

Abstract
Cellular therapies based on induced pluripotent stem cells (iPSCs) come out of age and an increasing number of clinical trials applying iPSC-based transplants are ongoing or in preparation. Recent studies, however, demonstrated a high number of small-scale mutations in iPSCs. Although the mutational load in iPSCs seems to be largely derived from their parental cells, it is still unknown whether reprogramming may enrich for individual mutations that could lead to loss of functionality and tumor formation from iPSC derivatives. 30 hiPSC lines were analyzed by whole exome sequencing. High accuracy amplicon sequencing showed that all analyzed small-scale variants pre-existed in their parental cells and that individual mutations present in small subpopulations of parental cells become enriched among hiPSC clones during reprogramming. Among those, putatively actionable driver mutations affect genes related to cell-cycle control, cell death, and pluripotency and may confer a selective advantage during reprogramming. Finally, a short hairpin RNA (shRNA)-based experimental approach was applied to provide additional evidence for the individual impact of such genes on the reprogramming efficiency. In conclusion, we show that enriched mutations in curated onco- and tumor suppressor genes may account for an increased tumor risk and impact the clinical value of patient-derived hiPSCs.
Keywords: pluripotent stem cells, reprogramming, whole-genome sequencing, genomic integrity, point mutations, INDELs, oncogenes, enrichment, parental cells, endothelial cells
Graphical abstract
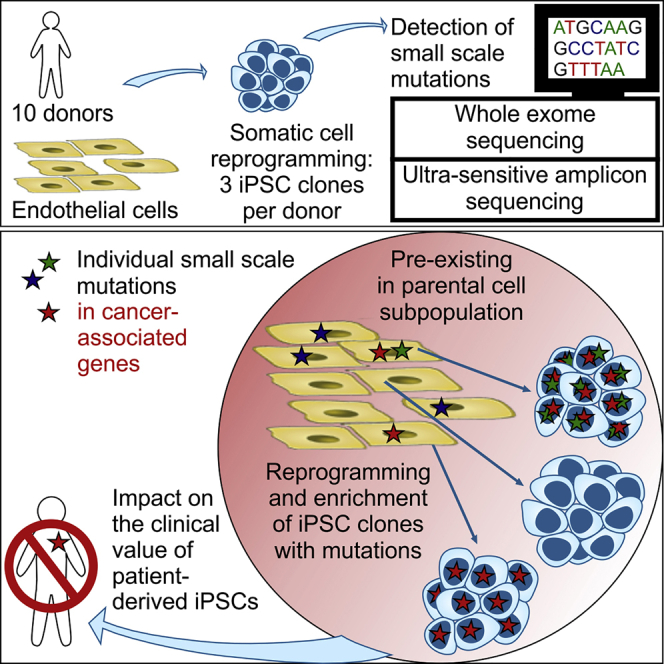
Kosanke et al. demonstrate enrichment of individual small-scale mutations among hiPSC clones during reprogramming. Those mutations pre-existed in small subpopulations of parental cells and affected (cancer-associated) genes of cell-cycle control, cell death, and pluripotency, which may confer a selective advantage but impact the clinical value of patient-derived hiPSCs.
Introduction
The availability of human induced pluripotent stem cells (hiPSCs)1 with their far-reaching potential for proliferation and differentiation offers novel opportunities for the development of tailored cellular therapies. Further research focusing on the genetic stability of reprogrammed cells is required, as considerable numbers of mutations in human iPSCs have been reported, and such genetic abnormalities might harbor the risk of tumor formation.2,3 In general, small and large scale aberrations in iPSCs are thought to have at least three origins: (1) mosaicism of pre-existing variants in the parental cell population, (2) mutagenicity of the reprogramming process itself, and (3) mutagenesis during prolonged culture.
Apparently, larger karyotype abnormalities and copy number variations arise in individual cells during culture expansion,4, 5, 6 while individual ones can provide selective advantages and eventually dominate the population (reviewed by Andrews et al.3 and Martin et al.7).
Besides larger aberrations, a high number of small genetic variants, including single nucleotide variants (SNVs) and insertions and deletions (INDELs), have been detected in iPSCs.8, 9, 10, 11, 12, 13, 14 Diverse experimental designs of recent studies, however, including the choice of somatic parental cell source, the passage number of analyzed iPSCs, the culture conditions, the type of abnormality examined, and the analytic method led to contradictory results and a limited informative value of published data.7 Moreover, while next generation sequencing platforms differ in their error rates and ultimately their detection sensitivities,15 applied computational strategies develop fast and are far from being standardized, which further contributes to different conclusions concerning number and origin of variants in iPSCs.15,16 Therefore, despite all efforts, selective clonal dynamics during reprogramming are poorly understood,17 and the extent, nature, and functional consequences of small genetic variants in iPSCs are still hardly characterized, preventing any adequate assessment of risks associated with cellular therapies.
Several reports have hypothesized that the reprogramming process itself is “mutagenic,” and that small-scale mutations are generated because of inefficient or inaccurate DNA repair mechanisms,18, 19, 20 by replicative or by oxidative stress.10,18,21, 22, 23 Others reported that a substantial part or even the majority of small genetic variants in iPSC lines originate from their individual parental somatic cell clones. The proportion of genetic variants found to pre-exist in the parental cell cultures, however, varies among studies, since the sensitivities of the applied analytic methods differ. While older reports propose a high contribution of 50%–74% of reprogramming-induced small-scale variants to the mutational load of iPSCs,8,10,18,22 more recent studies report that around 90% of variants originate from somatic mosaicism in the parental cell population.13,14 It is still unknown, however, whether the remaining substantial number of undetectable genetic variants represent de novo mutations that have emerged during the reprogramming process18,23 or whether those ones might also pre-exist in the founder cell population as very rare variants not detectable by the applied techniques.12,13
The finding that the majority of small genetic variants in iPSCs originate from their parental somatic cell clones led to the speculation as to whether iPSCs derived from aged donors that accumulated de novo mutations over a lifetime24 may contain increased numbers of genetic aberrations. This speculation could not be confirmed by D’Antonio et al.,14 who did not observe any correlation of donor age and mutational load in iPSCs. In contrast, Lo Sardo et al.11 reported a cumulative number of variants with progressive donor age, and Skamagki19 demonstrated an elevated genomic instability in iPSCs derived from aged donors due to reduction of DNA damage response by reactive oxygen species scavenge.
The parental origin of variants in iPSCs also raises the question whether specific mutations may provide a clonal selection advantage leading to enrichment of such mutations in iPSCs and to iPSC lines with altered cellular functions. In view of the various common characteristics of PSCs and cancer (stem) cells, it can also be presumed that mutations, which provide a selection advantage during the reprogramming process, may lead to an increased tumor-forming potential, in particular if cellular pathways are affected that regulate cell cycling, apoptosis, and pluripotency.25 Although the underlying molecular mechanisms remained largely unclear, this presumption was recently supported by a study of Shakiba et al.17 who observed that an “elite” subset of dominating mouse embryonic fibroblast-derived clones overtook the whole cell population during reprogramming. With that finding, the study challenged the concept of clonal equipotency where all clones have the same potential to attain iPSC state, and suggests that genetically encoded inequalities in cell fitness lead to dominance of otherwise hidden cells in the reprogramming niche.17 While Merkle et al.26 demonstrated a selective advantage of certain small-scale mutations in the p53 gene during culture expansion of PSCs, there is, however, so far no evidence for selection of small genetic variants in any other gene during culture expansion, and recent studies could not demonstrate enrichment of any genetic variants from the parental cells during the reprogramming process.9,11,13,18,27,28
For a more adequate investigation of risks associated with clinical application of iPSC derivatives, we have generated a series of 30 early passage iPSC clonal lines from neonatal and aged individuals under controlled and comparable conditions to allow a systematic analysis for small-scale variants via whole exome sequencing (WES). Importantly, we applied a sequencing technology with higher accuracy than the systems used in previous studies, and an ultra-sensitive amplicon sequencing approach to clarify the origin of detected variants and their frequency in parental cells. The most important aim of our study was to analyze whether individual variants and cell clones that carry such mutations in specific genes are enriched during the reprogramming process, and to what extent such variants are predicted to affect genes critical for cell function or cancer formation, which would call into question the general therapeutic usefulness of iPSCs.6
Results and discussion
Study design and characterization of small genetic variants in iPSCs
iPSC expansion-related enrichment of mutations26 was not in the focus of our study, so we therefore restricted our analyses to early passage (mean P8–P9, range P7–P12) clonal iPSC lines and their corresponding parental endothelial cell (EC) cultures. Overall, 30 iPSC clones (3 clones per donor to investigate inter-clonal variabilities) were analyzed (Figure 1; Table 1). Among them were 27 clones from human umbilical vein ECs (hUVECs) or human cord blood-derived ECs (hCBECs) of 6 neonatal donors, and from human saphenous vein ECs (hSVECs) of 3 aged donors. In case of aged donor D#37, 3 additional iPSC clones derived from human peripheral blood derived ECs (hPBECs) were analyzed (Table 1). For simplicity’s sake, hereinafter we will refer to D#37 hSVEC and D#37 hPBEC as individual “donors.” Direct WES was also performed for parental cells of two neonatal and two aged donors at the same passage as subjected to reprogramming (P4).
Figure 1.

Study design
30 iPSC clones were generated from early passage endothelial cells (ECs), derived from, in total, 6 neonatal (umbilical vein, hUVEC; cord blood, hCBEC) and 3 aged donors (64–88 years, saphenous vein, hSVEC; peripheral blood, hPBEC). iPSC clones in passages 7–12 were subjected to whole exome sequencing (WES). 3 single cell iPSC clones per donor (in case of donor D#37, 3 clones derived from hSVECs and 3 clones from hPBECs) were included to investigate inter-clonal variabilities. Moreover, for 4 donors, variant frequency in the corresponding parental cell population was determined by WES to discriminate between enrichment of pre-existing variants and de novo mutagenesis during reprogramming. Additionally, AFs of selected variants within parental cell populations were assessed by ultra-sensitive amplicon sequencing.
Table 1.
Summary and categorization of small genetic variants detected in iPSCs from neonatal and aged donors
| Variants |
|||||||
|---|---|---|---|---|---|---|---|
| Per donor |
Per iPSC clone |
||||||
| Donor | iPSC clone | Passage | Total number | Polymorphisms and common variants (categories A and B; %) | Donor-specific variants (category C) | Donor-specific variants (category C) | Potentially GAVs (category C GAVs) |
| iPSCs - Neonatal donors | |||||||
| D#1 hUVEC | C2 | P8 | 37106 | 98.7 | 481 | 471 | 249 |
| C5 | P9 | 475 | 252 | ||||
| C6 | P12 | 470 | 247 | ||||
| D#2 hUVEC | C2 | P7 | 36354 | 98.6 | 514 | 507 | 293 |
| C4 | P7 | 504 | 291 | ||||
| C5 | P7 | 503 | 288 | ||||
| D#3 hUVEC | C1 | P9 | 42815 | 98.2 | 762 | 754 | 368 |
| C2 | P10 | 749 | 361 | ||||
| C3 | P9 | 751 | 366 | ||||
| D#22 hCBEC | C15 | P9 | 41226 | 98.4 | 645 | 634 | 296 |
| C17 | P9 | 631 | 296 | ||||
| C21 | P10 | 631 | 295 | ||||
| D#23 hCBEC | C1 | P7 | 34512 | 98.4 | 539 | 533 | 302 |
| C5 | P7 | 530 | 299 | ||||
| C6 | P9 | 531 | 301 | ||||
| D#25 hCBEC | C11 | P8 | 37776 | 98.5 | 559 | 555 | 286 |
| C12 | P8 | 552 | 285 | ||||
| C13 | P8 | 555 | 286 | ||||
| Mean | 38298 | 98 | 583 | 574 | 298 | ||
| iPSCs - Aged donors | |||||||
| D#31 hSVEC (64 years) | C1 | P10 | 35509 | 98.7 | 465 | 454 | 251 |
| C3 | P9 | 452 | 250 | ||||
| C4 | P8 | 450 | 248 | ||||
| D#37 hSVEC (73 years) | C4 | P9 | 40296 | 98.4 | 626 | 594 | 292 |
| C8 | P9 | 600 | 292 | ||||
| C10 | P9 | 602 | 297 | ||||
| D#38 hSVEC (88 years) | C5 | P7+8 | 43292 | 97.8 | 946 | 920 | 291 |
| C6 | P9+10 | 920 | 290 | ||||
| C9 | P8 | 921 | 292 | ||||
| D#37 hPBEC (73 years) | C4 | P8 | 39588 | 98.4 | 620 | 613 | 301 |
| C14 | P9 | 616 | 304 | ||||
| C15 | P10 | 616 | 305 | ||||
| Mean | 39671 | 98 | 664 | 647 | 284 | ||
| iPSCs - All donors | |||||||
| Mean | 38847 | 98 | 616 | 603 | 292 | ||
Exomes from 30 early passage clonal iPSC lines of neonatal and aged donors were sequenced. Categories of variants are as follows: (A) polymorphisms described in any population of GnomAD and 1000 Genomes with minor allele frequency (MAF) ≥ 0.01; (B) potentially common variants not listed as polymorphisms, that were found in more than one donor; and C) donor-specific variants present in iPSCs of one donor only. Potential GAVs compromise all variants in substantial gene regions such as coding and non-coding transcript region, UTRs, and splice regions, after exclusion of intergenic and intron variants.
Importantly, our genome analyses were specifically designed to overcome limitations of recent studies with respect to detection and quantification of rare genetic variants in very small subpopulations of parental cells and to prove potential enrichment of such mutations in iPSCs. The necessity to quantify these rare events against a large background of non-mutated DNA sequences requires highly accurate amplification and sequencing techniques. In light of that requirement, the inaccuracy of the standard “Sequencing by Synthesis” NGS systems with error rates of ∼0.1%–1%15,16,29 is a major limitation for the detection of a specific mutation carried by only a few cells among thousands of non-mutated genomes. In contrast to all previous studies,8, 9, 10, 11, 12, 13, 14,18,20,22,28,30 we developed a study design based on a SOLiD 5500XL system with an Exact Chemistry Call (ECC) module. The SOLiD system is the only system to use “Sequencing by Ligation” technology and has a substantially lower error rate (SOLiD 5500XL with ECC module: lower than 0.01%; SOLiD 5500XL Manual). To further reduce the number of false-positive variants, we sequenced all analyzed iPSC clones twice, and only genomic variants which were detected in both runs, were included in the further analyses. Moreover, we built a variant refinement strategy based on orthogonal validation sequencing (amplicon and Sanger sequencing). Cross-validation of, in total, 133 variants (Tables S1 and S2) confirmed that our approach allowed reliable discrimination between sequencing artifacts and true variants. Although a number of true INDELs did not pass our filter criteria due to low mapping quality, in general INDELs up to 19 bp could be undoubtedly detected with our approach. Figure S1A depicts the whole workflow, while further details on variant calling and refinement strategy are described in Materials and methods. On average, 38,847 variants were detected in iPSCs per donor (Table 1). Direct WES of iPSCs resulted in three categories of detected genetic variants: (A) Polymorphisms described in any population of GnomAD and 1000 Genomes with minor allele frequency (MAF) ≥ 0.01, (B) potentially common variants not listed as polymorphisms that were found in more than one donor, and (C) donor-specific variants present in 1, 2, or 3 iPSC clones of the respective donor (Figure 2A).
Figure 2.

Distribution of small genetic variants in iPSC clones from neonatal and aged donors
(A) Averaged number of total genomic variants in 30 iPSC clones generated from neonatal and aged donors. Mean ± SD; n = 6 neonatal and 4 aged donors. For definition of variant categories, see Table 1. (B) Averaged number of donor-specific variants (category C) per iPSC clone. Data were analyzed with unpaired two-tailed t test; mean ± SD; n = 18 iPSC clones of neonatal donors, 12 of aged donors. (C) Averaged number of donor-specific, potentially gene affecting variants (GAVs, located in coding and non-coding transcripted regions, UTRs, and splice region) per iPSC clone derived of neonatal and aged donors. Data were analyzed with unpaired two-tailed t test; mean ± SD; n = 18 iPSC clones of neonatal donors, 12 of aged donors. (D) Venn diagrams illustrating distribution and intersections of donor-specific GAVs between individual iPSC clones of the different donors. (E) Averaged dispersion of donor-specific GAVs in iPSC clones. Mean ± SD; n = 6 neonatal and 4 aged donors.
In agreement with previous reports,8,9 about 98% of the detected genetic variants were common variants with 94% category A polymorphisms and 4% category B variants leaving 2% donor-specific category C variants (Table 1; Figures 2A and 2B). Despite enrichment for exomes, the pool of variants also included a substantial proportion of variants located in introns and intergenic regions. After elimination of such variants, on average, iPSC clones harbored 292 donor-specific, potentially gene affecting variants (GAVs) defined as variants located in the coding region, start or stop region, UTRs, splice regions, or non-coding transcript regions (Table 1; Figure 2C). While we cannot prove this for D#25 hCBEC C1 and C3, all other iPSC clones clearly represent independent, single-cell-derived clones as they all differ in terms of a number of unique GAVs (Figure 2D; Figure S2).
iPSCs from aged donors do not contain significantly increased levels of total SNVs and INDELs
Lo Sardo et al.,11 who have generated iPSCs from in vitro expanded erythroid progenitors isolated from peripheral blood of adult donors of different age, recently reported increased numbers of small-scale mutations in iPSCs derived from aged donors. While our experimental setting differed in the use of early passage ECs for reprogramming,31 we observed no significant difference in the number of variants in total (38,298 versus 39,671), donor-specific variants (574 versus 647), or GAVs (298 versus 284) per iPSC clone derived from neonatal or aged donors (Table 1; Figures 2A–2C). A slight trend toward a higher number of donor-specific GAVs in iPSCs from aged donors was observed only for those detectable in 1 or 2 iPSC clones, only (Figure 2E). These data are in general accordance with recent reports of D’Antonio et al.14 Interestingly, theoretical calculations demonstrated that the majority of cell divisions in humans already occur before birth:32 while there is an estimated number of 1013–1014 cells in a newborn32,33 that require at least 45 cell divisions to be generated, 1016 cells are produced in total over the whole lifetime,32 which corresponds to an average of only ∼10 additional divisions per cell after birth. On top of this calculation, one has to consider that the reprogrammed ECs, both from neonatal and aged donors, underwent at least 10 sequential cell divisions between isolation from donor tissue and reprogramming of the expanded early passage cultures. Altogether, this may explain why we did not observe significant differences in the total number of mutations between neonatal and aged individuals.
Whether the contrary findings of Lo Sardo et al.11 can be explained by the different sequencing technologies applied or have their basis in the different parental cell types that have been reprogrammed remains to be investigated. Actually, other studies indicated that the type of original somatic cell source and innate mosaicism may influence the mutational load of iPSCs.13,14,20
Highly sensitive amplicon sequencing argues against appreciable contribution of de novo mutagenesis to small-scale genetic variants detected in iPSCs
The majority of donor-specific variants in iPSCs (96.6%) including GAVs (95.1%) were detected in all 3 iPSC clones per donor representing donor-specific homo- or heterozygote variants (Figures 2D and 2E; Table S3). A minority of variants, however, were detected in only 1 or 2 out of 3 iPSC clones from the respective donors (Figures 2C and 2D; Table S3), a result that could be explained by three scenarios: (1) genetic heterogeneity among the primary parental cell populations, (2) de novo mutation during reprogramming, and (3) reversal of heterozygous genetic variants, e.g., by loss of heterozygosity (LoH)34 during reprogramming. Although the latter scenario cannot be entirely excluded, mitotic LoH is unlikely because it is a very rare event.35
Direct WES of primary parental EC populations of 2 neonatal (D#2 hUVEC and D#3 hUVEC) and 2 aged (D#37 hSVEC and D#38 hSVEC) donors was performed, in particular, to exemplify the origin of donor-specific category C GAVs detected in iPSCs and to obtain evidence for the potential enrichment of rare variants from the parental cells. The sensitivity of the applied direct WES approach reached down to 10% of the diploid parental cell population (equals AF 0.05, Materials and methods).
Overall, merely 7.1% of the donor-specific GAVs (80 SNVs and 5 INDELs, Figure 3A; Table S4) could not be detected in the parental cell populations using this approach. Beside 7 GAVs, pre-existence of all variants detected in 3 out of 3 iPSC clones of the 4 donors in the founder cell population could be demonstrated by WES (Figure 3B; Table S4). In contrast, it was not possible to statistically assure by WES the pre-existence in parental cells of any variant detected in 1 or 2 iPSC clones, only. According to these variants analyzed in parental cells from donors D#2 hUVEC, D#3 hUVEC, D#37 hSVEC, and D#38 hSVEC, also all variants detected in 1 or 2 out of 3 iPSC clones of the remaining 6 donors including 64 GAVs were generally considered as undetectable in the parental cells by WES with frequencies below 10% (Table S5). The variants undetectable by WES could be either absent in the founder cell population or present at allelic fractions below the sensitivity of our direct WES approach.
Figure 3.

Reprogramming enriches for pre-existing genetic variants
(A) The origin of donor-specific, potentially GAVs in iPSC clones was investigated in 4 donors by WES of the corresponding parental cell population. The majority of variants pre-existed in the parental cell population (detection limit of WES: 0.05 AF). Mean ± SD; n = 2 neonatal and 2 aged donors. (B) Number of donor-specific GAVs present in 1, 2, or all 3 iPSC clones that are pre-existent or not detectable by WES in parental cells. Mean ± SD; n = 2 neonatal and 2 aged donors. (C) The pre-existence of 100 GAVs (45 polymorphisms, blue), 6 common variants found in several donors (included in “Polymorphisms” in blue), and 49 donor-specific variants in the parental cell population was validated by amplicon sequencing (Table S2). The AF of GAVs in the parental cell population is plotted against their presence in 1, 2, or 3 iPSC clones. Donor-specific GAVs were found shared between 2 or 3 iPSC clones of one donor as well as being unique to only 1 clone. Donor-specific GAVs detected in all 3 iPSC clones were mostly frequent in the parental cell populations (black). Other donor-specific GAVs that were present in a small subpopulation in the respective parental cell population but were detected in 2 or 3 iPSC clones have evidently become enriched during reprogramming (“donor-specific rare GAVs, enriched” in red). Donor-specific rare GAVs that were unique to 1 iPSC clone are termed “donor-specific rare GAVs, potentially enriched” and are colored green.
To assess their pre-existence in the corresponding parental cells, we selected a representative choice of 100 genomic variants detected by WES in iPSCs. 45 of these variants were listed in GnomAD or 1000 Genomes as polymorphisms (category A; Table S2) and 6 were not listed as polymorphisms but detectable in several donors (category B), 49 were also not listed as polymorphisms and were donor-specific (category C). Among the 49 donor-specific GAVs, 21 SNVs and 3 INDELs had been detected in 3 out of 3 iPSC clones, 16 SNVs and 2 INDEL in 2 out of 3 iPSC clones, and 7 SNVs and 0 INDELs in 1 out of 3 iPSC clones from the respective donor (Table S2). An optimized amplicon sequencing assay was established using the SOLiD 5500XL with ECC module. To obtain a realistic detection limit, we determined the overall error rate experimentally. Our amplicon sequencing approach exhibited an averaged coverage of 615,274. Analysis of the non-mutated bases adjacent to the variant revealed an error rate of 0.12% and detection limit of 1 out of 2,149 (SD 2,564) reads to distinguish the existence of a SNV from average error (p value 0.1; Table S2A). Application of this ultra-sensitive amplicon sequencing approach enabled us to confirm the pre-existence of all analyzed variants in the parental cell population, comprising 93 SNVs and 7 INDELs (Figure 3C; Table S2). This finding strongly argues against appreciable de novo mutagenesis during reprogramming. In accordance with the reported high somatic mutation rates of 10−7 to 10−6 per gene per somatic cell division,36 our data indicate a substantial genetic heterogeneity with a multitude of rare SNVs and INDELs among the primary parental donor cell populations that are passed over to individual iPSC clones (Figure 3).
Nevertheless, we cannot entirely negate the possibility of mutagenic effects during early passages of the reprogrammed cell clones. Since our exome sequencing analyses of the iPSC clones were limited to variants that exist at least in one clone with a frequency >0.3, we would have excluded variants that might have developed after the single cell cloning step while the reprogramming process may not have been fully completed.
Reprogramming enriches for individual variants located in cancer-associated genes
The vast majority of variants were detected in 3 out of 3 iPSC clones (categories A or B, and most of the donor-specific variants) and showed a high AF in their parental cells, indicating homo- or heterozygosity in the majority of the respective cell population.
18 GAVs that had been detected in 2 of 3 iPSC clones of donors D#3 hUVEC, D#25 hCBEC D#31 hSVEC, and D#38 hSVEC (13 of those also analyzed and undetected by WES of parental cells) were further analyzed by amplicon sequencing. Remarkably, all these GAVs were detectable in the parental cells at low frequency (0.02%–4% of the diploid founder cell population, Table S2), strongly suggesting enrichment during the reprogramming process. It can be excluded that the presence of these 18 GAVs in 2 out of 3 iPSC clones simply represent a statistical phenomenon for two reasons: (1) Without enrichment, the likelihood of finding a rare variant with a frequency between 0.02% and 4% in 2 out of 3 iPSC clones of one donor is only between p = 4 × 10−7 and p = 1.5 × 10−2 (on average p = 2.6 × 10−4). (2) Supposing that appearance of these 18 GAVs in 2 out of 3 iPSC clones represent just a statistical phenomenon based on a very high number of rare variants in the parental cell populations, one would expect that a much higher number of such rare GAVs should be detectable in 1 out of 3 iPSC clones than in 2 out of 3 iPSC clones, which was not observed in our study (the likelihood P for presence in iPSC clone is on average 193-fold higher than for presence in 2 out of 3 iPSC clones). Therefore, the 18 GAVs mentioned above, as well as 2 GAVs that had been detected in 3 out of 3 iPSC clones derived from D#3 hUVEC and D#38 hSVEC and were confirmed to be rare in their parental cell population by amplicon sequencing (undetectable by WES; Table S2) are termed afterward “enriched GAV.” Since all analyzed 18 GAVs that were detected in 2 out of 3 iPSC clones proved to pre-exist only in a small subpopulation of parental cells, also all other 13 category C GAVs detected in 2 out of 3 iPSC clones were further considered generally as “enriched” (Table S5A), leading to a total number of 31 “enriched GAVs.” Based on that considerations, iPSCs overall comprise between 0 and 18 enriched GAVs per clone (Table S5A).
In addition, 108 donor-specific category C GAVs were only detected in 1 out of 3 iPSC clones of the 10 donors (Table S5B). As far as we analyzed, these GAVs were undetectable by direct WES but were shown to be present with low frequency in the parental cells by amplicon sequencing. Although our approach did not allow us to draw any direct conclusion about enrichment of individual GAVs of this group, it can be presumed that a considerable proportion of these variants had been enriched to a certain extent during reprogramming.
Furthermore, we analyzed whether the transition/transversion (Ts/Tv) ratio and mutation spectra of donor-specific and especially enriched GAVs may point to an in vitro or in vivo origin of the respective variants. In fact, increased oxidative stress can result in C > A transversions37 and is frequently observed during in vitro culture of somatic cells. An elevated frequency of C > A transversions is therefore considered as typical in vitro signature. Remarkably, we observed an overall increased proportion of C > A transversions among GAVs detected in 1 out of 3 iPSC clones (Figure 4A), suggesting that the in vitro expansion culture of ECs prior to reprogramming contributed to the mutational load.
Figure 4.

Reprogramming enriches for putatively actionable mutations in genes connected to cell cycling, cell death, or pluripotency, and in curated OG and TSGs
The nature of donor-specific category C, potentially GVAs detected in iPSC clones was characterized. (A) Mutational spectra of the collective of donor-specific GAVs, enriched GAVs, and GAVs unique to 1 iPSC clone. n = 10 donors. (B) Percentage of putatively actionable GAVs, defined by harmful designation predicted by a consensus of in silico prediction of Condel, FATHMM, CADD, and SnpEff impact (red), as well as proportion of neutral GAVs (blue). The percentage of variants that affect genes involved in control of cell cycling, cell death, and pluripotency is depicted in dark red or blue, respectively. The proportion of variants in genes with other cellular functions is plotted in light red or blue. n = 10 donors. (C) Total number of putative actionable enriched GAVs and GAVs unique to only 1 iPSC clone per iPSC clone. Mean ± SD; n = 18 iPSC clones of neonatal donors, 12 of aged donors; discrepancy of samples was assessed applying two-tailed Mann Whitney test. (D) Percentage of curated OGs or TSGs as found in the human genome (calculated based on COSMIC cancer gene census and OncoKB database), or affected by neutral or putatively actionable, donor-specific GAVs, enriched GAVs, or GAVs in 1 iPSC clone. n = 10 donors.
We also explored whether the mutation spectra of variants and especially enriched GAVs may correlate with specific mutational signatures of cancer. Actually, different mutational processes that include DNA damage and inaccurate maintenance mechanisms are considered to act with variable strength throughout the lineage specification and evolution of cancer cells.38,39 The overall Ts/Tv ratio of total variants, donor-specific variants, and donor-specific GAVs in the iPSC clones was 2.5, 2.2, and 2.7, respectively, which is consistent with the reported ratios of 2–2.1 for whole genome and 3.0 for human exonic regions.40,41 In agreement with the results of Kwon et al.,13 the entirety of donor-specific category C GAVs found in all 3 iPSC clones were dominated by C > T transition without strand-bias (Figure 4A). Such C > T transitions as principal nucleotide changes indicate spontaneous deamination of 5-methylcytosine and are a hallmark of signature 1 of “Signatures of Mutational Processes in Human Cancer” (COSMIC Catalogue of somatic mutations in cancer), which is the result of an endogenous mutational process present in most normal (and neoplastic) somatic cells. Interestingly, enriched GAVs were characterized by an increased T > C transition without strand-bias resembling mutational signatures 6, 15, 20, and 26, which are often found in different cancer types and are all caused by defective DNA mismatch repair during replication of somatic cells (COSMIC Catalogue of somatic mutations in cancer), supporting the parental cell origin of these mutations (Figure 4A).
Finally, we analyzed the entirety of donor-specific (category C) GAVs and all enriched GAVs for functional consequences on the affected genes. In general, it can be expected that enrichment of certain genetic variants in iPSCs implies that individual variants affect genes, pathways, and cellular functions that influence the reprogramming process. However, the observation that the analyzed iPSCs contain a substantial number of, on average, 2.8 (range 0–18) of enriched variants per clone suggests that only some of those variants actually operate as driver mutations, while others are passenger variants. Indeed, prediction of variant impact on gene functionality based on a consensus of Condel, FATHMM, CADD, and SnpEff in silico prediction tools revealed 53% neutral variants among the enriched GAVs, which most likely constitute passenger variants, and 47% putatively actionable enriched GAVs (Figure 4B; Table S5A). Remarkably, this proportion of putatively actionable GAVs among the enriched GAVs is much higher than within the entirety of category C GAVs found in 3 of 3 iPSC clones (23%), further supporting the hypothesized active role of putatively actionable GAVs in the enrichment process during reprogramming. While 18 putatively actionable enriched GAVs were detected in 4 of the donors, only (Table 2; Table S5A), D#38 hSVEC for instance harbors even 7 putatively actionable enriched GAVs including mutations in the curated oncogenes (OGs) and tumor suppressor genes (TSGs) JAK1 and XIAP (COSMIC Cancer gene census and OncoKB). Both mutations of the genes JAK1 and XIAP affect substantial protein domains thereof: the mutation in catalytic phosphotransferase domain (Uniprot, Pfam) of tyrosine-protein kinase JAK1 might affect diverse signaling pathways including interleukin, EGFR1, type II interferon, and TGF-β signaling pathways and alter cellular processes including proliferation, differentiation, or apoptosis (COSMIC, Reactome, WikiPathway). As an OG and TSG, JAK1 is reported to mediate “Escaping programmed cell death” and “Proliferative signaling” (COSMIC Hallmarks of Cancer). Similarly, the E3 ubiquitin-protein ligase XIAP harbors a putatively actionable mutation within its BIR1 domain, which is crucial for homodimerization with TAB1 and recruitment of TAK1, an important regulatory component of the NF-κB canonical pathway promoting cell survival.42,43 Moreover, mutation in the BIR1 domain (Pfam) might reduce SMAC binding, caspase release from XIAP, and induction of cell death.43,44 We therefore presume that these putatively actionable GAVs confer a selective advantage and drive the reprogramming of cells carrying such mutations, whereas the neutral GAVs more likely represent passenger mutations coincidentally coexisting in the somatic parental cells.
Table 2.
Reprogramming enriches for pre-existing genetic variants in curated OG and TSGs, and in genes connected to cell cycling, cell death, or pluripotency.
| Gene symbol | Donor | iPSC clone | Allele frequency | Consequence/variant type | GO processes | Mutations in affected genes previously detected in PSCs | ||
|---|---|---|---|---|---|---|---|---|
| iPSC clones | Parental cells | |||||||
| Exome sequencing | Exome sequencing | Amplicon sequencing | ||||||
| GOLGB1 | D#3 hUVEC | C1 | 0.41 | 0 | 0.0031 | missense | endomembrane system organization/establishment of localization in cell | |
| C2 | 0.33 | |||||||
| C3 | 0.43 | |||||||
| UACAb | D#3 hUVEC | C1 | 0.35 | 0 | nd | missense | cell death | |
| C2 | 0.36 | |||||||
| C3 | 0.36 | |||||||
| KIF1A | D#3 hUVEC | C1 | 0.42 | 0 | (0.0015) | missense | establishment of localization in cell/microtubule-based process | Lo Sardo et al.11 |
| C3 | 0.64 | |||||||
| HYOU1b | D#3 hUVEC | C1 | 0.36 | 0 | 0.0026 | frameshift | response to stress/establishment of localization in cell/cell death | |
| C3 | 0.28 | |||||||
| SLCO1B7 | D#3 hUVEC | C1 | 0.39 | 0 | 0.0001 | frameshift | anion transport | Ihry et al.45 |
| C3 | 0.35 | |||||||
| SLC8A3 | D#3 hUVEC | C1 | 0.39 | 0 | 0.0023 | missense | homeostasis/transmembrane transport/response to stress/establishment of localization in cell / ion transport | |
| C3 | 0.44 | |||||||
| PPP1CCb | D#38 hSVEC | C5 | 0.29 | 0 | (0.0002) | missense/stop gain | cell cycle/cell division/chromosome segregation/metabolic process/regulation of transport | Ihry et al.45 |
| C6 | 0.33 | |||||||
| C9 | 0.69 | |||||||
| JAK1a,b | D#38 hSVEC | C5 | 0.33 | 0 | (0.0003) | missense | catalytic activity/cell proliferation/response to stress/response to cytokine/signal transduction | |
| C6 | 0.48 | |||||||
| CEP350 | D#38 hSVEC | C5 | 0.39 | 0 | 0.0003 | stop gained | cytoskeleton organization / microtubule anchoring / microtubule based process | |
| C6 | 0.48 | |||||||
| MYO7A | D#38 hSVEC | C5 | 0.51 | 0 | 0.0004 | missense | cell morphogenesis involved in differentiation / establishment of localization in cell / protein transport / organelle assembly / phagocytosis | Lo Sardo et al.11; Rouhani et al.18 |
| C6 | 0.47 | |||||||
| SCEL | D#38 hSVEC | C5 | 0.38 | 0 | 0.0010 | missense; 3′ UTR | tissue development | |
| C6 | 0.32 | |||||||
| HIRIP3 | D#38 hSVEC | C5 | 0.10 | 0 | nd | splice site (acceptor) | chromatin assembly or disassembly/chromosome organization | |
| C6 | 0.33 | |||||||
| XIAPa,b | D#38 hSVEC | C5 | 1.00 | 0 | 0.0003 | missense | response to DNA damage stimulus/response to stress/cell cycle/spindle assembly/cell death/regulation of gene expression/response to growth factor stimulus/cell proliferation | Kilpinen et al.46 |
| C6 | 0.98 | |||||||
| ZBED9 | D#25 hCBEC | C11 | 0.39 | nd | nd | stop gained | DNA integration | |
| C13 | 0.41 | |||||||
| TRIM9 | D#25 hCBEC | C11 | 0.54 | nd | 0.0187 | missense | catabolic process/cell cell signaling/establishment of localization in cell / exocytosis / regulation of transport / proteolysis / vesicle mediated transport | |
| C13 | 0.44 | |||||||
| MMP9b | D#25 hCBEC | C11 | 0.57 | nd | 0.0058 | missense | catabolic process / cell death / signal transduction / ion transport / regulation of dna binding / mitochondrion organization / release of cytochrome c from mitochondria | |
| C13 | 0.36 | |||||||
| CSMD3a,b | D#31 hSVEC | C3 | 0.48 | nd | 0.0018 | missense | Interact with NEK4 (cell division / response to DNA damage stimulus / cell cycle / regulation of cellular senescence / regulation of gene expression) | Lo Sardo et al.11 |
| C4 | 0.50 | |||||||
| SALLb | D#31 hSVEC | C3 | 0.48 | nd | 0.0194 | missense | chromatin modification / chromosome organization / growth / maintenance of cell number / regulation of gene expression / biosynthetic process / response to stimulus / somatic stem cell population maintenance | Ihry et al.45; Bhutani et al.12; Gore et al.8 |
| C4 | 0.54 | |||||||
The table lists all putatively actionable GAVs found in 3 or 2 iPSC clones but not detected in parental cell population by WES (upper part) or found in 2 iPSC clones but were not analyzed in parental cell population via WES (lower part), and that have been defined as enriched variants. Ultra-sensitive amplicon sequencing of GAV spanning regions for precise determination of allelic frequencies in the corresponding parental cell population was performed for a representative choice of variants. Pre-existence of variants in parental cell population was confirmed (p value 0.1) taking local error rates into account (Table S2A). Functional consequence of potential GAVs was classified by a consensus of the in-silico prediction of Condel, FATHMM, CADD, and SnpEff impact. Pre-existence is very likely but not confirmed with statistical confidence.
Curated OGs or TSGs (annotation retrieved from OncoKB or COSMIC Cancer gene census).
Genes with function in control of cell cycling, cell death, or pluripotency.
Furthermore, we determined the proportion of GAVs that affect genes involved in control of cell cycling, cell death, and pluripotency networks (Gene Ontology [GO] process annotation-based classification), which can be expected to impact reprogramming efficiency. According to GO annotation ∼20.8% of all human genes belong to this group (calculated based annotation in GO biological process [C5BP] collection of MSigDB C5). The observed proportion of 20.2% (15.2% + 5.0%) of such GAVs among the entirety of category C GAVs correspond very well to that (Figure 4B, left graph). Remarkably, these genes are considerably overrepresented among the enriched GAVs with 26.3% (21.1% + 5.3%; Figure 4B, middle graph). Even more striking is their contribution within the putatively actionable enriched GAVs: here, 44.4% of variants (equals 21.1% of all enriched GAVs) affect genes involved in control of cell cycling, cell death, and pluripotency networks, while only 10.1% enriched neutral GAVs (equals to 5.3% of all enriched GAVs; Figure 4B, middle graph) affect such genes. In contrast, the entirety of putatively actionable category C GAVs contained only 21.6% of such genes (equals merely 5% of all donor-specific GAVs; Figure 4B, left graph).
Besides obviously enriched GAVs, we identified 44 putatively actionable GAVs unique to 1 out of 3 iPSC clones (Table S5B), which accounts for 40.7% (18.5% + 22.2%) of all GAVs found in 1 clone (Figure 4B, right graph). Remarkably, also in this group a disproportionately high number of genes involved in control of cell cycling, cell death, and pluripotency networks were identified (45.5%; equals to 18.5% of all GAVs unique to 1 iPSC clone), which is in contrast to their neutral counterpart (15.6%; equals to 9.3% of all GAVs unique to 1 iPSC clone; Figure 4B, right graph), supporting the presumption that also many of the GAVs unique to 1 out of 3 iPSC clones had been enriched during reprogramming.
Altogether, ∼87% of iPSC clones harbored 1–7 putatively actionable GAVs, in ∼67% of iPSC clones located in genes with pivotal function for cell cycling, cell death, or pluripotency (Table S5). Interestingly, in contrast to the overall number of donor-specific (category C) variants and GAVs, iPSC clones from aged donors harbored a higher proportion of putatively actionable GAVs in genes involved in cell cycling, cell death, or pluripotency (75%) than clones derived from neonatal donors (61%). Moreover, iPSCs derived from aged donors exhibited, on average, slightly more putatively actionable, biologically relevant mutations per clone (mean 2.1 and median 1 compared to 3.6 and 3.5 in iPSC clones of aged donors; Figure 4C).
In addition, we analyzed the proportion of variants in our iPSCs affecting OGs and TSGs. According to OncoKB or COSMIC Cancer gene census database, the human genome contains 942 curated OGs/TSGs representing ∼5% of all human genes. A very similar proportion of variants affecting OGs/TSGs (6.5%) were observed in the entirety of donor-specific GAVs detected in all 3 clones (Figure 4D). Enriched GAVs, however, contained a considerably higher percentage (17%) of putatively actionable GAVs located in curated OGs/TSGs, namely JAK1, XIAP, and CSMD3 (Figure 4D; Table 2; Table S5A shaded in red), suggesting that reprogramming also enriches for specific mutations in oncogenes.
Similar to the increased proportion of GAVs affecting genes involved in control of cell cycling, cell death, and pluripotency, GAVs unique to 1 iPSC clone also contained an increased percentage (9%) of variants in curated OGs/TSGs (4 variants in GRIN2A, TCF12, XPC, and SOX2; Figure 4D; Table S5B shaded in red).
Interestingly, some of the enriched mutations and potentially enriched mutations (detected in 1 out of 3 iPSC clones) that have been observed in our study affect genes that are also reported by previous studies to be mutated in iPSCs, although their relevance was, in general, not further recognized. In those studies, the respective variants had been considered as derived from a small subpopulation of the source cells during reprogramming, or, if not detectable in the parental cells, de novo mutations8, 9, 10, 11, 12, 13,18,46,47 or were reported to affected iPSC fitness.45 The recurrently affected genes comprise KIF1A,11 SLCO1B7,45 MYO7A,11,18 XIAP,46 CSMD3,11 and SALL1,8,12,45 all affected by enriched mutations in our work, and GRIN2A,11,18 CCDC180,45 TCF12,8 ATF7IP,18 HDAC3,45 PPRC1,45 TTN,9,10,47 SOX2,13 ABCB11,11 and PDE8A,11 which also carried mutations unique to 1 out of 3 of our iPSC clones. Some of those recurrently mutated genes represent curated OGs/TSGs (XIAP, CSMD3, GRIN2A, TCF12, and SOX2). Further genes, such as SOX2 or SALL1, are involved in the molecular control of pluripotency (Ractome.org; R-HSA-2972975.1: POU5F1 [OCT4], SOX2, NANOG Bind the SALL1 Promoter) or cell cycling (CCDC180, HDAC3, TTN; Table S5B). The predicted functional impact of these mutations and the biological relevance of affected genes suggest selective advantages as mechanism for enrichment of somatic cell clones carrying such a mutation during reprogramming.
Lastly, we aimed to experimentally confirm the impact of such genes, which were affected by enriched putatively actionable GAVs, on the reprogramming process. Among those genes (Table 2; Table S5A), in particular SALL1, a tumor suppressor,48 appeared of interest because mutations in SALL1 were also recently observed in hiPSCs by others8,12 or as a pluripotency-specific gene.45 Furthermore, SALL1 interacts with OCT4,49 SOX2,50 and NANOG51 as key factors of reprogramming.45 Hence, we investigated the effect of a knockdown of SALL1 and 15 other genes on the reprogramming efficiency by utilizing a lentiviral vector-based RNA interference system that delivers short hairpin RNAs (shRNAs) embedded in an optimized human microRNA-30 (miR-30) backbone (shRNAmiRs) to achieve stable and heritable sequence-specific gene knockdown.52 For this purpose, one of our parental cell populations, D#2 hUVECs, was transduced with a library containing 48 shRNAmiRs against those 16 genes (3 shRNAmiRs/gene). Besides SALL1, those 16 genes include ZFHX3, a curated oncogene, which is also directly interacting with OCT449 and was chosen as internal positive control, and a representative choice of genes that were identified in our iPSC clones to be affected by A (ALMS1, ACTR8), category B (CCDC14, KCTD8, KLHL14; Table S1), or enriched category C variants (putatively actionable: CEP350, SLCO1B7, MAGEB6, MYO7A, and SCEL, neutral or unconfirmed: SLC12A4, TMEM139, TMEM168, TNNI3K; Table S6). Next, 3 batches of the transduced cells were independently reprogrammed and the composition of shRNAmiRs in the cell population before and after reprogramming (P3) was quantified. While in the transduced parental cell population the distribution of all shRNAmiRs was homogeneous (mean frequency 0.019, SD 0.008), in the 3 iPSC batches only a few shRNAmiRs dominated the population (Figure 5A). These shRNAmiRs were found >2-fold enriched compared to their frequencies within the parental cell population (Figure 5B, red bar) in 1 or even 2 iPSC batches. In contrast, the frequency of most shRNAmiRs (77%) remained on a similar level or was reduced in the iPSC batches (fold change < 2; Figure 5B, black bars).
Figure 5.

Reprogramming leads to enrichment of individual shRNAmiRs directed against genes that were affected by enriched, putatively actionable GAVs
For a multiplexed shRNAmiR screen, D#2 hUVECs were transduced with a library containing shRNAmiRs against a choice of 16 genes (3 shRNAmiRs/gene) before cells were reprogrammed in 3 independent batches. The relative frequency of individual shRNAmiRs in the cell population before and after reprogramming was assessed via sequencing. (A) Frequency distribution of shRNAmiRs in transduced parental cells and the 3 iPSC batches. (B) Enrichment of shRNAmiRs displayed as fold change of shRNAmiR frequencies in iPSC batches compared to transduced starting EC population. Red marked shRNAmiRs significantly (p < 0.001) derivate from sample median (one sample, Wilcoxon signed rank test). Labels represent the names of those shRNAmiRs (Table S6) that were more than 2-fold enriched.
We observed 19.1- and 7.7-fold enrichment of one of the positive control shRNAmiRs ZFHX3.509 in 2 batches. Similarly, cells carrying shRNAmiRs SALL1.4587 and SALL1.110 were considerably enriched (5.7- and 6.4-fold), confirming our finding that actionable mutations in the SALL1 gene facilitate reprogramming. Also, KLHL14.3195 appeared enriched in 2 batches, but the underlying mechanism remained unclear since literature research did not reveal any obvious connection of KLHL14 to pluripotency, cell cycle, or apoptosis. In contrast, no positive selection (fold change < 2 in all batches) or enrichment in one batch only was detected for all other shRNAmiRs. Notably, in batch 3, one shRNAmiR SCEL.2743 took over almost the whole culture. However, we presume that this result rather represents a clonal event of insertional mutagenesis since enrichment is observed for one shRNAmiR in batch 3, only.
Conclusions
The high expectations for iPSCs as invaluable cell source for regenerative and personalized medicine provoked fast progress in this field. However, the extent, nature, and functional consequences of somatic variants are still largely unknown. Nevertheless, those mutations might compromise the safety and efficacy of iPSC-based cellular therapeutics, and the origin of genetic variants will impact further strategies for iPSC generation and expansion and preclinical risk assessment.3,7
Our results from EC-derived early passage iPSCs indicate that 98% of all iPSC small genetic variants are acceptable population polymorphisms that are passed over to iPSCs from founder cells. Among the remaining 2% of donor-specific variants, we detected on average 2.7 (range 0–8) putatively actionable iPSC-specific mutations per clonal iPSC line. Furthermore, our analyses show that all analyzed true genetic variants detected in early passage iPSC lines pre-existed in the corresponding parental cell populations at different frequencies and indicate a high level of genetic mosaicism in human tissue. Our results also substantiate that there is no appreciable contribution of a hypothesized inherent mutagenicity of the reprogramming process to the mutational load in iPSCs.
Most importantly, we demonstrate for the first time the enrichment of SNVs and small INDELs from rare subpopulations of parental cells during reprogramming. This finding confirms and extends recent observations by Shakiba et al.17 to human reprogramming. Apparently, mutated clones outperform other clones during reprogramming and ultimately take over cultures of early reprogrammed cells. Our observations substantiate the finding of Shakiba et al.17 by shedding light on the genetically encoded inequalities during the process of molecular reprogramming and by identifying potential driver genes affected by small-scale mutations. Among the group of enriched mutations, a substantial proportion of variants affected genes involved in control of cell cycling, cell death, and pluripotency. Although the numbers of variants were too low to reach statistical significance, putatively actionable mutations in OG/TSG genes were apparently overrepresented among the group of enriched variants. For the tumor suppressor and pluripotency-specific gene SALL1, which was affected by one of the identified, enriched, and putatively actionable donor-specific variants, we confirmed experimentally that downregulation can lead to clonal selection during reprogramming.
Although we did not observe a generally increased total number of SNVs and INDELs in iPSCs from elderly donors, our data show at least a trend for an increased number of putatively actionable variants in iPSCs from aged donors compared to neonatal cell sources, suggesting a lower biological quality of iPSCs from aged individuals.6,11 While we consider selection of putatively actionable variants a general phenomenon during reprogramming, more efficient reprogramming methods could help to reduce selection for potentially harmful mutations. A lower reprogramming efficiency of the lentiviral reprogramming approach, which was used in this study, compared to more recent techniques such as reprogramming with Sendai virus vectors, actually may have boosted selection for clones with putatively actionable mutations that facilitated the reprogramming. In this context, also the choice of the somatic cell source becomes again important as highly proliferative cell types, in particular from young donors, usually lead to higher reprogramming efficiencies, potentially associated with less selection pressure.
Apart from the donor age, the cell source itself is apparently a determining factor for enrichment of individual mutations. The in vivo somatic mutation rate in human is assumed to be around 10−7 to 10−6 per gene per cell division.36 However, the mutation rate differs among tissues with, for example, 3.5 × 10−9 for small intestine, 1.2 × 10−8 for neurons during early development, 1.6 × 10−7 for skin,53 and up to 4.9 × 10−6 to 10−3 in peripheral blood isolated cells.54, 55, 56 Hence, the rate and number of subsequent cell division, as well as variant mosaicism introduced by further factors like exposure to mutagens and radiation, are determining parameters for the likelihood to obtain iPSC lines with genetic aberrations. Noncancerous skin cells from aged donors, for example, were shown to harbor a comparable number of somatic mutations, including such in cancer-associated genes, to that in cell of skin cancer samples.57 Furthermore, around 45% of all iPSC clones derived of skin fibroblast were observed to carry a mutation caused by UV damage.14 Therefore, EC-derived iPSCs might offer a promising alternative compared to UV-exposed skin.
Our findings indicate the requirement for further research to clarify the clinical risks accompanied with mutations that become enriched during reprogramming, such as malignant transformation of iPSC transplants. In particular, this includes the establishment of comprehensive databases of putatively actionable, enriched genetic variants in iPSCs, and their linking to databases in cancer research.
To generate comprehensive databases of genes that facilitate, when mutated, reprogramming and potentially pathogenic transformation, the investigation of a large number of iPSC lines will be necessary. Furthermore, it will be important to verify experimentally the effect of all genes affected by enriched mutations, as done exemplarily for SALL1, and to further investigate molecular mechanisms of enrichment. Lastly, reprogramming methods and applied culture conditions should be optimized to avoid or at least minimize the selection of cell clones that carry cancer and disease-causing mutations, which was also not the focus of our study.
Materials and methods
Isolation and culture of parental cells
ECs were chosen as donor cells, since these were available from different sources from neonatal (hUVEC; hCBEC) and aged (64–88 years, hSVEC; hPBEC) individuals. hUVECs and hSVECs were isolated from umbilical veins, as well as hSVEC from saphenous veins using a standard enzymatic digestion protocol.58 hCBECs and hPBECs were isolated essentially as previously described.31 The ECs were cultured in EGM-2 medium (Lonza) for 4–5 passages, which equals to around 8–10 additional cell divisions after isolation, before reprogramming or subjecting to WES. Human material was collected after approval by the local ethics committee and following the donor’s written informed consent, or in the case of newborns, following parental consent.
Virus production
Plasmids pSIN-EF2-Lin-Pur, pSIN-EF2-Nanog-Pur, pSIN-EF2-Oct4-Pur, and pSIN-EF2-Sox2-Pur (OSLN vectors) were purchased from Addgene. Viruses were produced, concentrated, and titrated as described previously.31
Generation of hiPSCs
2 × 105 ECs of passage 4–5 were reprogrammed by lentiviral transduction (multiplicity of infection, MOI 20) and characterized essentially as previously described.31 The reprogramming efficiency ranged between 0.02 and 0.4 for neonatal cell sources and between 0.004 and 0.07 for aged donors. On day 6, the cells were transferred onto murine embryonic fibroblast (MEF) feeder layers and cultured with iPSC medium from day 7 onward. For single cell cloning, individual colonies were manually transferred into separate wells and further cultured on MEFs. In total, 30 EC-derived iPSC clones were generated from 9 donors equating to a number of 3 iPSC clones per donor, with exception of donor D#37 for which 6 iPSC clones were generated (3 iPSC clones from each hSVEC and hPBEC).
Genomic DNA extraction and WES
Genomic DNA (gDNA) was isolated from all 30 iPSC clones and EC parental cell population of 2 neonatal (D#2 hUVEC and D#3 hUVEC) and 2 aged (D#37 hSVEC and D#38 hSVEC) donors using QIAGEN Blood DNA mini kits. Exomes were enriched using the commercially available Agilent SureSelectXT2 Human All Exon v4 (Agilent Technologies). 2 μg of gDNA (>3 × 105 cells) of each sample were sheared and size selected to an average length of 200 bp via AMPure XP bead purification. After adaptor ligation, the fragments were amplified for 8 cycles, purified and hybridized for 36 h with an Exome capture library (Agilent, Santa Clara, USA; Human All Exon v4, consisting of biotinylated RNA probes). The DNA-biotinylated RNA hybrids were bound via streptavidin to magnetic beads, purified, and amplified for 8 cycles with barcode containing primers. The final concentration, fragment distribution, and quality of the exome libraries were assessed on Qubit fluorimeter (Invitrogen) and Agilent Bioanalyzer. The sequencing was performed on a SOLiD 5500XL (Applied Biosystems, USA) with an ECC module. We sequenced 2 technical replicates with separate exome DNA amplification, barcode binding, emulsion PCR, bead enrichment, and sequencing for each sample.
Read alignment and variant calling
Reads (75 bp, single-end) generated by the SOLiD 5500XL sequencer were aligned to the human reference genome hg19 using NovoalignCS (v1.04.02). Reads mapping equally well to more than one genomic location were discarded. The reads were trimmed during the alignment using the –H and –s 2 parameters of NovoalignCS. Reads with ≥4 nucleotides within the first 20 bp or with ≥20% of all bases of Phred quality score59 <10 were discarded and PCR duplicates eliminated. Mapping quality and coverage were assessed employing Qualimap (v2.2.1). Variants were called in multisample format (coverage ≥ 10) grouping all 6 or 8 samples per donor (3 iPSC clones as replicates and, if so, 2 replicates of parental cell population) utilizing FreeBayes (v1.0.2).
Variant validation and refinement
Variant call format (VCF) files with FreeBayes results were imported into Galaxy (v17.05)60 instance of the RCU Genomics, Hannover Medical School, Germany for variant refinement. Complex variants were parsed running vcfAllelicPrimitives (v1.0.0_rc1.0; Garrison 2015 [https://github.com/ekg/vcflib]) Galaxy built-in tool for simplification of variant refinement and annotation process.
The multisample variant calling allowed us to define variants with higher confidence also at a low coverage by applying a reverse variant refinement strategy. In detail, assuming inheritance from parental cell population, the most variants would exist in all 3 iPSC clones. Therefore, a variant was regarded as being present in all iPSC clones unless its absence in one iPSC clone was defined by distinctively lower variant AF in both replicates compared to the other samples of the same donor.
For developing our variant refinement strategy, we chose an empirical approach based on identification of true- and false-positive and -negative variants making use of variant phylogeny, meaning the dispersion of a variant in iPSC clones of one donor, and orthogonal validation sequencing (amplicon and Sanger sequencing). That means, the false-positive and -negative variants were identified by cross-validation sequencing or by the presence or absence in one iPSC clone in which it would be expected to be or not to be according to the distribution of most other variants among the iPSC clones. This strategy was designed with the pivotal goal of returning variants shared by 2 iPSC clones with confidence. For a first variant validation round, we applied no additional filter parameter. Polymorphisms were annotated utilizing Ensembl variant effect predictor (VEP; v95) for human GRCh37 (assembly GRCh37.p13) based at European Bioinformatics Institute (EMBL-EBI)61 with the GnomAD (gnomAD = 170228) and 1000 Genomes (dbSNP = 151) modules and SnpSift Annotate62 as Galaxy implemented tool with GnomAD (gnomad.exomes.r2.1) and dbSNP (149, reference GRCh37.p13). Polymorphisms defined by MAF ≥ 0.01 in any population of GnomAD or 1000 Genomes are termed in the following as “polymorphisms.” Moreover, a number of variants were found in more than one donor representing most likely either undescribed polymorphisms or sequencing error and barcode leakage and are hereafter termed “common variants.” We selected 84 variants (55 polymorphisms, 21 common variants, and 8 donor-specific variants of which some showed low multisample quality value, sample coverage, or alternative AF (Table S1) for cross-validation by amplicon sequencing. All primers used for amplicon assays are listed in Table S7. For a more detailed description of the amplicon sequencing approach, see below. The majority of polymorphisms were confirmed as homo- or heterozygote variants in iPSC clones and parental cell populations. However, a number of variants of the group of uncertain and common variants, especially SNVs, were found to be false-positive (Table S1A). By applying a filter on multisample quality (≥400 and 530 for 6 and 8 sample batches, respectively) and DP (≥24 and 32) only around 1/5 of those false-positive variants could be eliminated. To exclude further false-positive variants, we introduced AF of variants as a filter criterion. For calculation of AF, multiallelic variants were excluded and a minimum sample DP was set. With DP6 per sample, the allelic fractions of heterozygote variants followed roughly a binominal distribution with mean variant frequency of 0.45 (Figure S1B) as expected.26,63 In the next step, first variants that were not in at least one iPSC clone detected with AF ≥ 0.3 were excluded as such proved demanding in the evaluation process as they often fell in difficult genomic regions and were error prone. Then different AFs thresholds (0.3, 0.2, 0.1, and adaptable) were tested in D#3 hUVEC iPSC clones as parameters for exclusion. Variants detected in multisample calling were assumed to be present in all iPSC clones of the donor unless AF in both replicates of an iPSC clones were below the set AF threshold. For all different threshold AFs, the number of true- and false-positive and -negative variants was counted and error rates calculated. The assessment of true and false calls was based upon variant phylogeny (variants exist in iPSC clone 1 and 3 that originate apparently from same subpopulation, but not in iPSC clone 2), manual curation by differences in AF between samples, and supported by results of Sanger sequencing. While minimum AF 0.3 and AF 0.2 as threshold yielded many false-negative variants, a threshold of AF 0.1 left false-positive variants. Error rate was lowest with adaptable AF (Figure S1C), which considered differences between AFs of iPSC clones. In detail, a variant call in one clone was rejected if (1) AF in both replicates was < 0.1; (2) AF in one replicate was < 0.15, undetected in other replicate, and AF > 0.4 in both replicates of both other clones; (3) AF in both replicate < 0.15 and AF > 0.5 in both replicates of both other clones; (4) AF in one replicate < 0.2, and undetected in other replicate, and AF > 0.5 in both replicates of both other clones; or (5) AF in both replicates < 0.2 and AF > 0.6 in both replicates of both other clones.
After applying the new filter criteria, most false-positive variants were excluded (Table S1). However, a number of true-positive INDELs did not pass especially quality criterion (Table S1A). Although here all analyzed INDELs were common polymorphisms or variants and not interesting for our further analysis, INDEL detection remains more demanding and less sensitive. Lastly, 10 variants of the passing 61 were not confirmed in iPSC and/or parental cells and likely represented sequencing errors. Those belonged, in general, to the group of common variants (Table S1B).
Exemplarily for donor D#3 hUVEC histograms displaying only the category C variants found in 1, 2, or 3 iPSC clones, respectively (Figure S1D), demonstrated that there was no bias in the AF distribution between those. In all three groups (GAVs in all 3 iPSC clones, GAVs in 2 iPSC clones [=enriched GAVs], and GAVs in 1 iPSC clone) the variant frequency distribution was centered around 0.45. Of note, as one of our filter criterion for variants demanded that a variant has to be present in at least one iPSC clone with AF ≥ 0.3, the plot for the AF distribution of GAVs in 1 iPSC clone shows a cut at AF 0.3.
In the parental cell population, a variant was considered as not detectable by WES if AF < 0.05 in both samples. The variant frequency distribution in the parental cells of all variants including polymorphisms, the donor-specific variants (category C), and GAVs, that were detected in all 3 iPSC clones of the donor were also centered around 0.45 (heterozygote variants) or 1 (homozygote variants; Figure S1E) as expected for variants suspected to exist in all parental cells.26,63 The final workflow is displayed in Figure S1A.
Determination of variant allele fraction in parental cell population by amplicon sequencing
The error rate of around 0.1%–1% of “Sequencing by Synthesis” NGS systems15,16,29 hinders the identification of rare genomic variants against a large background of non-mutated DNA sequences. Scientists have tried to overcome this problem mathematically: statistical methods have been applied to find significant imbalances in the pattern of sequencing errors and, thus, to identify and quantify these rare events. Unfortunately, this method is biased. If a statistically significant anomaly is found, then this approach is robust and the analyzed variants can be considered verified. But the converse argument does not hold true: an insufficient significance is not proof for the absence of the respective variant. The smaller the mutated cell population, the higher the probability of missing this specific mutation event amid the vast statistical noise.
We developed an amplicon sequencing assay that combines amplification with high fidelity DNA polymerase and the highly accurate “Sequencing by Ligation” NGS technology to validate candidate variants in founder cell populations and to assess their allelic frequencies. PCR primers (Table S7) to amplify the regions spanning the variants were designed utilizing Primer3 (v0.4.0). In order to minimize the individual error rates for each sequenced base, we used the most accurate high fidelity DNA polymerase available for the initial amplification. 100 ng gDNA was used as template in the initial PCR amplification step, which corresponds to the genomes of > 1.6 × 104 cells. Amplicons were generated by running 25 PCR cycles.
For INDEL (>5 bp) validation, PCR products of 1,400–1,600 bp were generated by running 20 PCR cycles with a Q5 Hot Start High Fidelity DNA Polymerase (NEB GmbH, Frankfurt, Germany, error rates < 10-7 per base). PCR products were purified using gel electrophoresis, extracted (QIAquick Gel Extraction Kit; QIAGEN), and sheared on a Covaris sonifier according to manufacturer’s protocols to approximately 200 bp fragments. INDEL fragment libraries were generated using the Fragment Library Core Kit (Life Technologies) as described by the manufacturer.
Similarly, PCR products, 60–120 bp long, comprising SNV and INDELs ≤ 5 bp were generated and purified. Fragment end repair of PCR products and adaptor ligation were performed with NEBNext Ultra II DNA Library Prep Kit for Illumina according to the manufacturer. Subsequent steps of library preparation were performed using Fragment Library Core Kit.
Concentration, quality, and fragment length distribution of generated libraries was assessed on Qubit and Agilent Bioanalyzer. The sequencing of the amplicons was performed on a SOLiD 5500XL (Applied Biosystems, USA) with an ECC module. For SNVs, the error rate and detection limit of amplicon sequencing assay were determined experimentally for each genomic location spanning the variants. In a first step, to exclude false sequences and reduce background noise, reads with the individual PCR primer sequence of the primer closer located to the SNV location were directly extracted from fastq files. Within the extracted reads, the number of reads with the SNV enclosed by a 15 bp long sequence ranging from position −7 bp to +7 bp were counted, as well as the number of reads with the reference sequence. To assess the average error, we also determined the numbers of reads that comprised one of the other both nucleotides (not SNV or reference nucleotide). Furthermore, two additional 15 bp sequences, one directly upstream and one downstream of the SNV, were analyzed and numbers of reads for every non-reference nucleotide in the middle of the sequence were counted. The mean error rate and detection limit were calculated for every SNV. Thus, the assay allows a deep insight into rare variants without the need for statistical extrapolation beyond the resolution of the sequencing technique. Frequencies of INDELs were determined by extracting and counting reads for both INDEL and reference embedded by a 20 bp sequence directly from fastq files. In some rare cases, the determination of variant AF of SNVs and INDELs in parental cells was performed on Illumina MiSeq platform. Therefore, PCR fragments of 1,400–1,600 bp length were generated and sheared as described above, libraries were prepared via NEBNext Ultra II DNA Library Prep Kit for Illumina according to manufacturer, and then, due to higher error rate of the Illumina system, AF and errors were assed in an 11 bp genomic context instead of 15 bp as described above.
Variant validation in iPSC clones
As iPSC clones are derived from individual founder cells, a variant is theoretically homo- or heterozygous. Therefore, variant validation via amplicon sequencing in iPSC clones was performed with the same technical approach as described above for determination of variant fraction in parental cell population but the result interpretation was simplified. Only reads with the SNV enclosed by a 15 bp sequence and the reference sequence were quantified, but no error rate was estimated. Validation of INDELs was performed as described above.
For validation of variants via Sanger sequencing, PCR products were prepared as for amplicon sequencing. Purified PCR products were prepared according to the company’s requirement and sent together with the PCR primers to Microsynth Seqlab (Göttingen, Germany). Results were visualized via Chromas software (v1.45).
Variant annotation and prediction of functional consequences
Variant annotation was performed using SnpEff (hg19; v4.3)62 as Galaxy implemented tool and the web interface of Ensembl VEP (v95; assembly GRCh37.p13) based at European Bioinformatics Institute (EMBL-EBI).61 The functional consequence of a GAVs was classified by a consensus based on the in-silico predictions of Condel, FATHMM, CADD (Ensembl modules), and SnpEff impact. If a variant had a harmful designation by SnpEff (high impact) or at least by two of the algorithms (Condel, deleterious; FATHMM, damaging; CADD phred > 17), provided prediction tools returned a result, it was considered as putatively actionable.
The number of all nucleotide change events in the 3 collective of GAVs found in 1, 2, or 3 iPSC clones of all donors were determined and mutational spectra compared to Signatures of Mutational Processes in Human Cancer (v2) of COSMIC Catalogue of Somatic Mutations in Cancer hosted by Wellcome Trust Sanger Institute.
GO process annotation of genes was obtained from GO biological process (C5BP) collection of Molecular Signatures Database (MSigDB)64 maintained by Broad Institute. In case no GO process was curated, IntAct Molecular Interaction Database hosted by EMBL-EBI, BioGRID Biological General Repository for Interaction Datasets,65 and UniProt66 were consulted for interacting proteins and their GO processes. To estimate the oncogenic potential of each variant, we computed overlap of affected genes with MSigDB collection of computational gene sets (C4) and oncogenic signatures (C6), as well as Jensen DISEASE database67. Furthermore, OncoKB database68 as well as the COSMIC Cancer gene census69 were utilized for matching of curated OGs and TSGs.
Analysis of clonal enrichment of shRNAmiRs during reprogramming
Lentiviral vector construction of pLKO5d.SFFV.eGFP.miR30N.WPRE vector was described previously.52 Within the miR-N cloning cassette, the spleen focus forming virus (SFFV) promotor was replaced by a short cytomegalovirus enhancer and chiken beta-actin (CAG) promotor. shRNAmiR design, cloning and library construction, packaging into lentivirus, and test via reporter assay were performed as previously described.52,70,71 In short, for each target gene 3 shRNAmiRs were designed (Table S6). The 67-mer oligonucleotides encoding the shRNAmiR construct including passenger (22 bp), loop (19 bp), guide (22 bp), and overhang (4 bp) were purchased form Integrated DNA Technologies (IDT) and cloned into the pLKO5d.CAGs.eGFP.miR30N.WPRE backbone. The shRNAmiR constructs were then mixed equimolarly to form shRNAmiR libraries and virus containing the library was produced and tested. D#2 hUVEC cells were transduced with shRNAmiR library with MOI 2, cultured for 3 passages and then reprogrammed with monocistronic lentiviral Thomson factors as described above. 3 technical replicates (batches of iPSCs) were derived from the same transduced parental cell starting population with 4 × 105 transduced cells in each batch. 57, 53, and 64 iPSC colonies arose in the 3 batches, respectively, which equals to around 0.015% reprogramming efficiency. Reprogrammed cells were cultured for 3 passages and gDNA isolated (see above) from the entirety of reprogrammed cells (without sampling of cells during passaging).
Composition of the shRNAmiR library in the 3 iPSC batches compared to the transduced parental cell population was analyzed by sequencing. For this shRNAmiR sequencing, PCR product of 157 bp, spanning the shRNAmiR sequence and parts of the 3′ and 5′ miR30A flank were generated by running 25 cycles with Q5 Hot Start High Fidelity DNA Polymerase (forward primer: 5′-GTTAACCCAACAGAAGGCTAAAG-3′, reverse primer: 5′-TAATTGCTCCTAAAGTAGC CCCTTG-3′; annealing temperature 63°C). This PCR reaction amplified not only the shRNAmiR sequences but also the endogenous miRN30A of the cells. Sequencing library preparation was conducted on PCR fragments with NEBNext Ultra II DNA Library Prep Kit for Illumina according to the manufacturer’s instruction. The quality of the fragment library was assessed on Qubit fluorimeter and Agilent Bioanalyzer. The sequencing was performed on Illumina MiSeq.
Assessment of content of each individual shRNAmiR in iPSC batches and in transduced parental cells was performed by counting sequencing reads. For this, raw reads were filtered for 12 bp of PCR primer sequence allowing 1 mismatch, and then endogenous miR30A reads were excluded (filtering for 12 bp accepting 1 mismatch). Finally, the numbers of reads for each shRNAmiR were counted by filtering for a 15 bp sequence of the guide shRNA miR, allowing two mismatches within this 15 bp sequence.
In an initial experiment, other genes such as p53, CDKN1A, CDKN2A, and SUMO2 were tested as a potential internal positive control. However, those had very strong effects on the reprogramming efficiency, and we presumed that clones with knockdown in one of those genes might mask the effect of other shRNAmiRs with less pronounced effects. Hence, it was decided not to include shRNAmiR against genes such as p53 in subsequent experiments.
Instead, we have chosen ZFHX3 as an internal positive control in our approach. Although there are no studies addressing the reprogramming efficiency of somatic cells with ZFHX3 mutation or downregulation, ZFHX3 seems to be involved in induction of pluripotency, is an important regulator of cell proliferation, and represents a curated oncogene. ZFHX3 interacts with OCT4 and was described as a negative regulator of MYC expression in cancer. Loss of ZFHX3 increased cell proliferation and MYC expression.72 In human ESCs the ZFHX3 promoter is occupied by Polycomb-related factors, which are known to regulate self-renewal and pluripotency.73
Statistical analyses
GraphPad Prism (v6.07) or RStudio (v1.1.463) software were utilized for graph visualization and data statistics. Data are given as mean ± SD. Sample distribution was assessed employing D’Agostino and Pearson omnibus normality test. Unpaired two-tailed t tests were applied to compare number of variants found per iPSC clone between age groups. Nonparametric two-tailed Mann Whitney test was performed to compare number of putatively actionable variants found per iPSC clone between age groups.
For assessment of error rate of the amplicon sequencing assay, the mean value of incorrectly recorded nucleotides (n = 8) was calculated for every SNV. Distribution of errors for each SNV was evaluated via D’Agostino and Pearson omnibus normality test or, in rare cases, Shapiro-Wilk normality test if the first one was not applicable. In case given samples followed a normal distribution, a one-sample one-tailed t test was applied to compare read count of SNV as the hypothetical value against the background of incorrectly annotated bases. Otherwise, Wilcoxon one-tailed signed-rank test was employed. Null hypothesis, which proposes that read number of SNV lies within local error range, was rejected with p value 0.1.
Code availability
Variant refinement was performed on the internal Galaxy (v17.05)60 instance of the RCU Genomics, Hannover Medical School, Germany. All workflows that were used to process variants are available as a supplemental file (Data S2). The utilized script for processing and analysis of shRNAmiR sequencing data is available in Data S3.
Data availability
All data underlying the study are available on request and are currently deposited on servers of Hannover Medical School, Germany.
Acknowledgments
The authors are grateful to J. Gorenaja, K. Menge, M. Sievert, M. Tauscher, U. Opel, and S. Mielke for their technical support, to P. Chouvarine for initial support regarding bioinformatics, to N. McGuinness for a critical reading of the manuscript, and to T. Scheper for providing bFGF. Some of human cord blood samples were kindly provided by Vita34, Leipzig. Many thanks to C. Hess for the isolation of Vita34 CBECs and to P. Stiefel for several hSVEC isolations. We are also thankful to S. Rojas, T. Goecke, and other surgical colleagues, who provided human tissue samples. We thank J. Thomson for providing the reprogramming plasmids via Addgene and D. Trono for providing the lentiviral packaging and transfer plasmids psPAX2 and pMD2.G. This work was funded by the German Federal Ministry of Education and Research (CARPuD, 01GM1110A-C; iCARE 01EK1601A), the German Center for Lung Research (DZL; 82DZL00201 and 82DZL00401), the German Research Foundation (Cluster of Excellence REBIRTH, EXC 62/3), the Ministry for Science and Culture Lower Saxony (Niedersächsisches Vorab, REBIRTH, ZN 3440), and by the Sächsische AufbauBank/Vita34.
Author contributions
M.K. and K.O. performed experiments, collected and analyzed data, and contributed to writing the manuscript. C.D. and L.W. performed WES, analyzed and collected bioinformatics data, and contributed to writing the manuscript. F.A., A. Schwarzer, M.-J.K., and A. Schambach designed the shRNAmiR library. A.H., S. Merkert, and S.W. contributed to the conduction of experiments. S. Menke and M.D. provided technical assistance in cell culture, fragment library construction, and WES. U.M. designed and coordinated the study and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.04.007.
Supplemental information
References
- 1.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Weissbein U., Benvenisty N., Ben-David U. Quality control: Genome maintenance in pluripotent stem cells. J. Cell Biol. 2014;204:153–163. doi: 10.1083/jcb.201310135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews P.W., Ben-David U., Benvenisty N., Coffey P., Eggan K., Knowles B.B., Nagy A., Pera M., Reubinoff B., Rugg-Gunn P.J., Stacey G.N. Assessing the Safety of Human Pluripotent Stem Cells and Their Derivatives for Clinical Applications. Stem Cell Reports. 2017;9:1–4. doi: 10.1016/j.stemcr.2017.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayshar Y., Ben-David U., Lavon N., Biancotti J.C., Yakir B., Clark A.T., Plath K., Lowry W.E., Benvenisty N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 5.Hussein S.M., Batada N.N., Vuoristo S., Ching R.W., Autio R., Närvä E., Ng S., Sourour M., Hämäläinen R., Olsson C. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- 6.Ma H., Morey R., O’Neil R.C., He Y., Daughtry B., Schultz M.D., Hariharan M., Nery J.R., Castanon R., Sabatini K. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature. 2014;511:177–183. doi: 10.1038/nature13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin U. Genome stability of programmed stem cell products. Adv. Drug Deliv. Rev. 2017;120:108–117. doi: 10.1016/j.addr.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Gore A., Li Z., Fung H.L., Young J.E., Agarwal S., Antosiewicz-Bourget J., Canto I., Giorgetti A., Israel M.A., Kiskinis E. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young M.A., Larson D.E., Sun C.W., George D.R., Ding L., Miller C.A., Lin L., Pawlik K.M., Chen K., Fan X. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell. 2012;10:570–582. doi: 10.1016/j.stem.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji J., Ng S.H., Sharma V., Neculai D., Hussein S., Sam M., Trinh Q., Church G.M., McPherson J.D., Nagy A., Batada N.N. Elevated coding mutation rate during the reprogramming of human somatic cells into induced pluripotent stem cells. Stem Cells. 2012;30:435–440. doi: 10.1002/stem.1011. [DOI] [PubMed] [Google Scholar]
- 11.Lo Sardo V., Ferguson W., Erikson G.A., Topol E.J., Baldwin K.K., Torkamani A. Influence of donor age on induced pluripotent stem cells. Nat. Biotechnol. 2017;35:69–74. doi: 10.1038/nbt.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhutani K., Nazor K.L., Williams R., Tran H., Dai H., Džakula Ž., Cho E.H., Pang A.W.C., Rao M., Cao H. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat. Commun. 2016;7:10536. doi: 10.1038/ncomms10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon E.M., Connelly J.P., Hansen N.F., Donovan F.X., Winkler T., Davis B.W., Alkadi H., Chandrasekharappa S.C., Dunbar C.E., Mullikin J.C., Liu P. iPSCs and fibroblast subclones from the same fibroblast population contain comparable levels of sequence variations. Proc. Natl. Acad. Sci. USA. 2017;114:1964–1969. doi: 10.1073/pnas.1616035114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D’Antonio M., Benaglio P., Jakubosky D., Greenwald W.W., Matsui H., Donovan M.K.R., Li H., Smith E.N., D’Antonio-Chronowska A., Frazer K.A. Insights into the Mutational Burden of Human Induced Pluripotent Stem Cells from an Integrative Multi-Omics Approach. Cell Rep. 2018;24:883–894. doi: 10.1016/j.celrep.2018.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodwin S., McPherson J.D., McCombie W.R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lappalainen T., Scott A.J., Brandt M., Hall I.M. Genomic Analysis in the Age of Human Genome Sequencing. Cell. 2019;177:70–84. doi: 10.1016/j.cell.2019.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shakiba N., Fahmy A., Jayakumaran G., McGibbon S., David L., Trcka D., Elbaz J., Puri M.C., Nagy A., van der Kooy D. Cell competition during reprogramming gives rise to dominant clones. Science. 2019;364:eaan0925. doi: 10.1126/science.aan0925. [DOI] [PubMed] [Google Scholar]
- 18.Rouhani F.J., Nik-Zainal S., Wuster A., Li Y., Conte N., Koike-Yusa H., Kumasaka N., Vallier L., Yusa K., Bradley A. Mutational History of a Human Cell Lineage from Somatic to Induced Pluripotent Stem Cells. PLoS Genet. 2016;12:e1005932. doi: 10.1371/journal.pgen.1005932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skamagki M., Correia C., Yeung P., Baslan T., Beck S., Zhang C., Ross C.A., Dang L., Liu Z., Giunta S. ZSCAN10 expression corrects the genomic instability of iPSCs from aged donors. Nat. Cell Biol. 2017;19:1037–1048. doi: 10.1038/ncb3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su R.J., Yang Y., Neises A., Payne K.J., Wang J., Viswanathan K., Wakeland E.K., Fang X., Zhang X.B. Few single nucleotide variations in exomes of human cord blood induced pluripotent stem cells. PLoS ONE. 2013;8:e59908. doi: 10.1371/journal.pone.0059908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben-David U., Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer. 2011;11:268–277. doi: 10.1038/nrc3034. [DOI] [PubMed] [Google Scholar]
- 22.Sugiura M., Kasama Y., Araki R., Hoki Y., Sunayama M., Uda M., Nakamura M., Ando S., Abe M. Induced pluripotent stem cell generation-associated point mutations arise during the initial stages of the conversion of these cells. Stem Cell Reports. 2014;2:52–63. doi: 10.1016/j.stemcr.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshihara M., Araki R., Kasama Y., Sunayama M., Abe M., Nishida K., Kawaji H., Hayashizaki Y., Murakawa Y. Hotspots of De Novo Point Mutations in Induced Pluripotent Stem Cells. Cell Rep. 2017;21:308–315. doi: 10.1016/j.celrep.2017.09.060. [DOI] [PubMed] [Google Scholar]
- 24.Vijg J., Suh Y. Genome instability and aging. Annu. Rev. Physiol. 2013;75:645–668. doi: 10.1146/annurev-physiol-030212-183715. [DOI] [PubMed] [Google Scholar]
- 25.Hadjimichael C., Chanoumidou K., Papadopoulou N., Arampatzi P., Papamatheakis J., Kretsovali A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells. 2015;7:1150–1184. doi: 10.4252/wjsc.v7.i9.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruiz S., Gore A., Li Z., Panopoulos A.D., Montserrat N., Fung H.L., Giorgetti A., Bilic J., Batchelder E.M., Zaehres H. Analysis of protein-coding mutations in hiPSCs and their possible role during somatic cell reprogramming. Nat. Commun. 2013;4:1382. doi: 10.1038/ncomms2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng L., Hansen N.F., Zhao L., Du Y., Zou C., Donovan F.X., Chou B.K., Zhou G., Li S., Dowey S.N., NISC Comparative Sequencing Program Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell. 2012;10:337–344. doi: 10.1016/j.stem.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar K.R., Cowley M.J., Davis R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019;45:661–673. doi: 10.1055/s-0039-1688446. [DOI] [PubMed] [Google Scholar]
- 30.Li C., Klco J.M., Helton N.M., George D.R., Mudd J.L., Miller C.A., Lu C., Fulton R., O’Laughlin M., Fronick C. Genetic heterogeneity of induced pluripotent stem cells: results from 24 clones derived from a single C57BL/6 mouse. PLoS ONE. 2015;10:e0120585. doi: 10.1371/journal.pone.0120585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haase A., Olmer R., Schwanke K., Wunderlich S., Merkert S., Hess C., Zweigerdt R., Gruh I., Meyer J., Wagner S. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell. 2009;5:434–441. doi: 10.1016/j.stem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 32.Frank S.A. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. USA. 2010;107:1725–1730. doi: 10.1073/pnas.0909343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bianconi E., Piovesan A., Facchin F., Beraudi A., Casadei R., Frabetti F., Vitale L., Pelleri M.C., Tassani S., Piva F. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013;40:463–471. doi: 10.3109/03014460.2013.807878. [DOI] [PubMed] [Google Scholar]
- 34.Ryland G.L., Doyle M.A., Goode D., Boyle S.E., Choong D.Y., Rowley S.M., Li J., Bowtell D.D., Tothill R.W., Campbell I.G., Gorringe K.L., Australian Ovarian Cancer Study Group Loss of heterozygosity: what is it good for? BMC Med. Genomics. 2015;8:45. doi: 10.1186/s12920-015-0123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wijnhoven S.W., Kool H.J., van Teijlingen C.M., van Zeeland A.A., Vrieling H. Loss of heterozygosity in somatic cells of the mouse. An important step in cancer initiation? Mutat. Res. 2001;473:23–36. doi: 10.1016/s0027-5107(00)00163-9. [DOI] [PubMed] [Google Scholar]
- 36.Araten D.J., Golde D.W., Zhang R.H., Thaler H.T., Gargiulo L., Notaro R., Luzzatto L. A quantitative measurement of the human somatic mutation rate. Cancer Res. 2005;65:8111–8117. doi: 10.1158/0008-5472.CAN-04-1198. [DOI] [PubMed] [Google Scholar]
- 37.Viel A., Bruselles A., Meccia E., Fornasarig M., Quaia M., Canzonieri V., Policicchio E., Urso E.D., Agostini M., Genuardi M. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-defective Colorectal Cancer. EBioMedicine. 2017;20:39–49. doi: 10.1016/j.ebiom.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ju Y.S., Martincorena I., Gerstung M., Petljak M., Alexandrov L.B., Rahbari R., Wedge D.C., Davies H.R., Ramakrishna M., Fullam A. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–718. doi: 10.1038/nature21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandrov L.B., Stratton M.R. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014;24:52–60. doi: 10.1016/j.gde.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marth G.T., Yu F., Indap A.R., Garimella K., Gravel S., Leong W.F., Tyler-Smith C., Bainbridge M., Blackwell T., Zheng-Bradley X., 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang W., Ng H.W., Shu M., Luo H., Su Z., Ge W., Perkins R., Tong W., Hong H. Comparing genetic variants detected in the 1000 genomes project with SNPs determined by the International HapMap Consortium. J. Genet. 2015;94:731–740. doi: 10.1007/s12041-015-0588-8. [DOI] [PubMed] [Google Scholar]
- 42.Sorrentino L., Cossu F., Milani M., Malkoc B., Huang W.C., Tsay S.C., Ru Hwu J., Mastrangelo E. Structure-Activity Relationship of NF023 Derivatives Binding to XIAP-BIR1. ChemistryOpen. 2019;8:476–482. doi: 10.1002/open.201900059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu M., Lin S.C., Huang Y., Kang Y.J., Rich R., Lo Y.C., Myszka D., Han J., Wu H. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell. 2007;26:689–702. doi: 10.1016/j.molcel.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Attaran-Bandarabadi F., Abhari B.A., Neishabouri S.H., Davoodi J. Integrity of XIAP is essential for effective activity recovery of apoptosome and its downstream caspases by Smac/Diablo. Int. J. Biol. Macromol. 2017;101:283–289. doi: 10.1016/j.ijbiomac.2017.03.088. [DOI] [PubMed] [Google Scholar]
- 45.Ihry R.J., Salick M.R., Ho D.J., Sondey M., Kommineni S., Paula S., Raymond J., Henry B., Frias E., Wang Q. Genome-Scale CRISPR Screens Identify Human Pluripotency-Specific Genes. Cell Rep. 2019;27:616–630.e6. doi: 10.1016/j.celrep.2019.03.043. [DOI] [PubMed] [Google Scholar]
- 46.Kilpinen H., Goncalves A., Leha A., Afzal V., Alasoo K., Ashford S., Bala S., Bensaddek D., Casale F.P., Culley O.J. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017;546:370–375. doi: 10.1038/nature22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishikawa T. Next-generation sequencing traces human induced pluripotent stem cell lines clonally generated from heterogeneous cancer tissue. World J. Stem Cells. 2017;9:77–88. doi: 10.4252/wjsc.v9.i5.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma C., Wang F., Han B., Zhong X., Si F., Ye J., Hsueh E.C., Robbins L., Kiefer S.M., Zhang Y. SALL1 functions as a tumor suppressor in breast cancer by regulating cancer cell senescence and metastasis through the NuRD complex. Mol. Cancer. 2018;17:78. doi: 10.1186/s12943-018-0824-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pardo M., Lang B., Yu L., Prosser H., Bradley A., Babu M.M., Choudhary J. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell. 2010;6:382–395. doi: 10.1016/j.stem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karantzali E., Lekakis V., Ioannou M., Hadjimichael C., Papamatheakis J., Kretsovali A. Sall1 regulates embryonic stem cell differentiation in association with nanog. J. Biol. Chem. 2011;286:1037–1045. doi: 10.1074/jbc.M110.170050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopes Novo C., Rugg-Gunn P. Crosstalk between pluripotency factors and higher-order chromatin organization. Nucleus. 2016;7:447–452. doi: 10.1080/19491034.2016.1248013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams F.F., Heckl D., Hoffmann T., Talbot S.R., Kloos A., Thol F., Heuser M., Zuber J., Schambach A., Schwarzer A. An optimized lentiviral vector system for conditional RNAi and efficient cloning of microRNA embedded short hairpin RNA libraries. Biomaterials. 2017;139:102–115. doi: 10.1016/j.biomaterials.2017.05.032. [DOI] [PubMed] [Google Scholar]
- 53.Werner B., Sottoriva A. Variation of mutational burden in healthy human tissues suggests non-random strand segregation and allows measuring somatic mutation rates. PLoS Comput. Biol. 2018;14:e1006233. doi: 10.1371/journal.pcbi.1006233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Albertini R.J., Nicklas J.A., O’Neill J.P., Robison S.H. In vivo somatic mutations in humans: measurement and analysis. Annu. Rev. Genet. 1990;24:305–326. doi: 10.1146/annurev.ge.24.120190.001513. [DOI] [PubMed] [Google Scholar]
- 55.Bigbee W.L., Fuscoe J.C., Grant S.G., Jones I.M., Gorvad A.E., Harrington-Brock K., Strout C.L., Thomas C.B., Moore M.M. Human in vivo somatic mutation measured at two loci: individuals with stably elevated background erythrocyte glycophorin A (gpa) variant frequencies exhibit normal T-lymphocyte hprt mutant frequencies. Mutat. Res. 1998;397:119–136. doi: 10.1016/s0027-5107(97)00186-3. [DOI] [PubMed] [Google Scholar]
- 56.Rondelli T., Berardi M., Peruzzi B., Boni L., Caporale R., Dolara P., Notaro R., Luzzatto L. The frequency of granulocytes with spontaneous somatic mutations: a wide distribution in a normal human population. PLoS ONE. 2013;8:e54046. doi: 10.1371/journal.pone.0054046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martincorena I., Campbell P.J. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 58.Martin U., Kiessig V., Blusch J.H., Haverich A., von der Helm K., Herden T., Steinhoff G. Expression of pig endogenous retrovirus by primary porcine endothelial cells and infection of human cells. Lancet. 1998;352:692–694. doi: 10.1016/S0140-6736(98)07144-X. [DOI] [PubMed] [Google Scholar]
- 59.Ewing B., Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 60.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cingolani P., Patel V.M., Coon M., Nguyen T., Land S.J., Ruden D.M., Lu X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Genovese G., Kähler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oughtred R., Stark C., Breitkreutz B.J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019;47(D1):D529–D541. doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pletscher-Frankild S., Pallejà A., Tsafou K., Binder J.X., Jensen L.J. DISEASES: text mining and data integration of disease-gene associations. Methods. 2015;74:83–89. doi: 10.1016/j.ymeth.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 68.Chakravarty D., Gao J., Phillips S.M., Kundra R., Zhang H., Wang J., Rudolph J.E., Yaeger R., Soumerai T., Nissan M.H. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017;2017 doi: 10.1200/PO.17.00011. PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sondka Z., Bamford S., Cole C.G., Ward S.A., Dunham I., Forbes S.A. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer. 2018;18:696–705. doi: 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fellmann C., Hoffmann T., Sridhar V., Hopfgartner B., Muhar M., Roth M., Lai D.Y., Barbosa I.A., Kwon J.S., Guan Y. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 71.Schwarzer A., Emmrich S., Schmidt F., Beck D., Ng M., Reimer C., Adams F.F., Grasedieck S., Witte D., Käbler S. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat. Commun. 2017;8:218. doi: 10.1038/s41467-017-00212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu Q., Zhang B., Chen R., Fu C., A J., Fu X., Li J., Fu L., Zhang Z., Dong J.T. ZFHX3 is indispensable for ERβ to inhibit cell proliferation via MYC downregulation in prostate cancer cells. Oncogenesis. 2019;8:28. doi: 10.1038/s41389-019-0138-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang X.H., Tang F., Shin J., Cunningham J.M. Incorporating genomic, transcriptomic and clinical data: a prognostic and stem cell-like MYC and PRC imbalance in high-risk neuroblastoma. BMC Syst. Biol. 2017;11:92. doi: 10.1186/s12918-017-0466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data underlying the study are available on request and are currently deposited on servers of Hannover Medical School, Germany.