

ABSTRACT
High-molecular-mass penicillin-binding proteins (PBPs) are enzymes that catalyze the biosynthesis of bacterial cell wall peptidoglycan. The Gram-positive bacterial pathogen Streptococcus agalactiae (group B streptococcus [GBS]) produces five high-molecular-mass PBPs, namely, PBP1A, PBP1B, PBP2A, PBP2B, and PBP2X. Among these, only PBP2X is essential for cell viability, whereas the other four PBPs are individually dispensable. The biological function of the four nonessential PBPs is poorly characterized in GBS. We deleted the pbp1a, pbp1b, pbp2a, and pbp2b genes individually from a genetically well-characterized serotype V GBS strain and studied the phenotypes of the four isogenic mutant strains. Compared to the wild-type parental strain, (i) none of the pbp isogenic mutant strains had a significant growth defect in Todd-Hewitt broth supplemented with 0.2% yeast extract (THY) rich medium, (ii) isogenic mutant Δpbp1a and Δpbp1b strains had significantly increased susceptibility to penicillin and ampicillin, and (iii) isogenic mutant Δpbp1a and Δpbp2b strains had significantly longer chain lengths. Using saturated transposon mutagenesis and transposon insertion site sequencing, we determined the genes essential for the viability of the wild-type GBS strain and each of the four isogenic pbp deletion mutant strains in THY rich medium. The pbp1a gene is essential for cell viability in the pbp2b deletion background. Reciprocally, pbp2b is essential in the pbp1a deletion background. Moreover, the gene encoding RodA, a peptidoglycan polymerase that works in conjunction with PBP2B, is also essential in the pbp1a deletion background. Together, our results suggest functional overlap between PBP1A and the PBP2B-RodA complex in GBS cell wall peptidoglycan biosynthesis.
IMPORTANCE High-molecular-mass penicillin-binding proteins (HMM PBPs) are enzymes required for bacterial cell wall biosynthesis. Bacterial pathogen group B streptococcus (GBS) produces five distinct HMM PBPs. The biological functions of these proteins are not well characterized in GBS. In this study, we performed a comprehensive deletion analysis of genes encoding HMM PBPs in GBS. We found that deleting certain PBP-encoding genes altered bacterial susceptibility to beta-lactam antibiotics, cell morphology, and the essentiality of other enzymes involved in cell wall peptidoglycan synthesis. The results of our study shed new light on the biological functions of PBPs in GBS.
KEYWORDS: TraDIS, group B streptococcus, penicillin-binding proteins
INTRODUCTION
Streptococcus agalactiae (group B streptococcus [GBS]) is a Gram-positive bacterial pathogen that colonizes the female genital tract (1). It is the major cause of maternal and neonatal infections, including urinary tract infection, chorioamnionitis, postpartum endometritis, and bacteremia in pregnant women and sepsis, pneumonia, and meningitis in newborns (2–16). Like other Gram-positive bacteria, GBS cells are surrounded by thick layers of peptidoglycan, the major structural component of the cell wall (17, 18). Cell wall peptidoglycan protects GBS from osmotic lysis and provides the scaffold for the attachment of antiphagocytic polysaccharide capsule (19) and cell wall-anchored adhesins and virulence factors (20–24).
During late stages of peptidoglycan biosynthesis, transglycosylation (polymerization of nascent glycan strands) and transpeptidation (cross-linking of nascent glycan strands) are catalyzed in part by high-molecular-mass (HMM) penicillin-binding proteins (PBPs) (25). HMM PBPs are divided into two classes, A and B (26). Class A PBPs are bifunctional enzymes with both transglycosylase and transpeptidase activities, and class B PBPs are monofunctional enzymes with only transpeptidase activity (27). GBS produces three bifunctional class A PBPs (PBP1A, PBP1B, and PBP2A) and two monofunctional class B PBPs (PBP2X and PBP2B) (28, 29). With the exception of PBP2X, all other HMM PBPs are individually dispensable for the viability of GBS, as shown by Hooven et al. (30).
Previous functional studies of GBS PBPs using deletion mutagenesis have focused largely on PBP1A (also called PonA). A signature-tagged mutagenesis study revealed that a GBS pbp1a mutant was severely attenuated for virulence in a rat neonatal sepsis model of infection (31). Subsequent studies showed that the pbp1a gene is also required for GBS proliferation in the lungs of neonatal rats and for resistance to killing by neutrophils and alveolar macrophages (32). The role of PBP1A in evading the human innate immune response is likely mediated by its augmentation to GBS resistance to host antimicrobial peptides (33). Relative to PBP1A, the other GBS PBPs are not as well studied; thus, less is known about their biological function, including the contribution of each PBP to cell wall biosynthesis and the interactions between PBPs.
In this study, we individually deleted the pbp1a, pbp1b, pbp2a, and pbp2b genes from a well-characterized and clinically relevant serotype V GBS strain and studied the phenotypes of the four individual PBP isogenic mutant strains. We found that altered susceptibility to beta-lactam antibiotics and cell morphology resulted, depending on which PBP gene was deleted. To determine if deletion of one pbp gene affected the essentiality of other GBS genes, especially genes participating in cell wall biosynthesis, we performed saturated transposon mutagenesis studies on each of the four individual pbp deletion isogenic mutant strains and the wild-type parental strain. We discovered that deletion of pbp1a or pbp2b each changed the essentiality of other genes involved in peptidoglycan synthesis. The results of our study provide functional insights into PBPs from GBS.
RESULTS
Construction of isogenic PBP gene deletion mutant strains.
To study the function of HMM PBPs in GBS, we constructed isogenic, markerless, gene deletion mutant Δpbp1a, Δpbp1b, Δpbp2a, and Δpbp2b strains from the wild-type parental strain CNCTC10/84, a fully sequenced and well-characterized GBS capsule serotype V isolate (34). These four individual PBP gene deletion mutant strains were generated by allelic exchange as previously described (35, 36). The genomic context of the four deleted PBP genes is shown in Fig. 1. Whole-genome sequencing validated that each mutant strain had the appropriate gene deletion and lacked spurious mutations elsewhere in the genome.
FIG 1.

Genes flanking pbp1a, pbp1b, pbp2a, and pbp2b in GBS. PBP genes subjected to gene deletion studies are highlighted in red. Flanking genes are colored cyan. The locus tag numbers refer to the annotation for GBS strain CNCTC10/84.
Growth and susceptibility to β-lactam antibiotics.
We examined the growth of each PBP deletion mutant strain on blood agar plates (5% sheep blood in tryptic soy agar) and in rich medium, Todd-Hewitt broth supplemented with 2% yeast extract (THY). Compared to the wild-type parental strain, none of the isogenic PBP deletion mutant strains had a gross visual abnormality in colony size or hemolysis on blood agar (Fig. 2A). Moreover, none of the mutant strains had an apparent growth defect in THY medium (Fig. 2B). Because HMM PBP transpeptidase domains are the drug targets of β-lactam antibiotics, we first assessed the susceptibility of each PBP deletion mutant strain to penicillin G and ampicillin, two β-lactam antibiotics that are commonly used to treat streptococcal infections (Fig. 2C and D). Disk diffusion test results demonstrated that isogenic Δpbp1a and Δpbp1b mutant strains had a modest but statistically significant increase in susceptibility to penicillin and ampicillin (Fig. 2C and D). To validate this observation, we determined the MIC values for penicillin and ampicillin using the Etest (37–39). MIC values (Table 1) confirmed that isogenic Δpbp1a and Δpbp1b mutant strains had increased susceptibility to both penicillin and ampicillin. We also examined the susceptibility of each GBS pbp deletion mutant strain to two cephalosporins, ceftriaxone and cefepime. We found that all four pbp deletion mutant strains had a modest increase in susceptibility to ceftriaxone (Table 1); all but the Δpbp2a strain had increased susceptibility to cefepime (Table 1). Collectively, we found that individual deletion of GBS PBP-encoding genes did not affect growth in the rich medium; deletion of certain PBP-encoding genes altered GBS susceptibility to β-lactam antibiotics.
FIG 2.

In vitro phenotypes of PBP deletion mutant strains. (A) Colony morphology of GBS strains on sheep blood agar. (B) Growth of GBS strains in THY medium. (C) Sensitivity of GBS strains to penicillin G (10 units) and ampicillin (10 μg) measured by disk diffusion assay. Replicate data (n = 4) are expressed as means ± standard deviations (SDs). ****, P < 0.0001 versus wild-type (WT) group, one-way ANOVA.
TABLE 1.
In vitro susceptibility of each GBS strain to four β-lactams determined by Etest
| Genotype | MIC (μg/ml) of: |
|||
|---|---|---|---|---|
| Penicillin G | Ampicillin | Ceftriaxone | Cefepime | |
| WT | 0.064 | 0.125 | 0.094 | 0.25 |
| Δpbp1a | 0.032 | 0.032 | 0.064 | 0.19 |
| Δpbp1b | 0.047 | 0.094 | 0.064 | 0.19 |
| Δpbp2a | 0.064 | 0.125 | 0.064 | 0.25 |
| Δpbp2b | 0.064 | 0.125 | 0.064 | 0.19 |
Deletion of pbp1a or pbp2b alters the chain length of GBS grown in liquid medium.
Although none of the PBP deletion isogenic mutant strains had reduced growth in THY medium, we observed that Δpbp1a and Δpbp2b strains grown overnight in stationary culture formed visible cell pellets, whereas the wild-type parental strain remained in suspension (Fig. 3A). This observation stimulated us to investigate the microscopic morphology of each of the four isogenic mutant strains. Corresponding to their sedimented phenotype, Δpbp1a and Δpbp2b mutant strains formed significantly longer chains (∼65 cells/chain and 50 cells/chain, respectively) than the wild-type parental strain (∼6 cells/chain) in overnight culture (both P < 0.0001 versus wild type [WT]) (Fig. 3B and C; see also Fig. S1 in the supplemental material). In contrast, the chain length of the Δpbp2a mutant strain was only moderately longer than that of the wild-type parental strain (∼19 cells/chain, P = 0.13 versus WT), and the chain length of the Δpbp1b strain was indistinguishable from that of the wild-type parental strain (Fig. 3B and C; Fig. S1).
FIG 3.

Sedimentation and chaining phenotype of PBP deletion mutant strains. (A) Sedimentation of each GBS strain after overnight incubation in THY medium. (B) Microscopic visualization of bacterial cells from the overnight culture of each strain. (C) Chain length (number of cells in each chain) of each GBS strain. Replicate data (n = 20) are expressed as means ± SDs. ****, P < 0.0001 versus wild-type (WT) group, one-way ANOVA.
PBP gene deletion alters the essentiality of other cell wall biosynthesis genes.
To test the hypothesis that deletion of PBP genes affected the essentiality of other genes involved in cell wall biosynthesis, we generated highly saturated transposon insertion mutant libraries from the wild-type GBS strain and each of the four isogenic pbp deletion mutant strains (see Fig. S2). Each mutant library had between 180,261 and 249,719 unique transposon insertions (Fig. S2). For the saturating transposon insertion libraries generated from the wild type and each of the four individual PBP gene deletion mutants, we calculated the insertion index scores for each gene (Fig. 4A to E) and evaluated the essential gene content (Fig. 4F) in each of the five genetic backgrounds using the “tradis_gene_insert_sites” and “tradis_essentiality” scripts, respectively (40). We identified between 402 and 446 essential genes in each of the five strains (Fig. 4F; see also Tables S1 to S5). A common set of 350 genes was identified as essential in all five genetic backgrounds (Fig. 4F). We then focused on the essentiality of genes involved in cell wall biosynthesis. Although pbp1a, pbp1b, pbp2a, and pbp2b were individually dispensable for viability in the wild-type background (insertion index higher than the essential change point predicted by tradis_essentiality script) (Fig. 4A), pbp1a was essential for viability in the pbp2b deletion background (insertion index score lower than the essential change point) (Fig. 4B). Reciprocally, pbp2b was essential in the pbp1a deletion background (Fig. 4C). Moreover, rodA, encoding RodA, a SEDS family peptidoglycan polymerase that works in conjunction with PBP2B (41, 42), was additionally essential in the pbp1a deletion background (Fig. 4C). Investigation of transposon insertion profiles also confirmed that transposon insertion into pbp1a was not tolerated in the pbp2b deletion mutant genetic background, except for the last ∼10% of the gene (Fig. 5A). Similarly, transposon inactivation of pbp2b and rodA was not tolerated in the pbp1a deletion background (Fig. 5B and C).
FIG 4.

Determination of the essential genes of WT GBS and PBP deletion mutant strains. (A to E) Dot plots showing the insertion index of each gene in the GBS genome in the WT background and in each PBP deletion background. Insertion index scores of cell wall biosynthesis genes pbp1a, pbp1b, pbp2a, pbp2b, and rodA are highlighted in color. Pseudogene 1280 is highlighted in black as a control, with the assumption that insertion inactivation of a pseudogene should not affect bacterial fitness. The dotted lines denote the essential change point predicted by TraDIS. Data points below the dotted lines are predicted essential genes. (F) Venn diagram portraying the predicted essential genomes of WT GBS and PBP deletion mutant strains.
FIG 5.
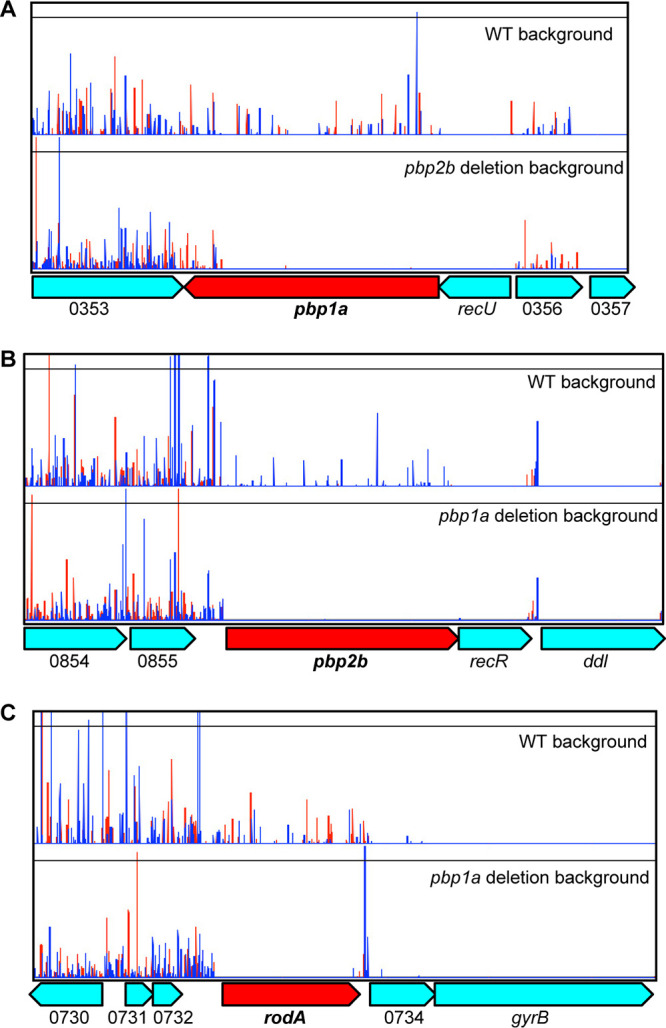
Transposon insertion profile showing the impact of deleting pbp2b or pbp1a on the essentiality of other peptidoglycan synthesis genes. (A) Transposon insertion into pbp1a is not tolerated in the pbp2b deletion background, except for the 3′ terminal last ∼10% of the gene. (B and C) Transposon inactivation of pbp2b or rodA is not tolerated in the pbp1a deletion background. The locus tag numbers refer to the annotation for GBS strain CNCTC10/84.
Unlike in the pbp1a or pbp2b deletion mutant backgrounds, we found that deletion of pbp2a or pbp1b had a more modest impact on the essentiality of other genes involved in cell wall biosynthesis (Fig. 4D and E and 6). Like in the WT GBS genetic background, in the pbp1b deletion background, pbp1a, pbp2b, rodA, and pbp2a individually were not essential (Fig. 4D). In the pbp2a knockout background, the insertion index scores of pbp1a and pbp2b approached but were slightly lower than the essential changepoint (Fig. 4E). Inspection of the transposon insertion profile showed that insertion into pbp1a and pbp2b was tolerated at a low level in the pbp2a deletion background (Fig. 6). Because the transposon insertion frequencies in pbp1a and pbp2b were much lower than the genome-wide average, they were predicted as essential genes by the tradis_essentiality script.
FIG 6.
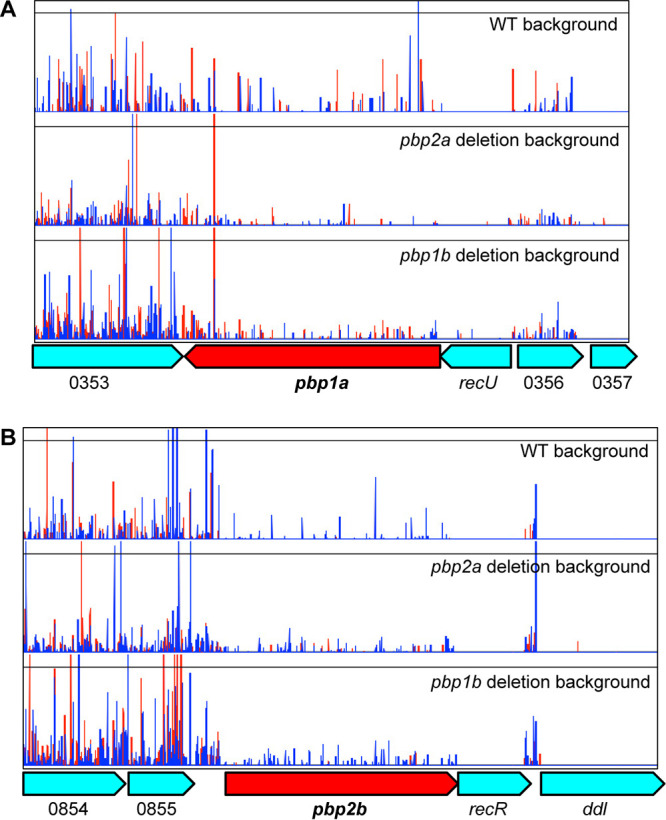
Transposon insertion profile showing the impact of deleting pbp2a or pbp1b on the essentiality of other peptidoglycan synthesis genes. In either the pbp2b deletion background or the pbp1b deletion background, transposon inactivation of pbp1a (A) or pbp2b (B) is tolerated. The locus tag numbers refer to the annotation for GBS strain CNCTC10/84.
Similarity of PBP gene essentiality between the GBS pbp2b deletion strain and Streptococcus pyogenes.
Streptococcus pyogenes is a human-specific Gram-positive bacterial pathogen causing a wide spectrum of infections (43). Unlike GBS, Streptococcus pyogenes lacks the pbp2b gene (44). This prompted us to investigate if there are essential gene content similarities between the GBS pbp2b deletion mutant strain and Streptococcus pyogenes strains. We and others have conducted comprehensive transposon mutagenesis studies in S. pyogenes strains and determined the essential genome of this organism (45, 46). We found the spectrums of essential high-molecular-mass PBP genes are identical between S. pyogenes and the GBS Δpbp2b mutant strain (Fig. 7A). Specifically, like the GBS Δpbp2b mutant strain, pbp1a is essential for viability in S. pyogenes (Fig. 7A). We previously generated two highly saturated transposon insertion mutant libraries in serotype M1 and M28 S. pyogenes strains (47). Insertion profiles of these two mutant libraries confirmed that (similarly to the GBS Δpbp2b mutant strain) transposon insertions in the S. pyogenes pbp1a gene were not tolerated, except for the 3′ terminal ∼10% of the gene (Fig. 7B).
FIG 7.
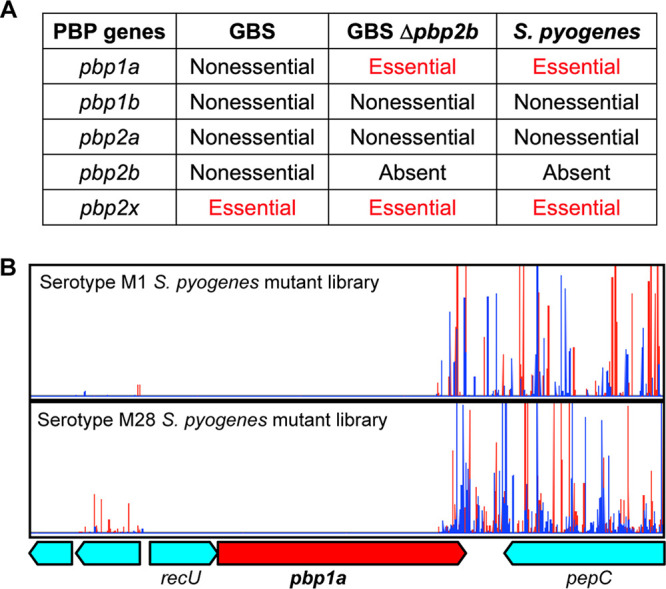
Similarity between S. pyogenes and GBS pbp2b deletion mutant strain. (A) Spectrum of essential PBP genes in WT GBS, GBS pbp2b deletion mutant, and S. pyogenes. (B) In both serotype M1 and M28 S. pyogenes, insertion inactivation of pbp1a is not tolerated, except for the last ∼10% of the coding sequence.
DISCUSSION
High-molecular-mass PBPs are essential for cell wall biosynthesis. The bacterial pathogen GBS produces five high-molecular-mass PBPs (28, 29). The relative contribution of each of these PBPs to cell wall biosynthesis and the interactions between the different PBPs are not well characterized in GBS. In this study, we performed a comprehensive deletion analysis of genes encoding high-molecular-mass PBPs in GBS. We found that depending on the PBP gene deleted, altered sensitivity to β-lactam antibiotics, cell morphology, and the essentiality of other genes involved in cell wall biosynthesis resulted.
Our results show that individual deletion of genes encoding PBP1A, PBP1B, PBP2A, or PBP2B had no apparent effect on GBS growth on blood agar or in THY medium (Fig. 2A and B). These results suggest the aforementioned PBPs are functionally redundant for catalyzing cell wall biosynthesis and resisting osmotic pressure under physiological conditions. This does not mean that the deleted PBPs are functionally equivalent and that their individual deletions had no effects. We showed that deletion of pbp1a or pbp1b resulted in enhanced sensitivity to penicillin and ampicillin (Fig. 2C and D), suggesting PBP1A and PBP1B play a larger role in transpeptidation, the cross-linking of peptidoglycan, than PBP2A or PBP2B when GBS cells are under stress imposed by these β-lactam antibiotics. Although β-lactam antibiotics inhibit all PBPs, the sensitivities of each of the PBPs to β-lactam inhibition are not equal. Kocaoglu et al. demonstrated that in Streptococcus pneumoniae, PBP2X is more sensitive than PBP1A, PBP1B, PBP2A, and PBP2B to the effects of several different β-lactams, including penicillin and ampicillin (48). It is likely that when PBP2X is partially inhibited by β-lactams, the remaining activities of PBP1A and PBP1B become crucial for maintaining cell viability. Detailed functional studies are needed to test this hypothesis. Nevertheless, our results indicate that although PBP1A, PBP1B, PBP2A, and PBP2B are functionally redundant in GBS, they are not functionally identical.
Bacterial cell wall synthesis and cell separation are intertwined processes (49–52). Many studies have demonstrated that PBP deletion mutant strains have separation defects (53–55). Here, we showed that the GBS pbp1a deletion strain and pbp2b deletion strain grew in significantly longer chains in the overnight culture (Fig. 3), suggesting these mutant strains had a cell separation defect at the stationary phase. pbp1a (also called ponA) encodes a bifunctional PBP with both transglycosylase and transpeptidase activities (27). The function of pbp1a (ponA) is well characterized in the model Gram-positive organism Bacillus subtilis (56–58). In line with our observation, Scheffers and Errington demonstrated that PBP1A is a component of the Bacillus subtilis cell division machinery (52), and mutant strains lacking PBP1A are filamentous or have a septation defect in media with low levels of divalent cations (56). Similarly, Wen et al. showed that PBP1A deficiency causes a major defect in cell division in Streptococcus mutans (59). These results suggest PBP1A may have a well-conserved role in cell division and separation in Gram-positive bacteria. pbp2b encodes a monofunctional PBP with only transpeptidase activity (60). Similar to our observation, Berg et al. demonstrated that depletion of PBP2B in S. pneumoniae gave rise to long chains of cells (61), suggesting that PBP2B also plays a role in efficient cell separation. However, it is noteworthy that although the chaining phenotype of Δpbp1a and Δpbp2b mutant strains is apparent in the overnight culture (after a 12-h incubation in THY medium), the chain lengths of Δpbp1a and Δpbp2b strains are not significantly different from that of the WT in the mid-exponential-phase culture (optical density at 600 nm [OD600] of 0.6) (data not shown), a finding consistent with a previous observation made by Jones et al. (62). We speculate that the nutrient-depleted stationary-phase culture triggered the long-chain formation by Δpbp1a and Δpbp2b strains. Further investigations are needed to test this hypothesis.
We found that although PBP2B and RodA are individually dispensable in WT GBS, both PBP2B and RodA are essential for GBS survival when PBP1A is absent. Reciprocally, PBP1A is essential for GBS survival when PBP2B is absent (Fig. 4 and 5). PBP2B is a monofunctional enzyme with transpeptidase activity (60). RodA is a well-characterized SEDS family protein with transglycosylase activity (41, 63). PBP2B and RodA are functionally related proteins (64, 65). In S. pneumoniae, PBP2B and RodA form a protein complex called the elongasome (64), which is required for lateral peptidoglycan synthesis (66). Intriguingly, PBP2B is a monofunctional transpeptidase, and RodA is a SEDS family transglycosylase (63). The PBP2B-RodA complex should have both transpeptidase activity and transglycosylase activity. We speculate that the PBP2B-RodA complex is functionally similar to PBP1A, a dual-functional enzyme with both transpeptidase and transglycosylase activities (Fig. 8). That is, the PBP2B-RodA complex could (at least partially) carry out PBP1A’s functions, and vice versa (Fig. 8). A detailed functional study of PBP1A and the PBP2B-RodA complex is needed to test this hypothesis.
FIG 8.

Schematic summary of findings. We showed that PBP1A, PBP2B, and RodA are individually dispensable for the growth of WT GBS. However, both PBP2B and RodA are essential for GBS survival when PBP1A is absent. Reciprocally, PBP1A is essential for GBS survival when PBP2B is absent. We also showed that GBS mutant strains lacking PBP1A or PBP2B formed significantly longer chains. Our results suggest the functional redundancy between PBP1A and the PBP2B-RodA complex in GBS cell wall peptidoglycan biosynthesis.
Our finding that PBP1A is essential in GBS when PBP2B is deleted is consistent with our previous saturating transposon mutagenesis studies that found that PBP1A is essential in S. pyogenes (Fig. 7), an organism that lacks PBP2B and RodA (44). This is consistent with the hypothesis that PBP1A and the PBP2B-RodA complex are functionally redundant (Fig. 8). Because S. pyogenes has fewer nonessential PBPs than GBS (Fig. 7A) (30, 45), it would be interesting to investigate if S. pyogenes has more functional constraints for acquiring PBP mutations that confer altered β-lactam susceptibility.
In summary, using targeted gene deletion and transposon mutagenesis, we performed a functional study of GBS genes encoding HMM PBPs. We found that deleting certain PBP-encoding genes alters bacterial susceptibility to β-lactam antibiotics, cell morphology, and the essentiality of other enzymes involved in cell wall peptidoglycan synthesis. The results of our study shed new light on the biological functions of PBPs in GBS.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Strain CNCTC10/84 is a well-characterized and fully sequenced type V GBS strain isolated from a septic neonate (67–84). Isogenic gene deletion mutant Δpbp1a, Δpbp1b, Δpbp2a, and Δpbp2b strains were derived from parental strain CNCTC10/84. Gene deletion was accomplished by allelic exchange as previously described (36). Briefly, two ∼1.5-kb fragments flanking the gene selected for deletion were amplified by PCR, using the genomic DNA purified from CNCTC10/84. The two flanking fragments were cloned into the suicide vector pBBL740 using the Gibson assembly (85). The resulting recombinant plasmid was then transformed into CNCTC10/84. The transformants (plasmid integrants) were isolated and passaged on nonselective agar to permit allelic exchange (36). After selecting GBS clones that were cured of the integrated pBBL740 plasmid, PCR was used to identify potential mutant candidates containing the desired deletion. Primers used for mutant construction are listed in Table S6 in the supplemental material. To rule out the introduction of spurious mutations, the genomes of all mutant strains were sequenced. Specifically, the genomes of GBS mutant strains were sequenced using an Illumina NextSeq 550 instrument. Sequence reads were quality filtered, adapter trimmed, and error corrected using Trimmomatic and Musket (86) and then mapped to the genome of CNCTC10/84 using SMALT (https://www.sanger.ac.uk/tool/smalt-0/). Single-nucleotide polymorphisms (SNPs) and insertions and deletions (indels) were identified using FreeBayes and Pilon (87). GBS strains used in this study were cultured in Todd-Hewitt broth supplemented with 0.2% yeast extract (THY broth) at 37°C with 5% CO2.
Antibiotic susceptibility determinations.
Disk diffusion assay and Etest were used to measure the sensitivity of each of the GBS strains to β-lactam antibiotics according to previously described methods (37–39, 88, 89). For the disk diffusion assay, GBS strains were evenly plated onto Mueller-Hinton agar using sterile swabs. Penicillin G disks (Thermo Scientific, CT0043B) or ampicillin disks (Thermo Scientific, CT0003B) were then placed onto the center of the agar. After overnight incubation at 37°C with 5% CO2, the zone of inhibition was measured with a digital caliper. MIC values of each of the GBS strains for β-lactam antibiotics (penicillin G, ampicillin, ceftriaxone, and cefepime) were determined by the gradient method (Etest strips; bioMérieux) using standard clinical laboratory procedures (37–39, 89).
Microscopic visualization of bacteria.
GBS mid-exponential-phase culture and overnight culture were used for microscopic analysis. After suspending GBS cells by gently inverting the liquid culture three times (avoiding disruption of the cellular chains), 10 μl of the cell suspension was added to the glass slide and air dried completely. Cellular morphology of each deletion mutant and wild-type GBS was observed using light microscopy (cells stained by Gram staining) (90) and phase-contrast microscopy (unstained cells) (91). To measure the chain length of each GBS strain, 20 chains from each strain were randomly selected and measured by counting the number of cells in each chain (Fig. 3) (92). One-way analysis of variance (ANOVA) was used to determine significant differences of chain lengths among GBS strains. A P value of less than 0.05 was considered statistically significant.
Generation of GBS transposon insertion mutant libraries.
Transposon insertion mutant libraries were generated in WT strain CNCTC10/84 and each of the isogenic gene knockout mutant strains using a well-established protocol (46). Briefly, transposon plasmid pGh9:ISS1 was transformed into each GBS strain by electroporation. Transformants were picked and grown overnight at 28°C (permissive temperature) in THY broth supplemented with 0.5 μg/ml erythromycin. The resulting culture was heat shocked at 40°C (nonpermissive temperature) for 3 h to permit random integration of pGh9:ISS1 into the GBS genome. After the heat shock, the GBS culture was plated on THY agar supplemented with 0.5 μg/ml erythromycin and incubated overnight at 37°C. The transposon mutant libraries were collected by scraping the colonies off the agar plates.
Preparing and sequencing the transposon insertion mutant libraries.
Preparation of genomic DNA and sequencing of transposon mutant libraries was performed as previously described (46, 93). Briefly, genomic DNA of each mutant library was isolated using the DNeasy blood and tissue kit (Qiagen). PCR was used to amplify the transposon-chromosome junction. The PCR-amplified libraries were sequenced with a NextSeq 550 instrument (Illumina) using a single-end 75-cycle protocol.
Statistical analysis.
Statistical significance was assessed by the one-way ANOVA (Prism 8; GraphPad Software, Inc.). A P value of less than 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by funds from the Methodist Hospital Research Institute.
L.Z., R.J.O., S.B.B., and J.M.M. generated, analyzed, and interpreted data. A.S.W. provided scholarly input on the TraDIS studies. P.Y., L.P., and A.M. provided expert technical assistance. L.Z. conceptualized and designed the study. All authors contributed to writing the manuscript. All authors reviewed and approved the final manuscript.
Footnotes
Supplemental material is available online only.
Contributor Information
Luchang Zhu, Email: lzhu2@houstonmethodist.org.
Michael J. Federle, University of Illinois at Chicago
REFERENCES
- 1.Campbell JR, Hillier SL, Krohn MA, Ferrieri P, Zaleznik DF, Baker CJ. 2000. Group B streptococcal colonization and serotype-specific immunity in pregnant women at delivery. Obstet Gynecol 96:498–503. 10.1016/s0029-7844(00)00977-7. [DOI] [PubMed] [Google Scholar]
- 2.Schuchat A. 1998. Epidemiology of group B streptococcal disease in the United States: shifting paradigms. Clin Microbiol Rev 11:497–513. 10.1128/CMR.11.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weston EJ, Pondo T, Lewis MM, Martell-Cleary P, Morin C, Jewell B, Daily P, Apostol M, Petit S, Farley M, Lynfield R, Reingold A, Hansen NI, Stoll BJ, Shane AL, Zell E, Schrag SJ. 2011. The burden of invasive early-onset neonatal sepsis in the United States, 2005–2008. Pediatr Infect Dis J 30:937–941. 10.1097/INF.0b013e318223bad2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoll BJ, Hansen NI, Sanchez PJ, Faix RG, Poindexter BB, Van Meurs KP, Bizzarro MJ, Goldberg RN, Frantz ID, III, Hale EC, Shankaran S, Kennedy K, Carlo WA, Watterberg KL, Bell EF, Walsh MC, Schibler K, Laptook AR, Shane AL, Schrag SJ, Das A, Higgins RD, Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. 2011. Early onset neonatal sepsis: the burden of group B streptococcal and E. coli disease continues. Pediatrics 127:817–826. 10.1542/peds.2010-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seale AC, Bianchi-Jassir F, Russell NJ, Kohli-Lynch M, Tann CJ, Hall J, Madrid L, Blencowe H, Cousens S, Baker CJ, Bartlett L, Cutland C, Gravett MG, Heath PT, Ip M, Le Doare K, Madhi SA, Rubens CE, Saha SK, Schrag SJ, Sobanjo-Ter Meulen A, Vekemans J, Lawn JE. 2017. Estimates of the burden of group B streptococcal disease worldwide for pregnant women, stillbirths, and children. Clin Infect Dis 65:S200–S219. 10.1093/cid/cix664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johri AK, Paoletti LC, Glaser P, Dua M, Sharma PK, Grandi G, Rappuoli R. 2006. Group B Streptococcus: global incidence and vaccine development. Nat Rev Microbiol 4:932–942. 10.1038/nrmicro1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nanduri SA, Petit S, Smelser C, Apostol M, Alden NB, Harrison LH, Lynfield R, Vagnone PS, Burzlaff K, Spina NL, Dufort EM, Schaffner W, Thomas AR, Farley MM, Jain JH, Pondo T, McGee L, Beall BW, Schrag SJ. 2019. Epidemiology of invasive early-onset and late-onset group B streptococcal disease in the United States, 2006 to 2015: multistate laboratory and population-based surveillance. JAMA Pediatr 173:224–233. 10.1001/jamapediatrics.2018.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. 2009. Trends in perinatal group B streptococcal disease - United States, 2000–2006. MMWR Morb Mortal Wkly Rep 58:109–112. [PubMed] [Google Scholar]
- 9.Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, Craig AS, Schaffner W, Zansky SM, Gershman K, Stefonek KR, Albanese BA, Zell ER, Schuchat A, Schrag SJ, Active Bacterial Core surveillance/Emerging Infections Program Network. 2008. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. JAMA 299:2056–2065. 10.1001/jama.299.17.2056. [DOI] [PubMed] [Google Scholar]
- 10.Kalin A, Acosta C, Kurinczuk JJ, Brocklehurst P, Knight M. 2015. Severe sepsis in women with group B Streptococcus in pregnancy: an exploratory UK national case-control study. BMJ Open 5:e007976. 10.1136/bmjopen-2015-007976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cape A, Tuomala RE, Taylor C, Puopolo KM. 2013. Peripartum bacteremia in the era of group B streptococcus prophylaxis. Obstet Gynecol 121:812–818. 10.1097/AOG.0b013e3182888032. [DOI] [PubMed] [Google Scholar]
- 12.Zaleznik DF, Rench MA, Hillier S, Krohn MA, Platt R, Lee ML, Flores AE, Ferrieri P, Baker CJ. 2000. Invasive disease due to group B streptococcus in pregnant women and neonates from diverse population groups. Clin Infect Dis 30:276–281. 10.1086/313665. [DOI] [PubMed] [Google Scholar]
- 13.Kankuri E, Kurki T, Carlson P, Hiilesmaa V. 2003. Incidence, treatment and outcome of peripartum sepsis. Acta Obstet Gynecol Scand 82:730–735. 10.1034/j.1600-0412.2003.00265.x. [DOI] [PubMed] [Google Scholar]
- 14.Krohn MA, Hillier SL, Baker CJ. 1999. Maternal peripartum complications associated with vaginal group B streptococci colonization. J Infect Dis 179:1410–1415. 10.1086/314756. [DOI] [PubMed] [Google Scholar]
- 15.Anderson BL, Simhan HN, Simons KM, Wiesenfeld HC. 2007. Untreated asymptomatic group B streptococcal bacteriuria early in pregnancy and chorioamnionitis at delivery. Am J Obstet Gynecol 196:524.e1–524.e5. 10.1016/j.ajog.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Weisman LE, Stoll BJ, Cruess DF, Hall RT, Merenstein GB, Hemming VG, Fischer GW. 1992. Early-onset group B streptococcal sepsis: a current assessment. J Pediatr 121:428–433. 10.1016/S0022-3476(05)81801-3. [DOI] [PubMed] [Google Scholar]
- 17.De Cueninck BJ, Shockman GD, Swenson RM. 1982. Group B, type III streptococcal cell wall: composition and structural aspects revealed through endo-N-acetylmuramidase-catalyzed hydrolysis. Infect Immun 35:572–581. 10.1128/iai.35.2.572-581.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pasquina-Lemonche L, Burns J, Turner RD, Kumar S, Tank R, Mullin N, Wilson JS, Chakrabarti B, Bullough PA, Foster SJ, Hobbs JK. 2020. The architecture of the Gram-positive bacterial cell wall. Nature 582:294–297. 10.1038/s41586-020-2236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng L, Kasper DL, Krick TP, Wessels MR. 2000. Characterization of the linkage between the type III capsular polysaccharide and the bacterial cell wall of group B Streptococcus. J Biol Chem 275:7497–7504. 10.1074/jbc.275.11.7497. [DOI] [PubMed] [Google Scholar]
- 20.Schubert A, Zakikhany K, Schreiner M, Frank R, Spellerberg B, Eikmanns BJ, Reinscheid DJ. 2002. A fibrinogen receptor from group B Streptococcus interacts with fibrinogen by repetitive units with novel ligand binding sites. Mol Microbiol 46:557–569. 10.1046/j.1365-2958.2002.03177.x. [DOI] [PubMed] [Google Scholar]
- 21.Lauer P, Rinaudo CD, Soriani M, Margarit I, Maione D, Rosini R, Taddei AR, Mora M, Rappuoli R, Grandi G, Telford JL. 2005. Genome analysis reveals pili in group B Streptococcus. Science 309:105. 10.1126/science.1111563. [DOI] [PubMed] [Google Scholar]
- 22.Rosini R, Rinaudo CD, Soriani M, Lauer P, Mora M, Maione D, Taddei A, Santi I, Ghezzo C, Brettoni C, Buccato S, Margarit I, Grandi G, Telford JL. 2006. Identification of novel genomic islands coding for antigenic pilus-like structures in Streptococcus agalactiae. Mol Microbiol 61:126–141. 10.1111/j.1365-2958.2006.05225.x. [DOI] [PubMed] [Google Scholar]
- 23.Baron MJ, Bolduc GR, Goldberg MB, Auperin TC, Madoff LC. 2004. Alpha C protein of group B Streptococcus binds host cell surface glycosaminoglycan and enters cells by an actin-dependent mechanism. J Biol Chem 279:24714–24723. 10.1074/jbc.M402164200. [DOI] [PubMed] [Google Scholar]
- 24.Mistou MY, Dramsi S, Brega S, Poyart C, Trieu-Cuot P. 2009. Molecular dissection of the secA2 locus of group B Streptococcus reveals that glycosylation of the Srr1 LPXTG protein is required for full virulence. J Bacteriol 191:4195–4206. 10.1128/JB.01673-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheffers DJ, Pinho MG. 2005. Bacterial cell wall synthesis: new insights from localization studies. Microbiol Mol Biol Rev 69:585–607. 10.1128/MMBR.69.4.585-607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghuysen JM. 1991. Serine beta-lactamases and penicillin-binding proteins. Annu Rev Microbiol 45:37–67. 10.1146/annurev.mi.45.100191.000345. [DOI] [PubMed] [Google Scholar]
- 27.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 28.Glaser P, Rusniok C, Buchrieser C, Chevalier F, Frangeul L, Msadek T, Zouine M, Couve E, Lalioui L, Poyart C, Trieu-Cuot P, Kunst F. 2002. Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol Microbiol 45:1499–1513. 10.1046/j.1365-2958.2002.03126.x. [DOI] [PubMed] [Google Scholar]
- 29.Tettelin H, Masignani V, Cieslewicz MJ, Eisen JA, Peterson S, Wessels MR, Paulsen IT, Nelson KE, Margarit I, Read TD, Madoff LC, Wolf AM, Beanan MJ, Brinkac LM, Daugherty SC, DeBoy RT, Durkin AS, Kolonay JF, Madupu R, Lewis MR, Radune D, Fedorova NB, Scanlan D, Khouri H, Mulligan S, Carty HA, Cline RT, Van Aken SE, Gill J, Scarselli M, Mora M, Iacobini ET, Brettoni C, Galli G, Mariani M, Vegni F, Maione D, Rinaudo D, Rappuoli R, Telford JL, Kasper DL, Grandi G, Fraser CM. 2002. Complete genome sequence and comparative genomic analysis of an emerging human pathogen, serotype V Streptococcus agalactiae. Proc Natl Acad Sci U S A 99:12391–12396. 10.1073/pnas.182380799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hooven TA, Catomeris AJ, Akabas LH, Randis TM, Maskell DJ, Peters SE, Ott S, Santana-Cruz I, Tallon LJ, Tettelin H, Ratner AJ. 2016. The essential genome of Streptococcus agalactiae. BMC Genomics 17:406. 10.1186/s12864-016-2741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones AL, Knoll KM, Rubens CE. 2000. Identification of Streptococcus agalactiae virulence genes in the neonatal rat sepsis model using signature-tagged mutagenesis. Mol Microbiol 37:1444–1455. 10.1046/j.1365-2958.2000.02099.x. [DOI] [PubMed] [Google Scholar]
- 32.Jones AL, Mertz RH, Carl DJ, Rubens CE. 2007. A streptococcal penicillin-binding protein is critical for resisting innate airway defenses in the neonatal lung. J Immunol 179:3196–3202. 10.4049/jimmunol.179.5.3196. [DOI] [PubMed] [Google Scholar]
- 33.Hamilton A, Popham DL, Carl DJ, Lauth X, Nizet V, Jones AL. 2006. Penicillin-binding protein 1a promotes resistance of group B streptococcus to antimicrobial peptides. Infect Immun 74:6179–6187. 10.1128/IAI.00895-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hooven TA, Randis TM, Daugherty SC, Narechania A, Planet PJ, Tettelin H, Ratner AJ. 2014. Complete genome sequence of Streptococcus agalactiae CNCTC 10/84, a hypervirulent sequence type 26 strain. Genome Announc 2:e01338-14. 10.1128/genomeA.01338-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu L, Yerramilli P, Pruitt L, Ojeda Saavedra M, Cantu CC, Olsen RJ, Beres SB, Waller AS, Musser JM. 2020. Genome-wide assessment of Streptococcus agalactiae genes required for survival in human whole blood and plasma. Infect Immun 88:e00357-20. 10.1128/IAI.00357-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu L, Olsen RJ, Nasser W, Beres SB, Vuopio J, Kristinsson KG, Gottfredsson M, Porter AR, DeLeo FR, Musser JM. 2015. A molecular trigger for intercontinental epidemics of group A Streptococcus. J Clin Invest 125:3545–3559. 10.1172/JCI82478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canton R, Livermore DM, Morosini MI, Diaz-Reganon J, Rossolini GM, Group PS, PREMIUM Study Group. 2017. Etest versus broth microdilution for ceftaroline MIC determination with Staphylococcus aureus: results from PREMIUM, a European multicentre study. J Antimicrob Chemother 72:431–436. 10.1093/jac/dkw442. [DOI] [PubMed] [Google Scholar]
- 38.Conceicao N, Rodrigues WF, de Oliveira KLP, da Silva LEP, de Souza LRC, da de Cunha Hueb Barata Oliveira C, de Oliveira AG. 2020. Beta-lactams susceptibility testing of penicillin-resistant, ampicillin-susceptible Enterococcus faecalis isolates: a comparative assessment of Etest and disk diffusion methods against broth dilution. Ann Clin Microbiol Antimicrob 19:43. 10.1186/s12941-020-00386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein FW, Ly A, Kitzis MD. 2007. Comparison of Etest with agar dilution for testing the susceptibility of Pseudomonas aeruginosa and other multidrug-resistant bacteria to colistin. J Antimicrob Chemother 59:1039–1040. 10.1093/jac/dkm046. [DOI] [PubMed] [Google Scholar]
- 40.Barquist L, Mayho M, Cummins C, Cain AK, Boinett CJ, Page AJ, Langridge GC, Quail MA, Keane JA, Parkhill J. 2016. The TraDIS toolkit: sequencing and analysis for dense transposon mutant libraries. Bioinformatics 32:1109–1111. 10.1093/bioinformatics/btw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emami K, Guyet A, Kawai Y, Devi J, Wu LJ, Allenby N, Daniel RA, Errington J. 2017. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat Microbiol 2:16253. 10.1038/nmicrobiol.2016.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sjodt M, Brock K, Dobihal G, Rohs PDA, Green AG, Hopf TA, Meeske AJ, Srisuknimit V, Kahne D, Walker S, Marks DS, Bernhardt TG, Rudner DZ, Kruse AC. 2018. Structure of the peptidoglycan polymerase RodA resolved by evolutionary coupling analysis. Nature 556:118–121. 10.1038/nature25985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin Microbiol Rev 27:264–301. 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Philippe J, Vernet T, Zapun A. 2014. The elongation of ovococci. Microb Drug Resist 20:215–221. 10.1089/mdr.2014.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Breton Y, Belew AT, Valdes KM, Islam E, Curry P, Tettelin H, Shirtliff ME, El-Sayed NM, McIver KS. 2015. Essential genes in the core genome of the human pathogen Streptococcus pyogenes. Sci Rep 5:9838. 10.1038/srep09838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu L, Charbonneau ARL, Waller AS, Olsen RJ, Beres SB, Musser JM. 2017. Novel genes required for the fitness of Streptococcus pyogenes in human saliva. mSphere 2:e00460-17. 10.1128/mSphereDirect.00460-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu L, Olsen RJ, Beres SB, Eraso JM, Saavedra MO, Kubiak SL, Cantu CC, Jenkins L, Charbonneau ARL, Waller AS, Musser JM. 2019. Gene fitness landscape of group A streptococcus during necrotizing myositis. J Clin Invest 129:887–901. 10.1172/JCI124994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kocaoglu O, Tsui HC, Winkler ME, Carlson EE. 2015. Profiling of beta-lactam selectivity for penicillin-binding proteins in Streptococcus pneumoniae D39. Antimicrob Agents Chemother 59:3548–3555. 10.1128/AAC.05142-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spratt BG. 1975. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc Natl Acad Sci U S A 72:2999–3003. 10.1073/pnas.72.8.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morlot C, Zapun A, Dideberg O, Vernet T. 2003. Growth and division of Streptococcus pneumoniae: localization of the high molecular weight penicillin-binding proteins during the cell cycle. Mol Microbiol 50:845–855. 10.1046/j.1365-2958.2003.03767.x. [DOI] [PubMed] [Google Scholar]
- 51.Pinho MG, Errington J. 2005. Recruitment of penicillin-binding protein PBP2 to the division site of Staphylococcus aureus is dependent on its transpeptidation substrates. Mol Microbiol 55:799–807. 10.1111/j.1365-2958.2004.04420.x. [DOI] [PubMed] [Google Scholar]
- 52.Scheffers DJ, Errington J. 2004. PBP1 is a component of the Bacillus subtilis cell division machinery. J Bacteriol 186:5153–5156. 10.1128/JB.186.15.5153-5156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haenni M, Majcherczyk PA, Barblan JL, Moreillon P. 2006. Mutational analysis of class A and class B penicillin-binding proteins in Streptococcus gordonii. Antimicrob Agents Chemother 50:4062–4069. 10.1128/AAC.00677-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Priyadarshini R, Popham DL, Young KD. 2006. Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J Bacteriol 188:5345–5355. 10.1128/JB.00476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang KN, Kazi MI, Biboy J, Gray J, Bovermann H, Ausman J, Boutte CC, Vollmer W, Boll JM. 2021. Septal class a penicillin-binding protein activity and ld-transpeptidases mediate selection of colistin-resistant lipooligosaccharide-deficient Acinetobacter baumannii. mBio 12:e02185-20. 10.1128/mBio.02185-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murray T, Popham DL, Setlow P. 1998. Bacillus subtilis cells lacking penicillin-binding protein 1 require increased levels of divalent cations for growth. J Bacteriol 180:4555–4563. 10.1128/JB.180.17.4555-4563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Popham DL, Setlow P. 1995. Cloning, nucleotide sequence, and mutagenesis of the Bacillus subtilis ponA operon, which codes for penicillin-binding protein (PBP) 1 and a PBP-related factor. J Bacteriol 177:326–335. 10.1128/jb.177.2.326-335.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McPherson DC, Popham DL. 2003. Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J Bacteriol 185:1423–1431. 10.1128/JB.185.4.1423-1431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wen ZT, Bitoun JP, Liao S. 2015. PBP1a-deficiency causes major defects in cell division, growth and biofilm formation by Streptococcus mutans. PLoS One 10:e0124319. 10.1371/journal.pone.0124319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pagliero E, Chesnel L, Hopkins J, Croize J, Dideberg O, Vernet T, Di Guilmi AM. 2004. Biochemical characterization of Streptococcus pneumoniae penicillin-binding protein 2b and its implication in beta-lactam resistance. Antimicrob Agents Chemother 48:1848–1855. 10.1128/AAC.48.5.1848-1855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berg KH, Stamsas GA, Straume D, Havarstein LS. 2013. Effects of low PBP2b levels on cell morphology and peptidoglycan composition in Streptococcus pneumoniae R6. J Bacteriol 195:4342–4354. 10.1128/JB.00184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones AL, Needham RH, Clancy A, Knoll KM, Rubens CE. 2003. Penicillin-binding proteins in Streptococcus agalactiae: a novel mechanism for evasion of immune clearance. Mol Microbiol 47:247–256. 10.1046/j.1365-2958.2003.03297.x. [DOI] [PubMed] [Google Scholar]
- 63.Meeske AJ, Riley EP, Robins WP, Uehara T, Mekalanos JJ, Kahne D, Walker S, Kruse AC, Bernhardt TG, Rudner DZ. 2016. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537:634–638. 10.1038/nature19331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Straume D, Stamsas GA, Berg KH, Salehian Z, Havarstein LS. 2017. Identification of pneumococcal proteins that are functionally linked to penicillin-binding protein 2b (PBP2b). Mol Microbiol 103:99–116. 10.1111/mmi.13543. [DOI] [PubMed] [Google Scholar]
- 65.Leclercq S, Derouaux A, Olatunji S, Fraipont C, Egan AJ, Vollmer W, Breukink E, Terrak M. 2017. Interplay between penicillin-binding proteins and SEDS proteins promotes bacterial cell wall synthesis. Sci Rep 7:43306. 10.1038/srep43306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsui HC, Zheng JJ, Magallon AN, Ryan JD, Yunck R, Rued BE, Bernhardt TG, Winkler ME. 2016. Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Mol Microbiol 100:1039–1065. 10.1111/mmi.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nizet V, Gibson RL, Chi EY, Framson PE, Hulse M, Rubens CE. 1996. Group B streptococcal beta-hemolysin expression is associated with injury of lung epithelial cells. Infect Immun 64:3818–3826. 10.1128/iai.64.9.3818-3826.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Randis TM, Gelber SE, Hooven TA, Abellar RG, Akabas LH, Lewis EL, Walker LB, Byland LM, Nizet V, Ratner AJ. 2014. Group B Streptococcus beta-hemolysin/cytolysin breaches maternal-fetal barriers to cause preterm birth and intrauterine fetal demise in vivo. J Infect Dis 210:265–273. 10.1093/infdis/jiu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patras KA, Doran KS. 2016. A murine model of group B streptococcus vaginal colonization. J Vis Exp 2016:54708. 10.3791/54708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Doran KS, Liu GY, Nizet V. 2003. Group B streptococcal beta-hemolysin/cytolysin activates neutrophil signaling pathways in brain endothelium and contributes to development of meningitis. J Clin Invest 112:736–744. 10.1172/JCI200317335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patras KA, Derieux J, Al-Bassam MM, Adiletta N, Vrbanac A, Lapek JD, Zengler K, Gonzalez DJ, Nizet V. 2018. Group B streptococcus biofilm regulatory protein A contributes to bacterial physiology and innate immune resistance. J Infect Dis 218:1641–1652. 10.1093/infdis/jiy341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kulkarni R, Randis TM, Antala S, Wang A, Amaral FE, Ratner AJ. 2013. β-Hemolysin/cytolysin of group B Streptococcus enhances host inflammation but is dispensable for establishment of urinary tract infection. PLoS One 8:e59091. 10.1371/journal.pone.0059091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin SM, Jang AY, Zhi Y, Gao S, Lim S, Lim JH, Song JY, Sullam PM, Rhee JH, Seo HS. 2017. Vaccination with a latch peptide provides serotype-independent protection against group B streptococcus infection in mice. J Infect Dis 217:93–102. 10.1093/infdis/jix565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gendrin C, Vornhagen J, Ngo L, Whidbey C, Boldenow E, Santana-Ufret V, Clauson M, Burnside K, Galloway DP, Adams Waldorf KM, Piliponsky AM, Rajagopal L. 2015. Mast cell degranulation by a hemolytic lipid toxin decreases GBS colonization and infection. Sci Adv 1:e1400225. 10.1126/sciadv.1400225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Banerjee A, Kim BJ, Carmona EM, Cutting AS, Gurney MA, Carlos C, Feuer R, Prasadarao NV, Doran KS. 2011. Bacterial pili exploit integrin machinery to promote immune activation and efficient blood-brain barrier penetration. Nat Commun 2:462. 10.1038/ncomms1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Sorge NM, Quach D, Gurney MA, Sullam PM, Nizet V, Doran KS. 2009. The group B streptococcal serine-rich repeat 1 glycoprotein mediates penetration of the blood-brain barrier. J Infect Dis 199:1479–1487. 10.1086/598217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang NY, Patras KA, Seo HS, Cavaco CK, Rosler B, Neely MN, Sullam PM, Doran KS. 2014. Group B streptococcal serine-rich repeat proteins promote interaction with fibrinogen and vaginal colonization. J Infect Dis 210:982–991. 10.1093/infdis/jiu151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alkuwaity K, Taylor A, Heckels JE, Doran KS, Christodoulides M. 2012. Group B Streptococcus interactions with human meningeal cells and astrocytes in vitro. PLoS One 7:e42660. 10.1371/journal.pone.0042660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cutting AS, Del Rosario Y, Mu R, Rodriguez A, Till A, Subramani S, Gottlieb RA, Doran KS. 2014. The role of autophagy during group B Streptococcus infection of blood-brain barrier endothelium. J Biol Chem 289:35711–35723. 10.1074/jbc.M114.588657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pritzlaff CA, Chang JC, Kuo SP, Tamura GS, Rubens CE, Nizet V. 2001. Genetic basis for the beta-haemolytic/cytolytic activity of group B Streptococcus. Mol Microbiol 39:236–247. 10.1046/j.1365-2958.2001.02211.x. [DOI] [PubMed] [Google Scholar]
- 81.Hooven TA, Bonakdar M, Chamby AB, Ratner AJ. 2019. A counterselectable sucrose sensitivity marker permits efficient and flexible mutagenesis in Streptococcus agalactiae. Appl Environ Microbiol 85:e03009-18. 10.1128/AEM.03009-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seo HS, Mu R, Kim BJ, Doran KS, Sullam PM. 2012. Binding of glycoprotein Srr1 of Streptococcus agalactiae to fibrinogen promotes attachment to brain endothelium and the development of meningitis. PLoS Pathog 8:e1002947. 10.1371/journal.ppat.1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maisey HC, Hensler M, Nizet V, Doran KS. 2007. Group B streptococcal pilus proteins contribute to adherence to and invasion of brain microvascular endothelial cells. J Bacteriol 189:1464–1467. 10.1128/JB.01153-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim BJ, Bee OB, McDonagh MA, Stebbins MJ, Palecek SP, Doran KS, Shusta EV. 2017. Modeling group B Streptococcus and blood-brain barrier interaction by using induced pluripotent stem cell-derived brain endothelial cells. mSphere 2:e00398-17. 10.1128/mSphere.00398-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO III. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 86.Liu Y, Schroder J, Schmidt B. 2013. Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 29:308–315. 10.1093/bioinformatics/bts690. [DOI] [PubMed] [Google Scholar]
- 87.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kimura K, Wachino J, Kurokawa H, Suzuki S, Yamane K, Shibata N, Arakawa Y. 2009. Practical disk diffusion test for detecting group B streptococcus with reduced penicillin susceptibility. J Clin Microbiol 47:4154–4157. 10.1128/JCM.02063-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sader HS, Pignatari AC. 1994. E test: a novel technique for antimicrobial susceptibility testing. Sao Paulo Med J 112:635–638. 10.1590/s1516-31801994000400003. [DOI] [PubMed] [Google Scholar]
- 90.Coico R. 2005. Gram staining. Curr Protoc Microbiol Appendix 3:Appendix 3C. 10.1002/9780471729259.mca03cs00. [DOI] [PubMed] [Google Scholar]
- 91.Poetschke G, Bommer W. 1950. Vital study of bacteria with the phase contrast microscope. Zentralbl Bakteriol Orig 156:128–136. (In undetermined language.) [PubMed] [Google Scholar]
- 92.Arrigucci R, Pozzi G. 2017. Identification of the chain-dispersing peptidoglycan hydrolase LytB of Streptococcus gordonii. PLoS One 12:e0176117. 10.1371/journal.pone.0176117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Charbonneau ARL, Forman OP, Cain AK, Newland G, Robinson C, Boursnell M, Parkhill J, Leigh JA, Maskell DJ, Waller AS. 2017. Defining the ABC of gene essentiality in streptococci. BMC Genomics 18:426. 10.1186/s12864-017-3794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download JB.00234-21-s0001.xlsx, XLSX file, 0.04 MB (36.7KB, xlsx)
Table S2. Download JB.00234-21-s0002.xlsx, XLSX file, 0.03 MB (35.4KB, xlsx)
Table S3. Download JB.00234-21-s0003.xlsx, XLSX file, 0.04 MB (36.6KB, xlsx)
Table S4. Download JB.00234-21-s0004.xlsx, XLSX file, 0.04 MB (37.4KB, xlsx)
Table S5. Download JB.00234-21-s0005.xlsx, XLSX file, 0.04 MB (38.2KB, xlsx)
Table S6. Download JB.00234-21-s0006.xlsx, XLSX file, 0.01 MB (10KB, xlsx)
Fig. S1 and S2. Download JB.00234-21-s0007.pdf, PDF file, 5.2 MB (5.2MB, pdf)