

Abstract
O-GlcNAcylation is a reversible post-translational protein modification that regulates fundamental cellular processes including immune responses and autoimmunity. Previously, we showed that hyperglycemia increases O-GlcNAcylation of the transcription factor, nuclear factor kappaB c-Rel at serine residue 350 and enhances the transcription of the c-Rel-dependent proautoimmune cytokines interleukin-2, interferon gamma and granulocyte macrophage colony stimulating factor in T cells. c-Rel also plays a critical role in the transcriptional regulation of forkhead box P3 (FOXP3)—the master transcription factor that governs development and function of Treg cells. Here we show that the regulatory effect of c-Rel O-GlcNAcylation is gene-dependent, and in contrast to its role in enhancing the expression of proautoimmune cytokines, it suppresses the expression of FOXP3. Hyperglycemia-induced O-GlcNAcylation-dependent suppression of FOXP3 expression was found in vivo in two mouse models of autoimmune diabetes; streptozotocin-induced diabetes and spontaneous diabetes in nonobese diabetic mice. Mechanistically, we show that both hyperglycemia-induced and chemically enhanced cellular O-GlcNAcylation decreases c-Rel binding at the FOXP3 promoter and negatively regulates FOXP3 expression. Mutation of the O-GlcNAcylation site in c-Rel, (serine 350 to alanine), augments T cell receptor-induced FOXP3 expression and resists the O-GlcNAcylation-dependent repression of FOXP3 expression. This study reveals c-Rel S350 O-GlcNAcylation as a novel molecular mechanism inversely regulating immunosuppressive FOXP3 expression and proautoimmune gene expression in autoimmune diabetes with potential therapeutic implications.
Keywords: autoimmunity, NF-kappaB, transcription, Treg cells, type 1 diabetes
Introduction
O-GlcNAcylation is a post-translational modification, which involves the enzymatic addition of an N-acetylglucosamine (GlcNAc) monosaccharide subunit to serine and threonine residues (Hart 2019). O-GlcNAcylation is a highly dynamic process that regulates protein-DNA and protein–protein interactions, transcription, translation and cellular functions. This modification has been identified on over 4000 proteins to date and can occur both in the cytoplasm and in the nucleus (Ma and Hart 2014). A unique feature of O-GlcNAcylation, compared with other post-translational modifications such as phosphorylation and ubiquitination, is that this process is catalyzed by a single set of conserved enzymes: O-GlcNAc transferase (OGT), which mediates addition, and O-GlcNAcase (OGA), which removes the modification (Hart et al. 2007; Hart and Akimoto 2009). Little is currently understood about the precise mechanisms that govern targeting of O-GlcNAcylation, but it has been widely speculated that adaptor/scaffold proteins direct OGT and OGA to their specific target residues (Hart et al. 2007; Janetzko and Walker 2014; Ma and Hart 2014). GlcNAc is a normal product of glucose metabolism with system-wide O-GlcNAcylation levels rising during conditions of hyperglycemia such as that in type 1 diabetes (Dennis et al. 2011; Levi et al. 2012; Makino et al. 2015).
Type 1 diabetes or autoimmune diabetes is a condition characterized by T cell mediated destruction of pancreatic β cells leading to failed insulin production and loss of blood glucose control (Melendez-Ramirez et al. 2010; Atkinson 2012). Though the overt symptoms of type 1 diabetes have been clearly identified, the underlying molecular mechanisms governing the autoimmune T cell activation and β cell destruction remain less clearly defined (Mathis et al. 2001; Gomez-Tourino et al. 2016). It has been demonstrated that activated CD4+ T cell in the pancreas drives a CD8+ cytotoxic T cell response that directly mediates β cell death (Arif et al. 2004; Coppieters et al. 2012). As in many T cell immune responses, this inflammatory cascade can be held in check by the activity of both resident and infiltrating FOXP3+ T regulatory (Treg) cells whose function is to suppress cytokine production and promote T cell anergy and/or death (Hull et al. 2017; Yu et al. 2018). Many of these T cell and Treg cell functions are controlled by the nuclear factor kappaB (NF-κB) family of transcription factors, which drives the expression of a large array of autoimmune and inflammatory genes (Oh and Ghosh 2013; Sun et al. 2013; Gerondakis et al. 2014). The NF-κB family is made up of five subunits—RelA, RelB, c-Rel, p105/p50 and p100/p52—that combinatorially form functional dimers capable of translocating into the nucleus and binding DNA (Ghosh et al. 1998). The c-Rel subunit in particular has been shown to be critical for T cell effector function (Hilliard et al. 2002; Visekruna et al. 2012). c-Rel is the predominant member of the NF-κB family activated following ligation of the T cell receptor (TCR) and controls the expression of several genes that regulate T cell activation and function (Liou et al. 1999; Rao et al. 2003; Bunting et al. 2007; Gilmore and Gerondakis 2011; Ramakrishnan et al. 2016). c-Rel has also been shown to be essential for the development of natural Treg cells, as it is required for expression of forkhead box P3 (FOXP3), the key transcription factor necessary for the development and function of Treg cells (Isomura et al. 2009; Grinberg-Bleyer et al. 2017).
The transcription of FOXP3 is dependent on multiple noncoding regulatory elements and multiple transcription factors binding to the 5′ regulatory region of FOXP3 gene (Lee and Lee 2018). It has a promoter region, and four conserved noncoding sequences (CNS0, CNS1, CNS2 and CNS3) located using comparative genomic alignment. The most recently discovered of the four conserved sequences is CNS0, which is located 5′ to the promoter and it is identified as a super enhancer element in an intron of a nearby gene (Kitagawa et al. 2017). The promoter, CNS1 and CNS2 are located within the first intron (Zheng et al. 2010), whereas CNS3 is located just after exon 1 (Zheng et al. 2010). c-Rel has been shown to bind to multiple regions within the FOXP3 promoter in T cells, two of which overlaps with the nuclear factor of activated T cells (NFAT) sites and another region near the transcription start site (Long et al. 2009; Ruan et al. 2009; Zheng et al. 2010). Additionally, c-Rel has been identified as an integral transcription factor in promoting the development of natural or thymic Treg cells (nTregs) and peripheral Tregs through binding conserved κB sites in the CNS3 region (Zheng et al. 2010).
c-Rel’s potential to regulate both T cell functions and Treg cell development poses it as a critical player in the molecular pathogenesis of T cell-mediated autoimmune conditions. Previously, we generated a c-Rel deficient nonobese diabetic (NOD) mouse model and showed the role of c-Rel in the development of autoimmune diabetes (Ramakrishnan et al. 2016). We found that c-Rel deficiency resulted in accelerated diabetes development in NOD mice, which was rescued by the supplementation of c-Rel competent Treg cells (Ramakrishnan et al. 2016). Our previous work also identified that c-Rel is modified by O-GlcNAcylation at serine residue 350 (Ramakrishnan et al. 2013). We found that hyperglycemia-induced and chemically enhanced O-GlcNAcylation of c-Rel at serine 350 increases the DNA binding capacity of c-Rel at CD28 responsive element (CD28RE), which is conserved in the promoters of key CD4+ T helper 1 (Th1) cytokines genes such as interleukin-2 (IL-2), interferon gamma (IFNG) and granulocyte macrophage colony stimulating factor (GM-CSF) (Ramakrishnan et al. 2013). Chemical inhibition of O-GlcNAcylation, or mutation of serine 350 to alanine, blocked the DNA binding of c-Rel at CD28RE and significantly decreased the expression of Th1 cytokine genes in T cells (Ramakrishnan et al. 2013).
In this study, because c-Rel has been shown to be critical for the expression of FOXP3 in T cells (Kim et al. 2007; Isomura et al. 2009; Long et al. 2009; Visekruna et al. 2010; Grigoriadis et al. 2011; Schuster et al. 2019), we studied how c-Rel O-GlcNAcylation affects FOXP3 expression. We provide in vitro, in vivo and detailed mechanistic studies to show that, unlike the positive regulatory action of c-Rel O-GlcNAcylation in Th1 proautoimmune cytokine expression, c-Rel negatively regulates FOXP3 expression. This suggests a dual, but reciprocal, role of serine 350 O-GlcNAcylation in transcriptional regulation by c-Rel in T cells.
Results
Enhanced O-GlcNAcylation reduces FOXP3 expression in T cells
Our prior study discovered the critical role of c-Rel S350 O-GlcNAcylation in enhancing TCR-induced expression of c-Rel-dependent proautoimmune genes, IL-2, IFNG and GM-CSF (Ramakrishnan et al. 2013). c-Rel has been shown to be critical for the expression of the transcription factor FOXP3, which regulates the development and function of immunosuppressive Treg cells. The role of O-GlcNAcylation in FOXP3 expression remains unknown. Here, we investigated the role of enhanced O-GlcNAcylation in the transcriptional control of FOXP3 expression. We chose the Treg-like cell line MT-2 with substantial expression of FOXP3 gene (Hamano et al. 2015). Because MT-2 cells do not express the CD3 receptor (Hamano et al. 2015), we used phorbol 12-myristate 13-acetate (PMA) and Ionomycin, termed (PMA/Ion) for stimulation, which bypass T cell surface receptors to activate PKC and downstream NF-κB (Chatila et al. 1989). We treated MT-2 cells with PMA/Ion and Thiamet-G (—a chemical inhibitor of the enzyme OGA) to enhance global O-GlcNAcylation (Yuzwa et al. 2008), and examined FOXP3 and IFNG expressions by quantitative polymerase chain reaction (qPCR). We observed a significant decrease in FOXP3 expression and a significant increase in IFNG expression under enhanced O-GlcNAcylation conditions (Figure 1A), suggesting an inverse regulatory role of O-GlcNAcylation at FOXP3 and IFNG promoters. We confirmed that the effect of Thiamet-G on gene expression was indeed mediated through OGA inhibition by genetic knockdown of OGA. We found that a lentiviral sh-OGA suppressed PMA/Ion-induced FOXP3 and enhanced IFNG expressions in MT-2 cells (Figure 1B). We also validated efficient suppression of OGA by western blotting (Figure 1C). To further validate this dual regulatory role of O-GlcNAcylation in gene expression, we also studied EL4 cells, a murine thymoma cell line known to express FOXP3 following TCR stimulation (Tone et al. 2008). We found that enhancing cellular O-GlcNAcylation by pretreatment of EL4 cells with Thiamet-G resulted in substantially diminished FOXP3 expression compared with TCR stimulation alone using anti-CD3 and anti-CD28 antibodies (Figure 1D, left). Consistent with our prior findings (Ramakrishnan et al. 2013), inhibition of OGA following Thiamet-G treatment significantly increased IFNG expression (Figure 1D, right). This inverse regulatory effect of O-GlcNAcylation was also found in EL4 cells stimulated with PMA/Ion and Thiamet-G (Figure 1E). Similar to MT-2 cells (Figure 1B and C), genetic suppression of OGA in EL4 cells using sh-OGA (Figure 1F), also showed significant decrease in FOXP3 expression and increase in IFNG expression (Figure 1G).
Fig. 1.
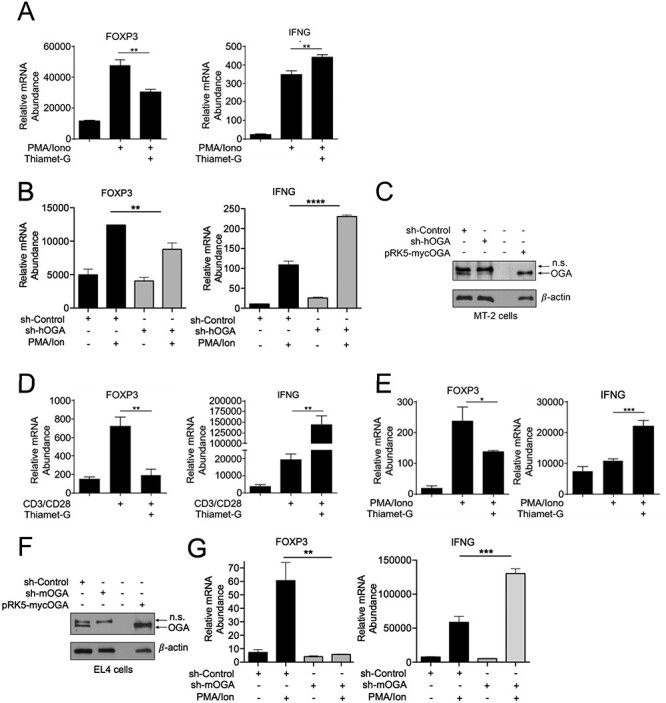
Increased O-GlcNAcylation reduces FOXP3 expression, (A) MT-2 cells (3 × 106) were treated with 50 μM Thiamet-G overnight in 6 cm plates and stimulated with 50 ng/mL PMA and 250 ng/mL Ionomycin for additional 18 h. Samples were then analyzed by qPCR to determine the abundance of indicated mRNAs relative to that of TFR. (B) MT-2 (3.0 × 106) were infected with lentivirus encoding human or mouse shOGA, respectively, or the control virus. Transduced cells were selected with 2 μg/mL puromycin for 48 h and then treated and gene expression was studied as in (A). (C) Suppression of O-GlcNAcase in MT-2 cells analyzed via western blotting. Transiently overexpressed OGA in HEK 293 T cells was used as a positive control to authenticate anti-OGA antibody and OGA detection. (D) EL4 thymoma cells (1 × 106) were treated with 50 μM Thiamet-G overnight in 6-well plates either uncoated or coated with 2 μg/mL anti-CD3 and anti-CD28 antibodies for 8 h. (E) EL4 thymoma cells were treated as in (D) and stimulated with 50 ng/mL PMA and 250 ng/mL Ionomycin for 18 h. (F–G) EL4 cells (3.0x106) were transduced with shOGA or control virus as in (B). (F) Suppression of OGA was assessed by western blotting. Transiently overexpressed OGA in HEK 293 T cells was used as a positive control to authenticate anti-OGA antibody and OGA detection. (G) Transduced EL4 thymoma cells were treated as in (E). (D, E, G) Samples were analyzed by qPCR to determine the abundance of indicated mRNAs relative to that of Ubiquitin Conjugating Enzyme E2 D2 (UBE2D2). Data are representative of three independent experiments, each performed in triplicates, presented as mean ± standard error of mean (SEM) (n = 3). P values were obtained by unpaired student t-test; **** P < 0.0001, *** P < 0.001, *#x002A; P < 0.01 and * P < 0.05.
c-Rel O-GlcNAcylation suppresses transcriptional activity at the FOXP3 promoter
To obtain direct evidence that the O-GlcNAcylation-mediated suppression of FOXP3 was due to c-Rel O-GlcNAcylation, first we utilized previously generated Jurkat T cells inducibly expressing wild-type or S350A mutant c-Rel (Ramakrishnan et al. 2013). We found that the S350A mutant c-Rel expressing cells showed higher basal and CD3/CD28-induced FOXP3 expression compared with wild-type cells (Figure 2A, left), while their TCR-induced IL-2 expression was entirely compromised (Figure 2A, right), despite equivalent levels of wild-type and mutant c-Rel (Figure 2B). Although initial studies on TCR-induced FOXP3 expression relied on Jurkat cells (Kim et al. 2007; Long et al. 2009; Dominguez-Villar et al. 2012), they exhibit diminutive transcriptional activation of FOXP3. Therefore, we studied transcriptional activation of FOXP3 in Treg like MT-2 cells. We transiently expressed wild-type and S350A mutant c-Rel with the luciferase reporter under CD28RE or FOXP3 promoter in MT-2 cells and examined PMA/Ion-induced reporter gene activity. We found that PMA/Ion stimulation enhanced CD28RE-dependent luciferase activity in MT-2 cells and that the addition of Thiamet-G, which increases global O-GlcNAcylation, further enhanced the PMA/Ion-induced luciferase activation (Figure 2C). Although the expression of wild-type c-Rel enhanced PMA/Ion-induced Thiamet-G-mediated luciferase activation, expression of S350A mutant c-Rel completely compromised the Thiamet-G-induced enhancement in CD28RE-dependent luciferase activation (Figure 2C). This suggests that c-Rel O-GlcNAcylation is critical in MT-2 cells to achieve peak CD28RE-dependent transcription. We also examined the effect of wild-type and S350A mutant c-Rel on FOXP3 promoter activity using a luciferase reporter gene assay (Zheng et al. 2010). Consistent with the O-GlcNAcylation-mediated decrease in FOXP3 expression (Figure 1), addition of Thiamet-G decreased the PMA/Ion-induced wild-type c-Rel-mediated transcriptional activation at FOXP3 promoter. Mutation of S350 in c-Rel enhanced its potential to transactivate FOXP3 promoter, which was further augmented following Thiamet-G treatment (Figure 2D).
Fig. 2.
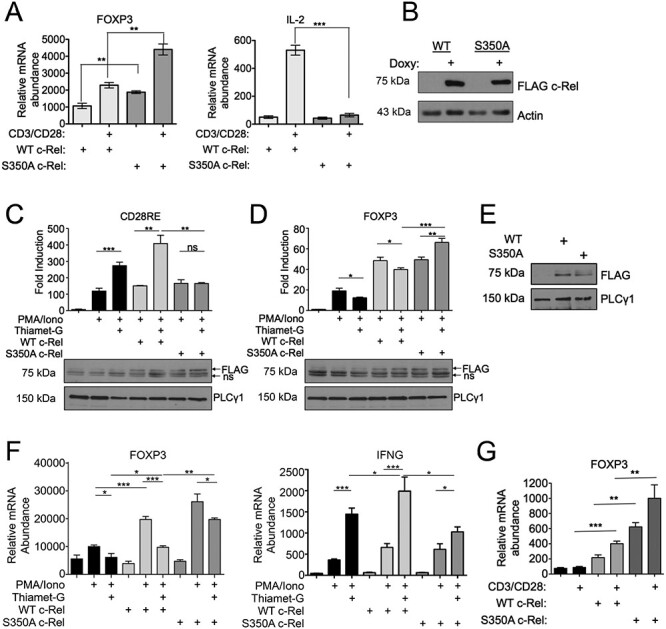
c-Rel S350 O-GlcNAcylation reduces FOXP3 expression, (A) Jurkat T-REx cells (1 × 106) were treated with 1 μg/mL doxycycline for 20 h to induce the expression of FLAG-tagged wild-type or S350A c-Rel. Cells were stimulated in 6-well plates coated with 2 μg/mL anti-CD3 and anti-CD28 antibodies for 3 h. Samples were then analyzed by qPCR to determine the abundance of indicated mRNAs relative to that of TFR. (B) Jurkat T-REx cells were treated with doxycycline as in (A) and FLAG-tagged wild-type or S350A c-Rel expression were examined by western blotting. (C and D) MT-2 cells (5 × 106) were electroporated with CD28RE (C) or FOXP3 CNS3 promoter (D) containing luciferase reporter plasmids and FLAG-tagged wild-type or S350A c-Rel as indicated. Twenty-four hours following transfection, cells were stimulated as indicated with 50 ng/mL PMA, 250 ng/mL Ionomycin and 50 μM Thiamet-G for an additional 24 h. Luciferase activity was assessed using dual luciferase assay system and values are presented as fold induction over luciferase vector alone-transfected sample. Data are representative of three independent experiments each done in triplicates and are presented as mean ± SEM. P values were obtained by unpaired student t-test. Ns—nonsignificant. Data in bar graphs are technical triplicates representative of three independent experiments. Bottom panels show western blotting of luciferase lysates probed for FLAG c-Rel expression. Ns—non specific. (E and F) MT-2 cells (5 × 106) were electroporated with plasmids containing empty vector, FLAG-tagged Wildtype c-Rel or FLAG-tagged S350A c-Rel. (E) Representative blot showing expression of FLAG-tagged wild-type or S350A c-Rel in electroporated MT-2 cell lysates. (F) Cells were stimulated 24 h post electroporation, with 50 ng/mL PMA and 250 ng/mL Ionomycin for 18 h. Samples were then analyzed by qPCR to determine the abundance of indicated mRNAs relative to that of Transferrin Receptor (TFR). (G) CD4+ T cells were isolated from c-Rel knockout C57BL/6J mice and 7.0 × 106 cells per condition were nucleofected with plasmids containing either empty vector, wild-type c-Rel, or S350A c-Rel. After 20 h, cells were stimulated with 2 μg/mL each of plate bound anti-CD3 and anti-CD28 antibodies for 6 h. Samples were then analyzed by qPCR to determine the abundance of FOXP3 mRNA relative to that of Transferrin Receptor (TFR). (A, F, G) Data are representative of three independent experiments, each performed in triplicates, presented as mean ± SEM (n = 3). P values were obtained by unpaired student t-test; *** P < 0.001, ** P < 0.01 * P < 0.05.
We also studied the effect of c-Rel S350A mutation in the expression of FOXP3 messenger RNA (mRNA) by qPCR. We expressed FLAG tagged wild-type c-Rel and S350A mutant c-Rel by lentiviral transduction in MT-2 cells and confirmed the expressions by anti-FLAG western blotting (Figure 2E). We treated the cells with PMA/Ion and found that the treatment increased FOXP3 expression in control and wild-type c-Rel expressing MT-2 cells and concurrent Thiamet-G treatment significantly decreased their FOXP3 expression. Interestingly, MT-2 cells expressing S350A mutant c-Rel showed substantial increase in PMA/Ion-induced FOXP3 expression, and treatment with Thiamet-G, showed only modest effect in suppressing FOXP3 expression (Figure 2F, left). This is in contrast to IFNG expression, where control and wild-type c-Rel expressing MT-2 cells showed a dramatic increase in IFNG expression over the S350A mutant expressing cells when co-treated with Thiamet-G (Figure 2F, right). To confirm that O-GlcNAcylation-dependent FOXP3 suppression is specifically regulated by O-GlcNAcylated c-Rel in primary T cells, we transiently expressed wild-type or S350A c-Rel in primary CD4+ T cells and assessed basal and TCR-induced FOXP3 expression. We found that S350A expressing primary CD4+ T cells exhibited increased FOXP3 expression, both basally and following TCR activation in comparison to cells expressing wild-type c-Rel illuminating the key role for c-Rel O-GlcNAcylation in suppressing FOXP3 expression (Figure 2G). These results show that c-Rel O-GlcNAcylation negatively regulates FOXP3 promoter activation and it has inverse transcriptional regulatory roles at CD28RE and FOXP3 CNS3 promoter regions.
Hyperglycemia-induced O-GlcNAcylation reduces FOXP3 expression in primary T cells in vitro and in vivo
To gain insight into the physiological significance of the O-GlcNAcylation-dependent suppression of FOXP3 expression, we studied the phenomenon in primary CD4+ T cells isolated from mice. CD4+ T cells were purified from spleen by magnetic sorting and subjected to TCR stimulation under physiological glucose (5 mM) and hyperglycemic conditions (30 mM) as well as high O-GlcNAcylation (100 μM PUGNAc—a chemical inhibitor of OGA) (Haltiwanger et al. 1998) conditions. We found that both hyperglycemia and the PUGNAc treatment, both of which enhance cellular O-GlcNAcylation, resulted in greatly reduced FOXP3 expression (Figure 3A). We used c-Rel knockout cells as a control for decreased FOXP3 expression, and as previously shown (Isomura et al. 2009; Visekruna et al. 2010) found greatly decreased FOXP3 expression in c-Rel deficient primary CD4+ T cells. To exclude the possible transient effects of acute high glucose treatment, we acclimatized the cells with high glucose for 48 h or longer and then studied its effect on FOXP3 expression. Similar to short-term high glucose treatment and OGA inhibition (Figure 3A), long-term high glucose treatment also significantly suppressed FOXP3 expression in primary CD4+ T cells (Figure 3B). To study the in vivo role of hyperglycemia-induced enhanced O-GlcNAcylation on FOXP3 suppression, first we utilized the streptozotocin (STZ)-induced diabetic mouse model. We assessed FOXP3 transcript levels in CD4+ T cells under pathological hyperglycemic conditions of STZ-diabetic mice and found that diabetic CD4+ T cells exhibited greatly diminished FOXP3 expression following ex vivo TCR stimulation (Figure 3C). This decrease in FOXP3 expression was recapitulated in freshly isolated splenocytes (Figure 3D) and CD4+ T cells (Figure 3E) from STZ-induced diabetic mice compared with normoglycemic controls.
Fig. 3.
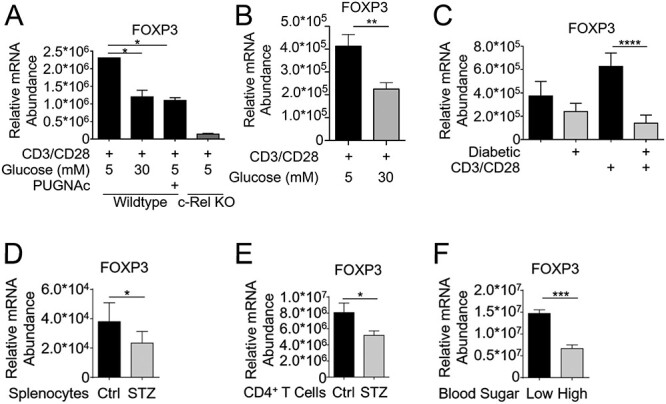
c-Rel O-GlcNAcylation reduces FOXP3 expression in primary T cells. (A) CD4+ T cells (1.5 × 106/condition) isolated from wild-type or c-Rel KO C57BL/6J mice were cultured for 6 h in either low-glucose media (5 mM glucose), low-glucose plus 100 μM PUGNAc or high-glucose (30 mM glucose) conditions. Cells were stimulated in 6-well plates coated with 2 μg/mL anti-CD3 and anti-CD28 antibodies for 6 h. (B) CD4+ T cells from wild-type or c-Rel KO C57BL/6J mice were cultured for 48 h in low or high glucose media and stimulated as in (A). (C) CD4+ T cells were isolated from normal glycemic or hyperglycemic (diabetic) NOD mice, and plated on 6-well plates at 1.5 × 106 cells/well either uncoated or coated with 2 μg/mL anti-CD3 and anti-CD28 antibodies for 6 h. (D) Whole splenocytes were isolated from control or STZ-treated mice and immediately processed for cDNA. (E) CD4+ T cells were isolated from C57BL/6 mice following 9 weeks of Streptozotocin treatment and immediately processed for cDNA. (F) CD4+ T cells were isolated from normal glycemic or hyperglycemic NOD mice, and immediately processed for cDNA. (A–F) Samples were analyzed by qPCR in triplicates to determine the abundance of FOXP3 mRNA relative to that of Ubiquitin Conjugating Enzyme E2 D2 (UBE2D2). Data are presented as mean ± SEM (A–E n = 3, F n = 6). P values were obtained by unpaired student t-test; **** P < 0.0001, *** P < 0.001, ** P < 0.01, * P < 0.05.
To determine if the CD4+ T cells isolated from a spontaneous type 1 diabetes model also had reduced FOXP3 expression, we examined the NOD mouse model. These mice have multiple mutations that result in the spontaneous lymphocyte infiltration into the pancreatic islets, and autoimmune destruction of the insulin-producing β cells (Wicker et al. 1987). Previously, we have shown that knockout of c-Rel on a NOD background results in over 80% reduction in FOXP3 expression and Treg cell numbers in NOD c-Rel−/− mice (Ramakrishnan et al. 2016). We isolated CD4+ T cells from NOD mice and processed for RNA without any culturing or stimulation. We observed that the endogenous FOXP3 expression in hyperglycemic c-Rel NOD mice was 3-fold lower compared with cells from normoglycemic NOD mice, suggesting that hyperglycemia suppressed FOXP3 transcription in diabetic NOD mice (Figure 3F).
Enhancement of c-Rel O-GlcNAcylation in diabetic mice correlates with reduction in FOXP3+ T cells
To study whether decreased FOXP3 transcription is translated to decrease in FOXP3+ T cells in vivo, we examined CD4+ FOXP3+ cells in diabetic NOD and STZ-treated mice. First, we compared FOXP3+ splenic Treg cells in age-matched normoglycemic (90–120 mg/dL) and hyperglycemic (500–600 mg/dL) NOD mice and found a statistically significant reduction in FOXP3+ cell numbers in hyperglycemic mice compared with normal glycemic mice (Figure 4A). We also utilized the STZ-induced diabetic mouse model to examine suppression of FOXP3+ cell numbers under hyperglycemic conditions. STZ-treated mice showing non-fasting blood glucose (NFBG) > 250 mg/dL were scored as diabetic. Mice were injected with insulin (Humulin—NPH, Eli Lilly and Co, Cambridge, MA, USA) thrice-weekly at 20 U/Kg starting 3 days post STZ-treatment to prevent catabolic weight loss. Similar to hyperglycemic NOD mice, we found a reduction in FOXP3+ splenic T cell numbers in the STZ-treated diabetic mice compared with control mice both at 4 and 9 weeks after STZ treatment (Figure 4B).
Fig. 4.
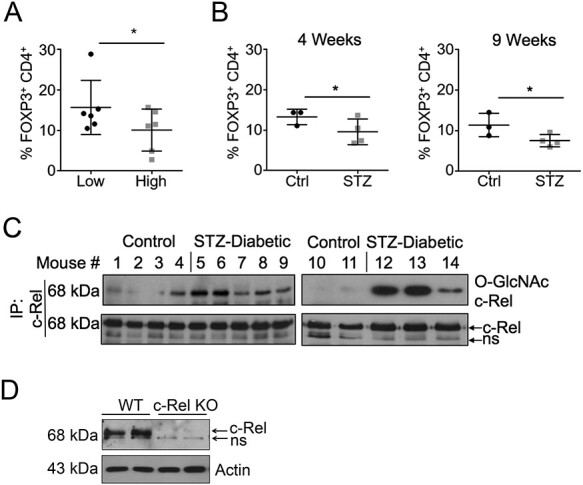
c-Rel O-GlcNAcylation is increased and Treg numbers are reduced in diabetic mice. (A) Splenocytes from normal or diabetic NOD mice were isolated and stained with anti-CD4 and anti-FOXP3 antibodies and analyzed by Flow Cytometry (n = 6) (B) Splenocytes from Control or C57BL/6 mice treated with STZ for 4 weeks (left) or 9 weeks (right) were isolated and stained as in (A). (C) Splenocytes were isolated from control or 9 weeks STZ-treated C57BL/6 mice were lysed, and c-Rel was immunoprecipitated from 25 to 30 × 106 cells. The O-GlcNAcylation state of c-Rel was probed using the anti-O-GlcNAc antibody RL2 and compared with measured mouse blood sugar: Mouse 1 = 130 mg/dL, Mouse 2 = 128 mg/dL, Mouse 3 = 157 mg/dL, Mouse 4 = 144 mg/dL, Mouse 5 = 509 mg/dL, Mouse 6 = 565 mg/dL, Mouse 7 = 531 mg/dL, Mouse 8 = 520 mg/dL, Mouse 9 = 479 mg/dL, Mouse 10 = 151 mg/dL, Mouse 11 = 129 mg/dL, Mouse 12 = 522 mg/dL, Mouse 13 = 511 mg/dL and Mouse 14 = 408 mg/dL. Bottom panel shows total c-Rel in the immunoprecipitates. ns—non specific. (D) Authenticity of anti-c-Rel antibody was validated using total lysates of control and c-Rel KO fibroblasts.
In order to confirm that c-Rel is O-GlcNAcylated during diabetes, we harvested the spleens from STZ-diabetic and nondiabetic controls at 9 weeks post development of diabetes. Single-cell suspensions of spleens were lysed and subjected to c-Rel Immunoprecipitation and probed with an anti-O-GlcNAc antibody. We observed enhanced c-Rel O-GlcNAcylation in the majority of the spleens of diabetic mice compared with nondiabetic mice (Figure 4C). The authenticity of c-Rel detection using anti-c-Rel antibody was confirmed using wild type and c-Rel KO cell lysates (Figure 4D). These data demonstrate that c-Rel O-GlcNAcylation is heightened during the pathological setting of type 1 diabetes, which correlates with decrease in FOXP3 expression and CD4+ FOXP3+ Treg cell numbers.
Hyper-O-GlcNAcylation reduces c-Rel binding to the FOXP3 promoter in vitro and in vivo
Our previous findings demonstrated that O-GlcNAcylation enhanced DNA binding of c-Rel to the CD28RE (Ramakrishnan et al. 2013). Based on this, we hypothesized that diminished FOXP3 expression under hyperglycemia and enhanced O-GlcNAcylation was due to decreased binding of O-GlcNAcylated c-Rel to the FOXP3 promoter. We studied the c-Rel binding site in the FOXP3 promoter (CBS-FP) before the transcription start site (TSS) and three κB-sites, κB2, κB1 and κB3, in the CNS3 region (schematic in Figure 5A and Supplementary Figure 1) for c-Rel binding at normal and hyper O-GlcNAcylation conditions (Zheng et al. 2010).
Fig. 5.
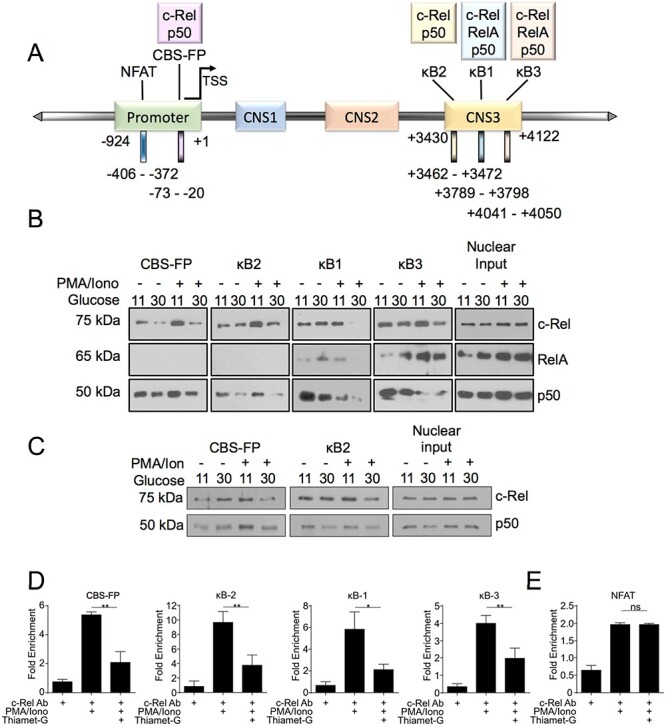
Hyper-O-GlcNAcylation reduces binding of c-Rel to FOXP3 promoter and enhancer elements. (A) Schematic representation of selected NF-κB binding sites and an NFAT binding site in the FOXP3 promoter and CNS3 region. The numbers below shows the locations of the promoter and CNS3 regions and binding sites examined (B) MT-2 cells were cultured in low (11 mM) or high glucose (30 mM) medium overnight and then treated with 50 ng/mL PMA and 250 ng/mL Ionomycin for 60 min. (C). MT2 cells were cultured in either low or high glucose medium for 48 h and then stimulated as in (B). (B, C) Nuclear and cytoplasmic extracts were prepared and 100 μg of nuclear proteins per sample was utilized in an in vitro pulldown assay using the indicated biotinylated oligonucleotides. The precipitated proteins as well as nuclear extracts as input controls were separated in SDS/PAGE gel and probed for c-Rel, RelA, and p50. (D–E) MT-2 cells (10 × 106) were pretreated overnight with Thiamet-G and then stimulated for 60 min with 50 ng/mL PMA and 250 ng/mL Ionomycin. Chromatin Immunoprecipitation was then carried out using anti-c-Rel antibody or nonspecific mouse IgG. The eluted chromatin was then analyzed by qPCR for the enrichment of indicated regions in anti-c-Rel precipitate compared with IgG precipitate as a control.
For the biochemical characterization of O-GlcNAcylated c-Rel binding to the FOXP3 promoter, we used Treg like MT-2 cells that express FOXP3 at high levels (Chen et al. 2006; Hamano et al. 2015). To assess how hyperglycemia and enhanced O-GlcNAcylation impacts c-Rel binding at the consensus NF-κB sequences in the FOXP3 promoter, we performed oligonucleotide pulldown assays using biotinylated minimal FOXP3 promoter regions. We found that hyperglycemia (treatment with 30 mM glucose, overnight) greatly decreased binding of c-Rel to all the sequences studied, CBS-FP and κB1, κB2, κB3 sites, following stimulation of MT-2 cells with PMA/Ion (Figure 5B, top panels). We did not find binding of NF-κB RelA subunit to CBS-FP and κB2 sites (Figure 5B, middle panels). Though hyperglycemia showed no effect on c-Rel nuclear translocation (Figure 5B, top panel, nuclear input), we found substantial increase in RelA nuclear translocation under hyperglycemic condition and PMA/Ion treatment further enhanced it (Figure 5B, middle panel, nuclear input). RelA showed modest binding to κB1 site that was completely abolished at high glucose and PMA/Ion treatment and its binding to κB3 region was found enhanced under hyperglycemic condition with minimal effect of hyperglycemia on PMA/Ion-induced RelA binding to κB3 site (Figure 5B, middle panels). We also found that hyperglycemia reduced binding of p50 at the CBS-FP and κB2 sites, suggesting the involvement of c-Rel: p50 heterodimers in driving FOXP3 expression in MT-2 cells through these sites. Both hyperglycemia and PMA/Ion treatment conditions decreased p50 binding to κB1 and κB3 sites, suggesting possible removal of the repressive p50 homodimers or p50/c-Rel heterodimers from these sites (Figure 5B, bottom panels). We also studied the binding of c-Rel to the FOXP3 promoters after acclimatizing the cells to hyperglycemic conditions for 48 h. Here we focused on DNA binding of c-Rel and studied two c-Rel binding sites (CBS-FP and κB2), where we saw predominantly c-Rel: p50 binding with no p65, by the oligonucleotide pull-down assay. Similar to overnight high glucose treatment, treatment of the cells with high glucose for 48 h also showed decreased c-Rel and p50 binding to these promoter sites (Figure 5C), further confirming hyperglycemia-induced suppression of c-Rel-dependent FOXP3 expression.
To further validate the differential c-Rel-DNA binding observed in the oligonucleotide pull-down assays under physiologically relevant endogenous conditions, we performed chromatin immunoprecipitation (ChIP) assays. We used an anti-c-Rel antibody as we described recently (de Jesus and Ramakrishnan 2020), to study binding of c-Rel to the FOXP3 promoter. Similar to oligonucleotide pulldown assays (Figure 5B and C), we found significant increase in PMA/Ion-induced c-Rel binding to all the four sites, κB1, κB2, κB3 and CBS-FP in ChIP assays (Figure 5D). Enhancement of global O-GlcNAcylation induced by Thiamet-G treatment significantly decreased PMA/Ion-induced c-Rel binding at all the sites studied (Figure 5D). Interestingly, NFAT site, which has been shown to bind to c-Rel (Ruan et al. 2009), while showing enrichment in PMA/Ion-induced c-Rel binding, showed no effect of Thiamet-G treatment, suggesting that c-Rel binding at this region is likely independent of O-GlcNAcylation (Figure 5E).
Mutation of the O-GlcNAcylation site S350 in c-Rel prevents its O-GlcNAcylation-mediated reduction in FOXP3 promoter binding
To confirm that O-GlcNAcylation-mediated reduction of c-Rel binding to the FOXP3 promoter is indeed due to c-Rel S350 O-GlcNAcylation, we performed ChIP assays using MT-2 cells expressing FLAG-tagged wild-type or S350A mutant c-Rel. MT-2 cells were transduced with lentivirus expressing wild-type or S350A c-Rel and their expression in transduced cells were confirmed by western blotting (Figure 6A). Transduced cells were treated with PMA/Ion and Thiamet-G and ChIP was performed using an anti-FLAG antibody.
Fig. 6.
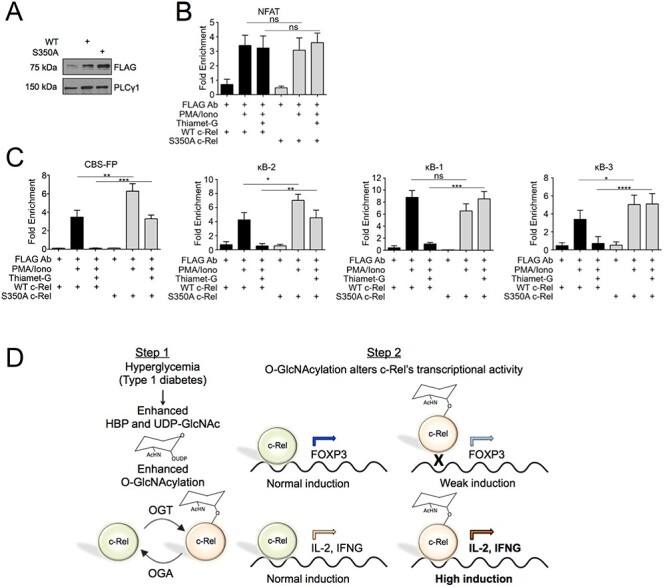
Unmodified c-Rel preferentially binds to FOXP3 promoter and enhancer elements (A–C) MT-2 cells (10 × 106) were infected with virus expression FLAG-tagged wild-type or FLAG-tagged S350A c-Rel (A) Expression of FLAG tagged c-Rel was examined by western blotting. (B and C) Forty-eight hours post transduction, cells were treated with 50 ng/mL PMA, 250 ng/mL Ionomycin and Thiamet-G as indicated in figure for 20 h. Chromatin Immunoprecipitation was then carried out using the M2 FLAG antibody and nonspecific mouse IgG. The eluted chromatin was then analyzed by qPCR for the enrichment of indicated regions in anti-c-Rel precipitate compared with IgG precipitate as a control. (D) Schematic model describing c-Rel O-GlcNAcylation-dependent dual regulation of T cell-dependent autoimmune gene expression in diabetes. Left. Genetic and environmental triggers result in autoimmune destruction of pancreatic β-cells and the development of hyperglycemia, which enhances hexosamine biosynthetic pathway, UDP-GlcNAc levels and O-GlcNAcylation. Right. Non-O-GlcNAcylated c-Rel-mediated transcription results in chromatin remodeling and gene expression at the FOXP3 and CD28RE gene loci. O-GlcNAcylation of c-Rel at S350 reduces FOXP3 induction, and highly induces CD28RE-dependent proautoimmune gene expression, that may promote autoimmunity.
Consistent with c-Rel ChIP (Figure 5D), we found no significant difference in FLAG-tagged wild-type c-Rel binding to the NFAT binding region following Thiamet-G treatment. The non-O-GlcNAcylatable S350A mutant c-Rel also showed no significant change in binding at the NFAT region both at basal and enhanced O-GlcNAcylation conditions, further suggesting that c-Rel binds to NFAT region independent of its O-GlcNAcylation status (Figure 6B). Enhanced O-GlcNAcylation induced by Thiamet-G addition caused a dramatic reduction in wild-type c-Rel binding to the CBS-FP site in the FOXP3 promoter, as well as to the κB2, κB1 and κB3 sites in the CNS3 region (Figure 6C). Non-O-GlcNAcylatable S350A mutant c-Rel S350A showed enhanced PMA/Ion-induced binding to the CBS-FP and κB2 sites. Addition of Thiamet-G showed a partial decrease in c-Rel S350A DNA binding, however, this decreased level of FOXP3 was comparable to the maximum level induced in wild-type c-Rel expressing cells (Figure 6C). We did not observe any suppressive effect of Thiamet-G addition on c-Rel S350A binding to the FOXP3 promoter’s κB1 or κB3 sites (Figure 6C). These results suggest that suppressive effect of enhanced O-GlcNAcylation on FOXP3 expression is mediated through c-Rel S350 O-GlcNAcylation. This identifies S350 O-GlcNAcylation of c-Rel as a novel mechanism that suppresses FOXP3 transcription under hyperglycemic and hyper O-GlcNAcylation conditions such as that in the diabetic mouse models studied here. Collectively, these data support our hypothesis that c-Rel S350 O-GlcNAcylation plays a dual inverse role in regulating T cell transcription by positively regulating proautoimmune gene expression and negatively regulating FOXP3 expression (Figure 6D, Schematic model).
Discussion
In this study, we discovered the novel negative regulatory role of c-Rel O-GlcNAcylation in the transcriptional control of FOXP3 expression in T cells. We found that hyperglycemic conditions enhance c-Rel S350 O-GlcNAcylation, which inhibits the binding of c-Rel to the FOXP3 promoter, leading to decreased FOXP3 expression. Supporting the pathophysiological relevance of these findings, we found decreased FOXP3 expression in two murine models of type 1 diabetes: chemically induced diabetes using STZ and the spontaneous diabetes in NOD mice, with reduced frequencies of FOXP3+ CD4+ T cells. Corroborating the direct role of c-Rel S350 O-GlcNAcylation in decreasing FOXP3 expression, our studies using non-O-GlcNAcylatable S350A mutant c-Rel showed no suppressive effect on FOXP3 expression even after treatment with Thiamet-G to enhance global O-GlcNAcylation.
Current literature on the subject of FOXP3 expression and Treg cell populations in type 1 diabetes models is incongruent. Some studies have observed no alteration of Treg cell numbers or FOXP3 expression in diabetes (Zavattari et al. 2004; Brusko et al. 2007; Mellanby et al. 2007), whereas others have noted a correlation between reduced FOXP3+ Treg cells numbers and diabetic progression in newly diagnosed children, latent type 1 diabetes in adults and cyclophosphamide-induced diabetic mice (Brode et al. 2006; Yang et al. 2006; Ryba-Stanislawowska et al. 2013; Aghili et al. 2015; Viisanen et al. 2019). Others have even noted elevation of Treg cell numbers in longer term diabetic studies (Zhen et al. 2012; Ryba-Stanislawowska et al. 2013). Despite the discrepancy in FOXP3 expression, in general, these studies are in agreement that the Treg cells present in the diabetic animals and patients show reduced suppressive function. Moreover, deficiency of FOXP3 resulting from its direct deletion (Chen et al. 2005) or through deletion of c-Rel (Ramakrishnan et al. 2016), results in enhanced diabetes incidence in animal models. Consistent with this suppressive role of FOXP3 expressing Treg cells in autoimmune diabetes, our data also show a substantial decrease in FOXP3 expression and Treg cell numbers in two mouse models of diabetes (Figures 3 and 4). Combined with our previous study (Ramakrishnan et al. 2013), it appears that O-GlcNAcylation of c-Rel may promote type 1 diabetes progression by: 1) enhancing proautoimmune cytokine production in conventional CD4+ T cells and 2) suppressing FOXP3 expression and development of Treg cells. Thus, this study uncovers a dual but inverse regulatory role of c-Rel O-GlcNAcylation that may lead to the acceleration of autoimmunity in type 1 diabetes, by augmenting autoreactive T cell function and diminishing FOXP3 expression and Treg cell function. This shows the intricacy of hyperglycemia-induced O-GlcNAcylation in the transcriptional control in a gene-dependent manner and links O-GlcNAcylation to the regulation of pathological signaling in the context of hyperglycemia.
This seminal study on the biochemistry and mechanism involving c-Rel O-GlcNAcylation in FOXP3 expression was mainly performed on mature T cells. However, it is plausible that O-GlcNAcylation may differentially impact c-Rel function in different T cell populations; i.e. developing thymocytes vs. naïve CD4+ T cells vs. antigen activated CD4+ T cells. In line with this, it has been shown that O-GlcNAcylation plays a role in T cell development and arrest of O-GlcNAc cycling decreases number of thymocytes (all sub populations; double negative, double positive and single positive) in a stem cell specific OGA knockout mouse model (Abramowitz and Hanover 2018; Abramowitz et al. 2019). Additional evidence for the complexity and context of O-GlcNAcylation comes from a study that discovered O-GlcNAcylation of the FOXP3 protein itself led to its stabilization (Liu et al. 2019). Thus, it is possible that heightened O-GlcNAcylation may initially augment Treg cell function by stabilizing preformed FOXP3 protein; yet eventually lead to a state of pathologically decreased Treg cells through the transcriptional repression of FOXP3 gene. It has also been shown that O-GlcNAcylation is essential for FOXP3 expression and inducible Treg cell differentiation in a rat model of autoimmune hepatitis (Hao et al. 2018). This warrants further comprehensive studies to dissect both transcriptional and translational regulation of FOXP3 expression by O-GlcNAcylation, especially in isolated naïve and inducible Treg cells as well as using transgenic mouse expressing S350A c-Rel.
Augmented T cell function serves as the core driver of a range of autoimmune and autoinflammatory diseases (Skapenko et al. 2005; Bluestone et al. 2015; Moulton and Tsokos 2015). Enhanced global O-GlcNAcylation leads to sustained activation of RAR-related orphan receptor γ t variant (RORγt) at the IL-17 gene promoter (Machacek et al. 2019) and increases NF-κB activity at IL-2, GMCSF and IFNG promoters in T cells (Ramakrishnan et al. 2013). Understanding such novel molecular mechanisms that regulate T cell-mediated autoimmunity is critical for developing new targeted therapies for type 1 diabetes for which the etiology is variant and diffuse at the population level (Maahs et al. 2010). Identifying common molecular mechanisms such as hyperglycemia induced enhanced c-Rel O-GlcNAcylation that occurs across the different etiologies represents an important step forward in finding new therapies for type 1 diabetes because eliminating the initial stimuli triggering the disease will likely prove difficult, if not impossible. Our previous study showing that c-Rel O-GlcNAcylation increases its binding to the promoters containing a CD28RE (Ramakrishnan et al. 2013) and this study showing that it suppresses c-Rel binding to FOXP3 promoter both in vitro and in vivo, reveals c-Rel O-GlcNAcylation as a unique molecular mechanism involved in both positive and negative regulation of c-Rel function. These data suggest that therapies targeting c-Rel O-GlcNAcylation may reduce autoimmune signaling and simultaneously enhance Treg cell function, ameliorating autoimmunity.
The findings of this study were focused on autoimmune diabetes as a model due to the link between hyperglycemia, enhanced global O-GlcNAcylation, augmented CD4+ T cell function and suppressed FOXP3 expression. It would be interesting to explore the role of c-Rel O-GlcNAcylation in other diseases involving hyperglycemia, such as type 2 diabetes and obesity, which may show that c-Rel S350 O-GlcNAcylation is a hyperglycemic condition-dependent, unified regulatory mechanism controlling transcription in T lymphocytes. Understanding such disease-specific molecular mechanism is critical to develop specific therapeutic agents mitigating side effects that may arise from global targeting of c-Rel.
Material and methods
Cells
Jurkat, EL4 and MT-2 cells were grown in RPMI media supplemented with 100 U/mL penicillin/streptomycin, 4 mM l-glutamine and 10% serum II plus (Sigma Aldrich, St. Louis, MO, USA). Primary CD4+ T Cells were treated in RPMI media supplemented with 100 U/mL penicillin/streptomycin, 4 mM l-glutamine and 10% fetal bovine serum (Sigma Aldrich, St. Louis, MO, USA).
Plasmids
The cDNAs for human wild-type c-Rel and the S350A mutant c-Rel with N-terminal FLAG tag were cloned into the pcDNA4 vector for transient expression. The S350A mutation was generated by PCR-based site-directed mutagenesis. The mammalian expression vector for human OGA, pRK5-myc-OGA, was kindly provided by Dr. Gerald W. Hart. The FOXP3 luciferase reporter construct was generously gifted by Alexander Rudensky’s lab (Zheng et al. 2010). Wild-type FLAG-c-Rel and S350A FLAG-c-Rel were cloned in pLM vector for lentiviral expression. Lentiviral expression plasmids encoding shRNA against OGA in pLKO.1 vector backbone was purchased from Sigma Aldrich, St. Louis, MO, USA (TRCN0000134040 for human OGA and TRCN0000248909 for mouseOGA).
Reagents and antibodies
Transfection of primary CD4+ T Cells was performed using a Nucleofector device (Lonza, Basel, Switzerland). Protein A and protein G agarose beads used for immunoprecipitation and Neutravidin beads for oligo pulldown were obtained from Thermo Fisher Scientific. Anti-O-linked N-acetylglucosamine antibodies, clone RL2, was purchased from Abcam, Cambridge, MA, USA and clone CTD110.6 was from Bio Legend, San Diego, CA, USA. Antibodies against RelA, p50 and PLCγ1 were obtained from Santa Cruz Biotechnology, Dallas, Texas, USA. Flow cytometry antibodies against mouse CD4 and FOXP3, intracellular permeabilizing and staining kit and anti c-Rel antibody were obtained from BioLegend. Anti-FLAG M2 antibody, anti-OGA, Thiamet-G, PMA and Ionomycin were from Sigma. PUGNAc was from Toronto Research Chemicals, Ontario, Canada.
Mice
The c-Rel knockout mouse line in C57BL/6 background was kindly provided by H.C. Liou (Weill Medical College of Cornell University, New York). C57BL/6J mice were from a colony maintained in-house. c-Rel NOD mice were generated and kept as described previously (Ramakrishnan et al. 2016). Mice were housed and handled in accordance with the National Institutes of Health (NIH) guidelines under protocols approved by the Institutional Animal Care and Use Committee.
Chemically induced diabetes mouse model
C57BL/6J mice were intraperitoneally (i.p.) injected with STZ (60 mg/kg) for 5 consecutive days after 6 hours fast each day. Insulin (Humulin—NPH, Eli Lilly and Co, Cambridge, MA, USA) treatments were performed at 20 U/kg 3 times a week for the duration of the study. Blood glucose was monitored for overt diabetes by tail vein puncture and sugar measurement was made using a glucometer. At the end of 2 months, mice were euthanized and splenocytes were collected for RNA analysis and flow cytometry.
Magnetic cell sorting
Total CD4+ T cells were purified from spleens of control and diabetic mice using L3T4 CD4 microbeads (Miltenyi Biotec, Auburn, CA, USA) following manufacturer’s instructions.
T cell stimulations
MT-2, Jurkat and EL4 cells were stimulated using 50 ng/mL PMA and 250 ng/mL Ionomycin at indicated time points. Jurkat, EL4 and purified primary CD4+ T cells were stimulated with 2 μg/mL anti-CD3 and anti-CD28 antibodies (BioLegend, San Diego, CA, USA) coated on Corning Costar six well plates.
Quantitative real time PCR
Total RNA was isolated from cells using the DNAaway RNA miniprep kit (Bio Basic, Markham, ON, Canada). RNA yields were quantified by NanoDrop spectrophotometer and 1 μg of total RNA was converted to cDNA using the High Capacity Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). qPCR was performed with cDNA corresponding to ~20–30 ng of RNA using Taqman gene expression assays (Thermo Fisher Scientific, Waltham, MA, USA). Experimental triplicate samples were run for biological replicates (n = 3) for all stimulation conditions. Gene expression was quantified as fold induction over control, and all values were normalized to the housekeeping genes transferrin receptor (TFR) for human samples or Ubiquitin Conjugating Enzyme E2 D2 (UBE2D2) for mouse samples.
Electroporation
MT-2 cells were suspended in plain RPMI medium with pcDNA4 empty vector, or vector encoding FLAG tagged wild-type c-Rel or S350A c-Rel. Cells were transfected by electroporation using Bio-Rad Electroporator set without resistance at 200v, and 960 μF.
Lentiviral production and transduction
Lentiviral packaging plasmids pMD2.G, psPAX2 and pLM vectors containing either wild-type FLAG-c-Rel or S350A FLAG-c-Rel, or pLKO.1 vectors containing mouse or human shOGA were transfected into HEK293 cells. After 48 h, supernatants were harvested and filtered using a 0.45 μM syringe filter. The virus was concentrated from 60 mL of supernatant by ultracentrifugation (16,500 RPM, 4°C, 90 min) and resuspended in 600 μL of PBS. MT-2 or EL4 cells were seeded at 1.5 million per well in a six well plate and spinfected with 100 μL/well of lentivirus in the presence of 10 μg/mL polybrene (2500 RPM, 30°C, 90 min). For shOGA transduction of MT-2 and EL4 cells, 48 h after infection, cells were selected with 2 μg/mL puromycin for 48 h. Suppression of O-GlcNAcase was confirmed via western blotting.
Immunoprecipitation and western blotting
Cells were lysed in a rotator at 4°C for 30 min in the lysis buffer (1.0% Triton-X100, 20 mM HEPES [pH 7.6], 0.1% SDS, 0.5% Sodium deoxycholate, 150 mM NaCl, 1 mM EDTA and complete protease inhibitor cocktail). Immunoprecipitation was carried out at 4°C for 2–3 hours using anti-c-Rel antibody. For western blot analysis, cell lysates as well as immunoprecipitates were resolved through 9% SDS–PAGE gels. Proteins from the gel were transferred onto nitrocellulose membranes, probed using relevant antibodies and visualized by enhanced chemiluminescence assay.
Oligonucleotide pulldown assay
Oligonucleotide pulldown assays were performed as previously described (Ramakrishnan et al. 2013). In brief, annealed, biotinylated oligos of key c-Rel binding sites within the FOXP3 promoter (CBS-FP) and CNS3 region (κB2, κB1 and κB3), were used to isolate active, DNA-binding NF-κB dimers from nuclear lysates using neutravidin beads (Thermo Fisher Scientific, Waltham, MA, USA). The following primer pairs were used:
CBS-FP: Fwd/Bio5′-TCC ACA CCG TAC AGC GTG GTT TTT CTT CTC GGT ATA AAA GCA AAG TTG TTT TT-3′
Rev/5′-AAA AAC AAC TTT GCT TTT ATA CCG AGA AGA AAA ACC ACG CTG TAC GGT GTG GA-3′
κB1: Fwd/Bio5′-TCA CCC CAC CTG GGC CAA GCC TGC TGC AGG ACA GGG CA-3′
Rev/5′-TGC CCT GTC CTG CAG CAG GCT TGG CCC AGG TGG GGT GA-3′
κB2: Fwd/Bio5′-CTT TCA GAT GGT TCC AAG GAG TTT GGA CAC CAG GGA CAC TGG CCT ACA C-3′
Rev/5′-GTG TAG GCC AGT GTC CCT GGT GTC CAA ACT CCT TGG AAC CAT CTG AAA G-3′
κB3: Fwd/Bio5′-ATT CAG CTT CTG AGA AAC CCA GTC AGA AAG GGA CGT CCC AAC AGA CAG TG-3′
Rev/5′-CAC TGT CTG TTG GGA CGT CCC TTT CTG ACT GGG TTT CTC AGA AGC TGA AT-3′
Chromatin immunoprecipitations
MT-2 cells were plated at 10 × 106 cells in 75 cm2 flasks per stimulus condition, and left untreated or treated with Thiamet-G and PMA/Ionomycin. Cells were pelleted and washed with warm PBS, then resuspended with 2 mM disuccinimidyl glutarate in PBS (+MgCl2) for cross-linking proteins in a 50 mL conical tube on a rocker for 45 min at room temperature. Cells were then washed with warm PBS and incubated for 15 min at room temperature in 1% formaldehyde for cross-linking DNA. Formaldehyde was quenched with 2.5 M glycine for 5 min. Cells were washed with PBS and pelleted at 2000 RPM for 5 min at 4°C. Cells were lysed in Farnham cell membrane lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, with protease inhibitors) for 15 min, and nuclear pellets were spun down at 10,000 RPM for 10 min at 4°C. The nuclear pellets were resuspended in 100 μL of RIPA buffer and chromatin was sheared at 4°C in a Sonicator® 3000 Ultrasonic Liquid Processor for 80 cycles of 8 s on and 40 s off at output 5. Magnetic beads were washed in 5 mg/mL BSA in PBS for blocking by pelleting beads using a magnetic block five times, and then incubated with 1 μg of c-Rel antibody per sample in an end-over-end rotator at 4°C overnight. Sonicated supernatants and antibody-coupled magnetic beads were incubated in an end-over-end rotator at 4°C overnight. Beads were pelleted and then washed with 1 mL LiCl IP wash buffer containing 10 mM Tris, pH 8.1, 0.25 M LiCl, 1% IGEPAL-CA 630, 1% Deoxycholic Acid and 1 mM EDTA, five times for 10 min at 4°C. Beads were then washed once with 1 mL TE buffer and resuspended in 200 μL IP elution buffer, and incubated on a heated shaker at 65°C at 900 RPM overnight. Extraction of DNA was performed using phenol/CHCl3/isoamyl alcohol and purified using Qiagen PCR Cleanup Kit. Quantitative RT-PCR was then performed on the eluates to amplify parts of the FOXP3 promoter. The following primer pairs were used:
NFAT: Fwd/5′-TTT CCC ATC CAC ACA TAG AG-3′
Rev/5′-AGA GGT GAG AGG TAT CAA TG-3′
CBS-FP: Fwd/5′-TGG ATT ATT AGA AGA GAG AGG TC-3′
Rev/5′-GAG AGC AGG GAC ACT CAC C-3′
κB2: Fwd/5′-AAG AGG CCC TAT GAA GCA GC-3′
Rev/5′-TCC TAG AAT CTC AAA ACC ACA-3′
κB1: Fwd/5′-GGG TCC CAG TAT CTG TGG AG-3′
Rev/5′-AGA GGG CAG ATA CCC CAC CC-3′
κB3: Fwd/5′-GTG TCC CAG AAA CAT CCC CC-3′
Rev/5′-GGA TGG CCT GAC TCA GCA AA-3′
Supplementary Material
Acknowledgements
We thank Dr. Alexander Rudensky for providing FOXP3 luciferase reporter plasmid and Dr. Gerald Hart for providing he mammalian expression vector for human OGA, pRK5-myc-OGA. The following reagent was obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH: MT-2 cells from Dr. Douglas Richman. We thank Dr. Keman Zhang for the pLM vector, Mr. Chieh Allen Lee for technical help, Dr. Sudhanshu Shukla for insightful discussions and Ms. Erin O’Kelly for proofreading the article.
Contributor Information
Tristan J de Jesus, Department of Pathology, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA.
Jeffrey A Tomalka, Department of Pathology, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA.
Joshua T Centore, Department of Pathology, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA.
Franklin D Staback Rodriguez, Department of Pathology, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA.
Ruchira A Agarwal, Department of Pathology, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA.
Angela R Liu, Department of Pathology, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA.
Timothy S Kern, Department of Ophthalmology, School of Medicine, University of California Irvine, 850 Health Sciences Road Irvine, CA 92697, USA.
Parameswaran Ramakrishnan, Department of Pathology, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA; Department of Biochemistry, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA; The Case Comprehensive Cancer Center, School of Medicine, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106, USA.
Funding
The National Institute of Health, NIH/The National Institute of Allergy and Infectious Diseases grants (R01 AI116730, R21 AI144264) and NIH/National Cancer Institute grant (R21 CA246194) to P.R. T.J.D. was supported by the National Institute of Health NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases T32 predoctoral training grant (T32AR007569) through the Department of Dermatology, Case Western Reserve University. J.T.C. was supported by the National Institute of Health NIH/National Eye Institute T32 predoctoral training grant (T32EY007157) through the Department of Ophthalmology, Case Western Reserve University. J.A.T. was supported by AAI Careers in Immunology Fellowship and the National Institute of Health NIH/NEI T32 postdoctoral training grant (T32EY007157) through the Department of Ophthalmology, Case Western Reserve University.
Authors’ contributions
T.J.D., J.A.T. and J.T.C. equally contributed to the work in planning and performing the experiments, analyzing the data and writing the manuscript. F.D.S., R.A. and A.R.L. performed the experiments. T.S.K. planned streptozotocin experiments and analyzed data. P.R. conceived the project, planned and performed the experiments, analyzed the data and edited and revised the manuscript.
Conflict of interest statement
P.R. has a patent on targeting c-Rel O-GlcNAcylation and uses thereof-US9696296B2. Other authors declare no competing financial interests.
Data and materials availability
All the data and methods necessary to reproduce this study are included in the manuscript and supplementary materials. Reagent request will be readily fulfilled following the materials transfer policies of Case Western Reserve University.
Abbreviations
CNS, conserved noncoding sequences; FOXP3, forkhead box P3, GlcNAc, N-acetylglucosamine; GM-CSF, granulocyte macrophage colony stimulating factor; IFNG, interferon gamma; IL-2, interleukin 2; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NFAT, nuclear factor of activated T cells; NOD, nonobese diabetic; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; PMA, phorbol 12-myristate 13-acetate; STZ, streptozotocin; TCR, T cell receptor; TG, Thiamet-G; Treg, T regulatory cells.
References
- Abramowitz LK, Hanover JA. 2018. T cell development and the physiological role of O-GlcNAc. FEBS Lett. 592(23):3943–3949. [DOI] [PubMed] [Google Scholar]
- Abramowitz LK, Harly C, Das A, Bhandoola A, Hanover JA. 2019. Blocked O-GlcNAc cycling disrupts mouse hematopoeitic stem cell maintenance and early T cell development. Sci Rep. 9(1):12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghili B, Amirzargar AA, Rajab A, Rabbani A, Sotoudeh A, Assadiasl S, Larijani B, Massoud A. 2015. Altered suppressor function of regulatory T cells in type 1 diabetes. Iran J Immunol. 12(4):240–251. [PubMed] [Google Scholar]
- Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, Roep BO, Peakman M. 2004. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. 113(3):451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA. 2012. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med. 2(11):a007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. 2015. T cells in the control of organ-specific autoimmunity. J Clin Invest. 125(6):2250–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brode S, Raine T, Zaccone P, Cooke A. 2006. Cyclophosphamide-induced type-1 diabetes in the NOD mouse is associated with a reduction of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 177(10):6603–6612. [DOI] [PubMed] [Google Scholar]
- Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, Haller M, Rockell J, Gottlieb P, Clare-Salzler M et al. 2007. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes. 56(3):604–612. [DOI] [PubMed] [Google Scholar]
- Bunting K, Rao S, Hardy K, Woltring D, Denyer GS, Wang J, Gerondakis S, Shannon MF. 2007. Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappa B transcription factor c-Rel. J Immunol. 178(11):7097–7109. [DOI] [PubMed] [Google Scholar]
- Chatila T, Silverman L, Miller R, Geha R. 1989. Mechanisms of T cell activation by the calcium ionophore ionomycin. J Immunol. 143(4):1283–1289. [PubMed] [Google Scholar]
- Chen S, Ishii N, Ine S, Ikeda S, Fujimura T, Ndhlovu LC, Soroosh P, Tada K, Harigae H, Kameoka J et al. 2006. Regulatory T cell-like activity of Foxp3+ adult T cell leukemia cells. Int Immunol. 18(2):269–277. [DOI] [PubMed] [Google Scholar]
- Chen Z, Herman AE, Matos M, Mathis D, Benoist C. 2005. Where CD4+CD25+ T reg cells impinge on autoimmune diabetes. J Exp Med. 202(10):1387–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, Roep BO, von Herrath. 2012. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 209(1):51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jesus TJ, Ramakrishnan P. 2020. NF-kappaB c-Rel dictates the inflammatory threshold by acting as a transcriptional repressor. iScience. 23(3):100876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MD, Schrufer TL, Bronson SK, Kimball SR, Jefferson LS. 2011. Hyperglycemia-induced O-GlcNAcylation and truncation of 4E-BP1 protein in liver of a mouse model of type 1 diabetes. J Biol Chem. 286(39):34286–34297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Villar M, Fernandez-Ponce C, Munoz-Suano A, Gomez E, Rodriguez-Iglesias M, Garcia-Cozar F. 2012. Up-regulation of FOXP3 and induction of suppressive function in CD4+ Jurkat T-cells expressing hepatitis C virus core protein. Clin Sci (Lond). 123(1):15–27. [DOI] [PubMed] [Google Scholar]
- Gerondakis S, Fulford TS, Messina NL, Grumont RJ. 2014. NF-kappaB control of T cell development. Nat Immunol. 15(1):15–25. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. 1998. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 16:225–260. [DOI] [PubMed] [Google Scholar]
- Gilmore TD, Gerondakis S. 2011. The c-Rel transcription factor in development and disease. Genes Cancer. 2(7):695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Tourino I, Arif S, Eichmann M, Peakman M. 2016. T cells in type 1 diabetes: Instructors, regulators and effectors: A comprehensive review. J Autoimmun. 66:7–16. [DOI] [PubMed] [Google Scholar]
- Grigoriadis G, Vasanthakumar A, Banerjee A, Grumont R, Overall S, Gleeson P, Shannon F, Gerondakis S. 2011. c-Rel controls multiple discrete steps in the thymic development of Foxp3+ CD4 regulatory T cells. PLoS One. 6(10):e26851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, Schmid RM, Klein U, Hayden MS, Ghosh S. 2017. NF-kappaB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell. 170(6):1096, e1013–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltiwanger RS, Grove K, Philipsberg GA. 1998. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-beta-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate. J Biol Chem. 273(6):3611–3617. [DOI] [PubMed] [Google Scholar]
- Hamano R, Wu X, Wang Y, Oppenheim JJ, Chen X. 2015. Characterization of MT-2 cells as a human regulatory T cell-like cell line. Cell Mol Immunol. 12(6):780–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao X, Li Y, Wang J, Ma J, Zhao S, Ye X, He L, Yang J, Gao M, Xiao F et al. 2018. Deficient O-GlcNAc glycosylation impairs regulatory T cell differentiation and notch signaling in autoimmune hepatitis. Front Immunol. 9:2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW. 2019. Nutrient regulation of signaling and transcription. J Biol Chem. 294(7):2211–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Akimoto Y. 2009. The O-GlcNAc modification. Essentials of Glycobiology. nd, A. Varki, R. D. Cummings et al. Cold Spring Harbor Laboratory Press; Cold Spring Harbor (NY). [Google Scholar]
- Hart GW, Housley MP, Slawson C. 2007. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 446(7139):1017–1022. [DOI] [PubMed] [Google Scholar]
- Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi SE, Liou HC, Hunter C, Chen YH. 2002. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest. 110(6):843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull CM, Peakman M, Tree TIM. 2017. Regulatory T cell dysfunction in type 1 diabetes: what's broken and how can we fix it? Diabetologia. 60(10):1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, Banerjee A, Proietto A, Gugasyan R, Wu L et al. 2009. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. 206(13):3001–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetzko J, Walker S. 2014. The making of a sweet modification: Structure and function of O-GlcNAc transferase. J Biol Chem. 289(50):34424–34432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kim HJ, Hurt EM, Chen X, Howard OMZ, Farrar WL. 2007. Functional and genomic analyses of FOXP3-transduced Jurkat-T cells as regulatory T (Treg)-like cells. Biochem Biophys Res Commun. 362(1):44–50. [DOI] [PubMed] [Google Scholar]
- Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, Yasuda K, Motooka D, Nakamura S, Kondo M et al. 2017. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat Immunol. 18(2):173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Lee GR. 2018. Transcriptional regulation and development of regulatory T cells. Exp Mol Med. 50(3):e456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi I, Segev Y, Priel E. 2012. Type 1 diabetes affects topoisomerase I activity and GlcNAcylation in rat organs: Kidney, liver and pancreas. Glycobiology. 22(5):704–713. [DOI] [PubMed] [Google Scholar]
- Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML. 1999. c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol. 11(3):361–371. [DOI] [PubMed] [Google Scholar]
- Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, Ball LE, Blazar BR, Farrar MA, Hogquist KA et al. 2019. The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nat Commun. 10(1):354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Park SG, Strickland I, Hayden MS, Ghosh S. 2009. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 31(6):921–931. [DOI] [PubMed] [Google Scholar]
- Ma J, Hart GW. 2014. O-GlcNAc profiling: From proteins to proteomes. Clin Proteomics. 11(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. 2010. Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am. 39(3):481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Saunders H, Zhang Z, Tan EP, Li J, Li T, Villar MT, Artigues A, Lydic T, Cork G et al. 2019. Elevated O-GlcNAcylation enhances pro-inflammatory Th17 function by altering the intracellular lipid microenvironment. J Biol Chem. 294(22):8973–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino A, Dai A, Han Y, Youssef KD, Wang W, Donthamsetty R, Scott BT, Wang H, Dillmann WH. 2015. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am J Physiol Cell Physiol. 309(9):C593–C599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis D, Vence L, Benoist C. 2001. Beta-cell death during progression to diabetes. Nature. 414(6865):792–798. [DOI] [PubMed] [Google Scholar]
- Melendez-Ramirez LY, Richards RJ, Cefalu WT. 2010. Complications of type 1 diabetes. Endocrinol Metab Clin North Am. 39(3):625–640. [DOI] [PubMed] [Google Scholar]
- Mellanby RJ, Thomas D, Phillips JM, Cooke A. 2007. Diabetes in non-obese diabetic mice is not associated with quantitative changes in CD4+ CD25+ Foxp3+ regulatory T cells. Immunology. 121(1):15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton VR, Tsokos GC. 2015. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest. 125(6):2220–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Ghosh S. 2013. NF-kappaB: Roles and regulation in different CD4(+) T-cell subsets. Immunol Rev. 252(1):41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan P, Clark PM, Mason DE, Peters EC, Hsieh-Wilson LC, Baltimore D. 2013. Activation of the transcriptional function of the NF-kappaB protein c-Rel by O-GlcNAc glycosylation. Sci Signal. 6(290):ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan P, Yui MA, Tomalka JA, Majumdar D, Parameswaran R, Baltimore D. 2016. Deficiency of nuclear factor-kappaB c-Rel accelerates the development of autoimmune diabetes in NOD mice. Diabetes. 65(8):2367–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Gerondakis S, Woltring D, Shannon MF. 2003. c-Rel is required for chromatin remodeling across the IL-2 gene promoter. J Immunol. 170(7):3724–3731. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. 2009. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 31(6):932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryba-Stanislawowska M, Skrzypkowska M, Mysliwiec M, Mysliwska J. 2013. Loss of the balance between CD4(+)Foxp3(+) regulatory T cells and CD4(+)IL17A(+) Th17 cells in patients with type 1 diabetes. Hum Immunol. 74(6):701–707. [DOI] [PubMed] [Google Scholar]
- Schuster M, Plaza-Sirvent C, Visekruna A, Huehn J, Schmitz I. 2019. Generation of Foxp3(+)CD25(−) regulatory T-cell precursors requires c-Rel and IkappaBNS. Front Immunol. 10:1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. 2005. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. 7(Suppl 2):S4–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC, Chang JH, Jin J. 2013. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. 34(6):282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. 2008. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 9(2):194–202. [DOI] [PubMed] [Google Scholar]
- Viisanen T, Gazali AM, Ihantola EL, Ekman I, Nanto-Salonen K, Veijola R, Toppari J, Knip M, Ilonen J, Kinnunen T. 2019. FOXP3+ regulatory T cell compartment is altered in children with newly diagnosed type 1 diabetes but not in autoantibody-positive at-risk children. Front Immunol. 10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visekruna A, Huber M, Hellhund A, Bothur E, Reinhard K, Bollig N, Schmidt N, Joeris T, Lohoff M, Steinhoff U. 2010. c-Rel is crucial for the induction of Foxp3(+) regulatory CD4(+) T cells but not T(H)17 cells. Eur J Immunol. 40(3):671–676. [DOI] [PubMed] [Google Scholar]
- Visekruna A, Volkov A, Steinhoff U. 2012. A key role for NF-kappaB transcription factor c-Rel in T-lymphocyte-differentiation and effector functions. Clin Dev Immunol. 2012:239368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker LS, Miller BJ, Coker LZ, McNally SE, Scott S, Mullen Y, Appel MC. 1987. Genetic control of diabetes and insulitis in the nonobese diabetic (NOD) mouse. J Exp Med. 165(6):1639–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZF, Zhou ZG, Tang WL, Huang G, Peng J, Li X, He L. 2006. Decrease of FOXP3 mRNA in CD4+ T cells in latent autoimmune diabetes in adult. Zhonghua Yi Xue Za Zhi. 86(36):2533–2536. [PubMed] [Google Scholar]
- Yu H, Paiva R, Flavell RA. 2018. Harnessing the power of regulatory T-cells to control autoimmune diabetes: Overview and perspective. Immunology. 153(2):161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ et al. 2008. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 4(8):483–490. [DOI] [PubMed] [Google Scholar]
- Zavattari P, Deidda E, Pitzalis M, Zoa B, Moi L, Lampis R, Contu D, Motzo C, Frongia P, Angius E et al. 2004. No association between variation of the FOXP3 gene and common type 1 diabetes in the Sardinian population. Diabetes. 53(7):1911–1914. [DOI] [PubMed] [Google Scholar]
- Zhen Y, Sun L, Liu H, Duan K, Zeng C, Zhang L, Jin D, Peng J, Ding W, Zhao Y. 2012. Alterations of peripheral CD4+CD25+Foxp3+ T regulatory cells in mice with STZ-induced diabetes. Cell Mol Immunol. 9(1):75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. 2010. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 463(7282):808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.