

Summary
The brain’s endocannabinoid system is a powerful controller of neurotransmitter release, shaping synaptic communication under physiological and pathological conditions. However, our understanding of endocannabinoid signaling in vivo is limited by the inability to measure their changes at timescales commensurate with the high lability of lipid signals, leaving fundamental questions of if, how, and which endocannabinoids fluctuate with neural activity unresolved. Using novel imaging approaches in awake behaving mice, we now demonstrate that the endocannabinoid 2-arachidonoylglycerol, not anandamide, is dynamically coupled to hippocampal neural activity with high spatiotemporal specificity. Furthermore, we show that seizures amplify the physiological endocannabinoid increase by orders of magnitude and drive the downstream synthesis of vasoactive prostaglandins that culminate in a prolonged stroke-like event. These results shed new light on normal and pathological endocannabinoid signaling in vivo.
Keywords: Endocannabinoid, prostanoid, COX-2, cyclooxygenase-2, 2-AG, AEA, postictal hypoxia, seizure, prostaglandin, MAGL, FAAH, PGE2, EP1
Graphical Abstract
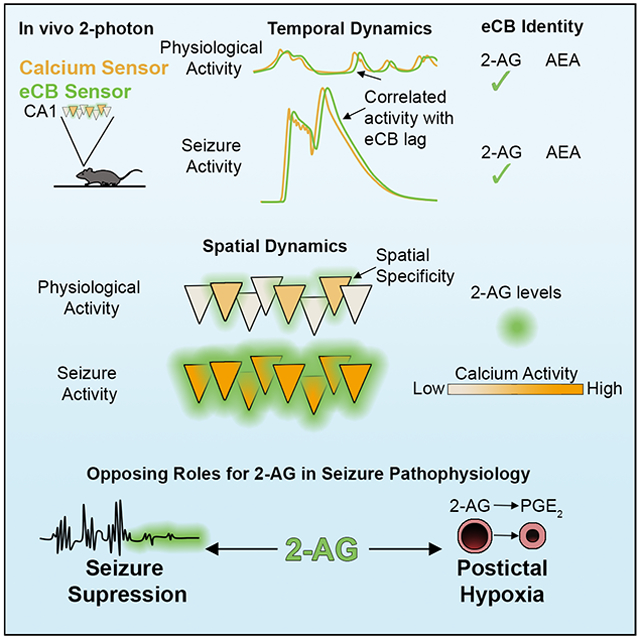
eTOC blurb
Farrell et al. resolve the molecular identity and spatiotemporal dynamics of hippocampal endocannabinoid signaling in vivo using a genetically encoded sensor. Compared to physiological activity, seizures result in excessive 2-AG levels, providing substrate for a prolonged stroke-like event.
Introduction
The cannabinoid type 1 receptor (CB1) is the most abundant G-protein coupled receptor in the brain and plays important roles in physiological and pathophysiological states, including epilepsy, mainly by reducing neurotransmitter release at pre-synaptic terminals (Katona and Freund, 2008; Kano et al., 2011; Soltesz et al., 2015). The main endogenous lipid ligands for CB1 (termed endocannabinoids: eCBs), anandamide (AEA) and 2-arachidonoyl glycerol (2-AG), are thought to be synthesized in an activity-dependent manner to provide negative feedback at the synapse (Stella et al., 1997; Di Marzo et al., 1994). However, the relative contributions of AEA and 2-AG are still debated (Di Marzo et al., 2011).
Until recently, our understanding of eCB dynamics in vivo has been limited by the inappropriately low spatiotemporal resolution of conventional biochemical approaches. While well-suited for measuring slow eCB changes, quantification of brain lipid levels post-mortem, the current gold standard, is unsuitable for resolving any rapid fluctuations (i.e. seconds to minutes) that would return to baseline during the time-consuming process of removing the brain and dissecting a sample. Thus, a fundamental understanding of how eCB levels fluctuate with neural activity is missing. We overcame this limitation by applying a recently developed genetically-encoded eCB sensor, GRAB-eCB2.0, which reports low micromolar eCB concentrations within seconds and is well-suited for in vivo investigation (Dong et al., 2020). We imaged this sensor in the hippocampus of awake mice (Figure 1A) to establish the contributions of AEA vs. 2-AG during physiological and pathological neural activity (i.e. seizures). This novel approach may also resolve the long-standing discrepancy between prior studies supporting crucial roles for eCBs in seizure control (Sugaya et al., 2016), but minimal eCB changes associated with seizure events (Wallace et al., 2003; Marsicano et al., 2003; Lerner et al., 2017).
Figure 1. Locomotion- and seizure-driven endocannabinoid increase depends on 2-AG, not AEA.

(A) Experimental set-up. 2-photon imaging of calcium and eCB signals from awake, head-fixed mice.
(B) Enzymes involved in eCB synthesis and hydrolysis and corresponding inhibitors (abbreviated) used in the study.
(C) eCB (green) and calcium (magenta) traces, averaged across cells, from a representative mouse during locomotion showing altered eCB signal with 2-AG perturbation.
(D) Representative traces during seizure activity showing a similar pharmacological sensitivity to that of eCB activity during locomotion C.
(E) Summary data for C reported as a percent change from vehicle. One-way ANOVAs were performed on the maximum ΔF/F for each session’s run-triggered average (see Figure S1B). n=12 for DO34, JZL184, and URB597 (eCB: F3,11=26.43, Dunnett post-test **p<0.01; calcium F3,11=3.18). n=6 for LEI401 (eCB: F4,5=8.61, calcium; F4,5=3.57, LEI non-significant for both).
(F) DO34 significantly prolonged electrographic seizure duration (n=12 group: Friedman test F3,11=9.65, Dunnett post-test *p<0.05; n=6 group: Friedman test F4,5=8.42, LEI non-significant).
(G) Summary data for D reported as a percent change in the area under curve for eCB (n=12 group: F3,11=156.0, Dunnett post-test ****p<0.0001, n=6 group: F4,5=49.73, LEI non-significant) and calcium (n=12 group: F3,11=2.00, n=6 group: F4,5=4.54).
See also Figure S1A.
Results
2-AG underlies activity-dependent eCB signaling in the hippocampus
Under physiological conditions, locomotion resulted in increased intracellular calcium activity in the CA1 pyramidal cell layer and rapid activation of the eCB sensor (Figure 1A,C; Figure S1A,B). Following cessation of locomotion, calcium and eCB activity returned to baseline within seconds (Figure 1C see VEH), which would not be captured by conventional methods and highlights the utility of GRAB-eCB2.0 for studying eCBs. Next, we assessed the coupling of neural activity and eCB production in the presence of specific inhibitors for the synthesis and breakdown of 2-AG and AEA to determine their relative roles as activity-dependent eCBs (Figure 1B). The locomotion-related eCB signal was suppressed by inhibiting 2-AG synthesis with a diacylglycerol lipase (DAGL) inhibitor (DO34, 50mg/kg i.p.) and enhanced by inhibiting 2-AG hydrolysis with a monoacylglycerol lipase (MAGL) inhibitor (JZL184, 16mg/kg, i.p.) (Figure 1B,C,E; Figure S1B). In contrast, it was unaffected by inhibiting AEA synthesis with a N-acylphosphatidylethanolamine phospholipase D (NAPE-PLD) inhibitor (LEI401, 30mg/kg i.p.) or inhibiting AEA hydrolysis with a fatty acid amide hydrolase (FAAH) inhibitor (URB597, 3mg/kg i.p.)(Figure 1B,C,E), supporting a specific role for 2-AG, not AEA, in activity-dependent eCB signaling.
Extracellular 2-AG is controlled in a spatiotemporally precise manner
We then determined the temporal offset and spatial specificity of eCB coupling to neural activity at the single cell level (Figure 2A). When averaged across cells and mice, the two signals had a strong, positive correlation (Figure S1C, see VEH in real time). This within-cell eCB vs. calcium correlation was selectively reduced by 2-AG, not AEA, synthesis and hydrolysis inhibition (Figure S1C). Cellular eCB traces were then shifted forwards and backwards in time to determine the amount of lag between eCB and calcium traces or vice versa (Figure 2B). The eCB signal had a lag of 1.55 +/− 0.14 seconds, which was significantly prolonged by the 2-AG hydrolysis inhibitor JZL184 (Figure 2C). After correcting for the time-offset, only 2-AG synthesis inhibition and not 2-AG hydrolysis inhibition reduced the correlation between cellular eCB and calcium signals (Figure S1C). These data support a crucial role for 2-AG synthesis in activity-dependent eCB signaling, whereas 2-AG hydrolysis selectively alters the kinetics of this coupling.
Figure 2. Spatiotemporal characteristics of activity-dependent 2-AG signaling.

(A) eCB and calcium traces were pulled from active neurons identified in the baseline period (no seizure). Inset shows neighboring cells with traces displayed in E.
(B) eCB traces from each cell were moved forward and backward in time to determine the optimal temporal offset (yielding maximum Pearson r value). Histogram shows an example from a vehicle locomotor session from one mouse with a mean lag of 1s across cells.
(C) The temporal offset, or eCB lag, was prolonged by JZL pre-treatment for locomotor sessions (F4,5=14.98, Dunnett post-test **p<0.01).
(D) Seizure traces similarly show a prolonged temporal offset by JZL (F4,5=26.13, Dunnett post-test ****p<0.0001)
(E) eCB traces from each cell were correlated to the calcium activity within its region of interest and to all other cells to determine spatial specificity (Pearson r values). Example traces from a vehicle session show stronger within cell coupling.
(F) Reduced correlation values were observed between cell relative to within for locomotor sessions (one-sample t-tests). No significant differences were found on the amount of reduction in correlation value across drug treatments (F4,5=1.67). Note the lower correlation values with DO34 (see Figure S1C).
(G) Reduced correlation values were observed between cell vs. within for seizure recordings (one-sample t-tests). No significant differences were found on the amount of reduction in correlation value across treatments (F4,5=0.39). Note the lower correlation values with JZL184 (see Figure S1D).
We also found that this coupling occurred in a spatially precise manner, since the eCB signal in a given cell was more correlated to its own calcium signal than that of other cells (Figure 2E,F; Figure S1A). The amount of reduction in correlation when moving from within-cell to between-cell was not significantly altered by the various drug treatments (Fig. 2F). These data suggest that the spatial precision of 2-AG signaling is retained even when its metabolism is inhibited, thereby prolonging its activity without significant spatial spillover.
Seizures amplify activity-dependent 2-AG synthesis
Relative to the locomotion-induced eCB signal change, electrically-induced seizures (1 second of kindling stimulation targeted to ventral hippocampus), on average, resulted in a 349 times greater signal increase (maximum dF/F of 572 +/− 34 % vs. 2.66 +/− 0.44 %, Figure 1Cvs.D; Figure S1A,B; Movie S1 vs. Movie S2). Based on controlled in vitro experiments, an additional 2 orders of magnitude increase in bath applied 2-AG to neuronal cultures would be needed to show similar changes in fluorescence to that observed during a seizure vs. locomotion (Dong et al., 2020). Moreover, this signal returned to baseline within seconds to minutes after seizure termination (Figure 1D; Figure S1A,B) and provides an explanation for why previous biochemical methods provided inconsistent assessments of the changes in the levels of eCBs with seizures. Like physiological activity, the seizure-induced eCB signal was also specifically sensitive to enzymatic manipulation of 2-AG, but not AEA (Figure 1G). Since electrographic seizure duration was prolonged by 2-AG synthesis inhibition (Figure 1F), these data provide further evidence for the role of 2-AG in seizure control (Katona and Freund, 2008; Sugaya et al., 2016).
At the single cell level, data obtained during seizures also supported a selective role for 2-AG, similar to the observations made during physiological locomotor activity. eCB traces during seizures had a time-offset of 1.12 +/− 0.20 seconds, which was significantly prolonged by 2-AG hydrolysis inhibition (Figure 2D). Inhibition of 2-AG hydrolysis also reduced the eCB vs. calcium correlation value in both real-time and adjusted traces (Figure S1D), which is expected due to the slower decay of the eCB signal under these conditions (Figure 1D; Figure S1B). The real time and shifted correlation values for 2-AG synthesis inhibition were not altered during seizures (Figure S1D). When comparing within cell and between cell correlations for eCB vs. calcium, seizures resulted in greater overall correlation and less spatial specificity than physiological conditions (Figure 2G vs. F), as expected during hyperactive and hypersynchronous activity.
This supraphysiological surge in 2-AG may indeed act as a seizure-disrupting circuit-breaker by suppressing glutamate release from excitatory fibers (Katona and Freund, 2008), but unusually high local levels of 2-AG may have other, less beneficial effects. Considering that 2-AG breakdown is coupled to prostaglandin synthesis (Nomura et al., 2011), we then assessed the potentially deleterious downstream pathophysiological effects of excessive substrate (2-AG) levels.
Pathological 2-AG production during seizures leads to elevated prostaglandins and a stroke-like event
The hydrolysis of 2-AG by MAGL produces arachidonic acid (AA), which is a substrate for the enzyme cyclooxygenase-2 (COX-2). COX-2, which is abundant in principal cells, oxygenates AA to produce vasoactive prostaglandins. This is particularly interesting in light of the recent finding that COX-2 is necessary for the induction of stroke-like hypoperfusion events that occur after seizures, both in animal models and persons with epilepsy (Farrell et al., 2016; Gaxiola-Valdez et al., 2017; Li et al., 2019). Based on the findings so far, we hypothesized that postictal hypoperfusion/hypoxia is initiated by excessive 2-AG during seizures leading to downstream engagement of a pathway involving COX-2 and prostaglandins.
To address this hypothesis, we determined how inhibitors of key endocannabinoid-prostaglandin targets modulate the expression of postictal hypoxia using chronically implanted oxygen-sensing probes in freely-behaving mice (Figure 3A). Without intervention, vehicle-treated mice displayed the previously reported robust postictal decreases in brain oxygen levels from the normoxic range of 20-30 mmHg to severely hypoxic levels (<10 mmHg) lasting for tens of minutes (Figure 3B) (Farrell et al., 2016). The 2-AG synthesis inhibitor, DO34 (50mg/kg i.p.), prevented the induction of severe postictal hypoxia (Figure 3B,C), demonstrating the requirement of 2-AG in this signaling cascade.
Figure 3. The endocannabinoid-prostaglandin pathway leads to severe postictal hypoxia.

(A) Experimental set-up (left) and molecular pathway with associated pharmacological inhibitors (right).
(B) Changes in oxygen profiles following drug pre-treatment. X and Y scales apply to all four graphs.
(C) All four drugs significantly reduced the area below 10mmHg (One sample t-test, DO34 – t4=7.51; JZL184 – t4=5.75; celecoxib – t4=22.73; ONO8130 – t4=8.03). **p<0.01, ****p<0.0001.
We next determined if MAGL is involved in postictal hypoxia, since MAGL is thought to hydrolyze the majority of brain 2-AG, thereby liberating AA for prostaglandin production (Nomura et al., 2011). Indeed, pre-treatment with JZL184 reduced the severity of postictal hypoxia (Figure 3B,C), further supporting a key role for 2-AG. These data are paralleled by the reduced severity of postictal hypoxia with the COX-2 inhibitor, celecoxib (50mg/kg i.p.) (Figure 3B,C). Together, these data highlight crucial roles for activity-dependent 2-AG synthesis, subsequent hydrolysis to AA, and oxygenation by COX-2 in severe postictal hypoxia.
We also found evidence for endocannabinoid-prostaglandin coupling when comparing changes in 2-AG and prostaglandin levels following single or four consecutive seizures (Figure S2A). We hypothesized that repeated seizure activity would diminish the levels of substrates in this pathway, resulting in prolonged seizures and reduced postictal hypoxia. Using 2-photon imaging in mice, we observed reduced eCB sensor activation and longer seizures after a fourth seizure in vivo (Figure S2B). Post-mortem levels of 2-AG, AA, and PGE2 were measured by mass spectrometry from rats immediately after a kindled seizure and showed a blunted response after a fourth seizure at each step of the endocannabinoid-prostaglandin pathway (Figure S2C-E). To determine if dampening of this molecular pathway is associated with a reduction in postictal hypoxia, we performed the same four-seizure protocol in rats with a chronically implanted oxygen sensor. If each seizure and the associated postictal hypoxia was independent of each other, their effects should summate and yield more severe hypoxia after a fourth seizure. Instead, the fourth seizure was associated with less severe postictal hypoxia (Figure S2F), consistent with reduced eCB-prostaglandin signaling. These results further support that activity-dependent 2-AG synthesis plays a role in restricting seizure activity but also provides substrate for downstream COX-2 signaling that drives severe postictal hypoxia.
Lastly, we investigated potential mechanisms downstream of COX-2, since little is known about how the oxygenation products of AA ultimately lead to postictal hypoperfusion/hypoxia. We previously observed that inhibition of PGE2 synthase ameliorated postictal hypoxia (Farrell et al., 2016). Here we observed that PGE2 was the most elevated prostanoid in the hippocampus of rats, measured immediately after a seizure (Figure S3). While PGE2 typically leads to vasodilation under physiological conditions via EP2 and EP4 receptors (Lacroix et al., 2015), lower affinity receptors, such as EP1 receptors, may only be activated by supra-physiological PGE2 concentrations, as observed here (1741 +/− 257% increase relative to baseline). Since parenchymal arterioles only constrict, rather than dilate, upon PGE2 exposure in an EP1 receptor-dependent manner (Dabertrand et al., 2013), this receptor is a prime candidate to mediate hypoperfusion/hypoxia after seizures. Indeed, pre-treatment of mice with a selective EP1 receptor antagonist (ONO8130, 25mgkg i.p.) inhibited postictal hypoxia (Figure 3B,C).
Discussion
These data provide evidence that 2-AG serves as the dominant activity-dependent endocannabinoid in the hippocampus and is coupled to physiological neural activity in a precise spatiotemporal manner. Moreover, 2-AG hydrolysis specifically affects the temporal dynamics of eCB signaling without resulting in spatial spillover. Seizures, however, result in a loss of eCB spatial specificity as neural activity becomes hyperactive and hypersynchronous.
GRAB-eCB2.0 has the potential to rapidly advance our knowledge of eCB dynamics across a range physiological and pathological settings. However, as this is a novel tool, possible limitations should be considered. For example, especially in the context of pathological changes during seizures (such as pH shifts), one should consider the possibility that some changes in sensor fluorescence might be unrelated to eCBs. It is noteworthy in this regard, therefore, that the scrambled control sensor did not report fluorescence changes during seizures (Dong et al., 2020), and the consistent negative or positive modulation of the sensor fluorescence with specific blockers of 2-AG synthesis or breakdown also argues against significant seizure-induced non-specific confounds (e.g., note that the 2-AG synthesis inhibitor DO34 strongly suppressed the sensor response in spite of the fact that it made the seizures worse). On the other hand, excessive sensor expression levels should be avoided in order to minimize the potential for eCB buffering effects that may restrict endogenous CB1 signaling.
For seizure pathophysiology, we found that 2-AG has two opposing roles. Mounting evidence supports that the eCB system is a prime target for seizure control (Katona and Freund, 2008; Soltesz et al., 2015) and these data provide clear in vivo evidence that 2-AG is produced in an activity-dependent manner and is necessary to suppress seizures. However, this previously undetected surge of 2-AG during seizures comes at the cost of producing extremely high levels of PGE2 that leads to prolonged hypoperfusion/hypoxia. Given that postictal stroke-like events result in long-lasting behavioral impairment (Farrell et al., 2016; Farrell et al., 2020), the finding that excessive 2-AG feeds this vasoconstriction pathway supports two opposing roles (anti-seizure vs. pro-hypoxia) that are fundamental to our understanding and treatment of epilepsy.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jordan S. Farrell (jsfarrel@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data are available upon reasonable request.
Experimental Model and Subject Details
Electrical Kindling Seizure Model in Mice
Experiments using mice were approved and performed in accordance with relevant guidelines and regulations by the Health Sciences Animal Care Committee at the University of Calgary (AC16-0272) and the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford University, respectively. Mice were group-housed in standard cages with unrestricted access to standard chow and water and all experiments were performed during the light cycle. 13 C57BL/6J (P90-120) male mice (Jackson Laboratories, Strain#000664) were used for In vivo 2-photon imaging. 12 C57BL/6J (P50-100) male mice (Jackson Laboratories, Strain#000664) were used in the pharmacological study in In vivo local tissue oxygen-sensing (note that some mice were used across more than one drug treatment with a >24 hour washout period). Procedures for electrical kindling are described below.
Electrical Kindling Seizure Model in Rats
Experiments using rats were approved and performed in accordance with relevant guidelines and regulations by the Health Sciences Animal Care Committee at the University of Calgary (AC16-0272). Rats were group-housed in standard cages with unrestricted access to standard chow and water and single-housed after surgery. All experiments were performed during the light cycle. 80 male Long-Evans hooded rats (weight 250g-350g, age 8-14 weeks, Charles River) were used for Liquid Chromatography/Mass Spectrometry. 5 male Long-Evans hooded rats (weight 250g-350g, age 8-14 weeks, Charles River) were used for the 4-seizure protocol in In vivo local tissue oxygen-sensing. Procedures for electrical kindling are described below.
Method Details
Seizure Model
Teflon-coated twisted bipolar electrodes (tip separation of 0.5mm) were chronically implanted to the ventral hippocampus of rats (4.5mm posterior, 4.5mm lateral, 7mm ventral to bregma) and mice (3.2mm posterior, 2.7mm lateral, 4.0mm ventral to bregma) as previously described (Farrell et al., 2016). Following recovery from surgery (1-2 weeks), seizures were elicited by kindling stimulation above seizure threshold (300-500μA of current delivered in 1ms biphasic pulses at 60Hz for 1 second).
In vivo 2-photon Imaging
400nL viral mixture of equal parts GRAB-eCB2.0 (prepared here) and jRGECO1a (both under control of a pan-neuronal promoter) were injected into the right CA1 (2.2mm posterior, 1.5mm lateral, 1.3 ventral to bregma) using a 25g Hamilton syringe. pAAV.Syn.NES-jRGECO1a.WPRE.SV40 was a gift from Douglas Kim & GENIE Project (Addgene viral prep 100854-AAV1; http://n2t.net/addgene:100854 ; RRID:Addgene_100854) (Dana et al., 2016). pAAV.hSyn.GRAB-eCB2.0.WPRE.hGH-polyA was constructed and AAV2/9.hSyn.GRAB-eCB2.0 was packaged at Vigene Biosciences, China with a titer of 9.5 x 10^13 viral genomes per mL. The overlying cortex was then aspirated and replaced with a 3mm diameter imaging cannula as previously described (Kaifosh et al., 2013). The kindling electrode was implanted in the contralateral ventral hippocampus and implants were secured with a thin layer of Metabond (Parkell) followed by dental cement (Lang) and a stainless steel headbar. Following recovery from surgery, mice underwent 1-2 habituation sessions to acclimate to head-fixation and to confirm good expression of the sensor and proper placement of the kindling electrode (presence of after-discharges consistent with ventral hippocampal kindling). The first experiment performed was the four-seizure protocol (Figure S2A). Mice then received i.p. injections, as described below, 45 minutes prior to seizure with at least 24 hours in between imaging sessions to ensure clearance of the previous days drug (mean interval = 3.78 +/− 0.71 days, see Drug Injections). Imaging sessions consisted of a 5 to 7-minute baseline period where locomotion on a 1-meter belt was tracked with a rotary encoder. Seizures were then initiated and monitored electrographically (amplified 1000X and digitized at 10kHz) and behaviorally (behavioral camera focused on the face and forelimbs) and imaging continued for 5 additional minutes. Statistical tests for changes in seizure duration were performed on raw electrographic seizure duration values using a non-parametric Friedman’s test, since the DO34 data was not gaussian distributed. 13 mice were used for data in Figure 1F,E and 12 mice were used for Figure 1D (one mouse was excluded because the calcium and eCB signals were too weak for reliable detection under physiological conditions).
Imaging data was collected on a Neurolabware 2-photon microscope (Neurolabware.com) controlled by Scanbox (scanbox.org), a GUI that runs in Matlab (mathworks.com). Data was collected at 15 fps with the laser tuned to 1000nm to simultaneously sample both sensors with low crosstalk between green and red channels. In a subset of data (n=6), we optimized the imaging parameters for single cell imaging, which involved separate laser power and photomultiplier tube gain settings for physiological vs. seizure recordings. Motion correction, ROI segmentation, and signal extraction was performed offline in python (python.org) using the SIMA package (Kaifosh et al., 2014) for broad pyramidal cell layer signal extraction or Suite2p (Pachitariu et al., 2017) for the data subset where single-cell analysis was performed. For analysis using SIMA, sensor signals were pulled from the pyramidal cell layer of CA1 by extracting the signal from several large polygons drawn around groups of approximately 40-80 cells and averaged across all ROIs. For suite2p, segmentation was based on calcium activity during the 5-7 minute baseline period to identify active neurons. Inactive neurons were excluded from analysis to avoid spurious eCB and calcium correlations due to a lack of signal change. Sensor signal was pulled from each non-overlapping ROI for baseline and seizure recordings and either analyzed separately or reported as the mean signal across cells where appropriate. For running-driven signal changes, the change in fluorescence (ΔF/F) across the pyramidal cell layer (averaged across individual cells for suite2p segmentation) was calculated using a baseline from 4 seconds to 1 second before the onset of running. Peak calcium and eCB signal change (maximum) were quantified for each session from gaussian-smoothed traces (sigma=0.5s) of the run-onset triggered average. Seizure-driven calcium and eCB signal changes were calculated using a baseline of 850 samples pre-seizure to derive ΔF/F and quantified as the total area under the curve from seizure onset to 1.5 minutes post-onset. We noted that there may have been a certain amount of fluorescence channel cross-talk during seizure recordings with JZL pre-treatment, where the bright and prolonged eCB fluorescence may have been detected by the red calcium channel (see Figure S1B top right). As apparent in the JZL data shown in Figure 1G, this may have occurred in a minority of subjects (likely due to slight differences in relative expression of each sensor). Note that the differences in temporal kinetics of each sensor suggests that crosstalk does not occur under physiologic conditions. For both locomotion- and seizure-driven changes, we performed within-subject ANOVAs and Dunnett’s test to correct for multiple comparisons between vehicle and drug conditions. Since only 6 of the animals were treated with LEI401, a separate ANOVA with Dunnett’s test for multiple comparisons was used on this subset. For display purposes, data are shown as a percent change from vehicle, but all statistical tests were performed on non-normalized data. For the 4-seizure experiment, we calculated a ratio of eCB AUC to calcium AUC to determine how much eCB is produced for a given seizure and performed a one-sample t-test on the difference between a 4th vs. 1st seizure.
For single cell eCB and calcium analysis, we first analyzed optimized recordings from mice treated with vehicle (n=6). Cellular ΔF/F across the entire recording was normalized to a 3rd order polynomial or the 10th percentile of the signal for baseline and seizure traces, respectively. To determine the general time-scale of eCB changes that reflect neural activity, we smoothed eCB and calcium traces from each cell with 1-dimensional gaussian filters ranging in widths to generate a plot of filter width vs. correlation value. We chose the filter width that achieved 80% of the maximal correlation value (17 frames or 1.1s), which captured most of the correlation between both signals without sacrificing temporal resolution. Optimally smoothed eCB traces from baseline and seizure recordings were shifted forwards and backwards relative to calcium traces for each cell to determine the time-shift parameter that yields maximal correlation. We reported the time-offset for each recording session, averaged across all cells in the session, and analyzed these data with a within-subject ANOVA and Dunnett’s post-test. We used the same statistical tests to determine if the average eCB vs. calcium correlation values were altered for real-time and optimally time-shifted traces. We also correlated a given cell’s eCB signal that of its own calcium signal or the calcium signal of all other cells. We reported the within and between cell comparison averaged across all cells for each session and performed a within subject ANOVA and Dunnett’s post-test on the difference between these two values. To determine if this with vs. between difference was significant, we performed one-sample t-tests on each treatment.
Liquid Chromatography/Mass Spectrometry
Rats received single, three, four, or no kindled seizure(s) (seizures spaced 30-minutes apart) according to the group they were assigned to. Rats were used in this experiment instead of mice, since we found more robust lipid level changes in preliminary experiments, perhaps relating to longer seizure durations in rats yielding higher levels of activity-dependent lipids. Rats were sacrificed at the time-point listed by decapitation without sedation/anesthesia. Brains were rapidly removed, hippocampi extracted, frozen on dry ice, and stored at −80C until analysis. The hippocampi contralateral to implantation (to avoid potential inflammation associated with implantation) were homogenized in methanol and lipid analysis was carried out on a SCIEX 6500 QTrap system as previously described (Shonesy et al., 2014).
In vivo local tissue oxygen-sensing
In addition to the implantation a kindling electrode, oxygen-sensing probes were implanted in the dorsal hippocampus (1.6mm posterior, 2.0mm lateral, 1.8mm ventral to bregma in mice; 3.5mm posterior, 3.5mm lateral to bregma and 3.5mm ventral to skull surface in rats). Following 1 week recovery, mice and rats were kindled approximately daily for 5-10 session to ensure that seizures were consistently elicited with postictal hypoxia, since we previously found that the first sessions are accompanied by shorter seizures and less severe hypoxia (Farrell et al., 2016). Once stable across consecutive days, oxygen recordings were obtained from freely-moving rats and mice sampled at 1Hz using the Oxylite system (Oxford Optronix), with a 100 second baseline period and 90-minutes of postictal recording. This oxygen-sensing system is based on the platinum fluorescence lifetime-quenching properties of oxygen and has several advantages over other methodologies (see Ortiz-Prado et al. 2010 for full validation), including measurement of absolute oxygen values, lack of oxygen consumption in the measurement process, ability to use chronically, and compatibility with freely-behaving recordings without movement artifact. Severity of hypoxia was quantified as the area below 10mmHg to take into account the depth and duration of hypoxia below this critical threshold (Farrell et a., 2016). Paired t-tests were used to determine statistical significance.
Drug Injections
Celecoxib, JZL184, ONO8130, and URB597 were obtained from Cayman Chemicals (Ann Arbor, MI), LEI401 was obtained from MedChemExpress (Monmouth Junction, NJ), and DO34 was obtained from Aobious (Gloucester, MA). Drugs were dissolved in dimethyl sulfoxide (DMSO) and stored at −20. A stock solution of each drug was further diluted to create a vehicle composing of 1:1:18 of DMSO:Tween80:saline and injected at a volume of 3mL/kg (intraperitoneal or i.p.) at 45 minutes pre-seizure. The minimum washout period for this study was 24 hours with a mean washout period of 3.78 +/− 0.71 days between previous drug treatments, to avoid potential lingering effects of previous drugs. This mean does not include the first drug exposure, as it is not associated with previous drug administration. The order that mice received drug treatments was varied to balance for the effect of repeated kindling and time.
Drugs that inhibit eCB synthesis or hydrolysis are often given up to several hours before lipid measurements are taken to allow enough time for substantial changes in lipid levels to accumulate (Fegley et al., 2005; Ogasawara et al., 2016; Nomura et al., 2011). In this study, our goal was to inhibit on-demand eCB synthesis/hydrolysis, not to measure slow changes that integrate over hours, and therefore chose a time-point when drug levels typically reach their maximum brain concentration. Since we required drug levels to be high in the brain during the baseline period (recording initiated at 10 minutes pre-seizure) and seizure, we chose an injection time-point of 45-minutes pre-seizure. While detailed pharmacokinetic data are not available for all drugs used in the study, this time-point is consistent with available data (Mock et al., 2020). Mock et al., (2020) reported brain drugs levels of LEI401, which reached a maximum between 30 minutes and 1 hour post-injection (intraperitoneal). Thus, the lack of drug effect in our study with LEI401 is not explained by inappropriate injection timing. Furthermore, the robust effects with JZL184 and DO34 are indicative of sufficient drug levels in the brain at the time of experimentation.
Quantification and Statistical Analysis
Statistical methods are described in the above Method Details section for each experiment and significance levels are reported in figure legends. All statistical tests were performed using Python 3 or Graphpad Prism 5.
Supplementary Material
Movie S1. eCB and calcium dynamics during locomotion. Related to Figure 1.
Raw fluorescence movies (composite and split) without motion correction showing fluorescence of each sensor. Fluorescence movies and mean eCB (green) and calcium (magenta) traces were down-sampled by a factor of 5 using the average of those 5 frames. Speed, behavioral camera, and LFP are also displayed. Image scale bar = 50μm. LFP scale bar = 1mV. Elapsed time in seconds is also displayed.
Movie S2. eCB and calcium dynamics during a seizure. Related to Figure 1.
Raw fluorescence movies (composite and split) without motion correction showing fluorescence of each sensor. Fluorescence movies and mean eCB (green) and calcium (magenta) traces were down-sampled by a factor of 5 using the average of those 5 frames. LFP is also displayed to show seizure activity. Image scale bar = 50μm. LFP scale bar = 1mV. Elapsed time in seconds is also displayed.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| pAAV.Syn.NES-jRGECO1a.WPRE. | Addgene.org | 100854-AAV1, RRID:Addgene_100854 |
| AAV2/9.hSyn.GRAB-eCB2.0 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Celecoxib | Cayman Chemicals | Cat#10008672 |
| JZL184 | Cayman Chemicals | Cat#13158 |
| ONO8130 | Cayman Chemicals | Cat#19118 |
| URB597 | Cayman Chemicals | Cat#10046 |
| LEI401 | MedChemExpress | Cat#HY-131181 |
| DO34 | Aobious | Cat#AOB8060 |
| Experimental models: organisms/strains | ||
| C57BL/6J mice | Jackson Laboratories | Strain#000664 |
| Long-Evans hooded rats | Charles River | Strain Code#006 |
| Software and algorithms | ||
| SIMA | https://pypi.org/project/sima/ | Kaifosh et al. 2014 |
| Suite2p | https://github.com/MouseLand/suite2p | Pachitariu et al., 2017 |
| Rastermap | https://github.com/MouseLand/rastermap | N/A |
| Python 3.6 & 3.7 | https://www.python.org/ | N/A |
Highlights.
2-AG, not AEA, is the main activity-dependent endocannabinoid in the CA1
In vivo 2-AG levels are under precise spatiotemporal regulation
Seizures amplify 2-AG production, which restricts seizure activity
Seizure-driven 2-AG synthesis, however, fuels a prolonged stroke-like event
Acknowledgements
The authors thank Bonita Gunning, Sylwia Felong, and Anna Ortiz for their technical support. K.A.S. holds the Crohn’s and Colitis Canada Chair in IBD Research at the University of Calgary. J.S.F. was supported by a Canadian Institutes for Health Research (CIHR) postdoctoral fellowship. R.C. was supported by an Eyes High postdoctoral fellowship funded by the University of Calgary. A.G.G. holds a graduate studentship from SUDEP Aware. K.A-O. was funded by a University of Calgary graduate scholarship. M.M. was funded by a CIHR postdoctoral fellowship and an Alberta Innovates postdoctoral fellowship. M.D.W. was funded by an Alberta Innovates graduate studentship. B.D. was funded by an American Epilepsy Society Postdoctoral Fellowship and is funded by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH) (#K99NS117795). The work in S.P., L.J.M. and I.S. labs was funded by NIH grants (#MH107435, #1S10OD017997-01A1 and #NS99457, respectively). Y.L. lab is funded by Beijing Municipal Science & Technology Commission (Z181100001318002, Z181100001518004), National Natural Science Foundation of China (31925017), NIH BRAIN Initiative (NS103558) and the State Key Laboratory of Membrane Biology at Peking University School of Life Sciences. G.C.T., M.N.H. and K.A.S. labs are funded by CIHR grants (# PJT-162204, # FDN 143329, #FDN-148380, respectively). M.N.H. holds a Tier 2 Canada Research Chair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Dabertrand F, Hannah RM, Pearson JM, Hill-Eubanks DC, Brayden JE and Nelson MT (2013). Prostaglandin E2, a postulated astrocyte-derived neurovascular coupling agent, constricts rather than dilates parenchymal arterioles. J. Cereb. Blood Flow Metab 33, 479–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D et al. (2016). Sensitive red protein calcium indicators for imaging neural activity. Elife 5, e12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V (2011) Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nat. Neurosci 14, 9–15. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC and Piomelli D (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372, 686–691. [DOI] [PubMed] [Google Scholar]
- Dong A, He K, Dudok B, Farrell JS, Guan W, Liput DJ, Puhl HL, Cai R, Duan J, Albarran E, et al. (2020). A fluorescent sensor for spatiotemporally resolved endocannabinoid dynamics in vitro and in vivo. bioRxiv, 10.1101/2020.10.08.329169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JS, Colangeli R, Dudok B, Wolff MD, Nguyen SL, Jackson J, Dickson CT, Soltesz I and Teskey GC (2020). In vivo assessment of mechanisms underlying the neurovascular basis of postictal amnesia. Sci. Rep 10, 14992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JS, Gaxiola-Valdez I, Wolff MD, David LS, Dika HI, Geeraert BL, Wang XR, Singh S, Spanswick SC, Dunn JF et al. (2016). Postictal behavioural impairments are due to a severe prolonged hypoperfusion/hypoxia event that is COX-2 dependent. Elife 5, e19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, and Piomelli D (2005). Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther 313, 352–358. [DOI] [PubMed] [Google Scholar]
- Gaxiola-Valdez I, Singh S, Perera T, Sandy S, Li E and Federico P (2017). Seizure onset zone localization using postictal hypoperfusion detected by arterial spin labelling MRI. Brain 140, 2895–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaifosh P, Lovett-Barron M, Turi GF, Reardon TR, and Losonczy A (2013) Septohippocampal GABAergic signaling across multiple modalities in awake mice. Nat. Neurosci 16, 1182–1184 (2013). [DOI] [PubMed] [Google Scholar]
- Kaifosh P, Zaremba JD, Danielson NB, and Losonczy A (2014) SIMA: Python software for analysis of dynamic fluorescence imaging data. Front. Neuroinform 8, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, and Watanabe M (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev 89, 309–380. [DOI] [PubMed] [Google Scholar]
- Katona I and Freund TF (2008). Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med 14, 923–930. [DOI] [PubMed] [Google Scholar]
- Lacroix A, Toussay X, Anenberg E, Lecrux C, Ferreirós N, Karagiannis A, Plaisier F, Chausson P, Jarlier F, Burgess SA et al. (2015). COX-2-derived prostaglandin E2 produced by pyramidal neurons contributes to neurovascular coupling in the rodent cerebral cortex. J. Neurosci 35, 11791–11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner R, Post J, Loch S, Lutz B, and Bindila L (2017). Targeting brain and peripheral plasticity of the lipidome in acute kainic acid-induced epileptic seizures in mice via quantitative mass spectrometry. Mol. Cell Biol. Lipids 1862, 255–267. [DOI] [PubMed] [Google Scholar]
- Li E, d’Esterre CD, Gaxiola-Valdez I, Lee TY, Menon B, Peedicail JS, Jetté N, Josephson CB, Wiebe S, Teskey GC et al. (2019). CT perfusion measurement of postictal hypoperfusion: localization of the seizure onset zone and patterns of spread. Neuroradiology 61, 991–1010. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, Van der Stelt M et al. (2003). CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88. [DOI] [PubMed] [Google Scholar]
- Mock ED, Mustafa M, Gunduz-Cinar O, Cinar R, Petrie GN, Kantae V, Di X, Ogasawara D, Varga ZV, Paloczi J et al. (2020). Discovery of a NAPE-PLD inhibitor that modulates emotional behavior in mice. Nat. Chem. Biol 16, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG, Ward AM, Hahn YK, Lichtman AH, Conti B and Cravatt BF (2011). Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334, 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara D, Deng H, Viader A, Baggelaar MP, Breman A, den Dulk H, van den Nieuwendijk AM, Soethoudt M, van der Wel T, Zhou J et al. (2016). Rapid and profound rewiring of brain lipid signaling networks by acute diacylglycerol lipase inhibition. Proc. Natl. Acad. Sci. U.S.A 113, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Prado E, Natah S, Srinivasan S and Dunn JF (2010). A method for measuring brain partial pressure of oxygen in unanesthetized unrestrained subjects: the effect of acute and chronic hypoxia on brain tissue PO2. J. Neurosci. Meth 193, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachitariu M, Stringer C, Dipoppa M, Schröder S, Rossi LF, Dalgleish H, Carandini M and Harris KD (2017). Suite2p: beyond 10,000 neurons with standard two-photon microscopy. BioRxiv 10.1101/061507. [DOI] [Google Scholar]
- Shonesy BC, Bluett RJ, Ramikie TS, Báldi R, Hermanson DJ, Kingsley PJ, Marnett LJ, Winder DG, Colbran RJ and Patel S (2014). Genetic disruption of 2-arachidonoylglycerol synthesis reveals a key role for endocannabinoid signaling in anxiety modulation. Cell Rep. 9, 1644–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltesz I, Alger BE, Kano M, Lee SH, Lovinger DM, Ohno-Shosaku T and Watanabe M, (2015). Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat. Rev. Neurosci 16, 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, and Piomelli D (1997). A second endogenous cannabinoid that modulates long-term potentiation. Nature 388, 773–778. [DOI] [PubMed] [Google Scholar]
- Sugaya Y, Yamazaki M, Uchigashima M, Kobayashi K, Watanabe M, Sakimura K and Kano M (2016). Crucial roles of the endocannabinoid 2-arachidonoylglycerol in the suppression of epileptic seizures. Cell Rep. 16, 1405–1415. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ (2003). The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J. Pharmacol. Exp. Ther 307, 129–137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. eCB and calcium dynamics during locomotion. Related to Figure 1.
Raw fluorescence movies (composite and split) without motion correction showing fluorescence of each sensor. Fluorescence movies and mean eCB (green) and calcium (magenta) traces were down-sampled by a factor of 5 using the average of those 5 frames. Speed, behavioral camera, and LFP are also displayed. Image scale bar = 50μm. LFP scale bar = 1mV. Elapsed time in seconds is also displayed.
Movie S2. eCB and calcium dynamics during a seizure. Related to Figure 1.
Raw fluorescence movies (composite and split) without motion correction showing fluorescence of each sensor. Fluorescence movies and mean eCB (green) and calcium (magenta) traces were down-sampled by a factor of 5 using the average of those 5 frames. LFP is also displayed to show seizure activity. Image scale bar = 50μm. LFP scale bar = 1mV. Elapsed time in seconds is also displayed.
Data Availability Statement
Data are available upon reasonable request.