

Abstract
Conventional randomized controlled trials (RCTs) compare treatment effectiveness to provide a basis for evidence-based treatments that can be generalized to the average patient. However, this information may not always be useful for treating individual patients. We present an alternative approach to identifying optimized treatments using experimental designs that focus on individuals. Personalized designs, or N-of-1 designs, provide both a functional analysis demonstrating that changes in patient symptoms are due to the treatment implemented and a comparative analysis of treatments. This approach is relevant in the zeitgeist of personalized medicine and provides clinicians with a paradigm for investigating optimal treatments for rare diseases in which RCTs are not feasible, identifying optimal treatments for patients with comorbidities who would be excluded from most RCTs, handling clinical situations in which patients respond idiosyncratically (either positively or negatively) to treatment, and shortening the gap between identification and implementation of an evidence-based treatment. These designs merge experimental analysis of behavior methods used for decades in psychology with new methodological and statistical advances to assess significance levels of change in individual patients, and they can be generalized to larger populations for meta-analytic purposes. This paper presents a case for why these models are needed, an overview of how to apply personalized designs for different types of clinical scenarios, and a brief discussion of issues associated with interpretation and implementation of personalized designs. Our goal is to empower pediatricians to take personalized trial designs into clinical practice to identify optimal treatments for their patients.
Keywords: single-case studies, personalized trials, research design, experimental medicine
Overview
One of the tenets of personalized experimental medicine is the idea that treatments that best benefit each patient should be identified. Parallel-group randomized controlled trials (RCTs) either (1) provide evidence on how groups of patients respond to a treatment versus a control or (2) compare alternative treatments, but they do not allow the identification of optimized treatments for an individual. An alternative experimental method and clinical decision approach is the personalized (N-of-1) trial, which is a multiple-time-period, active-comparator crossover trial that is frequently randomized and can be masked. Some consider such designs to be the pinnacle of the strength of evidence hierarchy (Table 1),1 as they provide evidence for the net benefit of treatments for individual patients. Educating clinicians and clinician-scientists about personalized trials provides a novel approach to improving pediatric care. Unique benefits of these designs are their usefulness in determining optimal therapy for patients with rare diseases, patients with comorbidities who do not meet criteria for usual RCTs, or patients with unusual side effects or idiosyncratic treatment responses. The approach may also be useful in accelerating the time from identification of evidence-based treatment to implementation of effective care.
Table 1.
Original Hierarchy of Strength of Evidence for Treatment Decisions (Guyatt et al 2002).1
| N-of-1 Randomized Trial |
|---|
| Systematic reviews of randomized trials |
| Single randomized trial |
| Systematic review of observational studies addressing patient-important outcomes |
| Single observational study addressing patient-important outcomes |
| Physiologic studies |
| Unsystematic clinical observations |
Parallel-group, randomized efficacy trials usually compare alternative treatments, or treatments to controls, and maximize the internal validity of findings across treatment groups. Parallel-group effectiveness trials are conducted with a wider range of participants, often in real-world settings, with the goal of bolstering external validity or extending the applicability of findings to broader populations. While different in their emphases, both types of RCTs compare the average response in the treatment versus comparator conditions—thereby accounting for variability in treatment response—with the goal of extrapolating optimal treatment for patients with characteristics similar to those of study participants.
If there is minimal variability in response to treatment in an RCT, the best prediction of the magnitude of treatment benefit for an individual patient will be that estimated from the overall trial—or the so-called main effect. However, there is almost always heterogeneity of treatment effects (HTE) or variability in the balance of benefits and harms of treatment found for different patients, both subgroups and individuals, included in the RCT.2 HTE may include some patients having idiosyncratic treatment responses to initial treatments, and these are difficult to ascertain from reports of parallel-group RCTs. In one empirical analysis, for example, only a minority of participants in RCTs experienced the average treatment benefit reported while the majority of participants did not receive the reported treatment benefit.3 This type of finding from parallel-group RCTs is ubiquitous and suggests that there is substantial uncertainty about extrapolating net treatment benefit for each patient.4
In addition to the frequency of HTE in clinical trials, there are several other reasons to consider alternative designs. First, RCTs often require hundreds of patients to have enough statistical power to precisely identify the treatment effect estimate, and they are thus conducted infrequently on patients with rare diseases. Second, many RCTs have strict entrance criteria, which a specific patient may not meet. This may be due to age, gender, race, or comorbidities and may force clinicians to extrapolate results from one population to another with little evidence to guide such decisions. Finally, there is a wide temporal window between identification of an evidence-based treatment and adoption of that treatment in practice. RCTs are often take extraordinary time to be completed.5 This means that discovery of potential benefits (or harms) are delayed for years before they are available for care. Personalized trials can provide tools for individual clinicians to test new treatments with patients under their care more rapidly and efficiently, and pediatricians may be more likely to adopt effective treatments if they can prove to themselves that these treatments work for their patients.
Introducing Personalized Trials
Personalized or N-of-1 trials are single-participant, multiple-time-period, active-comparator crossover trials that are frequently randomized and can be masked.6,7 As an alternative to extrapolating the results of a conventional trial to an individual, personalized trials provide a clinician with an empirical answer about an optimal treatment for a specific patient. This type of personalized trial has been considered more rigorous than a systematic review of multiple RCTs for making evidence-based treatment decisions, as systematic reviews still require clinicians to extrapolate results to their current patient.8
Personalized trials are part of a family of single-case designs that derive from the experimental analysis of behavior, and they have served as the platform for many current evidence-based treatments in psychology and medicine.9–11 Single-case designs use experimental methods to ensure that changes in outcome variables are caused by the intervention. These methodologies have been adapted into personalized trial designs, which are a specific form of randomized or balanced experiments, characterized by periodic and pre-planned switching from active treatment to placebo or between active treatments (“withdrawal-reversal” designs).12 Whereas conventional RCTs randomize persons, personalized trials randomize time periods within a patient. This type of design addresses concerns with regard to averaging treatment effects across many patients in the presence of known or unknown HTE, external validity-extrapolation issues, the exclusion of those with rare diseases or comorbidities, and the time lag between treatment discovery and implementation.
General Principles of Personalized (N-of-1) Designs
There are several common characteristics of personalized trial designs. Personalized designs trade collecting a few data points from many people for a detailed examination of the relationship between treatment and outcome for a single individual. Because of this, an outcome measure that can be measured frequently and shows rapid change when a treatment is implemented or withdrawn is needed. If a drug is studied, it should be administered under randomized, placebo-controlled conditions with appropriate return to baseline symptomatology or drug washout periods.
Personalized designs work well for both prevention and patients who have chronic, stable, or slowly deteriorating conditions. They can also be used to identify an optimized treatment plan for a patient as they begin a treatment regimen or when a change in an established, but ineffective, treatment is being considered. However, given that these are experimental designs and treatment comparators are delivered serially over randomly varying time periods, patients who require immediate treatment or have urgent medical needs may be inappropriate for this type of experiment.
Uses for a Personalized Trial
One broad category for use of a personalized trial is for patients with common conditions that do not have a universally beneficial or evidence-based treatment. Consider obesity, pain, asthma, irritable bowel syndrome, or a variety of behavioral problems—each of these is amenable to a personalized trial because there is clinical uncertainty as to the best treatment, conflicting evidence, known idiosyncratic responses to common treatments across individual patients, and clinically important endpoints that are easily measurable over a predictable timeframe.
A second broad use of personalized trials is for rare diseases for which (1) there are insufficient numbers for a clinical trial to provide stable findings for evidence-based solutions and (2) effective treatments are not known. As we later note, data from personalized trials can often be combined to provide an evidence base for treatments of children with rare diseases.
Third, personalized trials can be employed when a child has multiple comorbidities and consequently is on a polypharmaceutical regimen with suspected iatrogenic effects. In this case, the family and the clinician may want to investigate the safety of removing suspected medications from the regimen. Children with multiple morbidities are not often recruited for RCTs, and results of evidence-based treatments may not generalize to patients with multiple comorbidities. Finally, concerns about the efficacy of brand-name versus generic medications, or new adaptation of conventional treatments, can be tested with a personalized trial. While the goal of most experiments is to test whether treatments differ in their outcomes, there are instances in which a non-inferiority comparison would be useful, such as when evaluating a generic formulation or lower dose of a medication.
Fourth, personalized trials also can serve as pilot studies to test new interventions that speed up development of innovative treatments to test in RCTs. In this way, there can be beneficial feedback loops between personalized trials designed for discovery and the conduct of parallel group RCTs to ensure a generalizable treatment benefit. Personalized trials are thus recognized as important steps in translating basic science into new clinical interventions.13
Statistical Considerations for Analyzing a Personalized Trial
A basic step in analyzing data from a personalized trial is to examine the results graphically. This first step is useful for presenting results to non-clinicians to help them understand the relationship between treatment and outcome or the relative efficacy of various treatments. For some personalized trials, graphical analysis may provide such obvious results as to be sufficient to make conclusions about optimal treatment. In most instances, however, statistical analyses are needed to compare the treatments. Time series analyses can be used to leverage the frequent outcome assessments in a personalized trial.14 For example, autoregression models evaluate treatment effects while accounting for the correlation between successive outcomes and may be used to estimate the extent of carryover effects.15 Advanced time series methods have also been developed for trials with high-volume data measured via wearable devices16—such as actigraphs, step counters, heart rate monitors, and other wearables—as they become available. These analytic approaches provide valid treatment comparisons and contrast with comparative analyses in RCTs where the focus is on estimating the average treatment effect based on few data points collected from each individual.
Similar to parallel-group RCTs, statistical issues such as power and multiplicity adjustments need to be addressed when planning a personalized trial. The power to detect a treatment effect specific to an individual depends on the correlation across outcomes as well as the magnitude of treatment effect. Generally, a larger number of measurements in a time series is required with higher correlation across outcomes. In trials involving more than two treatments, it is necessary to adjust for multiple comparisons. Using a gate-keeping test can safeguard against false positive findings,17 while traditional methods such as Bonferroni’s adjustment is known to be over conservative and unnecessarily reduce power.
Finally, while a personalized trial focuses on developing optimized treatment for an individual patient, data can be combined across multiple patients to generate meta-analyses of personalized trials—just like conventional meta-analyses.9,11,18 Specifically, results from individual patients can be combined in random effects models and have the advantage of incorporating variability from various sources, including the overall treatment effect and patient-by-treatment interactions.19 In addition, when combining several personalized trials series that compare treatments, it is possible to eliminate time trend when treatments are introduced in a randomized sequence in different trials.
Cases for Personalized Trials
We illustrate the basics of personalized trial designs using three cases of attention deficit-hyperactivity disorder (ADHD). In the first case, the pediatrician is determining if pharmacological treatment is useful for a 12-year-old child newly diagnosed with ADHD based on daily parent ratings of the IOWA Conners Rating Scale (ICRS).20 To ensure that any improvements in ADHD symptoms are due to the pharmaceutical treatment, the pediatrician includes a baseline that precedes the experimental periods (drug or placebo), ideally randomizing experimental periods and masking parent and child to the type of pharmaceutical intervention (drug or placebo)—thus creating a multi-period crossover design. The baseline is critical because it serves as a control for the trial. If a drug or treatment that has a long half-life is being tested, unlike those typically used to manage ADHD, then additional washout periods may be needed to ensure a return to baseline symptom level in the interval between the periods. A graphical presentation of daily ICRS subscales in such an ABCCB trial are shown in Figure 1. With relatively high inattentive scores during baseline and placebo conditions, it is clear that the scores are consistently improved (i.e., lower) during the drug period. This is an example where visual inspection may be sufficient to determine that pharmaceutical treatment has benefits.
Figure 1.
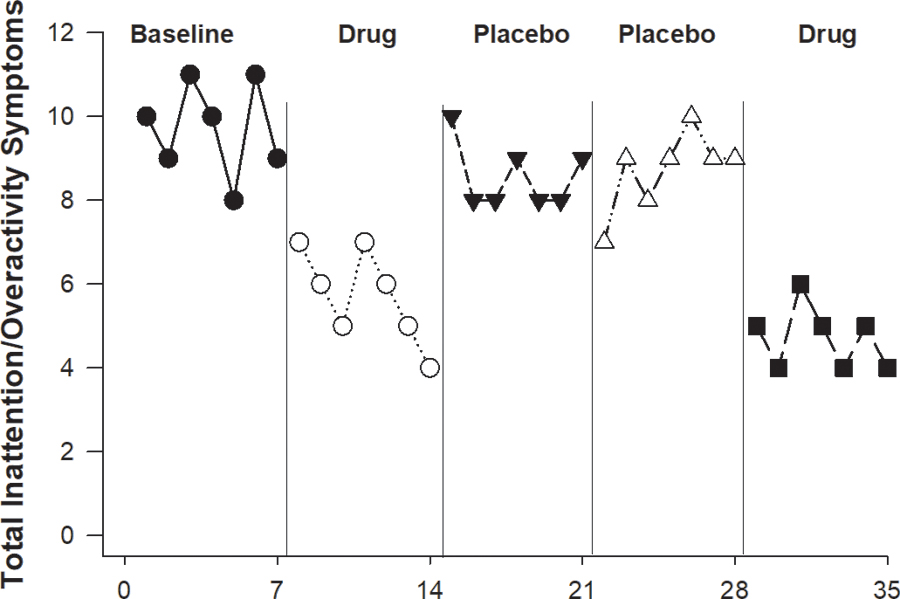
Inattention symptoms as assessed by Parental IOWA Conners Rating Scale20 scores during baseline (A) and repetitions of randomized drug (B) and placebo (C) time periods for an ABCCB design of a child to determine drug efficacy.
In the second case, a 9-year-old girl is recently diagnosed with ADHD following a history of school problems. However, this family has concerns about long-term use of pharmaceutical treatment and wants to determine if a behavioral intervention might be effective. To ensure that the behavioral intervention works as well or better than medication, the pediatrician chooses to use a similar design as before, and so includes a baseline and four experimental periods (i.e., drug, drug placebo, contingent reinforcement intervention, non-contingent reinforcement) and, ideally, randomizes said periods. In this case, masking or blinding to condition is not possible for the behavioral intervention, but it is possible for the drug-versus-placebo periods. As treatments should ideally be reversible, the pediatrician chooses a contingent reinforcement behavioral intervention and a non-contingent control. During the contingent reinforcement condition, when the child achieves a score of 6 or lower on the ICRS in a four-hour block, they are provided with a skipping rope and 20 minutes of physical activity reinforcement. In the non-contingent reinforcement condition, the child was provided with the physical activity reinforcer, independent of their behavior, on a randomly chosen schedule. In other periods of the trial, the activity reinforcer was not available. A sticker reward system could have been used as an alternative to physical activity reinforcers. Figure 2 depicts this trial wherein both drug and behavioral intervention clearly improve the over placebo. Fitting an autoregression model demonstrates significant effects of both drug and contingent reinforcement when compared to placebo with respective reductions in ICRS score of 2.8 and 3.2 (P < 0.001). Furthermore, a 95% confidence interval for comparing the scores during behavioral intervention and drug periods is −1.4 to 0.5, suggesting equivalence of the two modalities and providing empirical evidence for the family’s preference for contingency management for their child.
Figure 2.

Inattention symptoms as assessed by Parental IOWA Conners Rating Scale20 scores during randomized baseline (A), drug (B), drug placebo (C), contingent (D), and non-contingent (E) reinforcement (ABCDE) conditions.
Additionally, a few practical considerations should be noted in this case. First, since a behavioral program involves parent training for administration of a positively reinforcing contingency, plans for that education must be included in the overall treatment plan. Second, unlike testing drugs, it is impossible to test a behavioral technique in a double-blind design, and it will be clear to the provider and patient that a behavioral intervention in being implemented. Third, we have illustrated a relatively stable baseline, but in reality, symptoms may wax or wane prior to intervention, thereby requiring extending or reducing the duration of the baseline.
The third case is a 7.5-year-old patient who weighs 34 pounds and is diagnosed with nonorganic failure to thrive and ADHD with both inattention and oppositional defiant components. He is on 60 mg/day of methylphenidate, the FDA-approved maximum dosage. His pediatrician, recognizing methylphenidate may have the off-target effect of appetite suppression, wants to increase the methylphenidate dose as an off-label use. In the absence of a best course of treatment for this patient and a lack of RCT evidence due to comorbidities, a double-blind, randomized personalized trial dose finding trial is conducted. A personalized trial offers the flexibility to compare different modalities. Figure 3 shows the inattention scores and weights of the patient in a hypothetical trial of usual drug, a higher dose alternative, a placebo, and a behavioral contingency intervention. Since this child was already taking 60 mg/day of methylphenidate, that would be the baseline condition. Visual inspection demonstrates behavioral intervention as the optimal treatment for both inattention improvement and weight gain—after the drug was tested to be significantly worse than placebo baseline in terms of the inattention scores (1.5 and 1.2 respectively higher for 20 mg and 30 mg, with P<0.001) and had no impact on weight. Such a change in treatment for this child might not have been possible without the personalized trial, as he might have remained on a drug that had adverse impact on his weight while having little to no benefit for his attention symptoms. One of the strengths of personalized trials is that they can empirically inform the discontinuation of a current treatment that has no evidence for its effectiveness.21
Figure 3.
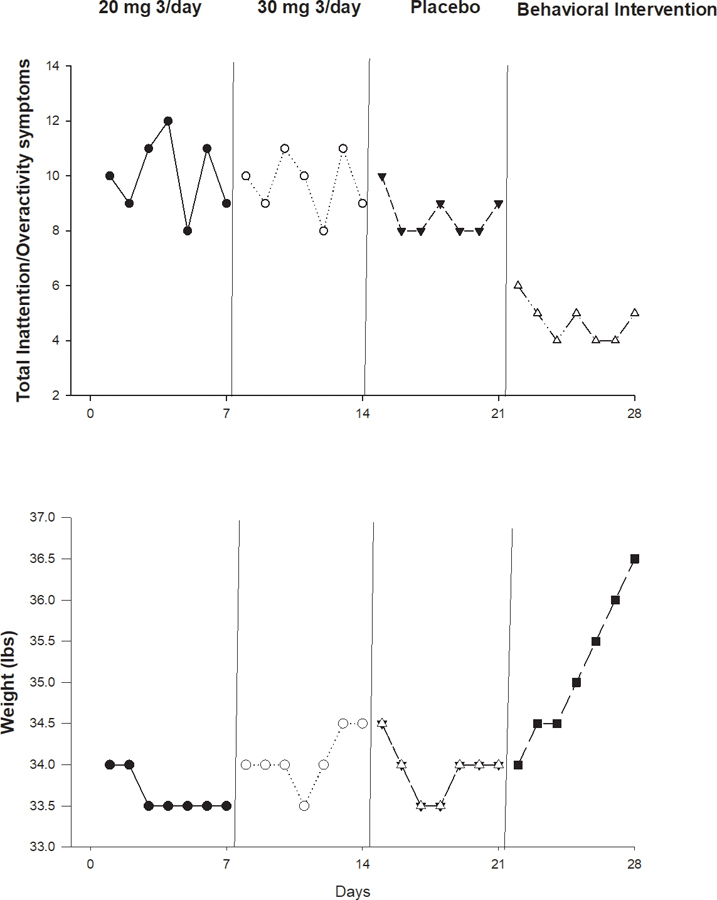
Inattention symptoms as assessed by teacher IOWA Conners Rating Scale20 scores and body weight during usual drug dose (A), randomized higher drug dose (B), placebo (C) and behavioral contingency (D) conditions (ABCD).
Implementation Considerations for Personalized Trials
Personalized trials require close coordination between a research-oriented pharmacy/pharmacist who can implement a randomized sequence of drug, drug doses, and placebo phases. Personalized trials also benefit from statistical analysis to determine whether differences in patient symptoms are significantly different. Having a biostatistician is useful, but many have not been trained to analyze individual subject data. Statistical programs or online statistical consultation that only require entry of an individual patient’s data, often without the needs for disclosing protected health information, can be developed to make analysis as easy as possible. Personalized trials also require collection of a lot of patient data, requiring staff who can collect data, check data quality control, and organize/present data. This challenge is not unique to personalized trials, but a greater use of transducers and reliable patient reporting methods to collect blood glucose, blood pressure, activity, sleep, and body weight as examples have been adapted for clinical work with the same requirements on staff.
In this type of trial, there are complex issues of patient consent for engagement that may be different depending on the motivation underlying the initiation of the trial. Treatment discovery is clearly considered research while optimal treatment identification may be considered best clinical practice. Questions of payment for engaging in a personalized trial may be appropriate in some use cases and not in others. Informed consent always is needed as is extended measurement.
Conclusion
Personalized or single-subject designs have been used for decades to identify effective interventions across disciplines, but they have not yet found a home in pediatrics. This approach is useful for rare diseases, for specialists who are working on developing new and innovative treatments for their patients, or for pediatricians in primary-care settings who want to ensure they are providing the best patient care given the current state of knowledge. It may also be useful for testing treatments in patients with comorbidities or suspected side-effect or idiosyncratic treatment responses. Using these designs in clinical practice can empower clinicians to test established and new evidence-based treatments for patients in their practices, thus accelerating the time from treatment-benefit discovery to clinical implementation.
Acknowledgment:
This work was supported by grants R01LM012836 from the National Library of Medicine of the National Institutes of Health; RO1HD080292 and RO1HD088131 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development; U01 HL131552 from the National Heart, Lung, and Blood Institute; and UH3 DK109543 from the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Conflicts of Interest: The views expressed in this paper are those of the authors and do not represent the views of the National Institutes of Health, the United States Department of Health and Human Services, or any other government entity. Drs. Davidson and Silverstein are members of the United States Preventive Services Task Force (USPSTF). This article does not represent the views and policies of the USPSTF.
References
- 1.Guyatt G, Haynes B, Jaeschke R, et al. Introduction: The philosophy of evidence-based medicine. In: Guyatt G, Rennie D, eds. Users’ Guides to the Medical Literature : A Manual for Evidence-based Clinical Practice Chicago, IL: American Medical Association; 2002:3–11. [Google Scholar]
- 2.Vijan S Evaluating heterogeneity of treatment effects. Biostat Epidemiol 2020;4(1):98–104. [Google Scholar]
- 3.Ioannidis JP, Lau J. The impact of high-risk patients on the results of clinical trials. J Clin Epidemiol 1997;50(10):1089–1098. [DOI] [PubMed] [Google Scholar]
- 4.Kent DM, Rothwell PM, Ioannidis JP, Altman DG, Hayward RA. Assessing and reporting heterogeneity in treatment effects in clinical trials: a proposal. Trials 2010;11:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lauer MS, D’Agostino RB Sr. The randomized registry trial--the next disruptive technology in clinical research? N Engl J Med 2013;369(17):1579–1581. [DOI] [PubMed] [Google Scholar]
- 6.Guyatt G, Sackett D, Taylor DW, Chong J, Roberts R, Pugsley S. Determining optimal therapy--randomized trials in individual patients. N Engl J Med 1986;314(14):889–892. [DOI] [PubMed] [Google Scholar]
- 7.McLeod RS, Taylor DW, Cohen Z, Cullen JB. Single-patient randomised clinical trial. Use in determining optimum treatment for patient with inflammation of Kock continent ileostomy reservoir. Lancet 1986;1(8483):726–728. [DOI] [PubMed] [Google Scholar]
- 8.Davidson KW, McGinn T, Wang YC, Cheung YK. Expanding the Role of N-of-1 Trials in the Precision Medicine Era: Action Priorities and Practical Considerations. NAM Perspectives 2018. 10.31478/201812d. [DOI] [Google Scholar]
- 9.Shaffer JA, Kronish IM, Falzon L, Cheung YK, Davidson KW. N-of-1 Randomized Intervention Trials in Health Psychology: A Systematic Review and Methodology Critique. Ann Behav Med 2018;52(9):731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kronish I, Falzon L, Hampsey M, Konrad B, Davidson K. Are N-of-1 trials useful for achieving therapeutic precision in the treatment of depression? A systematic review of N-of-1 trials for depressive symptoms. Paper presented at: American Psychosomatic Society: March 15–17, 2017; Sevilla, Spain. [Google Scholar]
- 11.Gabler NB, Duan N, Vohra S, Kravitz RL. N-of-1 trials in the medical literature: a systematic review. Med Care 2011;49(8):761–768. [DOI] [PubMed] [Google Scholar]
- 12.Kravitz RLDN, eds, and the DEcIDE Methods Center N-of-1 Guidance Panel (Duan N, Eslick I, Gabler NB, Kaplan HC, Kravitz RL, Larson EB, Pace WD, Schmid CH, Sim I, Vohra S),. Design and Implementation of N-of-1 Trials: A User’s Guide Rockville, MD; 2014. [Google Scholar]
- 13.Kravitz RL, Duan N, Niedzinski EJ, Hay MC, Subramanian SK, Weisner TS. What ever happened to N-of-1 trials? Insiders’ perspectives and a look to the future. Milbank Q 2008;86(4):533–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Box GEP, Jenkins GM, Reinsel GC. Time Series Analysis: Forecasting and Control 4th ed. Hoboken, NJ: John Wiley & Sons, Inc.; 2008. [Google Scholar]
- 15.Kronish IM, Cheung YK, Julian J, et al. Clinical Usefulness of Bright White Light Therapy for Depressive Symptoms in Cancer Survivors: Results from a Series of Personalized (N-of-1) Trials. Healthcare 2019;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung YK, Hsueh PS, Ensari I, Willey JZ, Diaz KM. Quantile Coarsening Analysis of High-Volume Wearable Activity Data in a Longitudinal Observational Study. Sensors (Basel) 2018;18(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong X, Cheng B, Qian M, Cheung YK. A gate-keeping test for selecting adaptive interventions under general designs of sequential multiple assignment randomized trials. Contemp Clin Trials 2019;85:105830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kronish IM, Hampsey M, Falzon L, Konrad B, Davidson KW. Personalized (N-of-1) Trials for Depression: A Systematic Review. J Clin Psychopharmacol 2018;38(3):218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jull A, Bennett D. Do n-of-1 trials really tailor treatment? Lancet 2005;365(9476):1992–1994. [DOI] [PubMed] [Google Scholar]
- 20.Pelham WE Jr., Milich R, Murphy D, Murphy HA. Normative data on the IOWA Conners Teacher Rating Scale. J Clin Child Psychol 1989;18(3):259–262. [Google Scholar]
- 21.Kronish IM, Cheung YK, Shimbo D, et al. Increasing the Precision of Hypertension Treatment Through Personalized Trials: a Pilot Study. J Gen Intern Med 2019;34(6):839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]