

Abstract
Chromatin Interaction Analysis Using Paired‐End Tag Sequencing (ChIA-PET) is an established method to map protein-mediated chromatin interactions. A limitation, however, is that it requires a hundred million cells per experiment, which hampers its broad application in biomedical research, particularly in studies in which it is intractable to obtain a large number of cells from rare samples. To reduce the required input cell number while retaining high data quality, we developed an in situ ChIA-PET protocol, which requires as few as 1 million cells. Here, we describe the detailed step-by-step procedures for performing in situ ChIA-PET from cultured cells, including both experimental protocols for sample preparation and data generation, and a computational protocol for data processing and visualization using the ChIA-PIPE pipeline. As the protocol significantly simplifies the experimental procedure, reduces the ligation noise, and decreases the required input cells compared to previous versions of ChIA-PET protocols, it can be applied to generate high-resolution chromatin contact maps mediated by various protein factors for a wide range of human and mouse primary cells.
Keywords: ChIA-PET, in-situ ChIA-PET, chromatin interaction
INTRODUCTION
Mammalian genomes are spatially organized into compartments, contact domains, and chromatin loops, and the three-dimensional (3D) chromosomal organization in the nucleus provides the structural framework for many fundamental biological processes such as transcription (Tang et al., 2015), DNA replication (Sima et al., 2019), development (Bonev et al., 2017), and cell fate (Dixon et al., 2015). Our understanding of the 3D spatial organization of chromatin relies mostly on high-throughput chromosome conformation capture assays such as Hi-C (Lieberman-Aiden et al., 2009), for genome-wide chromatin contacts, and ChIA-PET (Fullwood et al., 2009), for specific chromatin interactions mediated by protein factors. The original ChIA-PET protocol extracts only 20 bp tags from the interacting chromatin loci, for which tag mapping efficiency is limited. Because of this, an improved version of ChIA-PET that extracts longer reads (up to 150 bp) from interacting fragments, called “long-read ChIA-PET”, was developed, which increases the tag mapping efficiency and enables the identification of haplotype-specific chromatin interactions (Tang et al., 2015; Li et al., 2017).
To further uncover the important roles of 3D chromatin structure in different mammalian processes and disease, it is necessary to map chromatin interactions in vivo and identify functional elements, for instance, in primary cells and/or clinical samples, often from a limitednumbers of cells. Long-read ChIA-PET, however, cannot readily fulfill these needs because it requires 100 million cells per experiment, and it is challenging to obtain that number of cells from limited tissue material (for primary cell cultures) or clinical samples. Inspired by the in situ Hi-C protocol (Rao et al., 2014), we adopted in situ digestion and ligation steps into the ChIA-PET procedure, which significantly improvs the efficiency of proximity ligation for genuine chromatin interactions and greatly reduces random ligation noise. This modification not only significantly reduces the required cell number to as few as 1 million, but it also generates high-quality chromatin interaction data, comparable to that produced by the long-read ChIA-PET protocol using 100 million cells.
The in situ ChIA-PET protocol, described here, has been established as a standard data production protocol in the 4D Nucleome and ENCODE consortia, and has been successfully applied to generate hundreds of CTCF and RNAPII datasets from human cell lines and primary cells, as part of ENCODE phase 4 projects (https://www.encodeproject.org/). Here, we describe detailed step-by-step procedures for performing in situ ChIA-PET from cultured cells, including both a experimental protocol (Basic Protocol 1) for sample preparation and data generation, and a computational protocol (Basic Protocol 2) for data processing and visualization using the ChIA-PIPE pipeline (Lee et al., 2020). In Basic Protocol 1, we describe the detailed procedures of cell crosslinking (for both suspension and adherent cells) and in situ ChIA-PET library construction, followed by high-throughput sequencing. We use bridge linkers to connect two interacting chromatin fragments during proximity ligation. The preparation procedure for bridge linkers is described in Support Protocol 1. In Basic Protocol 2, we describe the necessary scripts and tools for data analysis, quality evaluation, and data visualization. A general outline of the complete ChIA-PET workflow is shown in Figure 1.
Figure 1:
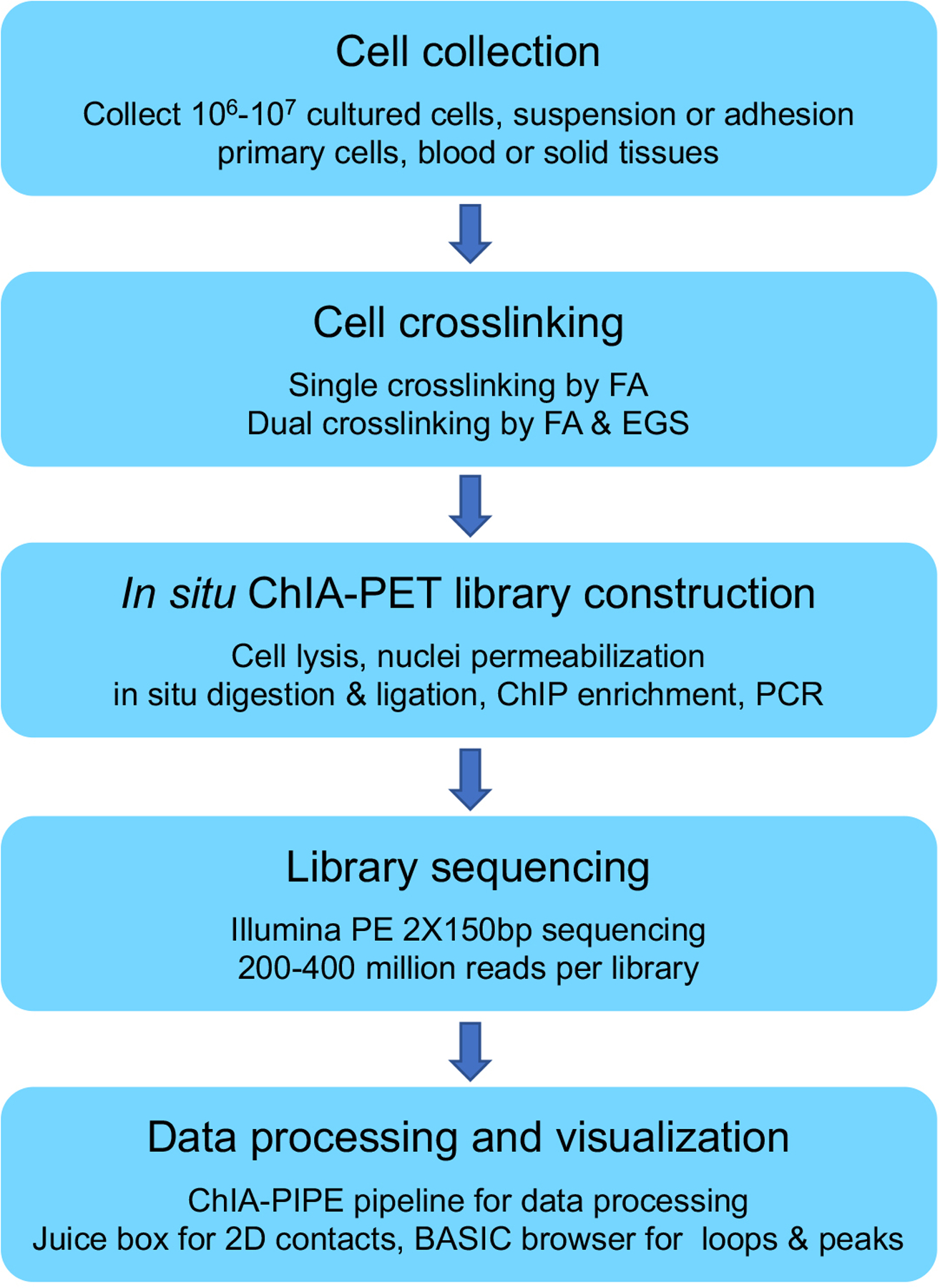
Outline of the in situ ChIA-PET workflow.
STRATEGIC PLANNING
When planning a research project that involves in situ ChIA-PET, there are several elements to be considered before starting, including the cells to be studied, sonication conditions for chromatin sample preparation, design and assembly of the bridge linker used in proximity ligation (Support Protocol 1), the protein factors of interest and the availability of antibodies against it, and capacity for high-throughput DNA sequencing and high-volume data analysis.
First, the current version of the in situ ChIA-PET protocol requires 106-107 cells as starting material. Most cell line cultures (suspension or adhesion) and some primary tissues (e.g. blood for immune cells, liver for hepatocytes, etc.) can provide sufficient numbers of cells for this. However, cell-type specific optimization for chromatin sample preparation may be required, for example, in the case of suspension vs. adhesion cells (see details in specific protocols below). The chromatin contact conformations within the nuclei of cells are often preserved by formaldehyde (FA) treatment, forming covalent bonds between DNA and associated proteins. To further capture chromatin associated protein-protein interactions, ethylene glycol bis (succinimidyl succinate) (EGS) has been used in the ChIA-PET protocol.
Second, the optimal sonication parameters for each individual cell type should be empirically determined. This step is critical to obtaining suitable chromatin fragments to maximize antibody capture efficiency during the chromatin immunoprecipitation (ChIP) step. Different sociation parameters, such as shearing time and power setting, should be optimized to get the ideal chromatin fragments (around 2–3 kb for this experiment) for each cell line.
Third, the most important consideration in a ChIA-PET experiment is the target protein factor to be studied and the availability of a robust antibody for it. Many proteins, such as CTCF, RNA polymerase II (RNAPII), cohesin, and transcription factors (Tang et al., 2015; Weintraub et al., 2017; Wang et al., 2020; Ji et al., 2016) can be studied by ChIA-PET for their roles in mediating chromatin interactions, as long as the target protein has a suitable antibody for ChIP-seq assays. Therefore, the quality and availability of a ChIP-grade antibody is critical. Another important consideration regarding the antibody is the specific “batch” of antibody used, due to quality variations in antibody production. Once a robust antibody batch is identified, what we typically do is stockpile enough of this specific batch for a given ChIA-PET project. The robustness of an antibody for its ChIP enrichment in a ChIA-PET experiment can be assessed by ChIP quantitative PCR (ChIP-qPCR) (Mukhopadhyay et al., 2008). In ChIP-qPCR, at least two positive and one negative target loci (binding sites) in a genome for the protein factor of interest should be selected, and pairs of PCR primers should be designed based on the specific sequences at the flanking regions of the selected loci. ChIP-enriched chromatin DNA samples along with the non-enriched input genomic DNA can then be quantitatively amplified using these primer pairs by qPCR, so to accurately measure the fold of ChIP enrichment by a specific antibody in a ChIA-PET experiment.
Fourth, once a quality in situ ChIA-PET library is generated, a large enough number of paired-end-tag (PET) reads (2×150 bp) by high-throughput DNA sequencing is required to provide enough data for genome-wide coverage, which ranges from 100 million to 1 billion paired-end-tag (PET) reads. Routinely, we produce 200–500 million PET reads per library, which should provide adequate coverage of protein binding peaks and chromatin loops between peak loci. Furthermore, for quantitative measurement, biological or technical replicates (at least 2 replicates) of in situ ChIA-PET data are desired for reliable data analysis and conclusions. Therefore, sufficient budget for DNA sequencing should be set aside.
Lastly, one should also be prepared for high-volume in situ ChIA-PET data analysis. There are several publicly available computational pipelines (Li et al., 2010a, 2017; Phanstiel et al., 2015; Lee et al., 2020), including the most recent computational pipeline, called ChIA-PIPE (Lee et al., 2020), for ChIA-PET data processing and result visualization, which we describe in this article.
BASIC PROTOCOL 1: Sample preparation and data generation
The following in situ ChIA-PET protocol describes the step-by-step procedures for the preparation of cellular samples and library construction for sequencing on the Illumina platform (Figure 2). For convenience, we describe the experimental protocol in two parts: cell crosslinking (Part 1) and library preparation (Part 2). In Part 1, to ensure the capturing of long-range chromatin contacts through protein-DNA and protein-protein interactions, we employ a dual crosslinking method, which we describe here for both suspension cells and adherent cells. The dual-crosslinking process uses formaldehyde (FA) and ethylene glycol bis (succinimidylsuccinate) (EGS) to maximize the capturing of chromatin interactions that involve DNA-protein and protein-protein contacts participating in chromatin interactions. Formaldehyde forms covalent bonds between DNA and associated proteins, and thus captures DNA-protein interactions, whereas EGS preserves protein-protein interactions by forming relatively weak but long-arm bonds. Although we describe the procedure for cultured cells, the protocols can be adapted for primary cells with minor modification. Once crosslinked, the nuclear conformation is preserved, and the cellular samples can be stored at −80 °C for months before proceeding to library construction. In Part 2, we describe detailed steps required for ChIA-PET library construction, which involve cell lysis, nuclei permeabilization, in situ digestion, in situ proximity ligation, chromatin sonication, ChIP enrichment, DNA tagmentation, and PCR amplification, which result in the final in situ ChIA-PET library that is ready for high throughput DNA sequencing.
Figure 2:
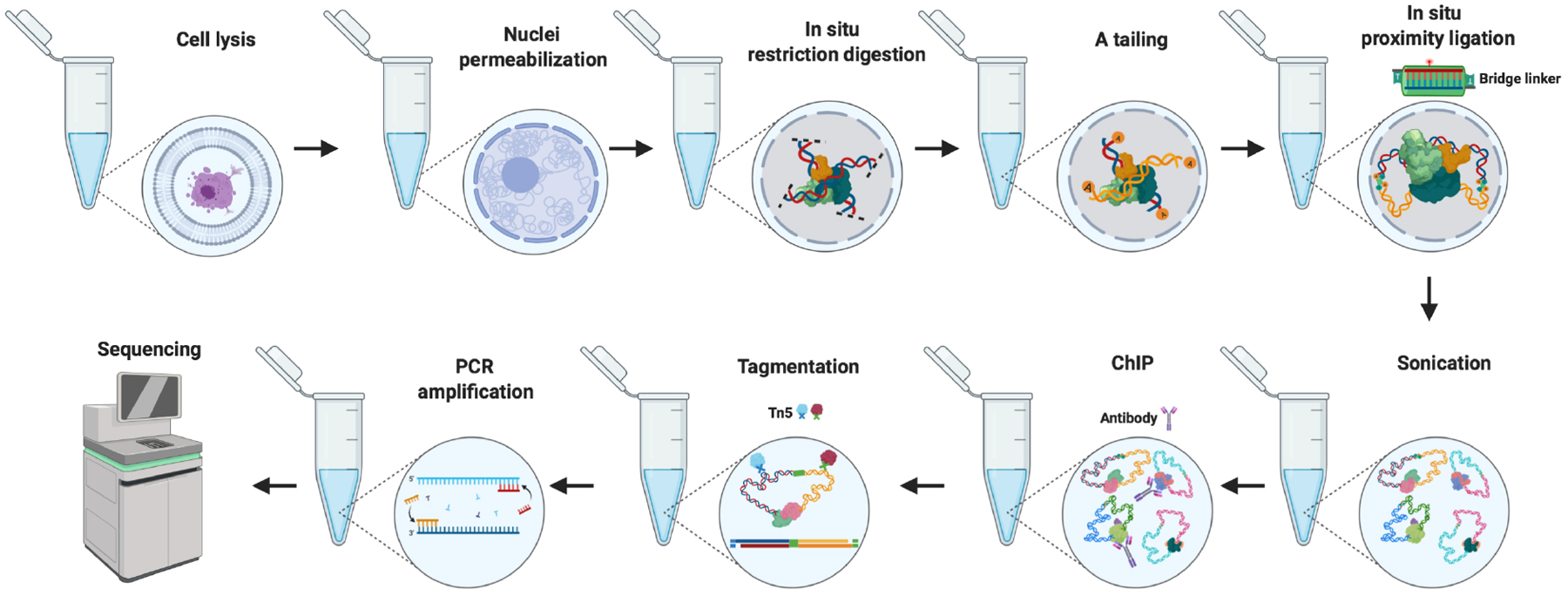
Schematic overview of Basic Protocol 1. The nuclei isolated from dual-crosslinked cells are permeabilized, and AluI (a four-base-pair cutter of restriction enzyme) is added to the reaction for in situ digestion. Proximity ligation is performed in situ, with a bridge linker after the A-tailing step. Chromatin immunoprecipitation (ChIP) against a specific protein factor is carried out after sonication of the DNA. The de-crosslinked ChIP DNA is tagmented by Tn5 transponase, and the ChIA-PET library is then amplified by PCR and is ready for sequencing analysis.
MATERIALS
Cultured suspension or adhesion cells (1–10 million cells/reaction).
Dulbecco’s Phosphate-Buffered Saline (DPBS) solution, calcium- and magnesium-free (Gibco, cat. no. 14190–250).
Dimethylsulfoxide (DMSO) (Sigma, cat. no. D2650).
Ethylene glycol bis (succinimidyl succinate) (EGS) (Thermo Fisher Scientific, cat. no. 21565).
Formaldehyde (FA) (36% (vol/vol); Sigma-Aldrich, cat. no. 47608–250ML-F).
Glycine (Sigma, cat. no. G7126).
Nuclease-free water (Thermo Fisher Scientific, cat. no. AM9932).
HEPES buffer, pH 7.3 (Fisher Scientific, cat. no. BP299–1).
Ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher Scientific, cat. no. 9261).
cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (PI) (Roche, cat. no. 11836170001).
Triton X-100, molecular biology grade (Sigma-Aldrich, cat. no. 648464).
Sodium dodecyl sulfate (SDS) solution (10%) (Thermo Fisher Scientific, cat. no. AM9822).
Alul restriction enzyme (NEB, cat. no. R0137L).
Proteinase K Solution (Thermo Fisher Scientific, cat. no. AM2548).
QIAquick PCR Purification Kit (Qiagen, cat. no. 28106).
Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific, cat. no. Q32854).
Agilent DNA High-Sensitivity Kit (Agilent Technologies, cat. no. 5067–4626).
BSA, Molecular Biology Grade, 20 mg/ml (NEB, cat. no. B9000S).
TE, pH 8.0, RNase-free (Thermo Fisher Scientific, cat. no. AM9858).
10 × PBS (Thermo Fisher Scientific, cat. no. AM9937).
dATP solution, 100 mM (NEB, cat. no. N0440S).
DNA Polymerase I, Large (Klenow) Fragment (NEB, cat. no. M0210L).
Bridge linker, 200 ng/μl (See Support Protocol 1).
NEBNext® Quick Ligation Reaction Buffer, 5× (NEB, cat. no. B6058S).
T4 DNA ligase (NEB, cat. no. M0202L).
Tween 20, molecular biology grade (Sigma-Aldrich, cat. no. P9416).
Dynabeads Protein G for Immunoprecipitation (Thermo Fisher Scientific, cat. no. 10009D).
Antibody against protein of interest - e.g. monoclonal antibody against RNA Polymerase II (8WG16) (BioLegend, cat. no. 664912).
Buffer EB (Qiagen, cat. no. 19086).
Tris-HCl, pH 7.5 (Thermo Fisher Scientific, cat. no. 15567027).
Tris-HCl, pH 8.0 (Thermo Fisher Scientific, cat. no. AM9856).
Lithium chloride solution (Sigma-Aldrich, cat. no. L7026).
Nonidet P40 Substitute (Sigma-Aldrich, cat. no. 11754599001).
DNA LoBind Tubes (1.5 ml, Eppendorf, cat. no. 022431021).
Centrifuge (Eppendorf 5810R, cat. no. 22628180).
DNA Clean & Concentrator-5 (Zymo Research, cat. no. D4014).
Qubit™ ssDNA Assay Kit (Thermo Fisher Scientific, cat. no. Q10212)
LightCycler® 480 SYBR Green I Master (Roche, 0–4707516001)
Illumina Tagment DNA Enzyme and Buffer Large Kit (Illumina, cat.no. 20034198).
Dynabeads M-280 Streptavidin (Thermo Fisher Scientific, cat. no.11205D).
iBlock Protein-Based Blocking Reagent (Thermo Fisher Scientific, cat. no. T2015).
20 × Saline-sodium citrate (SSC) solution (Thermo Fisher Scientific, cat. no. AM9763).
NEBNext® High-Fidelity 2 × PCR Master Mix (NEB, cat. no. M0541S).
Nextera XT Index Kit v2 Set A (Illumina, cat. no. FC-131–2001).
AmPure XP beads (60 ml; Beckman, cat. no. A63881).
Absolute ethanol.
Blue Pippin Cassette Kit (Sage Science, cat. no. BDF2010).
DNA LoBind Tubes (1.5 ml, Eppendorf, cat. no. 022431021).
Qubit 2.0 Fluorometer (Thermo Fisher Scientific, cat. no. Q32866).
Qubit® Assay tubes (Thermo Fisher Scientific, cat. no. Q32856).
Cell scraper (Corning, cat. no. 3011).
Falcon™ 15 mL Conical Centrifuge Tubes (Fisher Scientific, cat.no. 14–959-53A).
Falcon 50 mL Conical Centrifuge Tubes (Fisher Scientific, cat.no. 14–959-49A).
75 cm² (T-75) cell culture flask (Thermo Fisher Scientific, cat. no. 430641U).
500cm² Square TC-Treated culture dish (Corning, cat. no. 431110).
LightCycler® 480 Multiwell Plate 384 (Roche, # 04729749001).
LightCycler® 480 Sealing Foil (Roche, # 04729757001).
BUFFERS/SOLUTIONS (see Reagents and Solutions)
0.1% SDS Cell Lysis Buffer
1% FA-DPBS solution (50 ml)
0.55% SDS solution (10 ml)
10 mM dATP solution (200 μl)
1× PBST buffer (100 ml)
1× Binding & Wash Buffer
2× Binding & Wash Buffer
2× SSC/0.5% (w/v) SDS (100 ml)
2 mM EGS-DPBS solution (50 ml)
2.5 M Glycine solution (100 ml)
ChIP Elution Buffer
High Salt Buffer
LiCl Wash Buffer
iBlock buffer (100 ml)
Sheared genomic DNA mixture
EQUIPMENT
Eppendorf ThermoMixer® F1.5 (Eppendorf, cat. no. 5384000020).
ELMI RM-2M Intelli-Mixer Medium (ELMI, cat. no. IMIX-02).
Qubit 4 Fluorometer (Thermo Scientific).
LightCycler® 480 System (Roche).
Agilent 2100 Bioanalyzer (Agilent Technologies, cat. no. G2940CA).
DynaMag-2 Magnet (Thermo Fisher Scientific, cat. no. 12321D).
BluePippin instrument (Sage Science, BluePippin).
Bench top incubator (Thermo Scientific Heratherm, cat.no. IGS60 51028063).
Illumina NovaSeq 6000 high-throughput sequencing machine (Illumina).
Protocol Steps
Part 1: Dual crosslinking of Cells
In this section, we describe the optimized double-crosslinking procedures for both cultured suspension and adhesion cells, to highlight the differences in chromatin sample preparation for different cell types, which can be adapted for primary cells with minor modifications.
a. Crosslinking procedure for suspension cells:
1a. Grow cells to log phase (106 cells/ml maximum density) in a T75 flask. Count cells and measure viability before harvesting by staining an aliquot of the cells with trypan blue. If viability, as measured by exclusion of the blue dye, is >90%, count the cells with an haemocytometer and proceed with next steps. Otherwise, discard the cells and start over with new cells.
2a. Freshly prepare the FA-DPBS and EGS-DPBS solution and keep the EGS-DPBS solution in the 37°C incubator to facilitate dissolving of the EGS in solution. Also prepare another bottle of DPBS stored at the 4°C refrigerator for later use.
3a. After cell counting (1a), transfer cells and media from the flasks into either 50-ml conical tubes or centrifuge bottles. Spin down the cells at room temperature (RT) at 3000 rpm for 10 min, remove all supernatant, and add DPBS to resuspend the cell pellet to a final concentration of 107/ml. Transfer 10 million cells to one 1.5 ml Eppendorf DNA LoBind tube and record the cell number.
Use low binding tubes to prevent cell loss.
4a. Spin down the cells at room temperature (RT) at 3000 rpm for 10 min and remove all supernatant.
5a. Resuspend the cells in 1 ml of 1% FA-DPBS solution and mix well using a pipette tip. Incubate for 20 min at RT with rotation.
6a. Add 2.5 M Glycine to a final concentration of 0.2 M (87 μl for 1 ml 1% FA-DBPS solution). Incubate for 10 min at RT with rotation.
7a. Centrifuge the samples at RT at 2500 × g for 10 min. Remove supernatant and resuspend cells in 1 ml DPBS.
8a. Centrifuge the samples at RT at 2500 × g for 10 min. Remove supernatant and resuspend cells in 1 ml of 2 mM pre-warmed EGS-DPBS solution (pre-warmed solution in the 37°C incubator, Step 1a). Incubate for 45 min at RT with rotation.
9a. Add 2.5 M Glycine to a final concentration of 0.2 M (87 μl for 1ml EGS-DPBS solution). Incubate for 10 min at RT with rotation.
10a. Centrifuge the samples at RT at 2500 × g for 10 min. Remove supernatant and resuspend cells in 1 ml DPBS. Count the cells with an haemocytometer to ensure no significant loss of cells happened during the crosslinking steps.
11a. Centrifuge the samples at RT at 2500 × g for 10 min. Remove supernatant.
12a. Store the cell pellets at −80°C until ready to proceed to Part 2.
The crosslinked cells are safe to be stored for months.
b. Crosslinking procedure for adherent cells:
1b. Grow on tissue culture dishes rather than in flasks to facilitate cell harvesting and prepare an additional dish for counting the cells. For example, grow cells to a final cell density of 6–10 × 107 per 500cm² square in a TC-treated culture dish. Do not grow cells to confluency. Trypsinize the additional dish, count the number of cells, and test for viability by staining with trypan blue. If viability, as measured by exclusion of the blue dye, is >90%, count the cells with an haemocytometer and proceed to next steps with the original dishes. Meanwhile, freshly prepare the FA-DPBS and EGS-DPBS solution, and keep the EGS-DPBS solution in the 37°C incubator to facilitate dissolving of the EGS in solution (same as described in 2a). Prepare another bottle of DPBS and store it at 4°C.
2b. Remove the culture dishes from the incubator and place at room temperature on the bench.
3b. Remove the medium and rinse the monolayer twice with ~30 ml of RT DPBS.
4b. Add 50 ml of RT DPBS to each dish, and then add 1.429 ml of 36% formaldehyde (FA) to each dish. Shake the dishes on a rotator for 20 min at RT at 90 rpm.
5b. Add 4.47 ml of 2.5 M glycine (Glycine final concentration: 0.2 M) to each dish. Shake the dishes on a rotator for 10 min at RT at 90 rpm.
6b. Remove the FA crosslinking solution and wash with ~30 ml RT DPBS once. Then, try to take out all buffer with a pipette.
7b. Add 50 ml of 2 mM EGS-DPBS solution (pre-warmed solution in the 37°C incubator, step 1b) to each dish and shake for 45 min at RT at 90 rpm.
8b. Add 4.47 ml of 2.5 M glycine (Glycine final concentration: 0.2 M) to each dish. Shake the dishes on a rotator for 10 min at RT at 90 rpm.
9b. Remove the crosslinking solution and rinse the cells twice with 30 ml RT DPBS.
10b. Add 30 ml DPBS and store the dishes in the cold room (4°C) until ready to harvest.
11b. Bring dishes to the bench. Discard the 30 ml DPBS.
12b. Add 5 ml of pre-chilled (4 °C) DPBS (see 1b), scrape cells with a large scraper, and label a 50 ml collecting tube/ dish with date, passage number, and cell type. Collect cells in the 50ml tube. Repeat the scraping step twice with 10 ml DPBS to rinse the dishes, and collect these remaining cells into the same 50 ml tube to avoid cell loss.
13b. Centrifuge the tubes at 4 °C at 2000 rpm for 10 min.
14b. Remove supernatant and store the cell pellet at −80 °C until ready to proceed to Part 2.
Part 2: In situ ChIA-PET library construction
This section describes the procedures for in situ ChIA-PET library construction (Figure 2). It takes around 5 days to generate one in situ ChIA-PET library and we have divided the protocol steps by day, for convenience. Usually, it will be more efficient and economical to prepare multiple libraries simultaneously. At each major stop point, quality control (QC) measurements are included to evaluate the quality and quantity of chromatin DNA materials (Figure 3).
Figure 3:

Quality Control steps for Basic Protocol 1. Critical quality controls for each step are evaluated via Bioanalyzer. A. QC #1. DNA size distribution profiles of the chromatin DNA after In situ AluI digestions. The distribution usually peaks at around 5–8 kbps, as determined by Bioanalyzer profiling. B. QC #2. DNA size distribution profiles after proximity ligation of the AluI-digested chromatin sample. It is expected that after ligation, the size of chromatin DNA fragments would increase accordingly. Indeed, as shown in this sample data, the DNA size observed in QC #2 is approximately 2–5 kb larger than the DNA fragments in QC #1, indicating the proximity ligation was successful. C. QC #3. DNA size distribution profiles of sonication-sheared chromatin DNA fragments, showing obvious shift of peak from >10 kb to short chromatin fragments in the range of 2–3 kb. D. QC #4. After ChIP-enrichment, the enriched chromatin DNA is analyzed by Bioanalyzer for quality and quantity. The profile looks the same as in QC 3#, as expected. E. QC #5. DNA size distribution profiles of tagged-DNA fragments after tagmentation by Tn5 transposase step, showing the desired shifting of DNA fragments to the size range of 200–700 bp. F. QC #6. DNA size distribution profiles of PCR-amplified DNA fragments, showing that the majority of amplicons are in the 300–700 bp range. G. QC #7. DNA size distribution profiles of the final In situ ChIA-PET library after size selection for the 350–600 bp range, which is the ideal size range for Illumina paired-end-tag sequencing analysis.
Day 1: Cell lysis, nuclei permeabilization, and in situ restriction digestion
-
15.
Take out a 10 million dual crosslinked cell pellet tube from the −80 °C freezer (Step 12a or 14b), leave it on ice for 20 min to thaw the cells, centrifuge the tube at 2500 × g for 5 min at 4 °C, and discard the DPBS buffer.
-
16.
Prepare 10 ml of 0.1% SDS cell lysis buffer and 10 ml of 0.55% SDS solution in two separate 15 mL tubes. Add one cOmplete™, Mini, EDTA-free Protease Inhibitor (PI) Cocktail to each of the tubes, and dissolve completely. Leave the 0.1% SDS cell lysis buffer tube on ice and the 0.55% SDS solution tube at RT.
-
17.
Suspend the cell pellet with 1 ml of the 0.1% SDS cell lysis buffer (+PI) on ice. Incubate the tube for 1 hr at 4 °C with rotation.
-
18.
Centrifuge the tube at 2500 × g for 5 min at 4 °C and then carefully discard the supernatant.
-
19.
Re-suspend the cell pellet with 100 μl of the 0.55% SDS solution (+PI).
-
20.Incubate the tube at RT for 10 min and then at 62 °C in an Eppendorf ThermoMixer for 10 min, followed by an incubation at 37 °C for 10 min.The purpose of this step is to permeabilize the nuclear membranes, which will facilitate the entry of enzymes and reagents into the nuclear space for the following in situ digestion and ligation steps. The incubation temperature and duration can be modified according to the cell type used. These suggested parameters have been tested for Drosophila cells (S2 and KC167 cell) and a number of human cell lines for the ENCODE phase 4 Project (e.g., GM12878, MCF7, among others).
-
21.
Add 270 μl of double-distilled water (ddH2O) and 50 μl of 10% Triton X-100 solution to the tube and mix well. Incubate the tube at 37 °C for 15 min to quench SDS.
-
22.Add 50 μl of 10x NEB CutSmart buffer and 30 μl of AluI restriction enzyme.The tube will now have a final volume of 500 μl.
-
23.
Incubate the tube overnight with shaking on an Intelli-mixer (UU, 35rpm mode) in a 37 °C bench top incubator.
Day 2: A-tailing and Proximity ligation
-
24.
Perform the first quality control test (QC #1): After the overnight AluI digestion (Step 23), take 10 μl of the digestion sample to a new tube. To that, add 90 μl of TE pH 8.0 buffer and 5 μl of proteinase K, mix, and incubate for 1 hr at 65 °C. Purify the DNA with QIAquick PCR Purification Kit (following the manufacturer’s instructions) and check the DNA profile with a Bioanalyzer 2100 HS DNA chip (See QC1, Figure 3A). Proceed to the next step only if suitable digestion fragments (range around 5–8 kb) are visualized in the Bioanalyzer DNA profile.
-
25.Set up an A-Tailing reaction on ice. Add the reagents in the order shown below:
- 490 μl Restriction digested sample (from Step 23)
- 11 μl BSA (20 mg/ml)
- 11 μl 10 mM dATP
- 4 μl 10x CutSmart buffer
- 3 μl Nuclease-free water
- 11 μl Klenow Large fragment (3’→5’ exo-)
- Total: 530 μl
- Incubate the tube for 1 hr, with rotation, at 37°C.
-
26.Leave the sample tube at RT. Add proximity ligation reagents in the order shown below:
- 530 μl A-tailed sample (from Step 25)
- 257 μl Nuclease-free water
- 200 μl NEB 5x quick ligase buffer
- 3 μl Bridge linker (200 ng/μl, see Support Protocol 1).
- 10 μl T4 DNA ligase
- Total: 1000 μl
- Mix well and incubate for 1 hr at RT. Transfer the tube to the Intelli-mixer for overnight ligation (F1 rotation mode, 12 rpm) in a 16 °C incubator.
Thaw linkers in the pre-prepared bridge linker solution gently on ice, and mix well gently. Be sure to add bridge linker and ligase last. Additional details for the design of bridge linker nucleotide sequences and method for linker preparation are described in Support Protocol 1. -
27.
In the meantime, and to coat the antibody with Dynabeads protein G beads, thoroughly resuspend the protein G beads within the bottle and aliquot 100 μl of protein G beads in a 1.5 ml microfuge tube for the chromatin immunoprecipitation (ChIP) step.
-
28.
Wash the beads with 500 μl of cold PBST buffer, vortex briefly to mix well. Place the microfuge tube on the magnet rack, wait for 1–2 min, and remove the supernatant. Resuspend the beads in 500 μl of cold PBST buffer and repeat washing twice. Finally, re-suspend the beads in 500 μl of cold PBST buffer.
-
29.Add 20 μg of antibody to the washed protein G beads from Step 28, transfer the tube to the Intelli-mixer, and incubate the tube at 4 °C overnight, with rotation at 12rpm.Prepare the antibody binding to protein G beads ~ 6–24 hours before the ChIP experiment.
Day 3: Sonication and ChIP
-
30.Perform QC step #2: After the overnight chromatin proximity ligation step (Step 26), take 10 μl from the sample to a new tube and add 90 μl of TE (PH 8.0) buffer. Then, add 5 μl of proteinase K, mix, and incubate for 1 hr at 65°C. Purify the chromatin DNA with the QIAquick PCR Purification Kit (following the manufacturer’s instructions) and check the DNA profile with a Bioanalyzer 2100 HS DNA chip (See QC2, Figure 3B). Proceed to the next step only if suitable ligation fragments are visualized in the DNA profile.The fragment length of ligated DNA should be approximately 2–5 kb larger than the digested DNA fragments in QC #1, which is a good indication of a successful ligation.
-
31.
Spin the sample from step 26 at 5500 × g for 10 min at 4 °C, remove the supernatant, and gently resuspend the cell pellet in 0.5 ml of 0.1% SDS cell lysis buffer (+PI), for sonication.
-
32.
Shear the chromatin via sonication. The optimal sonication parameters for each individual cell type should be empirically determined (see Strategic Planning).
-
33.
Centrifuge the sonicated sample at 6500 × g for 10 min at 4 °C. Transfer the supernatant to a new tube.
-
34.
Perform QC step #3: Take out 5 μl of the sample from step 33 to a new tube and add 90 μl of TE pH 8.0 buffer. Then, add 5 μl proteinase K, mix, and incubate at 65 °C for 1 hr. Purify the chromatin DNA with the QIAquick PCR Purification Kit (following the manufacturer’s instructions) and check the DNA profile with a Bioanalyzer 2100 HS DNA chip (See QC3, Figure 3C). Proceed to the next step only if ideal sonication fragment sizes (2–3 kb) are visualized in the DNA profile.
-
35.
Take 100 μl of Protein-G beads per sample, transfer the beads to a microfuge tube on the magnet rack, wait for 1–2 min, and remove supernatant. Then add 500 ul of cold PBST and vortex briefly to mix well. Place the microfuge tube on the magnet rack again, wait for 1–2 min, and remove supernatant. Add 500 μl of cold PBST buffer and repeat the washing step twice.
-
36.Transfer the rest of the supernatant from step 33 to the washed Dynabeads Protein G beads from step 35 and incubate at 4 °C for at least 1 hr with rotation, 12 rpm.This step is for pre-clearing chromatin to reduce non-specific binding of chromatin materials with the magnetic beads.
-
37.
Take out the tube of antibody-coated-beads from step 29, place the microfuge tubes on the magnet rack, wait for 1–2 min, and remove the supernatant. Add 500 ul of cold PBST and vortex briefly to mix well. Place the microfuge tubes on the magnet rack again, wait for 1–2 min, and remove the supernatant. Add 500 μl of cold PBST buffer and repeat the washing step twice.
-
38.
Incubate the antibody-coated-beads (from step 37) with the pre-cleared chromatin materials (from step 36) overnight with rotation, at 4 °C.
Day 4: Wash chromatin DNA on beads after ChIP and reverse crosslinking
-
39.
Place the tube from Step 38 in the magnetic stand and remove supernatant.
-
40.
Wash the beads three times with 1 ml of 0.1% SDS Cell Lysis Buffer. For each wash, nutate for 5 min at 4°C and spin at 800 rpm for 1 min at 4°C.
-
41.
Wash the beads twice with 1 ml of High salt buffer. For each wash, nutate for 5 min at 4°C and spin at 800 rpm for 1 min at 4°C.
-
42.
Wash the beads once with 1 ml of LiCl buffer. Nutate for 5 min at 4°C and spin at 800 rpm for 1 min at 4°C.
-
43.
Wash the beads twice with 1 ml of TE (PH. 8.) buffer. For each wash, nutate for 5 min at 4°C and spin at 800 rpm for 1 min at 4°C.
-
44.
Elute DNA from the beads. For this, add 200 μl of ChIP Elution buffer and incubate the tube in the Thermomixer at 65 °C for 30 min with agitation at 900 rpm.
-
45.
Place the microfuge tube (from Step 44) on the magnet rack, wait for 1–2 min, and transfer the supernatant (200 μl) to a new tube (supernatant tube).
-
46.
Remove the tube with the beads from the magnetic rack and add 100 μl of EB buffer to re-suspend the beads. Mix well. Place the microfuge tube back on the magnet rack, wait for 1–2 min, and transfer the supernatant (100 μl of EB buffer) to the same “supernatant tube” as in Step 45, which has the 200 μl supernatant solution, for a final volume of ~300 uL.
-
47.
Add 10 μl of proteinase K (20 mg/ml) to the mixed solution and proceed to DNA de-crosslinking by incubating the tube in the Thermomixer at 65 °C overnight.
Day 5: ChIP DNA quantity and quality assessment, Tn5 tagmentation, biotin enrichment, and Illumina library preparation
-
48.Proceed with DNA purification: Purify the DNA from step 47 by using the Qiaquick PCR purification kit (following manufacturer’s instructions) and elute DNA twice with 11 μl of EB buffer (final volume ≈ 20 μl). Take 1 μl of the DNA to measure DNA concentration with Qubit (following the manufacturer’s instructions) and calculate the yield of ChIP-DNA.Routinely, we obtain ChIP-DNA in the range of 50–100 ng, depending on the nature of the protein factor and the robustness of the antibody used. In the case of RNAPII ChIA-PET, the yield of ChIP-DNA is usually > 50 ng, whereas for CTCF ChIA-PET the yield of ChIP-DNA is >100 ng.(Stopping point) ChIP DNA can be stored at −20 °C for two weeks.
-
49.Perform QC #4: First, check the ChIP DNA profile from the sample in Step 48 with a Bioanalyzer 2100 HS DNA chip (QC4, Figure 3D). The profile of the ChIP-DNA should look similar to that of the DNA before ChIP enrichment (QC3). Second, perform qPCR to check for ChIP-enrichment of a selected specific DNA region (binding region of the protein of interest) in ChIP-DNA. To do so, two pairs of PCR Test Primers for two known target binding sites and one Negative Primer pairs based on a non-target locus are used for ChIP-qPCR. Take 0.1 ng of the ChIP-DNA for each of the 3 PCR primer pairs to set up the qPCR reactions (Read, 2017) with at least 2 replicates, and use the same amount of non-enriched DNA from step 34 (QC3) as total input control, for a total of 12 qPCR reactions (2 template samples [ChIP-DNA and input DNA] × 3 primer pairs [two for 2 Test primers + one for negative primers] × 2 technical replicates = 12). After the qPCR reactions are completed, calculate the fold ChIP-enrichment based on qPCR Cycle threshold value (Ct) as follows:
- Calculate the average Ct for each Primer pair with 2 replicates.
- Calculate the average differential Ct (Δ Ct) for each primer pair using the formula below:
- Calculate the Final ΔΔ Ct for each Test Primer ChIP-DNA using formula:
A sample with a final ΔΔ Ct value > 5 is considered a successful ChIP enrichment for protein factors of interest.
Sample data can be found in Table 1. -
50.Proceed with tagmentation with ChIP-DNA: add the following reagents to a new 0.2 ml PCR tube, and mix well with a pipette after each addition:
- ___ μl ChIP DNA (add 50 ng)
- 25 μl 2 × NEXTERA Tagmentation Buffer
- ___ μl NEXTERA Transposase enzyme (TDE)
- ___ μl Nuclease-free water to adjust the final volume
- 50 μl total volume
Add enzyme last and mix well. Adjust the amount of Tn5 transposase enzyme according to the amount of ChIP-DNA. In this step, all ChIP-DNA will be used for tagmentation. Usually, a maximum of 50 ng ChIP-DNA is used for each reaction, and 8.5 μl of enzyme is added. The ratio of enzyme and DNA is around 0.17. If the amount of ChIP DNA is less than 50ng, adjust the enzyme amount to preserve the 0.17 ratio. -
51.
Short-spin the tube, incubate the PCR tube at 55°C for 5 min, and then at 10 °C for 10 min.
-
52.Purify tagmented ChIP DNA from step 51 by using the DNA Clean & Concentrator-5 kit (following the manufacturer’s instructions) and elute the DNA with 10 ul of EB buffer.(Stopping point). DNA can be stored at −20 °C for two weeks.
-
53.Perform QC #5: Measure DNA concentration with Qubit and check the DNA profile of the sample from step 52 with a Bioanalyzer 2100 HS DNA chip (QC5, Figure 3E). The expected size range of tagmented DNA fragments should be larger than 150 bp and less than 700 bp. Otherwise, the ratio of enzyme to DNA should be empirically adjusted.In the event of ChIP-DNA yield (Step 48) more than 50 ng, the remaining ChIP-DNA can also be processed for additional tagmentation reactions according to the abovementioned ratio, by repeating steps 50 to 52. Combine all purified tagmentation ChIP-DNA together in preparation for the biotin-DNA enrichment step.
-
54.
Equilibrate the M-280 streptavidin Dynabeads to RT for 30 min. Then, fully resuspend and transfer 30 μl of the suspended Dynabeads into a new 1.5 ml tube.
-
55.
Place the tube with the beads on the magnetic stand and wait for 1–2 min until the solution is clear. Discard the supernatant and wash twice with 150 μl 2 × Binding & Washing buffer.
-
56.
Resuspend beads in 100 μl iBlock Buffer, mix, and incubate at room temperature (RT) for 45 min on a rotating Intelli-mixer (UU, 50 rpm).
-
57.
Short spin the tube, place it on the magnet rack, discard the iBlock buffer, and then wash the beads twice with 200 μl of 1 × Binding & Washing buffer.
-
58.Discard the washing buffer, then add 100 μl of sheared genomic DNA mixture (consisting of 500 ng of genomic DNA in 50 μl nuclease-free water + 50 μl 2 × Binding & Washing buffer, see Reagents and Solutions) to the beads. Mix well and incubate on the Intelli-mixer with rotation for 30 minutes (UU, 50 rpm) at RT.The purpose of this step is to use total genomic DNA fragments to block non-specific affinity to M-280 beads, therefore, increasing the efficiency of specific streptavidin-biotin to enrich the tagmented DNA fragments containing the bridge linker with biotin and reducing the non-specific DNA (non-biotin DNA) binding to the M-280 beads.
-
59.
Place the tube with the beads and genomic DNA on the magnetic stand and wait for 1–2 minutes until the solution is clear. Discard the solution with the blocking DNA mixture, taking care not to eliminate the beads. Add 200 μl of 1 × Binding & Washing buffer to the beads and mix well. Discard supernatant and repeat the washing step with 200 μl of 1 × Binding & Washing buffer.
-
60.
Pool together all of the tagmented ChIP DNA products from step 52 and bring the volume to 50 μl with nuclease-free water. Then, add an equal volume of 2 × Binding & Washing buffer, mix well with the beads from step 59, and incubate at RT for 45 min using the Intelli-Mixer (UU, 50 rpm).
-
61.
Short-spin the tube, place tube on magnetic stand, and wait for 1–2 min until the solution are clear. Discard the supernatant, add 500 μl of 0.5% SDS/ 2 × SSC buffer, and mix well. Place the tube with the beads on the magnetic stand and wait for 1–2 min until the solution is clear. Discard the supernatant and repeat the washing of the beads 4 times with 500 μl 0.5% SDS/ 2 × SSC buffer.
-
62.
Short spin the tube, place tube on magnetic stand, and wait for 1–2 min until the solution is clear. Use a pipette tip to carefully remove and discard the supernatant. Wash the beads by adding 500 μl of 1 × Binding & Washing buffer, re-suspending the beads, placing the tube with the beads on the magnetic stand, waiting for 1–2 min until the solution is clear, and then carefully removing and discarding the supernatant. Repeat this washing with 500 μl of 1 × Binding & Washing buffer once more. Resuspend the washed beads with the immobilized DNA in 30 μl of EB buffer.
-
63.Prepare an Illumina library PCR reaction as follows:
- 10 μl DNA-coated beads (from Step 62)
- 5 μl Nuclease-free water
- 25 μl NEBNext® High-Fidelity 2 × PCR Master Mix
- 5 μl Index Primer 1 (i5)
- 5 μl Index Primer 2 (i7)
- Total: 50 μl
-
64.Set up the following PCR run:
- Initial denaturation at 72 °C for 3 min, then at 98 °C 30 s
- 12 cycles of 10 s 98 °C → 30 s 63 °C → 40 s 72 °C
- Final extension at 72 °C for 5 min
- Hold at 4 °CIn this step, tagmented ChIP DNA fragments immobilized on beads are directly amplified with the specific primer pair i5 and i7 (Nextera XT Index Kit v2 Set A, see Materials) by PCR into a library that is compatible with Illumina sequencing. Note that only 1/3 of the tagmented ChIP DNA from step 62 is used for PCR here.
-
65.
While the PCR is running, equilibrate AMPure XP beads to RT for 30 min before using.
-
66.
After the PCR reaction done, transfer the 50 μl of the PCR product in supernatant to a new tube and place it on the magnetic rack. Fully resuspend the AMPure XP beads by vortexing. Add an equal volume (1:1 ratio) of AMPure beads to the reaction tube in the rack (containing the PCR amplicons) and pipette-mix well ~10 times. Incubate the mixture at RT for 5 min using the Intelli-Mixer (F8, 30 rpm mode).
-
67.
Spin down the tube briefly, put the tube on the magnetic stand, and wait for the solution to clear (~3–5 minutes). Discard the supernatant, and then add 200 μl of freshly prepared 80% ethanol to resuspend the beads. Spin down the tube briefly, put the tube back on the magnetic stand, and wait for the solution to clear (~1–2 minutes). Remove ethanol and repeat the washing step with 200 μl ethanol. Let the tube sit on the magnetic stand for 1 min to allow any disturbed beads to settle, and carefully remove any residual ethanol. Leave the tubes open on the magnetic stand to air dry on the bench top for 10 min.
-
68.
Add 10 μl EB Buffer to the tube, vortex it, and mix well. Spin down the tube briefly, put the tube on the magnetic stand, and wait for the solution to clear (~3–5 min). Transfer the clear solution to a new 1.5 ml Eppendorf tube.
-
69.
Measure the concentration of the PCR product from step 68 with Qubit (following the manufacturer’s instructions) and check the DNA profile with a Bioanalyzer 2100 HS DNA chip (See QC #6, Figure 3F).
-
70.Based on the QC6 results (DNA yield and size profile of PCR product), adjust the numbers of PCR cycles (12) used in step 64 to as low as 9 cycles or up to 13 cycles, and repeat steps 64–69 to amplify the remaining 20 μl of DNA-beads. Pool the purified PCR products together in final volume of 30 μl for at least 100 ng of amplicons before library size-selection.(Stopping point). DNA can be stored at −20 °C for two weeks.The purpose of this step is to prepare a library with enough amplified ChIP DNA for library sequencing analysis but also avoiding over amplification of ChIP DNA that could lead to high redundancy of DNA fragments in a library. In general, lower cycle numbers (9–10) usually result in high library complexity, but higher cycle numbers (above 14) would result in low complexity library and high redundancy in sequencing reads.
-
71.Add 10 μl of V1 maker (from Blue Pippin Cassette Kit) to the library and use BluePippin (refer to the BluePippin’s manual for detailed operation instructions) for DNA size-selection in the range of 300 bp to 600 bp using a 2% agarose gel cassette.Usually, the yield of the final size-selected DNA is around 40–60 ng from the 100 ng input DNA (step 68). An alternate approach for DNA size selection for short DNA fragments (300–600 bp), if BluePippin is not available, is to perform AMPure XP beads purification twice for the PCR product.
-
72.Perform the final QC step #7: Measure the DNA concentration of the final library using Qubit and check the DNA size profile with a Bioanalyzer 2100 HS DNA chip (See QC #7, Figure 3G).At this point, the construction of an in situ ChIA-PET library is complete, and the library is ready for high throughput sequencing.
-
73.Perform paired-end sequencing of an in situ ChIA-PET library with 2×150bp module using an Illumina NovaSeq 6000 sequencer following the manufacturer’s instructions.Most of the DNA templates (300–600 bp) for sequencing within the library are composed of a bridge linker in the middle and two short DNA fragments, one on each side derived from interacting chromatin loci. Paired-end sequencing is done preferentially using 2×150bp module with the 300-cycle sequencing kit to read the paired-end-tags with the bridge linker, to capture chromatin interactions. The sequencing reads are processed by ChIA-PIPE, a fully automated pipeline for comprehensive ChIA-PET data analysis and visualization (Lee et al., 2020) (see Basic Protocol 2). For each library, we routinely perform a pilot small scale sequencing of about 1–10 million reads as a final confirmation of the quality of the in situ ChIA-PET library. The initial pilot sequencing data reveals if most of the reads possess the correct DNA template composition (tag-linker-tag), the degree of library complexity, and whether mapping data for ChIP-enrichment (peaks) and chromatin interactions (loops) are detectedat expected genomic loci. Once high quality of the library is confirmed, it can be further sequenced deeper with Illumina platform by 2×150bp module for at least 200 million reads for the final data production.
Table1.
An example of qPCR results from QC step #4
| qP CR Samples | Replicate 1 | Replicate 2 | Replicate Average | Δ Ct | Final Δ Ct | |
|---|---|---|---|---|---|---|
| Test Primer Set1 | Input | 23.76 | 23.53 | 23.645 | ||
| Test Primer Set 1 | ChIP | 18.53 | 18.75 | 18.64 | −5.005 | −7.645 |
| TestPrimerSet2 | Input | 23.11 | 23.04 | 23.075 | ||
| Test Primer Set2 | ChIP | 18.74 | 18.53 | 18.635 | −4.44 | −7 08 |
| Negative Primer Set | Input | 23.26 | 23.55 | 23.405 | ||
| Negative Primer Set | ChIP | 26.16 | 25.93 | 26.045 | 2.64 |
Note: numbers in Replicates are Ct values of PCR results for each test primer set in ChIP-DNA or input samples. Δ Ct: differential Ct value between two Ct numbers (ChIP vs. input or Test primer vs. negative primer). ΔΔ Ct: the final differential Ct values for the fold of ChIP enrichment as determined by ChIP-qPCR analysis after calibration with negative primer. According to our experience, a final ΔΔCt above 5 indicates good target enrichment during the ChIP process, directly reflected by sharp binding peaks of the protein in the genome.
Support Protocol 1: Bridge linker preparation
In a in situ ChIA-PET experiment, a DNA oligo linker, called the called bridge linker, is used to connect the two interacting chromatin fragments during proximity ligation. The bridge linker is composed of a double-strand DNA oligonucleotide (18 bp) with a “T” overhang at the 3’ end and phosphorylation at the 5’ end of both strands, and a modified “T” nucleotide with a biotin group in the middle of the top strand.

During proximity ligation, each bridge linker will be ligated to two chromatin fragments, each with an “A” overhang added via the A-tailing reaction after the restriction digestion during chromatin sample preparation. Therefore, during proximity ligation, only the bridge linker and chromatin fragments will be compatible for ligation, whereas self-ligation of bridge linkers and the ligation between chromatin fragments will be prevented. The bridge linkers should be prepared beforehand and can be stored at −20°C for several months.
Materials:
DNA oligonucleotides: (Ordered from IDT or similar, HPLC purification)
| Top oligo | 5’- /5Phos/CG CGA TAT C/iBIOdT/T ATC TGA CT −3’ |
| Bottom oligo | 5’- /5Phos/GT CAG ATA AGA TAT CGC GT −3’ |
TE buffer (Thermo Fisher Scientific, cat.no. AM9849)
RNase-free PCR tube (0.2 ml) (Thermo Fisher Scientific, cat.no. AM12225)
Novex™ TBE Gels, 4–20%, 10 well (Thermo Fisher Scientific, cat.no. EC6225BOX)
25 bp DNA Ladder (Thermo Fisher Scientific, cat.no. 10597–011)
TBE (10 x), RNase-free (Thermo Fisher Scientific, cat.no. AM9865)
Equipment:
PCR machine (Bio-rad, C1000 Touch Thermal cycler)
NanoDrop™ 8000 Spectrophotometer (Thermo Scientific)
SureLock™ Tandem Midi Gel Tank (Thermo Fisher Scientific, cat.no. STM1001)
Protocol Steps
Order the oligos from IDT (or similar) at 250 nmole scale, HPLC purified in desalted form.
Add TE buffer to dissolve the oligos to a final concentration of 100 μM.
Vortex to mix well, then place the tubes at 4°C overnight (without vortexing) to allow oligos to resuspend completely.
- Prepare 5 different ratios of top oligo : bottom oligo (1:1, 1.5:1, 2:1, 1:1.5, 1:2) for testing for best annealing efficiency. Ultimately, the selected ratio will be used for large scale annealing of bridge linkers.Example: for (1.5:1) mix together 7.5 μl of top oligo (100 μM) and 5 μl of bottom oligo (100 μM).
- Run on a PCR machine using the following program:
95 °C 2 min Ramp 95 °C to 75 °C (Rate: 0.1 °C/second) Hold at 75 °C 2 min Ramp 75 °C to 65 °C (Rate: 0.1 °C/second) Hold at 65 °C 2 min Ramp 65 °C to 50 °C (Rate of 0.1 °C/second) Hold at 50 °C 2 min Ramp 50 °C to 37 °C (Rate of 0.1 °C/second) Hold at 37 °C 2 min Ramp 37 °C to 20 °C (Rate of 0.1 °C/second) Hold at 20 °C 2 min Ramp 20 °C to 4 °C (Rate of 0.1 °C/second) Hold at 4 °C Indefinitely until collection Measure the concentration of the annealed bridge linkers using Nanodrop™ 8000 Spectrophotometer.
- Dilute annealed bridge linkers to 200 ng/μl.Important! Keep all annealed bridge linkers cold on ice.
Run 200 ng of each single stranded oligos and 200 ng of the annealed bridge linkers from each of the 5 different annealing conditions on the same 4–20% TBE gel. Usually, the best ratio of top oligo versus bottom oligo is the one that shows the maximal amount of annealed double stand linker with the minimal top or bottom oligo left on the gel. For example, in Figure 4, the best ratio of top versus bottom oligo is 1.5 to 1.
Perform a large-scale annealing reaction with the remaining top and bottom oligos according to the optimal ratio. For this, repeat step 5.
Perform Nanodrop quantification and dilute the annealed bridge linker to 200 ng/μl with TE buffer for use in ChIA-PET protocol (Basic Protocol 1). Store at −20°C.
Figure 4:
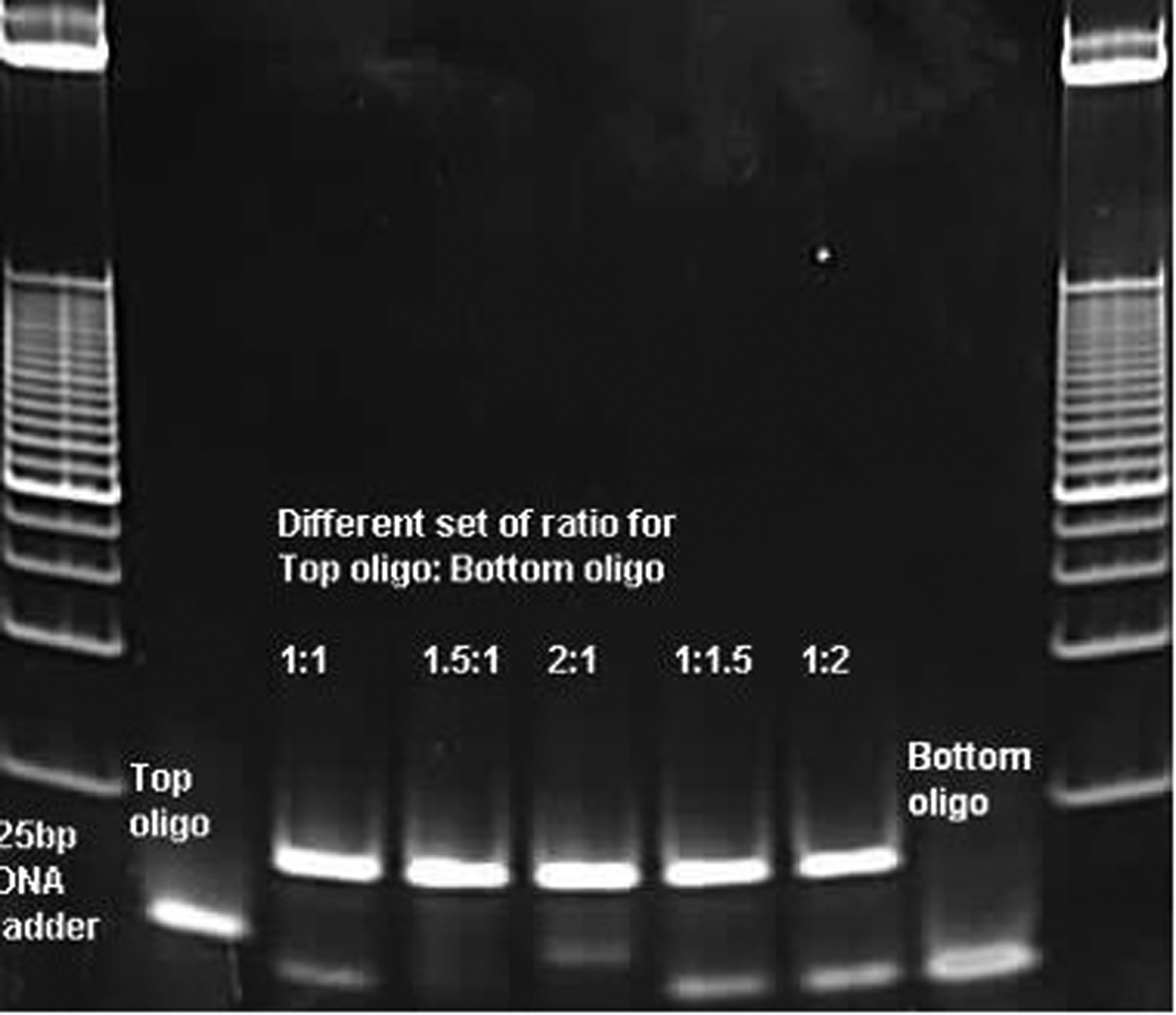
Bridge linker preparation described in Support Protocol 1. A gel picture of electrophoresis for the bridge linker DNA oligos. Sample Lanes 2 and 8 (by the ladders) are for the single strand top and bottom oligo, respectively. Lanes 3–7 are for the annealed bridge linkers under five different annealing ratio of top over bottom oligos, 1:1, 1.5:1, 2:1, 1:1.5, and 1:2, respectively. Lane 1 and 9 show the molecular markers of a 25 bp ladder. As shown in this example, the ratio of top oligo:bottom oligo at 1.5:1 has the best result to get the maximum double stranded linkers. All other ratios showed remaining single strand oligos that were not annealed into double stranded linkers.
BASIC PROTOCOL 2: Data processing and visualization
The workflow for sample preparation and library generation described in Basic protocol 1 results in paired-end raw sequencing reads that need to be further processed to gain biological insights. Here, we provide a step-by-step guide to convert raw sequencing data into processed files that can be visualized and analyzed. By running ChIA-PIPE (Lee et al., 2020), our fully automated ChIA-PET data processing pipeline, in a high-performance computing environment, the user should expect to obtain loops and protein binding profiles. Accompanying web browsers BASIC and Juicebox allow users to visualize the output files from ChIA-PIPE. An overview of the process is shown in Figure 5A. Here, we exemplify the protocol using a small test dataset.
Figure 5:
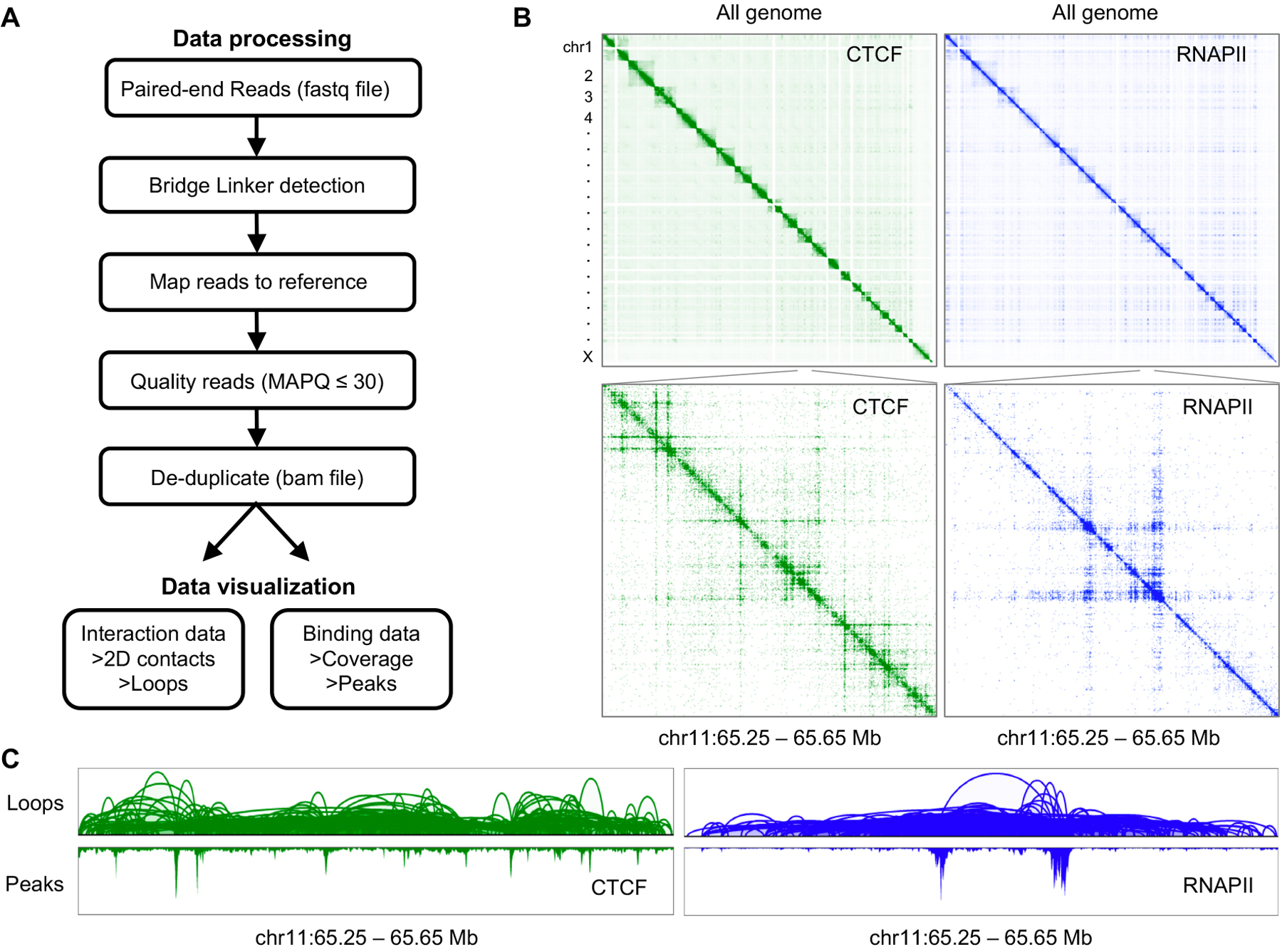
Overview of in situ ChIA-PET data processing and visualization. A. Data processing flowchart. B. Juicebox 2D contact map visualization for the whole genome (top) and a zoom-in genomic region in chr 11 (bottom) of GM10248 cell line for CTCF in situ ChIA-PET (green) and RNAPII in situ ChIA-PET (blue) data. C. BASIC browser view of in situ ChIA-PET loops and binding peak coverage in the region of chr11: 65.25 – 65.65 Mbp for the same proteins.
Materials
-High-performance computing environment, with an ability to allocate up to 60 GB of Random Access Memory (RAM) and 24 hours of runtime.
-A desktop computer or a laptop with internet access for data visualization.
-ChIA-PIPE package (https://github.com/TheJacksonLaboratory/ChIA-PIPE.git) v1.0
- Sequencing results (steps below are shown for a small test dataset, available at Zenodo, DOI: 10.5281/zenodo.4706038)
Protocol Steps
- Make a test directory in the home directory of the high-performance computing environment.
- $ mkdir -p testing_chia_pipe
- $ cd testing_chia_pipe
- Clone the ChIA-PIPE package (v1.0) from the github repository. If git is installed, type
- $ git clone https://github.com/TheJacksonLaboratory/ChIA-PIPE.gitIf the user does not have git installed, download ChIA-PIPE directly: $ wget git@github.com:TheJacksonLaboratory/chia_pipe.zip
- Install the dependencies for ChIA-PIPE.
- $ dep_dir=“dep_dir”
- $ bash local_install_chia_pipe_dependencies.sh -i ${dep_dir}
- Download test data from Zenodo: https://zenodo.org/record/4706038#.YIAx2R0pCHs
- $ mkdir -p fastq
- $ cp LDK0004-ds_*.fastq.gz fastq
Review the config file in chia_pipe-master/example_config_file.sh and ensure that bin_dir is specified according to the directory where ChIA-PIPE has been installed.
- Launch ChIA-PIPE.
- $ qsub -F “--conf chia_pipe-master/example_config_file.sh” chia_pipe-master/0.chia_pipe_hpc.pbs
Wait for the pipeline to finish running. Note that this is expected to take 5–10 hours depending on the user’s computing environment. After the run, there should be a 4.LDK0004-ds.extract_summary_stats.o and a LDK0004-ds.final_stats.tsv file in the /LDK0004-ds/ directory.
- Transfer the 7 key processed files in the /LDK0004-ds/ directory from HPC environment to the local desktop for downstream visualization and analyses.
-
LDK0004-ds.final_stats.tsv (similar examples with the same data format as provided in Table 2)→ A summary statistics table including the total read pairs, uniquely mapped read pairs, number of peaks, and number of PET clusters (loops).
-
LDK0004-ds.e500.clusters.cis.gz→ A list of intra-chromosomal loops in bedpe format, with 7th columns denoting the number of PETs contributing to a particular loop (7th column also referred to as PET count). In other words, this file can be considered as a table with 7 columns: chrom1, start1, end1, chrom2, start2, end2, PETcount.
-
LDK0004-ds.e500.clusters.cis.BE3→ A subset of LDK0004-ds.e500.clusters.cis.gz with 7th column >= 3.
-
LDK0004-ds.e500.clusters.trans.gz→ A list of inter-chromosomal loops in bedpe format, with 7th column denoting PET count.
-
LDK0004-ds.for.BROWSER.sorted.bedgraph→ The protein binding coverage file in a standard 4-column bedgraph format.
-
LDK0004-ds.no_input_all_peaks.narrowPeak→ A list of peaks called by MACS2 in a bed format.
-
ChIA-PET_dm3_Kc167_RNAPII_LDK0004-ds_miseq_pairs.hic→ A binary file that can be visualized through Juicebox (Durand et al., 2018)
-
- Visualize intra-chromosomal loops, binding coverage, and peaks.
- Download and install the dockerized version of the BASIC browser: https://github.com/TheJacksonLaboratory/basic-browser
- Follow the github instructions to upload LDK0004-ds.e500.clusters.cis.BE3 (loops), LDK0004-ds.for.BROWSER.sorted.bedgraph (binding coverage), and LDK0004-ds.no_input_all_peaks.narrowPeak (peaks).Alternatively, the data can be visualized via the WashU Epigenome browser: https://epigenomegateway.wustl.edu
- Visualize inter- and intra-chromosomal interactions through 2D contact maps.
- Transfer *.hic file from the high-performance computing environment to the local drive (e.g., Desktop).
- On a web browser, visit https://aidenlab.org/juicebox/
- Click on “Load Map”, “Local File” and locate the ChIA-PET_dm3_Kc167_RNAPII_LDK0004-ds_miseq_pairs.hic file.
Alternatively, 2D contact maps can be visualized via higlass (https://higlass.io) and the 3D genome browser (http://3dgenome.fsm.northwestern.edu).A summary of the computational data processing procedure is shown in Figure 5A. A 2D contact map visualization is shown in Figure 5B, and Basic browser visualization, in Figure 5C. Our data processing pipeline generates a statistic output (Table 2), which allows quality assessment of the in situ ChIA-PET library data. As an example, Table 2 shows a summary report for a ChIA-PET run for RNAPII and CTCF in GM10248 cells.
Table 2.
An example of two in situ ChIA-PET library statistics output
| Raw | QC matrix | In-house Library ID: LHG0047H | In-house Library ID: LHG0118V | Notes |
|---|---|---|---|---|
| 1 | Seq method (2X150) | Hiseq | Novaseq | |
| 2 | Reference genome | hg38 | hg38 | |
| 3 | Cell type | GM10248 | GM10248 | |
| 4 | Factor | CTCF | RNAPII | |
| 5 | Total read pair | 340,840,469 | 316,510,593 | Library seq depth & quality Higher fraction indicates higher quality of the library |
| 6 | Read pair with bridge linker | 320,834,185 | 286,944,905 | |
| 7 | Fraction of read pairs with linker | 0.94 | 0.91 | |
| 8 | Quality non-redundant tag | 441,205,365 | 74,642,122 | Tags used for peak calling # of peaks varies depending on factors |
| 9 | Protein factor binding peak | 929,221 | 222,016 | |
| 10 | Quality paired-end-tag (PET) | 218,869,947 | 170,109,710 | These PETs are used for interaction analysis Library complexity |
| 11 | Uniquely mapped PET | 162,534,265 | 127,491,848 | |
| 12 | Non-redundant PET | 142,662,939 | 16,107,717 | |
| 13 | PET redundancy | 0.12 | 0.87 | |
| 14 | Self-ligation PET (< 8 kb) | 27,534,605 | 10,575,144 | Chromatin fragment |
| 15 | Inter-ligation PET (> 8 kb) | 115,128,334 | 5,532,573 | Long range interaction |
| 16 | Intra-chr PET | 83,450,947 | 3,997,431 | Most useful data are intra-chr. Higher ratio (>1) indicates higher quality of Interaction data. |
| 17 | Inter-chr PET | 31,677,387 | 1,535,142 | |
| 18 | Ratio of intra/inter PET | 2.63 | 2.6 | |
| 19 | Singleton | 101,710,672 | 4,135,031 | |
| 20 | Intra-chr singleton | 70,457,789 | 2,909,661 | |
| 21 | Inter-chr singleton | 31,252,883 | 1,225,370 | |
| 22 | PET cluster | 4,453,925 | 498,026 | |
| 23 | Intra-chr PET cluster (≥2) | 4,245,707 | 379,818 | Indicator for good data |
| 24 | Inter-chr PET cluster (≥2) | 208,218 | 118,208 | Indicator for noise |
| 25 | Ratio of intra/inter PET cluster (≥2) | 20.39 | 3.21 | Higher ratio means higher quality of the data with more frequently recurrent PET cluster |
| 26 | Ratio of intra/inter PET cluster (≥5) | 1914.56 | 3.82 | |
| 27 | Ratio of intra/inter PET cluster (≥10) | 11409.57 | 9.01 | |
| 28 | Intra-chr PET cluster | |||
| 29 | PET number_2 | 3,097,854 | 263,388 | |
| 30 | PET number_3 | 606,301 | 54,569 | |
| 31 | PET number_4 | 214,162 | 24,180 | |
| 32 | PET number_5 | 101,160 | 13,720 | |
| 33 | PET number_6 | 56,565 | 8,343 | |
| 34 | PET number_7 | 35,610 | 5,186 | |
| 35 | PET number_8 | 23,968 | 3,290 | |
| 36 | PET number_9 | 17,347 | 2,180 | |
| 37 | PET number_10 | 12,873 | 1,298 | |
| 38 | PET number >10 | 79,867 | 3,664 | |
| 39 | Inter-chr PET cluster | |||
| 40 | PET number_2 | 201,273 | 86,018 | |
| 41 | PET number_3 | 6,234 | 15,427 | |
| 42 | PET number_4 | 540 | 6,887 | |
| 43 | PET number_5 | 99 | 3,942 | |
| 44 | PET number_6 | 45 | 2,354 | |
| 45 | PET number_7 | 11 | 1,483 | |
| 46 | PET number_8 | 7 | 871 | |
| 47 | PET number_9 | 2 | 514 | |
| 48 | PET number_10 | 0 | 308 | |
| 49 | PET number >10 | 7 | 404 |
Note: These two examples show a summarized statistical report for a ChIA-PET run for RNAPII or CTCF in GM10248 cells. The key parameters are in bold.
REAGENTS AND SOLUTIONS
0.1% SDS Cell Lysis Buffer
50 mM HEPES-KOH pH 7.5
150 mM NaCl, 1 mM EDTA
1% (w/v) Triton X-100
0.1% (w/v) Sodium Deoxycholate,
0.1% (w/v) SDS
Store at 4 °C for several months
1% FA-DPBS solution (50 ml)
Add 1429 μl of 36% formaldehyde to 50 ml DPBS. Prepare before use.
0.55% SDS solution (10 ml)
Add 55 μl of 10% (w/v) SDS solution to 10 ml of nuclease free water. Prepare before use.
10 mM dATP solution (200 μl)
Add 20 μl of dATP (100 mM) to 180 μl of nuclease free water. Store it at −20 °C freezer.
1× PBST buffer (100 ml)
Add 89.9 ml of ddH2O to 10 ml of 10× PBS (Life technologies, cat. no. AM9937) and 100 μl of Tween 20, and mix them well. Store at 4 °C for several months.
1× Binding & Wash Buffer
5 mM Tris-HCl pH 7.5
0.5 mM EDTA
1 M NaCl
Store at RT for several months.
2× Binding & Wash Buffer
5 mM Tris-HCl pH 7.5
1 mM EDTA
1 M NaCl
Store at RT for several months.
2× SSC/0.5% (w/v) SDS (100 ml)
Put 85 ml of ddH2O,10 ml of 20× SSC solution (Materials), and 5 ml of 10% (w/v) SDS solution (Thermo Fisher Scientific, cat. no. AM9822) together and mix them well. Store at RT for several months.
2 mM EGS-DPBS solution (50 ml)
Take EGS (ethylene glycol bis(succinimidyl succinate)) out of the 4 °C fridge and equilibrate to RT for at least 1 hr. Dissolve 45.63 mg of EGS in 250 μl of DMSO with vortexing. Add the EGS-DMSO solution to 50 ml pre-warmed (37 °C) DPBS. Prepare before use.
2.5 M Glycine solution (100 ml)
Add 27.89 g of glycine to 80 ml of ddH2O and mix well until the solution is clear. Bring the final solution volume to 100 ml. Store at RT for several months.
ChIP Elution Buffer
50 mM Tris-HCl pH 7.5
10 mM EDTA
1% SDS
Prepare before use.
High Salt Buffer
5 mM Tris-HCl pH 7.5
0.5 mM EDTA
1 M NaCl
Store at 4 °C for several months.
LiCl Wash Buffer
10 mM Tris-HCl pH 8.0
250 mM LiCl
1 mM EDTA
0.5% Nonident P-40
0.5% Sodium Deoxycholate
Store at 4 °C for several months.
iBlock buffer (100 ml)
Dissolve 2 g of iBlock Protein-Based Blocking Reagent (Thermo Fisher Scientific, cat. no. T2015) in 90 ml of ddH2O in a 65 °C water bath, add 5 ml of 10% (w/v) SDS (Thermo Fisher Scientific, cat. no. AM9822), and bring volume to 100 ml with ddH2O. Stored at RT for several months.
Sheared genomic DNA mixture
The sheared genomic DNA can be prepared from any species. Usually, genomic DNA is sheared to an approximate size range of 200–1000 kb using Covaris or Bioruptor. Measure the sheared DNA concentration with Qubit assay and store it at −20 °C for several months. Use 500 ng of sheared DNA for each ChIA-PET reaction.
COMMENTARY
Background Information
The original ChIA-PET method (Fullwood et al., 2009) was developed based on ChIP-PET (Wei et al., 2006), which enabled the genome-wide mapping of transcription factor (TF) occupancy. ChIA-PET added a proximity ligation step to the ChIPed chromatin material, thereby allowing users to observe the connectivity between chromatin fragments tethered together by protein factors. It employs dual linkers in proximity ligation to distinguish non-specific ligation, and a type IIS restriction enzyme digestion (MmeI) to generate a 20 bp tag of chromatin DNA on each side of the dual-linker ligation products. The paired-end-tag (PET, tag-linker-tag) templates are subjected to high-throughput paired end sequencing and mapping, to define long-range chromatin interactions. To increase PET mapping efficiency, we employed random digestion by Tn5 transposase of the proximity ligation products, to generate longer templates (tagmentation) for 2×150 bp PET sequencing (Li et al., 2017). This improved approach, called long-read ChIA-PET, significantly increases mapping efficiency and allows for the detection of allele-specific chromatin interactions at single-base-pair resolution (Li et al., 2017; Tang et al., 2015a)
One drawback of the long-read ChIA-PET protocol, however, is that it requires 100 million cells per experiment, thereby limiting its application in the study of primary cells or clinical samples, in which cells of interest are scarce, and tissue source, limited. Inspired by the in situ Hi-C protocol (Rao et al., 2014), we developed an in situ ChIA-PET protocol, in which chromatin materials are first fragmented by in situ restriction digestion, followed by in situ proximity ligation, chromatin immunoprecipitation (ChIP) against a protein factor of interest, and library preparation using Tn5 transposase. The in situ ChIA-PET procedure described in this article allows for a substantial reduction in the required number of cells to as few as 1 million cells per in situ ChIA-PET experiment, thereby making this mapping technique widely applicable to precious samples with low number of cells of interest. The key update in this new method is the improved efficiency of proximity ligation in the intact nuclei compared to ligation in solution. The in situ ChIA-PET data shows significant reduction of non-specific proximity ligations and increased intra- to inter- chromosomal interaction ratios compared to previous versions of the ChIA-PET protocols (Goh et al., 2012; Li et al., 2017). As a result, the in situ ChIA-PET library cost per sample (including library preparation and sequencing costs for 300–400 million reads) is approximately US $1000–1500, which is substantially lower than the previous library cost per sample of ~US $2500–3000.
As part of the ENCODE phase 4 project, the in situ ChIA-PET data generated from various cell lines and primary cells (e.g., human hematopoietic stem cells and primary immune cells) has provided high-resolution 3D chromatin topology on a global scale and helped to comprehensively map functional elements in the mammalian genome (https://www.encodeproject.org/).
Note that two other protocols, HiChIP (Mumbach et al., 2016) and PLAC-seq (Fang et al., 2016) share the same strategy as ChIA-PET for mapping chromatin interactions mediated by protein factors and similar procedures of in situ digestion/ligation as in situ ChIA-PET, but exhibit minor technical differences.
Critical Parameters
Cell number
In situ ChIA-PET is most robust with 10 million cells; it is also feasible, however, with fewer cells, with as few as 1 million cells, with the tradeoff of reduced library complexity and data quality.
Cell crosslinking conditions
Crosslinking is critical to capture chromatin interactions. However, there is a fine balance: too little formaldehyde (FA) could result in loss of expected chromatin interactions, and too much could lead to high background noise. The optimal concentration of FA for crosslinking should be empirically determined. We have used up to 2% FA with successful results. In addition, EGS, as a protein-protein crosslinker, will strengthen the robustness of chromatin complex mediated by proteins indirectly bound to DNA.
Nuclei Permeabilization
Nuclei permeabilization is critically important for successful in situ digestion and ligation, which ensures capturing bona fide chromatin contacts and avoiding non-specific ligation events. Equally important is the ChIP-enrichment for specific chromatin interactions mediated by a particular protein factor under study.
Antibody quality and ChIP
A ChIP-grade antibody of interest protein is required for in situ ChIA-PET. Usually, different assays are recommended to comprehensively characterize and assess the quality of an antibody such as western blot, immunoprecipitation (IP), and ChIP-Seq, with multiple cell lines. Those antibodies that exhibit the highest IP pull-down efficiency and cleanest background should be selected for in situ ChIA-PET experiments.
Quality controls
It is important to implement early QC steps in a ChIA-PET experiment. We have included several QC steps for the wet-lab part of the protocol, and data statistics & quality assessment metrics after data processing. During the experiment, we specifically check for the efficiency of enzyme digestion, ligation, and DNA size distribution after sonication, ChIP, tagmentation, PCR, and library size-selection, to ensure that each step is done correctly and generates the expected results (Figure 3).
Troubleshooting
There are a number of commonly encountered problems in ChIA-PET experiments as revealed by QC analysis during library construction and after library sequencing. In Table 3, we list some of these commonly observed problems, their possible causes, and potential solutions based on our experiences.
Table 3.
Troubleshooting guide for in situ ChIA-PET
| Troubleshooting during sample preparation and data generation (Basic protocol 1) | ||
|---|---|---|
| Problem | Possible reason | Solution |
| Large DNA fragments (>9–10kb) at step 24. | It indicates inefficient restriction digestion. Two possible reasons: a.) The restriction enzyme didn’t function well; b) incomplete nuclei permeabilization, so that the enzyme didn’t enter the nuclei for in situ digestion. |
a.) Check if the correct enzyme was used and its expiration date; b.) Optimize the conditions for nuclei permeabilization by increasing SDS concentration, temperature (up to 62 °C), and incubation time. Check the dynamic changes of nuclear morphology under microscopy to find out optimal conditions. |
| No significant size shift of DNA fragments (at least 2 kb) after the ligation step (step 30), indicating inefficient ligation reaction. | a.) Ligase didn’t work properly; b.) nuclei permeabilization might be too strong. | a.) Check and make sure you are using the ligase properly; b.) Try gentler conditions for nuclei permeabilization. Examine the nuclei under the microscope, and make sure most nuclei are still intact after the permeabilization step. |
| Low ΔΔCt < 5) in step 49, indicating poor ChIP enrichment by the antibody used in ChIA-PET | a.) Low quality (not ChIP-grade) antibody; b.) Crosslinking might not be strong enough, so that the target protein and DNA were not crosslinked; c.) Permeabilization conditions were too strong, so that they disrupted the protein-DNA interaction |
a.) Use fully characterized antibody for (ChIP grade), test ChIP efficiency, and identify the best batch of antibody for further use; b.) Increase the concentration of FA in the crosslinking step; c.) Try gentler conditions for nuclei permeabilization. |
| Too many large DNA fragments (≥ 1kb) in step 53, indicating improper tagmentation | The ratio of DNA vs. Tn5 transposase was suboptimal, too much DNA and not enough Tn5 transposase in the tagmentation reaction | Increase the amount of Tn5 transposase and perform a titration test to determine the best ratio in tagmentation for the optimal size of DNA fragment (300–800 bp). |
| Low DNA yield of PCR product for the final ChIA-PET library in step 69. (< 30 ng after 13 cycles PCR amplification) | a.) Not enough cells as starting material; b.) Poor ligation efficiency due to suboptimal conditions for nuclei permeabilization c). Poor ligation efficiency due to improper preparation of the bridge linke) |
a.) Increase cell numbers; b.) Try gentle conditions for permeabilization; c.) Change to a new prep of bridge linker. |
| Troubleshooting after DNA sequencing analysis based on ChIA-PET library statistic output (Table 2) | ||
| Low fraction of read pairs with bridge linker (<70%) in raw 7 (Fraction of read pairs with linker) | a.) The size of DNA templates in the final library was too big (>0.8 kb), so that the sequencing may not read through to the linker; b.) inefficient washing of streptavidin beads for the enrichment of ligated DNA fragments carrying the biotin-linker for ligation products. |
a.) Increase the amount of Tn5 transposase to optimize tagmentation reaction, to ensure that most DNA fragments in the final library are in the size range of 300–600 bp; b.) use freshly made I-block buffer and wash buffer to ensure the enrichment of ligation products (step 47) |
| Final library with high redundancy or low complexity in raw 13 (PET redundancy) | a.) Not enough starting material; b.) Too much PCR amplification (more than 13 PCR cycles); c.) The library was over sequenced. |
a.) Increase the number of cells for starting material; b.) Make sure you use the minimum number of PCR cycles (< 13 PCR cycles); c.) Do not over sequence. |
| Low ratio of intra-chr/inter-chr PET (< 1) in raw 18 (Ratio of intra/inter PET), indicating more non-specific interaction data than expected. Most inter-chr data were non-specific due to technical or biological noise | a.) The nuclei permeabilization conditions might be too disruptive, leading to the rupture of nuclei, resulting in most ligation happening in nuclei-free solution, instead of in situ inside intact nuclei. | a.) Use gentler conditions for nuclei permeabilization, including decreased concentration of SDS in solution and reduced temperature of incubation. |
Understanding Results
In situ ChIA-PET yields genome-wide chromatin contacts mediated by a specific protein (e.g. CTCF or RNAPII), along with the protein binding coverage. By running our data processing pipeline, ChIA-PIPE, with default parameters (Lee et al., 2020), detailed summary statistics of the in situ ChIA-PET library will be generated (example in Table 2, see below), and chromatin contacts are saved in a bedpe file format and are visualized in our BASIC Browser as arcs between two genomic loci with the loop height denoting the interaction frequency (Fig. 5C). The protein binding profile (akin to that from a ChIP-seq experiment) is saved as bedgraph and bigwig files, which can be visualized in any genome browser. Thus, ChIA-PET offers two layers of information: chromatin contacts associated with a specific protein, and that protein’s binding coverage. ChIA-PIPE also produces a 2D contact map file in .hic file format to be visualized on Juicebox (Fig. 5B), and a peak file (bed file format) generated by either SPP (Kharchenko et al., 2008) or MACS2 (Zhang et al., 2008) peak callers.
As mentioned, after the library is sequenced, ChIA-PIPE automatically processes the sequencing reads and generates a summary statistics report for quality assessment (Table 2). Key quality metric values are: (1) fraction of read pairs with linker, indicating the percentage of sequencing reads with bridge linkers; (2) redundancy, which quantifies the library complexity; and (3) number of intra-chromosomal and inter-chromosomal PETs. A high-quality library should have a high fraction of read pairs with linker, low redundancy, a low number of inter-chromosomal PETs, and a high number of intra-chromosomal PETs (Lee et al., 2020). We recommend performing a shallow QC sequencing (5–10 million sequencing reads) for quality assessment before a large-scale sequencing experiment (~200 million sequencing reads).
Below we comment on some of the parameters included in the summary statistics report and their meaning:
Total read pair: For decent coverage, we routinely aim for 200–500 million paired end reads (2X150 bp) for each library dataset.
Read pair with bridge linker: we use bridge linker to connect two chromatin fragments in the ChIA-PET protocol. The higher the fraction of read pairs containing the linker, the higher the number of chromatin interactions that will be captured. Usually, the fraction value is around 0.7–0.9.
Non-redundant PET: The most useful data for chromatin interaction is the uniquely mapped non-redundant PET. The more the better. Usually >10 million non-redundant PET per dataset will provide robust interactions data.
PET redundancy: The PET redundancy reflects the complexity of a library. Lower PET redundancy means higher library complexity. Most ChIA-PET libraries would reach plateau after total reads >500 million.
Intra-chr PET: The intra-chromosome PETs are the most useful data for discovery of cis-acting elements involved in chromatin interactions. The number of PETs varies in the range of millions to 100s of millions depending on library sequencing depth and protein factors tested.
Ratio of intra/inter PET: This is a very useful value to assess the noise level of proximity ligation. Most inter-chromosome PET are derived from random non-specific ligation, except for possible translocation events in cancer cells and likely rare specific inter-chr contacts, which will be analyzed separately according to your research interest (not included in this protocol). In our experience, most of the high-quality ChIA-PET library datasets have a ratio value higher than 2, meaning more intra-chromosome PETs than inter-chromosome PETs.
Intra-chr PET cluster: The cluster of PETs are a group of PETs that are overlapped in mapping alignment to the reference genome, indicating that these PETs are derived from chromatin interactions of the same loci and repeatedly captured in different cells. The minimal PET count of a cluster is 2, and could be up into the 1000s. The number of a PET cluster measures the relative frequency of chromatin interactions between two loci in a cell population. A higher PET count in a cluster suggests higher contact frequency and higher confidence in the interaction data.
Ratio of intra/inter PET cluster: The PET clusters derived from inter-chr PET data can be used as random background noise, and the ratio of intra/inter PET cluster (at least > 1) provides the best indicator of the quality of a ChIA-PET library.
Based on this, and as shown in Table 2, the quality of the CTCF ChIA-PET library data is higher, but that of the RNAPII ChIA-PET library is also high for capturing RNAPII-associated chromatin interactions.
Time Considerations
As shown in Figure 3, the experimental protocol can be completed in 5 days. The protocol recommends overnight restriction digestions and proximity ligations to achieve better results, even though incubation times can be as short as 2 hr at 37 °C for enzyme digestion and 4 hr at RT for ligation. Several stopping points are possible after the DNA is purified, and are highlighted in the protocol. The data processing time generally takes 1 – 3 hours for 10 million reads and 1 – 2 days for 300 million reads using a multi-threads high performance computer (Threads = 10 – 20, Memory = 20 – 60GB, Cluster OS = CentOS 6.5, CPU = Intel Xeon E5–2670 @ 2.60GHz).
Acknowledgements
This work is mainly supported by ENCODE Phase 4 project (UM1-HG009409).
Footnotes
CONFLICT OF INTEREST STATEMENT:
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT:
For research transparency, we make available the in-house ChIA-PET pipeline and BASIC browser scripts in the following Github repository:
https://github.com/TheJacksonLaboratory/ChIA-PIPE.git
https://github.com/TheJacksonLaboratory/basic-browser
The data used on Basic Protocol 2 is openly available in Zenodo, at http://doi.org/10.5281/zenodo.4706038,.
Literature Cited
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-P, Tanay A, et al. 2017. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171:557–572.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. 2015. Chromatin architecture reorganization during stem cell differentiation. Nature 518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, and Ren B 2016. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research 26:1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Han X, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. 2009. An Oestrogen Receptor α-bound Human Chromatin Interactome. Nature 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh Y, Fullwood MJ, Poh HM, Peh SQ, Ong CT, Zhang J, Ruan X, and Ruan Y 2012. Chromatin Interaction Analysis with Paired-End Tag Sequencing (ChIA-PET) for Mapping Chromatin Interactions and Understanding Transcription Regulation. Journal of Visualized Experiments:3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Dadon DB, Powell BE, Fan ZP, Borges-Rivera D, Shachar S, Weintraub AS, Hnisz D, Pegoraro G, Lee TI, et al. 2016. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 18:262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, and Park PJ 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature Biotechnology 26:1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Wang J, Cai L, Kim M, Namburi S, Tjong H, Feng Y, Wang P, Tang Z, Abbas A, et al. 2020. ChIA-PIPE: A fully automated pipeline for comprehensive ChIA-PET data analysis and visualization. Science Advances 6:eaay2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi H-S, Tennakoon C, et al. 2010a. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biology 11:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi H-S, Tennakoon C, et al. 2010b. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biology 11:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Sun T, Chang H, Cai L, Hong P, and Zhou Q Chromatin interaction analysis with updated ChIA- PET Tool (V3). 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Luo OJ, Wang P, Zheng M, Wang D, Piecuch E, Zhu JJ, Tian SZ, Tang Z, Li G, et al. 2017. Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nature Protocols 12:899–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Deplancke B, Walhout AJM, and Tissenbaum HA 2008. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nature Protocols 3:698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY 2016. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods 13:919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Heidari N, and Snyder MP 2015. Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics 31:3092–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read JE 2017. Chromatin Immunoprecipitation and Quantitative Real-Time PCR to Assess Binding of a Protein of Interest to Identified Predicted Binding Sites Within a Promoter. In Mammalian Synthetic Promoters (Gould D, ed.) pp. 23–32. Springer; New York, New York, NY: Available at: 10.1007/978-1-4939-7223-4_3. [DOI] [PubMed] [Google Scholar]
- Sima J, Chakraborty A, Dileep V, Michalski M, Klein KN, Holcomb NP, Turner JL, Paulsen MT, Rivera-Mulia JC, Trevilla-Garcia C, et al. 2019. Identifying cis Elements for Spatiotemporal Control of Mammalian DNA Replication. Cell 176:816–830.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. 2015a. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. 2015b. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Tang Z, Lee B, Zhu JJ, Cai L, Szalaj P, Tian SZ, Zheng M, Plewczynski D, Ruan X, et al. 2020. Chromatin topology reorganization and transcription repression by PML-RARα in acute promyeloid leukemia. Genome Biology 21:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C-L, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. 2006. A Global Map of p53 Transcription-Factor Binding Sites in the Human Genome. Cell 124:207–219. [DOI] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. 2017. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171:1573–1588.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]