

Abstract
In accordance with the comorbidity-inflammation paradigm, comorbidities and especially metabolic comorbidities are presumed to drive development and severity of heart failure with preserved ejection fraction (HFpEF) through a cascade of events ranging from systemic inflammation to myocardial fibrosis. Recently, novel experimental and clinical evidence emerged, that strengthens the validity of the inflammatory/profibrotic paradigm. This evidence consists among others of:
Myocardial infiltration by immunocompetent cells not only because of an obesity-induced metabolic load but also because of an arterial hypertension-induced hemodynamic load. The latter is sensed by components of the extracellular matrix like basal laminin, which also interact with cardiomyocyte titin;
Expression in cardiomyocytes of inducible nitric oxide synthase because of circulating proinflammatory cytokines. This results in myocardial accumulation of degraded proteins because of a failing unfolded protein response;
Definition by machine learning algorithms of phenogroups of HFpEF patients with a distinct inflammatory/profibrotic signature;
Direct coupling in mediation analysis between comorbidities, inflammatory biomarkers and deranged myocardial structure/function with endothelial expression of adhesion molecules already apparent in early preclinical HFpEF (HF stage A, B).
This new evidence paves the road for future HFpEF treatments such as biologicals directed against inflammatory cytokines, stimulation of protein ubiquitylation with phosphodiesterase 1 inhibitors, correction of titin stiffness through natriuretic peptide – particulate guanylyl cyclase – PDE9 signaling and molecular/cellular regulatory mechanisms that control myocardial fibrosis.
Keywords: Heart Failure, Ejection Fraction, Fibrosis, Inflammation
Subject Codes: Heart Failure, Cardiomyopathy, Pathophysiology, Fibrosis, Inflammation
Introduction
Heart failure with a preserved ejection fraction (HFpEF) is a growing public health problem with substantial morbidity and mortality which currently affects 9% of people older than 60 years. This implies that more than 6 million patients suffer from HFpEF in the US plus European Union combined1. The prevalence of HFpEF relative to heart failure with a reduced ejection fraction (HFrEF) has risen steadily as evident from the Framingham study which observed a reversal of the HFpEF / HFrEF ratio from 41/59 in the decade 1985–1994 to 56/44 in the decade 2005–20142. This evolution parallels concomitant trends in risk factors with a rise in the metabolic syndrome and a fall in obstructive coronary artery disease over the same decades. HFpEF is associated with a 5 year survival of 35% and the quality of life of HFpEF patients is as poor as in HFrEF patients.
Over the last decennium our understanding of HFpEF has evolved from mere left ventricular (LV) diastolic dysfunction to a multiorgan syndrome resulting from comorbidities like obesity, diabetes and arterial hypertension, which predispose to systemic inflammation affecting the myocardium, skeletal muscles, pulmonary vasculature and kidneys3. Although in distinct HFpEF subgroups, skeletal muscle fatigue and pulmonary hypertension impact clinical presentation, the vast majority of HFpEF patients suffer from invalidating dyspnea because of a brisk rise in LV filling pressures during exercise4. The exercise induced elevation in LV filling pressures mainly results from high diastolic LV myocardial stiffness with additional contributions in some patients of left atrial myopathy and of pericardial constriction because of epicardial fat accumulation. The present review will therefore first focus on pathophysiological mechanisms linking systemic inflammation to myocardial stiffness. It will subsequently address inflammatory/fibrotic signaling in distinct HFpEF phenotypes derived from machine-learning algorithms and finally apply concepts of inflammatory/fibrotic signaling to HFpEF therapy.
From Inflammation to LV Stiffness: Pathophysiological Mechanisms
Pathophysiological mechanisms linking systemic inflammation to myocardial stiffness are shown in Figure 1 and consist of: 1) Metabolic load induced proinflammatory signaling; 2) Hemodynamic load induced proinflammatory and fibrotic signaling; 3) Titin modifications and cardiomyocyte stiffness; 4) Myocardial collagen homeostasis; 5) Crosstalk between hemodynamic load, extracellular matrix and cardiomyocyte titin and 6) Myocardial accumulation of degraded proteins.
Figure 1. Pathophysiological mechanisms linking systemic inflammation to myocardial stiffness.
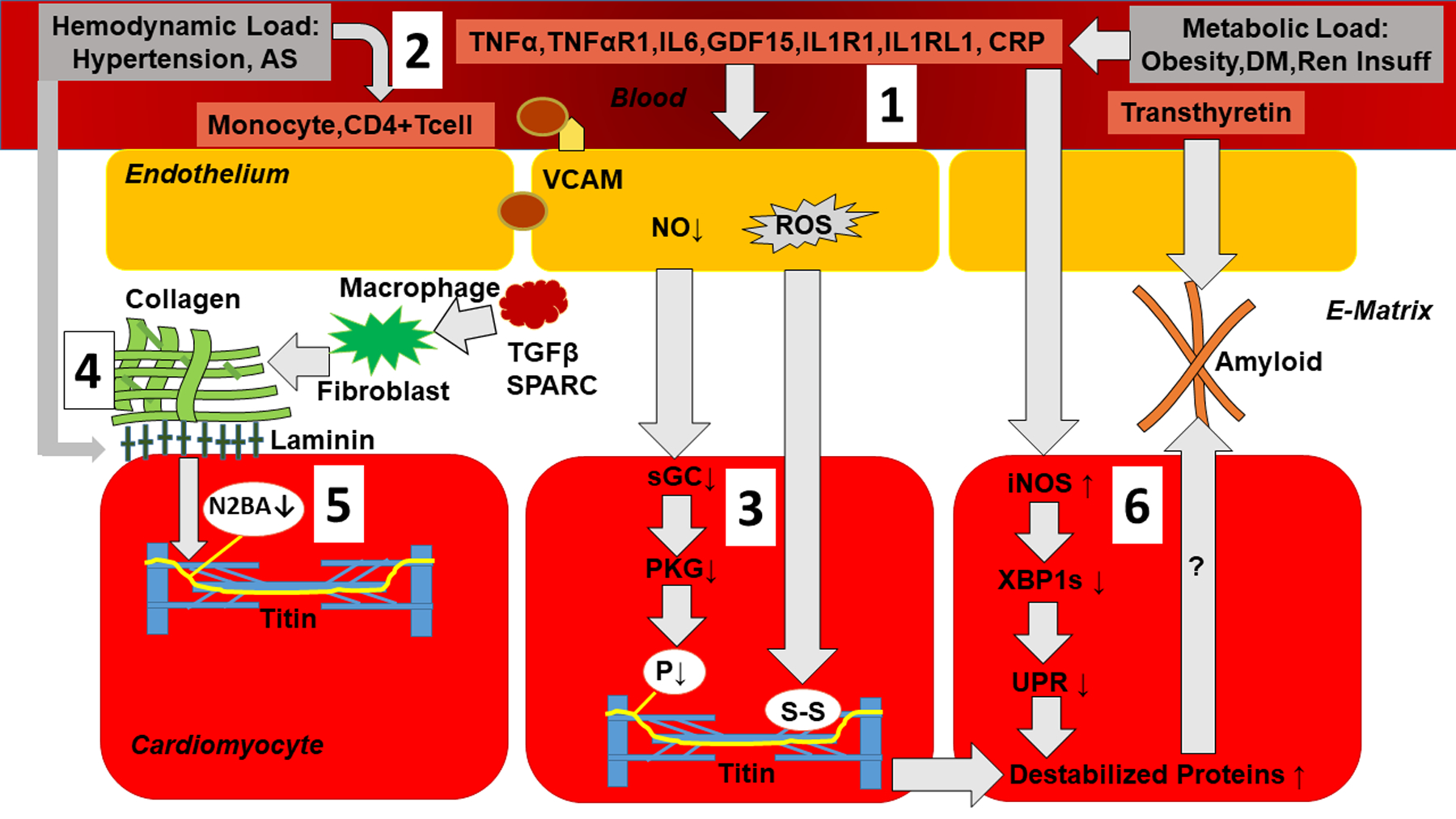
1) Metabolic load induces proinflammatory signaling: Metabolic load related to obesity, diabetes mellitus (DM) and renal insufficiency (Ren Insuff) triggers systemic inflammation evident from raised plasma levels of TNFα (Tumor Necrosis Factor α), TNFαR1 (Tumor Necrosis Factor α Receptor 1), IL6 (Interleukin 6), GDF15 (Growth Differentiation Factor 15), IL1R1 (Interleukin 1 Receptor 1), IL1RL1 (Interleukin 1 Receptor Like 1) and CRP (C-Reactive Protein). Systemic inflammation triggers endothelial expression of adhesion molecules (VCAM: Vascular Cell Adhesion Molecule), which attracts monocytes, lowers endothelial production of nitric oxide (NO) and raises endothelial production of Reactive Oxygen Species (ROS); 2) Hemodynamic load as occurs in arterial hypertension and aortic stenosis induces proinflammatory and fibrotic signaling evident from myocardial infiltration of monocytes and CD4+ T cells; 3) Low NO reduces activity of soluble Guanylyl Cyclase (sGC) and Protein Kinase G (PKG). This leads to hypophosphorylation of titin (P↓). ROS cause formation of disulfide bonds within titin. Both these titin modifications raise cardiomyocyte stiffness; 4) Myocardial collagen homeostasis: Infiltrating monocytes become macrophages with production of Transforming Growth Factor β (TGFβ) and secreted protein acidic and rich in cysteine (SPARC) which stimulate collagen production by fibroblasts; 5) Crosstalk between hemodynamic load, extracellular matrix basal laminin and cardiomyocyte titin results in changed titin isoform expression with less N2BA isoform (N2BA↓); 6) Myocardial accumulation of degraded proteins: Expression in cardiomyocytes of inducible Nitric Oxide Synthase (iNOS) lowers inositol-requiring enzyme1α (IRE1α), spliced X-box binding protein1 (XBP1s) and the Unfolded Protein Response (UPR). The latter leads to build-up of destabilized proteins which could potentially also accumulate in the extracellular matrix as occurs in transthyretin amyloidosis. (Illustration credit: Ben Smith)
Metabolic Load and Proinflammatory Signaling
Over the years, studies assessing biomarkers have analyzed an ever increasing number of circulating biomarkers in patients with HFpEF and HFrEF5–8 (initially just four5 and recently up to 2488). Initial evidence showed that elevated growth differentiation factor-15 (GDF-15) was associated with the presence and severity of HFpEF5. This finding was soon to be followed by similar observations that confirmed not only high levels of GDF-15 but also of soluble interleukin 1 receptor-like 1 (IL1RL1), C-reactive protein (CRP) and interleukin 6 (IL6) to be associated with HFpEF (Figure 2)6. The BIOSTAT-CHF program measured plasma levels of 92 biomarkers, analyzed protein-protein interactions, and overrepresentation of biological processes7. Integrin Subunit Beta 2, which is involved in adhesion of immune cells, was most strongly associated with HFpEF (Figure 2) whereas cAMP-dependent transcription factor ATF2, which is involved in cell growth, was most strongly associated with HFrEF. The distinct nature of HFpEF and HFrEF was evident from the analysis of protein-protein interactions, which revealed 6 interactions specific for HFpEF and 8 for HFrEF. Finally, pathways relating to inflammation and extracellular matrix organization were overrepresented in HFpEF in contrast to HFrEF, in which pathways relating to cellular proliferation and metabolism were overexpressed.
Figure 2. Mediations, associations and predictions involving inflammation markers in HFpEF.
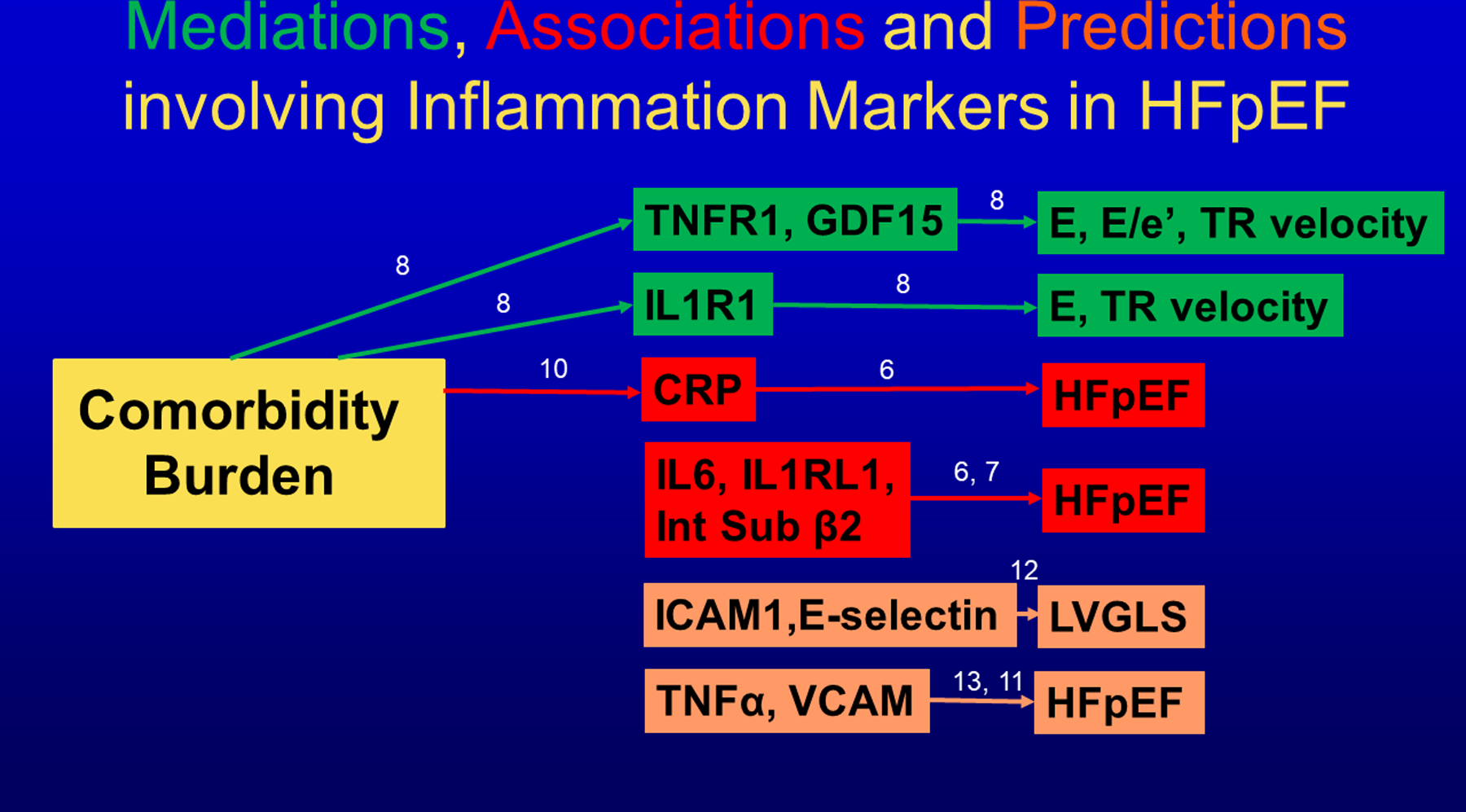
Mediations (Green) between comorbidity burden, inflammatory biomarkers and echocardiographic cardiac function (TNFR1: Tumor Necrosis Factor Receptor 1; GDF15: Growth Differentiation Factor 15; IL1R1: Interleukin 1 Receptor 1; E: Early diastolic mitral flow velocity; E/e’: ratio of Early diastolic mitral flow velocity over Early diastolic long axis lengthening velocity; TR: Tricuspid Regurgitation velocity). Associations (Red) between inflammatory biomarkers and presence of HFpEF (CRP: C-Reactive Protein; IL6: Interleukin 6; IL1RL1: Interleukin 1 Receptor Like 1; Int Sub β 2: Integrin Subunit Beta 2). Biomarkers predicting (Orange) myocardial function or HFpEF (ICAM1: Intercellular Adhesion Molecule 1; TNFα: Tumor Necrosis Factor α; VCAM: Vascular Cell Adhesion Molecule; LVGLS: Left Ventricular Global Longitudinal Strain). Numbers indicate corresponding reference.
In accordance to the comorbidity-inflammation paradigm, comorbidities and especially metabolic comorbidities were presumed to drive systemic inflammation in HFpEF3,9. This presumption was supported by a study that observed a progressive increase in plasma level of CRP with rising number of comorbidities in individual HFpEF patients (Figure 2)10. Among the comorbidities accounted for in this study were obesity (BMI>30 kg/m2), diabetes mellitus, anemia, and chronic kidney disease. Consecutive steps of the comorbidity-inflammation paradigm were recently evaluated in the PROMIS-HFpEF study8, which performed a mediation analysis based on associations of comorbidities with cardiac structure/function, of comorbidities with inflammation and of inflammation with cardiac structure/function (Figure 2). The strength of a mediation analysis is that it allows for causal inference within an observational study. Individual inflammatory biomarkers shown to mediate LV dysfunction were TNF receptor 1 (TNFR1), Interleukin1 receptor 1 (IL1R1) and GDF-15.
The importance of systemic inflammation for HFpEF is also evident from longitudinal observations that predicted HFpEF incidence in community based population samples (Figure 2). The earliest observation came from the Health ABC study, which observed an increased hazard ratio for incident HFpEF over a 9.4 year time span per doubling of baseline plasma level of tumor necrosis factor α (TNFα)11. In this study baseline TNFα predicted development of HFpEF (p<0.001) but not of HFrEF (p=0.08). In a similar experimental design, the CARDIA study demonstrated that high plasma levels of endothelial adhesion molecules E-Selectin and intercellular adhesion molecule 1 (ICAM-1) in early adulthood predicted that 15 to 23 years after the biomarker was assessed, patients would develop a depressed LV global longitudinal strain (GLS), a marker of incipient HFpEF-related LV dysfunction12. The MESA study looked at another endothelial adhesion molecule, vascular cell adhesion molecule (VCAM) and observed the highest incidence of HFpEF after 14.4 years in the highest quartiles of baseline VCAM13. Because of the early age at which baseline plasma samples were obtained, both studies suggested endothelial adhesion molecules to be relevant for the diagnosis of stage A or stage B HFpEF (i.e. asymptomatic with risk factors (=stage A) or with LV remodeling (=stage B)) in contrast to biomarkers of myocardial stress (natriuretic peptides) or damage (troponin-I), which only become abnormal in stage C HFpEF14. A similar predictive value has also been attributed to urinary albumin excretion (UAE), a renal consequence of endothelial activation and dysfunction. In a community-based, middle-aged cohort raised UAE was shown to predict incident clinical HFpEF after 11 years of follow-up15. These findings were confirmed in two studies, which respectively associated urinary albumin to creatinine ratio (UACR) with incident HFpEF and an increasing trajectory of high UACR over time with unfavorable hypertrophic LV remodeling16,17.
Hemodynamic Load, Proinflammatory and Profibrotic Signaling
In addition to metabolic load-induced activation of inflammatory signaling, the increased hemodynamic load resulting from arterial hypertension and aortic valve stenosis also activates proinflammatory and profibrotic signaling. Activation of these hemodynamic load-induced signaling cascades are evidenced by the alterations in myocardial and circulating biomarkers, including proteins, peptides and microRNAs. Within the myocardium, endothelial cells, fibroblasts, endogenous macrophages as well as cardiomyocytes are hemodynamic load sensitive and each may participate in this load-induced inflammatory and fibrosis signaling. Data from clinically relevant animal models of hemodynamic overload have shown that increased load alters the balance of histone acetylation (HDAC) induced control of microRNAs (miR) which in turn control myocardial specific proinflammatory and profibrotic signaling18. Murine transverse aortic constriction induced pressure-overload resulted in increased HDAC 1 and 2 with decreased miRs. MicroRNAs remain stable when circulating in plasma and can be assessed as biomarker evidence of epigenetic control of inflammatory and fibrotic signaling. Accordingly, decreased miR 1, 21, 29 and 133 have been identified in HFpEF patients. These hemodynamic load-induced changes are also believed to alter the cellular phenotype of resident cells such as fibroblasts and lead to fibroblast activation. In addition, increased hemodynamic load leads to proinflammatory and profibrotic specific cell recruitment into the myocardium such as activated macrophages and T-cells19–23. These cellular and molecular changes are summarized schematically in Figure 3.
Figure 3. Hemodynamic load-induced myocardial inflammatory/fibrotic signaling.
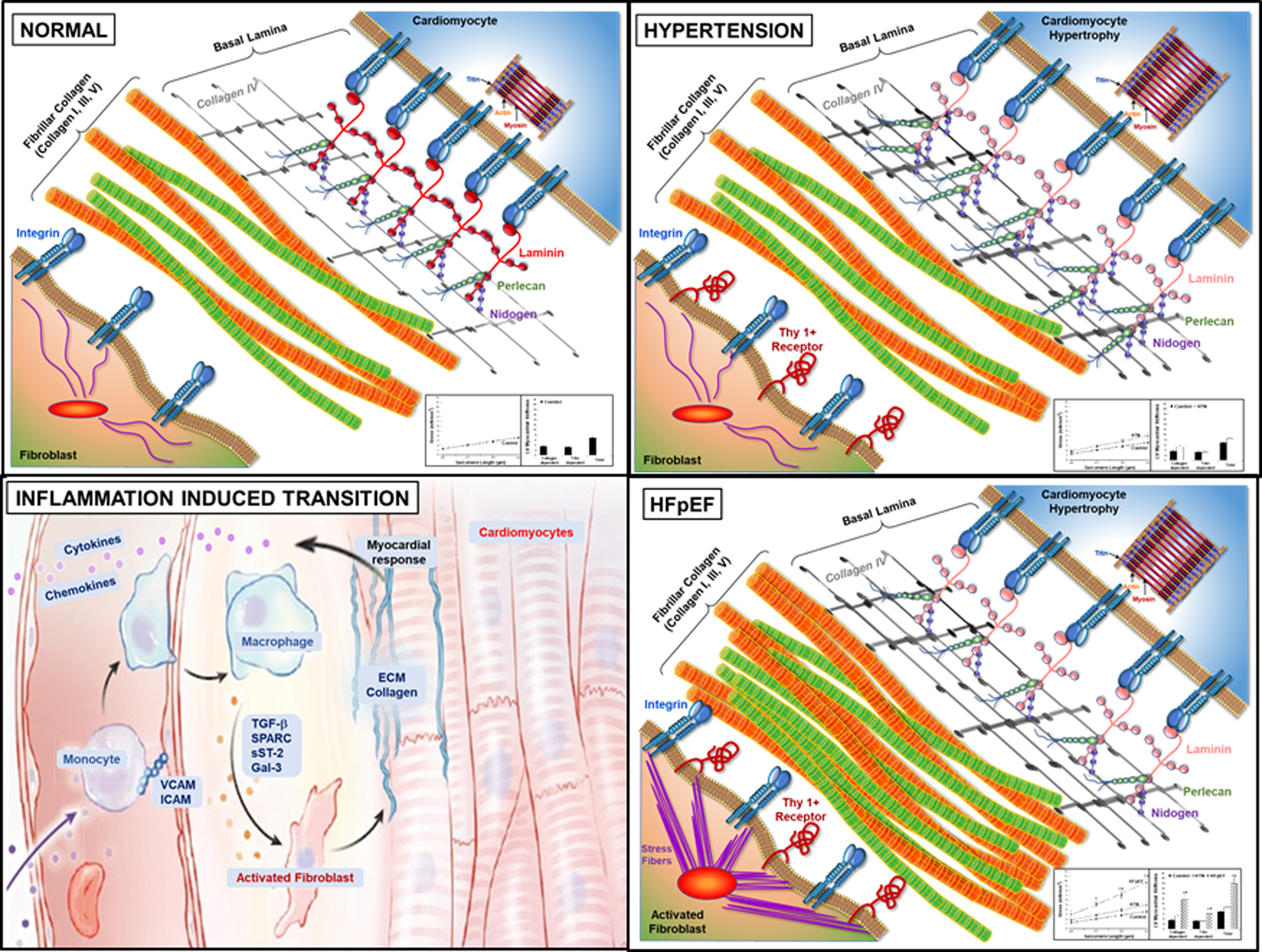
Sequential changes in myocardial cardiomyocytes, fibroblasts, fibrillar collagen, and basal lamina structures result from the imposition of increased hemodynamic load that lead to the development of HFpEF. (Illustration credit: Ben Smith).
Compared to Panel A, showing normal myocardium, Panel B depicts changes that result from increased hemodynamic load, such as that which occurs in systemic arterial hypertension. The change in load is sensed by cardiomyocytes, fibroblasts and resident macrophages and leads to alterations in the basal lamina structures. Basal lamina changes include changes in laminin isoform to a more compliant form, with increases in perlican, nitogen and collagen IV which may compliment and compensate for these changes in laminin. Cardiomyocytes undergo parallel addition of sarcomeres and increased cross-sectional area; these cellular changes lead to concentric LV hypertrophy. However, these changes in myocardial structure does not result in increased myocardial diastolic stiffness (inset). Panel C depicts the inflammation induced transition from hypertensive heart disease to HFpEF. This transition is led by proinflammatory signaling of increased cytokines and chemokines causing cell recruitment to the myocardium of macrophages and T/B cells. These cytokines and chemokines are secreted by the myocardium and enter the circulation. Circulating monocytes, both from the bone marrow and the spleen, migrate to myocardial endothelial cell surfaces, with attachment and extravasation into the interstitial space facilitated by vascular cell adhesion proteins and become activated macrophages. These macrophages both secrete matricellular proteins that facilitate procollagen processing and collagen fiber assembly and may further lead to fibroblasts activation. Panel D shows the results of this transition in HFpEF and the aggregate profibrotic changes in interstitial ECM and their resultant increase in myocardial diastolic stiffness (inset).
Some of the mechanisms displayed in this schematic are supported by clinical and basic studies, others await further study; however, it was our intent in this review to propose a comprehensive overview that not only summarizes past evidence but also guides future investigation. Increased hemodynamic load (Figure 3 Panel B), is sensed by cardiomyocytes, fibroblasts and resident macrophages and leads to alterations in the basal lamina (also termed basement membrane) structures that in part facilitate cardiomyocyte remodeling. Basal lamina changes include changes in laminin isoform to a more compliant form, with increases in perlican, nitogen and collagen IV which may compliment structural remodeling. In addition, cardiomyocytes undergo parallel addition of sarcomeres with increased cross-sectional area; these cellular changes lead to concentric LV hypertrophy.
Increased hemodynamic load also leads to proinflammatory signaling (Panel C) with cytokines or chemokines and inflammatory cell recruitment such as macrophages and T/B cells19–22. In relevant animal models and in patients with pressure-overload, myocardially produced cytokines and chemokines enter the circulation. Circulating monocytes, both from the bone marrow and the spleen, migrate to myocardial endothelial cell surfaces, with attachment and extravasation into the interstitial space facilitated by vascular cell adhesion proteins and become activated macrophages. These macrophages both secrete matricellular proteins that facilitate procollagen processing and collagen fiber assembly and may further activate fibroblasts19–22. These changes in aggregate alter the interstitial ECM in a profibrotic manner with concomitant increase in myocardial stiffness (Panel D)24.
Titin Modifications and Cardiomyocyte Stiffness
High LV myocardial diastolic stiffness as characteristically observed in HFpEF, results from both myocardial fibrosis and low cardiomyocyte and myocardial distensibility. These findings have been directly measured in cardiomyocytes retrieved from LV endomyocardial biopsies and LV myocardial muscle samples obtained from LV epicardial biopsies24,25,26. Myocardial fibrosis has been detected from the frequent presence of an elevated collagen content in the LV or septal myocardium of HFpEF patients27,28. The giant sarcomeric protein titin is responsible for the diastolic distensibility of cardiomyocytes and also plays a role in mechanical stress sensing. Titin spans Z disc, I band, A band and M band sarcomeric regions. Its elasticity resides in the I-band region, which consists of a series of segments (proximal IgG, N2B, PEVK, distal IgG), each of which can contribute to overall elasticity29,30. Deletion of the N2B segment increases titin stiffness and induces LV atrophy31 whereas deletion of the PEVK segment also increases titin stiffness but induces LV hypertrophy32. Titin modulates diastolic distensibility of cardiomyocytes through transcriptional and posttranslational modifications. Differential splicing gives rise to two titin isoforms of different size and stiffness: a short, less compliant N2B isoform and a long, more compliant N2BA isoform. RNA binding motif-20 (RBM20) is a major splicing factor of titin and its inhibition upregulates expression of long compliant isoforms capable of correcting diastolic LV compliance in HFpEF mouse models33,34. Posttranslational modifications of titin mainly consist of phosphorylation and oxidation. Phosphorylation of the N2B segment by PKA or PKG decreases and phosphorylation of the PEVK segment increases distensibility of titin35–37. Finally, reactive oxygen species modulate titin elasticity through formation of disulfide bonds, which interfere with protein folding and lead to titin aggregation in the distal IgG segment38.
All of the aforementioned mechanisms that modulate titin distensibility are involved in the high myocardual stiffness in HFpEF: 1) the N2BA/N2B titin isoform ratio is lower in HFpEF than in HFrEF25,39; 2) the N2B titin isoform is hypophosphorylated in clinical HFpEF39 and in the ZSF1 or Seipin-KO HFpEF rat models titin is respectively hypophosphorylated in the N2B segment or hyperphosphorylated in the PEVK segment40,41; 3) oxidative changes of titin account for the ageing-induced rise in cardiomyocyte stiffness and are paralleled by a protective build-up in the sarcomeres of anti-oxidant HSP27 or αB-crystallin42,43.
Two novel aspects of titin pathophysiology are especially relevant to HFpEF namely lack of stretch-induced titin phosporylation, which turns the left ventricle into a fixed filling pressure pump44 and modifications of titin as a result of diabetes mellitus45. In response to an acute volume load, the myocardium slowly adjusts its diastolic compliance over a 15 minute period. This gradually lowers LV filling pressures to values well below the initial values after imposition of the volume load. This myocardial property has previously been labelled stress relaxation, myocardial viscosity or creep. The adjustment of diastolic LV compliance results from PKG-induced phosphorylation of titin, disappears following inhibition of NO synthase or PKG and is absent in hypertrophied myocardium44 (Figure 4).
Figure 4. Stretch-induced titin phosporylation.
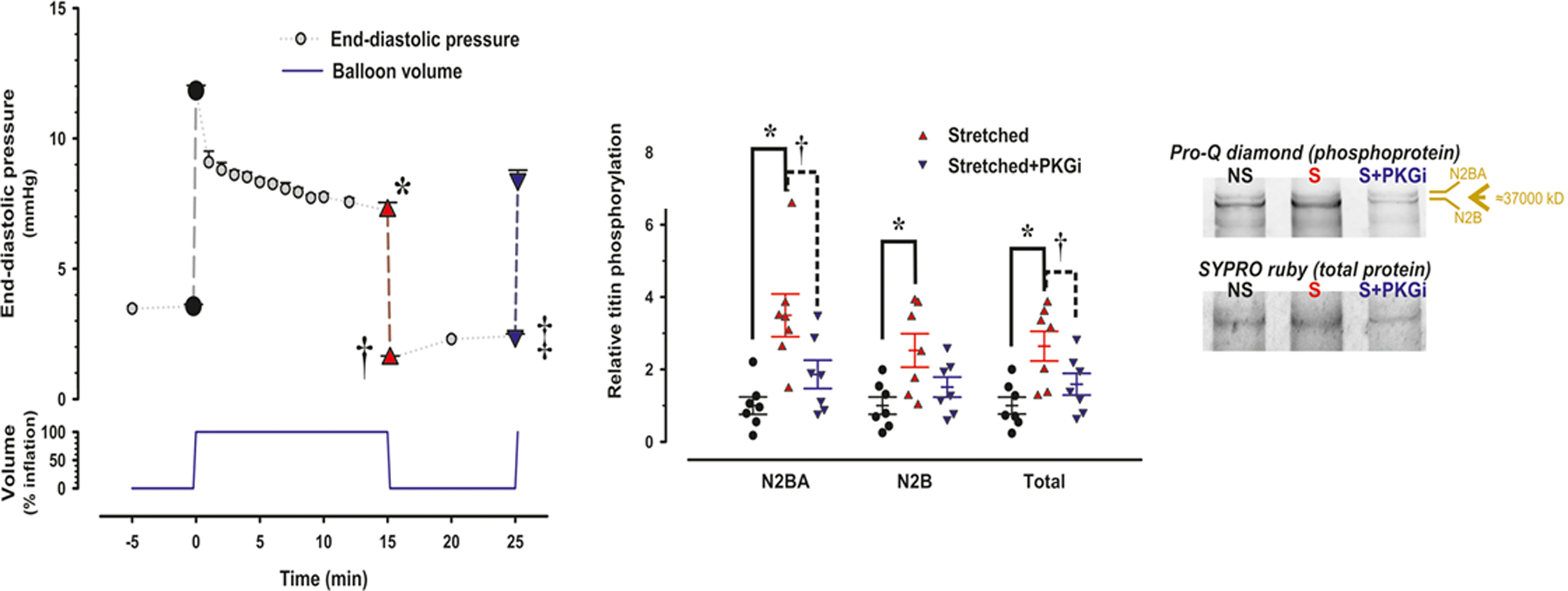
In a Langendorff preparation an acute volume load causes a brisk rise in LV end-diastolic pressure (LVEDP), which gradually falls over a 15 minute period. Subsequent removal of the volume load causes LVEDP to drop below baseline without full recovery over the next 10 minutes period (Left hand panel; *,†,‡: p<0.001). In stretched muscle strips, titin phosphorylation (total and isoform specific) was signficantly higher than in non-stretched muscle strips (Middle panel) as also evident from Pro-Q diamond staining (Right hand panel). Stretch induced effects disappeared following inhibition of PKG (PKGi) and were absent in hypertrophied myocardium.
In HFpEF, myocardial cGMP content and PKG activity are low mainly because of microvascular inflammation and rarefaction46,47 and the aforementioned adjustment of diastolic LV compliance following an acute volume load is therefore compromised. This results in a sustained elevation of LV filling pressures, which contributes to the poor tolerance of volume expanion in HFpEF. The same mechanism also affects exercise tolerance in HFpEF especially in the presence of chronotropic incompetence, as occurs with betablockade, when an inadequate rise in heart rate leads to LV end-diastolic volume expansion during the initial phase of exercise.
Diabetes mellitus worsens diastolic LV dysfunction in HFrEF, HFpEF and aortic stenosis (AS) through increased fibrosis in HFrEF, higher cardiomyocyte resting tension in HFpEF and both augmented fibrosis and cardiomyocyte resting tension in AS48,49. The mechanisms responsible for the higher cardiomyocyte resting tension have recently been unraveled and consist of hypophosphorylation of titin in the I-band N2B segment and hyperphosphorylation in the I-band PEVK segment50. Administration of insulin and metformin restored these posttranslational modifications of titin and a similar effect was recently also described for empagliflozin51.
Myocardial Collagen Homeostasis and Myocardial Stiffness
Both increased metabolic and hemodynamic load lead to abnormalities in the myocardial extracellular matrix (ECM) in HFpEF patients. These changes include altered fibrillar proteins such as increased fibrillar collagen content, a shift toward less compliant collagen types (increased collagen I vs III), increased collagen cross-linking, increase in collagen fiber size, changed basal lamina structures, glycosaminoglycans and proteoglycans (often referred as ground substance). In aggregate, these ECM changes in HFpEF patients result in both myocardial fibrosis and increased myocardial stiffness24,27. Myocardial biopsies in clinical studies and in animal model HFpEF studies have shown that a substantial portion of HFpEF patients have an increase in collagen volume fraction by histology and/or MRI using LGE and T1 mapping studies24,27.
Of the changes described above in the ECM, the most important in the development of HFpEF are changes in collagen. Collagen homeostasis (Figure 5) is dependent on procollagen synthesis by myocardial fibroblasts, post-synthetic procollagen processing in the interstitium facilitated by matricellular proteins (such as SPARC, thrombospondin, periostin, ADAMs, BMP), post translational collagen cross-linking (controlled by LOX enzymes, transglutaminase, AGEs) and collagen degradation (increased by MMPs, decreased by TIMPs). In clinically relevant animal models, transgenic overexpression, transgenic null expression and conditional constructs have demonstrated the causal relationship between these collagen homeostatic determinants and the development of fibrosis52–57. In each of these transgenic constructs and in clinical studies of HFpEF, a clear quantitative relationship between increased fibrillar collagen content and increased myocardial stiffness has been defined. The homeostatic regulatory proteins and peptides described above can be and have been measured in plasma/serum samples of HFpEF patients and shown to be altered58–62. In addition, collagen propeptides (reflective of synthesis) and telopeptides (reflective of degradations) have also been measured in HFpEF patients. The circulating levels of these biomarkers reflect the presence and severity of HFpEF, fibrosis and diastolic LV stiffness, provide prognostic information and are targets for therapy (Figure 5).
Figure 5. Molecular and cellular processes that control collagen homeostasis.
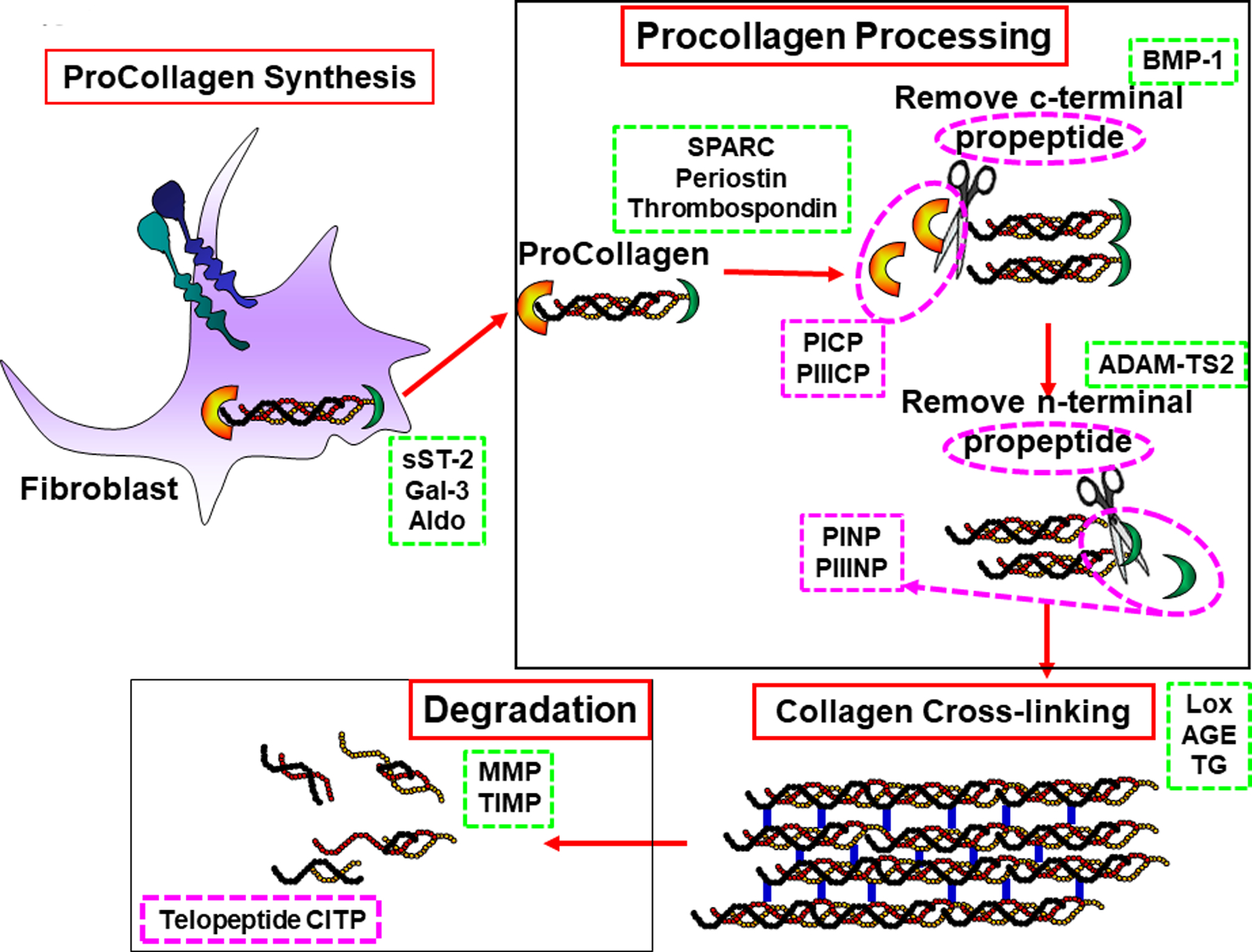
Panel A: Myocardial fibroblasts synthesize and secrete procollagen into the extracellular matrix (ECM) stimulated by sST-2, galectin-3 (gal-3), aldosterone (Aldo) and other proinflammatory and profibrotic factors (green dashed box). Panel B: Procollagen is processed in the ECM chaperoned by matricellular proteins (such as SPARC, periostin and thrombospondin) through sequential steps that remove c-terminal by bone morphometric peptide 1 (BMP-1) and n-terminal propeptides by ADAM-TS2. Plasma/serum concentrations of the resulting propeptides (PICP, PIIICP, PINP,PIIINP) reflect collagen synthesis rate. Panel C: Collagen cross-linking finalizes processing into mature insoluble collagen under the influence of lysyl oxidase (LOX), advanced glycation end products (AGE) and transglutaminase (TG). Panel D: Collagen degradation by matrix metalloproteinases (MMPs) into telopeptides (CITP) occurs under regulation of the endogenous tissue inhibitors of MMPs (TIMPs).
Each of the biomarkers within the green and purple dashed boxes can be measured in the circulation. (Illustration credit: Ben Smith).
The hemodynamic load dependent collagen homeostatic mechanisms described above play a role in the regression of myocardial fibrosis that occurs when hemodynamic overload is removed. The best example of regression of myocardial fibrosis occurs when the hemodynamic overload of aortic valve stenosis is removed by aortic valve replacement (AVR)63. Krayenbuehl and Hess used serial LV endocardial biopsies following AVR to document significant, albeit incomplete, regression of fibrosis that occurred over a 3–5 year time period64. The causal mechanisms, changes in collagen homeostasis, and changes in fibroblast and macrophage dependent inflammatory and fibrotic signaling that occur after removal of hemodynamic and / or metabolic load and that lead to regression of fibrosis remain however incompletely defined.
Crosstalk between Hemodynamic Load, ECM Laminin and Cardiomyocyte Titin
Hitherto, HFpEF related changes in cardiomyocyte titin were assessed independently from alterations in ECM components such as collagen and basal lamina. Recent evidence however suggests that hemodynamic load-induced changes in ECM laminin lead to changes in titin that affect cardiomyocyte and myocardial stiffness. In in-vitro and ex-vivo studies, treatment with polylaminin, a biomimetic polymer of the adult form of laminin, resulted in an increase in the compliant titin N2BA isoform expression and a decrease in the stiff titin N2B isoform65. In addition, polylaminin was shown to increase fibroblast secretion of MMP-2 and 9. This suggests a change in fibroblast phenotype to a less profibrotic de-activated state. Finally, polylaminin treatment was also associated with a less proinflammatory macrophage phenotype. These observations provide initial evidence on interdependence of load, laminin and titin determining overall diastolic myocardial stiffness in HFpEF.
Myocardial Accumulation of Degraded Proteins
Up till now high diastolic LV stiffness in HFpEF was presumed to result primarily from myocardial fibrosis and modifications of titin. Recently however, Schiattarella et al.66,67 unveiled a third mechanism responsible for high LV diastolic stiffness in HFpEF namely a deficient unfolded protein response (UPR) in cardiomyocytes. The UPR consists of three parallel enzyme systems in the endoplasmic reticulum that either correct misfolded proteins or transfer them for degradation to the ubiquitin/proteasome system or to lysosomes. Deficiency of the UPR in HFpEF is caused by systemic inflammation triggering expression of inducible nitric oxide synthase (iNOS) in cardiomyocytes (Figure 1). Induction of iNOS leads to reduced activity of inositol-requiring enzyme1α (IRE1α) which performs the initial step in one of the three UPR enzyme systems namely splicing of the messenger RNA of X-box binding protein1 (XBP1). Lower IRE1α activity in HFpEF cardiomyocytes modifies expression of XBP1s, which impairs the UPR and leads to cytoplasmic accumulation of degraded proteins. It remains unclear if sarcomeric proteins like troponin or titin are affected but some clinical observations support their involvement. Patients with HFpEF frequently have elevated plasma level of troponin68, which is more likely to result from cytoplasmic accumulation of destabilized troponin than from cardiomyocyte cell death because the latter was never observed in biopsy or autopsy LV specimens of HFpEF patients25,28. Elevated plasma levels of cleaved fragments of titin have recently also been observed and similarly to troponin related to worse HFpEF69. The accumulation of degraded proteins in cardiomyocytes could lead to accumulation in the myocardial interstitium as occurs in amyloidosis. In the latter condition however, the flux of proteins is reversed as interstitial accumulation results from high plasma levels of proteins such as immunoglobulin light chain, transthyretin or serum amyloid A. Apart from a deficient UPR, protein degradation by the ubiquitin/proteasome system also appears to be impaired in HFpEF myocardium. Overexpression of the ubiquitin ligase E3 WWP1 in mice, as occurs at higher age in humans, resulted in a two fold increase in myocardial collagen volume fraction and diastolic LV dysfunction70. Finally, protein ubiquitylation is facilitated by PKA or PKG and administration of a PDE1 inhibitor attenuated HFpEF in a mouse model of proteotoxic stress induced by mutated αB-Crystallin71.
Inflammatory/Fibrotic Signaling in HFpEF Phenogroups
Several groups of investigators applied a machine-learning based clustering strategy to identify distinct phenogroups within HFpEF populations derived either from local registries or from large trial populations72–76. Table 1 summarizes their findings. Despite large scatter in number of clinical and biomarker input variables, the 5 studies shared similar findings: 1) they all identified distinct phenogroups based on presence of metabolic comorbidities such as obesity (n=4), diabetes (n=5) and renal insufficiency (n=5); 2) phenogroups with CKD (n=4), high natriuretic peptides (NP) (n=4) or prominent diastolic LV dysfunction (n=3) had the poorest outcome. Studies differed widely in the incorporation of biomarker data. All studies used natriuretic peptides as input variable but only one study also included inflammatory biomarkers as input variables76. After establishing the distinct phenogroups, two studies analysed the distribution of biomarkers over the different phenogroups74,75.
Table 1:
HFpEF phenogroups derived from machine-learning clustering strategies.
| Shah SJ72 | Segar MW73 TOPCAT |
Hedman AK74 Karolinska-Rennes |
Cohen JB75 TOPCAT |
Sabbah M76 RELAX,NEAT,INDIE |
|
|---|---|---|---|---|---|
| Clin,ECG,Echo,Lab, Biomarkers | 66 1 (NP) |
60 1 (NP) |
43 1 (NP) |
60? 1 (NP) |
14 13 (inflammation) |
| Phenogroup 1 | Young Risk Low |
DM,CKD,NP↑↑ E/e’↑, Risk High |
DM,CKD,CAD NP↑, E/e’↑, Risk IM |
COPD, Risk Low, HFpEF? |
Male, NP↑↑, LAVI↑,Fibrosis Infl↑,Risk High |
| Phenogroup 2 | Obese, DM, NP↑, Risk IM |
Risk IM | COPD, NP↑↑ Risk High |
AF,NP↑,E/e’↑ Risk IM |
Old, Risk Low |
| Phenogroup 3 | CKD, NP↑↑, E/e’↑, Risk High |
CAD, Stroke, Risk IM |
Obese, Risk Low |
Obese, DM, CKD, Risk High |
Female, Obese, CRP↑, Risk IM |
| Phenogroup 4 | Obese, NP↑, Risk IM |
||||
| Phenogroup 5 | Female, NP↑, Risk IM |
||||
| Phenogroup 6 | Female, AF, NP↑, E/e’↑, Risk IM |
Clin, ECG, Echo, Lab: Number of clinical, electrocardiographic and laboratory characteristics used as input variables; Biomarkers: Number of biomarkers used as input variables; NP: Natriuretic Peptides; DM: Diabetes Mellitus; CKD: Chronic Kidney Disease; E/e’: ratio of early diastolic mitral flow velocity over early diastolic long axis lengthening velocity; CAD: Coronary Artery Disease; Risk IM: Intermediate Risk; COPD: Chronic Obstructive Pulmonary Disease; LAVI: Left Atrial Volume Index; Infl: Inflammatory biomarkers; CRP: C-Reactive Protein; AF: Atrial Fibrillation
When inflammatory biomarkers were used as input variable76, three distinct phenogroups appeared: 1) a phenogroup with a paninflammatory profile; 2) a phenogroup with selective elevation of CRP and Serum Amyloid A (SAA) and 3) a phenogroup with no evidence of inflammatory biomarkers. The phenogroup with the paninflammatory profile had the worst outcome and was the only one with elevated myocardial fibrosis biomarkers. The two studies74–75, which did not incorporate biomarkers as input variables but ‘a posteriori’ assessed their distribution in the phenogroups, confirmed these findings as they observed respectively high IL1RL1, also called solubleST2 (sST2), an inflammation/fibrosis marker74 and high TNFα75 in the phenogroup with the worst outcome. Finally, broad transcriptomic profiling of endomyocardial biopsies procured from HFpEF patients revealed three subgroups, one of which featured prominent proinflammatory signaling27.
Inflammatory/Fibrotic Signaling and HFpEF Therapy
Inflammatory/Fibrotic Biomarkers and Therapeutic Guidance
There is growing evidence that metabolic and hemodynamic load induced changes in inflammation and fibrosis that are reflected in circulating biomarkers provide significant improvement in diagnostic criteria for HFpEF, provide insights into the prognosis associated with the development of HFpEF and provide actionable targets for HFpEF therapy. Changes in natriuretic peptides (NPs), a direct reflection of hemodynamic load, have become pivotal to the accuracy of the diagnosis of HFpEF77,78. A circulating multibiomarker panel reflecting increased collagen synthesis and decreased degradation predicted the presence of HFpEF with an area under the curve of 0.79, performed better than any single biomarker including NT-proBNP and better than using clinical covariates alone61. In addition, baseline biomarkers and change from baseline levels provide prognostic value in HFpEF patients predicting both morbidity and mortality. The most studied of these is NPs77,78. In a similar manner, high sensitivity troponins, a biomarker reflecting load-dependent cardiomyocyte injury also predicts morbidity and mortality in HFpEF patients68. In two recent randomized HFpEF trials, PARAGON-HF and TOPCAT, biomarkers that reflect collagen homeostasis were altered58,59. These trials specifically observed: 1) increased plasma levels of aldosterone, galectin-3, and sST2; 2) increased plasma levels of biomarkers reflecting collagen synthesis/processing, such as PINP, PICP and PIIINP; 3) altered plasma levels of biomarkers that reflect collagen degradation such as lower MMPs, higher TIMPs and higher CITP. In addition, both baseline and change from baseline levels of these biomarkers provided prognostic value (Figure 6). For example, the higher the baseline value of TIMP-1, the higher the rate of HF hospitalization and CV mortality and if TIMP-1 increased during the trial, morbidity and mortality also increased. Higher TIMP-1 indeed reflects less collagen degradation therefore more collagen accumulation and increased myocardial fibrosis.
Figure 6. Prognostic value of biomarker data from PARAGON-HF Study59.
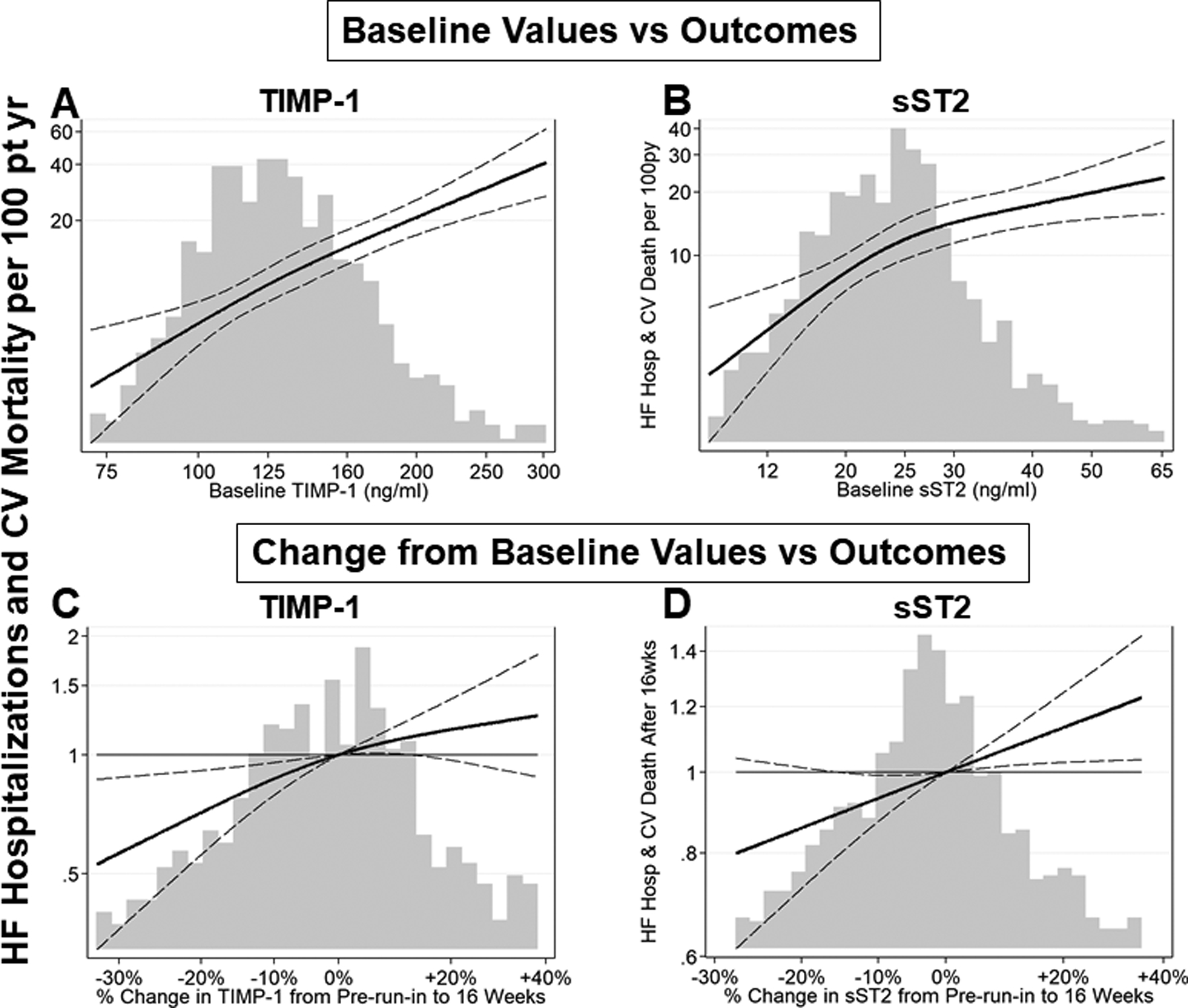
Biomarkers that reflect mechanisms that increase procollagen synthesis, such as soluble ST2, are increased in HFpEF patients; biomarkers that reflect mechanisms that decrease collagen degradation, such as increased tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) are increased in HFpEF patients. Both baseline and change from baseline levels of these profibrotic biomarkers provide prognostic value. Continuous relationships of TIMP-1 and sST2 baseline values and 16-week change from baseline values with incidence of subsequent heart failure (HF) hospitalization and cardiovascular (CV) death are plotted.
(A, B) The x-axis and histogram represent plasma biomarker level at baseline. The solid line represents estimated incidence rate of the primary endpoint, total HF hospitalizations and CV death. The dashed lines represent 95% confidence intervals for the estimated incidence rate. Incidence rate is displayed on the primary (left-sided) y-axis.
(C, D) The x-axis and histogram represent change in biomarker level between pre-run-in baseline visit and the week 16 visit. The solid line represents estimated incidence rate of the primary endpoint, total HF hospitalizations and CV death, that occurred after 16 weeks, relative to patients with no change in biomarker level, adjusted for log-transformed baseline value. The dashed lines represent 95% confidence intervals for the estimated incidence rate. Incidence rate ratio is displayed on the primary (left-sided) y-axis.
The higher the baseline value of TIMP-1 and sST2, the higher the rate of HF hospitalization and CV mortality. Over 16 weeks follow-up if TIMP-1 or sST2 decreased, the primary endpoints decreased.
Profibrotic Signaling as Therapeutic Target
Each of the profibrotic mechanisms discussed above represents a target for development of potential effective therapies for patients with HFpEF. The results from the TOPCAT Americas data support the efficacy of mineralocorticoid receptor antagonists (Spironolactone) that reduce aldosterone induced profibrotic signaling and decrease mortality and morbidity in HFpEF patients. Particularly in HFpEF patients with diabetes, spironolactone decreased collagen propeptides (decreased synthesis), increased collagen telopeptides (increased degradation) and altered upstream signaling that controls these processes58. The non-steroidal mineralocorticoid receptor antagonist finerenone improved cardiovascular outcomes in diabetic kidney disease (FIDELIO-DKD79) and is currently also being tested in HFpEF patients (FINEARTS-HF). The results from PARAGON-HF were similar to TOPCAT and support the efficacy of sacubitril/valsartan in reducing profibrotic signaling. Sacubitril/valsartan decreased collagen propetides (decreased synthesis), increased collagen telopeptides (increased degradation) and altered upstream signaling that controls these processes59.
Inflammatory Signaling as Therapeutic Target
Therapies that target proinflammatory signaling have been shown to hold promise. In HFrEF, therapy with a decoy TNFα receptor (etanercept) or a TNFα antibody (infliximab) has been disappointing with lack of benefit and higher risk of HF hospitalization80,81 probably because of loss of myocardial signaling of a key TNFα effector namely NFκB, which apart from deleterious effects also prevents mitophagy and cell death82,83. Prevention of cell death is relevant for the myocardium in HFrEF but not in HFpEF, which characteristically features myocardial hypertrophy25,27. Anti-TNFα therapy should therefore be reappraised in HFpEF. Support for a reappraisal is provided by use of anti-TNFα in chronic inflammatory joint diseases. Systemic sclerosis, systemic lupus erythematosus and rheumatoid arthritis have all been associated with an increased risk for heart failure84. In a Swedish registry on HF in rheumatoid arthritis, patients treated with corticosteroids had a higher incidence of non-ischemic heart failure but patients treated with biologicals showed a reverse trend namely a lower hazard ratio for non-ischemic heart failure85. Likewise, in rheumatoid arthritis patients with normal natriuretic peptides and LV diastolic function86, anti-TNFα therapy lowered natriuretic peptides (p=0.10) without effect on LV ejection fraction or LV global longitudinal strain (GLS).
In contrast to anti-TNFα therapy, IL-1β blockade with anakinra has already been tested in HFpEF patients. The DHART pilot trial (Diastolic Heart Failure Anakinra Response Trial) found anakinra to increase peak VO2 at 2 weeks87. The subsequent DHART2 trial however reported no significant change in either peak VO2 or the VE/VCO2 slope after 12 weeks but observed a significant fall in CRP and NT-proBNP after 4 weeks88. The most promising result on IL-1 blockade was observed in the CANTOS trial which observed less HF hospitalization when the IL-1 antibody canakinumab succeeded to lower hsCRP below 2mg/L89. Although the CANTOS trial did not discriminate between HFrEF and HFpEF, many patients likely suffered from HFpEF as they were old with a high prevalence of obesity, diabetes and hypertension. In line with IL-1 blockade, IL-6 blockade (e.g. with the monoclonal antibody tocilizumab) also deserves to be addressed in HFpEF. Apart from being an intermediate step between IL1 and hepatic CRP production, IL-6 activates the epithelial sodium channel and impairs natriuresis through reabsorption of sodium in the distal renal tubule. This leads to volume expansion, renal impairment and diuretic resistance90. Of relevance to use of IL-6 antagonism in HFpEF are the restored cardiomyocyte titin phosphorylation in experimental myocarditis91, the reduced myocardial fibrosis in rat pressure overload hypertrophy92 and the regression of LV hypertrophy in rheumatoid arthritis patients93.
The SATELLITE trial currently investigates efficacy of an orally available myeloperoxidase inhibitor (AZD4831) in HFpEF. Myeloperoxidase is abundantly present in neutrophile granulocytes and its release in the extracellular space can be triggered by presence of urate crystals. HFpEF patients frequently suffer of hyperuricemia, which can lead to deposition of urate crystals in the vasculature. The AMETHYST trial therefore investigates in HFpEF the use of Verinurad, a novel uric acid transporter 1 inhibitor. Both trials target the coronary microvasculature, which is known to be inflamed in HFpEF as evident from endothelial expression of adhesion molecules47 and to suffer from a blunted hyperemic response as convincingly demonstrated in the PROMIS-HFpEF study94.
Two landmark studies demonstrated SGLT2 inhibitors to favorably modify need for HF hospitalization in type 2 diabetes and a composite endpoint of cardiovascular death and worsening HF95,96. Their use in HFpEF patients is currently being assessed in the EMPEROR-PRESERVED97 and DELIVER trials. Two retrospective analyses not formally identifying HFpEF patients and only distinguishing HFrEF from nonHFrEF patients suggested canagliflozin and dapagliflozin to improve respectively new HF events and HF hospitalizations in HFpEF98,99. Two recent trials using sotagliflozin (SOLOIST-WHF, SCORED) identified HFpEF patients at baseline and observed a reduction of HF hospitalizations in the HFpEF subgroups100,101. Multiple reasons have been proposed for the beneficial effects of SGLT2 inhibitors in HF ranging from diuresis induced LV pre- and afterload reduction102, decreased intracellular sodium103, improved phosphorylation of titin104 and reduction of LV mass105,106. Lately, anti-inflammatory effects were also reported. In a co-culture experiment of human coronary endothelial microvascular cells and adult rat cardiomyocytes, addition of TNFα impaired cardiomyocyte shortening and relaxation kinetics, both of which were restored by subsequent addition of empagliflozin107. In this experimental set-up, the effect of empagliflozin related to alleviation of TNFα-induced oxidative stress which resulted in restored NO signaling from endothelial cells to cardiomyocytes. An anti-inflammatory effect of canagliflozin was also observed in lipopolysaccharide-stimulated human coronary artery endothelial cells and involved reduced upregulation of the glycolytic enzyme hexokinase108. Finally, in the DAPA-LVH trial, dapagliflozin reduced not only LV mass but also hsCRP106.
Statins have anti-inflammatory properties and lower CRP by 15 to 30%. These effects are largely independent of their cholesterol lowering action. In HFrEF, large outcome trials failed to observe a beneficial effect of statins on HF outcomes. In contrast, in HFpEF a series of small phase II studies or registry analyses observed positive outcomes109–111. Of specific interest is the reduced incidence of atrial fibrillation in HFpEF patients treated with statins112. Myocardial involvement in the positive outcome of statins was evident from endomyocardial biopsies, which revealed statin-treated HFpEF patients to have higher myocardial PKG activity, less cardiomyocyte hypertrophy and lower cardiomyocyte resting tension9.
Recognizing that myocardial cGMP levels are reduced in HFpEF, drugs targeting mechanisms that increase cGMP have been evaluated. These include agents that act through NO-soluble guanylyl cyclase (sGC)-phosphodiesterase-5 (PDE5) signaling and agents that act through natriuretic peptides (NP)-particulate guanylyl cyclase (pGC)-PDE9 signaling. Trials trying to manipulate cGMP through delivery of NO (INDIE-HFpEF113, NEAT-HFpEF114), stimulation of sGC (SOCRATES-Preserved115, VITALITY-HFpEF116, CAPACITY-HFpEF117) or inhibition of PDE5 (RELAX118) showed no benefit both overall or in specific subgroups. In contrast, sacubitril, which augments cGMP through NP signaling reached the primary outcome in women and in patients with LVEF<57%119. This disparity could result from compartimentalization in cardiomyocytes of both signaling routes and from counterregulatory mechanisms present in the NO-sGC-PDE5 route and absent in the NP-pGC-PDE9 route (Figure 7). Compartimentalization and counterregulatory mechanisms could explain why manipulation of the NO-sGC-PDE5 route was less successful to raise plasma or urinary cGMP in contrast to use of the NP-pGC-PDE9 route. In the INDIE-HFpEF trial, trough level of plasma cGMP was comparable in the inorganic nitrate and placebo groups and in SOCRATES-Preserved no trend in plasma cGMP was observed. In the RELAX trial a paired comparison of plasma cGMP in control and sildenafil groups showed higher plasma cGMP in the sildenafil group, which however failed to reach statistical significance (p=0.11). In contrast, in the PARAGON trial the sacubitril group had a significant increase over a 1 year period in urinary cGMP/creatinine ratio120 and in an ovine model of heart failure PDE9 inhibition significantly raised plasma cGMP/NP ratio121. The counterregulatory feedback in the NO-sGC-PDE5 signaling route consists among other mechanisms, of a cGMP-binding allosteric regulatory region in PDE5 capable of raising PDE5 activity at higher cytosolic cGMP122. A similar site is absent on PDE9123 and use of the NP-pGC-PDE9 route can therefore provide a sustained elevation of cGMP. Using biosensors, the elevation of cGMP was recently visualized in cardiomyocytes following NP dependent activation of pGC124. Elevated cGMP was specifically localized around the sarcoplasmic reticulum, myofilaments and outer mitochondrial membrane in contrast to stimulation of the NO-sGC-PDE5 signaling route, which elevates cGMP around the Z-disc. Especially the myofilamentary localization is of interest as it could relate to phosphorylation of the N2Bus segment of titin, which is known to occur following NP administration and to improve cardiomyocyte distensibility125. Furthermore, in an epidemiological study of a population free of cardiovascular disease or heart failure, plasma cGMP closely tracked NP signaling126. Finally, it is fair to conclude that the use of cGMP enhancing drugs in HFpEF has so far been disappointing. The outcome of studies using PDE9 inhibitors in HFpEF patients and sacubitril in hospitalized HFpEF patients (PARAGLIDE-HF) are therefore eagerly awaited to evaluate the importance of cGMP signaling for the inflammatory/profibrotic paradigm.
Figure 7. Compartimentalization and counterregulation of cardiomyocyte cGMP stimulation.
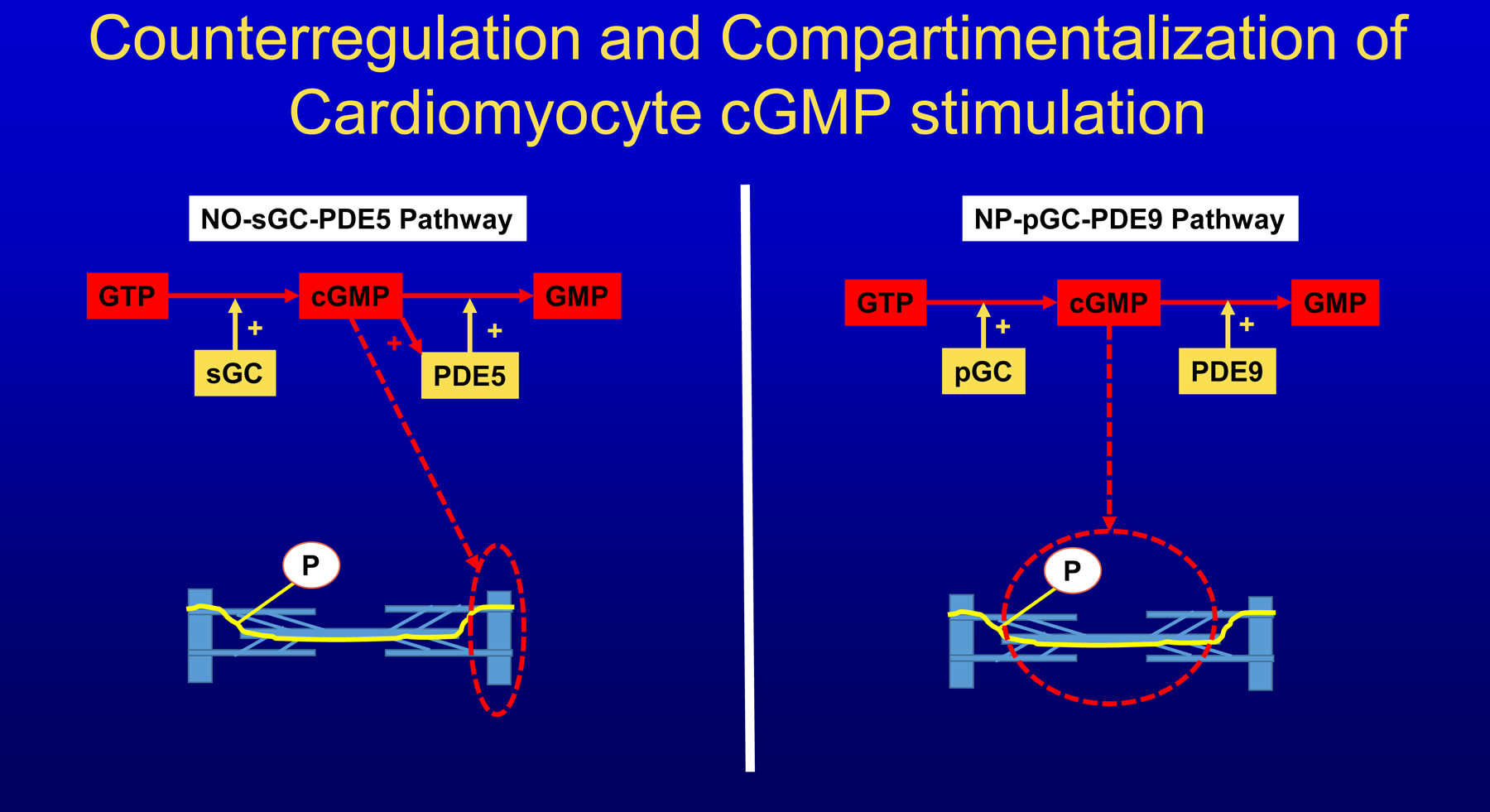
Cyclic guanosine monophosphate (cGMP) produced by soluble guanylyl cyclase (sGC) from guanosine triphosphate (GTP) is mainly localized in sarcomeres around Z discs and preferably degraded to guanosine monophosphate (GMP) by phosphodiesterase 5 (PDE5). PDE5 activity rises when cGMP is elevated, thus providing a counterregulatory feedback. cGMP produced by particulate guanylyl cyclase (pGC) is mainly localized around myofilamentary proteins like titin and preferably degraded by PDE9. PDE9 lacks a cGMP sensitive regulatory site and is therefore not subject to counterregulation by cGMP.
Conclusions and Future Directions
Over the past 10 years, novel experimental and clinical evidence emerged, that supports the inflammatory paradigm for HFpEF. This evidence consists of: 1) Hemodynamic overload-induced myocardial infiltration with immunocompetent cells triggered by components of the extracellular matrix like basal laminin; 2) Crosstalk between components of the extracellular matrix and cardiomyocyte titin resulting in altered titin isoform splicing; 3) Myocardial accumulation of degraded proteins because of failing UPR or ubiquitin/proteasome system; 4) Definition by machine learning algorithms of phenogroups of HFpEF patients with a distinct inflammatory/fibrotic signature; 5) Direct coupling in mediation analysis between comorbidities, inflammatory biomarkers and myocardial structure/function; 6) Endothelial expression of adhesion molecules in early HFpEF (stage A or B). In future clinical studies, the existing knowledge on the inflammatory/profibrotic paradigm in HFpEF needs to integrate new evidence from preclinical studies on metabolic substrate utilization, lipotoxicity and proteotoxicity127. Some specific topics which could be valuable subjects of future research efforts are listed in Table 2. Finally, insights gained from current evidence and future research efforts will pave the road for novel treatments of HFpEF such as antagonizing proinflammatory cytokines, promoting protein ubiquitylation with PDE1 inhibitors or correcting titin hypophosphorylation through stimulation of NP-pGC-PDE9 signaling.
Table 2:
Potential Topics for Future Research Efforts on Inflammatory/Fibrotic Mechanisms in HFpEF.
| 1 | Inflammatory signaling in obese, mostly female HFpEF patients with isolated CRP elevation |
| 2 | Involvement of myofilamentary proteins especially titin in nitrosative stress induced proteotoxicity |
| 3 | Gender related protective mechanisms against nitrosative stress induced proteotoxicity |
| 4 | Proinflammatory effects of microvascular endothelial upregulation of glycolytic enzymes |
| 5 | Blunted microvascular upregulation of adhesion molecules in the presence of elevated plasma high density lipoproteins |
| 6 | Linkage between overload induced basal lamina deformation and secretion of proinflammatory cytokines in cardiomyocytes |
| 7 | Molecular/cellular regulatory mechanisms that control myocardial fibrosis |
Sources of Funding:
WJP: None; MRZ: Medical Research Funds from the Department of Veterans Affairs (Middleton Award and Merit review grant - CSRD 1 I01 CX001608), NIH-NHLBI: 1R01HL123478-01A1, R01HL144927-01, Department of Defense: W81XWH-16-1-0592, South Carolina SMARTSTATE Research Centers of Economic Excellence/Endowed Professorship Program.
Footnotes
Disclosures: WJP: None; MRZ: Consultancy fees of Novartis, Merck, Ironwood, Eli Lilly, Capricor, Astra Zeneca, Myokardia, Gate House Bio.
References
- 1.Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation. 2020;141:1001–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasan RS, Xanthakis V, Lyass A, Andersson C, Tsao C, Cheng S, Aragam J, Benjamin EJ, Larson MG. Epidemiology of Left Ventricular Systolic Dysfunction and Heart Failure in the Framingham Study: An Echocardiographic Study Over 3 Decades. JACC Cardiovasc Imaging. 2018;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation. 2016;134:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borlaug BA. Evaluation and management of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2020;17:559–573. [DOI] [PubMed] [Google Scholar]
- 5.Santhanakrishnan R, Chong JP, Ng TP, Ling LH, Sim D, Leong KT, Yeo PS, Ong HY, Jaufeerally F, Wong R et al. Growth differentiation factor 15, ST2, high-sensitivity troponin T, and N-terminal pro brain natriuretic peptide in heart failure with preserved vs. reduced ejection fraction. Eur J Heart Fail 2012;14:1338–1347. [DOI] [PubMed] [Google Scholar]
- 6.Sanders-van Wijk S, van Empel V, Davarzani N, Maeder MT, Handschin R, Pfisterer ME, Brunner-La Rocca HP; TIME-CHF investigators. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail. 2015;17:1006–14. [DOI] [PubMed] [Google Scholar]
- 7.Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, Metra M, Anker SD, Cleland JG, Dickstein K et al. Identifying Pathophysiological Mechanisms in Heart Failure With Reduced Versus Preserved Ejection Fraction. J Am Coll Cardiol. 2018;72:1081–1090. [DOI] [PubMed] [Google Scholar]
- 8.Sanders-van Wijk S, Tromp J, Beussink-Nelson L, Hage C, Svedlund S, Saraste A, Swat SA, Sanchez C, Njoroge J, Tan RS et al. Proteomic Evaluation of the Comorbidity-Inflammation Paradigm in Heart Failure with Preserved Ejection Fraction: Results from the PROMIS-HFpEF Study. Circulation. 2020. October 9. doi: 10.1161/CIRCULATIONAHA.120.045810. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paulus WJ, Tschoepe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 10.DuBrock HM, AbouEzzeddine OF, Redfield MM. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS One. 2018;13:e0201836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, Liu Y, Hoffmann U, Bauer DC, Newman AB et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol. 2010;55:2129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel RB, Colangelo LA, Reiner AP, Gross MD, Jacobs DR Jr, Launer LJ, Lima JAC, Lloyd-Jones DM, Shah SJ. Cellular Adhesion Molecules in Young Adulthood and Cardiac Function in Later Life: The CARDIA Study. J Am Coll Cardiol 2020;75:2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel RB, Colangelo LA, Bielinski SJ, Larson NB, Ding J, Allen NB, Michos ED, Shah SJ, Lloyd-Jones DM. Circulating Vascular Cell Adhesion Molecule-1 and Incident Heart Failure: The Multi-Ethnic Study of Atherosclerosis (MESA). J Am Heart Assoc. 2020;9:e019390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulus WJ. Adhesion Molecules in Early Adulthood Predict Heart Failure With Preserved Ejection Fraction at Older Age. J Am Coll Cardiol. 2020;75:2166–2168. [DOI] [PubMed] [Google Scholar]
- 15.Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, Hillege HL, van Veldhuisen DJ, van Gilst WH. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J. 2013;34:1424–31. [DOI] [PubMed] [Google Scholar]
- 16.de Boer RA, Nayor M, deFilippi CR, Enserro D, Bhambhani V, Kizer JR, Blaha MJ, Brouwers FP, Cushman M, Lima JAC et al. Association of Cardiovascular Biomarkers With Incident Heart Failure With Preserved and Reduced Ejection Fraction. JAMA Cardiol. 2018;3:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel RB, Colangelo LA, Reis JP, Lima JAC, Shah SJ, Lloyd-Jones DM. Association of Longitudinal Trajectory of Albuminuria in Young Adulthood With Myocardial Structure and Function in Later Life: Coronary Artery Risk Development in Young Adults (CARDIA) Study. JAMA Cardiol. 2020;5:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renaud L, Harris LG, Mani SK, Kasiganesan H, Chou JC, Baicu CF, Van Laer A, Akerman AW, Stroud RE, Jones JA et al. HDACs Regulate miR-133a Expression in Pressure Overload Induced Cardiac Fibrosis. Circulation Heart Failure. 6:1094–1104, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riley HJ, Kelly RR, Van Laer AO, Neff LS, Dasgupta S, Baicu CF, McDonald LT, LaRue AC, Zile MR, Bradshaw AD. SPARC Production by Bone Marrow-Derived Cells Contributes to Myocardial Fibrosis in Pressure-Overload. Am J Physiol Heart Circ Physiol. December 11, 2020. doi: 10.1152/ajpheart.00552.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien M, Baicu CF, Van Laer AO, Zhang Y, McDonald LT, LaRue AC, Zile MR, Bradshaw AD. Pressure overload generates a cardiac-specific profile of inflammatory mediators. Am J Physiol Heart Circ Physiol. 319:H331–H340, 2020. doi: 10.1152/ajpheart.00274.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald LT, Zile MR, Zhang Y, Van Laer AO, Baicu CF, Stroud RE, Jones JA, LaRue AC, Bradshaw AD. Increased Macrophage-derived SPARC Precedes Collagen Deposition in Myocardial Fibrosis. Am J Physiol Heart Circ Physiol. 315:H92–H100, 2018. DOI: 10.1152/ajpheart.00719.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 215:423–440, 2018. doi: 10.1084/jem.20171274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallet R, de Couto G, Simsolo E, Valle J, Sun B, Liu W, Tseliou E, Zile MR, Marbán E. Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC- Basic and Translational Research, 1:14–28, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Heerebeek L, Borbély A, Niessen HWM, Bronzwaer JGF, Van Der Velden J, Stienen GJM, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–73. [DOI] [PubMed] [Google Scholar]
- 26.Borbély A, van der Velden J, Papp Z, Bronzwaer JGF, Edes I, Stienen GJM, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005; 111:774–781. [DOI] [PubMed] [Google Scholar]
- 27.Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZJ, Riley SJ, Subramanya V, Brown EE, Hopkins CD et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020;8:712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation. 2013;127:938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linke WA. Titin Gene and Protein Functions in Passive and Active Muscle. Annu Rev Physiol. 2018;80:389–411. [DOI] [PubMed] [Google Scholar]
- 31.Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschöpe C, McNabb M et al. Truncation of titin’s elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ Res. 2009;105:557–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radke MH, Peng J, Wu Y, McNabb M, Nelson OL, Granzier H, Gotthardt M. Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc Natl Acad Sci U S A. 2007;104:3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Methawasin M, Strom JG, Slater RE, Fernandez V, Saripalli C, Granzier H. Experimentally Increasing the Compliance of Titin Through RNA Binding Motif-20 (RBM20) Inhibition Improves Diastolic Function In a Mouse Model of Heart Failure With Preserved Ejection Fraction. Circulation. 2016;134:1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bull M, Methawasin M, Strom J, Nair P, Hutchinson K, Granzier H. Alternative Splicing of Titin Restores Diastolic Function in an HFpEF-Like Genetic Murine Model (TtnΔIAjxn). Circ Res. 2016;119:764–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90:1181–1188. [DOI] [PubMed] [Google Scholar]
- 36.Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, Dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2008;104:87–94. [DOI] [PubMed] [Google Scholar]
- 37.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loescher CM, Breitkreuz M, Li Y, Nickel A, Unger A, Dietl A, Schmidt A, Mohamed BA, Kötter S, Schmitt JP et al. Regulation of titin-based cardiac stiffness by unfolded domain oxidation (UnDOx). Proc Natl Acad Sci U S A. 2020;117:24545–24556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borbély A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J et al. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780–6. [DOI] [PubMed] [Google Scholar]
- 40.Hamdani N, Franssen C, Lourenço A, Falcão-Pires I, Fontoura D, Leite S, Plettig L, López B, Ottenheijm CA, Becher PM et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–49. [DOI] [PubMed] [Google Scholar]
- 41.Bai B, Yang W, Fu Y, Foon HL, Tay WT, Yang K, Luo C, Gunaratne J, Lee P, Zile MR et al. Seipin Knockout Mice Develop Heart Failure With Preserved Ejection Fraction. JACC Basic Transl Sci. 2019;4:924–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bódi B, Tóth EP, Nagy L, Tóth A, Mártha L, Kovács Á, Balla G, Kovács T, Papp Z. Titin isoforms are increasingly protected against oxidative modifications in developing rat cardiomyocytes. Free Radic Biol Med. 2017;113:224–235. [DOI] [PubMed] [Google Scholar]
- 43.Franssen C, Kole J, Musters R, Hamdani N, Paulus WJ. α-B Crystallin Reverses High Diastolic Stiffness of Failing Human Cardiomyocytes. Circ Heart Fail. 2017;10(3):e003626. [DOI] [PubMed] [Google Scholar]
- 44.Leite-Moreira AM, Almeida-Coelho J, Neves JS, Pires AL, Ferreira-Martins J, Castro-Ferreira R, Ladeiras-Lopes R, Conceição G, Miranda-Silva D, Rodrigues P et al. Stretch-induced compliance: a novel adaptive biological mechanism following acute cardiac load. Cardiovasc Res. 2018;114:656–667. [DOI] [PubMed] [Google Scholar]
- 45.Methawasin M, Granzier H. Softening the Stressed Giant Titin in Diabetes Mellitus. Circ Res. 2018;123:315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–9. [DOI] [PubMed] [Google Scholar]
- 47.Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschöpe C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016;4:312–24. [DOI] [PubMed] [Google Scholar]
- 48.van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, Ijsselmuiden AJ, Schalkwijk CG, Bronzwaer JG, Diamant M et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. 2008;117:43–51. [DOI] [PubMed] [Google Scholar]
- 49.Falcão-Pires I, Hamdani N, Borbély A, Gavina C, Schalkwijk CG, van der Velden J, van Heerebeek L, Stienen GJ, Niessen HW, Leite-Moreira AF et al. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. 2011;124:1151–9. [DOI] [PubMed] [Google Scholar]
- 50.Hopf AE, Andresen C, Kötter S, Isić M, Ulrich K, Sahin S, Bongardt S, Röll W, Drove F, Scheerer N et al. Diabetes-Induced Cardiomyocyte Passive Stiffening Is Caused by Impaired Insulin-Dependent Titin Modification and Can Be Modulated by Neuregulin-1. Circ Res. 2018;123:342–355. [DOI] [PubMed] [Google Scholar]
- 51.Pabel S, Wagner S, Bollenberg H, Bengel P, Kovács Á, Schach C, Tirilomis P, Mustroph J, Renner A, Gummert J et al. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail. 2018;20:1690–1700. [DOI] [PubMed] [Google Scholar]
- 52.Toba H, de Castro Brás LE, Baicu CF, Zile MR, Lindsey ML, Bradshaw AD. Increased ADAMTS1 mediates SPARC-dependent collagen deposition in the aging myocardium. Am J Physiol Endocrinol Metab. 2016;310:E1027–E1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toba H, de Castro Brás L, Baicu C, Zile MR, Lindsey M, Bradshaw AD. Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization AJP-Cell Physiology. 2015;308:C972–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Boggs J, Lacy JM, Zile MR. Pressure-Overload Induced Alterations in Fibrillar Collagen Content and Myocardial Diastolic Function: Role of SPARC in Post-Synthetic Procollagen Processing. Circulation. 2009;119:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baicu CF, Zhang Y, Van Laer AO, Renaud L, Zile MR, Bradshaw AD. Effects of the Absence of Procollagen C-Endopeptidase Enhancer-2 (PCOLCE-2) On Myocardial Collagen Accumulation in Chronic Pressure-Overload. Am J Physiol Heart Circ Physiol. 2012;303:H234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baicu CF, Li J, Zhang Y, Kasiganesan H, Cooper G, Zile MR, Bradshaw AD. Time Course of Right Ventricular Pressure-Overload Induced Myocardial Fibrosis: Relationship to Changes in Fibroblast Dependent Post-synthetic Procollagen Processing. Am J Physiol Heart Circ Physiol. 2012; 303:H1128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zile MR, Baicu CF, Stroud RE, Van Laer AO, Jones JA, Patel R, Mukherjee R, Spinale FG. Mechanistic relationship between membrane type-1 matrix metalloproteinase and the myocardial response to pressure overload. Circ Heart Fail. 2014;7:340–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Marco C, Claggett BL, de Denus S, Zile MR, Huynh T, Desai AS, Sirois MG, Solomon SD, Pitt B, Rouleau JL et al. Impact of diabetes on serum biomarkers in heart failure with preserved ejection fraction: insights from the TOPCAT trial. ESC Heart Fail. 2021. January 12. doi: 10.1002/ehf2.13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cunningham JW, Claggett BL, O’Meara E, Prescott MF, Pfeffer MA, Shah SJ, Redfield MM, Zannad F, Chiang LM, Rizkala AR et al. Effect of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFpEF. J Am Coll Cardiol. 2020;76:503–514. doi: 10.1016/j.jacc.2020.05.072 [DOI] [PubMed] [Google Scholar]
- 60.Chow SL, Maisel AS, Anand I, Bozkurt B, de Boer RA, Felker GM, Fonarow GC, Greenberg B, Januzzi JL Jr, Kiernan MS et al. American Heart Association Clinical Pharmacology Committee of the Council on Clinical Cardiology; Council on Basic Cardiovascular Sciences; Council on Cardiovascular Disease in the Young; Council on Cardiovascular and Stroke Nursing; Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation; Council on Epidemiology and Prevention; Council on Functional Genomics and Translational Biology; and Council on Quality of Care and Outcomes Research. Role of Biomarkers for the Prevention, Assessment, and Management of Heart Failure: A Scientific Statement From the American Heart Association. Circulation. 2017;135:e1054–e1091. [DOI] [PubMed] [Google Scholar]
- 61.Zile MR, DeSantis SM, Baicu CF, Stroud RE, Thompson SB, McClure CD, Mehurg SM, Spinale FG. Plasma Biomarkers That Reflect Determinants of Matrix Composition Identify the Presence of Left Ventricular Hypertrophy and Diastolic Heart Failure. Circ Heart Fail. 2011;4:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zile MR, Jhund PS, Baicu CF, Claggett BL, Pieske B, Voors AA, Prescott MF, Shi V, Lefkowitz M, McMurray JJ et al. Prospective Comparison of ARNI With ARB on Management of Heart Failure With Preserved Ejection Fraction (PARAMOUNT) Investigators. Plasma Biomarkers Reflecting Profibrotic Processes in Heart Failure With a Preserved Ejection Fraction: Data From the Prospective Comparison of ARNI With ARB on Management of Heart Failure With Preserved Ejection Fraction Study. Circ Heart Fail. 2016;9:e002551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yarbrough WM, Mukherjee R, Ikonomidis JS, Zile MR, Spinale FG. Myocardial Remodeling With Aortic Stenosis and Following Aortic Valve Replacement-Mechanisms and Future Prognostic Implications. J Thorac Cardiovasc Surg. 2012;143:656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krayenbuehl HP, Hess OM, Monrad ES, Schneider J, Mall G, Turina M. Left ventricular myocardial structure in aortic valve disease before, intermediate, and late after aortic valve replacement. Circulation. 1989;79:744–55. [DOI] [PubMed] [Google Scholar]
- 65.Hochman-Mendez C, Curty E, Taylor DA. Change the Laminin, Change the Cardiomyocyte: Improve Untreatable Heart Failure. Int J Mol Sci. 2020;21:6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tong D, Schiattarella GG, Jiang N, May HI, Lavandero S, Gillette TG, Hill JA. Female Sex Is Protective in a Preclinical Model of Heart Failure With Preserved Ejection Fraction. Circulation. 2019;140:1769–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fudim M, Ambrosy AP, Sun JL, Anstrom KJ, Bart BA, Butler J, AbouEzzeddine O, Greene SJ, Mentz RJ, Redfield MM et al. High-Sensitivity Troponin I in Hospitalized and Ambulatory Patients With Heart Failure With Preserved Ejection Fraction: Insights From the Heart Failure Clinical Research Network. J Am Heart Assoc. 2018;7:e010364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Palau P, Reese-Petersen AL, Domínguez E, Ramón JM, López L, Mollar A, Chorro FJ, Sanchis J, Núñez J. Matrix metalloproteinase-12 cleaved fragment of titin as a predictor of functional capacity in patients with heart failure and preserved ejection fraction. Scand Cardiovasc J. 2020. October 8:1–6. doi: 10.1080/14017431.2020.1831052. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 70.Matesic LE, Freeburg LA, Snyder LB, Duncan LA, Moore A, Perreault PE, Zellars KN, Goldsmith EC, Spinale FG. The ubiquitin ligase WWP1 contributes to shifts in matrix proteolytic profiles and a myocardial aging phenotype with diastolic heart. Am J Physiol Heart Circ Physiol. 2020;319:H765–H774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang H, Pan B, Wu P, Parajuli N, Rekhter MD, Goldberg AL, Wang X. PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci Adv. 2019;5:eaaw5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC, Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2015;131:269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Segar MW, Patel KV, Ayers C, Basit M, Tang WHW, Willett D, Berry J, Grodin JL, Pandey A. Phenomapping of patients with heart failure with preserved ejection fraction using machine learning-based unsupervised cluster analysis. Eur J Heart Fail. 2020;22:148–158. [DOI] [PubMed] [Google Scholar]
- 74.Hedman ÅK, Hage C, Sharma A, Brosnan MJ, Buckbinder L, Gan LM, Shah SJ, Linde CM, Donal E, Daubert JC et al. Identification of novel phenogroups in heart failure with preserved ejection fraction using machine learning. Heart. 2020;106:342–349. [DOI] [PubMed] [Google Scholar]
- 75.Cohen JB, Schrauben SJ, Zhao L, Basso MD, Cvijic ME, Li Z, Yarde M, Wang Z, Bhattacharya PT, Chirinos DA et al. Clinical Phenogroups in Heart Failure With Preserved Ejection Fraction: Detailed Phenotypes, Prognosis, and Response to Spironolactone. JACC Heart Fail. 2020;8:172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sabbah MS, Fayyaz AU, de Denus S, Felker GM, Borlaug BA, Dasari S, Carter RE, Redfield MM. Circ Heart Fail. Obese-Inflammatory Phenotypes in Heart Failure With Preserved Ejection Fraction. 2020;13:e006414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cunningham JW, Vaduganathan M, Claggett BL, Zile MR, Anand IS, Packer M, Zannad F, Lam CSP, Janssens S, Jhund PS et al. Effects of Sacubitril/Valsartan on N-Terminal Pro-B-Type Natriuretic Peptide in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 8:372–381, 2020. doi: 10.1016/j.jchf.2020.03.002 [DOI] [PubMed] [Google Scholar]
- 78.Ibrahim NE, Burnett JC Jr, Butler J, Camacho A, Felker GM, Fiuzat M, O’Connor C, Solomon SD, Vaduganathan M, Zile MR et al. Natriuretic Peptides as Inclusion Criteria in Clinical Trials: A JACC: Heart Failure Position Paper. JACC Heart Fail. 8:347–358, 2020. doi: 10.1016/j.jchf.2019.12.010 [DOI] [PubMed] [Google Scholar]
- 79.Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, Kolkhof P, Nowack C, Schloemer P, Joseph A et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N Engl J Med. 2020;383:2219–2229. [DOI] [PubMed] [Google Scholar]
- 80.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004;109:1594–602. [DOI] [PubMed] [Google Scholar]
- 81.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003;107:3133–40. [DOI] [PubMed] [Google Scholar]
- 82.Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. 2020;17:269–285. [DOI] [PubMed] [Google Scholar]
- 83.Murphy SP, Kakkar R, McCarthy CP, Januzzi JL Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020;75:1324–1340. [DOI] [PubMed] [Google Scholar]
- 84.Prasada S, Rivera A, Nishtala A, Pawlowski AE, Sinha A, Bundy JD, Chadha SA, Ahmad FS, Khan SS, Achenbach C et al. Differential Associations of Chronic Inflammatory Diseases With Incident Heart Failure. JACC Heart Fail. 2020;8:489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mantel Ä, Holmqvist M, Andersson DC, Lund LH, Askling J. Association Between Rheumatoid Arthritis and Risk of Ischemic and Nonischemic Heart Failure. J Am Coll Cardiol. 2017;69:1275–1285. [DOI] [PubMed] [Google Scholar]
- 86.Baniaamam M, Handoko ML, Agca R, Heslinga SC, Konings TC, van Halm VP, Nurmohamed MT. The Effect of Anti-TNF Therapy on Cardiac Function in Rheumatoid Arthritis: An Observational Study. J Clin Med. 2020;9:3145–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, Jahangiri A, Falcao RA, Kontos MC, Shah KB et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol 2014;113:321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, Del Buono MG, Billingsley H, Wohlford G, Viscusi M et al. IL-1 Blockade in Patients With Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2018;11:e005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, Ridker PM. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation. 2019;139:1289–1299. [DOI] [PubMed] [Google Scholar]
- 90.Hanberg JS, Rao VS, Ahmad T, Chunara Z, Mahoney D, Jackson K, Jacoby D, Chen M, Wilson FP, Tang WHW et al. Inflammation and cardio‐renal interactions in heart failure: a potential role for interleukin‐6. Eur J Heart Fail. 2018;20:933–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Savvatis K, Müller I, Fröhlich M, Pappritz K, Zietsch C, Hamdani N, Grote K, Schieffer B, Klingel K, Van Linthout S et al. Interleukin-6 receptor inhibition modulates the immune reaction and restores titin phosphorylation in experimental myocarditis. Basic Res Cardiol. 2014;109(6):449. [DOI] [PubMed] [Google Scholar]
- 92.Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A et al. Deletion of Interleukin-6 Attenuates Pressure Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Circ Res. 2016;118:1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kobayashi H, Kobayashi Y, Giles JT, Yoneyama K, Nakajima Y, Takei M. Tocilizumab treatment increases left ventricular ejection fraction and decreases left ventricular mass index in patients with rheumatoid arthritis without cardiac symptoms: assessed using 3.0 tesla cardiac magnetic resonance imaging. J Rheumatol 2014;41:1916–21. [DOI] [PubMed] [Google Scholar]
- 94.Shah SJ, Lam CSP, Svedlund S, Saraste A, Hage C, Tan RS, Beussink-Nelson L, Ljung Faxén U, Fermer ML, Broberg MA et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. 2018;39:3439–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373:2117–28. [DOI] [PubMed] [Google Scholar]
- 96.McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med. 2019;381:1995–2008. [DOI] [PubMed] [Google Scholar]
- 97.Anker SD, Butler J, Filippatos GS, Jamal W, Salsali A, Schnee J, Kimura K, Zeller C, George J, Brueckmann M et al. Evaluation of the effects of sodium-glucose co-transporter 2 inhibition with empagliflozin on morbidity and mortality in patients with chronic heart failure and a preserved ejection fraction: rationale for and design of the EMPEROR-Preserved Trial. Eur J Heart Fail. 2019;21:1279–1287. [DOI] [PubMed] [Google Scholar]
- 98.Figtree GA, Rådholm K, Barrett TD, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Matthews DR, Shaw W, Neal B. Effects of Canagliflozin on Heart Failure Outcomes Associated With Preserved and Reduced Ejection Fraction in Type 2 Diabetes Mellitus Circulation. 2019;139:2591–2593. [DOI] [PubMed] [Google Scholar]
- 99.Kato ET, Silverman MG, Mosenzon O, Zelniker TA, Cahn A, Furtado RHM, Kuder J, Murphy SA, Bhatt DL, Leiter LA et al. Effect of Dapagliflozin on Heart Failure and Mortality in Type 2 Diabetes Mellitus. Circulation. 2019. May;139:2528–2536. [DOI] [PubMed] [Google Scholar]
- 100.Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, Lewis JB, Riddle MC, Voors AA, Metra M et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N Engl J Med. 2021;384:117–128. [DOI] [PubMed] [Google Scholar]
- 101.Bhatt DL, Szarek M, Pitt B, Cannon CP, Leiter LA, McGuire DK, Lewis JB, Riddle MC, Inzucchi SE, Kosiborod MN et al. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N Engl J Med. 2021;384:129–139. [DOI] [PubMed] [Google Scholar]
- 102.Connelly KA, Zhang Y, Visram A, Advani A, Batchu SN, Desjardins JF, Thai K, Gilbert RE. Empagliflozin Improves Diastolic Function in a Nondiabetic Rodent Model of Heart Failure With Preserved Ejection Fraction. JACC Basic Transl Sci. 2019;4:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baartscheer A, Schumacher CA, Wüst RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ. Empagliflozin decreases myocardial cytoplasmic Na + through inhibition of the cardiac Na +/H + exchanger in rats and rabbits. Diabetologia. 2017;60:568–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pabel S, Wagner S, Bollenberg H, Bengel P, Kovács Á, Schach C, Tirilomis P, Mustroph J, Renner A, Gummert J et al. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail. 2018;20:1690–1700. [DOI] [PubMed] [Google Scholar]
- 105.Verma S, Mazer CD, Yan AT, Mason T, Garg V, Teoh H, Zuo F, Quan A, Farkouh ME, Fitchett DH et al. Effect of Empagliflozin on Left Ventricular Mass in Patients With Type 2 Diabetes Mellitus and Coronary Artery Disease: The EMPA-HEART CardioLink-6 Randomized Clinical Trial. Circulation. 2019;140:1693–1702. [DOI] [PubMed] [Google Scholar]
- 106.Brown AJM, Gandy S, McCrimmon R, Houston JG, Struthers AD, Lang CC. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: the DAPA-LVH trial. Eur Heart J. 2020;41:3421–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Juni RP, Kuster DWD, Goebel M, Helmes M, Musters RJP, van der Velden J, Koolwijk P, Paulus WJ, van Hinsbergh VWM. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC Basic Transl Sci. 2019;4:575–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Uthman L, Kuschma M, Römer G, Boomsma M, Kessler J, Hermanides J, Hollmann MW, Preckel B, Zuurbier CJ, Weber NC. Novel Anti-inflammatory Effects of Canagliflozin Involving Hexokinase II in Lipopolysaccharide-Stimulated Human Coronary Artery Endothelial Cells. Cardiovasc Drugs Ther. 2020. October 13. doi: 10.1007/s10557-020-07083-w. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fukuta H, Sane DC, Brucks S, Little WC. Statin therapy may be associated with lower mortality in patients with diastolic heart failure: a prelimininary report. Circulation 2005;112:357–363. [DOI] [PubMed] [Google Scholar]
- 110.Nochioka K, Sakata Y, Miyata S, Miura M, Takada T, Tadaki S, Ushigome R, Yamauchi T, Takahashi J, Shimokawa H et al. Prognostic impact of statin use in patients with heart failure and preserved ejection fraction. Circ J. 2015; 79:574–582. [DOI] [PubMed] [Google Scholar]
- 111.Alehagen U, Benson L, Edner M, Dahlström U, Lund LH. Association between use of statins and mortality in patients with heart failure and ejection fraction of ≥50. Circ Heart Fail. 2015;8:862–870. [DOI] [PubMed] [Google Scholar]
- 112.Zakeri R, Chamberlain AM, Roger VL, Redfield MM. Temporal relationship and prognostic significance of atrial fibrillation in heart failure patients with preserved ejection fraction: a community-based study. Circulation. 2013; 128:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Borlaug BA, Anstrom KJ, Lewis GD, Shah SJ, Levine JA, Koepp GA, Givertz MM, Felker GM, LeWinter MM et al. Effect of Inorganic Nitrite vs Placebo on Exercise Capacity Among Patients With Heart Failure With Preserved Ejection Fraction: The INDIE-HFpEF Randomized Clinical Trial. JAMA. 2018;320:1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Redfield MM, Anstrom KJ, Levine JA, Koepp GA, Borlaug BA, Chen HH, LeWinter MM, Joseph SM, Shah SJ, Semigran MJ et al. Isosorbide Mononitrate in Heart Failure with Preserved Ejection Fraction. N Engl J Med. 2015;373:2314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pieske B, Maggioni AP, Lam CSP, Pieske-Kraigher E, Filippatos G, Butler J, Ponikowski P, Shah SJ, Solomon SD, Scalise AV et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES-PRESERVED) study. Eur Heart J. 2017;38:1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Armstrong PW, Lam CSP, Anstrom KJ, Ezekowitz J, Hernandez AF, O’Connor CM, Pieske B, Ponikowski P, Shah SJ, Solomon SD et al. Effect of Vericiguat vs Placebo on Quality of Life in Patients With Heart Failure and Preserved Ejection Fraction: The VITALITY-HFpEF Randomized Clinical Trial. JAMA. 2020;324:1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Udelson JE, Lewis GD, Shah SJ, Zile MR, Redfield MM, Burnett J Jr, Parker J, Seferovic JP, Wilson P et al. Effect of Praliciguat on Peak Rate of Oxygen Consumption in Patients With Heart Failure With Preserved Ejection Fraction: The CAPACITY HFpEF Randomized Clinical Trial. JAMA. 2020;324:1522–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL et al. RELAX Trial. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, Martinez F, Packer M, Pfeffer MA, Pieske B et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N Engl J Med. 2019;381:1609–1620. [DOI] [PubMed] [Google Scholar]
- 120.McMurray JJV, Jackson AM, Lam CSP, Redfield MM, Anand IS, Ge J, Lefkowitz MP, Maggioni AP, Martinez F, Packer M et al. Effects of Sacubitril-Valsartan Versus Valsartan in Women Compared With Men With Heart Failure and Preserved Ejection Fraction: Insights From PARAGON-HF. Circulation. 2020;141:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Scott NJA, Rademaker MT, Charles CJ, Espiner EA, Richards AM. Hemodynamic, Hormonal, and Renal Actions of Phosphodiesterase-9 Inhibition in Experimental Heart Failure J Am Coll Cardiol. 2019;74:889–901. [DOI] [PubMed] [Google Scholar]
- 122.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fisher DA, Smith JF, Pillar JS, St Denis SH, Cheng JB. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J Biol Chem. 1998;273:15559–64. [DOI] [PubMed] [Google Scholar]
- 124.Calamera G, Li D, Ulsund AH, Kim JJ, Neely OC, Moltzau LR, Bjørnerem M, Paterson D, Kim C, Levy FO et al. FRET-based cyclic GMP biosensors measure low cGMP concentrations in cardiomyocytes and neurons. Commun Biol. 2019;2:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, Brozovich FV, Burnett JC Jr, Linke WA, Redfield MM. Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation. 2011;124:2882–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ying W, Zhao D, Ouyang P, Subramanya V, Vaidya D, Ndumele CE, Guallar E, Sharma K, Shah SJ, Kass DA et al. Associations Between the Cyclic Guanosine Monophosphate Pathway and Cardiovascular Risk Factors: MESA. J Am Heart Assoc. 2019;8:e013149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2021. doi: 10.1038/s41569-020-00480-6. Online ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]