

Abstract
Sialuria, a rare inborn error of metabolism, was diagnosed in a healthy 12-year-old boy through whole exome sequencing. The patient had experienced mild delays of speech and motor development, as well as persistent hepatomegaly. Identification of the 8th individual with this disorder, prompted follow-up of the mother-son pair of patients diagnosed over 15 years ago. Hepatomegaly was confirmed in the now 19-year-old son, but in the 46-year-old mother a clinically silent liver tumor was detected by ultrasound and MRI. The tumor was characterized as an intrahepatic cholangiocarcinoma (IHCC) and DNA analysis of both tumor and normal liver tissue confirmed the original GNE mutation. As the maternal grandmother in the latter family died at age 49 years of a liver tumor, a retrospective study of the remaining pathology slides was conducted and confirmed it to have been an IHCC as well. The overall observation generated the hypothesis that sialuria may predispose to development of this form of liver cancer. As proof of sialuria in the grandmother could not be obtained, an alternate cause of IHCC cannot be ruled out. In a series of 102 patients with IHCC, not a single instance was found with the allosteric site mutation in the GNE gene. This confirms that sialuria is rare even in a selected group of patients, but does not invalidate the concern that sialuria may be a risk factor for IHCC.
Synopsis:
Sialuria is a rare inborn error of metabolism characterized by excessive synthesis and urinary excretion of free sialic acid with only minimal clinical morbidity in early childhood, but may be a risk factor for intrahepatic cholangiocarcinoma in adulthood.
Keywords: Sialuria, Sialic acids, UDP acetylglucosamine-2-epimerase, Hepatomegaly, Intrahepatic cholangiocarcinoma
1. Introduction
Sialuria (also called ‘French type sialuria: MIM 269921) is a rare autosomal dominant inborn error that results in overproduction of free sialic acid (N-acetyl-neuraminic acid, Neu5Ac). It was first observed and delineated in a French patient [1,2] and has since been reported in only four other unrelated single children [3–6] and in a mother-and-son pair in a fifth family [7] (Table 1).
Table 1.
Reported sialuria patients.
| # | Gender | GNE mRNA Varianta | GNE Protein Variantb | refSNP number in dbSNPc | Inheritance | Age in original report | Estimated Current Age (2015) | References |
|---|---|---|---|---|---|---|---|---|
| 1 | Maled | c.788G>T | p.R263L | rs121908623 | Unknown | 3 y | Deceasedd | [2,1] |
| 2 | Female | c.798C>T | p.R266W | rs121908621 | Unknown | 2 y | 30 y | [3,11] |
| 3 | Male | c.797G>A | p.R266Q | rs121908622 | Unknown | 10 mo | 27 y | [10,13,11] |
| 4 | Male | c.788G>T | p.R263L | rs121908623 | Unknown | 4.5 y | 27 y | [5,11] |
| 5 | Female | c.797G>A | p.R266Q | rs121908622 | Unknown | 7.5 y | 24 y | [6] |
| 6 | Male | c.797G>A | p.R266Q | rs121908622 | Familial | 4.5 y | 19.5 y | [7]; Patient 2 in current report |
| 7 | Female | c.797G>A | p.R266Q | rs121908622 | Unknown | 34 y | 47 y | [7]; Patient 3 in current report |
| 8 | Male | c.797G>A | p.R266Q | rs121908622 | De novo | 12 y | 14 y | Patient 1 in current report |
Abbreviations: y, years; mo, months.
GenBank Accession number NM_005476.5 (GNE mRNA Variant 2). This GenBank Accession number was used for reporting al previous sialuria variants. Note that recent GNE nomenclature changes based on the longest mRNA variant have been proposed and may be used to report future sialuria variants [22].
GenBank Accession number NP_005467 (hGNE1 protein isoform).
As reported in dbSNP http://www.ncbi.nlm.nih.gov/SNP/.
Patient died at age 30 (car accident).
Sialuria is characterized metabolically by excessive urinary excretion of free sialic acid (> 1 g/day), the consequence of failing feedback-inhibition of epimerase enzymatic activity in the rate limiting bifunctional enzyme, uridine diphosphate (UDP)-N-acetylglucosamine (GlcNAc) 2-epimerase/N-acetylmannosamine (ManNAc) kinase (GNE: EC 5.1.3.14/EC 2.7.1.60) by the downstream intermediate compound, cytidine monophosphate (CMP)-sialic acid [8–10].
The GNE gene (9p13.3) encodes the bifunctional GNE enzyme, which contains a small, but still incompletely outlined allosteric site, the binding site for CMP-sialic acid that loses its wild-type function in the heterozygous mutant genotype of sialuria [11,12]. Consequently, a heterozygous missense variant in the allosteric site of GNE in individuals with sialuria results in loss of feedback inhibition, causing overproduction, cytoplasmic accumulation and urinary excretion of large amounts of free sialic acid.
The clinical onset of sialuria in infancy is hardly recognizable because of the inconsistent, nonspecific and rather subtle features that may include mild neuromotor and cognitive developmental delay, hypotonia, slightly coarse facies, recurrent respiratory infections, transient mild anemia and equivocal or mild hepatomegaly [13,14]. Moreover, assaying urinary free sialic acid is not a routine laboratory test. Therefore, individuals with sialuria may go undiagnosed. Increased use of exome sequencing for diagnostic purposes may lead more frequently to making the diagnosis.
The purpose of this report is dual. First, we report on a newly diagnosed eighth patient with sialuria, identified by exome sequencing. Second, evaluation of the new patient prompted follow-up attention to the previously reported family, which found markedly increased hepatomegaly in the son (patient 2 in this report) and was of critical value to the diagnosis and treatment of intrahepatic cholangiocarcinoma (IHCC) in his affected mother (patient 3 in this report). The hepatic findings in the only adult with proven sialuria may also explain the nature and cause of the IHCC with fatal outcome at age 49 years in the maternal grandmother (patient 4 in this report).
2. Materials and methods
2.1. Patients
Patient 1 and his family were clinically examined at Greenwood Genetic Center (Greenwood, SC, USA). Patient 1 was also enrolled in the “Genomic Study of Medical Development or Congenital Problems of unknown Etiology in Pediatric Patients” at Duke University Medical Center (Division of Medical Genetics, Durham, NC, USA).
Patients 2, 3 and 4 were clinically examined at Ghent University Hospital and School of Medicine (Ghent, Belgium). Hepatic analysis for these patients was performed at Delta General Hospital (Department of Gastroenterology & Hepatology, Department of Pathology, Roeselare, Belgium) and University Hospital Leuven (Department of Hepatology, Leuven, Belgium).
The Institutional Review Boards of each respective center approved the described studies and the patients and/or their parents gave written informed consent.
2.2. Pathology
The liver biopsy of patient 2 and the liver resection specimen of patient 3 were processed for light microscopy and immunohistochemistry according to standard procedures. For patient 2, material was fixed in glutaraldehyde for electron microscopy. Hematoxylin and eosin (H&E) sections from a small incisional biopsy of patient 4, the mother of patient 3, prepared in 1989 were available for review; additionally, on this limited amount of tumor tissue immunohistochemistry for cytokeratin 7 (CK7) and cytokeratin 19 (CK 19) was performed, using standard methods.
2.3. Biochemical assays
Urinary sialic acid was measured according to Hommes et al. [15]. Briefly, free sialic acid was derivatized with 1.2-diamino-4.5-methylenedioxybenzene dihydrochloride (DMB). The fluorescent complex was separated by high pressure liquid chromatography (HPLC) using a ZORBAX column SB-C18 (4.6 × 100 mm × 3.5 OD). The amount of free sialic acid was quantitated using a standard curve. Urinary creatinine was calculated using the Jaffe method and final free sialic acid concentration was expressed as nmol sialic acid per mg creatinine.
2.4. GNE mutation analysis
DNA was extracted from peripheral blood samples from patient 1, his parents and two unaffected siblings. Exome sequencing was performed on all samples. To capture the coding regions, the 65-Mb Illumina TruSeq Exome Enrichment Kit (Illumina, San Diego, CA) was used. Sequencing was performed on the Illumina HiSeq 2000 platform (Illumina). Primers were designed to amplify the region of the GNE gene containing the c.797G>A (p.R266Q) alteration. Sequencing was performed by the Sanger dideoxy method using the Big Dye Terminator Cycle Sequencing Kit v3.1 on a ABI 3730xl automated sequencer (Applied Biosystems, Life Technologies, Foster City, CA). Data were collected and analyzed using Sequencher 4.5 DNA sequence assembly software (Gene Codes Corporation, Ann Arbor, MI).
2.5. Oncoscan assays
DNA was isolated from formalin-fixed, paraffin-embedded (FFPE) liver tissues from patient 3, both from the tumor tissue and adjacent normal tissue. The Oncoscan® assays (Affymetrix Inc., Santa Clara, CA) were performed on the normal and tumor liver DNA samples following the protocol recommended by the manufacturer. Briefly, the DNA was annealed with molecular inversion probes (MIP) that detect both the copy number and somatic mutations of specific genes at 58 °C overnight (16–18 h) after an initial denaturation at 95 °C for 5 min. Each sample was then split into two reactions and a gap fill reaction was performed for the (A/T) probes by adding dATP (A) and dTTP (T) in the first reaction and dGTP (G) and dCTP (C) for the (G/C) probes in the second reaction. Subsequently, exonuclease treatment digested the uncircularized MIP probes and genomic DNA. Linearization of the remaining circular MIP probes was then carried out using a cleavage enzyme and amplified by PCR. A second round of PCR amplification resulted in amplicons of 120-bp size, which were then digested with the HaeIII restriction enzyme. The smaller 44-bp digestion products were hybridized onto OncoScan® arrays for 16–18 h. Following hybridization, the arrays were stained and washed using the GeneChip® Fluidics Station 450 and loaded into the GeneChip® Scanner 3000 7G (Affymetrix Inc) where array fluorescence intensity was scanned to generate array images (DAT file). Array fluorescence intensity (CEL) files were automatically generated from DAT files by the Affymetrix® GeneChip® Command Console® (AGCC) Software version 4.0. The analysis of the .oschp files generated in Chromosome Analysis Suite 3.0 was performed on the tumor sample matched to the normal control.
2.6. Exome analysis of 102 Chinese IHCC samples
Exome analysis on IHCC tumor/control paired samples isolated from 103 IHCC Chinese patients was previously described [16]. This exome database was analyzed for presence of one of three germline dominant missense variants in the GNE gene (NM_001128227.2; GeneID 10020) associated with sialuria; rs121908623, rs121908622, or rs121908621 (Table 1). While the total number of patients was 103, GNE variant analysis was performed on 102 samples, since one sample had a high general mutation rate (including mutations in DNA repairing genes).
3. Results
3.1. Clinical reports
3.1.1. Patient 1
A male born at term to a 36-year-old G4P2 mother and 35-year-old father. The pregnancy was complicated by nausea in the 2nd trimester and maternal edema during the 3rd trimester. Level II ultrasound showed an increased nuchal fold. Amniocentesis revealed a normal 46,XY karyotype. At birth the weight was 3147 g and length 51 cm. Occipitofrontal head circumference (OFC) was not recorded. There were no delivery or newborn complications other than mild jaundice that did not require treatment. Mild hypotonia was observed throughout infancy and childhood. Neuromotor development was slightly delayed, particularly when compared to his two older healthy brothers. He held his head up at 1 month, rolled over at 6 months, sat upright by 7 months and walked unaided at 13 months. His expressive speech development was also delayed. He had limited babbling as an infant and only a few single words by two years. He was enrolled in speech therapy and diagnosed with a verbal dyspraxia. There was no hearing deficit.
Routine pediatric examination at 3 years identified liver enlargement and firmness in addition to mild elevation in plasma of aspartate transaminase (AST) (85 U/L), alanine aminotransferase (ALT) (131 U/L) and alkaline phosphatase (4018 U/L). The latter result gradually returned to normal. The radiographic skeletal survey showed that the skeletal age was lagging about 6 months behind the chronological age. The calvaria was rather thick and the lateral spine film showed an ovoid rather than a rectangular shape of all vertebrae with the ventral rim being taller than the dorsal one. Tubulation of the large long bones was normal. The coarsely trabeculated metacarpals were shorter than expected for age as were the terminal phalanges that had prominent terminal tufting. Similar changes were observed in the corresponding foot bones.
Study of the liver biopsy specimen taken at 3 4/12 years yielded inconclusive results, but showed increased “cytoplasmic glycogen” in hepatocytes. There was neither histochemical nor biochemical evidence for fibrotic changes within the liver parenchyma. Biochemical and molecular studies failed to provide confirmation of either glycogenosis type III or type IX.
As a toddler he had recurrent upper respiratory infections. Adenoidectomy was performed in the second year of life and tonsillectomy about 5 years. In later childhood his overall health was good. He attended normal grade school and speech therapy was discontinued at 7 years. He has actively participated in competitive sports.
Because of lack of a specific diagnosis at 11 years, he was enrolled in the “Genomic Study of Medical Development or Congenital Problems of unknown Etiology in Pediatric Patients” at Duke University. Exome sequencing detected the heterozygous pathogenic missense mutation [NM_005476: c.797G>A; p.R266Q] in the GNE gene (MIM 603824). He was examined clinically at the GGC at the age of 12 6/12 years. Stature was 148 cm (25–50th centile), weight 41.9 kg (50th centile) and OFC 56.9 cm (90–97th centile), consistent with earlier data recorded on cumulative growth curves. Hair was red-brown and curly. Eyes were slightly proptotic with fullness of the upper and lower eyelids. Inner and outer canthal measurements were within 2 SDs of the mean for age, but interpupillary measurement was >3 SDs above the mean. The lens in the right eye had a small-black appearing dot, later identified as a remnant of the embryonic lenticular vascular tunic (Mittendorf's dot). The earlobes were uplifted bilaterally (Fig. 1A). The midface was notably flattened (Fig. 1A). The nasal bridge was low. The philtrum was long and N2 SDs above the mean. Heart auscultation revealed an early systolic murmur (3/6) best audible at the apex. The liver edge was 6 cm below the right costal margin, firm in texture, but without nodularity. The musculoskeletal exam showed looseness of the interphalangeal joints of both thumbs and mild hyperextensibility at the elbows. Deep tendon reflexes were symmetrically 1 + at the elbows, but 2 + elsewhere. Gait was normal but associated with flattening of the pedal arches, eversion of the feet and widened sandal gap. During the exam, he was noted to be intelligent with normal verbal communication.
Fig. 1.
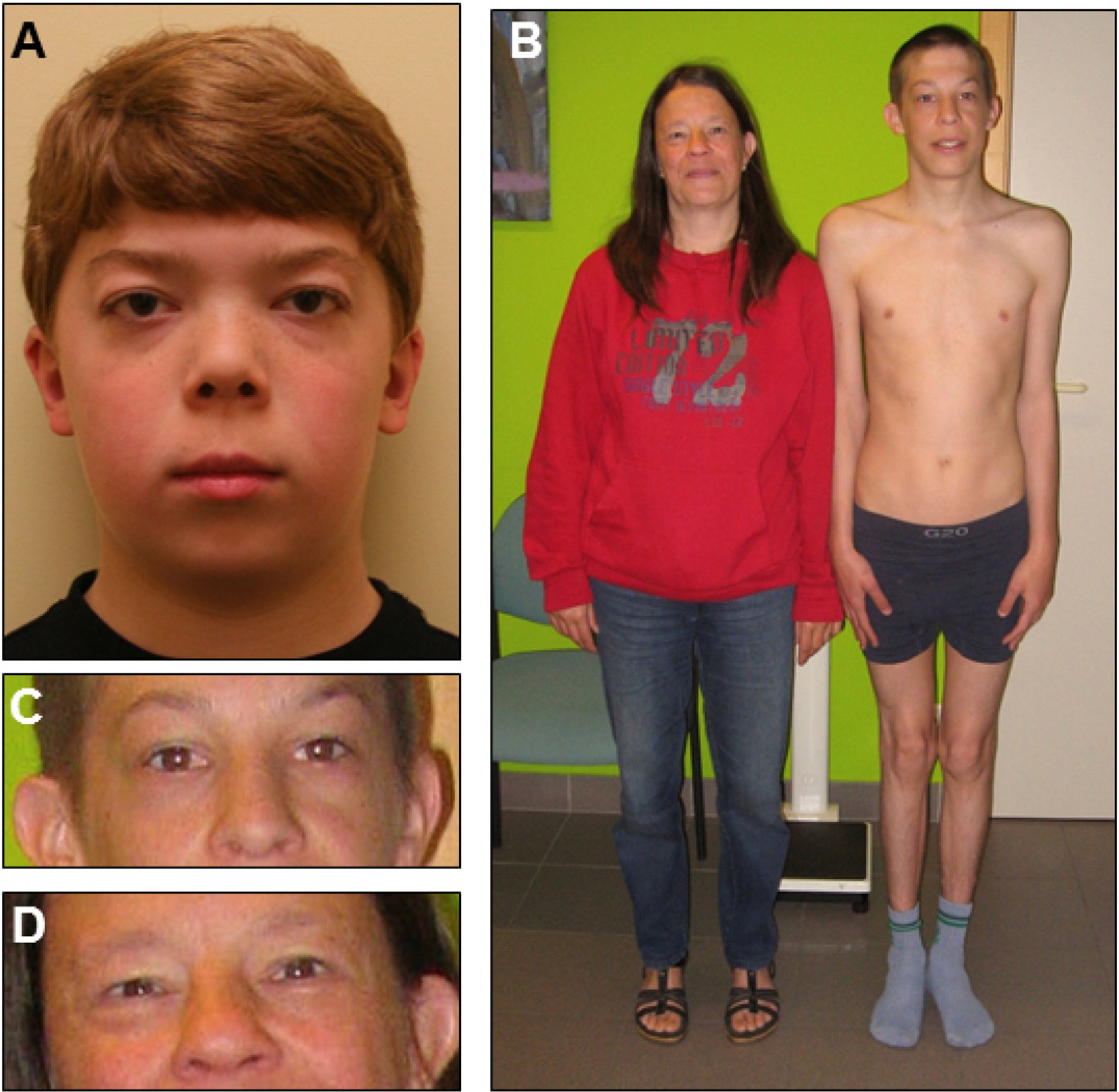
Features of sialuria patients. A. Patient 1 at 12 years of age. B. Patient 2 and his mother, patient 3, shown at ages 17 and 45 respectively. C. Enlarged facial image of patient 2. D. Enlarged facial image of patient 3. Note facial features reported for other sialuria patients: mild coarse facial features, widened, low nasal bridge and epicanthal folds.
He was reexamined at the GGC at 14 6/12 years. Updates to his medical history revealed he developed a large nasal polyp in his right maxillary sinus that was surgically removed and was positive for aspergillus. He was also noted to have a deceleration of growth and pubertal delay. Endocrinology evaluation noted bone growth delay of one year with growth factors in the lower range of normal suggestive of constitutional growth delay. Academically, he was doing very well in advanced classes. His stature was 156 cm (5–10th centile), weight 49 kg (30th centile) and OFC 57.3 cm (90–97th centile). His physical exam was unchanged from previous other than the liver edge was 5 cm below the right costal margin and less firm on palpation. Recent blood work was remarkable for a normal complete blood count and mildly elevated transaminases (AST 42 U/L and ALT 77 U/L).
3.1.2. Patient 2
A male, whose clinical and family history findings up to the age of 4 8/12 years have been reported earlier [7]. Infrequent routine pediatric examinations did not record any health problems, but the mild, apparently stable hepatomegaly remained a constant finding. His speech development lagged and he attended a special education grade school program until he enrolled in a special vocational school at age 13 years. He is currently 19 years of age completing his last year of schooling with plans to graduate as a welder. He has been in good overall health, likes to ride his bicycle and has several friends, whom he prefers to meet at home instead of going out. He has always had a calm disposition without any behavioral difficulties.
When examined clinically by one of the co-authors at the age of 17 3/12 years, he was a thin, lean young man with normal stature, 170 cm (50th centile), weight 47.3 kg (<3rd centile) and OFC 54 cm (25th centile) in good general health (Fig. 1B). There was slight edema of the lower eyelids, but no other facial or intraoral abnormality (Fig. 1C). Vision and hearing were normal. He tended to hold his neck in mild flexion. Examination of heart and lungs found no abnormalities. The liver edge was nearly 6 cm below the right costal margin and on palpation more solid than soft to the touch but without nodularity. The spleen could not be palpated. He was a normal postpubertal male. He had a slight kyphosis of the upper back but also a reduced anteroposterior thoracic diameter. Gait had a mildly hypotonic character with exaggeration of the genua valga type axis deviation. Passive extension of the knees was incomplete. The knee joints were never fully extended when walking or standing. The upper limbs and their major joints functioned normally except for radial deviation and some hyperextension in the elbows. The neurologic examination including the deep tendon reflexes, was normal. Ultrasound examination of the liver at 17 7/12 years confirmed the liver enlargement, but did not reveal any textural abnormality. It also revealed the slightly increased size of the spleen. The transaminases were slightly increased above the upper limit of normal. At age 17, the urinary free sialic acid was 3475 μmol/mmol creatinine (normal <74 μmol/mmol creatinine) and 4100 μmol/mmol creatinine in a later sample.
The patient was seen by the hepatologist at the regional general hospital. Magnetic resonance imaging (MRI) confirmed the hepatomegaly and mildly enlarged spleen. The enlarged liver had a normal outline. Follow-up evaluation 18 months later revealed a significant liver enlargement. A cylindrical liver biopsy specimen was obtained for light microscopic and electromagnetic (EM) study. There was no sign of either cirrhosis or any other general or localized tissue abnormality. Currently 19 6/12 years of age, he is in good health with unchanged physical characteristics.
3.1.3. Patient 3
A female, mother of patient 2, is currently 47 years of age and the only adult ever reported with sialuria [7]. This diagnosis was excluded in all other close relatives of patient 2 by assay of free sialic acid in the urine. Patient 3 was in apparent good general health when accompanying her son to the clinic visit (Fig. 1B). Edema in the lower eyelids was the only clinical abnormality readily noticeable (Fig. 1C). Ultrasonographic examination of the abdomen was performed and revealed a normal sized liver with a hyperechogenic lesion in the dorsocranial part of the right lobe with maximal diameters, 3.7 × 4.8 cm. The lesion was surrounded by a hyperechogenic halo. There was no dilatation of the intrahepatic bile channels. No gallstones were in the gall bladder and no abnormality was detected in any of the other abdominal organs.
MRI examination of the liver performed one month later confirmed the presence of a poorly outlined lesion of heterogeneous texture, located predominantly in liver segment 7 with maximal diameters of 4.0 × 4.1 × 4.7 cm. The lesion extended cranially into the subcapsular region. Either rupture of the liver capsula or perihepatic collection of liquid could not be demonstrated. The appearance of contrast substance within the lesion was not homogeneous, but highlighted its hypervascular periphery. MRI examination performed three months hence showed that the main lesion had not changed in size or composition during the intervening period. Hemihepatectomy and removal of the gall bladder were subsequently performed. Several tumor biopsy specimens were taken, all of them containing zones of normal appearing liver parenchyma (Fig. 2A). Preoperative elevation of ALT and gamma-glutamyl transpeptidase (GGT) was minimal and alpha-fetoprotein was 6.5 kU/L. There was no preoperative value of carbohydrate/cancer antigen (CA) 19–9.
Fig. 2.
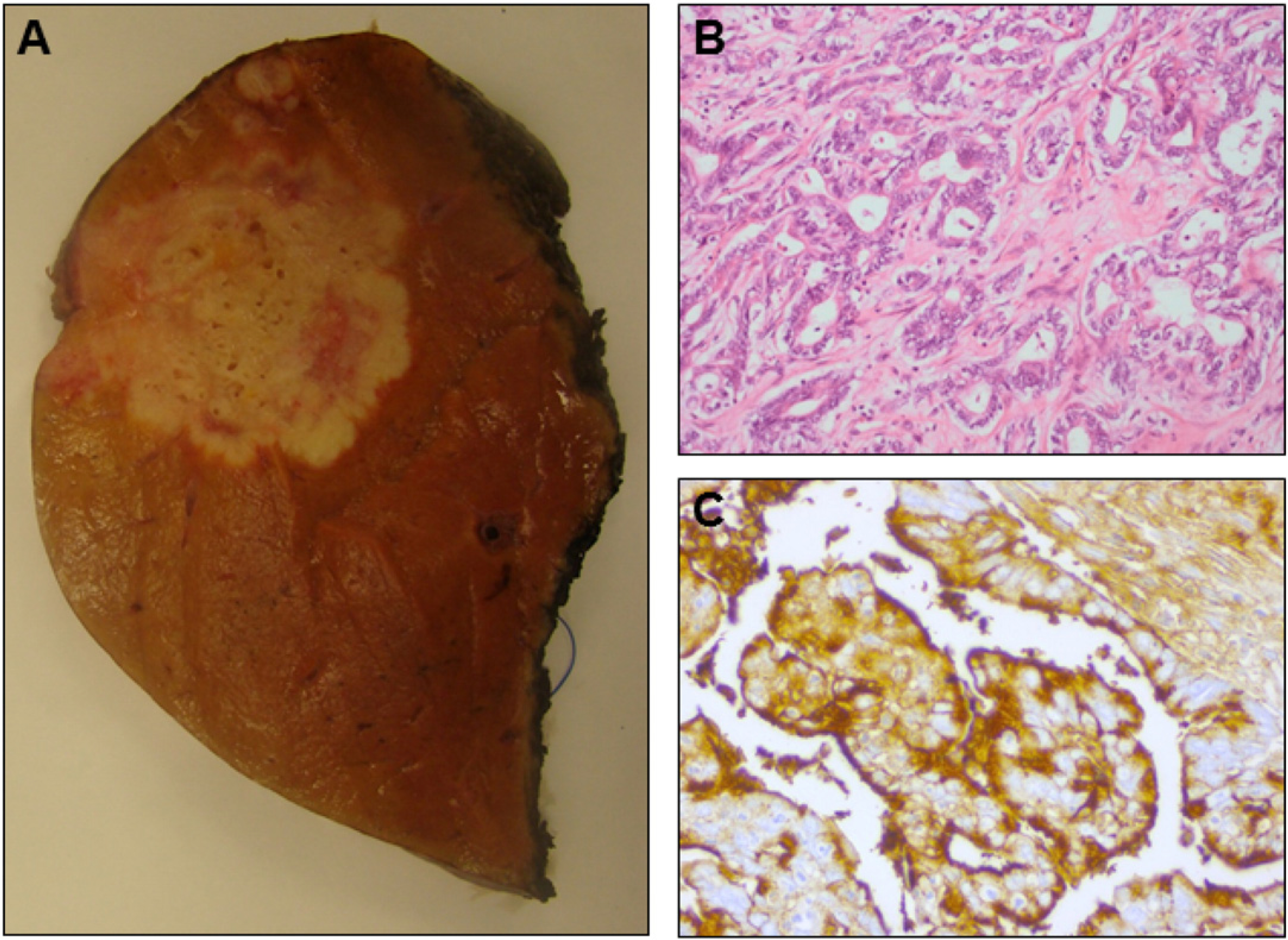
Macroscopic and microscopic features of the intrahepatic cholangiocarcinoma in patient 3. A. Macroscopic aspect of the liver lesion removed per hemihepatectomy from patient 3: the irregular, ill-defined white mass differs significantly from the normal liver tissue. B. Photomicrograph of the intrahepatic cholangiocarcinoma in the liver resection specimen of patient 3 shows irregular tumoral glands in a desmoplastic stroma (H&E: × 200). C. The intrahepatic cholangiocarcinoma removed from patient 3 expresses CA19–9 (immunohistochemistry for CA19–9; × 400).
Postoperatively the clinical course was without significant complications or setbacks. Because of focal perforation of the visceral peritoneum, lymphovascular invasion and the patient’s relatively young age, adjuvant chemotherapy with gemaitabine and cisplatin was prescribed and applied for six months. Postoperatively the expected elevation of liver tests has been noticed. ALT and GGT remained slightly elevated throughout chemotherapy, but normalized afterwards. Frequent hospital follow-up showed that the treatment was well tolerated other than complaints of pain irradiating from the back towards the right side hypochondrium. MRI one year after surgery showed no tumor recurrence.
3.1.4. Patient 4
The mother of patient 3 (maternal grandmother of patient 2) died of liver cancer in 1989 at the age of 49. She complained about dyspepsia and postprandial bloating beginning in the autumn of 1988. Her appetite decreased and anorexia, nausea and occasional bouts of vomiting occurred. She had no fever, jaundice, signs of dysphagia or stool abnormalities. Examination in early December 1988 found a significantly enlarged liver. There was a small umbilical hernia, but no other abdominal or general abnormal clinical findings. Neither signs of any preceding chronic disease nor of sialuria had been recorded in the clinical history. She had been a homemaker and the mother of five girls and one boy. Except for patient 3 and her unaffected son, all of her children successfully attended schools of higher education.
3.2. GNE mutation analysis
A heterozygous de novo nucleotide transition, (NM_005476: c.797G>A; p.R266Q) in the GNE gene was found by exome sequencing in the DNA sample of patient 1. This mutation was not found in the parents or the two older siblings, suggesting a de novo pathogenic variant in this individual. In silico study showed that this missense mutation had previously been found to be associated with sialuria (MIM 269921). This mutation is identical to the single mutation detected in patient 2 N15 years earlier [7]. This same mutation was confirmed in genomic DNA extracted from the normal liver tissue as well as in that from the tumor samples obtained from the liver specimen obtained by hemihepatectomy in patient 3. Unfortunately the quantity and quality of the DNA extracted from the 1988 stained histologic slides from patient 4 was suboptimal, such that a molecular diagnosis of sialuria in this patient could not be made.
3.3. Biochemical confirmation of sialuria
Patient 1 urine testing at the Greenwood Genetic Center (Greenwood, SC) revealed highly excessive urinary excretion of free sialic acid (32,617 nmol/mg creatinine; normal 155–352 nmol/mg creatinine). This finding of ~130-fold increased urinary excretion of free sialic acid confirmed the diagnosis of sialuria in patient 1.
3.4. Light- and electron microscopy of liver biopsies
Light microscopic examination of the liver biopsy from patient 2 revealed minimal periportal fibrosis. There was no inflammation. The cytoplasm of hepatocytes did not show vacuolation or abnormal inclusions. Interlobular bile ducts appeared normal. The biopsy specimen did not show evidence of dysplasia or any premalignant lesion. EM confirmed the absence of abnormal inclusions in the cells. Mitochondria, lysosomes and endoplasmatic reticulum.
appeared normal. Intracellular abnormalities suggestive of a storage disease could not be demonstrated (results not shown).
The resection specimen removed from patient 3 showed a moderately differentiated adenocarcinoma with a CK7+/CK20− cytokeratin profile (Fig. 2B) and expression of CA19–9 at the apical side of the cells (Fig. 2C). This profile is highly suggestive of a carcinoma of pancreatico-biliary origin and compatible with an intrahepatic cholangiocarcinoma (IHCC). Random biopsies from the liver parenchyma at a distance from the tumor did not show significant abnormalities. No pre-malignant lesions were found. There was no cirrhosis.
The incisional liver biopsy from the liver tumor of patient 4 was reviewed. The tumor was composed of irregular infiltrating glands and trabecular structures set in sclerotic stroma. Obvious nuclear atypia was present in the tumor cells. At immunohistochemistry, the tumor cells showed strong expression of CK7 and CK19, compatible with the diagnosis of a poorly differentiated IHCC.
3.5. Oncoscan assays results
The molecular comparison of the genomic DNA from normal liver tissue and the liver tumor from patient 3 achieved by the Affymetrix Oncoscan technology revealed mosaic copy number gains, losses and loss of heterozygosity (LOH) in the tumor sample only. Significant genomic changes in the tumor included mosaic deletion of the distal part of chromosomal 1p, mosaic deletion of the entire 6q, mosaic deletion of the distal 8p, mosaic duplication of to 8qter, mosaic LOH of chromosome 10p and mosaic deletion of the entire 8q, and mosaic deletion of the distal half of 14q (Fig. 3). Since this platform also can detect 74 somatic mutations from 9 genes (BRAF, KRAS, EGFR, IDH1, IDH2, PTEN, PIK3CA, NRAS and TP53) implicated in various cancers, we also were able to detect the heterozygous KRAS gene missense mutation, c.34G>T; p.G12C/S, which could not be confirmed by Sanger sequencing. The KRAS mutation was not observed in the normal sample by either Oncoscan or Sanger sequencing.
Fig. 3.
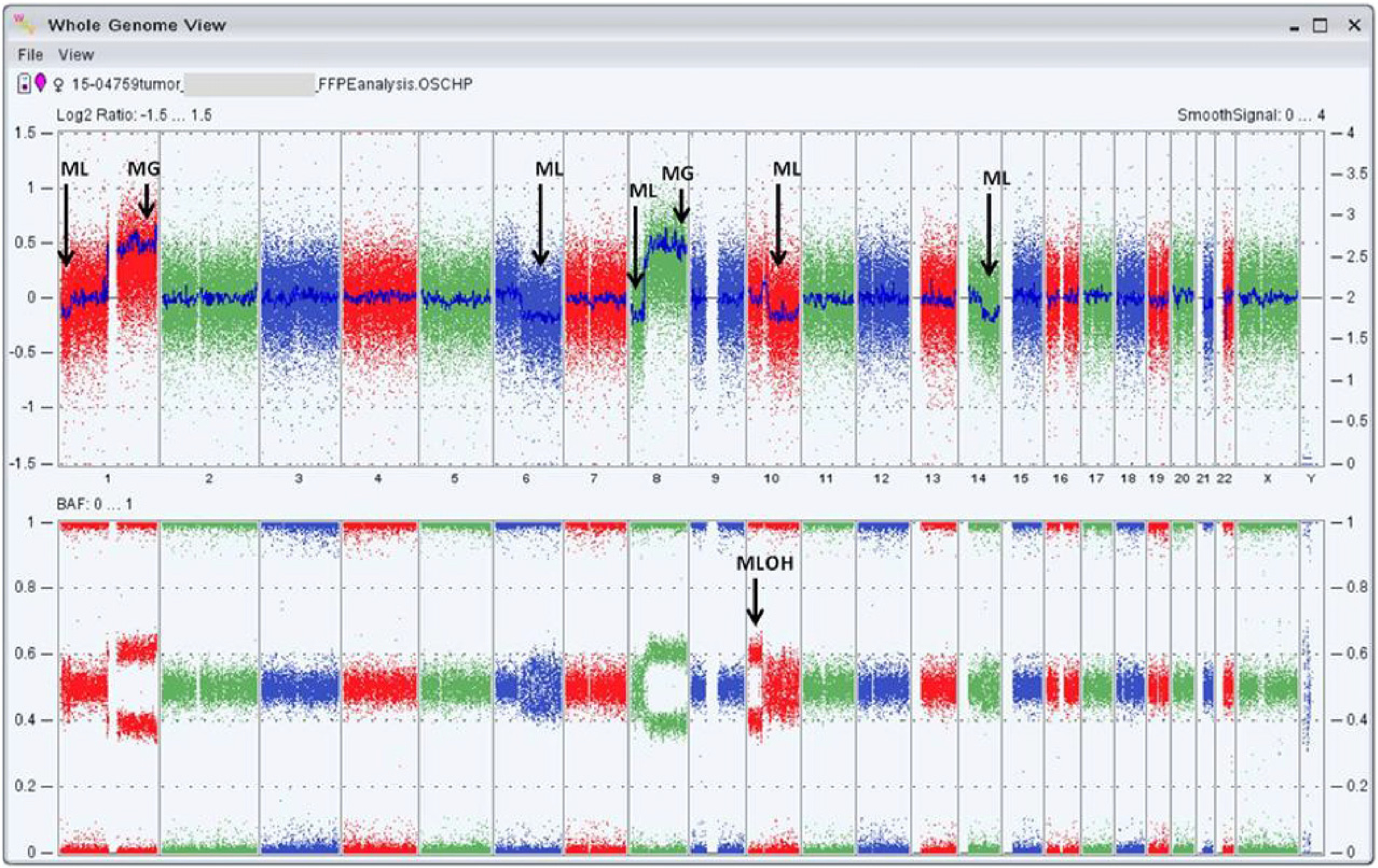
OncoScan results from IHCC tumor from patient 3. The Whole Genome View figure of the IHCC tumor from patient 3 (15–04759) is captured by the Affymetrix Chromosome Analysis Suite 3.0. The top panel shows the log2 ratio of the entire genome where the blue line shows the copy number state of each chromosome (represented as smooth signal track). Analysis revealed mosaic gains of chr1q and 8q. Also, mosaic losses of chr 1p, 6q, 8p, 10q and 14q were observed. Of note of the copy neutral mosaic LOH on chr 10p (copy number state = 2). The bottom panel shows the B allele frequency (BAF) plots (1 = BB, 0.5 = AB and 0 = AA). ML = mosaic copy loss, MG = mosaic copy gain, MLOH = mosaic loss of heterozygosity.
4. Discussion
The diagnosis of sialuria in the 12-year-old boy (patient 1) is by itself worthwhile to record as he becomes only the 8th individual known with this rare metabolic disorder. The new observation confirms that throughout childhood and adolescence sialuria is clinically a mild disorder. In contrast with the few previously reported patients, the diagnosis in this new proband with de novo dominant sialuria was made by exome sequencing methodology, providing one more example of the diagnostic power of this method. The diagnosis in the new patient has subsequently been confirmed by the demonstration of highly excessive urinary excretion of free sialic acid.
The apparent rarity of sialuria is the first reason that more than a decade has passed since the penultimate diagnosis and report of sialuria [7], which also reported its autosomal dominant mode of inheritance. Secondly, the apparent minor or barely recognizable morbidity of sialuria during childhood, adolescence and even in a few subsequent decades of life, not only postpones the diagnosis in some instances but may preclude it altogether. And third, assaying urinary free sialic acid is not a routine laboratory procedure, but should be considered as part of the metabolic screening of young children with mild developmental delay and in patients with a phenotype suggestive of mucopolysaccharidosis or oligosaccharidosis [13].
Screening of free sialic acid would not only detect patients with sialuria, but also diagnose Infantile Free Sialic Acid Storage Disorder (ISSD, MIM 269920) with early fatal outcome, or the allelic Salla disease (MIM 604369) with slightly later onset and slower progression, both with autosomal recessive mode of inheritance and caused by mutations in the lysosomal sialic acid transporter SLC17A5 [17]. However, in the latter disorders, the excess of urinary free sialic acid is rarely twentyfold the normal urinary level. These storage disorders are consistently associated with a more severe and progressive clinical phenotype of significant developmental delay and severe neuromotor and intellectual disability [14].
The observation of the new sialuria proband prompted interest in a follow-up study of all previously studied and reported cases (Table 1). It is known that the first reported sporadic patient, subject No. 1 [1,2] has died at the age of 30 years in a car accident. The mutation in this subject listed in Table 1, is reported here for the first time. Unfortunately written contact with the physicians of the remaining subjects (No. 2, No. 4, No. 5 in Table 1), presumed to be sporadic instances and known to be currently in their third decades of life, has not been successful. In most instances only indirect information of the subjects being lost to follow-up, became available. Subject No. 3 in the chronological list had his last consultations in the local pediatric clinic at the ages of 19 and 23 years and was found consistently in good general health. Currently 27 years old, he too has since been lost to follow-up.
Contrary to the unproductive attempts at gathering follow-up information on nearly all previously reported subjects, follow-up of the family of patients No. 6 and No. 7 in Table 1 (patients 2 and 3 in this report) has yielded new data that are medically of interest and worrisome. They were seen again after 12 years covered by only two short intermediate reports stating good health in both except for mild and stable hepatomegaly in the son. Neither reported complaints nor worries about their health. This understandable attitude tends to increase the chances of losing contact with the attending physician and may contribute to the loss to follow-up in the earlier reported subjects.
The finding of IHCC in patient 3 and the family history of a fatal liver tumor in her mother at age 49 years (patient 4 in this report) kindled the hypothesis that the liver tumor may be a mid-life complication in the natural course of sialuria. Recent studies of the 1989 post-mortem pathology slides on patient 4 characterized the tumor as an IHCC. We have been unable to genetically confirm that patient 4 also had sialuria. Detection of excessive amounts of free sialic acid in these tissues was equally unsuccessful. A priori reasoning would suggest that it is not likely that patient 4 was heterozygous for the germinal GNE mutation, because only one out of six of her offspring was diagnosed with the metabolic disorder. Patient 4 may, however, have acquired an early somatic mutation resulting in a mosaic genotype, which could explain both the IHCC and the hereditary transmission to only one of her children.
Pathologically identical liver tumors in two consecutive generations could also be due to some genetic predilection unrelated to sialuria or its effects. Even though our follow-up study cannot fully prove that the IHCC in patient 3 is a middle age complication in the natural course of sialuria, it may be prudent and heuristic to assume association of hepatic malignancy and the long-term exposure to largely excessive amounts of free sialic acid. Hypersialylation of proteins in sialuria subjects is likely, as indicated by significant hypersialylation of serum apoC-III and hypersialylation of serum transferrin by isoelectric focusing studies [18]. Hypersialylation of proteins is also indicated in multiple aspects of tumor growth and behavior [19].
Several well-known liver diseases are known to be associated with hepatocellular carcinoma (HCC), the most common intrinsic liver tumor. However, only few specific causes of IHCC outside parasite infection in Southeast Asia, are currently known. Contrary to HCC, little is known about genomic/cytogenetic alterations in IHCC [20]. The Affymetrix Oncoscan study amply illustrates the existence of such changes in the IHCC of our patient 3 (Fig. 3). Therefore, this report also adds information to the molecular characterization of IHCC.
The genomic alterations in IHCC tissue in patient 3 are impressive and include large and small chromosomal zones of duplication and deletion in addition to a mosaic LOH covering almost the entire 10p. These findings may serve as a reference to future analogous observations. However, as probable end-stage genomic changes in this IHCC, they can hardly contribute to sorting the assumed multi-step changes that played a role in the tumor's earlier stages of pathogenesis [21]. Our suspicion is that the KRASG12S oncogene mutation identified by microarray in the tumor genome is present at a low mosaic level (<20%) and hence could not be confirmed by Sanger sequencing. The normal liver tissue did not implicate the presence of this mutation by either the microarray or sequencing platforms. Pathologic evidence of presumably precancerous changes in the epithelial cells of some bile ducts supports the hypothesis that continuous handling of high concentrations of free sialic acid may ultimately result in a KRASG12S mutant allele, which could be the initial driving force in this pathogenesis. In fact, in a recent IHCC exome sequencing study, codon 12 of the KRAS oncogene was identified as a significantly frequent somatic mutation in IHCC tumor tissue [16].
This follow-up study suggests considering undiagnosed sialuria as a potential cause in subjects with IHCC without previous health problems or liver disease. Genetic testing for sialuria-related GNE variants and/or biochemical assay of urinary free sialic acid is recommended for such individuals. However, our analysis of an existing exome sequence database of DNA isolated from IHCC tumor/control tissue of 102 Chinese patients [16] did not identify any sialuria-related GNE variants in this cohort. These exome analysis results not only support our impression that sialuria is a rare disease, but also emphasize that sialuria mutations are not frequent among patients with IHCC.
The frequency of IHCC in sialuria patients remains unknown, but ultimately may be answered by the diagnosis of additional middle-aged sialuria patients. We recommend regular, at least yearly hepatological follow-up for all known sialuria patients. We also suggest considering the diagnosis of sialuria in any IHCC patients without a clinical history of any major medical disorder.
Acknowledgments
The authors would like to acknowledge the skillful laboratory contributions of Miss Joshni Simon and of Dr. Jennifer A. Lee, PhD, FACMG. We also thank Dr. S. Packman for providing the more recent, postpubertal information on sialuria patient No. 3 (Table 1). And the serious but unfortunately unsuccessful efforts by Drs J. and P. Frias (Atlanta, GA) to trace sialuria patient No. 4 (Table 1) are also gratefully acknowledged
Funding
This study was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA (to MH) and the Greenwood Genetic Center Foundation.
Abbreviations:
- ALT
alanine aminotransferase
- AST
aspartate transaminase
- CA
carbohydrate antigen
- CMP
cytidine monophosphate
- CK
cytokeratin
- DMB
dihydrochloride
- EM
electromagnetic
- FFPE
formalin-fixed, paraffin-embedded
- GGC
Greenwood Genetic Center
- GlcNAc
N-acetylglucosamine
- GNE
UDP-GlcNAc 2-epimerase/ManNAc kinase enzyme
- GNE
gene encoding the UDP-GlcNAc 2-epimerase/ManNAc kinase enzyme
- GT
glutamyl transpeptidase
- HCC
hepatocellular carcinoma
- H&E
hematoxylin and eosin
- HPLC
high pressure liquid chromatography
- IHCC
intrahepatic cholangiocarcinoma
- ISSD
infantile free sialic acid storage disorder
- LOH
loss of heterozygosity
- ManNAc
N-acetylmannosamine
- MG
mosaic copy gain
- MIM
Mendelian Inheritance in Man
- MIP
molecular inversion probe
- ML
mosaic copy loss
- MOHL
mosaic loss of heterozygosity
- MRI
magnetic resonance imaging
- OFC
occipitofrontal head circumference
- SD
standard deviation
- UDP
uridine diphosphate
Footnotes
Conflict of interest
None.
References
- [1].Fontaine G, Biserte G, Montreuil J, Dupont A, Farriaux JP, Sialuria: an original metabolic disorder, Helv. Paediatr. Acta 23 (Suppl. 17) (1968) 1–32. [PubMed] [Google Scholar]
- [2].Montreuil J, Biserte G, Strecker G, Spik G, Fontaine G, Farriaux JP, Description of a new type of melturia called sialuria, Clin. Chim. Acta 21 (1968) 61–69. [DOI] [PubMed] [Google Scholar]
- [3].Wilcken B, Don N, Greenway R, Hammond J, Sosula L, Sialuria: a second case, J. Inherit. Metab. Dis. 10 (1987) 97–102. [DOI] [PubMed] [Google Scholar]
- [4].Seppala R, Tietze F, Krasnewich D, Weiss P, Ashwell G, Barsh G, Thomas GH, Packman S, Gahl WA, Sialic acid metabolism in sialuria fibroblasts, J. Biol. Chem. 266 (1991) 7456–7461. [PubMed] [Google Scholar]
- [5].Krasnewich DM, Tietze F, Krause W, Pretzlaff R, Wenger DA, Diwadkar V, Gahl WA, Clinical and biochemical studies in an American child with sialuria, Biochem. Med. Metab. Biol. 49 (1993) 90–96. [DOI] [PubMed] [Google Scholar]
- [6].Ferreira H, Seppala R, Pinto R, Huizing M, Martins E, Braga AC, Gomes L, Krasnewich DM, Sa Miranda MC, Gahl WA, Sialuria in a Portuguese girl: clinical, biochemical and molecular characteristics, Mol. Genet. Metab. 67 (1999) 131–137. [DOI] [PubMed] [Google Scholar]
- [7].Leroy JG, Seppala R, Huizing M, Dacremont G, De Simpel H, Van Coster RN, Orvisky E, Krasnewich DM, Gahl WA, Dominant inheritance of sialuria, an inborn error of feedback inhibition, Am. J. Hum. Genet. 68 (2001) 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kornfeld S, Kornfeld R, Neufeld E, O'Brien PJ, The feedback control of sugar nucleotide biosynthesis in liver, Proc. Natl. Acad. Sci. U. S. A. 52 (1964) 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sommar KM, Ellis DB, Uridine diphosphate N-acetyl-D-glucosamine-2-epimeras from rat liver. I. Catalytic and regulatory properties, Biochim. Biophys. Acta 268 (1972) 581–589. [DOI] [PubMed] [Google Scholar]
- [10].Weiss P, Tietze F, Gahl WA, Seppala R, Ashwell G, Identification of the metabolic defect in sialuria, J. Biol. Chem. 264 (1989) 17635–17636. [PubMed] [Google Scholar]
- [11].Seppala R, Lehto VP, Gahl WA, Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme, Am. J. Hum. Genet. 64 (1999) 1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yarema KJ, Goon S, Bertozzi CR, Metabolic selection of glycosylation defects in human cells, Nat. Biotechnol. 19 (2001) 553–558. [DOI] [PubMed] [Google Scholar]
- [13].Enns GM, Seppala R, Musci TJ, Weisiger K, Ferrell LD, Wenger DA, Gahl WA, Packman S, Clinical course and biochemistry of sialuria, J. Inherit. Metab. Dis. 24 (2001) 328–336. [DOI] [PubMed] [Google Scholar]
- [14].Aula P, Gahl WA, in: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonorakis SE, Ballabio A, Gibson K, Mitchell G (Eds.), Disorders of free sialic acid storage, McGraw-Hill, New York, NY, 2014. (OMMBID Web http://ommbid.mhmedical.com/context.aspx?bookid=971&Sectionid=6265581). [Google Scholar]
- [15].Hommes FA, Determination of bound and free sialic acid in urine, in: Hommes F (Ed.), Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual, Wiley-Liss, New York: 1991, pp. 219–232. [Google Scholar]
- [16].Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JS, Zhao X, Li Y, Li Q, Wang H, Hu J, Kong G, Wu M, Ding C, Chen N, Hu H, Mutational landscape of intrahepatic cholangiocarcinoma, Nat. Commun. 5 (2014) 5696. [DOI] [PubMed] [Google Scholar]
- [17].Strehle EM, Sialic acid storage disease and related disorders, Genet. Test. 7 (2003) 113–121. [DOI] [PubMed] [Google Scholar]
- [18].Wopereis S, Abd Hamid UM, Critchley A, Royle L, Dwek RA, Morava E, Leroy JG, Wilcken B, Lagerwerf AJ, Huijben KM, Lefeber DJ, Rudd PM, Wevers RA, Abnormal glycosylation with hypersialylated O-glycans in patients with sialuria, Biochim. Biophys. Acta 1762 (2006) 598–607. [DOI] [PubMed] [Google Scholar]
- [19].Büll C, Stoel MA, den Brok MH, Adema GJ, Sialic acids sweeten a tumor's life, Cancer Res. 74 (2014) 3199–3204. [DOI] [PubMed] [Google Scholar]
- [20].Shibata T, Aburatani H, Exploration of liver cancer genomes, Nat. Rev. Gastroenterol. Hepatol. 11 (2014) 340–349. [DOI] [PubMed] [Google Scholar]
- [21].O'Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N, Hezel AF, KRAS G12D and p53 mutation cause primary intra-hepatic cholangiocarcinoma, Cancer Res. 72 (2012) 1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huizing M, Carrillo-Carasco N, Malicdan MC, Noguchi S, Gahl WA, Mitrani-Rosenbaum S, Argov Z, Nishino I, GNE myopathy: new name and new mutation nomenclature, Neuromuscul. Disord. 24 (2014) 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]