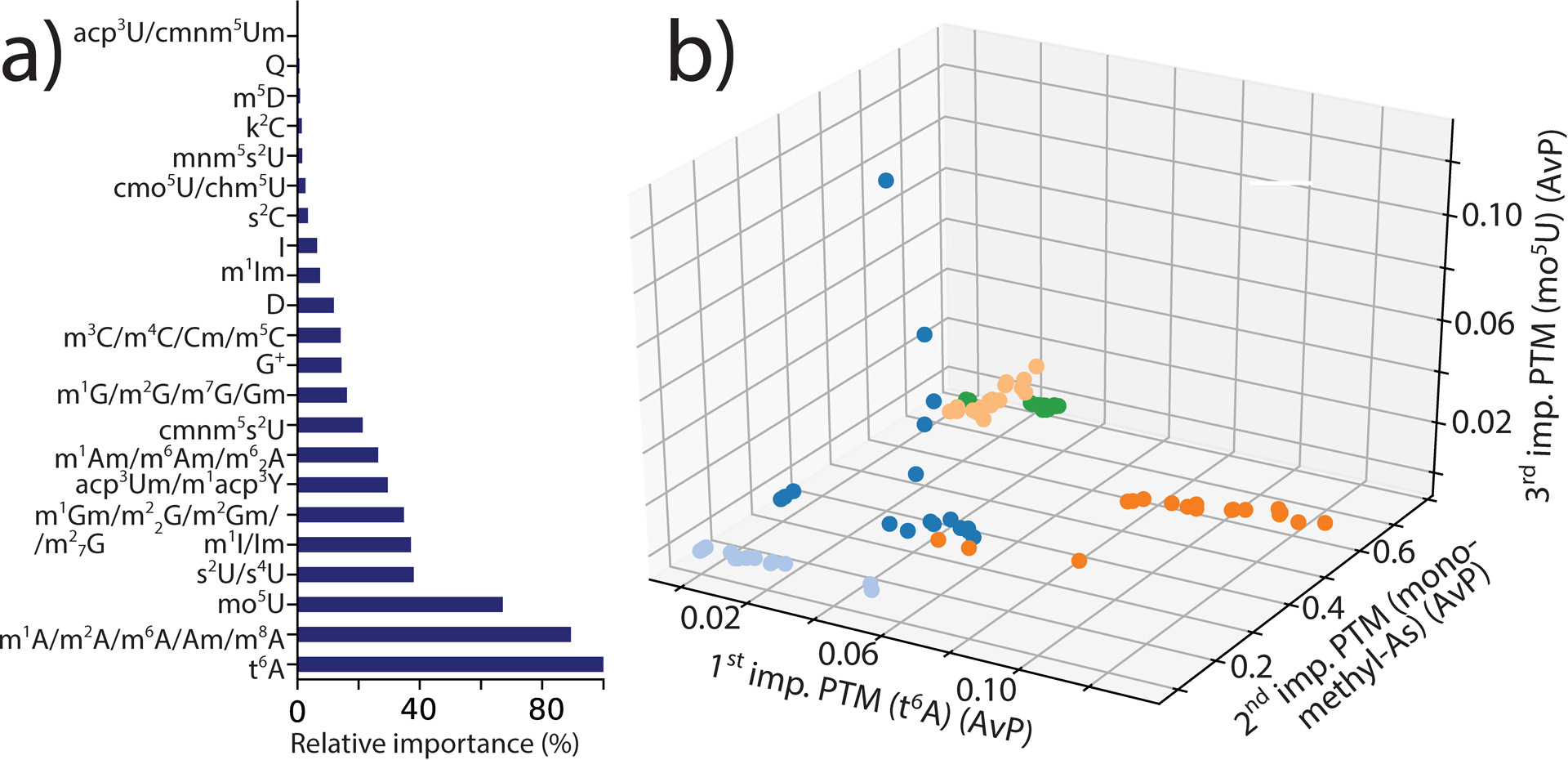
a) Relative importance of rPTMs on GB predictions and b) correlation between the three more important rPTMs in the various profiles. The bars in panel a) convey the weight of each rPTM in differentiating the various classes, which was obtained by activating the “feature importance” attribute of the GB classifier (see Experimental of S.I.). In panel b), the weighty rPTMs are plotted on different axes to visualize correlations across classes. E. coli OR:H48:K data points are shown as dark blue dots; H. salinarum light blue; S. cerevisiae BY4741 dark orange; A. thaliana light orange; and H. sapiens HeLa green.
Differentiating power of rPTM profiles.
The five organisms selected for the initial experiments represented very diverse phylogenetic domains that have diverged significantly during evolution and, thus, would be expected to exhibit significantly different profiles. In subsequent experiments, we analyzed organisms present in taxonomy groups that were progressively closer to one another, with the goal of evaluating the ability of an rPTM-based platform to recognize them as separate classes. For example, we examined samples obtained from S. typhimurium, K. aerogenes, and E. coli OR:H48:K, which are members of the same family of Enterobacteriaceae bacilli. The microorganisms were grown in the same type of medium under identical conditions to minimize any source of variability (see Experimental). The ML classifiers were trained and tested on separate 70:30 datasets, following the procedures described above. As shown in Table 1a, the various rPTM profiles were correctly assigned with accuracies of 99% or better by all ML algorithms. The observed precision and recall followed similar trends, with values exceeding 95% for all classifiers. The importance plot generated by the GB classifier indicated that ms2io6A and monomethyl-adenosines shared comparable weights in differentiating these classes (Figure S3a of S.I.). Consistent with this observation, the correlation plot based on these PTMs displayed excellent clustering between classes (Figure S3b.).
Table 1.
Average accuracy, precision, and recall afforded by the ML classifiers in separate experiments.
| KNN | NB | GB | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Acc | Prc | Rec | Acc | Prc | Rec | Acc | Prc | Rec | |
| S. typh. | 1.00 | 1.00 | 1.00 | 0.99 | 0.96 | 1.00 | 0.99 | 0.96 | 1.00 |
| K. aero. | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |||
| OR:H48:K | 1.00 | 1.00 | 1.00 | 0.95 | 1.00 | 0.96 | |||
| OR:H48:K | 1.00 | 1.00 | 1.00 | 0.98 | 1.00 | 0.96 | 1.00 | 1.00 | 1.00 |
| O157:H7 | 1.00 | 1.00 | 0.96 | 1.00 | 1.00 | 1.00 | |||
| BY4741 | 0.88 | 1.00 | 0.88 | 0.85 | 0.75 | 0.75 | 0.96 | 0.89 | 1.00 |
| set1Δ | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |||
| trf4Δ | 0.73 | 1.00 | 0.80 | 1.00 | 1.00 | 0.88 | |||
| rit1Δ | 1.00 | 0.71 | 1.00 | 0.71 | 1.00 | 1.00 | |||
| HeLa | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| HEK 293T | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |||
| U-251 MG | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |||
| astrocytes | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| microglia | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |||
| neurons | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |||
Differentiation of a) members of the Enterobacteriaceae family (i.e., S. typhimurium, K. aerogenes, and E. coli OR:H48:K, each with testing N = 8); b) E. coli serotypes (i.e., OR:H48:K and O157:H7, N = 8 ea.); c) wildtype and deletion mutants of S. cerevisiae (i.e., BY4741, trf4Δ, and rit1Δ N = 8 ea., set1Δ N=3); d) immortalized human cell lines from different organs (i.e., HEK 293T and HeLa N = 8 ea., and U-251 MG N = 10); and e) primary human cell lines from the central nervous system (i.e., astrocytes N = 3, microglia N = 15, and neurons (N = 5). N indicates the number of rPTM profiles utilized exclusively during the testing phase (i.e., 30% of the entire set available for each class, see Experimental).
Analogous experiments were carried out to test the ability to differentiate distinct serotypes, mutants, and cell lines derived from different organs of the same species. The first experiment compared E. coli OR:H48:K and O157:H7. The former is a benign laboratory strain, whereas the latter is a pathogenic version responsible for deadly food poisoning and expensive product recalls.22 Consistent with the fact that these serotypes differ by a sizeable 30% of their total genes, their respective profiles enabled unambiguous differentiation (Table 1b). According to the representative GB classifier, mnm5U, monomethyl-adenosines, and cmo5U were the three most important features in determining differentiation (Figure S4 of S.I.). A closer look at the respective profiles revealed that mnm5U was absent in the benign OR:H48:K serotype, whereas was consistently detected in the pathogenic O157:H7. Conversely, the former displayed elevated levels of mnm5s2U that is absent in the latter. It should be noted that mnm5U is an essential precursor in the biogenesis of mnm5s2U and is typically found on the wobble position 34 of E. coli tRNA,23 which is involved in the mechanism of transcriptional recoding. Based on these observations, one could speculate that such rPTM might play a role in regulating the production of Shiga toxin responsible for the hemorrhagic colitis and hemolytic-uremic syndrome caused by O157:H7.24
The next experiment examined three deletion mutants of S. cerevisiae S288C lacking the rit1, trf4, or set1 gene, which were obtained from the Yeast Knock-Out collection25 and grown under identical conditions (see Experimental). The mutants were compared also with S. cerevisiae BY4741, a common laboratory strain with a genome that differs by less than 0.1% from that of S288C (https://www.yeastgenome.org/). The very close similarities between these organisms proved to be particularly challenging to the classifiers, which produced mixed results (Table 1c). In this case, di-methyl-adenosines, ac4C/f5Cm, and I6A emerged as the most important differentiating features in spite of the absence of any apparent relationship with the deleted genes (Figure S5 of S.I.). Among them, only the rit1 gene has known implications in rPTM biogenesis through the activity of the corresponding protein product, a methionine 2’-O-ribosyl phosphate transferase responsible for introducing an essential modification necessary to distinguish the initiator tRNAMet from its elongator tRNAMet analog.26 In contrast, the products of trf4 and set1 are involved in processes that directly or indirectly affect the levels of specific RNAs in the cell. Deleting the former can adversely affect RNA degradation in the exosome and to reduce its overall turnover rate. The set1 protein is part of a histone methyltransferase complex involved in modulating the efficient termination of snoRNAs to produce cryptic unstable transcripts (CUTs).27 The effects of such deletions could be thus ascribed to the significant variations of the levels of specific classes of RNAs enacted by these genes, which were reflected in the levels of the respective rPTMs.
The analysis of HEK 293T, HeLa, and U-251 MG cells cultured in the laboratory was carried out to evaluate the classifiers’ performance with cells that shared a similar H. sapiens genetic background, but differed in the types of organs (i.e., embryonic kidney, mature cervix, and central nervous system, respectively) and putative immortalization processes.28–30 In spite of extensive commonalities expected from these types of cells, the various ML algorithms were once again able to distill rigorous differentiating rules that led to correct assignments across the board (Table 1d). The GB classifier identified monomethyl-uridines, I6A, and acp3U/cmnm5Um as the most important differentiating features (Figure S6 of S.I.). Analogous predictive outcomes were observed for classifiers built to differentiate primary cells obtained from the human central nervous system (CNS), such as astrocytes, microglia, and neurons (Table 1e). In this case, acp3U, monomethyl-uridines, and monomethyl-cytosines exhibited the greater weight on GB classification (Figure S7 of S.I.). The distinctive profiles of these CNS cells belied a common embryonal precursor and exposure to a common environment, while reflecting instead the different developmental trajectories necessary to creating their highly specialized infrastructures. These observations reinforced the notion that rPTM profiles are not determined exclusively by the genetic makeup of a cell, but result from a combination of factors working in concert to shape their pertinent regulatory pathways.
Increasing the complexity of the classification problem.
The above classification experiments provided an excellent measure of the putative resolving power of rPTM-based classifiers by considering classes that were progressively closer to one another on a typical phylogenetic tree. These experiments, however, provided no indication of whether the classifiers would retain similar capabilities when the number of classes under consideration were significantly increased, to the point where the chance of partial profile overlaps might inadvertently increase the probability of erroneous assignments. For this reason, a total of 20 different types of samples were examined, which included organisms from closely related groups or distal regions of the phylogenetic tree (see Experimental). The average rPTM profiles obtained from these samples are reported in Table S3. Different numbers of data points per class were utilized to evaluate the ability of the ML algorithms to perform in unbalanced statistical situations. As described above, the entire available dataset was randomly split into training and testing subsets of 70:30 proportions. Training was again carried out according to a LOO cross validation process, the outcome of which was visualized by matrix plots like the representative one displayed in Figure 3a for the GB classifier (those of KNN and NB are provided in Figure S8a and S9a of S.I.).
Figure 3.
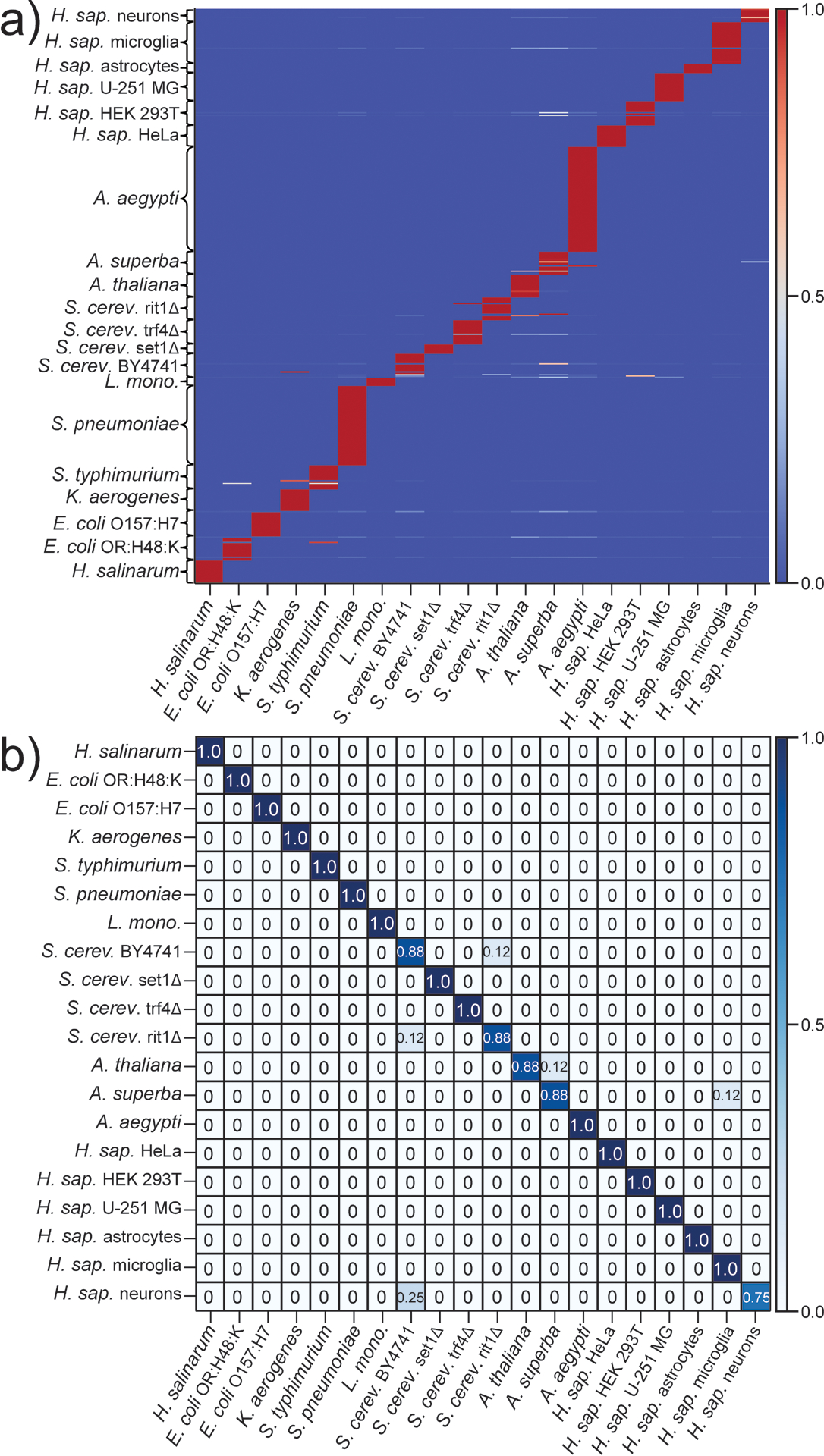
Representative results obtained by training a) and testing b) the gradient boosting (GB) algorithm on rPTM profiles from 20 different classes (see Experimental). Predicted and actual classes are reported on the x- and y-axis, respectively. Color gradients express the frequency by which a data point was assigned to a given class. The following classes were considered: H. salinarum (N = 25 for both training/testing); E. coli OR:H48:K (N = 25) and O157:H7 (N = 25); K. aerogenes (N = 25); S. typhimurium (N = 25); S. pneumoniae (N = 85); L. monocytogenes (N = 10); S. cerevisiae samples BY4741 (N = 25), set1Δ (N = 10), trf4Δ (N = 25) and rit1Δ (N = 25); A. thaliana (N = 25); A. superba (N = 25); A. aegypti (N = 110); and H. sapiens samples HeLa (N = 25), HEK 293T (N = 25), U251 MG (N = 30), astrocytes (N = 10), microglia (N = 45), and neurons (N = 15).
The results obtained from these classifiers were in line with those generated from more limited number of classes. The LOO plots once again provided an immediate appreciation of the performance of the respective ML algorithm during cross validation and highlighted possible issues with individual datapoints. For example, the representative GB plot showed that the vast majority of the training samples were consistently assigned to the correct class, as exemplified by all the 18 samples of H. salinarum, the 60 S. pneumoniae, or the 77 A. aegypti (Figure 3a). Only 23 out of the 434 rPTM profiles used for training exhibited either ambiguous or erroneous assignments with any frequency. Among the former, one of the 18 samples of S. cerevisiae BY4741 was assigned with comparable frequencies to a series of different classes. Among the latter, a sample of E. coli OR:H48:K was incorrectly assigned as S. typhimurium 92% of the times, whereas a different one was mistaken for A. aegypti 13% of the times. Overall, the various S. cerevisiae samples (i.e., the BY4741 laboratory strain and three knockout variants) incurred in the most ambiguities, accounting for 10 out of the 23 total misidentifications observed in the entire LOO matrix. These challenges could be attributed to the higher degree of similarity possessed by such classes as compared to the other ones. The fact that no classification ambiguities were observed for S. pneumoniae and A. aegypti, which were among the classes with the larger datasets under consideration (i.e., N = 60 and 77, respectively, Figure 3a), should not be considered as fortuitous. Indeed, this observation substantiated the fact that, in the absence of overfitting, increasing the number of datapoints per class tends to increase a classifier’s predictive power.
The performance of the predictive models was then evaluated by using the testing subset (i.e., 30% of the 620 total datapoints), which had been excluded from the training process to allow for an unbiased determination of assignment accuracy. The results were displayed by a confusion matrix in which predicted and actual labels were plotted on the x- and y-axis, respectively (Figure 3b). The confusion matrix obtained from the GB classifier showed that samples belonging to 15 out of the 20 classes considered here were predicted correctly 100% of the times (Figure 3b). Samples belonging to 4 additional classes were correctly predicted 88% of the times, whereas those in the remaining class 75% of the times. It should be noted, however, that the testing sets for the former (i.e., S. cerevisiae BY4741, S. cerevisiae rit1Δ, A. thaliana, and A. superba) comprised 8 datapoints each, whereas the latter (i.e., primary H. sapiens neurons) comprised only 4. When relatively small sets of datapoints are considered, even a single erroneous prediction can have significant effects on figures of merit expressed in percentage form. Significantly, the close match between the accuracies obtained from LOO cross-validation and those produced by the corresponding testing process indicated the absence of any significant data overfitting. This implies that the classifier’s performance could be further improved by increasing the overall dataset size. The frequent mis-assignment of a H. sapiens neurons profile as S. cerevisiae BY4741 could be ascribed to another instance of possible contamination (Figure 3b). This profile was among the 30% that had not been used for training and, thus, was not properly recognized during testing. This ambiguous outcome would be also expected when testing any class that was not part of training. Overall, the GB classifier reported 5 false negatives, while the corresponding KNN and NB classifiers produced totals of 9 and 17, respectively. The comprehensive figures of merit (i.e., average accuracy, precision, and recall) for all algorithms are summarized in Table S4 of S.I.
rPTM-defined class relationships.
Owing to the agnostic nature of the selected algorithms and to the complexity of the differentiation rules generated by the training process, the classifiers were not immediately capable of providing useful insights into the relationships between classes and their putative organization. We thus explored the possibility of utilizing the ML classifiers to curate the initial dataset and then applying a more traditional clustering approach to visualize putative relationships between classes. For instance, the GB classifier identified 28 ambiguous or mis-assigned datapoints out of the 620-total included in Figure 3. The remaining 592 datapoints were employed to obtain average rPTM profiles for the various organisms (Table S3), which were in turn utilized to calculate actual Euclidean distance between classes. An agglomerative hierarchical clustering approach was then utilized to generate the dendrogram shown in Figure 4 (see Experimental of S.I.). In this type of representation, the length of horizontal lines represents the average Euclidean distance between classes or nodes connected by corresponding vertical lines. Therefore, the dendrogram provided a comprehensive view of hierarchical relationships between classes based on their respective rPTM profiles. In most instances, such relationships reflected very closely the evolutionary trajectory and established taxonomic classification of the various organisms. For example, it was not surprising to observe that H. Sapiens cell lines, S. cerevisiae variants, and Enterobacteriaceae bacilli formed distinct recognizable clusters. Within the H. Sapiens cluster, the CNS cells were closer to one another than to the other cell lines derived from different human organs. The close grouping between S. typhimurium, E. coli, and K. aerogenes could be attributed to the fact that these bacteria are all members of the same Enterobacteriaceae family. In contrast, the Gram-positive S. pneumoniae and L. monocytogenes were not only far removed from the Gram-negative Enterobacteriaceae, but also from one another, in agreement with their classification in distinct Streptococcaceae and Listeriaceae families, respectively. The position of H. salinarum - the only Archaea in the study- was significantly closer to Eukaryotes than Bacteria, consistent with recent genomic evidence that advanced Archaea as possible precursors or close evolutionary relatives of the former.31,32 Within the S. cerevisiae group, the wildtype strain BY4741 clustered closer to the trf4Δ and rit1Δ mutants than set1Δ did, in spite of the fact that the knockout variants differed from one another by just a single gene (they were all derived from the S288C strain), whereas they differed from BY4741 by a more sizeable 0.1% portion of the entire genome. This apparent discrepancy was consistent with the fact that rPTM profiles are not defined exclusively by the genetic makeup, which determines the entire complement of rPTM biogenetic enzymes in a cell, but also by the activation state of their specific regulatory pathways. This could help explain the subtle variations present between the dendrogram based on rPTM profiles and typical phylogenetic trees based on more established features, such as the sequence of SSU rRNA.33
Figure 4.
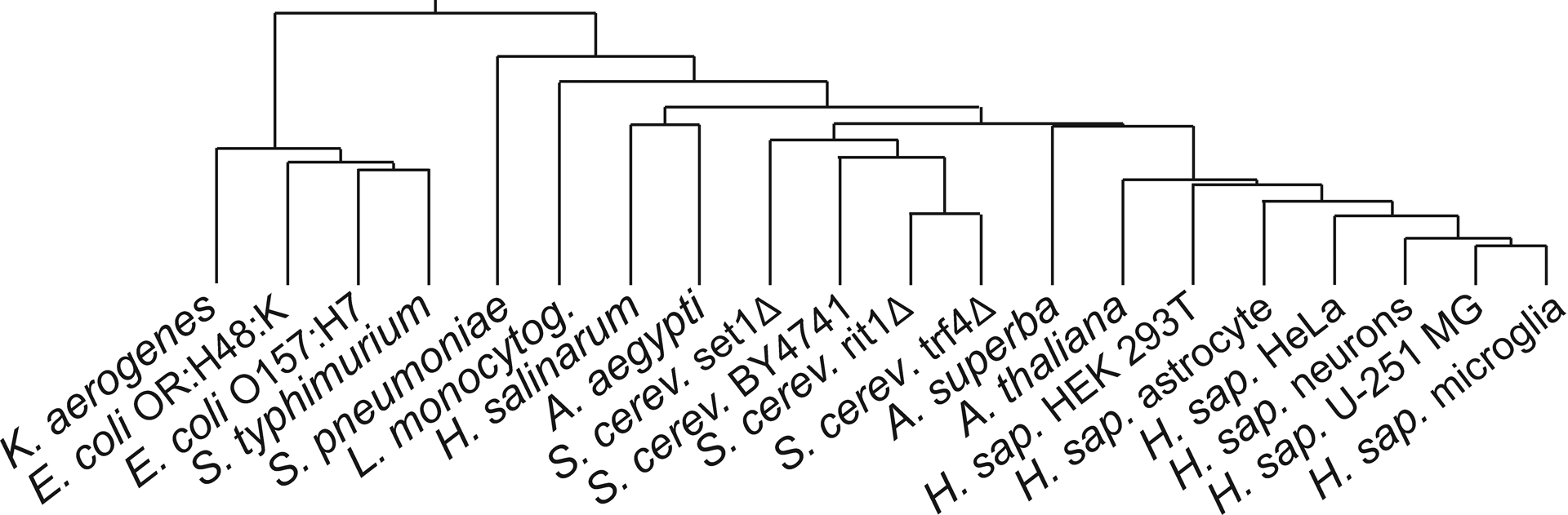
Dendrogram generated from Euclidean distances that were calculated from 592 curated PTM profiles of 20 different classes (see Experimental of S.I.).
CONCLUSIONS
The evidence presented here supports the creation of a new classification/identification framework based on RNA PTMs. The excellent reproducibility of rPTM profiles obtained from each individual class, in terms of number, identity, and relative abundance of RNA modifications, fulfills an essential requirement for supporting meaningful comparisons between classes, while also accounting for possible sample-to-sample variability. At the same time, the diversity of rPTM profiles exhibited by individual classes affords the ability to achieve unambiguous differentiation. Sample reproducibility was promoted by examining samples obtained under constant growth/environmental conditions, which minimized the uncertainties associated with one of the major factors affecting the complexion of rPTM profiles. Class diversity stems instead from the combination of unique genetic and regulatory factors that make an rPTM profile into a very specific characteristic of a given organism. The possibility to capitalize on these very favorable features was realized by applying advanced MS technologies to enable the simultaneous analysis of all rPTMs present in the sample, which ensured the acquisition of comprehensive information on the incidence of RNA modifications.
The application of supervised ML algorithms proved to be essential for handling the multidimensional problem posed by comprehensive rPTM analysis. The study examined 620 individual profiles corresponding to both biological and technical replicates of samples representing 20 individual classes. On average, each profile comprised ~30 different types of rPTMs with associated abundance information. Overall, 102 out of the >160 known types of natural rPTMs were detected across all samples considered. The classifiers exhibited no significant drops in performance when the number of classes considered increased ten-fold from 2 to 20, thus assuaging any notion that increasing the complexity of the problem would necessarily lead to a greater incidence of errors. As expected from effective learning processes, the predictive power increased with the number of datapoints per class used for training. Barring overfitting, the ability to distill more stringent decision rules from progressively larger datasets represents a strength of ML algorithms. The outcomes observed here afforded an excellent measure of the ability of rPTM profiles to differentiate organisms with very similar genetic makeups in the context of a large number of classes with varying degrees of mutual separation. At the same time, the outcome demonstrated also the capacity to achieve the correct assignment of unlabeled samples within a large field of possible matches.
The actual observations afforded by the selected organisms indicated that approximately two thirds of the rPTMs in an average profile were present in all classes considered in the study, including Y, methyl-cytosines, methyl-guanosines, methyl-uridines, methyl-adenosines, ac4C and f5Cm. Increasing evidence suggests that most rPTMs do not constitute mere manifestations of the cellular response to changing environmental conditions, but rather essential actors in the regulation of specific response mechanisms. For this reason, the sets of rPTMs detected across the board are likely involved in fundamental processes that are common to all the organisms examined here. Conversely, the weighty rPTMs that contributed the most to class differentiation could represent more specific processes that set different organisms apart. Both possible aspects of rPTM function were clearly recognizable in the outcomes produced by the ML classifiers, regardless the agnostic data-driven nature of the classification. As demonstrated by the dendrogram, this approach led to an overall hierarchical organization that replicated very closely those afforded by established taxonomic systems. At the same time, it also produced finer clustering patterns that reflected the effects of the smallest variation between organisms that would be generally considered as intimately related. In this context, it was surprising to note that the deletion of a single pivotal gene, such as the S. cerevisiae set1, could have a more significant impact on rPTM expression than varying approximately 0.1 of the entire genome. This type of information will be essential to understand the interplay between rPTM pathways and to decipher the epitranscriptomics code.
Finally, the ubiquitous presence of RNA in virtually all known organisms (with DNA viruses arguably representing the only exception) will be expected to promote the broadest possible range of applications for rPTM-based classification/identification outside familiar research settings. In particular, the ability to unambiguously recognize pathogenic bacteria, such as S. pneumoniae, L. monocytogenes, S. typhimurium, and E. coli O157:H7, as well as to differentiate mammalian cells infected by distinct RNA viruses,12 foreshadows the possible development of a new family of diagnostic approaches. The ability to achieve positive identification from mixed samples containing multiple classes will be essential to the success of such applications. In this direction, the possibility to base identification on the detection of an entire panel of features, rather than just one specific biomarker, will be expected to play a significant role in increasing the confidence level in the results of the analysis.
Supplementary Material
ACKNOWLEDGMENTS
Funding provided by the University at Albany-SUNY (to D.F.); NIH-NIAID [R21 AI133617 to D.F. and C.P.]; NIH-NIGMS [R01 GM123050 to D.F. and C.P.]; NIH-NIGMS [R01 GM121844 to D.F.]; and NIH- NIDA [R01 DA046113 to D.F.]. We thank H. Yu and J. Hua of Cornell University for samples of A. superba.
Footnotes
S.I.
S.I. is available free of charge on the ACS Publications website.
The authors declare no competing financial interests.
REFERENCES
- (1).Linné C. von; Salvius L Caroli Linnaei … Species Plantarum :Exhibentes Plantas Rite Cognitas, Ad Genera Relatas, Cum Differentiis Specificis, Nominibus Trivialibus, Impensis Laurentii Salvii, Holmiae, 1753, 1–572. [Google Scholar]
- (2).Woese CR Bacterial Evolution. Microbiol. Rev 1987, 51 (2), 221–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).D’Argenio V; Salvatore F The Role of the Gut Microbiome in the Healthy Adult Status. Clin. Chim. Acta 2015, 451, 97–102. [DOI] [PubMed] [Google Scholar]
- (4).Kitahara K; Miyazaki K Revisiting Bacterial Phylogeny: Natural and Experimental Evidence for Horizontal Gene Transfer of 16S RRNA. Mob. Genet. Elem 2013, 3 (1), e24210, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cantara WA; Crain PF; Rozenski J; McCloskey JA; Harris KA; Zhang X; Vendeix FAP; Fabris D; Agris PF The RNA Modification Database: 2011 Update. Nucleic Acids Res 2011, 39 (Suppl 1), D195–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Boccaletto P; Machnicka MA; Purta E; Piatkowski P; Baginski B; Wirecki TK; de Crécy-Lagard V; Ross R; Limbach PA; Kotter A; Helm M; Bujnicki JM MODOMICS: A Database of RNA Modification Pathways. 2017 Update. Nucleic Acids Res. 2018, 46 (D1), D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Roundtree IA; Evans ME; Pan T; He C Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169 (7), 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Suzuki T; Nagao A; Suzuki T Human Mitochondrial tRNAs: Biogenesis, Funct., Struct. Aspects, and Diseases. Annu. Rev. Genet 2011, 45 (1), 299–329. [DOI] [PubMed] [Google Scholar]
- (9).Brandmayr C; Wagner M; Brückl T; Globisch D; Pearson D; Kneuttinger AC; Reiter V; Hienzsch A; Koch S; Thoma I; Thumbs P; Michalakis S; Müller M; Biel M; Carell T Isotope-Based Analysis of Modified TRNA Nucleosides Correlates Modification Density with Translational Efficiency. Angew. Chem. Int. Ed 2012, 51 (44), 11162–11165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rose RE; Quinn R; Sayre JL; Fabris D Profiling Ribonucleotide Modifications at Full-Transcriptome Level by Electrospray Ionization Mass Spectrometry. RNA 2015, 21, 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rose RE; Pazos MA; Curcio MJ; Fabris D Global Profiling of RNA Post-Transcriptional Mods as an Effective Tool for Investigating the Epitranscriptomics of Stress Response. Mol. Cell. Prot. MCP 2016, 15 (3), 932–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McIntyre W; Netzband R; Bonenfant G; Biegel JM; Miller C; Fuchs G; Henderson E; Arra M; Canki M; Fabris D; Pager CT Positive-Sense RNA Viruses Reveal the Complexity and Dynamics of the Cellular and Viral Epitranscriptomes during Infection. Nucl Acids Res 2018, 46 (11), 5776–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dominissini D; Moshitch-Moshkovitz S; Schwartz S; Salmon-Divon M; Ungar L; Osenberg S; Cesarkas K; Jacob-Hirsch J; Amariglio N; Kupiec M; Sorek R; Rechavi G Topology of the Human and Mouse M6A RNA Methylomes Revealed by M6A-Seq. Nature 2012, 485 (7397), 201–206. [DOI] [PubMed] [Google Scholar]
- (14).Motorin Y; Muller S; Behm-Ansmant I; Branlant C Identification of Modified Residues in RNAs by Reverse Transcription-Based Methods. Methods Enzymol. 2007, 425, 21–53. [DOI] [PubMed] [Google Scholar]
- (15).Trixl L; Rieder D; Amort T; Lusser A Bisulfite Sequencing of RNA for Transcriptome-Wide Detection of 5-Methylcytosine. In Epitranscriptomics: Methods and Protocols; Wajapeyee N, Gupta R, Eds.; Methods in Molecular Biology; Springer: New York, NY, 2019, 1–21. [DOI] [PubMed] [Google Scholar]
- (16).Crain PF Mass Spectrometric Techniques in Nucleic Acid Research. Mass Spectrom Rev 1990, 9, 505–554. [Google Scholar]
- (17).Limbach PA; Paulines MJ Going Global: The New Era of Mapping Modifications in RNA. Wiley Interdiscip. Rev. RNA 2017, 8 (1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jora M; Borland K; Abernathy S; Zhao R; Kelley M; Kellner S; Addepalli B; Limbach PA Amination/Imination of Carbonothiolated Nucleosides During RNA Hyd.s. Angew. Chem. Int. Ed 2021, 60 (8), 3961–3966. [DOI] [PubMed] [Google Scholar]
- (19).Bay SD Combining Nearest Neighbor Classifiers Through Multiple Feature Subsets. In ICML; 1998, 1–8. [Google Scholar]
- (20).Islam MJ; Wu QMJ; Ahmadi M; Sid-Ahmed M Investigating the Performance of Naive- Bayes Classifiers and K- Nearest Neighbor Classifiers. JCIT 2010, 5, 133–137. [Google Scholar]
- (21).Friedman JH Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat 2001, 29 (5), 1189–1232. [Google Scholar]
- (22).Currie A; Honish L; Cutler J; Locas A; Lavoie M-C; Gaulin C; Galanis E; Tschetter L; Chui L; Taylor M; Jamieson F; Gilmour M; Ng C; Mutti S; Mah V; Hamel M; Martinez A; Buenaventura E; Hoang L; Pacagnella A; Ramsay D; Bekal S; Coetzee K; Berry C; Farber J; Team OBOTNI Outbreak of Escherichia Coli O157:H7 Infections Linked to Mechanically Tenderized Beef and the Largest Beef Recall in Canada, 2012. J. Food Prot 2019, 82 (9), 1532–1538. [DOI] [PubMed] [Google Scholar]
- (23).Watanabe K; Hayashi N; Oyama A; Nishikawa K; Ueda T; Miura K Unusual Anticodon Loop Structure Found in E.Coli Lysine TRNA. Nucleic Acids Res. 1994, 22 (1), 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Joseph A; Cointe A; Mariani Kurkdjian P; Rafat C; Hertig A Shiga Toxin-Ass.d Hemolytic Uremic Syndrome: A Rev. Toxins 2020, 12 (2), 1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Brachmann CB; Davies A; Cost GJ; Caputo E; Li J; Hieter P; Boeke JD Designer Deletion Strains Derived from Saccharomyces Cerevisiae S288C: A Useful Set of Strains and Plasmids for PCR-Mediated Gene Disruption and Other Applications. Yeast 1998, 14 (2), 115–132. [DOI] [PubMed] [Google Scholar]
- (26).Kolitz SE; Lorsch JR Eukaryotic Initiator tRNA: Finely Tuned and Ready for Action. FEBS Lett. 2010, 584 (2), 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Boa S; Coert C; Patterton H-G Saccharomyces Cerevisiae Set1p Is a Methyltransferase Specific for Lysine 4 of Histone H3 and Is Required for Efficient Gene Expression. Yeast Chichester Engl. 2003, 20 (9), 827–835. [DOI] [PubMed] [Google Scholar]
- (28).Graham FL; Smiley J; Russell WC; Nairn R Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol 1977, 36 (1), 59–72. [DOI] [PubMed] [Google Scholar]
- (29).Landry JJM; Pyl PT; Rausch T; Zichner T; Tekkedil MM; Stütz AM; Jauch A; Aiyar RS; Pau G; Delhomme N; Gagneur J; Korbel JO; Huber W; Steinmetz LM The Genomic and Transcriptomic Landscape of a HeLa Cell Line. G3 Genes Genomes Genet. 2013, 3 (8), 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Torsvik A; Stieber D; Enger PØ; Golebiewska A; Molven A; Svendsen A; Westermark B; Niclou SP; Olsen TK; Enger MC; Bjerkvig R U-251 Revisited: Genetic Drift and Phenotypic Consequences of Long-Term Cultures of Glioblastoma Cells. Cancer Med. 2014, 3 (4), 812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Spang A; Saw JH; Jørgensen SL; Zaremba-Niedzwiedzka K; Martijn J; Lind AE; van Eijk R; Schleper C; Guy L; Ettema TJG Complex Archaea That Bridge the Gap between Prokaryotes and Eukaryotes. Nature 2015, 521 (7551), 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Eme L; Spang A; Lombard J; Stairs CW; Ettema TJG Archaea and the Origin of Eukaryotes. Nat. Rev. Microbiol 2017, 15 (12), 711–723. [DOI] [PubMed] [Google Scholar]
- (33).Wu D; Hartman A; Ward N; Eisen JA An Automated Phylogenetic Tree-Based Small Subunit RRNA Taxonomy and Alignment Pipeline (STAP). PloS One 2008, 3 (7), e2566, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.