

Abstract
The result of a deprivation of oxygen and glucose to the brain, hypoxic–ischemic encephalopathy (HIE), remains the most common cause of death and disability in human neonates globally and is mediated by glutamate toxicity and inflammation. We have previously shown that the enzyme glutamate carboxypeptidase (GCPII) is overexpressed in activated microglia in the presence of inflammation in fetal/newborn rabbit brain.
We assessed the therapeutic utility of a GCPII enzyme inhibitor called 2-(3-Mercaptopropyl) pentanedioic acid (2MPPA) attached to a dendrimer (D-2MPPA), in order to target activated microglia in an experimental neonatal hypoxia-ischemia (HI) model using superoxide dismutase transgenic (SOD) mice that are often more injured after hypoxia-ischemia than wildtype animals.
SOD overexpressing and wild type (WT) mice underwent permanent ligation of the left common carotid artery followed by 50 min of asphyxiation (10% O2) to induce HI injury on postnatal day 9 (P9). Cy5-labeled dendrimers were administered to the mice at 6 h, 24 h or 72 h after HI and brains were evaluated by immunoflu orescence analysis 24 h after the injection to visualize microglial localization and uptake over time. Expression of GCPII enzyme was analyzed in microglia 24 h after the HI injury. The expression of pro- and anti-inflammatory cytokines were analyzed 24 h and 72 h post-HI. Brain damage was analyzed histologically 7 days post-HI in the three randomly assigned groups: control (C); hypoxic-ischemic (HI); and HI mice who received a single dose of D-2MPPA 6 h post-HI (HI+D-2MPPA).
First, we found that GCPII was overexpressed in activated microglia 24 h after HI in the SOD overexpressing mice. Also, there was an increase in microglial activation 24 h after HI in the ipsilateral hippocampus which was most visible in the SOD+HI group. Dendrimers were mostly taken up by microglia by 24 h post-HI; uptake was more prominent in the SOD+HI mice than in the WT+HI. The inflammatory profile showed significant increase in expression of KC/GRO following injury in SOD mice compared to WT at 24 and 72 h. A greater and significant decrease in KC/GRO was seen in the SOD mice following treatment with D-2MPPA. Seven days after HI, D-2MPPA treatment decreased brain injury in the SOD+HI group, but not in WT+HI. This reduced damage was mainly seen in hippocampus and cortex.
Our data indicate that the best time point to administer D-2MPPA is 6 h post-HI in order to suppress the expression of GCPII by 24 h after the damage since dendrimer localization in microglia is seen as early as 6 h with the peak of GCPII upregulation in activated microglia seen at 24 h post-HI. Ultimately, treatment with D-2MPPA
Keywords: Neuroprotection, Glutamate carboxypeptidase, Nanoparticle, Dendrimer, Neonatal brain injury, Microglia, N-acetyl-aspartyl-glutamate (NAAG)
1. Introduction
Hypoxic–ischemic encephalopathy (HIE) results from a deprivation of oxygen and glucose to the brain, and remains the most common cause of death and disability in neonates globally. HIE is mediated by oxidative stress, glutamate excitotoxicity and inflammation (McLean and Ferriero, 2004). After hypoxia-ischemia (HI) insult, excitatory amino acid neurotransmitters are released, causing reactive oxygen species (ROS)-dependent changes in blood-brain barrier (BBB) permeability that allow immune cells to enter and stimulate an inflammatory response (Moretti et al., 2015). Brain resident microglia activate (Arteaga et al., 2017; Li et al., 2011; Mike et al., 2020) and initiate ROS generation, antigen presentation, phagocytosis, and the production of inflammatory mediators (Tsuji et al., 2020). The neonatal brain is highly susceptible to oxidative stress because of its high concentration of unsaturated fatty acids, high rate of oxygen consumption, availability of redox-active iron (Halliwell, 1992) and because developing endogenous antioxidant mechanisms are rapidly overwhelmed. We have previously demonstrated that transgenic mice with the human Cu/Zn superoxide dismutase 1 enzyme (SOD) have greater brain damage than their non-transgenic littermates after HI (Ditelberg et al., 1996; Sheldon et al., 2002, 2017) due to hydrogen peroxide accumulation (H2O2), similar to that observed in human newborns (Fullerton et al., 1998). Accumulated H2O2 coupled with an inability to adequately scavenge H2O2 by increasing antioxidant activity is likely responsible for the greater susceptibility to oxidative stress in the neonatal brain (Epstein et al., 1987; Fullerton et al., 1998). Similarly, neuroinflammation mediated by activated microglia and astrocytes (Bhalala et al., 2015), extracellular glutamate buildup and over-activation of glutamate receptors (Jabaudon et al., 2000), are also implicated in the pathogenesis of HIE. Developmental changes in microglial phenotype, distribution and numbers can increase vulnerability of the neonatal brain to inflammatory insults. A transient increase in amoeboid microglia in white matter tracts is normally seen in the fetal and perinatal period (Mallard et al., 2018).
Glutamate is the major excitatory neurotransmitter in the central nervous system (CNS). Glutamate excitotoxicity can lead to excessive calcium intake (Fern and Möller, 2000), cause severe impairment of oxidative metabolism (Johnston et al., 2011), induce progressive cell loss, and exacerbate neurological dysfunction (Choi, 1992). Although activation of glutamatergic systems have been implicated in a number of neurologic disorders, glutamate-mediated neurotransmission is also critical for normal brain development and injury response (Cotman et al., 1987). Glutamate receptor function is tightly regulated under normal conditions for maintenance of homeostasis (Bai and Zhou, 2017). Expression of the glutamate receptors N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) is greater in the neonate than the adult which makes the neonatal brain more prone to excitotoxic injury (Azzopardi et al., 2009). Glutamate is primarily synthesized from glutamine by the enzyme glutaminase in neurons and microglia. However, it can also be produced by hydrolysis of N-acetylaspartylglutamate (NAAG), an abundant peptide neurotransmitter in the brain that has been shown to have neuroprotective effects (Bratek et al., 2020; Cai et al., 2002; Van Hemelrijck et al., 2005), including its action as a metabotropic glutamate receptor 3 (mGLuR3) agonist (Spillson and Russell, 2003). Activation of mGluR3 through NAAG a short time after HI leads to neuroprotective mechanisms that act through the inhibition of oxidative stress and ROS production (Bratek et al., 2018, 2020). Hydrolysis of NAAG by the enzyme glutamate carboxypeptidase II (GCPII) leads to the formation of NAA and glutamate (Neale et al., 2011; Tsukamoto et al., 2007). GCPII was previously thought to be exclusively expressed in astrocytes (Šácha et al., 2007). However, we have recently shown that GCPII is constitutively expressed at very low levels in microglia, and in the presence of inflammation, GCPII activity and expression increases. Microglial cells treated in vitro with LPS show an increase in GCPII enzyme activity (Zhang et al., 2016a, 2016b). Further, microglial cells overexpress GCPII enzyme in the fetal/newborn rabbit brain in a maternal inflammation model of cerebral palsy (Zhang et al., 2016a, 2016b). Therefore, strategies to specifically target the upregulated GCPII enzyme in activated microglia may decrease glutamatergic injury, potentially slow disease progression, and increase the therapeutic window while enabling normal brain development.
GCPII inhibitors have been used to reduce the deleterious effects of glutamate excitotoxicity in a variety of neurological diseases (Ghadge et al., 2003; Long et al., 2005; Potter et al., 2014; Rahn et al., 2012; Slusher et al., 1999; Zhong et al., 2005). Infusion of NAAG in a neonatal model of white matter injury resulted in neuroprotection (Cai et al., 2002), indicating that GCPII inhibitors may be effective. In the present study we investigated the potential neuroprotective effects of a GCPII enzyme inhibitor called 2-(3-Mercaptopropyl) pentanedioic acid (2MPPA) attached to a poly (amidoamine) dendrimer (D-2MPPA), to target activated microglia in an experimental neonatal HI model using superoxide dismutase 1 transgenic mice. Dendrimer conjugation of 2MPPA has the potential to improve delivery to affected areas of the brain and affected cells, improving drug efficacy and reducing side effects. In addition, dendrimers are non-toxic, non-immunogenic and cleared intact through the kidneys. We have previously demonstrated that these dendrimers selectively localize in activated microglia in the retina and the brain in small and large animal models and can deliver drugs specifically to these cells (Arteaga Cabeza et al., 2019; Smith et al., 2019). Here we demonstrate that HI leads to overexpression of GCPII in activated microglia in the neonatal mouse brain and treatment with D-2MPPA leads to decreased brain injury.
2. Material and methods
2.1. Animals and HI procedures
Transgenic mice overexpressing human SOD1 (SOD) and their wildtype (WT) littermates (C57/Bl6) were used for this study (Epstein et al., 1987). All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at UCSF, in accordance with NIH guidelines for the Care and Use of Laboratory Animals. Genotyping was performed by gel electrophoresis and protein staining for SOD1 as previously described (Elroy-Stein et al., 1986) when animals were between 5 and 7 days old (P5-P7). Animals were maintained and experiments were conducted in accordance with ARRIVE (Animal Research: Reporting In Vivo Experiments). All efforts were made to minimize suffering (all surgery was performed under anesthesia) and to avoid unnecessary animal testing.
Hypoxic-ischemic brain injury was induced in perinatal mouse pups on postnatal day 9 (P9) by the Vannucci method (Rice et al., 1981). Under isoflurane anesthesia administered at 3%, the left common carotid artery was permanently ligated by electrocoagulation and laceration. After a 1 h recovery period with the dam, the mice were placed in chambers floating in a water bath at 36.5 °C and exposed to 10% oxygen and 90% nitrogen for 50 min. Bupivacaine was used as an analgesic. After hypoxic exposure, pups were returned to their biological mothers until they were euthanized at the different times of study. If mice showed signs of increased or decreased respiratory rate, decrease of activity, loss of appetite, or isolation from littermates (which may indicate pain or distress in the animal), they were euthanized; but no animals showed any signs of pain. After weaning, animals were housed in individual cages, and maintained in a climate-controlled environment on a 12-h light/dark cycle where they had free access to food and water.
2.2. Experimental groups
Pups were randomly assigned to three experimental groups. 1) Control (C) had neither common carotid artery ligation nor a period of hypoxia; they were only anesthetized and the left carotid was exposed. 2) Hypoxic-ischemic injured with no additional treatment (HI). 3) Hypoxic-ischemic injured that also received a single dose of D-2MPPA (10 mg/kg on a 2MPPA basis) injected intraperitoneally 6 h after HI injury (HI+D-2MPPA). The dendrimers were diluted in sterile normal saline.
The experimental design is presented in Fig. 1. The sample size differed in each experiment type. For morphological evaluation by damage score: WT+HI n = 16, WT+HI+D-2MPPA n = 13, SOD+HI n = 20, SOD+HI+D-2MPPA n = 19.
Fig. 1.
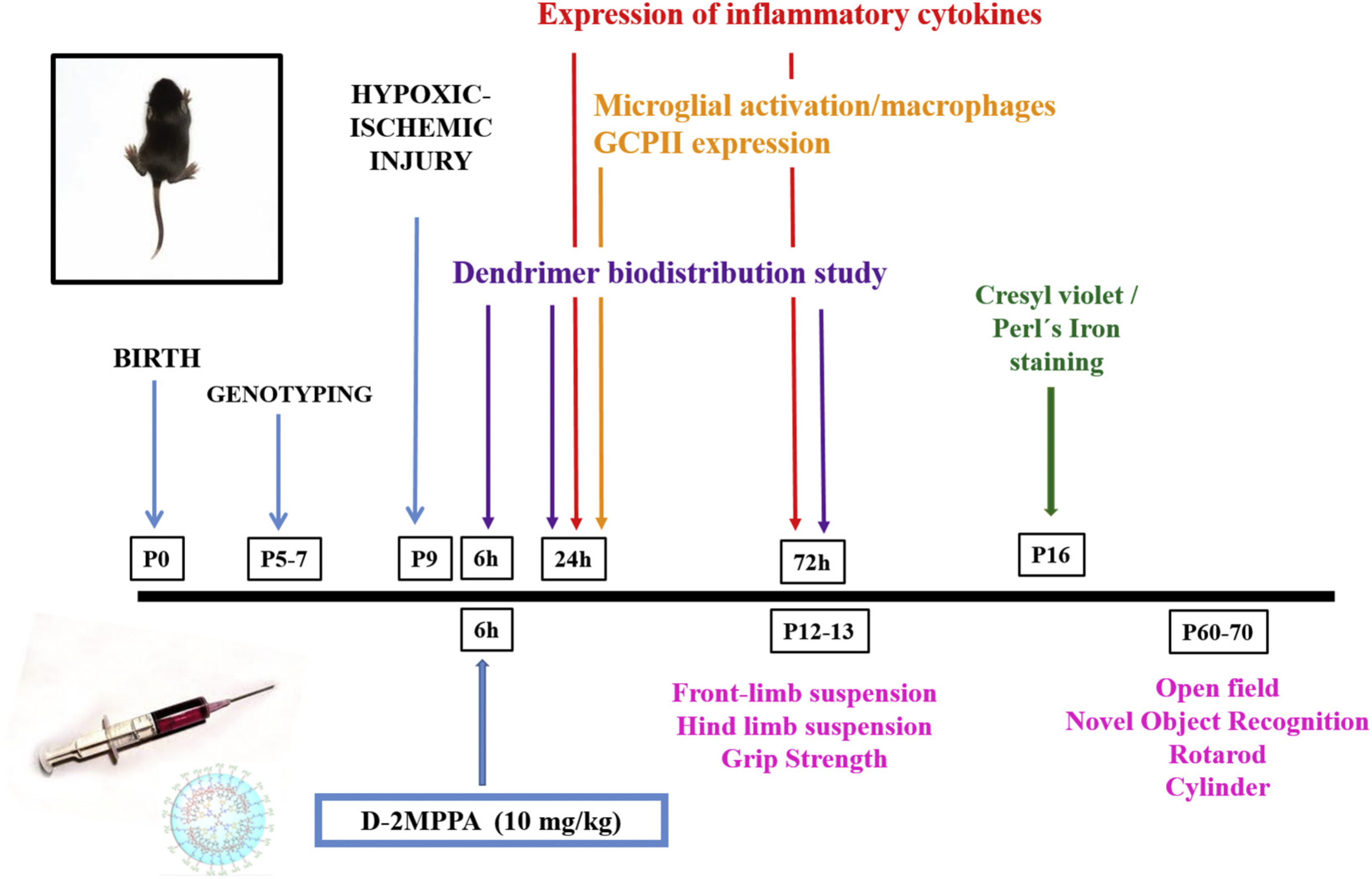
Experimental Timeline. HI was induced on postnatal day 9 (P9 mouse pups (C57/Bl6)) by the Vannucci method. For the dendrimer biodistribution studies, dendrimers were administered 6 h, 24 h and 72 h after the HI, and brains were analyzed 24 h after the injections by immunofluorescent histochemistry for microglial activation/macrophages expression and the GCPII expression. HI mice were treated with D-2MPPA 6 h post-HI. Inflammatory profiling was done on fresh frozen brain at 24 h and 72 h after the HI. Animals were sacrificed 7 days after the HI event, at P16, and brain sections were stained with cresyl violet and Perl’s iron and evaluated using a semi-quantitative neuropathological scoring system. Behavioral studies were performed in neonates (P12–13) and adults (P60–70).
For cytokine analysis: 24 h and 72 h after HI: WT C n = 9–10, WT+HI n = 9–10, WT+HI+D-2MPPA n = 9–10, SOD C n = 9–10, SOD+HI n = 9–10, SOD+ HI+D-2MPPA n = 9–10. For behavioral tests: WT C n = 5, WT+HI n = 5, WT+HI+D-2MPPA n = 6, SOD C n = 5, SOD+HI n = 5, SOD+HI+D-2MPPA n = 5. Detailed methods and results for the behavioral studies are provided in supplement.
2.3. Synthesis and characterization of dendrimer conjugates (D-2MPPA and D-Cy5)
We used generation 4 hydroxyl polyamidoamine (PAMAM) dendrimers (~4 nm, neutral) for the conjugation of the GCPII enzyme inhibitor 2MPPA. Ethylenediamine-core PAMAM dendrimer (generation 4, 64 –OH groups, pharmaceutical grade) was received from Dendritech (Midland, MI, USA) in the form of a methanolic solution. The solvent was removed from the as-received dendrimer solution by evaporation on a rotary evaporator to afford white hygroscopic solid. The thiol group of 2MPPA was utilized for its covalent conjugation on the dendrimer surface through glutathione sensitive disulfide linkages to enable intracellular drug release. The synthesis and characterization of D-2MPPA conjugate was carried out using our previously published protocol for the synthesis of dendrimer-N-acetyl cysteine conjugate with minor modifications (Kannan et al., 2012, (Nance et al., 2015)). Briefly, the surface hydroxyl groups of PAMAM dendrimer were partially modified to have protected thiol groups for later conjugation of 2MPPA through its thiol group forming disulfide linkages. The structure and purity of the intermediates and the final conjugate were analyzed using nuclear magnetic resonance (NMR) spectroscopy and high-performance liquid chromatography (HPLC). The drug loading was calculated using proton integration method based on NMR. On an average, 13 molecules of 2MPPA were conjugated with a loading of 11% w/w. The conjugate demonstrated a purity of >98% as analyzed by HPLC. The theoretical molecular weight of D-2MPPA is ~19 kDa. The D-2MPPA conjugate demonstrated a hydrodynamic diameter of 5.9 ± 0.6 nm as analyzed by dynamic light scattering and exhibited a near neutral zeta potential of +3.4 ± 5.5 mV. For imaging purposes, fluorescently labeled dendrimers (D-Cy5) were synthesized and characterized using a previously reported procedure (Lesniak et al., 2013; Sharma et al., 2018a, (Nance et al., 2016)).
2.4. Immunofluorescence analysis for Cy5-labeled dendrimer (D-Cy5) biodistribution
Cyanine 5 (Cy5)-labeled dendrimer (D-Cy5) were administered intraperitoneally (20 mg/kg) to newborn mice at 6, 24 or 72 h after the hypoxic-ischemic brain injury. Animals were euthanized at 24 h after D-Cy5 administration and perfused with 10% formalin. Coronal brain sections (30 μm, 1:6 series) were cut using a Leica cryostat.
To evaluate the co-localization of D-Cy5 and astrocytes or microglia, brain sections were incubated overnight at 4 °C with chicken anti-glial fibrillary acidic protein (GFAP) (1:250, Abcam, MA. U.S.A.) and goat anti- calcium binding adaptor molecule 1 (IBA1) (1:250, Abcam, MA. U. S.A.). Sections were subsequently washed and incubated with fluorescent secondary antibodies (1:250; Life Technologies, MA, U.S.A.) for 2 h at room temperature. Next, the sections were incubated with 4',6-dia-midino-2-phenylindole (DAPI) (1:1000, Invitrogen) for 15 min to stain the nuclei. After washing, the slides were dried and cover slipped with mounting medium (Dako, Carpinteria, CA, USA). Confocal images were acquired with Zeiss ZEN LSM 710 (Zeiss, CA, U.S.A.) and processed with ZEN software.
2.5. Immunofluorescence analysis for glutamate carboxypeptidase II (GCPII)
To evaluate the expression of GCPII enzyme, brain sections were incubated overnight at 4 °C with mouse anti-GCPII (1:250, Abcam, MA. U.S.A.) and goat anti-IBA1 (1:250, Abcam, MA. U.S.A.). Sections were subsequently washed and incubated with fluorescent secondary antibodies (1:250; Life Technologies, MA, U.S.A.) for 2 h at room temperature. Next, the sections were incubated with DAPI (1:1000, Invitrogen) for 15 min. After washing, the slides were dried and cover slipped with mounting medium (Dako, Carpinteria, CA, USA). Confocal images were acquired with Zeiss ZEN LSM 710 (Zeiss, CA, U.S.A.) and processed with ZEN software.
2.6. Morphological evaluation
2.6.1. Tissue processing
Brains were morphologically evaluated on postnatal day 16 (P16), seven days after the HI insult (WT+HI n = 16, WT+HI+D-2MPPA n = 13, SOD+HI n = 20, SOD+HI+D-2MPPA n = 19). Animals were sacrificed with a sodium pentobarbital overdose and transcardially perfused with saline-heparin followed by cold 4% paraformaldehyde (Sigma-Aldrich) in 0.1 M phosphate-buffered saline (PBS) (pH 7.3). Brains were removed and immersed in the same fixative at 4 °C overnight. Sections were cut using a vibrotome (50 μm). Alternate sections were stained with Nissl (cresyl violet) and Perl’s iron histological stains to assess the degree of injury.
2.6.2. Histology and injury analysis
To assess the severity of injury, coronal sections stained with cresyl violet (Sigma-Aldrich Co.) and Perl’s iron (10–15 sections each) were evaluated by a researcher who was blinded to the conditions of treatment, using a semi-quantitative injury scoring system previously described (Sheldon et al., 2017). 11 brain regions (anterior, middle and posterior cortex; CA1, CA2, CA3 and dentate gyrus of the hippocampus; anterior, middle and posterior striatum; and thalamus) were analyzed with a Nikon Optiphot II light microscope. Injury in each region was graded from 0 to 3, with 0 = no injury, 1 = small focal areas of cell loss and iron deposition, 2 = patchy areas of cell loss in multiple areas of the region and 3 = cystic infarction. Thus, injury in the hippocampus was graded from 0 to 12, in the cortex and striatum from 0 to 9 and in the thalamus from 0 to 3, for a total score 0–33 for the whole ipsilateral hemisphere.
2.7. Immunofluorescence analysis for activated microglia
Microglia were visualized using IBA1, which is a microglia/macrophage-specific calcium-binding protein immunohistochemistry. To this end, perfused brains were carefully removed and post-fixed by 4% paraformaldehyde overnight, equilibrated in 30% sucrose in 0.1 M phosphate-buffered saline and left at 4 °C. 50-μm coronal sections using a vibratome (Leica 1325 Biosystems). After washing three times in PBS, a permeabilization solution (0.25% Triton X-100 in 0.5% BSA in PBS) was applied for 15 min. Sections were then blocked for one hour at room temperature with a blocking solution containing normal serum from the same host species as the labeled secondary antibody. Sections were incubated overnight at 4 °C with a polyclonal goat primary anti-IBA-1 antibody (1:1000, Abcam, Cambridge, UK) diluted in 0.25% Triton X-100 in 0.5% BSA in PBS. The next day, slides were washed three times in PBS and incubated for 1 h in donkey anti-goat secondary antibody conjugated with AlexaFluor 594 (1:200; Invitrogen, The Netherlands). The sections were also counterstained with DAPI (1:1000; Invitrogen). Negative controls received the same treatment omitting the primary antibodies and showed no specific staining. Images from the the hippocampus were taken with an Olympus Fluoview FV500 Confocal Microscope. A quantitative analysis of area measurement of histological fluorescence was performed using Fiji software.
2.8. Cytokine study
Pro- and anti-inflammatory cytokine levels were evaluated 24 h and 72 h after HI with V-PLEX Mesoscale cytokine panel (Meso Scale Discovery MULTI-SPOT Assay System, Proinflammatory Panel 1 mouse kits, Rockville, MD, USA). Animals were sacrificed with pentobarbital sodium overdose. Brains were rapidly removed and the ipsilateral cortex and hippocampus were dissected on a cold surface, frozen immediately, and stored at −80 °C. The expression of pro-inflammatory cytokines (IL-1β, TNF-α, IFN-γ, IL-12P70 and IL-6) and anti-inflammatory cytokines (IL-10, IL-4, KC/GRO, IL-2 and IL-5) in the ipsilateral cortex from the different experimental groups (n = 9–10 per group) were evaluated 24 h and 72 h after HI.
As per the manufacturer’s instructions, standards and samples (50 μL) were incubated in the plate for 2 h at room temperature with shaking. After washing, the plate was incubated in a detection antibody solution mix for all 10 analytes (IFN-γ, IL-1β, IL-2, IL-4, IL-5, IL-6, KC/GRO, IL-10, IL-12p70, TNF-α) for 2 h at room temperature with shaking. The plate was washed again three times, read buffer was pipetted into all wells, and then the plate was placed on a QuickPlex SQ 120 plate reader (Meso Scale Discovery, Rockville, MD, USA) and electro-chemoluminescence from each analyte was measured sequentially.
2.9. Statistical analyses
Statistical analysis was performed using GraphPad Prism version 7 (Graph Pad Software, San Diego, CA, USA). Data were analyzed for the presence of outliers using Grubbs’ outlier test. All data were expressed as the mean ± standard error of the mean (SEM) and were analyzed using a one-way analysis of variance followed by Bonferroni-Dunn correction. Nonparametric data was analyzed using Fisher’s exact test. A p value less than or equal to 0.05 was considered statistically significant.
3. Results
3.1. Microglial activation
Representative images from brain sections stained with IBA1 show that there is an increase in microglial activation in the animals that underwent HI brain injury 24 h after HI in the ipsilateral hippocampus and it is most pronounced in the SOD+HI group. The results from the quantification of the average fluorescent area also demonstrate that there is a statistically significant increase in expression of activated microglia in the ipsilateral hippocampus in comparison to the SOD control group as observed in the Fig. 2.
Fig. 2.
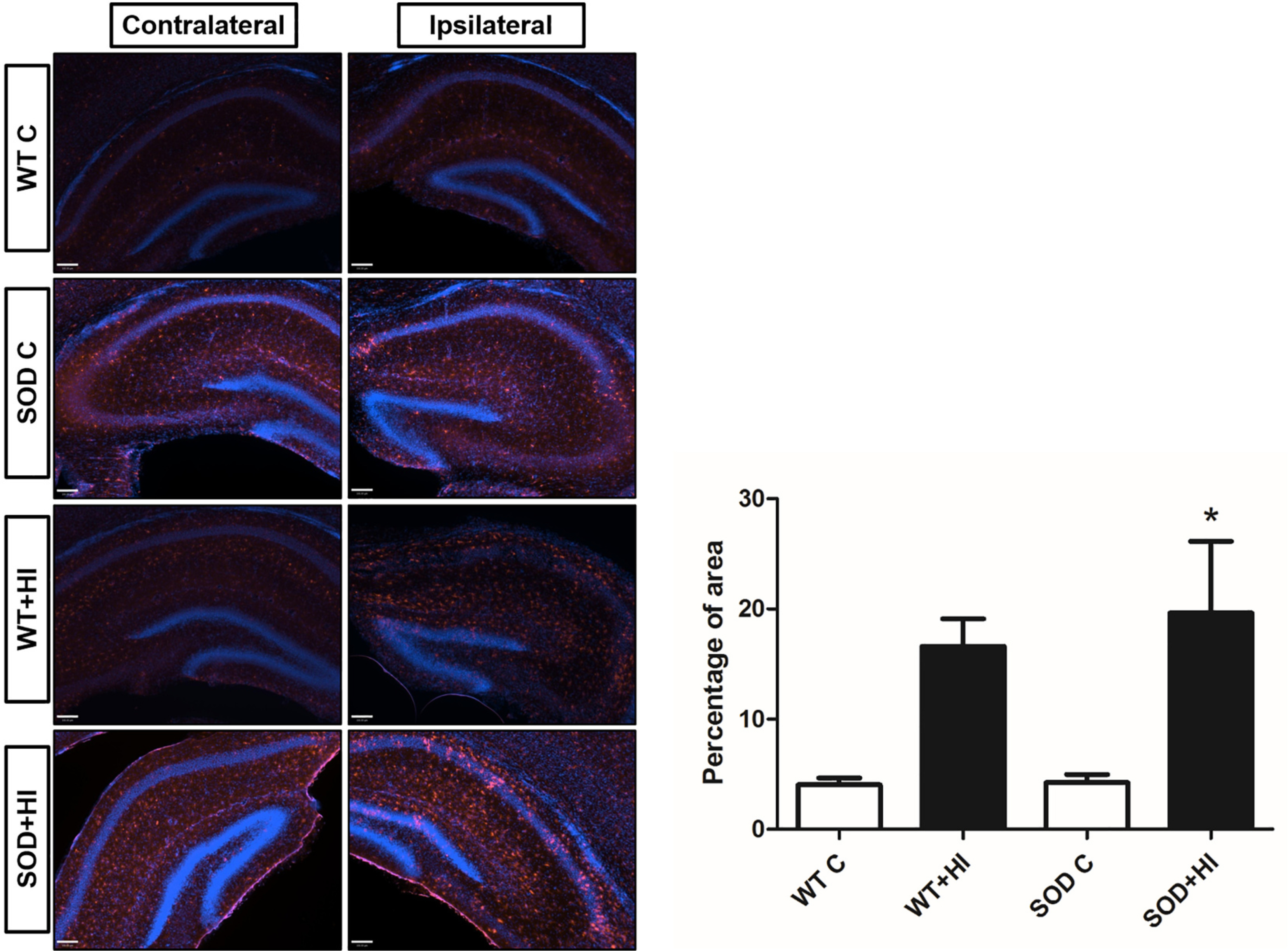
Activated microglia presence in the ipsilateral hippocampus 24 h after the hypoxic-ischemic brain injury. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI) (Blue) and microglia were labeled using IBA1 (red). Scale bar: 100 μm. The representative images and the corresponding histogram show an increase in the expression of activated microglia/macrophages in the pups that underwent HI, and particularly in the SOD+HI 24 h after HI (*P < 0.05 vs. SOD C, n = 3–4).
3.2. Trajectory of dendrimer cellular uptake
3.2.1. Immunofluorescence analysis for Cy5-labeled dendrimer (D-Cy5) biodistribution study
To determine the dendrimer uptake over time after HI injury, mice were injected with dendrimer conjugated with the fluorophore Cy5 (D-Cy5) at 6 h, 24 h or 72 h after the hypoxic-ischemic insult. Animals were sacrificed 24 h after D-Cy5 administration, at which time point all the D-Cy5 has been shown to have cleared from plasma (Sharma et al., 2018b). We find that D-Cy5 co-localizes in microglia at all these time points post-HI, as shown in Fig. 3. Although both SOD+HI and WT+HI groups show microglial uptake of D-Cy5, it is more prominent in the SOD+HI mice. Similarly, D-Cy5 uptake by microglia is most visible at 24 h post-HI in the SOD overexpressing mice. Dendrimer uptake was seen as early as 6 h and appeared to peak at 24 h after injury and is more prominent in the SOD+HI mice than in the WT+HI. This appears to be related to the timing of peak microglial activation and the degree of microglial activation which is greater in the SOD mice when compared to the WT mice following HI (as seen in Fig. 3).
Fig. 3.
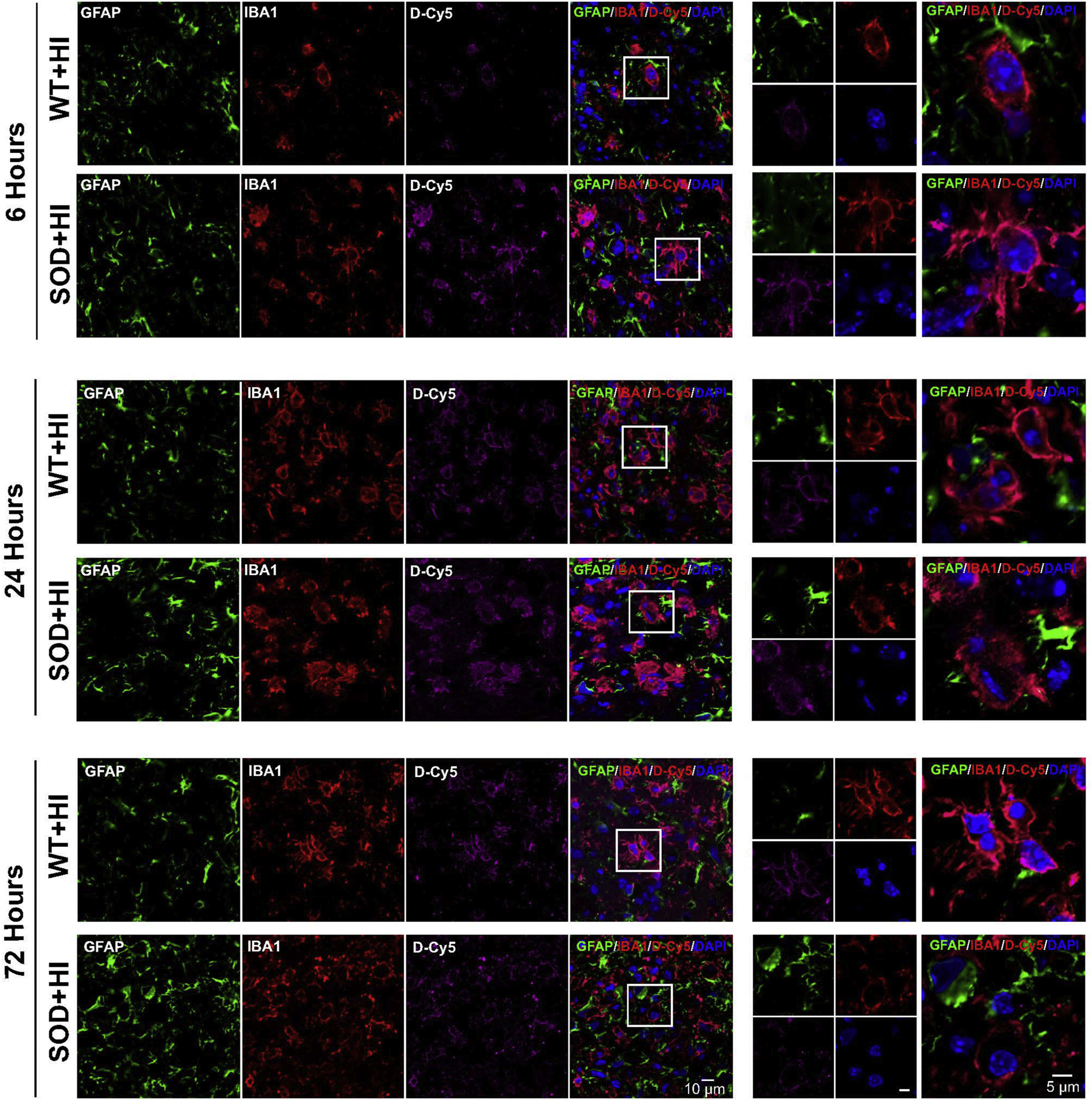
Dendrimer (magenta) co-localization 6 h, 24 h and 72 h after the hypoxic-ischemic brain injury in astrocytes (green) and in microglia (red) in WT+HI and SOD+HI pups in the ipsilateral internal capsule/striatum. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI) (blue). The images in the right panels are higher magnification images indicated by the boxes on the merged images. Dendrimers were taken up by activated microglia/macrophages as early as 6 h post-HI and are more prominent in the SOD+HI mice than WT+HI.
3.2.2. Glutamate carboxypeptidase (GCPII) is overexpressed in microglia following HI
We evaluated the expression of GCPII enzyme in the different experimental groups 24 h after the HI injury by immunofluorescence. We found that GCPII enzyme is constitutively expressed in the microglial cells in SOD control mice but not in WT control mice, as seen in Fig. 4. Following HI, increased expression of GCPII is seen in activated microglia both in SOD and WT mice but is much more prominent in the SOD mice. D-Cy5 localized predominantly in activated microglial cells that over-express GCPII enzyme. There is also overexpression of GCPII in activated microglia 24 h after the damage in the SOD mice in the ipsilateral corpus callosum.
Fig. 4.
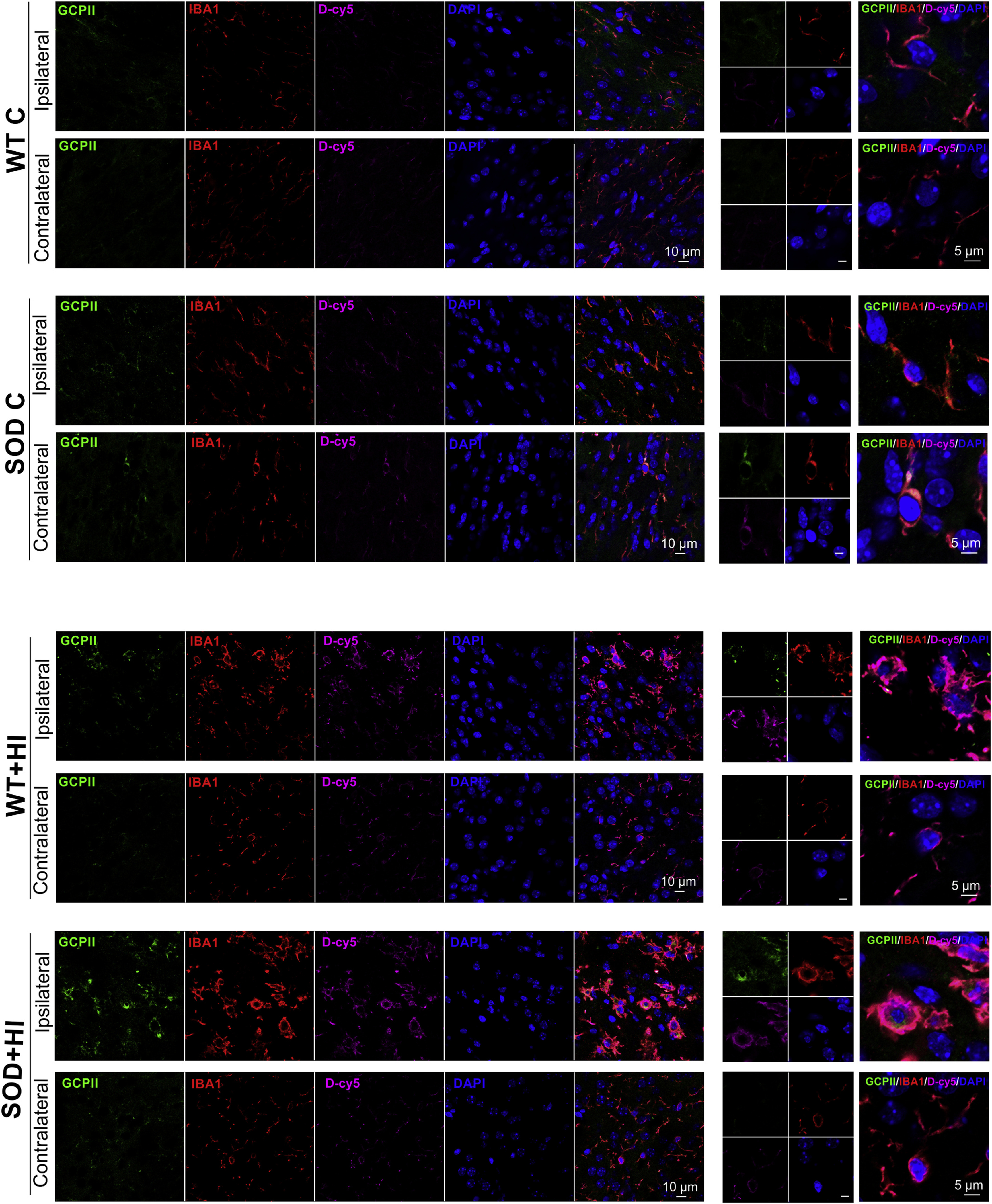
Glutamate carboxypeptidase II enzyme expression (green) 24 h after the hypoxic-ischemic brain injury and co-localization with microglia (red) and with dendrimer localization (magenta) in the different experimental groups (WT C, WT+HI, SOD C and SOD+HI) in the contralateral and ipsilateral corpus callosum. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI, blue). Dendrimers were labeled with Cy5 fluorochrome. The images in the right panels are the higher magnification images indicated by the boxes on the merged images. Scale bar: 10 μm and 5 μm. There was an overexpression of GCPII 24 h after the damage on the ipsilateral side of the injury in the corpus callosum in the mice transgenic for SOD that underwent HI brain injury.
3.3. Hippocampal evaluation
As shown in Nissl stained brain sections observed in Fig. 5 and supplementary S1, hypoxia-ischemia caused significant cell loss in HI mouse pups. HI animals show swollen and deformed neurons in the ipsilateral areas of hippocampus. In iron stained brain sections, there are patches of iron deposits in response to HI injury. In contrast, for D-2MPPA treated pups, there is mild cell loss and fewer damaged neurons in Nissl stained sections and not as much iron deposition in Perl’s Iron stained sections, demonstrating a remarkable conservation of cellularity. We propose that this improvement in in hippocampal morphology and iron deposits is due to D-2MPPA uptake in after HI. We see indeed that GCPII expression is present in hippocampal IBA1+ microglia in SOD+HI mouse brains, and further that there is profound colocalization of dendrimer (labeled with Cy5) with GCPII+IBA1+cells (Fig. 6).
Fig. 5.
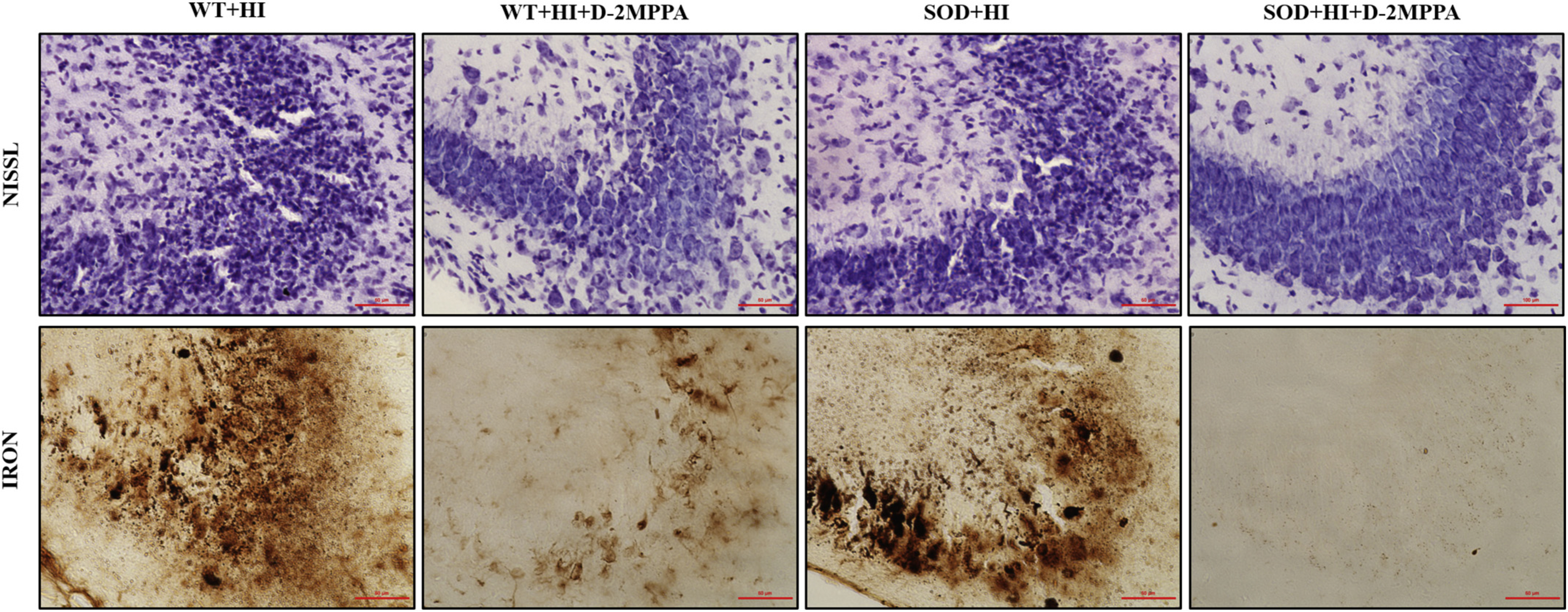
Representative photographs of the CA2–3 area of the ipsilateral hippocampus stained with cresyl violet and Perl’s iron in the different experimental groups: WT+HI, WT+HI+D-2MPPA, SOD+HI and D) SOD+HI+D-2MPPA. Pups underwent hypoxic-ischemic brain injury at P9 and some of them were treated with D-2MPPA (10 mg/kg on a 2MPPA basis) 6 h after HI (P9). Scale bar: 50 μm.
Fig. 6.
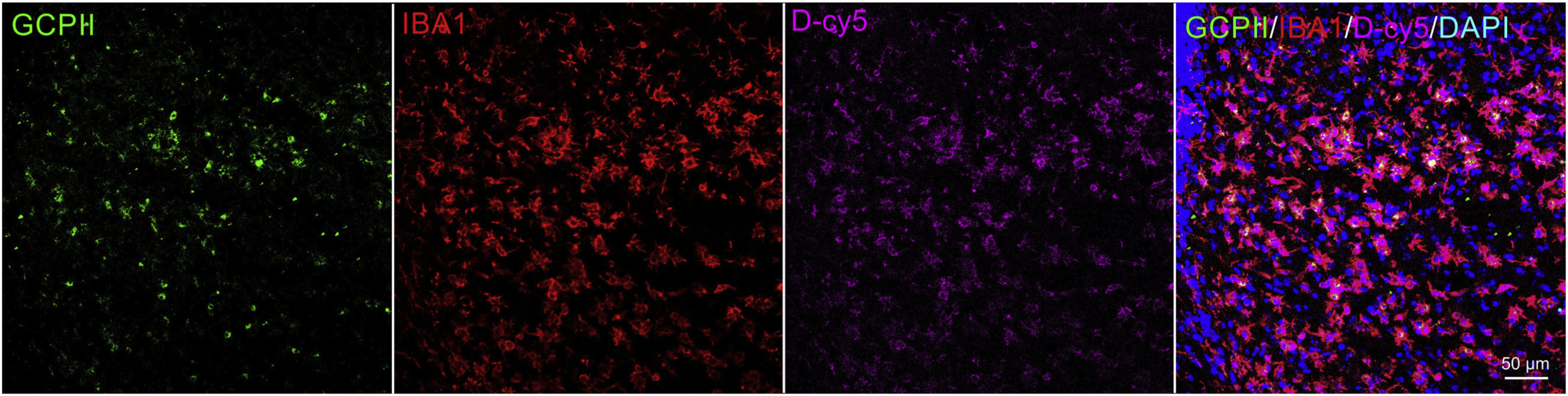
Glutamate carboxypeptidase II enzyme expression (green) 24 h after the hypoxic-ischemic brain injury and co-localization with microglia (red) and with dendrimer localization (magenta) in the hippocampus of SOD+HI mice. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI, blue). Dendrimers were labeled with Cy5 fluorochrome. Scale bar: 50 μm.
3.3.1. Neuropathological score of damage
Results obtained from the neuropathological scoring system reveal that D-2MPPA treatment significantly decreases brain injury in SOD+HI group seven days after injury, as shown in Fig. 7. The total score for the injured hemisphere is significantly reduced in the SOD+HI+D-2MPPA (12.79 ± 1.90) compared with the SOD+HI group (18.5 ± 0.77). Likewise, WT+HI+D-2MPPA (12.54 ± 2.01) shows decreased brain damage compared to WT+HI (16.88 ± 1.15), but does not reach statistical significance. There is also a shift in global injury severity in SOD+HI mice treated with D-2MPPA. Fewer SOD+HI+D-2MPPA mice exhibit a moderate-severe neuropathology score than SOD+HI mice treated with saline (Table 1). When analyzed based on area of injury, the greatest improvement is seen in the ipsilateral hippocampus in the SOD+HI+D-2MPPA group. Neuropathological scoring revealed that D-2MPPA treatment significantly decreases brain injury in SOD+HI group seven days after damage in the ipsilateral hippocampus, but did not reach significance in the rest of the evaluated areas (Table 2).
Fig. 7.
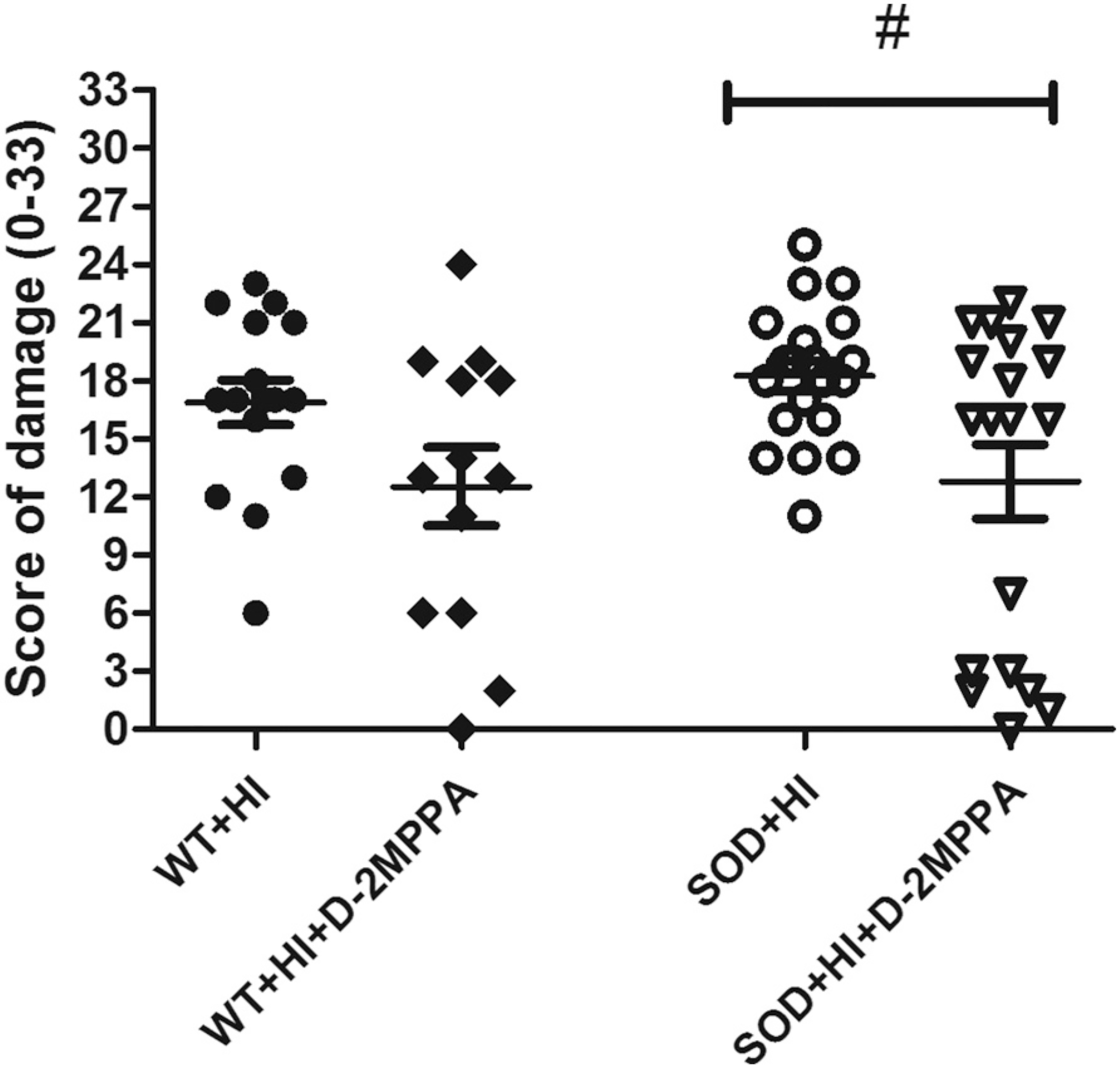
Neuropathological score of damage (0· 33) at P16. Pups underwent HI at P9 and some were treated with a dendrimer-based GCPII inhibitor (D-2MPPA) 6 h after HI. Ipsilateral brains areas were evaluated at P16 using a semi-quantitative neuropathological scoring system. Mean ± SEM damage score is significantly lower in SOD+HI+D-2MPPA treated mice than SOD+HI mice (#P < 0.05) (n = 13–20).
Table 1.
Injury severity distribution. Neuropathological injury score: Summed scores less than 11 are considered mild and scores 12–33 are considered moderate/severe. SOD+HI+D-2MPPA mice have a significant decrease in the percentage of moderate/severe injury scores.
| Injury severity distribution | |||
|---|---|---|---|
| Group | Mild (n) | Moderate/Severe (n) | Moderate/Severe (%) |
| WT+HI | 2 | 14 | 87.50% |
| WT+HI+D2MPPA | 5 | 8 | 61.50% |
| SOD+HI | 1 | 19 | 95% |
| SOD+HI+D2MPPA | 7 | 12 | 63.2%* |
(*p < 0.05 as compared to SOD+HI). No significant differences in neuropathological severity distribution are seen with WT mice.
Table 2.
Neuropathological score of damage in the brain subregions A) Hippocampus (0–12), B) Cortex (0–9), C) Striatum (0–9) and D) Thalamus (0–3). Ipsilateral brains areas were evaluated at P16 using a semi-quantitative neuropathological scoring system, expressed as the mean ± SEM. D-2MPPA shows a protective effect after HI (i.e. lower injury score) in the hippocampus of SOD mice
| Neuropathological injury score in each area | ||||
|---|---|---|---|---|
| Group | Hippocampus (0–12) | Cortex (0–9) | Striatum (0–9) | Thalamus (0–3) |
| WT+HI | 7.06 ± 0.39 | 3.87 ± 0.35 | 4.37 ± 0.40 | 1.56 ± 0.15 |
| WT+HI+D2MPPA | 5.30 ± 0.74 | 3.23 ± 0.50 | 2.92 ± 0.62 | 1 ± 0.22 |
| SOD+HI | 7.65 ± 0.26 | 4.60 ± 0.30 | 4.15 ± 0.32 | 1.65 ± 0.10 |
| SOD+HI+D2MPPA | 5.10 ± 0.80* | 3.31 ± 0.39 | 3.26 ± 0.53 | 1.15 ± 0.21 |
(*P < 0.05 vs. SOD+HI).
3.3.2. Sex difference in neuropathology score
We also analyzed the animals based on sex to determine if there are any sex differences noted for the neuroprotective effects of GCPII inhibition. According to the neuropathological score of damage there are statistically significantly differences in female mice between SOD+HI and SOD+HI+D-2MPPA in the score of damage analysis in the whole brain and in the hippocampus, as shown in Fig. 8. There is a significant decrease in the neuropathological score following D-MPPA treatment in females (WT+HI n = 6, WT+HI+D-2MPPA n = 5, SOD+HI n = 11, SOD+HI+D-2MPPA n = 14) vs males (WT+HI n = 10, WT+HI+D-2MPPA n = 8, SOD+HI n = 9, SOD+HI+D-2MPPA n = 6). This improvement is most profound in the hippocampus with similar sex differences observed.
Fig. 8.
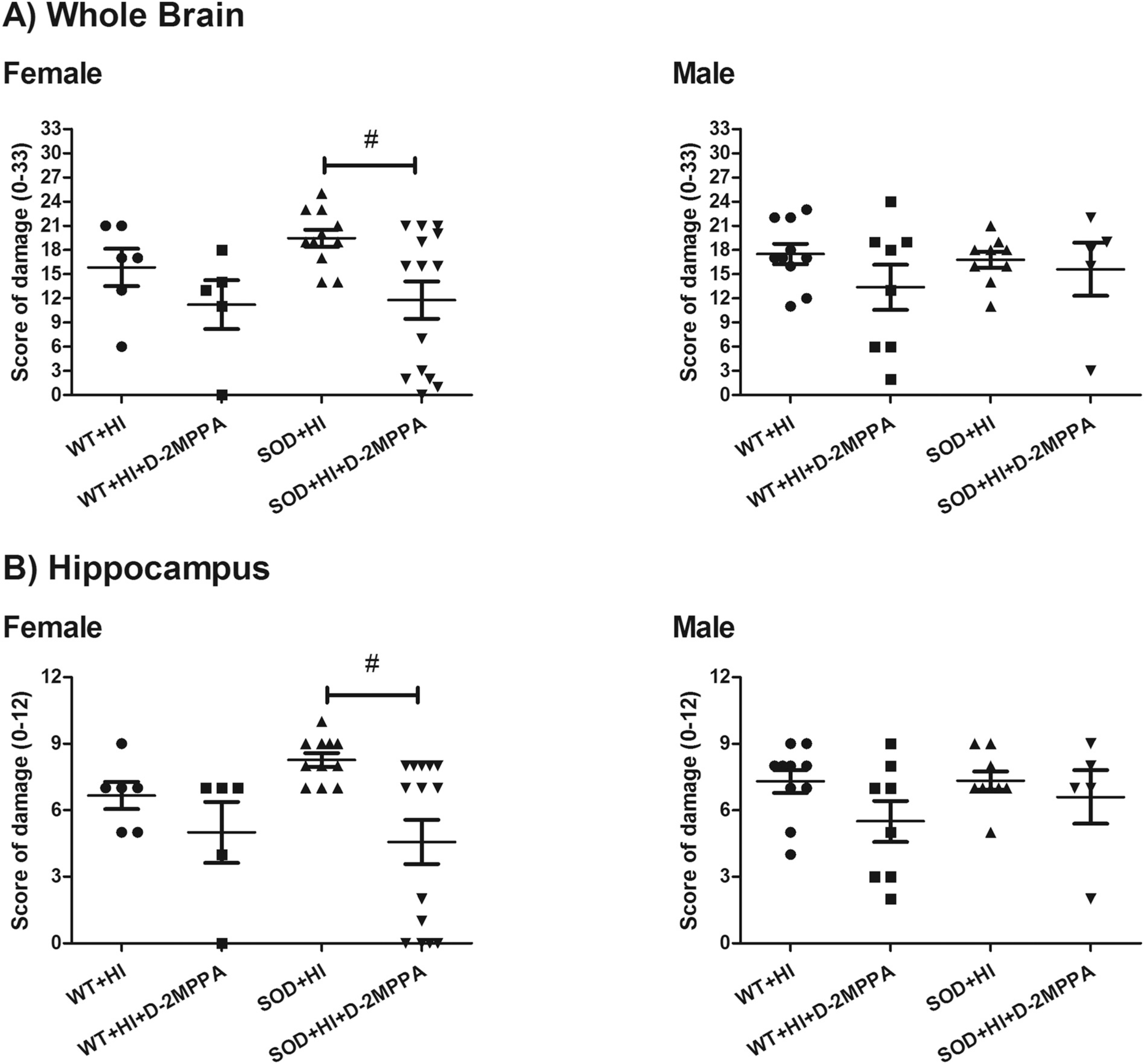
Sex differences in brain damage and protection by D-2MPPA in accordance with the semi-quantitative neuropathological scoring system. Results from the A) whole brain in females (F) and males (M) (0–33) and in the b) hippocampus in females and males (0–12) in the different experimental groups: WT+HI (n = 16; F: n = 6 and M: n = 10), WT+HI+D-2MPPA (n = 13; F: n = 5 and M: n = 8), SOD+HI (n = 20; F: n = 11 and M: n = 9) and SOD+HI+D-2MPPA (n = 19; F: n = 14 and M: n = 6).
3.4. Cytokine expression
We evaluated the inflammatory response of these mice after the HI injury, as shown in Fig. 9 and supplementary fig. S2. To this end, the expression of pro- and anti-inflammatory cytokines in the ipsilateral cortex of fresh frozen brains were analyzed 24 h and 72 h after the HI injury using a multiplex cytokine panel (Meso Scale Discovery).
Fig. 9.
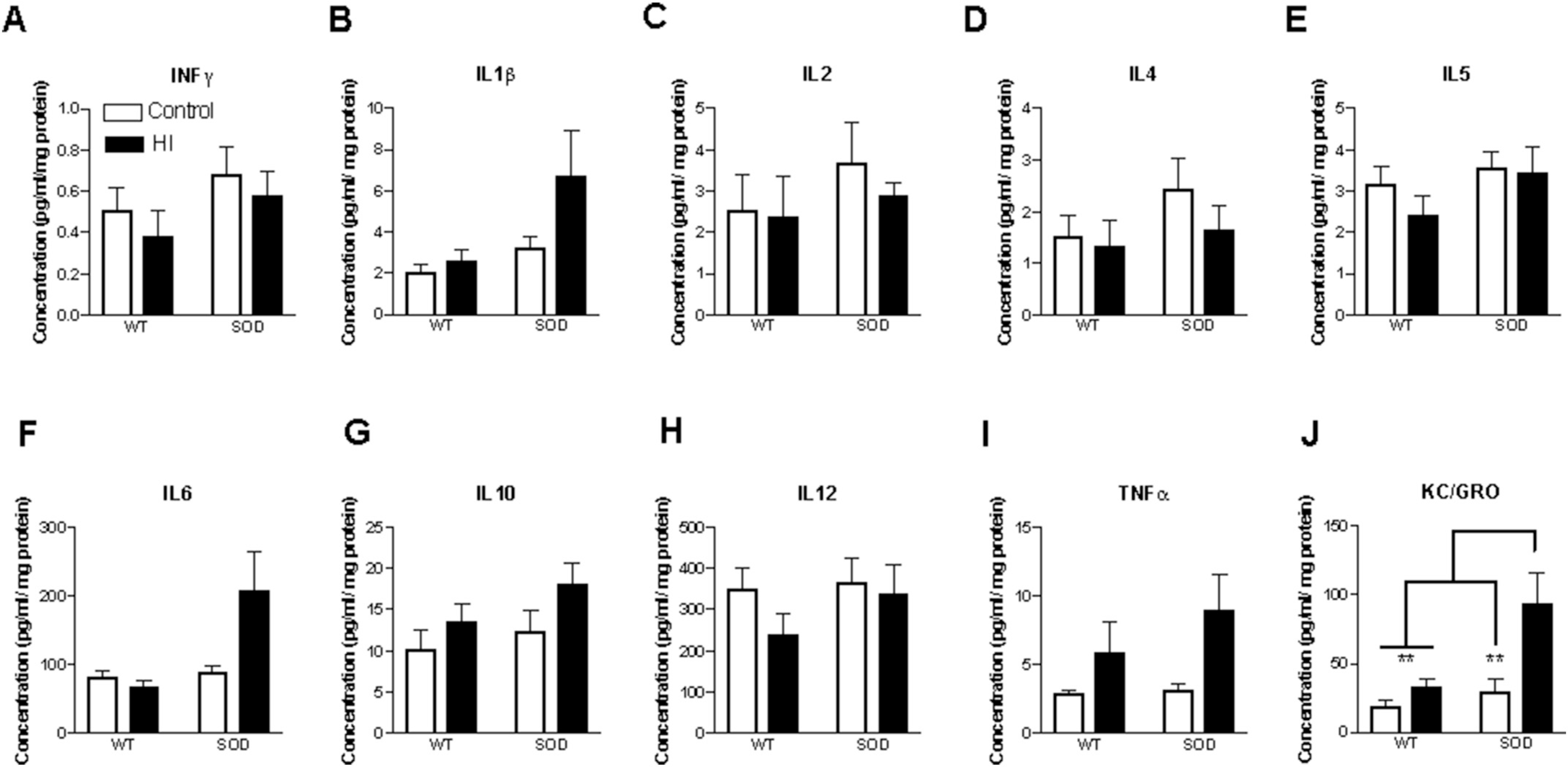
Cytokine profile in ipsilateral cortex of SOD mice 24 h after HI. Transient changes in pro and anti-inflammatory cytokines were noted following HI at 24 h in the SOD mice. However, a consistent increase in KC/GRO (CXCL1) was seen in the SOD mice undergoing HI at 24 h. Cytokine levels were evaluated with V-PLEX cytokine panel (Meso Scale Discovery). n = 9–10 animals per group with 4–5 males and females per group.
Significant differences in expression following injury were seen in SOD mice when compared to WT at 24 h and 72 h for KC/GRO and at 24 h for TNF-α (Fig. 9 and S2). D-2MPPA does not have a significant effect on the KC/GRO cytokine response in WT mice (data not shown). However, a greater and significant decrease in CXCL1 (KC/GRO) is seen in the SOD mice following treatment with D-2MPPA (Fig. 10). D-2MPPA has a greater effect in inhibiting GCPII enzyme in the SOD mice. No significant difference is seen between the groups for the other cytokines.
Fig. 10.
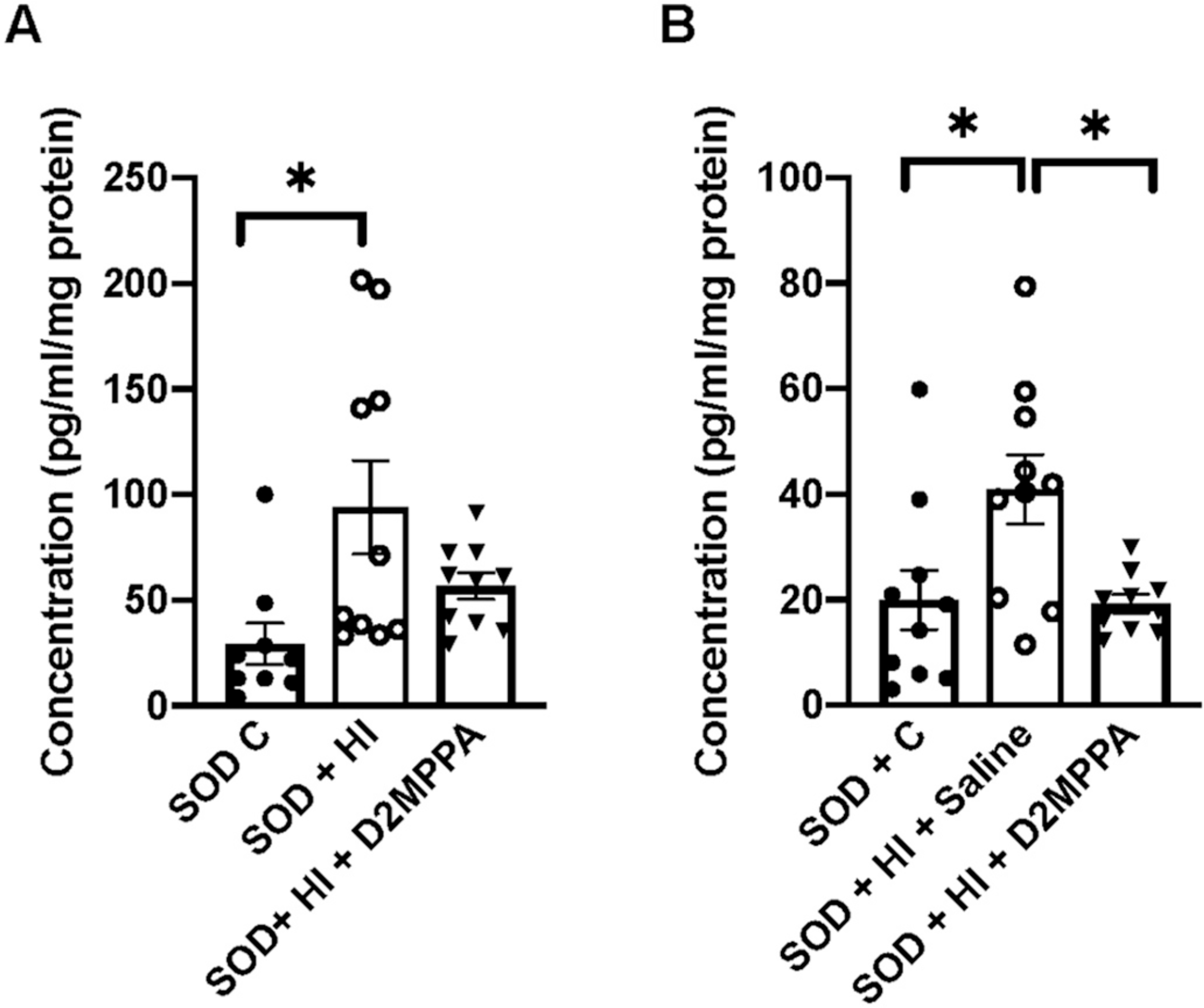
Cortical KC/GRO (CXCL1) is improved with D-2MPPA 24 h (A) and 72 h (B) after injury. Treatment with D-2MPPA resulted in a decrease in KC/GRO to control levels at both time points. n = 9–10 animals per group with 4–5 males and females per group.
4. Discussion
In this study, we demonstrate that GCPII is constitutively expressed at low levels in microglia in the developing brain but is upregulated in microglia following HI both in transgenic SOD and WT mice. Previously, astrocytes were thought to exclusively express GCPII in the CNS (Šácha et al., 2007). However, we have recently demonstrated that intrauterine inflammation leads to increase in GCPII expression in the microglia in the newborn rabbit brain (Zhang et al., 2016a, 2016b). Inflammation may also lead to an increase in enzyme activity of GCPII. We have found that LPS stimulation of primary rodent microglia lead to increased GCPII enzyme activity (Zhang et al., 2016a, 2016b).
SOD mice after HI displayed a greater neuroinflammatory response with greater increase in activated microglia, qualitatively higher GCPII expression, greater damage score and increased inflammatory cytokine response following injury when compared to WT mice after HI. This is consistent with previous data from our group demonstrating that SOD overexpressing mice demonstrated greater oxidative stress and a greater degree of injury when compared to WT mice when injury occurred at P7 (Ditelberg et al., 1996; Fullerton et al., 1998; Sheldon et al., 2002) and at P9 (Sheldon et al., 2017). We also demonstrate here that the SOD overexpressing mice have a greater response to treatment with D-2MPPA when compared to the WT animals. This indicates that the GCPII pathway may play a greater role in the injury in the SOD transgenic mice.
We also observed that D-2MPPA specifically localizes to activated microglia as early as 6 h post-injury, and through 72 h post-injury. It is also plausible that invading macrophages also take up D-2MPPA en route to the site of damage/inflammation. Previously we have demonstrated that activated microglia/macrophages are the predominant cell type to take up dendrimer in a mouse model of HI (Nance et al., 2015; Nemeth et al., 2017). Further, dendrimer uptake is dependent on disease severity with more dendrimer being taken up in animals experiencing more severe injury/inflammation (Nemeth et al., 2017). In severely injured HI mice, 60% of microglia in the hippocampus were shown to have dendrimer colocalized. These findings imply that dendrimers allow for (1) effective targeting of microglia/macrophages and (2) a broader window of treatment from 6 to 24 h following HI. This is relevant for patients for whom therapeutic hypothermia (TH) is not implemented early enough or is contraindicated, and also for patients with severe injury where adjunct therapies with TH are critically required.
Both oxidative stress and glutamate excitotoxicity are known to play a major role in brain injury during the neonatal period and may potentiate each other (Bratek et al., 2018). This is exaggerated in the neonatal brain where anti-oxidant defenses are lower making the brain more vulnerable to oxidative stress (Ferriero, 2004; Halliwell, 1992). HI has been shown to lead to decreased glutamate clearance/uptake and that enhancing glutamate clearance by activating Group II or Group III metabotropic glutamate receptors leads to neuroprotection due to improved glutamate clearance by astrocytes and by decreasing ROS production (Bratek et al., 2018). In the SOD model of amyotrophic lateral sclerosis (ALS), selective mGluR3 glutamate receptor agonists are neuroprotective (Battaglia et al., 2015). NAAG, the substrate for GCPII, is a potent activator of mGluR3, and has been shown to exert its neuroprotective effects by both inhibiting pre-synaptic release of glutamate and decreasing ROS production (Neale and Yamamoto, 2020). NAAG and other mGluR2/3 agonists have been shown to be neuroprotective in models of neonatal HI (Bratek et al., 2018, 2020; Cai et al., 2002; Van Hemelrijck et al., 2005).
Rat pups exposed to intermittent hypoxia from P2 to P12 demonstrated increased inflammatory cytokines including increases in KC/GRO and a significant decrease in N-acetylaspartate/choline (NAA/Cho) in the hippocampus and brainstem (Darnall et al., 2017) and ipsilateral cortex at P14 and P37 in rats that suffered HI at P7–10 (Pazos et al., 2013), which reflects loss of neural cells (Li et al., 2010). Likewise, thalamic ratio of NAA/Cho was significantly lower in babies suffering mild HIE at 6 h of age compared to ones that were treated with therapeutic hypothermia (TH) (Montaldo et al., 2019). Moreover, during TH, neonates with severe HIE had significantly lower concentrations of NAA and NAA+ NAAG in the basal ganglia compared to neonates with moderate HIE (Lucke et al., 2019). There is an over-expression of NMDA glutamate receptors in selective regions like the basal ganglia in term infants, which is the predominant mediator of excitotoxicity (Ferriero and Miller, 2010).
Chemokines such as CXCL1 (also known as KC/GRO in rodents) are small molecule chemotactic cytokines that are integral for migration of inflammatory cells to areas of injury in the body. Chemokines also have a role to play in normal CNS development. Both GRO (growth related oncogene) and its cognate receptor CXCR2 are expressed by oligodendrocyte progenitors and are responsible for oligodendrocyte proliferation and migration. CXCL1 is an ERL (glutamic acid-arginine-leucine) CXC chemokine defined by potent CXCR2 receptor-dependent neutrophil chemoattractant activity (Biondo et al., 2014; Murphy, 1997). CXCL1 is the dominant CXCR2 ligand expressed in the inflamed CNS (Roy et al., 2012). In the presence of neuroinflammation, CXCL1 and CXCR2 are upregulated rapidly (Carlson et al., 2008; Kerstetter et al., 2009; Kielian et al., 2001; Liu et al., 2010; Roy et al., 2012). Studies in neonatal brain injury and in adult neurodegenerative disorders have shown that inhibiting the CXCR2 receptor is neuroprotective and can attenuate white matter and axonal injury (Yellowhair et al., 2019). This neuroprotective effect is felt to be related to a decrease in neutrophil infiltration in the brain (Yellowhair et al., 2018, 2019) which is often a secondary response following injury and can exacerbate the initial injury. Therefore limiting neutrophil infiltration will attenuate the injury. In this study we see a significant increase in KC/GRO expression both in the WT and SOD over-expressing mice following HI and this increase is attenuated by treatment with D-MPPA, albeit to a greater extent in the SOD overexpressing mice. Inhibition of CXCL1/CXCR2 signaling not only decreases inflammation but also has been shown to reduce microgliosis, similar to what we see in our model.
Interestingly, female SOD over-expressing mice experienced substantial neuroprotection after D-2MPPA treatment, indicated by decreased injury scores noted at 7 days post-injury when compared to males. It is possible that this sex difference in response to D-2MPPA treatment may be due to having several fewer males than females treated with D-2MPPA. Regardless, this finding is surprising since inflammatory microglial responses have been shown to be greater in males when compared to females (Mirza et al., 2015; Rosenkrantz et al., 2019). In fact, in rodent models of neonatal HI, greater microglial activation, peripheral immune activation and neutrophil infiltration has been seen in males when compared to females (Mirza et al., 2015; Rosenkrantz et al., 2019). Glutamate excitotoxicity and oxidative stress have also been found to be greater in males vs females. Interestingly, cell death pathways have been found to be different in males vs females with females demonstrating caspase dependent programmed cell death whereas male cells appear to be more susceptible to oxidative stress and glutamate excitotoxicity mediated apoptosis-inducing factor (AIF) and caspase independent cell death. Caspase activation and increased caspase-3 levels have been shown in female rat pups following HI (Netto et al., 2017; Weis et al., 2014; Zhu et al., 2006). NAAG, through its action on mGluR3 has been shown to prevent caspase mediated programmed cell death. Treatment with NAAG decreased cleaved caspase-3 levels in neuronal cultures and was also shown to decrease ROS production (Spillson and Russell, 2003). It is possible that the significant protection in females seen with D-2MPPA treatment may be related to the neuroprotective effects of NAAG and its effects on caspase mediated cell death that is more prominent in females. Although the cytokine analysis included both males and females, these tests were underpowered to detect consistent sex differences in injury and response to treatment.
Our broad based motor and cognitive behavioral assessments did not show any long-term deficits due to injury (HI compared to sham) or to SOD overexpression (SOD compared to WT). Further, no effects of D-2MPPA were observed (Supplementary fig. 3). This is likely due to the study being underpowered to detect differences in these mice. Previously we have demonstrated that strain differences exist in injury severity and pattern with CD1 mice showing more consistent, severe injury after neonatal HI than C57/BL6 mice (Sheldon et al., 2019). This may suggest that behavioral deficits may be more variable in C57/BL6 mice requiring larger group sizes to detect deficits as well as evaluate treatment effects.
5. Conclusion
GCP-II is upregulated in microglia following HI, both in the SOD and WT mice. Targeted systemic therapy with dendrimer-2MPPA as early as 6 h could provide significant treatment benefits. Dendrimer localization in microglia is seen as early as 6 h with a peak of GCPII upregulation in activated microglia seen at 24 h post-HI. Treatment with D-2MPPA at 6 h post-HI leads to a decrease in KC/GRO (CXCL1) by 24 h and reduction in brain injury in the SOD overexpressing mice exposed to HI.
Supplementary Material
Acknowledgements
Research reported in this publication was supported in part by the National Institute of Neurological Disorders and Stroke NIH, DHHS, under Award Number R35NS097299 (DMF), NS093416-04 (SK) and by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, DHHS award HD076901 (RMK). Olatz Arteaga Cabeza was partially supported by a research fellowship from Fundacioón Alfonso Martin Escudero (FAME).
The authors would like to thank Caroline Chen for her help during behavioral tests. The authors also thank the Wilmer Core Module for Microscopy and Imaging for use of the Zeiss LSM710 confocal. The authors would also like to thank Dr. Allen Everett for the use of the MESO QuickPlex SQ 120 plate reader.
Footnotes
Declaration of Competing Interest
Under license agreements involving Ashvattha Therapeutics Inc., and its subsidiary, Orpheris, Inc., and the Johns Hopkins University, Drs. Kannan, Rangaramanujam, Slusher and the University are entitled to royalty distributions from patents related to technology involved in the study discussed in this publication. Drs. Kannan (Co-founder), Rangaramanujam (Co-founder) and Slusher (Board Member) hold equity in Ashvattha Therapeutics Inc., and serve on the Board of Directors of Ashvattha Therapeutics Inc. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. AS and FZ are co-inventors of patents licensed by Ashvattha, relating to the dendrimer platform. All other authors declare that there are no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.105201.
References
- Arteaga Cabeza O, Mikrogeorgiou A, Kannan S, Ferriero DM, 2019. Advanced nanotherapies to promote neuroregeneration in the injured newborn brain. Adv. Drug Deliv. Rev. 10.1016/j.addr.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Arteaga O,Revuelta M, Urigüen L, Martínez-Millán L, Hilario E, Álvarez A, 2017. Docosahexaenoic acid reduces cerebral damage and ameliorates long-term cognitive impairments caused by neonatal hypoxia–ischemia in rats. Mol. Neurobiol. 54 10.1007/s12035-016-0221-8. [DOI] [PubMed] [Google Scholar]
- Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, Thoresen M, Whitelaw A, Brocklehurst P, 2009. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N. Engl. J. Med. 361, 1349–1358. 10.1056/NEJMoa0900854. [DOI] [PubMed] [Google Scholar]
- Bai W, Zhou YG, 2017. Homeostasis of the intraparenchymal-blood glutamate concentration gradient: maintenance, imbalance, and regulation. Front. Mol. Neurosci. 10.3389/fnmol.2017.00400. [DOI] [PMC free article] [PubMed]
- Battaglia G, Riozzi B, Bucci D, Di Menna L, Molinaro G, Pallottino S, Nicoletti F, Bruno V, 2015. Activation of mGlu3 metabotropic glutamate receptors enhances GDNF and GLT-1 formation in the spinal cord and rescues motor neurons in the SOD1 mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 74, 126–136. 10.1016/j.nbd.2014.11.012. [DOI] [PubMed] [Google Scholar]
- Bhalala US, Koehler RC, Kannan S, 2015. Neuroinflammation and neuroimmune dysregulation after acute hypoxic-ischemic injury of developing brain. Front. Pediatr. 10.3389/fped.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Lanza Cariccio V, Mohammadi N, Venza M, Venza I, Teti G, Beninati C, 2014. The interleukin-1ß/CXCL1/2/neutrophil axis mediates host protection against group B streptococcal infection. Infect. Immun. 82, 4508–4517. 10.1128/IAI.02104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratek E, Ziembowicz A, Bronisz A, Salinska E, 2018. The activation of group II metabotropic glutamate receptors protects neonatal rat brains from oxidative stress injury after hypoxia-ischemia. PLoS One 13, e0200933. 10.1371/journal.pone.0200933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratek E, Ziembowicz A, Salinska E, 2020. N-Acetylaspartylglutamate (NAAG) pretreatment reduces hypoxic-ischemic brain damage and oxidative stress in neonatal rats. Antioxidants 9, 877. 10.3390/antiox9090877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Lin S, Rhodes PG, 2002. Neuroprotective effects of N-acetylaspartylglutamate in a neonatal rat model of hypoxia-ischemia. Eur. J. Pharmacol. 437, 139–145. 10.1016/S0014-2999(02)01289-X. [DOI] [PubMed] [Google Scholar]
- Carlson T, Kroenke M, Rao P, Lane TE, Segal B, 2008. The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 205, 811–823. 10.1084/jem.20072404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, 1992. Excitotoxic cell death. J. Neurobiol. 23, 1261–1276. 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Monaghan DT, Ottersen OP, Storm-Mathisen J, 1987. Anatomical organization of excitatory amino acid receptors and their pathways. Trends Neurosci. 10.1016/0166-2236(87)90172-X. [DOI] [Google Scholar]
- Darnall RA, Chen X, Nemani KV, Sirieix CM, Gimi B, Knoblach S, McEntire BL, Hunt CE, 2017. Early postnatal exposure to intermittent hypoxia in rodents is proinflammatory, impairs white matter integrity, and alters brain metabolism. Pediatr. Res. 82, 164–172. 10.1038/pr.2017.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditelberg JS, Sheldon RA, Epstein CJ, Ferriero DM, 1996. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr. Res. 39, 204–208. 10.1203/00006450-199602000-00003. [DOI] [PubMed] [Google Scholar]
- Elroy-Stein O, Bernstein Y, Groner Y, 1986. Overproduction of human Cu/Zn-superoxide dismutase in transfected cells: extenuation of paraquat-mediated cytotoxicity and enhancement of lipid peroxidation. EMBO J. 5, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein CJ, Avraham KB, Lovett M, Smith S, Elroy-Stein O, Rotman G, Bry C, Groner Y, 1987. Transgenic mice with increased cu/Zn-superoxide dismutase activity: animal model of dosage effects in down syndrome. Proc. Natl. Acad. Sci. U. S. A. 84, 8044–8048. 10.1073/pnas.84.22.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fern R, Möller T,2000.Rapidischemiccelldeathinimmature oligodendrocytes: a fatal glutamate release feedback loop. J. Neurosci. 20, 34–42. 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferriero DM, 2004. Neonatal brain injury. N. Engl. J. Med. 351, 1985–1995. 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- Ferriero DM, Miller SP, 2010. Imaging selective vulnerability in the developing nervous system. J. Anat. 217, 429–435. 10.1111/j.1469-7580.2010.01226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton HJ, Ditelberg JS, Chen SF, Sarco DP, Chan PH, Epstein CJ, Ferriero DM, 1998. Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann. Neurol. 44, 357–364. 10.1002/ana.410440311. [DOI] [PubMed] [Google Scholar]
- Ghadge GD, Slusher BS, Bodner A, Dal Canto M, Wozniak K, Thomas AG, Rojas C, Tsukamoto T, Majer P, Miller RJ, Monti AL, Roos RP, 2003. Glutamate carboxypeptidase II inhibition protects motor neurons from death in familial amyotrophic lateral sclerosis models. Proc. Natl. Acad. Sci. U. S. A. 100, 9554–9559. 10.1073/pnas.1530168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, 1992. Reactive oxygen species and the central nervous system. J. Neurochem. 59, 1609–1623. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Scanziani M, Gähwiler BH,Gerber U, 2000.Acutedecreaseinnet glutamate uptake during energy deprivation. Proc. Natl. Acad. Sci. U. S. A. 97, 5610–5615. 10.1073/pnas.97.10.5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, Fatemi A, Wilson MA, Northington F, 2011. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 10.1016/S1474-4422(11)70016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan S, Dai H, Navath RS, Balakrishnan B, Jyoti A, Janisse J, Romero R, Kannan RM, 2012. Dendrimer-based postnatal therapy for neuroinflammation and cerebral palsy in a rabbit model. Sci. Transl. Med. 4 (130) 10.1126/scitranslmed.3003162,130ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH, 2009. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp. Neurol. 220, 44–56. 10.1016/j.expneurol.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Barry B, Hickey WF, 2001. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses 1. J. Immunol. 166, 4634–4643. 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Lesniak WG, Mishra MK, Jyoti A, Balakrishnan B, Zhang F, Nance E, Romero R, Kannan S, Kannan RM, 2013. Biodistribution of fluorescently labeled PAMAM dendrimers in neonatal rabbits: effect of neuroinflammation. Mol. Pharm. 10, 4560–4571. 10.1021/mp400371r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YK, Liu GR, Zhou XG, Cai AQ, 2010. Experimental hypoxic-ischemic encephalopathy: comparison of apparent diffusion coefficients and proton magnetic resonance spectroscopy. Magn. Reson. Imaging 28, 487–494. 10.1016/j.mri.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Li H, Li Q, Du X, Sun Y, Wang X, Kroemer G, Blomgren K, Zhu C, 2011. Lithium-mediated long-term neuroprotection in neonatal rat hypoxia-ischemia is associated with antiinflammatory effects and enhanced proliferation and survival of neural stem/progenitor cells. J. Cereb. Blood Flow Metab. 31, 2106–2115. 10.1038/jcbfm.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LP, Darnall L, Hu T, Choi K, Lane TE, Ransohoff RM, 2010. Myelin repair is accelerated by inactivating CXCR2 on nonhematopoietic cells. J. Neurosci. 30, 9074–9083. 10.1523/JNEUROSCI.1238-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JB, Yourick DL, Slusher BS, Robinson MB, Meyerhoff JL, 2005. Inhibition of glutamate carboxypeptidase II (NAALADase) protects against dynorphin A-induced ischemic spinal cord injury in rats. Eur. J. Pharmacol. 508, 115–122. 10.1016/j.ejphar.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Lucke AM, Shetty AN, Hagan JL, Walton A, Stafford TD, Chu ZD, Rhee CJ, Kaiser JR, Sanz Cortes M, 2019. Early proton magnetic resonance spectroscopy during and after therapeutic hypothermia in perinatal hypoxic–ischemic encephalopathy. Pediatr. Radiol. 49 10.1007/s00247-019-04383-8. [DOI] [PubMed] [Google Scholar]
- Mallard C, Tremblay M-E, Vexler ZS, 2018. Microglia and neonatal brain injury. Neuroscience. 10.1016/j.neuroscience.2018.01.023. [DOI] [PMC free article] [PubMed]
- McLean C, Ferriero D, 2004. Mechanisms of hypoxic-ischemic injury in the term infant. Semin. Perinatol. 28, 425–432. [DOI] [PubMed] [Google Scholar]
- Mike JK, Pathipati P, Sheldon RA, Ferriero DM, 2020. Changes in arginase isoforms in a murine model of neonatal brain hypoxia–ischemia. Pediatr. Res. 1–8 10.1038/s41390-020-0978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza MA, Ritzel R, Xu Y, McCullough LD, Liu F, 2015. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J. Neuroinflammation 12. 10.1186/s12974-015-0251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaldo P, Lally PJ, Oliveira V, Swamy R, Mendoza J, Atreja G, Kariholu U, Shivamurthappa V, Liow N, Teiserskas J, Pryce R, Soe A, Shankaran S, Thayyil S, 2019. Therapeutic hypothermia initiated within 6 hours of birth is associated with reduced brain injury on MR biomarkers in mild hypoxic-ischaemic encephalopathy: a non-randomised cohort study. Arch. Dis. Child. Fetal Neonatal Ed. 104, F515–F520. 10.1136/archdischild-2018-316040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti R, Pansiot J, Bettati D, Strazielle N, Ghersi-Egea J-F, Damante G, Fleiss B, Titomanlio L, Gressens P, 2015. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 9, 40. 10.3389/fnins.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PM, 1997. Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin. Hematol. [PubMed]
- Nance E, Porambo M, Zhang F, Mishra MK, Buelow M, Getzenberg R, Johnston M, Kannan RM, Fatemi A, Kannan S, 2015. Systemic dendrimer-drug treatment of ischemia-induced neonatal white matter injury. J. Control. Release 214, 112–120. 10.1016/j.jconrel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance E, Zhang F, Mishra MK, Zhang Z, Kambhampati SP, Kannan RM, Kannan S, 2016. Nanoscale effects in dendrimer-mediated targeting of neuroinflammation. Biomaterials 101, 96–107. 10.1016/j.biomaterials.2016.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale JH, Olszewski RT, Zuo D, Janczura KJ, Profaci CP, Lavin KM, Madore JC, Bzdega T, 2011. Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J. Neurochem. 10.1111/j.1471-4159.2011.07338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale H Joseph, Yamamoto Tatsuo, January 2020. N-acetylaspartylglutamate (NAAG) and glutamate carboxypeptidase II: An abundant peptide neurotransmitter-enzyme system with multiple clinical applications. Prog Neurobiol. 184 10.1016/j.pneurobio.2019.101722. [DOI] [PubMed] [Google Scholar]
- Nemeth CL, Drummond GT, Mishra MK, Zhang F, Carr P, Garcia MS, Doman S, Fatemi A, Johnston MV, Kannan RM, Kannan S, Wilson MA, 2017. Uptake of dendrimer-drug by different cell types in the hippocampus after hypoxic–ischemic insult in neonatal mice: effects of injury, microglial activation and hypothermia. Nanomed. Nanotechnol. Biol. Med. 13, 2359–2369. 10.1016/j.nano.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netto CA, Sanches E, Odorcyk FK, Duran-Carabali LE, Weis SN, 2017. Sex-dependent consequences of neonatal brain hypoxia-ischemia in the rat. J. Neurosci. Res. 10.1002/jnr.23828. [DOI] [PubMed] [Google Scholar]
- Pazos MR, Mohammed N, Lafuente H, Santos M, Martínez-Pinilla E, Moreno E, Valdizan E, Romero J, Pazos A, Franco R, Hillard CJ, Alvarez FJ, Martínez-Orgado J, 2013. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: role of 5HT(1A) and CB2 receptors. Neuropharmacology 71, 282–291. 10.1016/j.neuropharm.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Potter MC, Wozniak KM, Callizot N, Slusher BS, 2014. Glutamate carboxypeptidase II inhibition behaviorally and physiologically improves pyridoxine-induced neuropathy in rats. PLoS One 9. 10.1371/journal.pone.0102936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn KA, Watkins CC, Alt J, Rais R, Stathis M, Grishkan I, Crainiceau CM, Pomper MG, Rojas C, Pletnikov MV, Calabresi PA, Brandt J, Barker PB, Slusher BS, Kaplin AI, 2012. Inhibition of glutamate carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 109, 20101–20106. 10.1073/pnas.1209934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JE, Vannucci RC, Brierley JB, 1981. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 9, 131–141. 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Rosenkrantz TS, Hussain Z, Fitch RH, 2019. Sex differences in brain injury and repair in newborn infants: clinical evidence and biological mechanisms. Front. Pediatr. 7, 211. 10.3389/fped.2019.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy M, Richard JF, Dumas A, Vallières L, 2012. CXCL1 can be regulated by IL-6 and promotes granulocyte adhesion to brain capillaries during bacterial toxin exposure and encephalomyelitis. J. Neuroinflammation 9. 10.1186/1742-2094-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šácha P, Zámečník J,Bařinka C, Hlouchová K, Vícha A,Mlčochová P,Hilgert I, Eckschlager T, Konvalinka J, 2007. Expression of glutamate carboxypeptidase II in human brain. Neuroscience 144, 1361–1372. 10.1016/j.neuroscience.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Sharma A, Liaw K, Sharma R, Zhang Z, Kannan S, Kannan RM, 2018a. Targeting mitochondrial dysfunction and oxidative stress in activated microglia using dendrimer-based therapeutics. Theranostics 8, 5529–5547. 10.7150/thno.29039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Porterfield JE, Smith E, Sharma R, Kannan S, Kannan RM, 2018b. Effect of mannose targeting of hydroxyl PAMAM dendrimers on cellular and organ biodistribution in a neonatal brain injury model. J. Control. Release 283, 175–189. 10.1016/J.JCONREL.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, Almli L, Ferriero DM, 2002. Copper/zinc superoxide dismutase transgenic brain in neonatal hypoxia-ischemia. In: Methods in Enzymology. Academic Press Inc., pp. 389–397. 10.1016/S0076-6879(02)53063-9 [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Windsor C, Lee BS, Arteaga Cabeza O, Ferriero DM, 2017. Erythropoietin treatment exacerbates moderate injury after hypoxia-ischemia in neonatal superoxide dismutase transgenic mice. Dev. Neurosci. 39, 228–237. 10.1159/000472710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, Windsor C, Ferriero DM, 2019. Strain-related differences in mouse neonatal hypoxia-ischemia. In: Developmental Neuroscience. Karger SAG, pp. 490–496. 10.1159/000495880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XCM, Tortella FC, Wozniak KM, Yudkoff M, Potter BM, Jackson PF, 1999. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 5, 1396–1402. 10.1038/70971. [DOI] [PubMed] [Google Scholar]
- Smith ES, Porterfield JE, Kannan RM, 2019. Leveraging the interplay of nanotechnology and neuroscience: designing new avenues for treating central nervous system disorders. Adv. Drug Deliv. Rev. 10.1016/j.addr.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillson AB, Russell JW, 2003. Metabotropic glutamate receptor regulation of neuronal cell death. Exp. Neurol. 184, 97–105. 10.1016/j.expneurol.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Tsuji Shunichiro, Di Martino E, Mukai T, Tsuji, Shoko, Murakami T, Harris RA, Blomgren K, Åden U, 2020. Aggravated brain injury after neonatal hypoxic ischemia in microglia-depleted mice. J. Neuroinflammation 17, 111. https://doi. org/10.1186/s12974–020-01792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto T, Wozniak KM, Slusher BS, 2007. Progress in the discovery and development of glutamate carboxypeptidase II inhibitors. Drug Discov. Today. 10.1016/j.drudis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Van Hemelrijck A, Hachimi-Idrissi S, Sarre S, Ebinger G, Michotte Y, 2005. Neuroprotective effect of N-acetyl-aspartyl-glutamate in combination with mild hypothermia in the endothelin-1 rat model of focal cerebral ischaemia. J. Neurochem. 95, 1287–1297. 10.1111/j.1471-4159.2005.03450.x. [DOI] [PubMed] [Google Scholar]
- Weis SN, Toniazzo AP, Ander BP, Zhan X, Careaga M, Ashwood P, Wyse ATS, Netto CA, Sharp FR, 2014. Autophagy in the brain of neonates following hypoxia-ischemiashowssex-and region-specificeffects.Neuroscience 256, 201–209. 10.1016/j.neuroscience.2013.10.046. [DOI] [PubMed] [Google Scholar]
- Yellowhair TR, Noor S, Maxwell JR, Anstine CV, Oppong AY, Robinson S, Milligan ED, Jantzie LL, 2018. Preclinical chorioamnionitis dysregulates CXCL1/CXCR2 signaling throughout the placental-fetal-brain axis. Exp. Neurol. 301, 110–119. 10.1016/j.expneurol.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Yellowhair TR, Newville JC, Noor S, Maxwell JR, Milligan ED, Robinson S, Jantzie LL, 2019. CXCR2 blockade mitigates neural cell injury following preclinical chorioamnionitis. Front. Physiol. 10, 324. 10.3389/fphys.2019.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Bassam B, Thomas AG, Williams M, Liu J, Nance E, Rojas C, Slusher BS, Kannan S, 2016a. Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiol. Dis. 94, 116–128. 10.1016/j.nbd.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Nance E, Alnasser Y, Kannan R, Kannan S, 2016b. Microglial migration and interactions with dendrimer nanoparticles are altered in the presence of neuroinflammation. J. Neuroinflammation 13, 65. 10.1186/s12974-016-0529-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Zhao X, Sarva J, Kozikowski A, Neale JH, Lyeth BG, 2005. NAAG peptidase inhibitor reduces acute neuronal degeneration and astrocyte damage following lateral fluid percussion TBI in rats. J. Neurotrauma 22, 266–276. 10.1089/neu.2005.22.266. [DOI] [PubMed] [Google Scholar]
- Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H, 2006. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J. Neurochem. 96, 1016–1027. 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.