

Abstract
From the CH2Cl2 extract of the Antarctic sponge Dendrilla antarctica we found spongian diterpenes, including previously reported aplysulphurin (1), tetrahydroaplysulphurin-1 (2), membranolide (3), and darwinolide (4), utilizing a CH2Cl2/MeOH extraction scheme. However, the extracts also yielded diterpenes bearing one or more methyl acetal functionalities (5–9), two of which are previously unreported, while others are revised here. Further investigation of diterpene reactivity led to additional new metabolites (10–12), which identified them as well as the methyl acetals as artifacts from methanolysis of aplysulphurin. The bioactivity of the methanolysis products, membranoids A–H (5–12), as well as natural products 1–4, were assessed for activity against Leishmania donovani-infected J774A.1 macrophages, revealing insights into their structure/activity relationships. Four diterpenes, tetrahydroaplysulphurin-1 (2) as well as membranoids B (6), D (8), and G (11), displayed low micromolar activity against L. donovani with no discernible cytotoxicity against uninfected J774A.1 cells. Leishmaniasis is a neglected tropical disease that affects one million people every year and can be fatal if left untreated.
Graphical Abstract
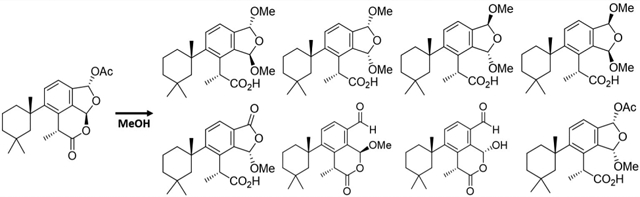
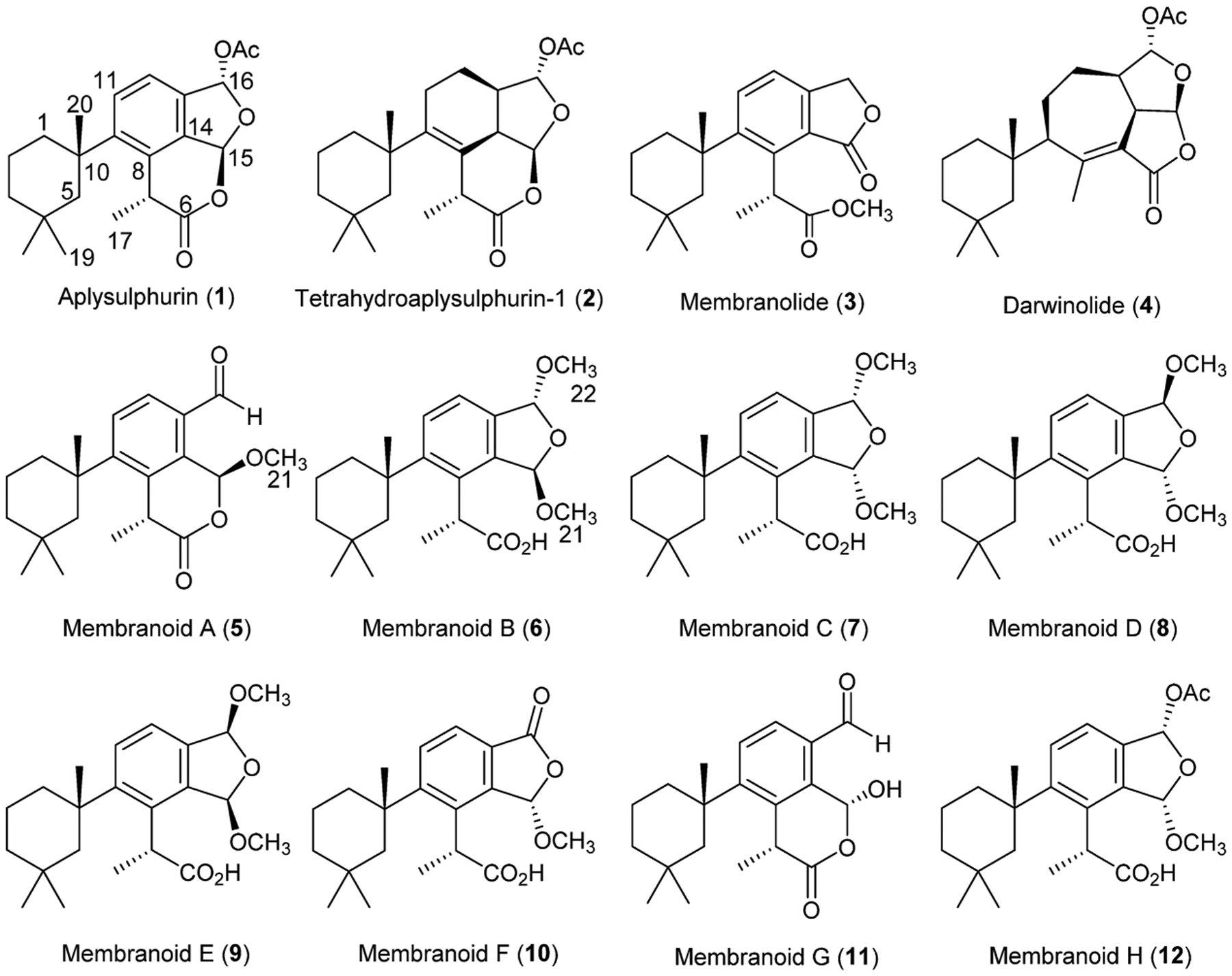
The sponge Dendrilla membranosa has been the subject of a number of chemical investigations resulting in reports of over a dozen rearranged and oxidized spongian diterpenes.1–6 Though there are numerous reports of chemistry isolated from individuals collected in Antarctica, it has recently been suggested that these belong to a distinct species endemic to the southernmost continent, which we will herein refer as Dendrilla antarctica.7 Some evidence of location-dependent chemodiversity variation is found in these reports with diterpene profiles seemingly representative of collection site around the continent. We have previously reported that specimens found near Palmer Station, Antarctica, produce primarily spongian diterpenes aplysulphurin (1), tetrahydroaplysulphurin-1 (2), and membranolide (3).2 More recently we reported darwinolide (4),5 a rearranged spongian diterpene from the same D. antarctica population near Palmer Station, demonstrating that chemodiversity plasticity is in fact phenotypic. Our 2001 collection noted2 three minor bis(methyl acetal) derivatives (5, 7, 9) of aplysulphurin which have demonstrated variable concentrations in subsequent studies of the sponge.
Spongian diterpenes display a variety of pharmacological properties, including cytotoxicity and antibiotic and anti-inflammatory activity,8 though they are rarely potent or selective. In a screening campaign for natural products active against the leishmaniasis parasite, Leishmania donovani, we found four diterpenoids from a D. antarctica extract, tetrahydroaplysulphurin-1 (2) as well as membranoids B (6), D (8), and G (11), that are both very potent and highly selective for parasite-infected cells over host mammalian cells. L. donovani is a neglected tropical disease spread by sandflies affecting millions of people per year and can be fatal in its visceral form if left untreated.9 Intravenous amphotericin is the only currently approved treatment for visceral leishmaniasis in the US,9 though interest in new treatments is strong.10–13 The bis(methyl acetal) membranoid scaffold seen in membranoids B–E (6–9) represents an intriguing model in functionality that could serve as the basis of future studies to enhance their therapeutic potential.
D. antarctica collected near Palmer Station, Antarctica, was reported in 2004 to produce diterpene metabolites including aplysulphurin (1), tetrahydroaplysulphurin-1 (2), membranolide (3), and the three methyl acetal membranolide derivatives (5, 7, 9) termed membranolides B–D.2 When our L. donovani screening program identified D. antarctica extract as active, we began the analysis of additional specimens of this sponge and report here revisions of stereochemical assignments and NMR shifts of the two originally published bis(methyl acetal) compounds (7 and 9) as well as further methyl acetal and hemiacetal derivatives of membranolide (6, 8, 10–12).
Besides previously characterized bis(methyl acetal) derivatives 7 and 9, the new sponge specimens yielded two additional bis(methyl acetal) derivatives, 6 and 8, representing all four possible diastereomeric bis(methyl acetal) derivatives. Such a display of methyl acetals led us to reinvestigate the reactivity of D. antarctica spongian diterpenes toward alcoholic solvents. We found aplysulphurin alone among D. antarctica diterpenes is sensitive to MeOH, but not other alcohols.2 That diterpenes 5–12 can be obtained by treatment of aplysulphurin with MeOH leads us to conclude these D. antarctica methyl acetals (and one hemiacetal) are artifacts, derived from using methanolic solvent mixtures in the extraction protocol. In the current study, spongian diterpenes 1–5 were characterized based on comparison of their spectroscopic data with published data.2,3,14,15 X-ray crystal structures obtained of the previously isolated bis(methyl acetal) derivatives 7 and 9 forced us to re-examine and revise their configurational assignments and spectroscopic data. We introduce the name membranoids for all MeOH adducts, which requires renaming membranolides B–D2 (5, 7, 9) as membranoids A, C, and E, while new derivatives 6, 8, and 10–12 are referred to as membranoids B, D, F, G, and H, respectively. Characterization of compounds 6–12 is reported here along with biological evaluation of natural and artifactual diterpenes for the control of the leishmaniasis parasite, L. donovani.
RESULTS AND DISCUSSION
Chemical Analysis.
This study began with a 1:1 CH2Cl2/MeOH extract derived from 330 g of freeze-dried D. antarctica collected in 2010. The 20% EtOAc/hexanes fraction from MPLC analysis was subjected to reversed-phase HPLC (60% acetonitrile in water), providing previously unreported bis(methyl acetal) membranoids B (6) (5.3 mg) and D (8) (22.2 mg). For comparison, the same MPLC fraction, upon further HPLC, also yielded membranolide (3, 19.3 mg), membranoids C and E (7, 4.8 mg; 9, 19.9 mg), and tetrahydroaplysulphurin-1 (2, 52.3 mg).
Membranoid B (6) was obtained as a clear oil with spectroscopic data very similar to that of the previously reported bis(methyl acetal) metabolites.2 A formula of C22H32O5 was established from HRESIMS. Key 1H NMR signals (Table 1) include a gem-dimethyl group, H3-18 (δH 0.36) and H3-19 (δH 0.90), and a singlet methyl at H3-20 (δH 1.44), all of which are shown to be substituents on a trimethylcyclohexyl ring via HMBC correlations of H3-18, H3-19, and H3-20 to alkyl carbon C-5 (δC 51.0) and correlations of H3-18 and H3-19 to C-3 (δC 39.6), which is connected to C-1 (δC 41.2) through a COSY spin system involving H2-3 (δH 1.27), H-2α (δH 1.64), H-2β (δH 1.78), H-1α (δH 1.45), and H-1β (δH 2.32). Further HMBC correlations of H3-20 to C-1 and C-9 (δC 148.5) established the trimethylcyclohexyl ring as a substituent on a phenyl ring that also bears two COSY linked adjacent aromatic protons, H-11 (δH 7.58) and H-12 (δH 7.24). HMBC correlations of a doublet methyl at H3-17 (δH 1.58; J = 7.2) and its coupled quartet methine at H-7 (δH 4.64, J = 7.0) to both the carboxylic acid C-6 (δC 178.8) and aromatic carbon C-8 (δC 135.7) verifies a propanoic acid substituent on the phenyl ring, and additional HMBC correlations of H-7 to C-9 and H-11 to C-8 establish it adjacent on the phenyl ring to the trimethylcyclohexyl group. Two acetal methine signals, H-15 (δH 6.32) and H-16 (δH 6.24), along with two overlapping methoxy groups, H3-21 (δH 3.40) and H3-22 (δH 3.40), are characteristic of the bis(methyl acetal)-fused furano-membranoids. The coincident methoxy signals are resolved in CD3CN at H3-21 (δH 3.26) and H3-22 (δH 3.30), respectively (Figure S7). HMBC correlations of H3-21 to C-15 (δC 106.2) and H3-22 to C-16 (δC 105.9), as well as H-15 to C-16, establish the methyl acetal positions about the dihydrofuran ring and are supported in their locations as substituents on the phenyl ring by HMBC correlations of H-15 to C-13 (δC 137.1) and H-16 to C-12 (δC 121.4) and C-14 (δC 138.4).
Table 1.
NMR Data From Membranoids B–E (6–9)a
| membranoid B (6) | membranoid C (7) | membranoid D (8) | membranoid E (9) | |||||
|---|---|---|---|---|---|---|---|---|
| pos | δC, typeb | δHd (J in Hz) | δC, typec | δHe (J in Hz) | δC, typeb | δHd (J in Hz) | δC, typec | δHe (J in Hz) |
| 1α | 41.2, CH2 | 1.45, ov dt | 41.3, CH2 | 1.46, ov dt | 41.3, CH2 | 1.47, ov dt | 41.2, CH2 | 1.47, ov, m |
| β | 2.32, d (14.0) | 2.31, d (14.0) | 2.30, d (14.2) | 2.33, d (14.5) | ||||
| 2α | 20.0, CH2 | 1.64, ov m | 20.0, CH2 | 1.66, ov m | 20.0, CH2 | 1.67, ov m | 19.9, CH2 | 1.64, ov, m |
| β | 1.78, ov dd | 1.82, ov dd | 1.80, ov dd | 1.77, ov, m | ||||
| 3 | 39.6, CH2 | 1.27, ov m | 39.9, CH2 | 1.31, ov m | 39.9, CH2 | 1.29, ov m | 39.6, CH2 | 1.27, ov, m |
| 4 | 31.7, C | 31.8, C | 31.8, C | 31.7, C | ||||
| 5α | 51.0, CH2 | 1.47, d (14.2) | 50.6, CH2 | 1.51, d (14.1) | 50.5, CH2 | 1.51 d, (14.1) | 50.9, CH2 | 1.47, ov, m |
| β | 2.00, d (14.2) | 2.10, d (14.1) | 2.08, d (14.1) | 1.99, d (13.6) | ||||
| 6 | 178.8, C | 180.1, C | 179.8, C | 179.8, C | ||||
| 7 | 40.0, CH | 4.64, q (7.0) | 41.2, CH | 4.71, q (7.0) | 41.3, CH | 4.68, q (7.2) | 40.0, CH | 4.63, q (7.1) |
| 8 | 135.7, C | 135.7, C | 135.8, C | 135.6, C | ||||
| 9 | 148.5, C | 148.6, C | 148.8, C | 148.2, C | ||||
| 10 | 39.3, C | 39.5, C | 39.5, C | 39.3, C | ||||
| 11 | 129.3, CH | 7.58, d (8.3) | 128.9, CH | 7.58, d (8.3) | 128.9, CH | 7.57, d (8.3) | 129.0, CH | 7.56, d (8.3) |
| 12 | 121.4, CH | 7.24, d (8.3) | 121.6, CH | 7.26, d (8.3) | 121.4, CH | 7.23, d (8.3) | 121.3, CH | 7.21, d (8.3) |
| 13 | 137.1, C | 137.3, C | 137.5, C | 136.5, C | ||||
| 14 | 138.4, C | 139.5, C | 139.6, C | 138.4, C | ||||
| 15 | 106.2, CH | 6.32, d (1.8) | 105.4, CH | 5.89, s | 106.2, CH | 6.12, d (1.5) | 105.6, CH | 6.02, s |
| 16 | 105.9, CH | 6.24, d (1.8) | 105.1, CH | 5.89, s | 105.6, CH | 6.31, d (1.5) | 104.5, CH | 6.02, s |
| 17 | 15.1, CH3 | 1.58, d (7.2) | 16.8, CH3 | 1.73, d (7.2) | 17.0, CH3 | 1.71, d (7.2) | 14.6, CH3 | 1.57, d (7.2) |
| 18 | 27.3, CH3 | 0.36, s | 27.4, CH3 | 0.49, s | 27.5, CH3 | 0.49, s | 27.3, CH3 | 0.34, s |
| 19 | 32.9, CH3 | 0.90, s | 32.8, CH3 | 0.94, s | 32.7, CH3 | 0.94, s | 32.9, CH3 | 0.90, s |
| 20 | 32.9, CH3 | 1.44, s | 33.0, CH3 | 1.41, s | 32.9, CH3 | 1.41, s | 32.8, CH3 | 1.46, s |
| 21 | 55.6, CH3 | 3.40, s | 54.7, CH3 | 3.49, s | 54.6, CH3 | 3.45, s | 55.6, CH3 | 3.37, s |
| 22 | 54.2, CH3 | 3.40, s | 54.8, CH3 | 3.52, s | 53.4, CH3 | 3.33, s | 52.8, CH3 | 3.30, s |
CDCl3.
125 MHz.
150 MHz.
500 MHz.
600 MHz; ov = overlapped.
The three additional bis(methyl acetal) membranoids (7–9) shared 1H and 13C shifts (Table 1) and connectivity (Figure 1) with membranoid B (6). Membranoid C (7) was obtained as a clear oil with spectroscopic data (Table 1) consistent with that previously published as membranolide C.2 A formula of C22H32O5 was established based on HRESIMS. The 1H and 13C NMR data and 2D NMR connectivity and correlations unambiguously established the planar structure for membranoid C as that of the bis(methyl acetal) fused furano-membranoids as depicted for membranoid B in Figure 1 and supports the previously reported two-dimensional structure.2 Membranoid D (8) was likewise obtained as a clear oil with a formula of C22H32O5 established based on HRESIMS. The 1H and 13C NMR data and 2D NMR connectivity and correlations also established the planar structure for membranoid D as that of the bis(methyl acetal)-fused furano-membranoids (Figure 1). The last of the four possible diastereomers, membranoid E (9), was similarly obtained as a clear oil with NMR data (Table 1) nearly identically matched with that of previously published membranolide D, with the exception of the acetal methine 1H NMR shifts of H-15 and H-16, which were originally reported as overlapping singlets at 6.20 ppm in CDCl3.2 We have reviewed the data and note the same overlapping acetal methine signals in membranoid E (9) occurring at 6.02 ppm and have determined the original report incorrectly reported the 6.02 ppm shift as 6.20 ppm. As with 6–8, a formula of C22H32O5 was established for 9 based on HRESIMS, and the 1H and 13C NMR data and 2D NMR connectivity and correlations definitively support the previously reported two-dimensional structure (Figure 1).2
Figure 1.
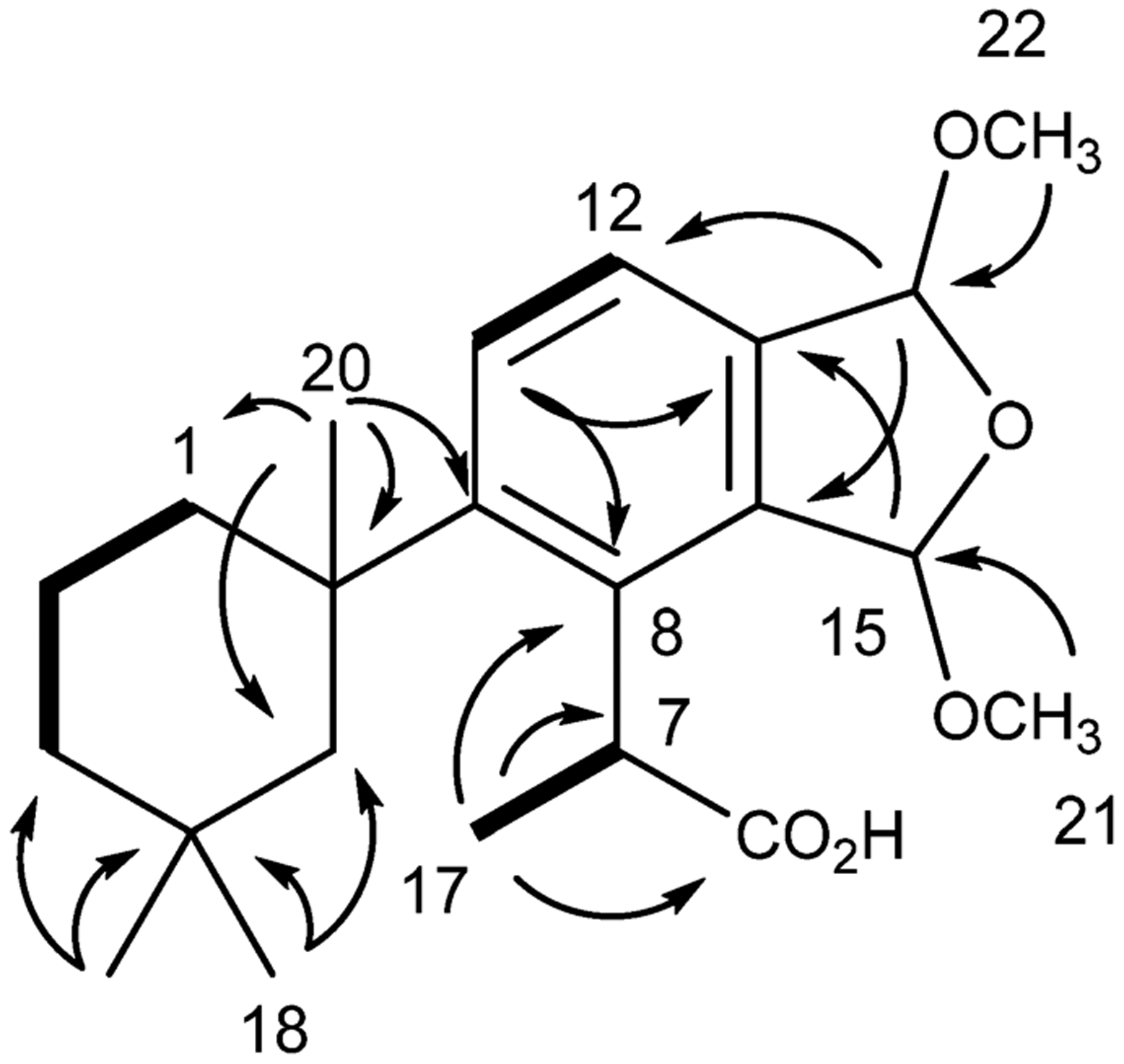
Key HMBC (→) and COSY (―) correlations establishing the planar structure for membranoids B–E (6–9).
The relative and absolute configurations of the four bis(methyl acetal) fused furano-membranoids were ascertained using a combination of X-ray crystallography and 1D and 2D NMR data and have resulted in the reassignment of some of the originally reported structures.2 Slow evaporation from 4:1 hexane/EtOAc at −80 °C over 9 months afforded crystals of membranoid C (7) suitable for X-ray crystallographic analysis. The crystallographic metadata (Flack parameter −0.03(7)), Bijvoet-pair analysis, and Bayesian statistics method (P2 (true) = 1; P3 (true) = 1; P3 (false) = 0.2 × 10−58; P3 (rac-twin) = 0.1 × 10−14 (Table S4)) supported the assignment of the configuration depicted in Figure 2 as the absolute configuration of membranoid C (7). This configuration revises the original published structure for membranolide C from an (R,R) configuration about stereocenters C-15 and C-16, respectively,2 to (S,R), establishing a new α-cis orientation of the methoxy substituents and β-cis orientation of the acetal methines about the dihydrofuran ring for membranoid C (7).
Figure 2.
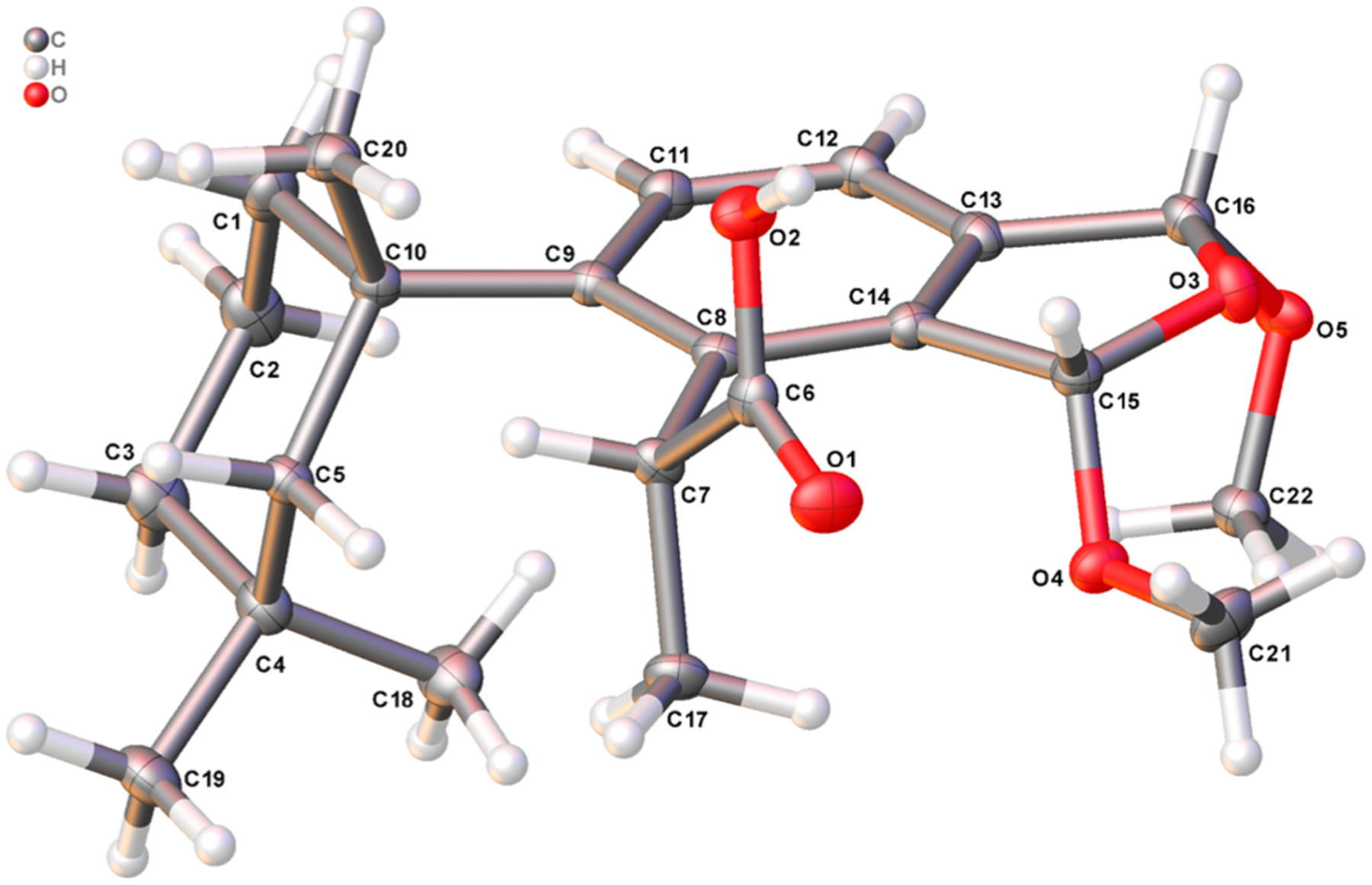
X-ray crystal structure depicting the absolute configuration of membranoid C (7). Anisotropic displacement parameters are drawn at 50% probability.
Similarly, slow evaporation from 4:1 hexane/EtOAc at −80 °C, this time over 12 months, afforded crystals of membranoid E (9) for X-ray analysis. The crystallographic metadata (Flack parameter −0.01(8)), Bijvoet-pair analysis, and Bayesian statistics method (P2 (true) = 1; P3 (true) = 1; P3 (false) = 0.7 × 10−37; P3 (rac-twin) = 0.4 × 10−9 (Table S4)) established the configuration depicted in Figure 3 as the absolute configuration of membranoid E (9). The asymmetric unit found by X-ray spectroscopy includes two molecules of 9 disposed as H-bonded mirror-images about the carboxylic acid groups. The data support the originally reported (R,S) configuration about stereocenters C-15 and C-16, respectively, and β-cis orientation of the methoxy substituents about the dihydrofuran ring.2
Figure 3.
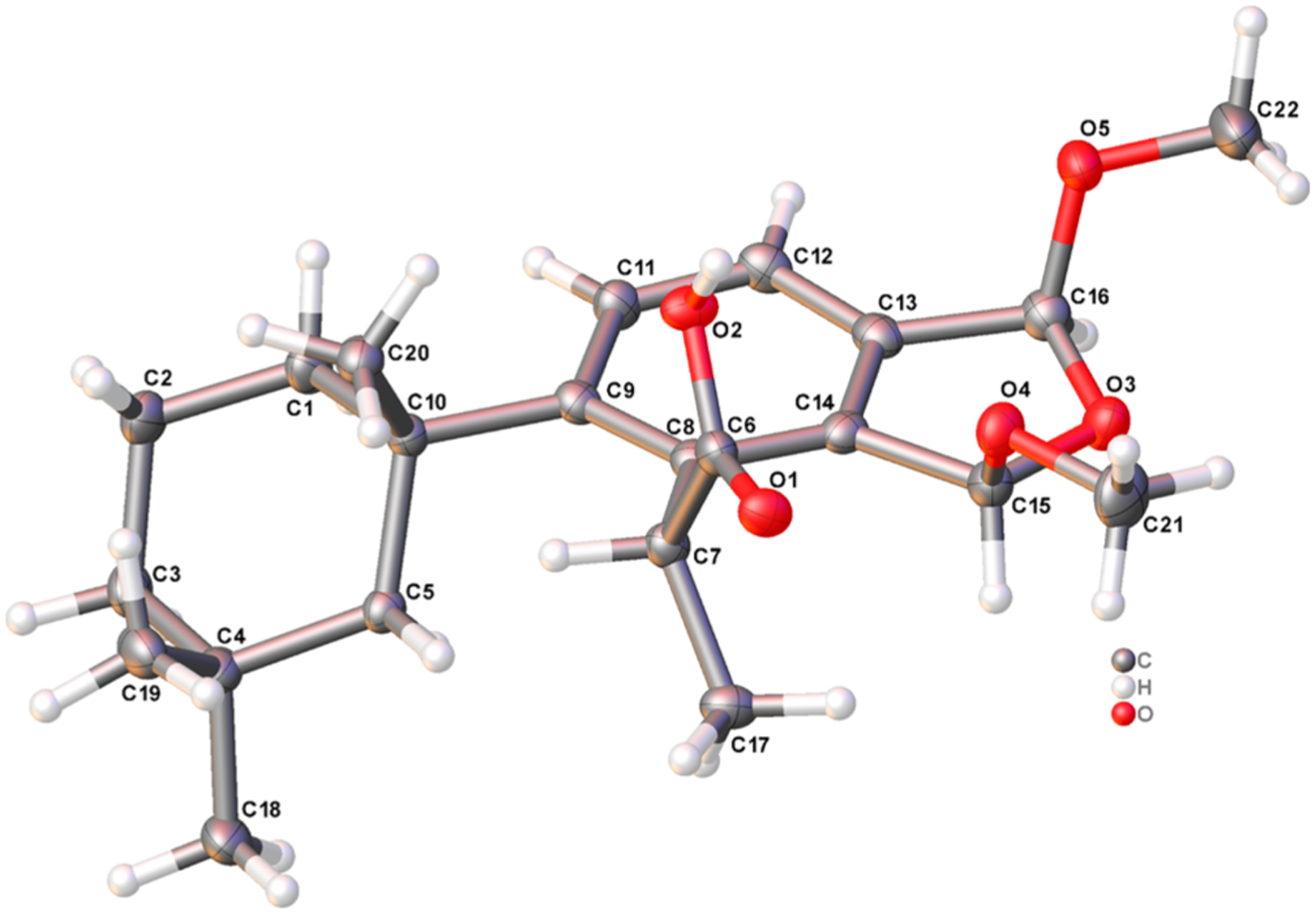
X-ray crystal structure depicting the absolute configuration of membranoid E (9). Anisotropic displacement parameters are drawn at 50% probability.
Because both of the cis-bis(methyl acetal) diastereomers are accounted for by membranoids C (7) and E (9), both membranoid B (6) and membranoid D (8) must display a trans-orientation about the dihydrofuran ring. Examination of the overlapping 1H NMR shifts of the H-15 and H-16 acetal methines of the cis diastereomers reveals a trend in which the β-orientation of the acetal methines as seen in 7 results in a more shielded shift for both positions (δH 5.89) relative to the α-orientation seen in 9, which are more deshielded (δH 6.02). The acetal methine 1H NMR shifts seen in membranoid B (6) and membranoid D (8) reflect this trend and support a trans configuration about the dihydrofuran ring for both compounds (Table 1). In membranoid B (6), H-15 (δH 6.32) appears more deshielded relative to H-16 (δH 6.24), indicating an (R,R) conformation about stereocenters C-15 and C-16, respectively, with an α-orientation for H-15 and β-orientation for H-16. Observation of a NOESY correlation obtained in CD3CN between H-15 (δH 6.26) and H3-17 (δH 1.52) supports the α-orientation for H-15 (Figure 4). Conversely membranoid D (8) displays H-15 (δH 6.12) appearing more shielded relative to H-16 (δH 6.31), representing an (S,S) conformation about stereocenters C-15 and C-16, respectively, with an β-orientation for H-15 and α-orientation for H-16. Observation of a ROESY correlation between H3-21 (δH 3.45) and H3-17 (δH 1.71) supports the α-orientation for H3-21 (Figure 4).
Figure 4.
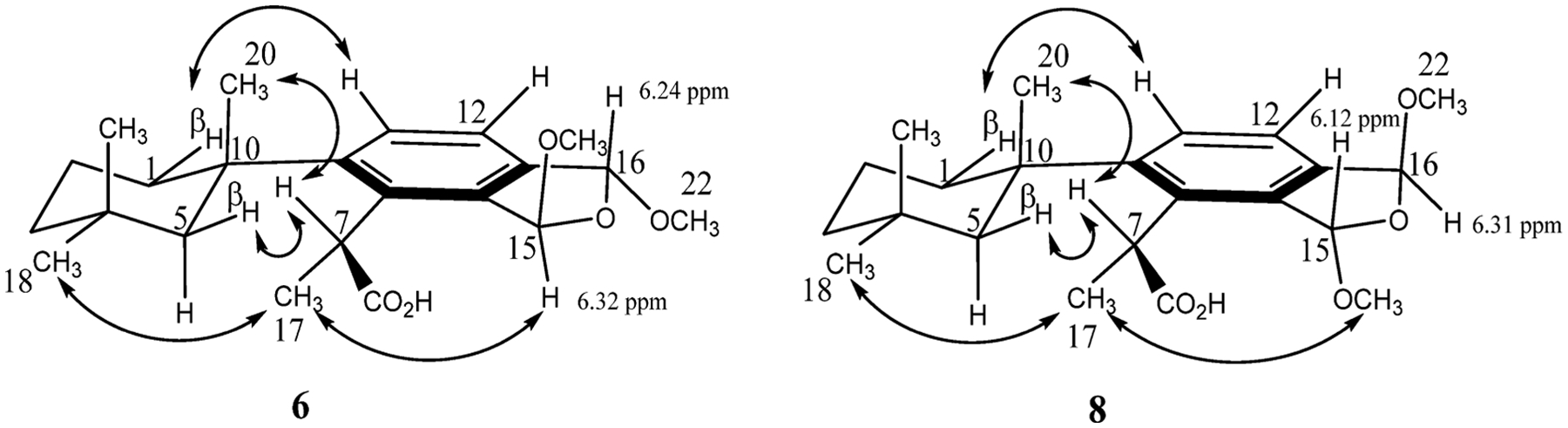
NOESY correlations (CD3CN) and 1H NMR shifts (CDCl3) establishing the relative configurations of membranoids B (6) and D (8).
Treatment of Spongian Diterpenes with MeOH.
Isolation of the four bis(methyl acetal) diastereomers led us once again2 to consider the artifactual nature of the compounds. Extraction of D. antarctica in the absence of MeOH, using just CH2Cl2, produced an extract containing only aplysulphurin (1), tetrahydroaplysufphurin-1 (2), membranolide (3), and darwinolide (4). That extract, when stirred with MeOH for 12 h, displayed new products with masses and retention times consistent with the membranoids. Similarly, purified aplysulphurin, the only natural product present in the extract with the same oxidation state as the membranoids, was similarly treated with MeOH and demonstrated (Figure 5) to convert to a mixture of new compounds bearing appropriate masses (m/z 345.2064 [M + H]+; m/z 399.2148 [M + Na]+; 361.1982 [M + H]+) of membranoids 5–10.
Figure 5.
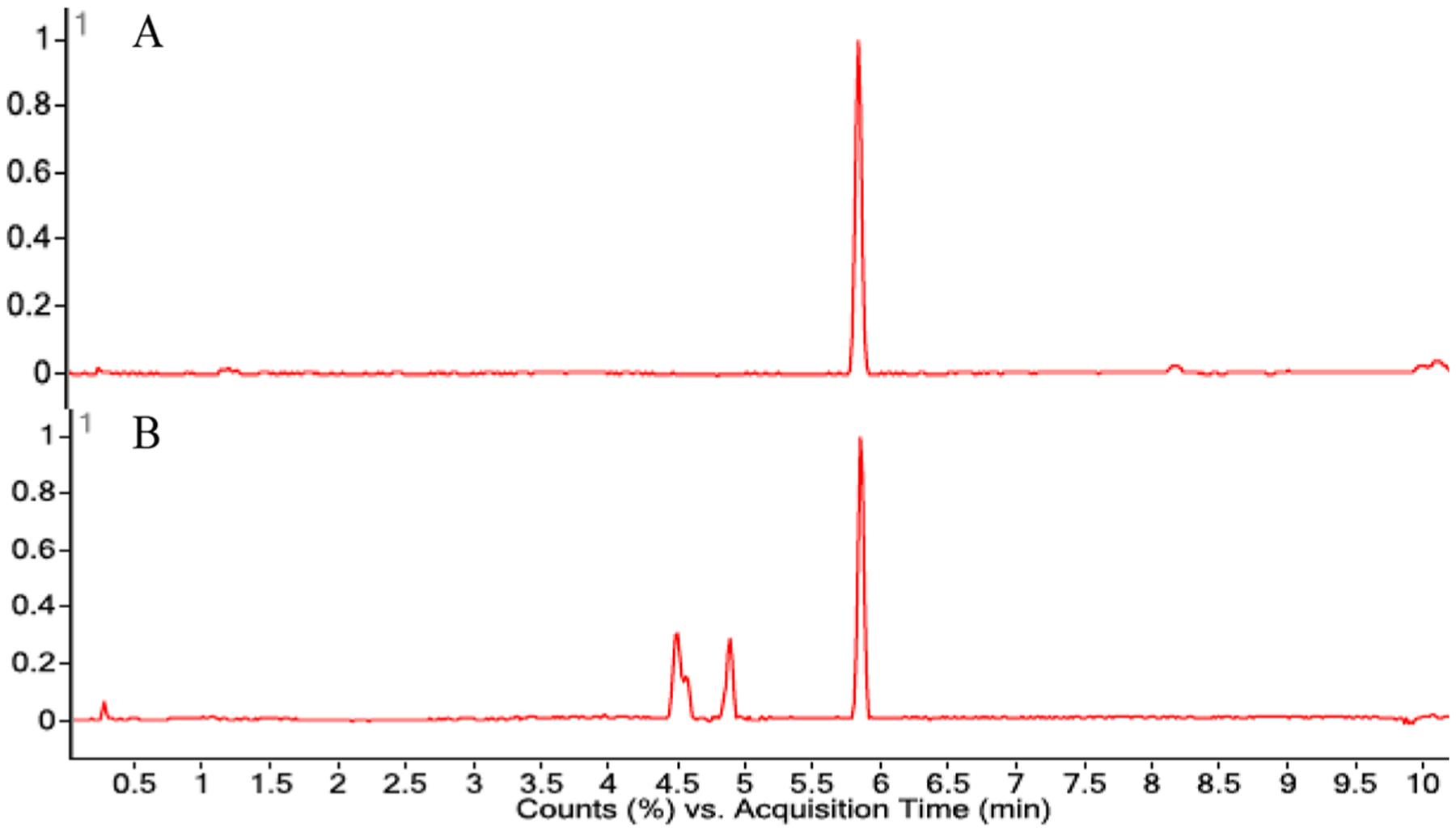
LCMS chromatograms of (A) aplysulphurin (1) and (B) the emergence of new peaks (4.5–5.0 min) with masses matching the membranoids (5–10), after 12 h in MeOH.
Purification of the reaction products from methanolysis of aplysulphurin resulted in the isolation of membranoids A (5, 20.8%), B (6, 4.2%), C (7, 1.3%), D (8, 3.8%), and E (9, 2.9%), along with new compounds membranoids F (10, 2.1%) and G (11, 0.8%). Unreacted aplysulphurin (60%) was also recovered. Membranoid F (10) was isolated as a clear oil with a molecular formula established as C21H28O5 through HRESIMS. The 1H NMR spectrum (Table 2) exhibited several key signals characteristic of the membranoid series including two doublet aromatic protons at δH 7.73 and δH 7.76 (H-11; H-12; J = 8.3 Hz) consistent with a tetrasubstituted phenyl ring, as well as doublet methyl H3-17 (δC 1.73; J = 7.3 Hz), gem-dimethyl H3-18 (δH 0.48) and H3-19 (δH 0.95), and singlet methyl H3-20 (δH 1.42) groups. The anomalous shielded chemical shift for H3-18 again identified it as the axial methyl signal on C-4 of the cyclohexane ring, which is characteristic of the 1′,3′,3′-trimethylcyclohexylbenzene system due to the proximity of the axial methyl in the anisotropic shielding zone of the aromatic ring.15 Assignment of the membranoid F planar structure was completed via HMBC correlations and comparison of 13C NMR data to that of previously reported compounds.2,3,15 For example, connectivity to the aromatic ring from C-10 (δC 40.3) was established via HMBC correlations from H3-20 to aromatic C-9 (δC 154.9) (Figure 6). Methine singlet H-15 (δH 6.14) displays HMBC correlations to a lactone-type C-16 (δC 168.6) as well as C-21 (δC 56.5) and C-13 (δC 126.0), establishing the connectivity of the methoxy furanone moiety to the aromatic ring. Consistent with previous studies, spatial arrangement of methyl doublet H3-17 and methine quartet H-7 (δH 4.73; J = 6.7) was established via ROESY correlations of H-7 to H-5β (δH 2.13) (e.g., Figure 6).14 The configuration of the methoxy furanone was established by observation of a ROESY correlation between H3-17 and H3-21 (δH 3.59), establishing an α-orientation of the C-15 methyl acetal.
Table 2.
| membranoid F (10) | membranoid G (11) | membranoid H (12) | ||||
|---|---|---|---|---|---|---|
| pos | δC, typec | δHd (J in Hz) | δC, typec | δHd (J in Hz) | δC, typec | δHd (J in Hz) |
| 1α | 41.2, CH2 | 1.50, ov m | 39.8, CH2 | 1.52, m | 41.9, CH2 | 1.45, ov m |
| β | 2.32, d (13.7) | 2.23, m | 2.33, d (14.3) | |||
| 2α | 19.9, CH2 | 1.69, ov m | 19.9, CH2 | 1.73, m | 20.8, CH2 | 1.67, m |
| β | 1.80, m | 1.80, m | 1.79, m | |||
| 3α | 39.7, CH2 | 1.33, ov m | 39.8, CH2 | 1.33, m | 40.5, CH2 | 1.31, m |
| 4 | 31.8, C | 31.8, C | 32.4, C | |||
| 5α | 50.6, CH2 | 1.55, d (14.2) | 50.5, CH2 | 1.59, m | 50.9, CH2 | 1.52, m |
| β | 2.13, d (14.2) | 2.06, m | 2.16, d (14.2) | |||
| 6 | 177.6, C | 174.3, C | 175.6, C | |||
| 7 | 40.8, CH | 4.73, q (6.7) | 39.5, CH | 4.56, q (7.3) | 41.7, CH | 4.64, q (7.2) |
| 8 | 137.5, C | 138.2, C | 137.9, C | |||
| 9 | 154.9, C | 154.4, C | 150.1, C | |||
| 10 | 40.3, C | 40.5, C | 40.4, C | |||
| 11 | 130.3, CH | 7.76, d (8.3) | 128.4, CH | 7.76, d (8.3) | 122.4, CH | 7.65, d (8.3) |
| 12 | 123.9, CH | 7.73, d (8.3) | 134.1, CH | 7.73, d (8.3) | 130.2, CH | 7.30, d (8.3) |
| 13 | 126.0, C | 130.5, C | 137.5, C | |||
| 14 | 145.1, C | 134.0, C | 140.8, C | |||
| 15 | 103.6, CH | 6.14, s | 92.9, CH | 7.26, s | 107.4, CH | 5.86, s |
| 16 | 168.6, C | 192.5, CH | 10.1, s | 97.7, CH | 6.98, s | |
| 17 | 17.7, CH3 | 1.73, d (7.3) | 24.2, CH3 | 1.87, d (7.1) | 17.6, CH3 | 1.63, d (7.1) |
| 18 | 27.3, CH3 | 0.48, s | 28.2, CH3 | 0.58, s | 27.5, CH3 | 0.47, s |
| 19 | 32.7, CH3 | 0.95, s | 32.2, CH3 | 0.97, s | 33.2, CH3 | 0.93, s |
| 20 | 32.7, CH3 | 1.42, s | 32.4, CH3 | 1.32, s | 33.3, CH3 | 1.38, s |
| 21 | 56.5, CH3 | 3.59, s | 55.3, CH3 | 3.39, s | ||
| 22 | 171.2, C | |||||
| 23 | 21.5, CH3 | 2.06, s | ||||
| OH | 5.00, bs | |||||
CDCl3.
CD3CN.
125 MHz.
500 MHz; ov = overlapped.
Figure 6.
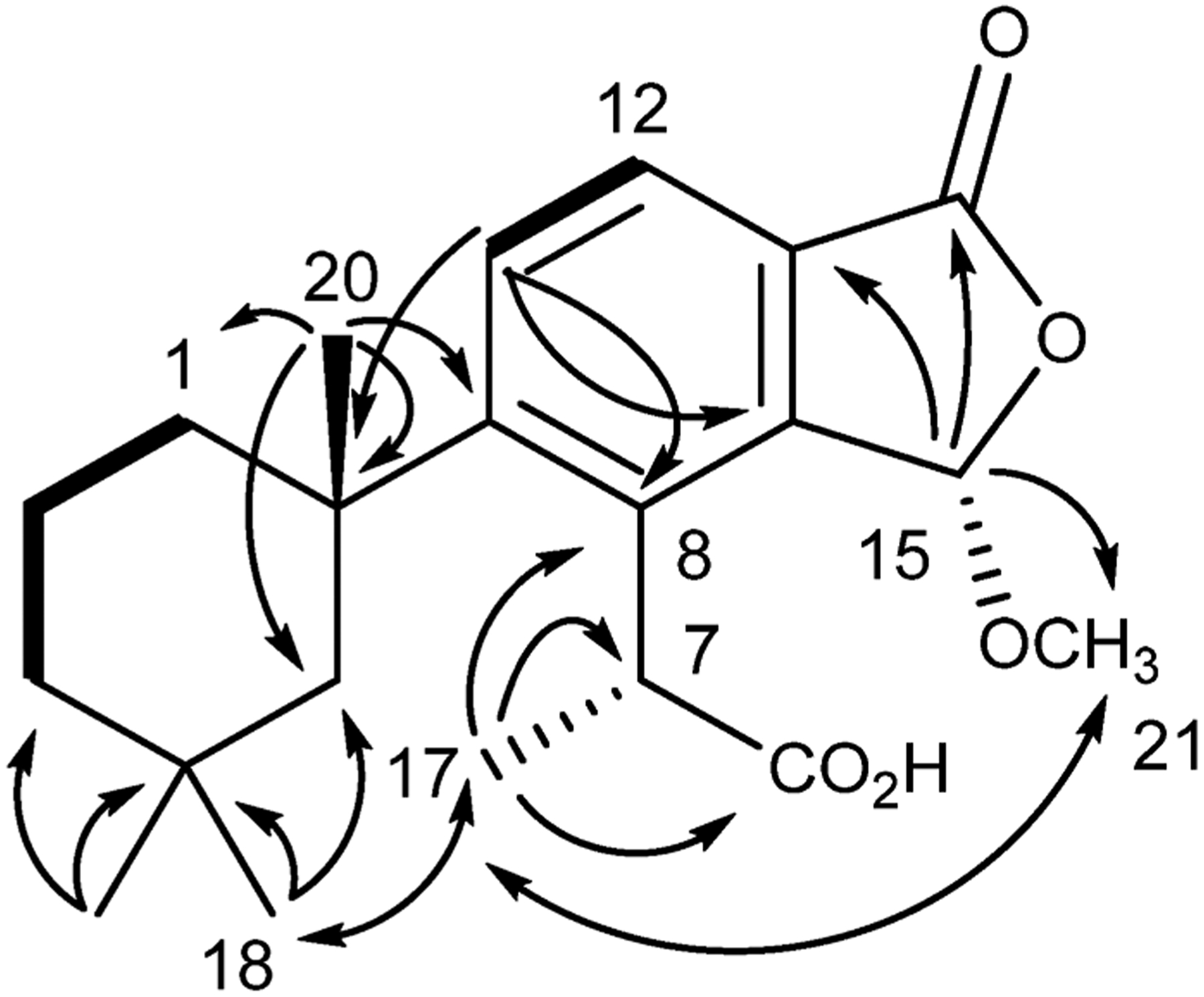
Key HMBC (→), COSY (―), and ROESY (↔) correlations for membranoid F (10).
Membranoid G (11) was isolated as a clear oil with a molecular formula of C20H26O4 established by HRESIMS. The 1H and 13C NMR data (Table 2) exhibited general features similar to those observed for 5 including an aldehyde singlet H-16 (δH 10.1), doublet methyl H3-17 (δH 1.87; J = 7.1 Hz), gem-dimethyl H3-18 (δH 0.58) and H3-19 (δH 0.97), and a singlet methyl H3-20 (δH 1.32). It was, however, lacking the H3-21 methoxy group and instead bears a hydroxy group (δH 5.00). The only other significant difference observed in the 1H NMR data was at position C-15, where the methine shift at δH 7.26 (H-15) differs by 0.46 ppm compared to 5, suggesting 11 is similar but contains a hemiacetal at C-15 instead of a methyl acetal. This is supported by HMBC correlations of H-15 to C-6 (δC 174.3), C-8 (δC 138.2), and C-13 (δC 130.5), establishing the hemiacetal at the C-15 position within the lactone ring. Additional correlations of H-12 to C-16 (δC 192.5) and H-16 to C-13 (δC 130.5) and C-14 (δC 134.0) place the aldehyde as a substituent on the benzene ring at the C-16 position. In 1984 Karuso et al. in fact reported14 the hemiacetal equivalent structure to 5 as a semisynthetic derivative obtained by heating aplysulphurin in wet DMSO and purified using HPLC; however, the aldehyde reported in that paper differs from 11 in its doublet methyl shift (H3-17) by 0.19 ppm, suggesting that membranoid G (11) could be a diastereomer, a notion that is supported by observation of a NOESY correlation between H3-17 and the proton of the hydroxy group (Figure 7).
Figure 7.
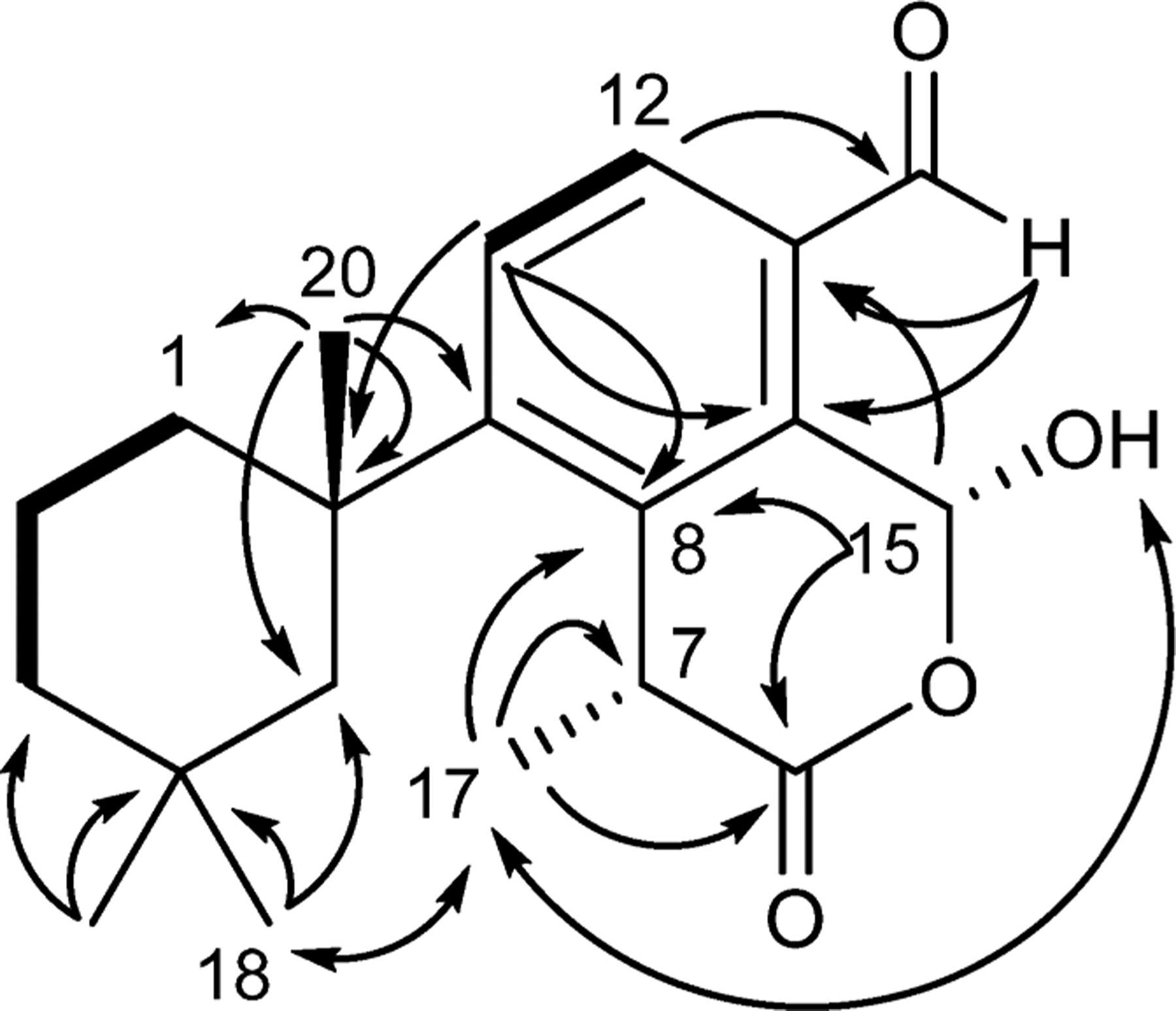
Key HMBC (→), COSY (―), and NOESY (↔) correlations for membranoid G (11).
Acquisition of an X-ray crystal structure confirmed membranoid G (11) to be epimeric at the C-15 center to the compound originally described by Karuso et al., 1984, with the difference in the doublet methyl shift (H3-17) likely due to the deshielding effect of its closer three-dimensional proximity to the hemiacetal hydroxy group (Figure 8). Large pristine crystals ideal for X-ray crystallographic analysis were grown by slow cooling of a saturated solution in a 9:1 mixture of hexane/CH2Cl2 from 25 °C down to a temperature of 10 °C over a period of 10 days. The crystallographic metadata (Flack parameter −0.13(15)), Bijvoet-pair analysis, and Bayesian statistics method (P2 (true) = 1; P3 (true) = 1; P3 (false) = 0.3 × 10−17; P3 (rac-twin) = 0.4 × 10−5 (Table S4)) established the configuration depicted in Figure 8 as the absolute configuration of membranoid G, displaying an (R) conformation about stereocenter C-15, with the hemiacetal methine in the β-orientation.
Figure 8.
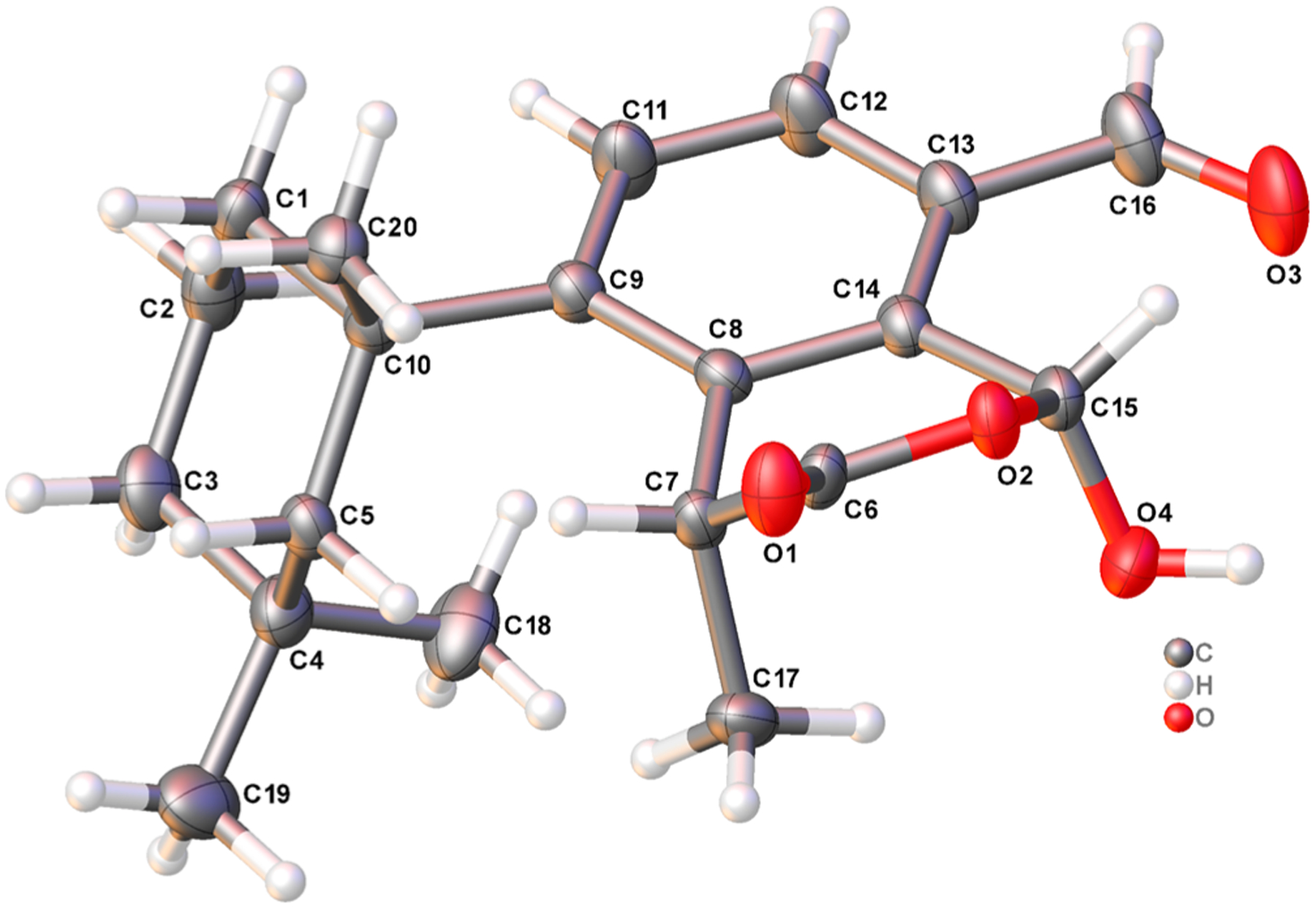
X-ray crystal structure depicting the absolute configuration of membranoid G (11). Anisotropic displacement parameters are drawn at 50% probability.
A subsequent methanolysis reaction, increased by an order of magnitude to a 250 mg scale, produced similar yields of membranolides A–G, except that no membranoid F was recovered and a new membranoid, membranoid H (12, 7.1%), was found. We have noted in subsequent observations of the methanolysis reaction that membranoid H is not isolated from the mixture if the reaction is allowed to stir for long periods (>48 h). Furthermore, membranoid H degraded to a binary mixture of membranoids A (5) and G (11) upon standing in chloroform for 72 h, leading us to believe membranoid H is a reactive intermediate that is formed early in the reaction and eventually progresses to the other products. Similarly, if left in chloroform over several months, the bis(methyl acetal) membranoids (6–9) will also degrade to a mixture of the two aldehydes 5 and 11.
Membranoid H (12) was obtained as a clear oil with spectroscopic data similar to those of membranoids B–E (6–9) but containing an additional carbon and oxygen. A formula of C23H32O6 was established from HRESIMS. Key 1H NMR signals (Table 2) include a gem-dimethyl group at H3-18 (δH 0.47) and H3-19 (δH 0.93), a singlet methyl at H3-20 (δH 1.38), a doublet methyl at H3-17 (δH 1.63; J = 7.1), a quartet methine at H-7 (δH 4.64, J = 7.2), and the pair of aromatic protons at H-11 (δH 7.65) and H-12 (δH 7.30) and establish this structure as the membranoid scaffold. Two acetal methine signals at H-15 (δH 5.86) and H-16 (δH 6.98) indicate this as a fused furano-membranoid supported in their positions by HMBC correlations of H-15 to C-13 (δC 137.5) and H-16 to C-12 (δC 130.2) and C-14 (δC 140.8). Signals establishing the substituents about the dihydrofuran ring include one methoxy group at H3-21 (δH 3.39) and one acetate methyl at H3-23 (δH 2.06), supported in their positions by HMBC correlations of H-15 to C-21 (δC 55.3), and H-16 to C-22 (δC 171.2) as well as H3-23 to C-22. The relative configuration of the fused dihydrofuran ring of 12 was established by observation of a NOESY correlation between H3-17 and H3-21 (δH 3.39), establishing the same α-relationship of the C-15 methyl acetal as was observed in membranoid F (10). An additional NOESY correlation was observed between H3-21 and H3-23, indicating a cis conformation and α-orientation of the methoxy and acetate groups about the dihydrofuran ring (Figure 9).
Figure 9.
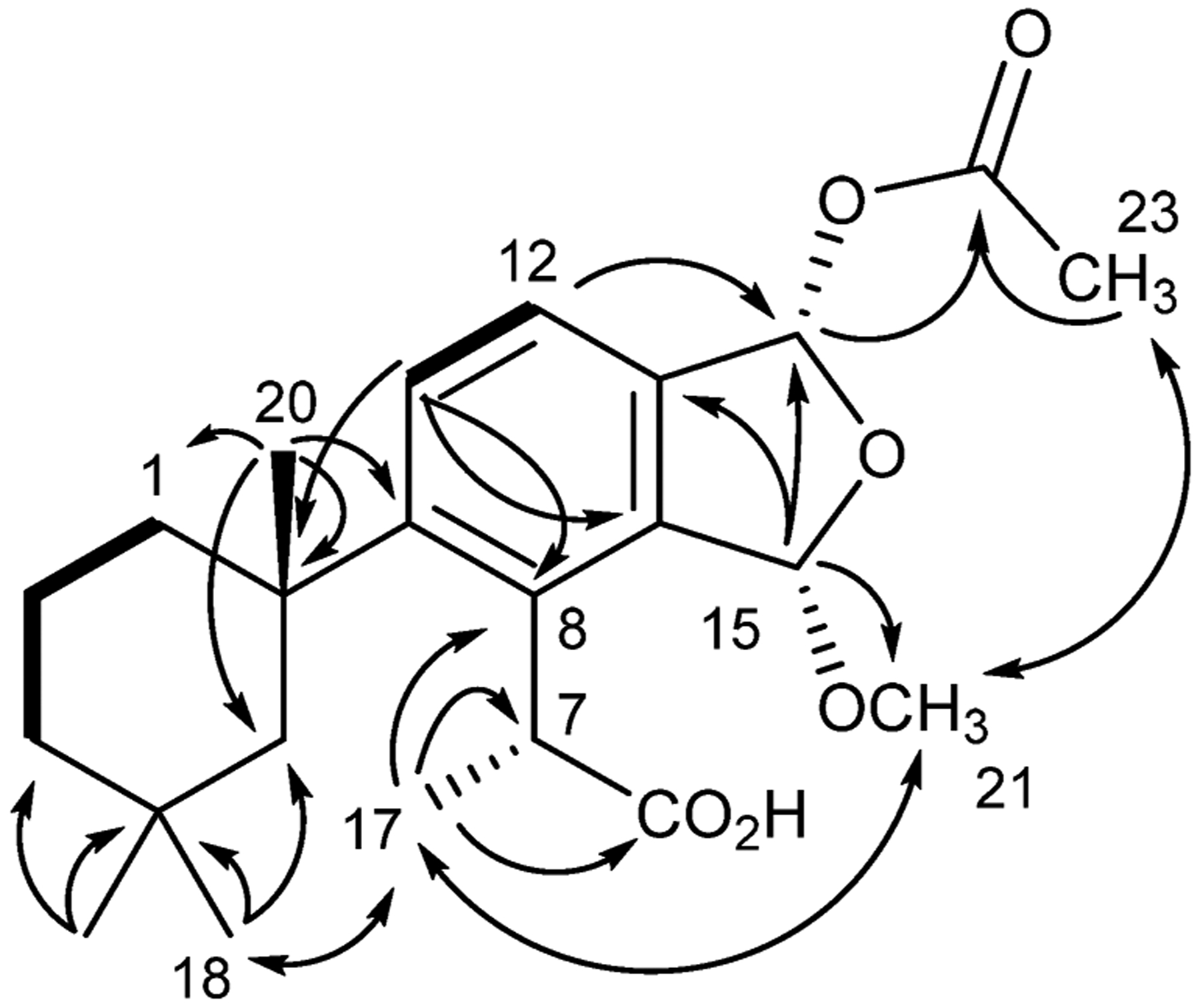
Key HMBC (→), COSY (―), and NOESY (↔) correlations for membranoid H (12).
The reactivity of aplysulphurin (1) is enigmatic, likely driven by the densely functionalized central aromatic ring in a constrained trifused 6/6/5 system. The X-ray structure16 depicts a congested central aromatic ring in the axial position of a trimethylcyclohexane ring, a concave orientation that largely blocks the α-face and produces anisotropic NMR shifts described previously.14 That MeOH, but not EtOH,2 induces solvolysis emphasizes the congestion. Further studies of the mechanistic aspects of solvolysis that explain the cascade of products are currently ongoing.
Leishmaniasis Activity of D. antarctica-Derived Compounds.
The full suite of D. antarctica-derived compounds were tested against L. donovani-infected macrophages. Companion cytotoxicity measurements were conducted using the host mammalian J774A.1 cell line. The results (Table 3) suggest modest activity (lower activity than the miltefosine positive control) for membranolide (3, 9.7 μM), darwinolide (4, 11.2 μM), and membranoids C (7, 6.5 μM), E (9, 6.6 μM), and H (12, 12.0 μM), but potent bioactivity (greater activity than the miltefosine positive control) for aplysulphurin (1, 3.10 μM) and tetrahydroaplysulphurin-1 (2,3.50 μM), as well as membranoids B (6, 0.8 μM), D (8, 1.40 μM), and G (11, 2 μM). In addition, a high selectivity index (SI, mammalian cytotoxicity/antiprotozoal activity) was observed for 2 (>38), 6 (>166), 8 (>95), and 11 (>100). Three of these potent and selective hits (6, 8, 11) are newly reported artifacts, showing greater antileishmanial activity than the standard treatment of miltefosine (2.9 μM), and are more active and far more selective than the aplysulphurin precursor (1, 3.10 μM, SI 4.0). To our surprise, both of the cis methyl acetals membranoids C (7) and E (9) were less active against L. donovani-infected macrophages compared with their diastereomers 6 and 8, which are both trans about the dihydrofuran ring. This makes the functionality and orientation of these substituents at positions C-15 and C-16 crucial for L. donovani bioactivity and could represent a feasible starting point for further SAR studies utilizing the membranoid backbone. Another interesting trend is observed in the stark contrast between the activity and selectivity of membranoids A (5) and G (11). While they are structurally very similar, each bearing an aldehyde at the C-16 position and differing only in the relative configuration and functionality of the oxidized C-15 carbon, membranoid A (5) was inactive against L. donovani-infected macrophages at the highest concentrations tested and mildly toxic to the J774A.1 cells, while membranoid G showed potent activity against the infection with no discernible cytotoxicity against healthy mammalian cells. This could also represent a promising motif practicable for optimization through SAR.
Table 3.
Bioactivity of D. antarctica-Derived Spongian Diterpenes and Their Methanolysis Products (IC50, μM)
| compound | Leishmania donovani IM | cytotoxicity (J774A.1) |
|---|---|---|
| aplysulphurin (1) | 3.1 | 12.3 |
| tetrahydroaplysulphurin-1 (2) | 3.5 | >133 |
| membranolide (3) | 9.7 | 76.8 |
| darwinolide (4) | 11.2 | 73.4 |
| membranoid A (5) | >29.0 | 54.6 |
| membranoid B (6) | 0.8 | >133 |
| membranoid C (7) | 6.5 | >133 |
| membranoid D (8) | 1.4 | >133 |
| membranoid E (9) | 6.6 | 95.0 |
| membranoid F (10) | 26.7 | 42.4 |
| membranoid G (11) | 1.9 | >150 |
| membranoid H (12) | 12.0 | 62.9 |
Positive control, miltefosine = 2.9 μM (IM), >120 μM (Cytotox).
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were measured on a Rudolph Research Analytical AUTOPOL IV digital polarimeter. UV absorptions were measured by an Agilent Cary 60 UV–vis spectrophotometer in MeOH or MeCN, while IR spectra were recorded with an Agilent Cary 630 FTIR. All NMR spectra were acquired in CDCl3 or CD3CN with residual solvent referenced as an internal standard (7.27 and 1.94 ppm for 1H and 77.0 and 1.29 ppm for 13C, respectively). All 1H NMR spectra were recorded on a Varian 500 or 600 MHz Direct Drive instrument equipped with cold-probe detection, and 13C NMR spectra were recorded at 125 or 150 MHz, respectively. Analytical LC/MS was performed on a Phenomenex Kinetex C18 column (50 × 2.1 mm, 2.6 μm) on either an Agilent 6120 single quadrupole, an Agilent 6230 LC/ToF-MS, or an Agilent 6540 LC/QToF-MS with electrospray ionization detection, the latter two of which were utilized for HRESIMS. All HPLC analysis was performed on a Shimadzu LC20-AT system equipped with a photodiode array detector (M20A) using semipreparative [Phenomenex Luna C18 (250 × 10 mm, 5 μm)] or analytical [Phenomenex Luna C18 (250 × 4.6 mm, 5 μm) and Phenomenex Luna Silica (250 × 4.6 mm, 5 μm)] conditions. All solvents were obtained from Fisher Scientific and were HPLC grade (>99% purity) unless otherwise stated.
Collection of D. antarctica.
Sponge samples were collected from various sites around Palmer Station, Antarctica, in the Austral summers of 2010, 2011, and 2016. The collection sites chosen were Norsel Point (64°45.674′ S, 64°05.467′ W), Bonaparte Point (64°46.748′ S, 64°02.542′ W), Gamage Point (64°46.345′ S 64°02.915′ W), and Laggard Island (64°48.568′ S, 64°00.984′ W) at depths between 5 and 35 m. Samples were frozen and transported back to the University of South Florida at −70 °C, where tissues were lyophilized and stored at −80 °C until further processing. Specimens examined by Professor Rob van Soest, then at the University of Amsterdam, were previously2,6 identified as Dendrilla membranosa. More recent consideration7 suggests specimens of this sponge originating in Antarctica should be identified as D. antarctica. Previous reports of chemistry from Antarctic D. membranosa should be considered as originating from the same sponge species as studied here.
Extraction and Isolation of Natural Products.
A typical CH2Cl2/MeOH (1:1) extraction was conducted on the 2010 collection. From 330 g of freeze-dried D. antarctica, 12.2 g of CH2Cl2/MeOH was obtained and subsequently fractionated by NP MPLC into 11 fractions using a linear gradient from hexanes to EtOAc to MeOH. The third through fifth fractions, eluting roughly between 20% and 50% EtOAc in hexanes, contained diterpenes. The diterpene fractions were combined, and a 400 mg aliquot separated on RP HPLC using 60% MeCN in water to 100% MeCN. In order of elution were obtained membranoid E (9, 4.8 mg), B (6, 19.9 mg), D (8, 5.3 mg), and C (7, 22.2 mg), as well as natural products membranolide (3, 19.3 mg) and tetrahydroaplysulphurin-1 (2, 52.3 mg).
Similarly, a typical CH2Cl2 extraction is illustrated by a 25.7 g sample of freeze-dried D. antarctica, which was extracted three times with CH2Cl2 (ACS grade), and the combined extracts were concentrated in vacuo. The lipophilic extract (994 mg) was absorbed onto Waters Sep-Pak C18 cartridges and eluted with MeCN. The dried eluate (205 mg) was separated by isocratic semipreparative HPLC using 60% MeCN in H2O for 35 min and ramping up to 100% MeCN after 50 min to afford (in retention time order) membranolide (3, 8.7 mg), aplysulphurin (1, 10.2 mg), tetrahydroaplysulphurin-1 (2, 1.5 mg), and darwinolide (4, 2.0 mg).
Methanolysis of Aplysulphurin.
Aplysulphurin (1) was dissolved to 1 mg/mL in MeOH; 10 μL aliquots were taken at time zero and then every 3 h. Those aliquots were transferred to LCMS vials, dried under nitrogen, and resuspended in 1 mL of acetonitrile. Sample analysis was performed via LC/QToF-MS with a gradient of 40–60% acetonitrile in water (0.1% formic acid) over 7 min, before a ramp to 100% acetonitrile for 10 min.
Isolation of Membranoids.
Previously purified aplysulphurin (1, 24.0 mg) treated with MeOH for 48 h was subject to semipreparative RP HPLC using a 60–65% MeCN in H2O gradient over 30 min followed by NP analytical HPLC with 10–100% EtOAc in hexane, yielding membranoid A (5, 5.0 mg), membranoid B (6, 0.7 mg), membranoid C (7, 0.3 mg), membranoid D (8, 0.9 mg), membranoid E (9, 1.0 mg), membranoid F (10, 0.5 mg), and membranoid G (11, 0.2 mg). No membranoid H (12) was isolated.
The methanolysis was then scaled up by an order of magnitude, stirring 250 mg of aplysulphurin (1) in MeOH at 1 mg/mL for 24 h. The MeOH was then removed under reduced pressure, and the mixture was subjected to semipreparative NP HPLC, this time using a 10–50% EtOAc in hexane gradient over 30 min, which was sufficient to achieve separation of the products. This yielded membranoid A (5, 27.3 mg), membranoid B (6, 8.8 mg), membranoid C (7, 19.9 mg), membranoid D (8, 23.5 mg), membranoid E (9, 10.5 mg), membranoid G (11, 0.7 mg), and membranoid H (12, 17.7 mg). The membranoid H (12) isolated from the scale up degraded to a binary mixture of membranoids A (5, 7.3 mg) and G (11, 7.1 mg) upon solvation in CHCl3 over a 72 h period, and each was purified under the same NP HPLC conditions and confirmed by NMR data.
Membranoid B (6):
clear oil; [α]20D −87 (c 0.6, CHCl3); UV (MeOH) λmax (log ε) 269 nm (3.57); IR ν (thin film) 2940, 1733, 1698, 1585, 1467, 1382 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 399.2139 [M + Na]+ (calcd for 399.2142, C22H32O5Na).
Membranoid C (7):
clear oil that crystallized on long-standing; [α]20D −100 (c 0.6, CHCl3); UV (MeOH) λmax (log ε) 267 nm (3.31); IR ν (thin film) 2940, 1733, 1698, 1585, 1467, 1382 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 399.2141 [M + Na]+ (calcd for 399.2142, C22H32O5Na).
Membranoid D (8):
clear oil; [α]20D +33 (c 0.6, CHCl3); UV (MeOH) λmax (log ε) 267 nm (3.41); IR ν (thin film) 2940, 1733, 1698, 1585, 1467, 1390 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 399.2145 [M + Na]+ (calcd for 399.2142, C22H32O5Na).
Membranoid E (9):
clear oil that crystallized on long-standing; [α]20D −37 (c 0.6, CHCl3); UV (MeOH) λmax (log ε) 269 nm (3.48); IR ν (thin film) 2940, 1733, 1698, 1585, 1467, 1382 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 399.2140 [M + Na]+ (calcd for 399.2142, C22H32O5Na).
Membranoid F (10):
clear oil; [α]20D +135 (c 0.14, CHCl3); UV (MeOH) λmax (log ε) 223 nm (3.32); 1H and 13C NMR data, Table 2; HRESIMS m/z 383.1821 [M + Na]+ (calcd for 383.1829, C21H28O5Na).
Membranoid G (11):
clear oil that could be crystallized on long-standing; [α]20D −50 (c 0.14, CHCl3); UV (MeOH) λmax (log ε) 259 nm (3.71); IR ν (thin film) 3340, 2940, 2865, 1740, 1698, 1585, 1462, 1390 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 353.1723 [M + Na]+ (calcd for 353.1723, C20H26O4Na).
Membranoid H (12):
clear oil; [α]25D +50 (c 0.17, MeCN); UV (MeCN) λmax (log ε) 264 nm (3.38); IR ν (thin film) 2940, 1740, 1704, 1472, 1385 cm−1; 1H and 13C NMR data, Table 2; HRESIMS m/z 427.2089 [M + Na]+ (calcd for 427. 2091 C23H32O6Na).
X-ray Crystallography.
Crystals of 7 and 9 were obtained from EtOAc/hexane, while crystals of 11 were grown in CH2Cl2/hexane. The X-ray diffraction data were measured on a Bruker D8 Venture PHOTON II CPAD diffractometer equipped with a Cu Kα INCOATEC ImuS microfocus source (λ = 1.541 78 Å). Indexing was performed using APEX3 (Difference Vectors method).17 Data integration and reduction were performed using SaintPlus.18 Absorption correction was performed by the multiscan method implemented in SADABS.19 Space group was determined using XPREP implemented in APEX3.17 Structure was solved using SHELXT and refined using SHELXL-2018 (full-matrix least-squares on F2) through the OLEX2 interface program.20–22 All molecules are conformationally strained: corresponding hydrogen atoms were refined with restraints and with Uiso = 1.2Ceq (1.5 for the −CH3 group). All other hydrogen atoms were placed in geometrically calculated positions and were included in the refinement process using a riding model with isotropic thermal parameters. Absolute configuration for all compounds was established based on the Flack parameter value and verified additionally with Bijvoet-pair analysis and Bayesian statistics methods (Table S4).23,24 P2 = 1 for all cases and is the probability that the current model is correct assuming two possibilities: one out of two enantiomers present. Crystal data and refinement conditions are shown in Tables S1–S3.
Crystallographic Data for Membranoid C (7):
C22H32O5, M = 376.47, crystal size 0.14 × 0.079 × 0.054 mm3, orthorhombic, a = 7.8794(2) Å, b = 13.8188(3) Å, c = 18.3689(4) Å, α = β = γ = 90°, V = 2000.08(8) Å3, T = 100 K, space group P212121, Z = 4, Dcalcd = 1.250 g/cm3, μ = 0.704 mm−1, F(000) = 816.0, 4201 independent reflections (Rint = 0.0477, Rsigma = 0.0300). The final R1 = 0.0305 (I ≥ 2σ(I)), wR2 = 0.0733 (I ≥ 2σ(I)), R1 = 0.0332 (all data), wR2 = 0.0756 (all data), S = 1.064. The Flack parameter was −0.03(7).
Crystallographic Data for Membranoid E (9):
C22H32O5, M = 376.47, crystal size 0.097 × 0.09 × 0.037 mm3, monoclinic, a = 13.4041(3) Å, b = 9.8471(2) Å, c = 16.3030(4) Å, α = 90°, β = 105.2120(10)°, γ = 90°, V = 2076.46(8) Å3, T = 100 K, space group P21, Z = 4, Dcalcd = 1.204 g/cm3, μ = 0.678 mm−1, F(000) = 816.0, 8629 independent reflections (Rint = 0.0636, Rsigma = 0.0437). The final R1 = 0.0363 (I ≥ 2σ(I)), wR2 = 0.0826 (I ≥ 2σ(I)), R1 = 0.0439 (all data), wR2 = 0.0868 (all data), S = 1.046. The Flack parameter was −0.01(8).
Crystallographic Data for Membranoid G (11):
C20H26O4, M = 330.41, crystal size 0.19 × 0.12 × 0.12 mm3, orthorhombic, a = 10.9817(4) Å, b = 11.0246(4) Å, c = 14.6564(5) Å, α = β = γ = 90°, V = 1773.23(11) Å3, T = 134.98 K, space group P212121, Z = 4, Dcalcd = 1.238 g/cm3, μ = 0.684 mm−1, F(000) = 712.0, 3513 independent reflections (Rint = 0.0463, Rsigma = 0.0450). The final R1 = 0.0385 (I ≥ 2σ(I)), wR2 = 0.0961 (I ≥ 2σ(I)), R1 = 0.0394 (all data), wR2 = 0.0971 (all data), S = 1.056. The Flack parameter was −0.13(15).
Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1942687 for 7, CCDC 1942688 for 9, and CCDC 1942689 for 11. Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (tel: (+44) 1223-336-408; fax: (+44) 1223-336-033; or email: deposit@ccdc.cam.ac.uk).
Biological Assays.
Leishmania donovani Cell Line Infected Macrophage Assay.
MHOM/SD/75/1246/130 L. donovani axenic amastigotes were cultured in RPMI 1640 at a pH of 5.5 with 7.5 g/L HEPES (Invitrogen Corp.), 5.86 g/L MES (Sigma-Aldrich), 2 g/L sodium bicarbonate (Fisher Scientific), 10 mg/L hemin (Sigma-Aldrich), 100 μM xanthine (Sigma-Aldrich), 40 mg/L Tween-80 (Sigma-Aldrich), 1% penicillin–streptomycin, 5 g/L Trypton-Peptone (BD Bioscience), and 20% 24 h heat-inactivated FBS. L. donovani was incubated at 37 °C. All culturing was done using nonvented 25 cm2 tissue culture flasks (Corning). J774A.1 macrophages (ATCC) were cultured using RPMI 1640 (Gibco), 1% penicillin–streptomycin (Gibco, 10,000 u/mL), and 10% 24 h heat-inactivated fetal bovine serum (Gibco) at a pH of 7.2. All culturing was done using vented 75 cm2 tissue culture flasks (Corning) and incubated at 37 °C, 5% CO2.
In a 384-well plate (CellCarrier-384 black, optically clear bottom, tissue culture treated, sterile), 2000 J774A.1 cells were seeded. L. donovani axenic amastigotes (MHOM/SD/75/1246/130 cell line) were then added to the plate at a ratio of 10:1 and incubated at 37 °C, 5% CO2 for 24 h. The excess extracellular amastigotes were then washed away using prewarmed media. Compounds 1–12, in addition to the positive control miltefosine, were prepared in a 384-well plate (Thermo Scientific Nunc 384-well clear polystyrene plates (non/treated surfaces)) with a starting concentration of 10 μg/mL and serially diluted at 1:2. Drugs were then added to the assay plate and incubated at 37 °C, 5% CO2 for 72 h. Cells were then fixed with 2% paraformaldehyde (Alfa Aesar paraformaldehyde, 16% w/v aqueous solution, MeOH free) and incubated for 15 min at room temperature, then stained with 5 μM Draq5 (Thermo Scientific DRAQ5 fluorescent probe) and incubated for 5 min at room temperature. A PerkinElmer Operetta (high-content imager) was used to capture images for each well and find macrophage and amastigote nuclei within macrophage cytoplasm using Harmoney software that counts the number of amastigotes per 500 macrophages in each well and generated IC50 values.
Cytotoxicity Assay.
Cytotoxicity was determined by using a colorimetric assay where compounds 1–12 and the positive control miltefosine were tested against J774.A1 macrophages using the CellTiter 96 AQueous One solution cell proliferation assay (Promega). In a 96-well drug plate (Costar, assay plate, 96-well with low-evaporation lid, flat bottom, nontreated) drugs were diluted in a series of six 2-fold dilutions in media to produce a concentration range from 500 to 15.625 μg/mL. A 10 μL amount from each well was transferred to another 96-well plate (Costar, assay plate, 96-well with low-evaporation lid, flat bottom, tissue culture treated), and 90 μL of macrophages in media in a 50 000 cell per well concentration was added to produce a final concentration range from 50 to 1.6 μg/mL. The plates were then incubated at 37 °C, 5% CO2 for 72 h. A 20 μL amount of 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium (MTS: Promega) solution was added to each well and incubated for an additional 4 h. A Spectra Max M2e (Molecular Devices) was used to measure optical density at 490 nm. Nonlinear regression via Trifox software was used to determine IC50 values.
Supplementary Material
Chart 1.
ACKNOWLEDGMENTS
This work was funded by the National Science Foundation awards ANT-0838773 and PLR-1341333 (C.D.A., J.B.M.) and ANT-0838776 and PLR-1341339 (B.J.B.) from the Antarctic Organisms and Ecosystems program and a Center of Excellence award from the State of Florida to support the Center for Drug Discovery and Innovation, whose facilities made much of the chemical analysis possible. We greatly appreciate the outstanding logistical support of the employees and subcontractors of Raytheon Polar Services Company and Antarctic Support Contract. We are appreciative of Dr. E. Rivera for assistance in obtaining high-quality NMR spectra and Dr. L. Wojtas, G. Verma, and C. Shan for assistance with X-ray crystallography. J.B.M. acknowledges the support of an Endowed Professorship from the University of Alabama at Birmingham.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.0c00025.
1H, 13C, 2D NMR, and HRESIMS spectra for membranoids B–H (6–12) (PDF)
X-ray crystallographic metadata for membranoid C (7) (CIF)
X-ray crystallographic metadata for membranoid D (9) (CIF)
X-ray crystallographic metadata for membranoid G (11) (CIF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jnatprod.0c00025
The authors declare no competing financial interest.
Contributor Information
Andrew J. Shilling, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States
Christopher G. Witowski, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States
J. Alan Maschek, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Ala Azhari, Department of Global Health, University of South Florida, Tampa, Florida 33612, United States;; Department of Microbiology and Medical Parasitology, King Abdulaziz University, Jeddah 22252, Saudi Arabia
Brian A. Vesely, Department of Global Health, University of South Florida, Tampa, Florida 33612, United States
Dennis E. Kyle, Department of Global Health, University of South Florida, Tampa, Florida 33612, United States
Charles D. Amsler, Department of Biology, University of Alabama at Birmingham, Birmingham, Alabama 35294, United States
James B. McClintock, Department of Biology, University of Alabama at Birmingham, Birmingham, Alabama 35294, United States
Bill J. Baker, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
REFERENCES
- (1).Fontana A; Scognamiglio G; Cimino G J. Nat. Prod 1997, 60, 475–477. [Google Scholar]
- (2).Ankisetty S; Amsler CD; McClintock JB; Baker BJ J. Nat. Prod 2004, 67, 1172–1174. [DOI] [PubMed] [Google Scholar]
- (3).Molinski TF; Faulkner DJ J. Org. Chem 1987, 52, 296–298. [Google Scholar]
- (4).Manriquez V; San-Martin A; Rovirosa J; Darias J; Peters K Acta Crystallogr., Sect. C: Cryst. Struct. Commun 1990, C46, 2486–2487. [Google Scholar]
- (5).von Salm JL; Witowski CG; Fleeman RM; McClintock JB; Amsler CD; Shaw LN; Baker BJ Org. Lett 2016, 18, 2596–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Baker BJ; Kopitzke RW; Yoshida WY; McClintock JB J. Nat. Prod 1995, 58, 1459–1462. [Google Scholar]
- (7).van Soest R Personal communication.
- (8).Keyzers RA; Northcote PT; Davies-Coleman MT Nat. Prod. Rep 2006, 23, 321–334. [DOI] [PubMed] [Google Scholar]
- (9).van Griensven J; Diro E Infect. Dis. Clin. North Am 2019, 33, 79–99. [DOI] [PubMed] [Google Scholar]
- (10).Joice AC; Yang S; Farahat AA; Meeds H; Feng M; Li J; Boykin DW; Wang MZ; Werbovetz KA Antimicrob. Agents Chemother 2018, 62, DOI: 10.1128/AAC.01129-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kanwar A; Eduful BJ; Barbeto L; Carletti Bonomo P; Lemus A; Vesely BA; Mutka TS; Azhari A; Kyle DE; Leahy JW ACS Med. Chem. Lett 2017, 8, 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Demers DH; Knestrick MA; Fleeman R; Tawfik R; Azhari A; Souza A; Vesely B; Netherton M; Gupta R; Colon BL; Rice CA; Rodriguez-Perez MA; Rohde KH; Kyle DE; Shaw LN; Baker BJ Mar. Drugs 2018, 16, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tchokouaha Yamthe LR; Appiah-Opong R; Tsouh Fokou PV; Tsabang N; Fekam Boyom F; Nyarko AK; Wilson MD Mar. Drugs 2017, 15, No. e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Karuso P; Skelton BW; Taylor WC; White AH Aust. J. Chem 1984, 37, 1081–1093. [Google Scholar]
- (15).Karuso P; Bergquist PR; Cambie RC; Buckleton JS; Clark GR; Rickard CEF Aust. J. Chem 1986, 39, 1643–1653. [Google Scholar]
- (16).Graham SK; Garson MJ; Bernhardt PV J. Chem. Crystallogr 2010, 40, 468–471. [Google Scholar]
- (17).Bruker. APEX3 2015.9 ed; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- (18).Bruker. SAINT 8.35A ed; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- (19).Sheldrick GM SADABS, Program for Empirical Absorption Correction; University of Gottingen, 1996. [Google Scholar]
- (20).Sheldrick GM Acta Crystallogr., Sect. A: Found. Crystallogr 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- (21).Sheldrick GM Acta Crystallogr., Sect. A: Found. Crystallogr 2015, C71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dolomanov OV; Bourhis LJ; Gildea RJ; Howard JAK; Puschmann H J. Appl. Crystallogr 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Spek AL Acta Crystallogr., Sect. D: Biol. Crystallogr 2009, D65, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hooft RWW; Straver LH; Spek AL J. Appl. Crystallogr 2008, 41, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.