

Abstract
Cilia and flagella are evolutionarily conserved eukaryotic organelles involved in cell motility and signaling. In humans, mutations in Radial Spoke Head Component 4A (RSPH4A) can lead to primary ciliary dyskinesia (PCD), a life-shortening disease characterized by chronic respiratory tract infections, abnormal organ positioning, and infertility. Despite its importance for human health, the location of RSPH4A in human cilia has not been resolved, and the structural basis of RSPH4A–/– PCD remains elusive. Here, we present the native three-dimensional structure of RSPH4A–/– human respiratory cilia using samples collected noninvasively from a PCD patient. Using cryo–electron tomography (cryo-ET) and subtomogram averaging, we compared the structures of control and RSPH4A–/– cilia, revealing primary defects in two of the three radial spokes (RSs) within the axonemal repeat and secondary (heterogeneous) defects in the central pair complex. Similar to RSPH1–/– cilia, the radial spoke heads of RS1 and RS2, but not RS3, were missing in RSPH4A–/– cilia. However, RSPH4A–/– cilia also exhibited defects within the arch domains adjacent to the RS1 and RS2 heads, which were not observed with RSPH1 loss. Our results provide insight into the underlying structural basis for RSPH4A–/– PCD and highlight the benefits of applying cryo-ET directly to patient samples for molecular structure determination.
INTRODUCTION
Cilia and flagella are highly conserved, microtubule-based organelles that extend from the surfaces of eukaryotic cells and play critical roles in cell motility, sensing, and signaling. In mammals, motile cilia are required for a number of physiological processes, including embryonic development, cerebrospinal fluid circulation, mucus clearance, and sperm motility (Satir and Christensen, 2007; Drummond, 2012). Ciliary dysfunction causes diverse disorders collectively known as ciliopathies, including the severe inherited disorder PCD (OMIM#244400), also known as immotile cilia syndrome (Fliegauf et al., 2007; Brown and Witman, 2014). PCD is usually an autosomal recessive, heterogeneous genetic disorder characterized by chronic respiratory disease, organ laterality defects, and infertility (Afzelius, 1976). PCD is caused by mutations in genes involved in the biogenesis, assembly, and/or function of motile cilia (Afzelius and Stenram, 2006; Kennedy et al., 2007; Escudier et al., 2009; Knowles et al., 2013). Although the number of known PCD-associated genes/mutations continues to grow (Zariwala et al., 2007–2019), approximately one-third of PCD cases have no clear genetic basis (Kurkowiak et al., 2015; Horani and Ferkol, 2018). Furthermore, abnormalities within the ciliary ultrastructure that contribute to PCD clinical phenotypes remain poorly understood.
Most motile cilia are built on a “9+2″ axonemal core with nine peripheral doublet microtubules (DMTs) surrounding two central singlet microtubules known as the central pair complex (CPC; Figure 1A). The axoneme consists of 96-nm repeat units containing regularly spaced protein complexes, including dynein motors and their regulators. Each axonemal repeat contains four identical outer dynein arms (ODAs) and six unique inner dynein arms (IDAs) that are arranged in two rows along the length of the A-tubule (Figure 1, A–C). The dynein motors “walk” along the neighboring B-tubule to generate interdoublet sliding, which is transferred into bending by interdoublet linkers that resist the sliding (Porter and Sale, 2000). Dynein activity is coordinated to generate the undulating motion typical of cilia (Lin and Nicastro, 2018), which requires regulatory signals to be transmitted from the CPC to the radial spokes (RSs) and to two regulatory hubs: the double-headed I1/f dynein and the nexin–dynein regulatory complex (N-DRC) (Figure 1, A–C; Rupp and Porter, 2003; Wirschell et al., 2007 , 2009; Heuser et al., 2009). In humans, each axonemal repeat contains three RSs that are anchored to the A-tubule and project to the axoneme center, where they interact with the CPC (Figure 1, A and B; Warner and Satir, 1974; Goodenough and Heuser, 1985; Smith and Lefebvre, 1997; Mitchell and Sale, 1999; Omoto et al., 1999; Nicastro et al., 2005). Cryo–electron tomography (cryo-ET) has revealed RS heterogeneity; that is, RS1 and RS2 share a similar protein composition and structure, whereas RS3 has a different and mostly unknown proteome (Dymek et al., 2011; Pigino et al., 2011; Barber et al., 2012; Lin et al., 2012). Chlamydomonas reinhardtii, a biflagellated unicellular alga commonly used as a ciliary model system, has a similar RS arrangement but lacks a full-length RS3, containing instead a shortened RS3S stump (Barber et al., 2012). Full-length RSs are structurally composed of a stalk, an arch, and a head region, which interacts with the CPC projections (Figure 1C; Pigino et al., 2011; Barber et al., 2012; Lin et al., 2012). Though exact mechanisms remain elusive, RSs are involved in the signaling that regulates ciliary motility (Smith and Lefebvre, 1997; Smith and Yang, 2004), and mutations in RS proteins result in severe motility defects or paralyzed flagella (Warner and Satir, 1974; Piperno et al., 1977; Mitchell and Sale, 1999).
FIGURE 1:
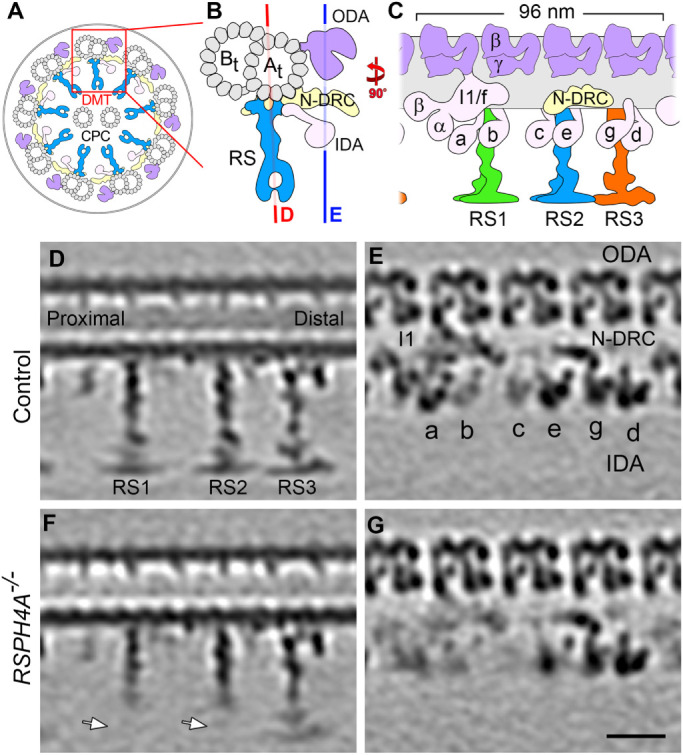
Structural defects in RSPH4A–/– cilia. (A–C) Schematic cross view of a human motile cilium (A) and zoomed-in cross-sectional (B) and longitudinal (C) views of one of the nine doublet microtubules, highlighting major components of the 96-nm repeat. Colored lines in B indicate the locations of the tomographic slices shown in D and E. (D–G) Longitudinal tomographic slices of averaged 96-nm repeats from control HAE (D, E) and RSPH4A–/– (F, G) cilia. White arrows in F denote the missing distal regions of RS1 and RS2 in RSPH4A–/– cilia. RS3, I1/f, and IDAs a, b, c, and e also exhibit lower densities in RSPH4A–/– cilia than the control. Labels: CPC, central pair complex; At and Bt, A- and B-tubules; RS1/2/3, radial spoke1/2/3; a–e and g, inner dynein arm isoforms; I1, I1 dynein complex; N-DRC, nexin dynein regulatory complex; ODA, outer dynein arm; IDA, inner dynein arm. Panels B, D, and E were generated from previously reported HAE control data (Lin et al., 2014). Scale bar: D–G, 20 nm.
Biallelic pathogenic variants (termed mutations hereafter) in the gene encoding putative RS head protein RSPH4A are a relatively frequent cause of PCD in Eastern European populations (Castleman et al., 2009; Zietkiewicz et al., 2012) and in patients of Puerto Rican descent (Daniels et al., 2013). However, conventional electron microscopy studies of RSPH4A (as well as RSPH1 and RSPH9) mutant cilia have failed to detect clear RS defects, finding instead that up to 60% of the cilia contained heterogeneous defects in the CPC, such as “9+0″ and “8+1″ axonemes (Castleman et al., 2009; Zietkiewicz et al., 2012; Daniels et al., 2013; Kott et al., 2013; Knowles et al., 2014; Onoufriadis et al., 2014). Electron tomography using chemically fixed and plastic-embedded RSPH4A mutant cilia also revealed CPC defects, but no RS defects (Burgoyne et al., 2014). Examinations of ciliary motility in nasal biopsies from PCD subjects with RSPH4A mutations have reported a range of motility defects, including reduced cilia beat frequency, dyskinesia, and rotational, instead of planar, beating patterns (Castleman et al., 2009; Daniels et al., 2013; Frommer et al., 2015). In contrast, mutating the orthologous RSP4 gene in C. reinhardtii causes clear loss of RS head structures and complete paralysis of ciliary motion (Piperno et al., 1977; Yang et al., 2006; Pigino et al., 2011; Barber et al., 2012). These studies raise questions about the location and function of RSPH4A in human cilia as well as the underlying basis for PCD caused by RSPH4A mutations.
Here, we use procedures previously established in our laboratories (Lin et al., 2014) to collect RSPH4A–/– cilia noninvasively from a PCD patient and characterize their structures. We present the native three-dimensional (3D) structure of RSPH4A–/– cilia, identifying primary defects in RS1 and RS2, as well as secondary CPC and IDA defects. By comparing RSPH4A–/– and RSPH1–/– cilia structures from two PCD patients, we find that the RSPH4A mutation causes more severe RS defects, characterized by both shorter RSs and a higher proportion of affected neighboring structures. Our findings suggest that RSPH4A localizes to the radial spoke heads, similar to RSPH1. However, in contrast to RSPH1, RSPH4A also plays a role in stabilizing the arch domain of RS1 and RS2. Together, these results provide insights into PCD etiology in patients with RSPH4A mutations and lay the foundation for structure-based improvements in diagnosis and treatment of PCD.
RESULTS
Cryo-ET reveals structural defects in RSPH4A –/– cilia
Previous work has characterized the structural defects underlying PCD in a patient with a mutation in RSPH1 (Lin et al., 2014). However, the structural basis for the phenotypically and clinically more severe RSPH4A–/– PCD has not been investigated (Knowles et al., 2014). To address this, we cultured control HAE cells and respiratory epithelial cells obtained from a patient with homozygous pathogenic mutations in RSPH4A by nasal scrape biopsy as previously reported (Lin et al., 2014). We then isolated ciliary axonemes from each sample and visualized their 3D structures using cryo-ET and subtomogram averaging. The 96-nm axonemal repeat of the control HAE cilia contained four dimeric ODAs, six monomeric IDAs (a–e and g), the dimeric I1/f dynein, the N-DRC, and three full-length RSs (Figure 1, D–E; Lin et al., 2014).
In contrast to the control cilia, we found that RSPH4A–/– cilia consistently lacked densities in the distal RS-head regions of RS1 and RS2 (Figure 1F, white arrows). RS3 remained intact in RSPH4A–/– cilia, although its density was lower than in the control (compare Figure 1, D and F). In addition to the RS head defects, we observed lower densities for IDAs a, b, c, e, and I1/f dynein in the averaged axonemal repeat of the mutant than in control cilia (compare Figure 1, E and G). We also observed heterogeneous CPC defects; that is, in most axonemes, the CPC was completely missing (9+0)–sometimes with one DMT moving to the axoneme center instead ((8+1)+0), and occasionally a partial CPC (9+1) was still present (Supplemental Figure S1).
RSPH4A mutation causes primary defects in the distal regions of RS1 and RS2
To determine which abnormalities were primary defects caused by the RSPH4A mutation rather than secondary defects from destabilization of neighboring structures or the axoneme isolation procedure, we next performed classification analyses (Heumann et al., 2011) using masks focused on the axonemal complexes with observed defects, RS1-3, IDAc, and the I1 dynein, which appear to be missing or reduced in the subtomogram averages (Figure 2; Supplemental Figures S2 and S3). Automated classification separates the axonemal repeats into classes based on structural similarities, determining whether a structure is missing from all axonemal repeats, missing from only a subset of the repeats, or present but positionally flexible. After careful analyses, we report that the RS head-containing distal regions of RS1 and RS2 were missing in 100% of axonemal repeats of RSPH4A–/– cilia (Figure 2, A–D, white arrows). In addition, RS1 and RS2 were further shortened, meaning the arch and distal RS stalk region were also reduced, in 37% and 30% of repeats, respectively (Figure 2, B and D). Similarly shortened radial spokes were also found in the classification analyses from control and RSPH1 PCD cilia, albeit with much lower frequency (RS1: 8% and 11%, respectively; RS2: 6% and 18%, respectively; see Supplemental Figure S2, Class 2). In contrast, the RS3 head was missing in only about one-third of axonemal repeats (Figure 2, E and F), consistent with the RSPH1–/– phenotype (Supplemental Figure S2). Classification analyses on individual IDAs in RSPH4A–/– cilia revealed a loss of IDA c and I1/f dynein in 84% and 57% of axonemal repeats, respectively (Supplemental Figure S3, B and D). Consistent with the weaker densities of IDA c and I1/f observed in the initial subtomogram averages (Figure 1, E vs. G), the partial loss of the RS3 head, IDA c, and I1/f in the classification analyses indicates that these defects are likely secondary in contrast to the primary 100% loss of distal RS1 and RS2 in RSPH4A–/– cilia. Alternatively, some of the IDA defects in RSPH4A–/– cilia could also be caused by extraction during the sample preparation, as suggested by our previous classification analyses on axonemal structures from wild-type human and Tetrahymena cilia (Lin et al., 2014). Taken together, these data strongly suggest that loss of the distal regions of RS1 and RS2 are the primary defects in RSPH4A–/– cilia.
FIGURE 2:
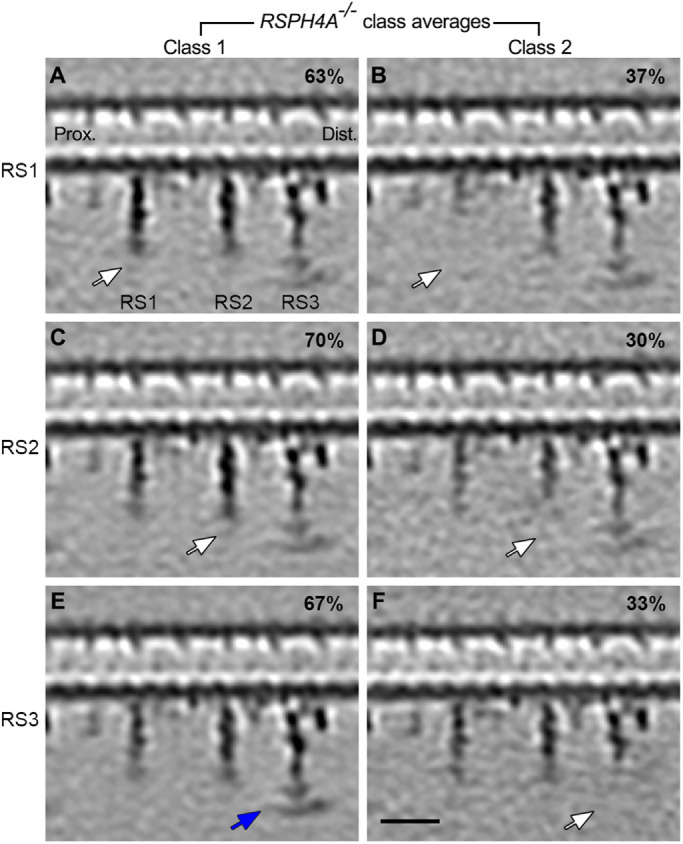
RSPH4A–/– cilia exhibit primary defects in RS1 and RS2. (A–F) Longitudinal tomographic slices of two class averages focusing on RS1 (A and B), RS2 (C and D), and RS3 (E and F) from RSPH4A–/– patient cilia. The location of the slice is the same as that shown in Figure 1D. White arrows highlight the missing distal regions of radial spokes, while blue arrows indicate their presence. Note: the distal regions of RS1 and RS2 were missing in all RSPH4A–/– 96-nm repeats, with remnant stalk regions varying in length between classes, whereas RS3 was only absent in a subset of repeats. Scale bar: A–F, 20 nm.
RSPH4A mutation affects the arch domain of RS1 and RS2
Similar to the RSPH4A–/– cilia presented here, RSPH1–/– cilia exhibit primary defects in the head domains of RS1 and RS2 (Figure 3, A–F, and Supplemental Figure S2; Lin et al., 2014). This is consistent with the finding that the Chlamydomonas homologues of both RSPH4A and RSPH1, that is, RSP4 and RSP1, were reported as RS head proteins (Yang et al., 2006; Kohno et al., 2011). However, PCD patients with mutations in RSPH4A have more severe symptoms than patients with mutations in RSPH1, suggesting potential differences between RSPH1 and RSPH4A localization and function (Knowles et al., 2014). To better understand the relative positions of RSPH1 and RSPH4A in human cilia, we performed local alignments focusing on each RS to improve the resolution and therefore the comparison of RS structures between control, RSPH1 PCD, and RSPH4A PCD cilia (Figure 3, G and H; Supplemental Movie S1). Isosurface renderings of the axonemal averages clearly reveal the similarities between the primary defects in the 96-nm repeat of RSPH4A–/– and RSPH1–/– cilia, with densities missing in the distal regions of RS1 and RS2 (Figure 3, D–F). However, the remnant structures of the RS1 and RS2 stalks are approximately 2 nm shorter in RSPH4A–/– cilia than in RSPH1–/–, corresponding to a volume of about 100–150 kDa (Figure 3, G and H; see direct comparison in the last column; Supplemental Movie S1). This suggests that RSPH4A localizes to the RS head domain of RS1 and RS2 close to or reaching into the RS arch region between the stalk and head. These differences in the primary defects between RSPH4A–/– and RSPH1–/– cilia may reflect a functional significance of the arch region in ciliary motility.
FIGURE 3:

RS1 and RS2 defects are more severe in RSPH4A–/– cilia than RSPH1–/– cilia. (A–C) Longitudinal tomographic slices show the averaged 96-nm repeat from control HAE (A), RSPH1–/– patient (B), and RSPH4A–/– patient cilia (C; the class average with I1 dynein is shown). White arrowheads indicate loss of RS1/RS2 head regions, while black arrows indicate full length RS3 structures. (D–F) Isosurface renderings of the averaged 96-nm repeat from control (D), RSPH1–/– (E), and RSPH4A–/– cilia (F; the class average with I1 dynein is shown). (G, H) Isosurface renderings of the averaged radial spokes viewed from the longitudinal back (G) and the proximal side (H). RS1, RS2, and RS3 structures are shown in green, blue, and orange, respectively. Difference maps were obtained by superimposing the control HAE or RSPH1–/– patient structures (gray) over the RSPH4A–/– patient structures (green, blue, or orange), highlighting the structural defects in the RSPH4A–/– patient cilia and the differences between the two PCD patient defects. Images of control and RSPH1–/– cilia were reproduced from previously reported data (Lin et al., 2014). Labels: RS1/2/3, radial spoke1/2/3; a–e and g, inner dynein arm isoforms; I1, I1 dynein complex; N-DRC, nexin dynein regulatory complex; ODA, outer dynein arm; IDA, inner dynein arm. Scale bar: A–C, 20 nm.
Movie S1.
Animated visualization comparing averaged axonemal repeats from control HAE, RSPH1-/-, and RSPH4A-/- cilia. Each isosurface rendering begins from a back view and rotates clockwise 360 degrees around its longitudinal axis. The first part of the video displays (from left to right) RS1 renderings from control, RSPH1-/-, and RSPH4A-/- cilia (green) and difference maps obtained by superimposing control or RSPH1-/- structures (gray) over the RSPH4A-/- patient structures (green). The second part of the video includes difference maps between RSPH1-/- (gray) and RSPH4A-/- structures of RS1 (green), RS2 (blue), and RS3 (orange). Images of control and RSPH1-/- cilia were generated from previously reported data (Lin, et al., 2014).
DISCUSSION
Diagnosis and treatment of PCD is challenging due to the heterogeneity of underlying genetic defects and their resulting phenotypes. Understanding abnormalities in the ciliary ultrastructure of PCD-associated mutations using conventional methods, which rely on model organisms that do not fully recapitulate human phenotypes, as well as chemical fixation methods that may damage ciliary structures, has been difficult. However, directly studying axonemal structure and function within human tissues presents its own challenges, particularly with preparation and collection of sufficient quantities of high-quality patient sample material. Our previous work has substantially improved methods for collecting and preparing axonemes from human respiratory cells for cryo-ET analysis by optimizing noninvasive cell sampling, in-culture expansion and differentiation, and a gentler method of cilia isolation (Lin et al., 2014). These advances, combined with the application of robust image processing and classification methods (Heumann et al., 2011), make it possible to reconstruct high-quality human axonemal structures and to detect and dissect the heterogeneous structural defects observed in patient samples. The latter is critical to distinguish sample variation and preparation artifacts from biologically meaningful defects resulting from genetic mutations.
Here, we show that the primary defect of the RSPH4A mutation is the complete lack of the distal regions, that is, of the entire RS head and at least part of the arch, of RS1 and RS2. In contrast, the distal region of RS3 was reduced in only one-third of the axonemal repeats, demonstrating that RSPH4A is not a RS3 subunit (Figure 2, E and F). This is consistent with previous conclusions that RS3 has a different subunit composition than RS1 and RS2 (Dymek et al., 2011; Pigino et al., 2011; Barber et al., 2012; Lin et al., 2012). Therefore, the observed RS3 defects are secondary and likely caused by structural destabilization of the RS3 head without its connection to the head of RS2 (Lin et al., 2014). A similar, partial reduction of the RS3 distal region was observed in RSPH1–/– cilia that also lack the RS heads of RS1 and RS2 (Supplemental Figure S2L; Lin et al., 2014). The RS2–RS3 head connection is not only present in control human cilia (Figure 3D), but was also observed in all previously studied organisms with three full-length ciliary radial spokes (e.g., Tetrahymena cilia and sea urchin sperm flagella; Lin et al., 2014). In addition to the RS3 defect, we observed heterogeneous secondary CPC defects in both RSPH4A–/– and RSPH1–/– cilia with disrupted RS1 and RS2 heads, suggesting a common underlying mechanism in which RS head interactions with CPC projections are critical for assembly and/or maintenance of the CPC (Oda et al., 2014).
Although the overall axonemal structure of motile cilia is broadly conserved between species, cryo-ET studies have also revealed species-specific structural differences in major axonemal complexes, making the correlation between structure and axonemal function more challenging. Within the RSs, in particular, the head regions are structurally dichotomous between unicellular protists (e.g., Chlamydomonas, Tetrahymena) and multicellular metazoans, including humans (Lin et al., 2012, 2014; Pigino et al., 2012). In protists, the RS heads (especially of RS1 and RS2) are considerably larger, with multiple projections that form an extended surface for interactions with CPC projections, whereas the RS heads in metazoans are two small parallel densities resembling ice skates (Lin et al., 2014). Furthermore, in Chlamydomonas, which is commonly used as a model organism to study cilia structure and motility, the distal half of RS3 is completely absent (Lin et al., 2014). These observations warrant caution in using Chlamydomonas or other nonhuman species as cellular models for PCD and other ciliopathies. Interestingly, mutating RSP4, the Chlamydomonas ortholog of RSPH4A, results in complete paralysis of Chlamydomonas flagella, whereas RSPH4A mutations in humans result in milder and less penetrant motility defects (Piperno et al., 1977; Pigino et al., 2011; Barber et al., 2012; Knowles et al., 2014). The increased severity of the RSP4 mutant motility defects in Chlamydomonas compared with human cilia suggests that the presence of a full-length RS3 in human cilia can mitigate some of the deleterious effects of the RSPH4A mutation on ciliary motility. In our analyses of human RSPH4A, RS3 remained intact in approximately two-thirds of axonemal repeats, which may functionally compensate for the loss of RSPH4A in human ciliary motility, though not completely. Interestingly, the angle of the RS3 head compared with its stalk is slightly altered in the RSPH1–/–, but not RSPH4A–/– cilia, compared with the control (compare Figure 2E, Supplemental Figures S2I and S2K). It is unclear whether this angular difference reflects a functional significance. Previous studies demonstrated that the RS3 proteome differs from that of RS1 and RS2 (Dymek et al., 2011; Pigino et al., 2011; Barber et al., 2012; Lin et al., 2012), but the actual protein composition of the stalk and head region of RS3 and specific protein interactions between RS2 and RS3 are so far unknown. The difference in RS3 head tilt between RSPH1–/– and RSPH4A–/– cilia could mean that one of these proteins either is present in or interacts with RS3. However, future studies are needed to investigate the RS3 proteome and the mechanisms by which RS3 contributes to ciliary motility.
Classical EM studies of RPSH4A mutant cilia have reported a heterogeneous set of CPC defects in about 50% of the cilia, whereas the remaining cilia appeared structurally intact (Castleman et al., 2009; Zietkiewicz et al., 2012; Daniels et al., 2013; Kott et al., 2013; Burgoyne et al., 2014; Knowles et al., 2014; Onoufriadis et al., 2014). Despite their apparent structural integrity, however, all RSPH4A mutant cilia exhibited lower mean ciliary beat frequencies as well as abnormal waveforms, with decreased range of motion and lack of coordinated movement (dyskinesis) between adjacent cilia (Daniels et al., 2013). Our cryo-ET data suggest that these motility defects are in fact due to RS head defects that were previously not visible in chemically fixed EM samples. RSPH4A is either 600 or 716 amino acids in length, depending on isoform, and it contains no known functional domains (Yang et al., 2006). Although it is possible that RSPH4A regulates ciliary motility through an unknown signaling function, our data strongly support the conclusion that RSPH4A is critically important for the assembly of the head and likely the arch domains of RS1 and RS2, as well as assembly and/or maintenance of a regular CPC complex with two singlet microtubules. The RS heads interact with the CPC projections, and the integrity of RSs, as well as RS-CPC interactions, is critical for stable CPC assembly and function, as they distribute and resist internal mechanical forces during bending that cause local cilia compression (Oda et al., 2014). Cilia from mice deficient in RSPH4A showed similar CPC defects, and were more sensitive to the destabilizing effects of Taxol (Shinohara et al., 2015). Interestingly, in the Rsph4a–/– mouse model, the radial spoke heads were missing in all 3 radial spokes (Yoke et al., 2020). In human RSPH4A–/– cilia, RS1 and RS2 heads were completely missing, but the RS3 heads were missing from only one-third of the axonemal repeats. This is higher than in control cilia (9%) and suggests that the RS2–RS3 head connection is important for RS3 head stability. Although we cannot exclude that RS3 from mouse and human respiratory cilia may contain different proteins, it is more likely that the reduction of RS3 heads in Rsph4a–/– mouse cilia is also a secondary effect caused by the primary RS2 head defect. The observed heterogeneous CPC abnormalities suggest that when the RS heads are not intact, the increased central cylinder space within the axoneme could cause abnormal assembly or arrangement between the CPC and the DMTs in some, but not necessarily all of the patient’s cilia. Given these findings, PCD diagnosis could be improved by directly visualizing the primary mutant defects in the axonemal structure instead of relying on secondary defects in CPC morphology and functional phenotypes, which may be heterogeneous (Supplemental Figure S1; Daniels et al., 2013; Knowles et al., 2014).
PCD patients with RSPH1 mutations show milder clinical manifestations than those with RSPH4A mutations, as characterized by a lower incidence of neonatal respiratory distress, a later onset of daily year-round wet cough, a delayed onset of pulmonary symptoms, significantly higher levels of nasal nitric oxide, and better overall lung function (Knowles et al., 2014). Similar findings have been reported in mice, in which Rsph1–/– animals exhibit a low level of mucociliary clearance and a less-severe PCD phenotype than those lacking dynein axonemal intermediate chain 1 (Yin et al., 2019). In contrast, patients with RSPH4A mutations exhibit symptoms similar to those of PCD patients with “classic” dynein arm defects, including a high prevalence of neonatal respiratory distress and low nasal nitric oxide (Noone et al., 2004). This raises the question: why do RSPH4A mutations lead to poorer PCD outcomes than mutations in RSPH1? Our comparative analyses suggest that RPSH4A–/– cilia exhibit more severe RS defects, with the remnant of the stalk structures of RS1 and RS2 appearing shorter than those of RSPH1–/– cilia in a larger proportion of axonemal repeats (Figures 2 and 3, G and H; Supplemental Figure S2; Supplemental Movie S1). These data support a model in which RSPH4A is more centrally located within the RS head than RSPH1, potentially reaching into the arch region or interacting strongly with arch proteins, in agreement with recent findings (Zheng et al., 2021; Grossman-Haham et al., 2020; Gui et al., 2020). This is consistent with IHC data that demonstrate loss of RSPH4A results in the loss of RSPH1; while the loss of RSPH1 does not result in the loss of RSPH4A (Frommer et al., 2015). Of note, a recent study using chemical cross-linking followed by mass spectrometry in Chlamydomonas has established putative interactions between RSPH4A homologue RSP4 and arch protein RSP2 (Poghosyan et al., 2020). Therefore, arch destabilization due to RSPH4A loss may cause RSPH4A PCD to present a more severe clinical phenotype than RSPH1 PCD (Knowles et al., 2014). In addition, the differences in the extent and location of RS defects clearly correspond to increased severity and prevalence of CPC defects; classical EM studies of patient cilia showed the axonemal organization deviating from the standard 9+2 arrangement in approximately 50% of RSPH4A mutant cilia compared with 20% of RSPH1 mutant cilia (Daniels et al., 2013; Knowles et al., 2013). Our cryo-ET studies of isolated axonemes also show that 77% of RSPH4A–/– cilia completely lacked the CPC without any remnant density in the axoneme center (9+0), whereas only 30% in RSPH1–/– cilia lacked any density in the center (Supplemental Figure S1B). Although usually the RS heads (and not the arches) are interacting with the CPC, it is possible that the remaining arch region of RS1 and RS2 in RSPH1–/– cilia provides some support to withstand forces exerted during ciliary beating, thus maintaining more of the ciliary functionality. Alternatively, one could invoke a mechanism by which RSPH4A itself or RS arch proteins that require RPSH4A for proper assembly maintain important signaling functions necessary for proper ciliary motility. Future studies might consider replacing RSPH1 and RSPH4A with altered versions (more flexible or rigid, mutations in key residues) to determine their effects on CPC morphology and overall ciliary motility. By better understanding how mutations within axonemal structures contribute to ciliary disease, we can work toward improving the diagnosis and treatment of PCD and other ciliopathies.
MATERIALS AND METHODS
Human nasal epithelial cell culture
Non-PCD, non–cystic fibrosis control human airway epithelial (HAE) cells were obtained from the UNC Cell and Tissue Culture Core Facility under protocols approved by the Institutional Review Board for the Protection of the Rights of Human Subjects at UNC. All human tissues were obtained with informed consent. Cells were cultured as previously described (Fulcher and Randell, 2013). Briefly, human airway epithelial cells (approximately 106) were plated on human type IV collagen–coated (250 μg, Sigma) Millicell culture inserts 4.2 cm2 in area (Millipore). Then an air–liquid interface was established on confluent cells (approximately 3 days after plating) by removing the apical media and feeding from the basolateral side. Cultures were fed three times per week with ALI media (Fulcher and Randell, 2013; 50:50 mixture of LHC-basal and DMEM-H media) for 2 weeks, after which feedings were reduced to twice per week. Along with medium changes, phosphate-buffered saline (PBS) was used to wash the apical surfaces of the cultures to remove mucus and cell debris. Phase contrast microscopy was used to monitor ciliated-cell differentiation, and cultures became heavily ciliated at the air–liquid interface after approximately 4 weeks.
We identified a PCD patient with a classic PCD phenotype, including neonatal respiratory distress despite term birth, along with daily nasal congestion and year-round daily wet cough from infancy, chronic recurrent sinusitis, severe otitis media and chronic mastoiditis, bronchiectasis, and low nasal nitric oxide (21 nl/min). The patient had normal situs and homozygous pathogenic mutations in the RSPH4A gene (c. 921+3-6delAAGT [p.splice]; Daniels et al., 2013). Nasal epithelial cells were obtained via nasal scrape biopsy under protocols approved by the Institutional Review Board for the Protection of the Rights of Human subjects at UNC (Knowles et al., 2014). As previously described (Suprynowicz et al., 2012), the cells were initially expanded on plastic dishes containing fibroblast-conditioned medium and Rho kinase inhibitor (Y-27632). Then, as described above, the expanded cells were cultured at an air–liquid interface on collagen-coated culture inserts until ciliated cell differentiation occurred (∼4 weeks). Consistent with in vivo observations (Daniels et al., 2013), ciliary beat in the RSPH4A–/– cultures was dyskinetic, and ciliary beat frequency was reduced (3.6 Hz; n = 6). PCR and Western blot analyses confirmed that no full-length mRNA or protein for RSPH4A could be detected in the cultured RSPH4A–/– cells or cilia, respectively.
Isolation of human ciliary axonemes
Axonemes from respiratory cilia were isolated from human nasal epithelial cells as previously described (Lin et al., 2014). Briefly, well-differentiated cells were gently washed with PBS to remove mucus and cell debris. The air–liquid interface culture dishes were then filled (both apical and basal compartments) with ice-cold PBS and placed on a bed of ice for approximately 5 min to reduce ciliary beating until it nearly ceased. The PBS was then removed from the apical surface of the culture insert and replaced with 50 μl of ice-cold deciliation buffer (10 mM Tris pH 7.5, 50 mM NaCl, 10 mM CaCl2, 1 mM EDTA, 0.1% Triton X-100, 7 mM β-mercaptoethanol, 1% protease inhibitor cocktail [Sigma P8340]) per 12-mm insert. After 2 min incubation without shaking, the apical solution containing cilia was transferred to a microcentrifuge tube and gently centrifuged at 4°C for 1 min at 500 × g to remove mucus and cellular debris. The cilia-containing supernatant was then centrifuged at 4 °C for 5 min at 5000 × g to collect the ciliary axonemes. Finally, the axonemal pellet was gently dispersed with resuspension buffer (30 mM HEPES pH 7.3, 1 mM EGTA, 5 mM MgSO4, 0.1 mM EDTA, 25 mM NaCl, 1 mM dithiothreitol, 1% protease inhibitor cocktail, and 100 gml-1 soybean trypsin inhibitor [Sigma T9128]) and kept on ice until cryo-sample preparation.
Cryo-sample preparation and cryo–electron tomography
Quantifoil holey carbon grids (R2/2, Quantifoil Micro Tools GmbH, Germany) were glow discharged and loaded into a homemade plunge-freezing device. Isolated axoneme samples (3 μl) and fivefold concentrated 10-nm colloidal gold (1 μl, Sigma-Aldrich) were added to the grid. The grid was blotted with filter paper for 1.5–2.5 s and immediately plunge-frozen in liquid ethane that was cooled by liquid nitrogen. The vitrified samples were then stored in liquid nitrogen until data collection by cryo-ET.
Single-axis tilt series were collected using a Tecnai F30 transmission electron microscope (FEI, Hillsboro, OR) equipped with a postcolumn energy filter (Gatan, Pleasanton, CA). Cryo-samples were transferred into the microscope with a cryo-holder (Gatan) and kept below the devitrification temperature (approximately –170°C). The samples were imaged at 300 kV, with –6 or –8 μm defocus, under low-dose conditions and in the zero-loss mode of the energy filter (slit width 20 eV). Axonemes that appeared well preserved and not obviously compressed by EM inspection were selected for cryo–electron tomography. Tilt series images were recorded automatically over a tilt range of –65° to +65° with 1.5–2.5° angular increments using the SerialEM image acquisition software (Mastronarde, 2005). The cumulative electron dose was limited to 100 e/Å2 for individual tilt series. All images were recorded on a 2k × 2k charge-coupled device camera (Gatan) at 13,500× magnification, resulting in a pixel size of approximately 1 nm.
Image processing
Tilt series images with sufficient and evenly distributed gold fiducial markers were aligned and then reconstructed by weighted back projection into tomograms using the IMOD software (Kremer et al., 1996). Tomograms with compressed axonemes (caused by insufficient ice thickness during cryo-sample preparation) were excluded from further subtomogram averaging and analysis. Subtomograms of axonemal repeats were extracted from the raw tomograms, aligned, and averaged (with missing wedge compensation) using the PEET software (Nicastro et al., 2006) to enhance the signal-to-noise ratio and improve resolution (Table 1). Two different alignment strategies were employed to optimize alignment accuracy: 1) for the entire 96-nm axonemal repeat, global alignment with relatively large particle volumes was used (Figures 1, 2, and 3, A–F; Supplemental Figures S1, S2, and S3); 2) to resolve the RSs with greater detail, smaller subtomograms centered on the structure of interest were used for local alignment and then averaged (Figure 3, G and H). Subtomogram averaging of positionally flexible structures or structures that are present in only a subset of averaged particles causes blurring out of the structure signal in the average. Therefore, we used the unsupervised classification tool built into the PEET program (Heumann et al., 2011) to analyze the heterogeneity of axonemal structures that appeared blurred in the subtomogram averages. Briefly, after alignment, soft-edged masks were applied to focus the classification on the structures of interest, that is, areas with lower electron density. The classification uses principal component analysis and clustering to divide the particles (axonemal repeats) into different classes based on structural similarity. If a structure is positionally flexible, the class averages will show the unblurred structure in different positions in the class averages, whereas reduced structures will result in classes that either contain or lack the structure of interest (Figure 2; Supplemental Figures S2 and S3). The resolution of the resulting averages was estimated in a 20 nm3 subvolume in the 96-nm axonemal repeat using the Fourier shell correlation method and the 0.5 criterion (Harauz and van Heel, 1986). The IMOD software (Kremer et al., 1996) and the UCSF Chimera package (Pettersen et al., 2004) were used for visualization of tomographic slices and 3D isosurface renderings, respectively.
TABLE 1:
Image processing information for specimens used in this study.
| Specimen | Tomograms included | Averaged repeats | Resolution (nm)a | Used in figure(s) |
|---|---|---|---|---|
| Control ciliab | 12 | 857 | 3.44 | 1,3,S1,S2 |
| RSPH1–/– ciliab | 20 | 3044 | 3.36 | 3,S1,S2 |
| RSPH4A–/– cilia | 13 | 1637 | 4.18 | 1,2,3,S1,S3 |
The 0.5 Fourier shell correlation criterion was used to estimate the resolution of the averaged repeats at the site of the doublet microtubule.
Data have been previously published (Lin et al., 2014).
Supplementary Material
Acknowledgments
We thank Chen Xu for providing EM training and management of the electron microscopes in the Louise Mashal Gabbay Cellular Visualization Facility at Brandeis University. We thank David Mastronarde and John Heumann (University of Colorado at Boulder) for continued development of image processing tools, including PEET classification. We also thank the University of North Carolina Cystic Fibrosis Cell and Tissue Culture Core (supported by R026-CR11 from the Cystic Fibrosis Foundation and NIH DK065988) for providing samples and expertise. This research benefitted from the computational resources provided by the BioHPC supercomputing facility located in the Lyda Hill Department of Bioinformatics at UT Southwestern Medical Center. This work was supported by funding from the National Institutes of Health (GM083122 to D.N., 2R01HL117836 to L.E.O. and M.A.Z., 2U54HL096458 and 5R01HL071798 to M.R.K. and M.A.Z., and F32GM137470 to J.P.). The Genetic Disorders of Mucociliary Clearance Consortium (U54HL096458) is part of the National Center for Advancing Translational Sciences (NCATS) Rare Diseases Clinical Research Network (RDCRN) and is supported by the RDCRN Data Management and Coordinating Center (DMCC) (U2CTR002818). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR) funded through a collaboration between NCATS and the National Heart, Lung, and Blood Institute (NHLBI). Additional support was provided by the Cancer Prevention and Research Institute of Texas Grant RR140082 (D.N), by the March of Dimes Foundation (to D.N.), and by a private donation from Arnold Cohen (to D.N.). Cryo-ET data for the RSPH1 and RSPH4A patient samples have been deposited with the EM Data Bank under accession codes EMD-22874 and EMD-22875, respectively.
Abbreviations used:
- CPC
central pair complex
- cryo-ET
cryo-electron tomography
- 3D
three-dimensional
- DMT
doublet microtubule
- EM
electron microscopy
- HAE cells
human airway epithelial cells
- IDA
inner dynein arm
- N-DRC
nexin-dynein regulatory complex
- ODA
outer dynein arm
- PCD
primary ciliary dyskinesia
- RS
radial spoke
- RSPH1
radial spoke head component 1
- RSPH4A
radial spoke head component 4A
- RSPH9
radial spoke head component 9.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-12-0806) on April 14, 2021.
REFERENCES
- Afzelius BA (1976). A human syndrome caused by immotile cilia. Science 193, 317–319. [DOI] [PubMed] [Google Scholar]
- Afzelius BA, Stenram U (2006). Prevalence and genetics of immotile-cilia syndrome and left-handedness. Int J Dev Biol 50, 571–573. [DOI] [PubMed] [Google Scholar]
- Barber CF, Heuser T, Carbajal-Gonzalez BI, Botchkarev VV Jr, Nicastro D (2012). Three-dimensional structure of the radial spokes reveals heterogeneity and interactions with dyneins in Chlamydomonas flagella. Mol Biol Cell 23, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Witman GB (2014). Cilia and diseases. Bioscience 64, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne T, Lewis A, Dewar A, Luther P, Hogg C, Shoemark A, Dixon M (2014). Characterizing the ultrastructure of primary ciliary dyskinesia transposition defect using electron tomography. Cytoskeleton (Hoboken) 71, 294–301. [DOI] [PubMed] [Google Scholar]
- Castleman VH, Romio L, Chodhari R, Hirst RA, de Castro SC, Parker KA, Ybot-Gonzalez P, Emes RD, Wilson SW, Wallis C, et al. (2009). Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am J Hum Genet 84, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels ML, Leigh MW, Davis SD, Armstrong MC, Carson JL, Hazucha M, Dell SD, Eriksson M, Collins FS, Knowles MR, Zariwala MA (2013). Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia. Hum Mutat 34, 1352–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond IA (2012). Cilia functions in development. Curr Opin Cell Biol 24, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymek EE, Heuser T, Nicastro D, Smith EF (2011). The CSC is required for complete radial spoke assembly and wild-type ciliary motility. Mol Biol Cell 22, 2520–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudier E, Duquesnoy P, Papon JF, Amselem S (2009). Ciliary defects and genetics of primary ciliary dyskinesia. Paediatr Respir Rev 10, 51–54. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H (2007). When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 8, 880–893. [DOI] [PubMed] [Google Scholar]
- Frommer A, Hjeij R, Loges NT, Edelbusch C, Jahnke C, Raidt J, Werner C, Wallmeier J, Grosse-Onnebrink J, Olbrich H, et al. (2015). Immunofluorescence analysis and diagnosis of primary ciliary dyskinesia with radial spoke defects. Am J Respir Cell Mol Biol 53, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher ML, Randell SH (2013). Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol Biol 945, 109–121. [DOI] [PubMed] [Google Scholar]
- Goodenough UW, Heuser JE (1985). Substructure of inner dynein arms, radial spokes, and the central pair/projection complex of cilia and flagella. J Cell Biol 100, 2008–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman-Haham I, Coudray N, Yu Z, Wang F, Zhang N, Bhabha G, Vale RD (2020). Structure of the radial spoke head and insights into its role in mechanoregulation of ciliary beating. Nat Struct Mol Biol 28, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui M, Ma M, Sze-Tu E, Wang X, Koh F, Zhong ED, Berger B, Davis JH, Dutcher SK, Zhang R, Brown A (2020). Structures of radial spokes and associated complexes important for ciliary motility. Nat Struct Mol Biol 28, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, van Heel M (1986). Exact filters for general geometry three dimensional reconstruction. Optik 73, 146–156. [Google Scholar]
- Heumann JM, Hoenger A, Mastronarde DN (2011). Clustering and variance maps for cryo–electron tomography using wedge-masked differences. J Struct Biol 175, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser T, Raytchev M, Krell J, Porter ME, Nicastro D (2009). The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J Cell Biol 187, 921–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horani A, Ferkol TW (2018). Advances in the genetics of primary ciliary dyskinesia: clinical implications. Chest 154, 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, Robinson BV, Minnix SL, Olbrich H, Severin T, et al. (2007). Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation 115, 2814–2821. [DOI] [PubMed] [Google Scholar]
- Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW (2013). Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med 188, 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MR, Ostrowski LE, Leigh MW, Sears PR, Davis SD, Wolf WE, Hazucha MJ, Carson JL, Olivier KN, Sagel SD, et al. (2014). Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am J Respir Crit Care Med 189, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Wakabayashi K, Diener DR, Rosenbaum JL, Kamiya R (2011). Subunit interactions within the Chlamydomonas flagellar spokehead. Cytoskeleton (Hoboken) 68, 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kott E, Legendre M, Copin B, Papon JF, Dastot-Le Moal F, Montantin G, Duquesnoy P, Piterboth W, Amram D, Bassinet L, et al. (2013). Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects. Am J Hum Genet 93, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Kurkowiak M, Zietkiewicz E, Witt M (2015). Recent advances in primary ciliary dyskinesia genetics. J Med Genet 52, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Heuser T, Carbajal-Gonzalez BI, Song K, Nicastro D (2012). The structural heterogeneity of radial spokes in cilia and flagella is conserved. Cytoskeleton (Hoboken) 69, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Nicastro D (2018). Asymmetric distribution and spatial switching of dynein activity generates ciliary motility. Science 360, eaar1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Yin W, Smith MC, Song K, Leigh MW, Zariwala MA, Knowles MR, Ostrowski LE, Nicastro D (2014). Cryo–electron tomography reveals ciliary defects underlying human RSPH1 primary ciliary dyskinesia. Nat Commun 5, 5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mitchell DR, Sale WS (1999). Characterization of a Chlamydomonas insertional mutant that disrupts flagellar central pair microtubule-associated structures. J Cell Biol 144, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D, McIntosh JR, Baumeister W (2005). 3D structure of eukaryotic flagella in a quiescent state revealed by cryo–electron tomography. Proc Natl Acad Sci USA 102, 15889–15894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR (2006). The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313, 944–948. [DOI] [PubMed] [Google Scholar]
- Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, Zariwala MA, Knowles MR (2004). Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med 169, 459–467. [DOI] [PubMed] [Google Scholar]
- Oda T, Yanagisawa H, Yagi T, Kikkawa M (2014). Mechanosignaling between central apparatus and radial spokes controls axonemal dynein activity. J Cell Biol 204, 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omoto CK, Gibbons IR, Kamiya R, Shingyoji C, Takahashi K, Witman GB (1999). Rotation of the central pair microtubules in eukaryotic flagella. Mol Biol Cell 10, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoufriadis A, Shoemark A, Schmidts M, Patel M, Jimenez G, Liu H, Thomas B, Dixon M, Hirst RA, Rutman A, et al. (2014). Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects. Hum Mol Genet 23, 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pigino G, Bui KH, Maheshwari A, Lupetti P, Diener D, Ishikawa T (2011). Cryoelectron tomography of radial spokes in cilia and flagella. J Cell Biol 195, 673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Maheshwari A, Bui KH, Shingyoji C, Kamimura S, Ishikawa T (2012). Comparative structural analysis of eukaryotic flagella and cilia from Chlamydomonas, Tetrahymena, and sea urchins. J Struct Biol 178, 199–206. [DOI] [PubMed] [Google Scholar]
- Piperno G, Huang B, Luck DJ (1977). Two-dimensional analysis of flagellar proteins from wild-type and paralyzed mutants of Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 74, 1600–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poghosyan E, Iacovache I, Faltova L, Leitner A, Yang P, Diener DR, Aebersold R, Zuber B, Ishikawa T (2020). The structure and symmetry of the radial spoke protein complex in Chlamydomonas flagella. J Cell Sci 133. [DOI] [PubMed] [Google Scholar]
- Porter ME, Sale WS (2000). The 9 + 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J Cell Biol 151, F37–F42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp G, Porter ME (2003). A subunit of the dynein regulatory complex in Chlamydomonas is a homologue of a growth arrest-specific gene product. J Cell Biol 162, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P, Christensen ST (2007). Overview of structure and function of mammalian cilia. In: Annu Rev Physiol, 69, 377–400. [DOI] [PubMed] [Google Scholar]
- Shinohara K, Chen D, Nishida T, Misaki K, Yonemura S, Hamada H (2015). Absence of radial spokes in mouse node cilia is required for rotational movement but confers ultrastructural instability as a trade-off. Dev Cell 35, 236–246. [DOI] [PubMed] [Google Scholar]
- Smith EF, Lefebvre PA (1997). The role of central apparatus components in flagellar motility and microtubule assembly. Cell Motil Cytoskeleton 38, 1–8. [DOI] [PubMed] [Google Scholar]
- Smith EF, Yang P (2004). The radial spokes and central apparatus: mechano-chemical transducers that regulate flagellar motility. Cell Motil Cytoskeleton 57, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X, Yuan H, Cheluvaraju C, Clapp PW, Boucher RC Jr, et al. (2012). Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci USA 109, 20035–20040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner FD, Satir P (1974). The structural basis of ciliary bend formation. Radial spoke positional changes accompanying microtubule sliding. J Cell Biol 63, 35–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirschell M, Hendrickson T, Sale WS (2007). Keeping an eye on I1: I1 dynein as a model for flagellar dynein assembly and regulation. Cell Motil Cytoskeleton 64, 569–579. [DOI] [PubMed] [Google Scholar]
- Wirschell M, Yang C, Yang PF, Fox L, Yanagisawa H, Kamiya R, Witman GB, Porter ME, Sale WS (2009). IC97 is a novel intermediate chain of I1 dynein that interacts with tubulin and regulates interdoublet sliding. Mol Biol Cell 20, 3617–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Diener DR, Yang C, Kohno T, Pazour GJ, Dienes JM, Agrin NS, King SM, Sale WS, Kamiya R, et al. (2006). Radial spoke proteins of Chlamydomonas flagella. J Cell Sci 119, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Livraghi-Butrico A, Sears PR, Rogers TD, Burns KA, Grubb BR, Ostrowski LE (2019). Mice with a deletion of Rsph1 exhibit a low level of mucociliary clearance and develop a primary ciliary dyskinesia phenotype. Am J Respir Cell Mol Biol 61, 312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoke H, Ueno H, Narita A, Sakai T, Horiuchi K, Shingyoji C, Hamada H, Shinohara K (2020). Rsph4a is essential for the triplet radial spoke head assembly of the mouse motile cilia. PLoS Genet 16, e1008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zariwala MA, Knowles MR, Leigh MW (2007. –2019). Primary Ciliary Dyskinesia, eds. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A, Seattle, WA: University of Washington, Available from: https:// www.ncbi.nlm.nih.gov/books/NBK1122/. [PubMed] [Google Scholar]
- Zheng W, Li F, Ding Z, Liu H, Zhu L, Xu C, Li J, Gao Q, Wang Y, Fu Z, et al. (2021). Distinct architecture and composition of mouse axonemal radial spoke head revealed by cryo-EM. Proc Natl Acad Sci USA 118, e2021180118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zietkiewicz E, Bukowy-Bieryllo Z, Voelkel K, Klimek B, Dmenska H, Pogorzelski A, Sulikowska-Rowinska A, Rutkiewicz E, Witt M (2012). Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS One 7, e33667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.