

Abstract
EUK-207 is a synthetic superoxide dismutase/catalase mimetic that has been shown to reverse age-related learning deficits and brain oxidative stress in mice. In the present experiments, we tested the effects of EUK-207 on oxygen/glucose deprivation (OGD)-induced cell death in cultured hippocampal slices and on several mechanisms that have been postulated to participate in this process. Cultured hippocampal slices were subjected to 1 h OGD followed by 3 or 24 h recovery in regular medium with glucose and oxygen. Lactate dehydrogenase (LDH) release in culture medium and propidium iodide (PI) uptake in slices were used to evaluate cell viability. When EUK-207 was applied either 1 or 2 h before OGD, OGD-induced LDH release was significantly reduced. When EUK-207 was applied 1 h before OGD and during 24 h recovery, PI uptake was also reduced. OGD-induced accumulation of reactive oxygen species (ROS) was evaluated with the fluorescent probe DCF. DCF fluorescence in slices increased steadily during OGD treatment, rapidly disappeared following return to regular medium before slowly increasing again during the 24 h recovery period. When measured 3 h after OGD, increased ROS levels were significantly reduced by EUK-207. OGD also increased lipid peroxidation levels and this effect was also reduced by EUK-207 6 h following OGD. Cytosolic cytochrome c and nuclear apoptosis-inducing factor (AIF) were increased 3 h after OGD, and the translocation of AIF from mitochondria to nucleus was partly blocked by treatment with EUK-207. In conclusion, EUK-207 provides neuroprotection against OGD-induced cell death in cultured hippocampal slices. As EUK-207 prevents free radical formation and lipid peroxidation, the neuroprotection is related to elimination of free radical generation and lipid peroxidation, as well as to decreased activation of pro-apoptotic factors. Our data support the further clinical evaluation of this class of molecules for the prevention of ischemic cell damage.
Keywords: EUK-207, oxygen/glucose deprivation, ischemia, reactive oxygen species, cultured hippocampal slices, lipid peroxidation, apoptosis
1. Introduction
Cerebral ischemia triggers a variety of pathological events, including release of glutamate followed by overactivation of glutamate receptors, increased intracellular calcium concentration, activation of calcium-dependent protease, excessive generation of reactive-oxygen species (ROS), and ischemic cell death by necrosis and/or apoptosis (Koistinaho and Koistinaho, 2005; Zipp and Aktas, 2006; Won et al., 2002). Ischemia followed by reperfusion induces a number of structural and biochemical changes, and the complex interactions between these changes have hampered progress in understanding the respective roles of the different processes postulated to participate in hypoxia/ischemia-induced neuronal damage (Lipton, 1999; Banasiak et al., 2000). Different experimental models have been used to study ischemia-induced damage, each with their own advantages and drawbacks. In vitro models generally use primary cortical cultures or hippocampal slice cultures that are exposed to N2/CO2 equilibrated bath solution with (anoxia or hypoxia) or without glucose (oxygen/glucose deprivation). While primary neuronal cultures might be somewhat easier to use, organotypic hippocampal slice cultures (OHSC) have the advantage that they represent a more mature preparation and might better mimic the in vivo conditions (Strasser and Fischer, 1995).
Superoxide, and hydrogen peroxide are two important reactive oxygen species (ROS) that are produced mainly within the mitochondria from both complex I and complex III of the electron transport chain (Cadenas and Sies, 1998). At moderate concentrations, ROS are involved in defense against infectious bacteria and in a number of cellular signaling pathways (Valko et al., 2004). However, overproduction of ROS results in damage to cellular lipids, proteins, and DNA, and the generation of ROS has been associated with both necrosis and apoptosis (Henderson et al., 2006; Noh et al., 2006). Free radicals, such as superoxide and hydroxyl radicals, are involved in ischemic cell death, and increased free radical formation has been shown both in global and focal ischemia (Chabrier et al., 1999). A number of traditional antioxidants scavenging various types of ROS have been tested in different diseases associated with increased ROS formation, including ischemia; however, the results obtained in most experiments are rarely convincing, as it has been difficult to establish causal relationships between prevention of ROS formation/accumulation and prevention of neuronal damage.
In the present experiments, we tested the contribution of increased ROS formation in OGD-induced neuronal damage by a detailed analysis of the effects of a superoxide dismutase/catalase mimetic, EUK-207, in cultured hippocampal slices. EUK-207 is a salen-manganese complex of the EUK family that includes EUK-8, the prototype molecule, EUK-134, with significantly higher catalytic activities than EUK-8, EUK-189 and EUK-207, which exhibit similar catalase activity and higher lipophilicity and neuroprotective activity when compared to EUK-134, and some other EUK compounds (Liu et al., 2003; Baker et al., 1998). These compounds have shown efficacy in different disease models associated with increased ROS formation, including reduction of 1-methyl-4-phenylpyridinium (MPP+)-induced toxicity in primary dopaminergic neuronal cultures (Pong et al., 2000), prolongation of survival and reduction of oxidative stress in a mouse model of amyotrophic lateral sclerosis (Jung et al., 2001), protection of cultured cortical neurons from staurosporine-induced apoptosis (Pong et al., 2001), extension of lifespan in Caenorhabditis elegans and superoxide dismutase 2 (sod2) nullizygous mice (Melov et al., 2000; Melov et al., 2001), and reversal of cognitive deficits, protein oxidation, and lipid peroxidation in 11-month-old (Liu et al., 2003) and 23-month-old mice (Clausen et al., 2008). Protective effects of EUK-134 in a rat focal ischemia model have also been reported (Baker et al., 1998). However, the exact mechanisms involved in neuroprotection in the majority of these models need to be further studied.
Ischemia induces cell death either through necrosis or apoptosis, and it has been suggested that mild ischemia induces caspase-3 activation and apoptosis, while severe ischemia elicits mitochondria cytochrome c release and necrosis (Li et al., 1997; Saikumar et al., 1998). Necrosis is a rapid form of cell death that is due to the breakdown of ionic homeostasis, while apoptosis is generally considered as a form of delayed cell death involving activation of genetic programs, although there is no distinct time point during reperfusion following hypoxia/ischemia to separate necrotic and apoptotic cell death (Banasiak et al., 2000). Under normal conditions, the apoptosis-inducing factor (AIF) is localized in mitochondria, and AIF translocation from mitochondria to nucleus in OGD-treated neuronal cultures and in focal and global ischemia induces chromatin condensation, DNA fragmentation and caspase-independent apoptosis (Cao et al., 2003; Komjati et al., 2004; Daugas et al., 2000). In transient and permanent cerebral ischemia, cytochrome c is also released from mitochondria to cytosol (Perez-Pinzon et al., 1999; Sugawara et al., 1999), where it binds to Apaf-1, recruits procaspase-9 to form the apoptosome, and initiates further apoptotic cascades (Li et al., 1997). These mechanisms were studied in the present report and our results indicate that increased ROS formation, especially during the OGD period, plays an important causal role in ischemic cell death in cultured hippocampal slices.
2. Results
2.1. EUK-207 attenuates OGD-induced cell death in organotypic hippocampal slice cultures
To test the effects of EUK-207 on OGD-induced cell death, cultured hippocampal slices were subjected to 30, 40, or 60 min of OGD followed by 3 or 24 h recovery in regular medium with glucose and oxygen, in the absence or presence of EUK-207 (40 μM, applied 1 h before OGD and during OGD), and LDH release was measured at the end of the recovery periods. Compared to control, 30, 40, or 60 min OGD followed by 3 h recovery induced a 2.50 ± 0.32, 4.25 ± 0.46, or 12.0 ± 1.2 fold increase in LDH release respectively; LDH release elicited by 60 min OGD was significantly reduced by EUK-207 (7.12 ± 0.74, p <0.01, Fig. 1A, n=6). Compared to control, 30, 40, or 60 min OGD followed by 24 h recovery induced a 3.09 ± 0.24, 8.64 ± 0.71, or 20.0 ± 1.6 fold increase in LDH release respectively; LDH release elicited by 60 min OGD was also attenuated when hippocampal slices were pre-treated with EUK-207 (15.0 ± 1.0, p <0.05, Fig. 1B, n=6). In view of these results, we selected 60 min OGD for the remaining of the experiments, except when otherwise indicated. Cultured hippocampal slices were subjected to OGD followed by 3 h or 24 h recovery in the absence or presence of various EUK-207 concentrations (1 μM, 10 μM, 40 μM, or 100 μM), applied 1 h before and during OGD), and LDH release was measured at the end of the recovery periods. Euk-207 treatment produced a concentration-dependent decreased in OGD-induced LDH release, although only the effect of 40 μM EUK-207 was statistically significant (Fig. 2).
Figure 1:
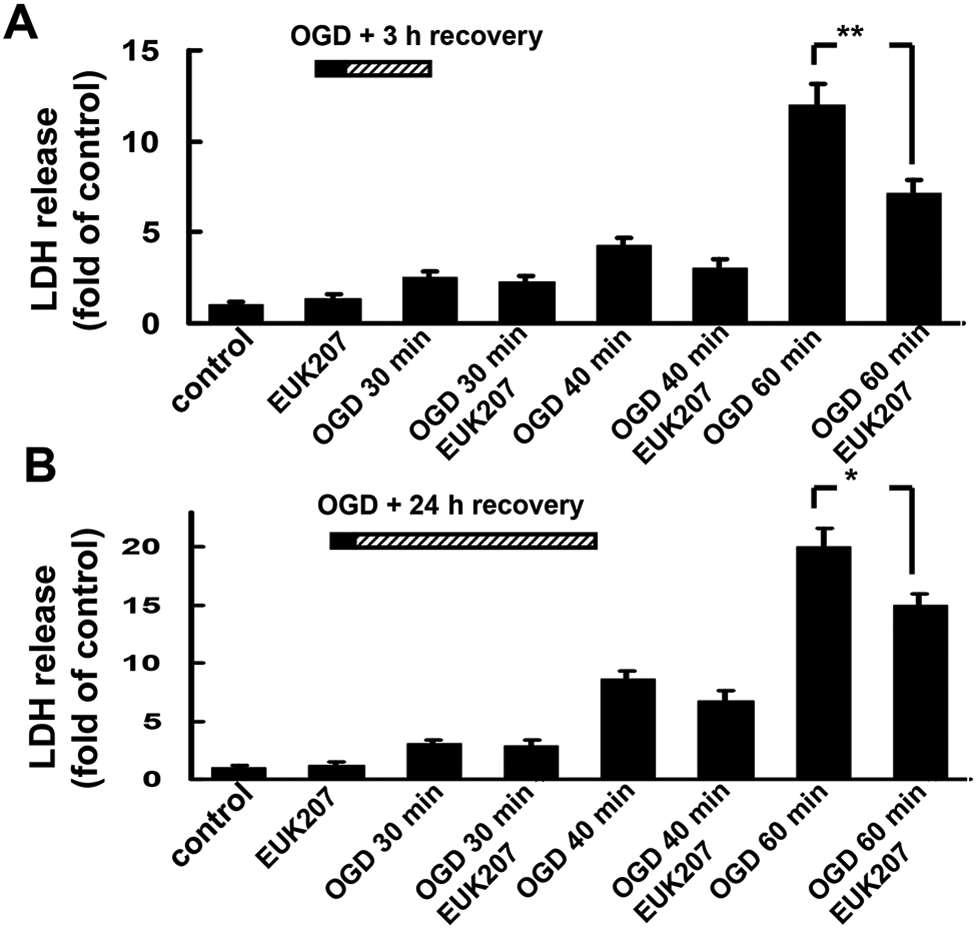
Effects of EUK-207 on LDH release elicited by different periods of OGD in organotypic hippocampal slice cultures.
(A) Hippocampal slices were subjected to 30, 40, or 60 min of OGD followed by 3 h recovery in regular medium with glucose and oxygen, in the absence or presence of EUK-207 (40 μM, applied 1 h before and during OGD). LDH release was measured at the end of 3 h recovery.
(B) Hippocampal slices were subjected to 30, 40, or 60 min of OGD followed by 24 h recovery in regular medium, in the absence or presence of EUK-207 (40 μM, applied 1 h before and during OGD). LDH release was measured at the end of 24 h recovery.
Results are expressed as fold of increase over the respective control values and are means ± S.E.M. of 6 experiments. Statistical analysis was done by student’s t-test (* p < 0.05, ** P < 0.01).
Figure 2:
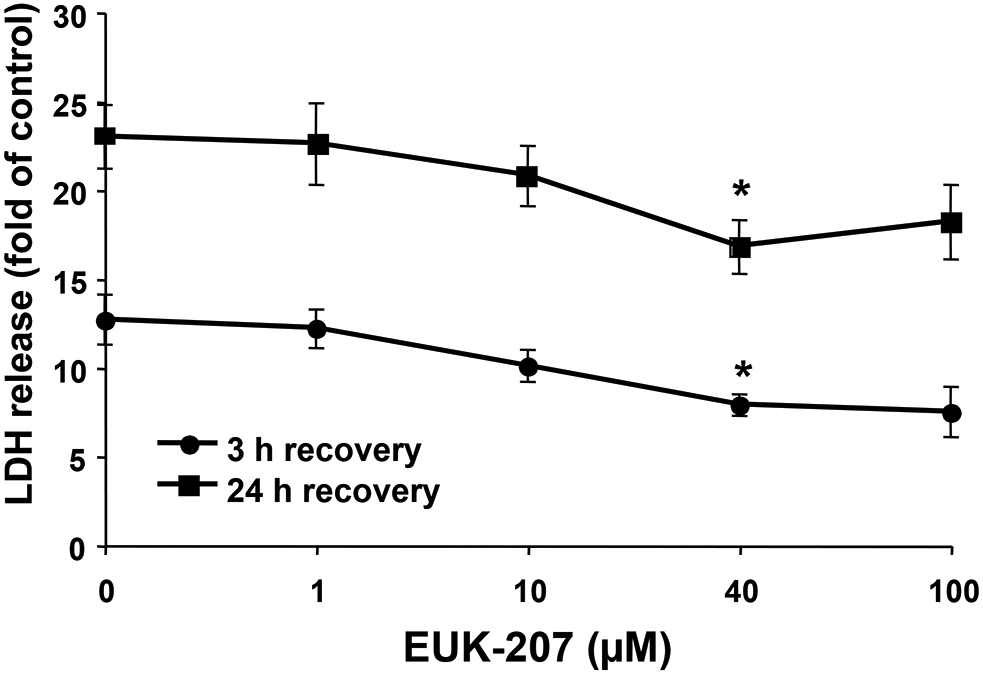
Effects of different concentrations of EUK-207 on LDH release elicited by 1 h OGD in organotypic hippocampal slice cultures.
Hippocampal slices were subjected to 1 h OGD followed by 3 h or 24 h recovery, in the absence or presence of EUK-207 (1 μM, 10 μM, 40 μM, or 100 μM, applied 1 h before and during OGD). LDH release was measured at the end of 3 h or 24 h recovery.
Results are expressed as fold of increase over the respective control values and are means ± S.E.M. of 5 experiments. Statistical analysis was done by one-way ANOVA followed by Tukey’s post hoc test (* P < 0.05 as compared to control slices treated with OGD and no EUK-207).
To determine the best protocol for EUK-207 treatment, cultured hippocampal slices were pretreated with 40 μM EUK-207 for 1 or 2 h before OGD, during OGD, or during the 24 h recovery period (EUK-207 concentration was decreased to 20 μM). Although treatment with EUK-207 during the 24 h recovery period slightly decreased OGD-induced LDH release (86% of OGD alone), only the addition of EUK-207 for 1 or 2 h before OGD significantly reduced OGD-induced LDH release to 70% or 75% of OGD alone (Fig. 3A, n=6). It was of interest to determine the pattern of LDH release during and after OGD. Therefore, we calculated the rate of LDH release (expressed in LDH release/h) during various periods starting at the end of the OGD period and until 24 h of recovery (Fig. 3B). The highest rate of release, and presumably the highest rate of neuronal damage, was observed between 0 and 3 h after OGD. By 6 and 24 h after OGD, the rate of LDH release decreased, but was still significantly higher than in control slices (Fig. 3B).
Figure 3:

Effects of pre- or post-treatment with EUK-207 on OGD-induced LDH release and rate of LDH release at various times after OGD in organotypic hippocampal slice cultures.
(A) Hippocampal slices were subjected to 1 h OGD followed by 24 h recovery in regular medium, in the absence or presence of EUK-207. EUK-207 (40 μM) was applied under various conditions: (1) 2 h before OGD, (2) 1 h before OGD, (3) during OGD, or (4) during 24 h recovery (EUK-207, 20 μM). LDH release was measured at the end of 24 h recovery (* p < 0.05, ** p < 0.01, as compared to OGD group, Student’s t-test).
(B) Rate of LDH release at various times after OGD treatment (* p < 0.001 as compared to 3 h control; † p < 0.01 as compared to 3 h recovery after OGD; Student’s t-test).
Results are expressed as fold of increase over the respective control values and are means ± S.E.M. of 6 experiments (A) or of 8 to 12 experiments (B).
The protective effects of EUK-207 against OGD-induced cell death were also determined by analyzing propidium iodide (PI) uptake. Cultured hippocampal slices were subjected to OGD followed by 24 h recovery in the absence or presence of EUK-207 (40 μM EUK-207 applied 1 h before OGD, and 20 μM EUK-207 applied during 24 h recovery). Compared to control, OGD followed with 24 h recovery significantly increased propidium iodide uptake throughout the hippocampus and this increase was significantly reduced by EUK-207 (Fig. 4). Treatment with EUK-207 alone did not modify PI uptake.
Figure 4:

Effects of EUK-207 on OGD-induced increase in PI uptake in organotypic hippocampal slice cultures.
(A) Cultured hippocampal slices were subjected to OGD for 1 h followed by 24 h recovery in the absence or presence of EUK-207 (40 μM EUK-207 applied 1 h before OGD, and 20 μM EUK-207 applied during 24 h recovery).
(B) Quantification of PI staining. Results are expressed as fold of increase over control values and are means ± S.E.M. of 12 experiments (** p < 0.01, Student’s t-test).
2.2. EUK-207 protects organotypic hippocampal slice cultures from OGD-induced cell death by attenuation of OGD-induced ROS generation, lipid peroxidation and AIF translocation
To determine whether EUK-207 protected cultured hippocampal slices from OGD-induced cell death by eliminating free radical formation, ROS accumulation was evaluated with the fluorescent probe DCF. The amount of free radicals generated during OGD and recovery was studied by measuring DCF fluorescence in hippocampal slices subjected to OGD for 0 (control), 10, 30, or 60 min, as well as in slices observed 5, 10, 30 min, 1 h, 3 h, 6 h, or 24 h after 1 h OGD (Fig. 5A,B). Compared to control, DCF fluorescence increased steadily and markedly with time of OGD treatment; interestingly DCF fluorescence decreased sharply at the beginning of the recovery period, before increasing slowly with further recovery. When hippocampal slices were subjected to OGD followed by 3 h recovery in the absence or presence of EUK-207 (40 μM, 1 h pretreatment before OGD), EUK-207 significantly reduced DCF fluorescence intensity from (Fig. 5C, n=12).
Figure 5:
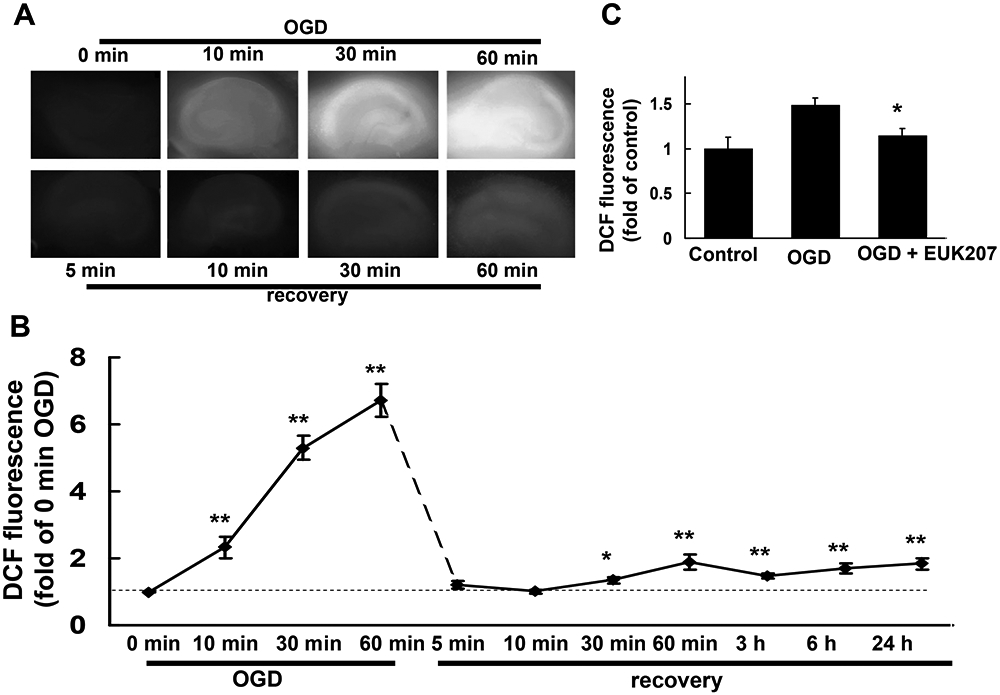
ROS levels, measured with DCF fluorescence during and after OGD.
Cultured hippocampal slices were subjected to OGD for different times, or to 1 h OGD followed by different times of recovery.
(A) Representative images of DCF fluorescence in cultured hippocampal slices subjected to OGD for 0 (control), 10, 30, or 60 min, or 1 h OGD followed by 5, 10, 30, or 60 min recovery.
(B) Quantification of DCF fluorescence intensity (including 3 h, 6 h and 24 h recovery after 1 h OGD). Results are expressed as fold of increase over control (0 min OGD) values and are means ± S.E.M. of 8 to 12 experiments (* p < 0.05, ** p < 0.01 as compared to control, Student’s t-test).
(C) Hippocampal slices were subjected to 1 h OGD followed by 3 h recovery in the absence or presence of EUK-207 (40 μM, applied 1 h before OGD). Results are expressed as fold of increase over control values and are means ± S.E.M. of 12 experiments (* p < 0.05 as compared to OGD group, Student’s t-test).
When excess ROS is generated during ischemia or OGD, free radicals react strongly with unsaturated lipids and cause lipid peroxidation, which decreases cell membrane fluidity and induces cell death. To test the effects of OGD and EUK-207 on lipid peroxidation, cultured hippocampal slices were subjected to OGD and lipid peroxidation was measured with the TBARS assay at 0, 3, 6, or 24 h following OGD. Lipid peroxidation results were normalized to protein levels in slices to minimize differences between slices. Compared to control, lipid peroxidation was increased immediately after OGD treatment (120% of control level), further increased after 6 h of recovery (147% of control level), and remained slightly higher than control at 24 h of recovery (112% of control level, Fig. 6A). EUK-207 (40 μM, applied 1 h before and during OGD) significantly reduced lipid peroxidation measured at 6 h recovery (Fig. 6B, n=6).
Figure 6:
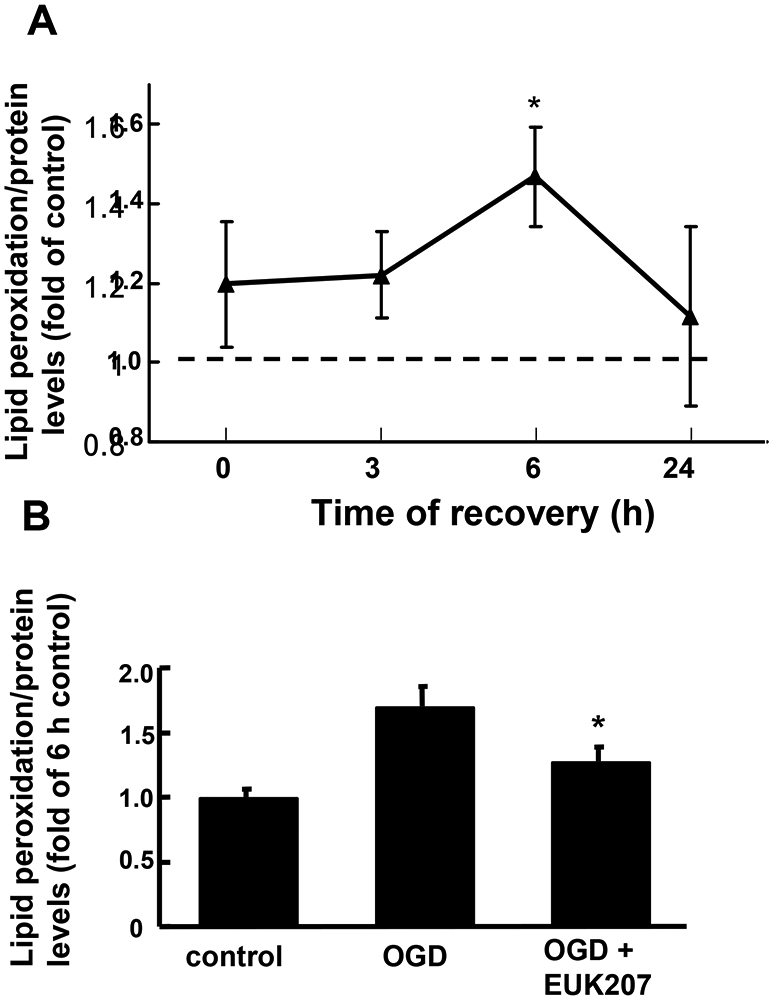
Effects of OGD and EUK-207 on lipid peroxidation in cultured hippocampal slices.
Cultured hippocampal slices were subjected to 1 h OGD followed by different times of recovery. Lipid peroxidation was measured with the TBARS assay and normalized to protein levels.
(A) Lipid peroxidation levels in hippocampal slices subjected to OGD followed by 0, 3, 6, or 24 h recovery. Results are expressed as fold of increase over control (w/o OGD treatment) values and are means ± S.E.M. of 6 experiments.
(B) Hippocampal slices were subjected to 1 h OGD followed by 6 h recovery in the absence or presence of EUK-207 (40 μM, applied 1 h before OGD and during OGD).
Results are expressed as fold of increase over the 6 h control values and are means ± S.E.M. of 6 experiments (* p < 0.05 as compared to OGD group, Student’s t-test).
Hippocampal slices subjected to OGD treatment might undergo cell death either by necrosis or by apoptosis. To determine whether apoptosis contributes to OGD-induced cell death, we evaluated AIF translocation from mitochondria into nucleus, and cytochrome c release from mitochondria into cytosol after OGD. Cultured hippocampal slices were subjected to OGD followed by 3 h recovery in the absence or presence of EUK-207 (40 μM, applied 1 h before and during OGD). At the end of recovery, slices were homogenized and centrifuged, and the nuclear fraction was processed for immunoblotting with an antibody against AIF, while the cytosolic fraction was probed with an antibody against cytochrome c. Compared to control, OGD and 3 h recovery induced an increase in nuclear AIF levels, an effect which was significantly blocked by EUK-207 (Fig. 7A). Levels of cytosolic cytochrome c were also significantly increased 3 h after OGD, and although this effect was reduced by EUK-207, the decrease did not reach statistical significance (p >0.05, Fig. 7B).
Figure 7:
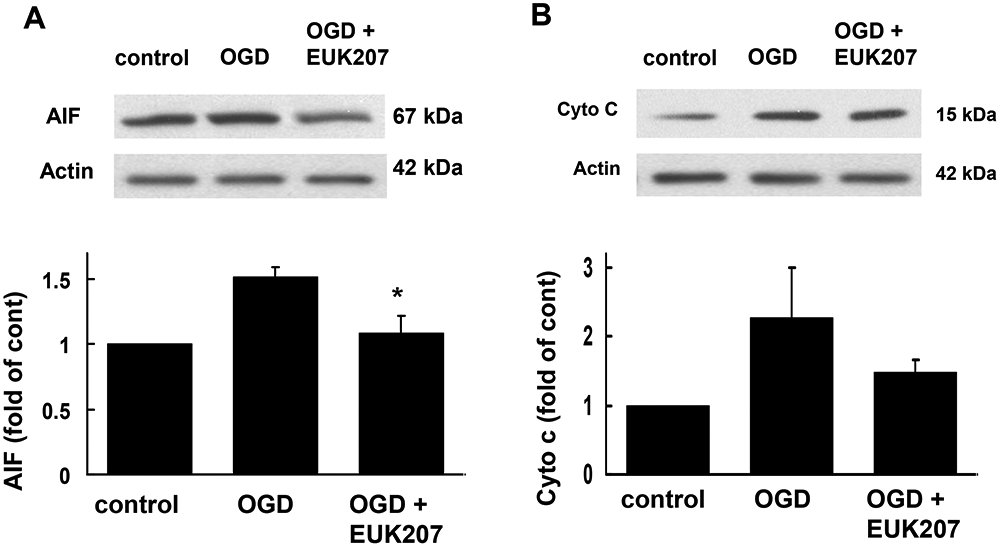
Effects of OGD and EUK-207 on apoptosis-inducing factor (AIF) and cytochrome c (cyto c) levels in different subcellular fractions in cultured hippocampal slices.
Cultured hippocampal slices were subjected to 1 h OGD followed by 3 h recovery in the absence or presence of EUK-207 (40 μM, applied 1 h before OGD and during OGD). At the end of recovery, slices were homogenized, and different subcellular fractions were prepared by differential centrifugation. The nuclear fraction was processed for immunoblotting with an antibody against AIF, and the cytosolic fraction was probed with an antibody against cytochrome c.
(A) Representative images and quantitative analysis of western blots probed with an AIF antibody.
(B) Representative images and quantitative analysis of western blots probed with a cytochrome c antibody.
Results are expressed as fold of increase over the respective control values and are means ± S.E.M. of 3 experiments (A) and of 4 experiments (B) (* p < 0.05 as compared to OGD group, Student’s t-test).
3. Discussion
Both acute and cultured hippocampal slices have been used as in vitro ischemic models because they provide better approximations of in vivo conditions as compared to dissociated neuronal cultures (Strasser and Fischer, 1995). We previously reported that EUK-207 and EUK-189 protected acute hippocampal slices from both 2-month-old and postnatal day 10rats from OGD and recovery-induced cell death (Zhou et al., 2007). We proposed that the protective effects of these synthetic SOD/catalase mimetics involved the elimination of free radicals and the partial reversal of ATP depletion; EUK-207 provided better protection than EUK-189, an effect which could be due to chemical structure differences between the compounds, as EUK-207 has similar catalytic activities but greater biological stability than EUK-189 (Liu et al., 2003). Although not as widely used as cultured slices, acute hippocampal slices represent a good model to study ischemic damage because they can be prepared from both neonatal and mature rats, thus providing a model to study developmental as well as age-related changes in ischemic damage. On the other hand, the use of acute slices prevent the possibility of studying the mechanisms involved in delayed ischemic damage taking place withiin hours to days following the ischemic episode. Therefore, cultured slices represent a better model to study the cellular and functional changes taking place over prolonged periods of recovery that would better reflect events taking place during reperfusion in in vivo models. Moreover, cultured slices exhibit many features of in vivo tissues including morphological organization, receptor expression, and synaptic function (Frotscher et al., 1995; Gahwiler et al., 1997; Stoppini et al., 1991).
Under normal conditions, the rate of ROS formation is equal to that of their elimination. However, during ischemia and reperfusion, this balance is perturbed either due to increased ROS production or decreased activity of cellular defense systems (Valko et al., 2007). Our results indicate that excessive ROS production plays an important role in OGD- and recovery-induced cell death in cultured hippocampal slices, as incubation of cultured slices with EUK-207 for 1 or 2 h before OGD significantly reduced cell death assessed with either LDH release or PI uptake. When EUK-207 was applied only during the recovery period, LDH release was slightly but not significantly decreased as compared to control. Similarly, when EUK-207 was applied only during OGD, little protection was observed. The optimal degree of protection was obtained when EUK-207 was applied before the OGD period, suggesting that the compound needs to be present in the tissue before OGD to exhibit neuroprotection. It is also important to stress that the maximal degree of protection, determined by either LDH release or PI uptake, represented only 30-40%. These results suggest that ROS accumulation is likely to represent only one of the critical factors involved in ischemic cell death, and that although ROS are generated throughout the OGD and the recovery period, there is a critical period at the beginning of the OGD period that triggers events leading to irreversible neuronal damage. This result has obvious consequences regarding the potential use of ROS scavengers for the treatment of stroke in humans, and could account for the recent failure of NXY-059 in clinical trials (Savitz and Fisher, 2007).
While increased free radical formation was found during both 15 min global ischemia (Piantadosi and Zhang, 1996a) and 30 min focal ischemia (Kinuta et al., 1989), it has been generally assumed that more free radicals are generated during reperfusion (Piantadosi and Zhang, 1996; Dirnagl et al., 1995; Oliver et al., 1990). In our experiments, ROS levels measured with DCF fluorescence were increased within 5 min of OGD (data not shown) and increased steadily with increased OGD duration. These results are in good agreement with several recent reports indicating that ROS are generated during period of hypoxia and hypoglycemia (Abramov et al., 2007; Yu et al., 2008; McGowan et al., 2006; Moro et al., 2005). Our results are also consistent with the fact that it is necessary to apply EUK-207 before OGD treatment to achieve maximal protection. Interestingly, enhanced DCF fluorescence dropped sharply at the beginning of recovery and increased again slowly during recovery. Although DCF fluorescence is widely used as an index of oxidative stress due to its high sensitivity, it has been reported that under aerobic conditions, the production of fluorescent DCF from DCFH generates the DCF semiquinone free radical, which could produce superoxide and hydrogen peroxide, thus further increasing DCFH oxidation and leading to more DCF fluorescence (Marchesi et al., 1999; Bonini et al., 2006). In our experiment, DCF fluorescence was generated with OGD treatment in an anaerobic environment, and thus superoxide and hydrogen peroxide formation as by-products should be much smaller than under aerobic conditions. However, due to its superoxide dismutase/catalase activity, EUK-207 appears to interfere with the DCF assay, possibly by generating hydrogen peroxide as an interim substrate. To avoid this interference, cultured slices were treated with EUK-207 1 h before OGD, and DCF fluorescence was measured after 3 h recovery; under these conditions, DCF fluorescence was significantly lower than in untreated slices. This result strongly suggests that EUK-207 protects cultures hippocampal slices from OGD-induced cell death by elimination of ROS accumulation.
The protective effects of EUK-207 against OGD were measured with both LDH release and PI uptake, and these two assays are generally considered as markers of necrotic cell death due to lethal membrane injury (Dursun et al., 2006). After cultured slices were subjected to OGD, LDH release reached its highest level in the first 3 h of recovery, before gradually decreasing at 6 h and 24 h of recovery, suggesting that after 1 h OGD treatment in cultured slices, more necrotic cell death took place in the early period of recovery. In contrast to early cell death through necrosis, apoptosis has generally been considered to be involved in delayed ischemic cell death (Banasiak et al., 2000). In our experiments, activation of pro-apoptotic factors was detected relatively early after OGD treatment. In particular, AIF translocation to the nucleus as well as cytochrome c release from mitochondria was observed at 3 h of recovery. EUK-207 partly reduced OGD-induced cytochrome c release and significantly decreased AIF translocation into nucleus. By inhibition of early pro-apoptotic factor activation, further activation of downstream apoptotic pathways might be attenuated by EUK-207, suggesting that EUK-207 might also reduce apoptosis-induced cell death. Conceivably, the early increased in ROS formation triggered by OGD might lead to activation of apoptotic cascades involving release of pro-apoptotic factors from mitochondria.
In transient forebrain ischemia and permanent focal ischemia, lipid peroxidation levels have been reported to increase in ischemia-sensitive regions such as hippocampus and striatum (Bromont et al., 1989; Sharma and Kaundal, 2007). Both the highly reactive hydroxyl and peroxynitrite radicals can initiate lipid peroxidation. Once generated, lipid peroxidation can break membrane integrity, change membrane permeability, fluidity and ion transport, and induce further ROS production with mitochondrial damage (Green and Reed, 1998; Nigam and Schewe, 2000). In cultured hippocampal slices, when assessed with the thiobarbituric acid assay, lipid peroxidation levels increased after 1 h OGD treatment and peaked at 6 h recovery. EUK-207 significantly reduced OGD-induced increase of lipid peroxidation, a result consistent with the notion that lipid peroxidation is downstream from ROS generation.
In summary, by inhibition of ROS generation during OGD and recovery, EUK-207 attenuated OGD-induced lipid peroxidation and activation of pro-apoptotic cascades, and protected cultured hippocampal slices from ischemic cell death. The partial protection provided by EUK-207 against OGD-induced cell death also indicates that factors other than increased ROS formation are involved in ischemic/hypoxic cell death. Importantly, our results indicate that the critical source of ROS formation in ischemia and reperfusion occurs very early during the ischemic period, and that the clinical use of ROS scavengers might be much more restricted than previously thought.
4. Experimental procedures
4.1. Chemicals
EUK-207 was generously provided by Eukarion, Inc (now Proteome Systems, Ltd, Woburn, MA). Antibodies were obtained from Cell Signaling Technology, Inc. All other chemicals used were purchased from Sigma Chemical Co. (St. Louis, MO, USA), unless indicated otherwise.
4.2. Organotypic hippocampal slice culture
Organotypic hippocampal slice cultures (OHSC) were prepared from postnatal day 7-9 Sprague-Dawley rats according to Stoppini et al. (1991). Hippocampi were rapidly dissected and transverse slices (400 μm thick) were prepared with a McIlwain tissue chopper and placed on porous Millipore membrane inserts in a 6-well plate with each well containing 1 ml culture medium (50% basal medium eagle (BME), 25% horse serum (HS), 25% Earle’s Balanced Salts (EBSS), 1 mM glutamine, 25 mM HEPES, 15 mM glucose, 2 mM NaHCO3, 100 U/ml penicillin and 100 μg/ml streptomycin, pH 7.2). Culture medium was changed three times a week and slices were cultured for 2 weeks at 35 °C in a humidified incubator with 5% CO2 before being used for OGD.
4.3. OGD treatment of cultured hippocampal slices
Cultured hippocampal slices were washed twice with sterile PBS, and then transferred into new six-well plates with each well containing 1 ml of glucose-free and serum-free DMEM pre-saturated with 95% N2–5% CO2 for 60 min. Hippocampal slices were incubated at 36 °C for 1 h in an anaerobic chamber saturated with 95% N2–5% CO2. At the end of the OGD period, hippocampal slices were washed twice in regular serum-free medium and returned to serum-free culture medium for the indicated periods of time.
4.4. Cytosolic and nuclear fraction preparation
Hippocampal slices were manually homogenized using a Kontes pellet pestle in a buffer containing: 20 mM HEPES (pH 7.5), 250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 0.5 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulphonyl fluoride (PMSF), 2 mg/mL leupeptin, and 1:200 protease inhibitor cocktail (Sigma). Homogenates were centrifuged at 500 g for 5 min at 4 °C and the pellet was designated as the nuclear fraction and used for western blots to analyze nuclear AIF. The supernatants were centrifuged at 1,000 g for 10 min at 4 °C, and the pellets were discarded. The resulting supernatants were centrifuged again at 8,000 g for 30 min at 4 °C, and the pellets were again discarded. The resulting supernatants were further centrifuged at 100,000 g for 60 min at 4 °C and the resulting supernatants were designated as the cytosolic fraction for analysis of cytosolic cytochrome c.
4.5. Western blots
Hippocampal slices were homogenized in a lysis buffer containing: 150 mM NaCl, 5 mM EDTA, 1% SDS, 10 mM Tris-HCl (pH 7.4), 0.5 mM phenylmethylsulphonyl fluoride (PMSF), 2 mg/mL leupeptin, and 1:200 protease inhibitor cocktail. After sample preparation, proteins were loaded to each lane of 8% or 12% SDS-PAGE gels and, after separation, proteins were transferred onto PVDF membranes. The PVDF membranes were blocked with 5% non-fat milk at room temperature for 1 h and probed with different primary antibodies (actin, 1:10,000 dilution; AIF and cytochrome c, 1:1,000 dilution) at 4 °C overnight. Membranes were then incubated with secondary antibodies for 1 h and developed with ECL solutions. Western blots were scanned and analyzed quantitatively by densitometry with ImageJ software. Data were generally calculated as fold of control and expressed as means ± SEM. Student’s t-test was used for statistical analyses and only p values < 0.05 were considered as statistically significant.
4.6. Cell viability assays
-LDH assay:
Neuronal damage was assessed by measurement of lactate dehydrogenase (LDH) released into the medium (Bruce et al., 1995; Koh and Choi, 1987). At the end of the recovery, 0.3 ml of medium solution was mixed with 0.7 ml potassium phosphate buffer (100 mM K2HPO4, adjusted to pH 7.5 with KH2PO4). After 20 min, 0.5 ml freshly made sodium pyruvate/NADH solution was added to this solution immediately followed by measuring absorbance at 340 nm at 1 min interval. LDH release was normalized to protein levels and expressed as fold of control.
-PI uptake assay:
Neuronal damage was also assessed with propidium iodide (PI) uptake as previously described (Laake et al., 1999). PI (4.6 μg/ml) was added to culture medium at the beginning of different treatments. PI uptake was visualized using a 5X objective with a microscope fitted with fluorescence detection, and images of PI-labeled slices were captured with a CCD camera; at this magnification, one image was sufficient to analyze an entire hippocampal slice. Fluorescence intensity was estimated by the following method: first, images were adjusted to gray levels and captured with Adobe Photoshop, with the background of images in white and PI-stained structures in black; second, modified images were analyzed quantitatively by densitometry with ImageJ software. Data are generally shown as means ± SEM from the indicated number of independent experiments.
4.7. Evaluation of ROS production in slices
ROS production in slices was determined with the fluorescent probe 2’,7’-dichlorofluorescin diacetate (DCFH-DA). A 20 mM DCFH-DA stock solution was prepared and stored at −20 °C. DCFH-DA was added to culture medium 1 h before the end of treatment, at a final concentration of 10 μM. For the time course experiment during OGD (10, 30, 60 min), DCFH-DA was added to slices before OGD treatment to ensure that all slices were incubated with DCFH-DA for a total time of 1 h. DCF fluorescence was visualized using a 5X objective with a microscope fitted with fluorescence detection, and all images were quickly captured with a CCD camera to prevent changes of OGD-induced DCF fluorescence after light illumination. DCF fluorescence intensity was estimated by the following method: first, images were adjusted to gray levels and captured with Adobe Photoshop, with the background of images in white and DCF fluorescence in black; second, modified images were analyzed quantitatively by densitometry with ImageJ software. Data are generally shown as means ± SEM from the indicated number of independent experiments.
4.8. TBARS assay
Lipid peroxidation was measured with the TBARS assay. After different treatments, hippocampal slices were homogenized in 250 μl of TBARS Homogenization Buffer (THB) containing: 2.5% SDS, 6.25 μM deferoxamine, and 2.5 μM probucol. Then 300 μl of TBA reagent was added to 200 μl of homogenate and the mixture was vortexed for 15 s. The mixed samples were incubated at 95 °C for 1 h, cooled at room temperature for 20 min, and 300 μl 15:1 n-butanol/pyridine solution was added to each sample and vortexed for 30 s. The mixed samples were centrifuged at 4,000 g for 10 min at room temperature. The organic layer (top layer) was collected and 200 μl of each sample was added into a 96-well plate and absorbance was measured at 532 nm.
Acknowledgements
This study was supported by grant NS048521 from NINDS to MB. We thank Anna Knize for experimental help, and Aaron Clausen and Hussam Jourdi for methodological support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abramov AY, Scorziello A, Duchen MR, 2007. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci 27, 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Marcus CB, Huffman K, Kruk H, Malfroy B, Doctrow SR, 1998. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: a key role for reactive oxygen species in ischemic brain injury. J. Pharmacol. Exp. Ther 284, 215–221. [PubMed] [Google Scholar]
- Banasiak KJ, Xia Y, Haddad GG, 2000. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol 62, 215–249. [DOI] [PubMed] [Google Scholar]
- Bonini MG, Rota C, Tomasi A, Mason RP, 2006. The oxidation of 2',7'-dichlorofluorescin to reactive oxygen species: a self-fulfilling prophesy? Free Radic. Biol. Med 40, 968–975. [DOI] [PubMed] [Google Scholar]
- Bromont C, Marie C, Bralet J, 1989. Increased lipid peroxidation in vulnerable brain regions after transient forebrain ischemia in rats. Stroke 20, 918–924. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Sakhi S, Schreiber SS, Baudry M, 1995. Development of kainic acid and N-methyl-D-aspartic acid toxicity in organotypic hippocampal cultures. Exp. Neurol 132, 209–219. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Sies H, 1998. The lag phase. Free Radic. Res 28, 601–609. [DOI] [PubMed] [Google Scholar]
- Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, Graham SH, Chen J, 2003. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J. Cereb. Blood Flow Metab 23, 1137–1150. [DOI] [PubMed] [Google Scholar]
- Chabrier PE, Auguet M, Spinnewyn B, Auvin S, Cornet S, Demerle-Pallardy C, Guilmard-Favre C, Marin JG, Pignol B, Gillard-Roubert V, Roussillot-Charnet C, Schulz J, Viossat I, Bigg D, Moncada S, 1999. BN 80933, a dual inhibitor of neuronal nitric oxide synthase and lipid peroxidation: a promising neuroprotective strategy. Proc. Natl. Acad. Sci. U. S. A 96, 10824–10829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen A, Doctrow S, Baudry M, 2008. Prevention of cognitive deficits and brain oxidative stress with superoxide dismutase/catalase mimetics in aged mice. Neurobiol. Aging [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prevost MC, Leber B, Andrews D, Penninger J, Kroemer G, 2000. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 14, 729–739. [PubMed] [Google Scholar]
- Dirnagl U, Lindauer U, Them A, Schreiber S, Pfister HW, Koedel U, Reszka R, Freyer D, Villringer A, 1995. Global cerebral ischemia in the rat: online monitoring of oxygen free radical production using chemiluminescence in vivo. J. Cereb. Blood Flow Metab 15, 929–940. [DOI] [PubMed] [Google Scholar]
- Dursun B, He Z, Somerset H, Oh DJ, Faubel S, Edelstein CL, 2006. Caspases and calpain are independent mediators of cisplatin-induced endothelial cell necrosis. Am. J. Physiol Renal Physiol 291, F578–F587. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Zafirov S, Heimrich B, 1995. Development of identified neuronal types and of specific synaptic connections in slice cultures of rat hippocampus. Prog. Neurobiol 45, 143–164. [DOI] [PubMed] [Google Scholar]
- Gahwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM, 1997. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 20, 471–477. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC, 1998. Mitochondria and apoptosis. Science 281, 1309–1312. [DOI] [PubMed] [Google Scholar]
- Henderson D, Bielefeld EC, Harris KC, Hu BH, 2006. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 27, 1–19. [DOI] [PubMed] [Google Scholar]
- Jung C, Rong Y, Doctrow S, Baudry M, Malfroy B, Xu Z, 2001. Synthetic superoxide dismutase/catalase mimetics reduce oxidative stress and prolong survival in a mouse amyotrophic lateral sclerosis model. Neurosci. Lett 304, 157–160. [DOI] [PubMed] [Google Scholar]
- Kinuta Y, Kikuchi H, Ishikawa M, Kimura M, Itokawa Y, 1989. Lipid peroxidation in focal cerebral ischemia. J. Neurosurg 71, 421–429. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW, 1987. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J. Neurosci. Methods 20, 83–90. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J, 2005. Interactions between Alzheimer's disease and cerebral ischemia--focus on inflammation. Brain Res Brain Res Rev 48, 240–250. [DOI] [PubMed] [Google Scholar]
- Komjati K, Mabley JG, Virag L, Southan GJ, Salzman AL, Szabo C, 2004. Poly(ADP-ribose) polymerase inhibition protect neurons and the white matter and regulates the translocation of apoptosis-inducing factor in stroke. Int. J. Mol. Med 13, 373–382. [PubMed] [Google Scholar]
- Laake JH, Haug FM, Wieloch T, Ottersen OP, 1999. A simple in vitro model of ischemia based on hippocampal slice cultures and propidium iodide fluorescence. Brain Res. Brain Res. Protoc 4, 173–184. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X, 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489. [DOI] [PubMed] [Google Scholar]
- Lipton P, 1999. Ischemic cell death in brain neurons. Physiol Rev. 79, 1431–1568. [DOI] [PubMed] [Google Scholar]
- Liu R, Liu IY, Bi X, Thompson RF, Doctrow SR, Malfroy B, Baudry M, 2003. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc Natl Acad Sci U S A 100, 8526–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi E, Rota C, Fann YC, Chignell CF, Mason RP, 1999. Photoreduction of the fluorescent dye 2'-7'-dichlorofluorescein: a spin trapping and direct electron spin resonance study with implications for oxidative stress measurements. Free Radic. Biol. Med 26, 148–161. [DOI] [PubMed] [Google Scholar]
- McGowan JE, Chen L, Gao D, Trush M, Wei C, 2006. Increased mitochondrial reactive oxygen species production in newborn brain during hypoglycemia. Neurosci. Lett 399, 111–114. [DOI] [PubMed] [Google Scholar]
- Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B, 2001. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J Neurosci 21, 8348–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ, 2000. Extension of life-span with superoxide dismutase/catalase mimetics. Science 289, 1567–1569. [DOI] [PubMed] [Google Scholar]
- Moro MA, Almeida A, Bolanos JP, Lizasoain I, 2005. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic. Biol. Med 39, 1291–1304. [DOI] [PubMed] [Google Scholar]
- Nigam S, Schewe T, 2000. Phospholipase A(2)s and lipid peroxidation. Biochim. Biophys. Acta 1488, 167–181. [DOI] [PubMed] [Google Scholar]
- Noh HS, Hah YS, Nilufar R, Han J, Bong JH, Kang SS, Cho GJ, Choi WS, 2006. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J. Neurosci. Res 83, 702–709. [DOI] [PubMed] [Google Scholar]
- Oliver CN, Starke-Reed PE, Stadtman ER, Liu GJ, Carney JM, Floyd RA, 1990. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. U. S. A 87, 5144–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Xu GP, Born J, Lorenzo J, Busto R, Rosenthal M, Sick TJ, 1999. Cytochrome C is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J. Cereb. Blood Flow Metab 19, 39–43. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J, 1996. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke 27, 327–331. [DOI] [PubMed] [Google Scholar]
- Pong K, Doctrow SR, Baudry M, 2000. Prevention of 1-methyl-4-phenylpyridinium- and 6-hydroxydopamine-induced nitration of tyrosine hydroxylase and neurotoxicity by EUK-134, a superoxide dismutase and catalase mimetic, in cultured dopaminergic neurons. Brain Res. 881, 182–189. [DOI] [PubMed] [Google Scholar]
- Pong K, Doctrow SR, Huffman K, Adinolfi CA, Baudry M, 2001. Attenuation of staurosporine-induced apoptosis, oxidative stress, and mitochondrial dysfunction by synthetic superoxide dismutase and catalase mimetics, in cultured cortical neurons. Exp Neurol 171, 84–97. [DOI] [PubMed] [Google Scholar]
- Saikumar P, Dong Z, Weinberg JM, Venkatachalam MA, 1998. Mechanisms of cell death in hypoxia/reoxygenation injury. Oncogene 17, 3341–3349. [DOI] [PubMed] [Google Scholar]
- Savitz SI, Fisher M, 2007. Future of neuroprotection for acute stroke: in the aftermath of the SAINT trials. Ann. Neurol 61, 396–402. [DOI] [PubMed] [Google Scholar]
- Sharma SS, Kaundal RK, 2007. Neuroprotective effects of 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox), an antioxidant in middle cerebral artery occlusion induced focal cerebral ischemia in rats. Neurol. Res 29, 304–309. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D, 1991. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182. [DOI] [PubMed] [Google Scholar]
- Strasser U, Fischer G, 1995. Quantitative measurement of neuronal degeneration in organotypic hippocampal cultures after combined oxygen/glucose deprivation. J. Neurosci. Methods 57, 177–186. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH, 1999. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J. Neurosci 19, RC39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J, 2004. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell Biochem 266, 37–56. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J, 2007. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 39, 44–84. [DOI] [PubMed] [Google Scholar]
- Won SJ, Kim DY, Gwag BJ, 2002. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol 35, 67–86. [DOI] [PubMed] [Google Scholar]
- Yu S, Liu M, Gu X, Ding F, 2008. Neuroprotective Effects of Salidroside in the PC12 Cell Model Exposed to Hypoglycemia and Serum Limitation. Cell Mol. Neurobiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Dominguez R, Baudry M, 2007. Superoxide dismutase/catalase mimetics but not MAP kinase inhibitors are neuroprotective against oxygen/glucose deprivation-induced neuronal death in hippocampus. J. Neurochem 103, 2212–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipp F, Aktas O, 2006. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci 29, 518–527. [DOI] [PubMed] [Google Scholar]