

Abstract
Omics analyses are powerful methods to obtain an integrated view of complex biological processes, disease progression, or therapy efficiency. However, few studies have compared different disease forms and different therapy strategies to define the common molecular signatures representing the most significant implicated pathways. In this study, we used RNA sequencing and mass spectrometry to profile the transcriptomes and proteomes of mouse models for three forms of centronuclear myopathies (CNMs), untreated or treated with either a drug (tamoxifen), antisense oligonucleotides reducing the level of dynamin 2 (DNM2), or following modulation of DNM2 or amphiphysin 2 (BIN1) through genetic crosses. Unsupervised analysis and differential gene and protein expression were performed to retrieve CNM molecular signatures. Longitudinal studies before, at, and after disease onset highlighted potential disease causes and consequences. Main pathways in the common CNM disease signature include muscle contraction, regeneration and inflammation. The common therapy signature revealed novel potential therapeutic targets, including the calcium regulator sarcolipin. We identified several novel biomarkers validated in muscle and/or plasma through RNA quantification, western blotting, and enzyme-linked immunosorbent assay (ELISA) assays, including ANXA2 and IGFBP2. This study validates the concept of using multi-omics approaches to identify molecular signatures common to different disease forms and therapeutic strategies.
Keywords: genetic disease, congenital myopathy, centronuclear myopathy, myotubular myopathy, XLMTM, MTM1, myotubularin, omics, biomarker, myostatin, RNA interference
Graphical abstract
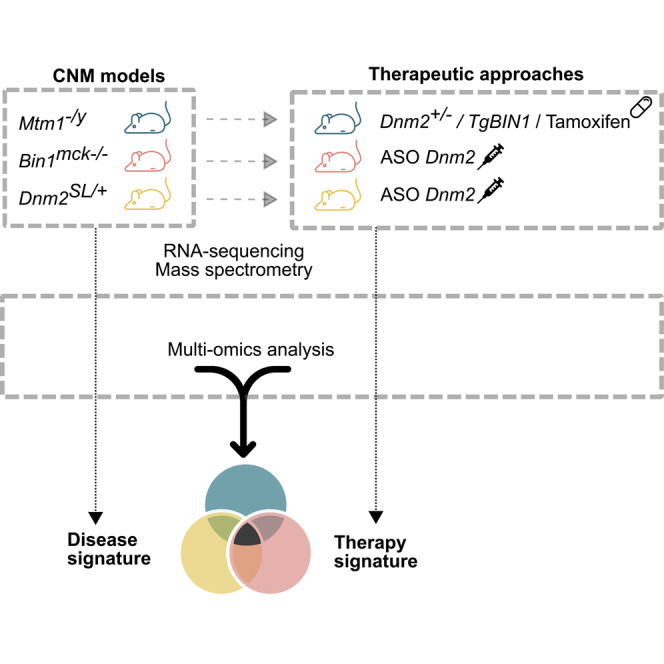
A multi-omics analysis is performed on transgenic mouse lines faithfully modeling different forms of centronuclear myopathies treated or not with several therapeutic approaches. The common disease and therapy signatures are described at the molecular level. Potential circulating biomarkers are identified and await confirmation in patients.
Introduction
In recent years, omics strategies (e.g., transcriptome, proteome) have become powerful methods to obtain an unbiased and integrated view of complex biological processes, disease progression, or therapy efficiency.1,2 Most previous studies focused on a single disease or therapeutic approach. However, omics have the potential to identify molecular signatures common to different disease forms or to several therapeutic strategies. In this study, we performed omics analyses in mouse models faithfully mimicking different forms of centronuclear myopathies (CNMs) and myotubular myopathies treated or not with different therapeutic strategies to identify common disease and therapy signatures.
CNMs and myotubular myopathies are a sub-group of congenital myopathies whose clinical signs develop from birth to adulthood. They are rare genetic diseases with a strong medical impact on patient survival and quality of life.3,4 CNMs are characterized by generalized muscle weakness and hypotonia impairing breathing capacity.5 Histological hallmarks of patients’ muscle include the presence of internal or central nuclei that are normally at the fiber periphery, aggregation of oxidative staining, hypotrophy of myofibers that also present with a rounder shape, predominance of oxidative type I fiber, and structural disorganization of sarcomeres and triads.6 Several genes were found mutated in different forms of CNM. The most common and severe form, X-linked CNM, which is also called myotubular myopathy (MIM: 310400), is due to loss-of-function mutations in MTM1 coding for the lipid phosphatase myotubularin.7 Dominant forms are linked to mutations in DNM2 (dynamin 2; MIM: 160150) coding the large GTPase DNM2 implicated in membrane trafficking and fission.8 Some DNM2 mutations lead to a neonatal form, such as the Ser619Leu missense mutation, while others are associated with later onset.9,10 Recessive and dominant forms are also due to mutations in BIN1 (amphiphysin 2; MIM: 255200), coding for the membrane curvature remodeling protein BIN1.11,12 These three forms represent the main forms of CNM. Nevertheless, additional genes are implicated in phenotypes overlapping with CNM such as RYR1, TTN, SPEG, CACNA1S, or PYROXD1.3,13,14 SPEG is linked to CNM with cardiomyopathy, while the histopathology associated with the other genes combines internal nuclei with additional defects such as cores or protein inclusions.
Previous studies in cellular and animal models and in patients’ muscle biopsies for the canonical CNM forms suggested several pathomechanisms in skeletal muscle, including defects in triad structure and deficient excitation-contraction coupling, altered organelle positioning and function, abnormal neuromuscular junction (NMJ), deficient satellite cells, and dysregulation of autophagy.4,15, 16, 17
Defects in the genes implicated in the three canonical CNM forms were modeled in vivo in different organisms, ranging from yeast to C. elegans, Drosophila, zebrafish, and mice.17,18 In addition, spontaneous mutations in either MTM1 or BIN1 were found in dogs developing CNM.19, 20, 21, 22 In mice, the Mtm1−/y knockout mouse develops a progressive myopathy with a histopathology mimicking patient hallmarks.23 Additional Mtm1 knockout lines were generated and showed a similar phenotype, while the Mtm1RC/y knockin led to a milder phenotype.24, 25, 26 Concerning Dnm2, knockin mice for the most common mutations in the mildest form (Arg465Trp; Dnm2RW/+) or the severe neonatal form (Ser619Leu; Dnm2SL/+) were generated and reproduce a mild or severe muscle weakness, respectively, with CNM-like histopathology without centralized nuclei.27,28 For Bin1, full loss of BIN1 in Bin1−/− mice is perinatally lethal, preventing the comparison with the other CNM models.29,30 We recently created a mouse model with a skeletal muscle-specific Bin1 deletion that is viable and faithfully reproduces the decreased muscle force and most histopathological hallmarks of CNM (Bin1mck−/−; unpublished data). Herein, we focus on omics analysis of Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ mice, since they represent faithful models for the three canonical CNM forms and the mice share a similar skeletal muscle organization with patients.
Several therapeutic proofs of concepts were recently validated in different CNM models, including the three CNM mice investigated in the present study.17 Adeno-associated virus (AAV) transduction of MTM1 or its closer homolog MTMR2 rescued the Mtm1−/y mouse, and AAV-MTM1 was further validated in the MTM1 Labrador model and recently injected in patients in a clinical trial.31, 32, 33 DNM2 level was found increased at least in Mtm1−/y mice and MTM1 patients, whereas normalization of DNM2 level rescued the Mtm1−/y mouse, the Bin1−/− and Bin1mck−/− mice, and both Dnm2RW/+ and Dnm2SL/+ mice.28,34, 35, 36 DNM2 was reduced through three methods: genetic cross with a Dnm2+/− mouse, short hairpin RNA (shRNA), or antisense oligonucleotides (ASOs).34,37,38 Overexpression of BIN1 through genetic cross with a TgBIN1 mouse or AAV-BIN1 expressing human BIN1 rescued Mtm1−/y and Bin1−/− mice.39 In addition, treatment with tamoxifen, an estrogen modulator already used in clinic for breast cancer, partially rescued the Mtm1−/y mouse, potentially representing a drug repurposing strategy.40,41 Additional potential therapies have been tested in mice and other CNM models, and they include Dnm2 allele-specific silencing or trans-splicing, Pik3c2b downregulation, mTOR or acetylcholine esterase inhibitors.17
To identify the main pathomechanisms, potential biomarkers, and novel therapeutic targets for different forms of CNM, we performed transcriptome and proteome analyses of muscles from Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ mice either developing the disease or treated with three different therapies, including two methodological approaches for one of the targets. We identified disease and therapeutic molecular signatures common to the three main forms of CNM.
Results
Animal models, treatments, and omics strategies
In mice, muscle embryonic development proceeds from embryonic day (E)10.5 to birth that happens at about E19.42 Then, muscle growth during postnatal maturation follows two phases: one based on satellite cells fusion up to 2–3 weeks, and a second based on growth factor signaling from weaning (around 3 weeks of age) to adulthood at 7 weeks (Figure 1A).43,44 Mtm1−/y mice develop a progressive myopathy from 2 to 3 weeks, leading to a strong muscle hypotrophy and decreased locomotor activity, and death by about 8 weeks. Bin1mck−/− mice have near normal locomotor activity and decreased muscle force at 8 weeks. Dnm2SL/+ mice have some feeding defects at birth, correlating with decreased body weight, strong muscle atrophy, and decreased locomotor activity, and survive in adulthood. All of these mice have CNM-like histopathology by 7–8 weeks. Based on the key steps of muscle maturation and on the disease progression in the different models, we performed RNA sequencing (RNA-seq) for transcriptome analysis in tibialis anterior (TA) muscle from the three CNM mice at 7 weeks. In addition, RNA-seq was done at E18.5 and 2 weeks for a longitudinal follow-up of Mtm1−/y mice. Mass spectrometry for proteome analysis was performed in TA at E18.5, 2 weeks, and 7 weeks for a longitudinal follow-up in Mtm1−/y mice (Table S1).
Figure 1.

Experimental design
(A) Timeline of the different steps occurring during muscle development in mice. The green arrow represents the normal lifespan of control mice (WT). The phenotype of the three mouse models (Mtm1−/y, Bin1mck−/−, Dnm2SL/+) used in this study is illustrated by the colored arrows with a color gradient ranging from green for non-affected mice, yellow for the onset of myopathy, and red for affected mice. (B) Molecular analyses were performed on different mouse models, Mtm1−/y, Bin1mck−/−, Dnm2SL/+, untreated or treated by different therapeutic approaches (overexpression of BIN1, tamoxifen supplementation, or downregulation of Dnm2 either by genetic cross or by ASO injection). Disease signature refers to the common dysregulated genes in the three mouse models compared to WT littermates, while the therapy signature refers to the common genes rescued following the different treatments.
Several therapeutic approaches were applied to Mtm1−/y mice and consisted of either BIN1 overexpression by crossing with TgBIN1 mice (Mtm1−/yTgBIN1), tamoxifen supplementation in food, or DNM2 downregulation by crossing with Dnm2+/− mice (Mtm1−/yDnm2+/−) (Figure 1B; Table S1). DNM2 was also downregulated with another method, systemic injection of ASOs (ASO Dnm2), in Bin1mck−/− and Dnm2SL/+ mice and compared to injection of PBS or control ASO. All of these treated cohorts were analyzed by RNA-seq in TA at 7 weeks and compared to the above untreated mice. In addition, RNA-seq and mass spectrometry of TA from Mtm1−/yDnm2+/− mice were performed at E18.5, 2 weeks, and 7 weeks. Muscle samples were obtained from our previous studies reporting therapeutic efficacy.28,34,39,40
In addition, in order to identify dysregulated muscle proteins that are potentially circulating, mass spectrometry was performed in sera from wild-type (WT) mice at 7 weeks and compared with the above muscle transcriptome and proteome data.
Influence of the genetic and environment backgrounds
To assess the impact of the genetic background and animal housing on the transcriptome, we analyzed different RNA-seq data from different cohorts of the same Mtm1−/y mouse line on different genetic backgrounds and raised in different animal houses. Four different cohorts were evaluated (Table 1): cohort MTM1-a was crossed on the 129Pas background and bred in France, cohort MTM1-b was on a 50% 129Pas and 50% C57BL/6N background and raised in the same animal facility in France, cohort MTM1-c was raised in Switzerland on the 129Pas background, and cohort MTM1-d was bred in Canada on a C57BL/6J background. Muscles used for transcriptomics for the first three cohorts were TA analyzed at 7 weeks. The transcriptome for cohort MTM1-d was previously published and done from quadriceps at 5 weeks.41 We compared differentially expressed genes between Mtm1−/y and WT mice for the different cohorts. The threshold used to define dysregulated genes was set at log2fold change (log2FC) ±1 and a p value of <0.05. The number of dysregulated genes in Mtm1−/y mice ranged from 1,275 to 1,981 (Figure 2A). A total of 287 genes were found commonly dysregulated across the different cohorts. These genes correspond to the disease signature following MTM1 loss, and their expression is not impacted by any environmental or housing parameters, the genetic background, or the muscle analyzed (Table S2). As expected, the most divergent transcriptome was from cohort 4 that differs from the other cohorts by both the genetic background and the muscle, as 56% of dysregulated genes are specific to this cohort versus 23%–33% for the other cohorts.
Table 1.
Description of the mouse cohorts used in the study, including country, age, background, muscle and sequencer used
| Country | Age (weeks) | Background | Muscle | Sequencer | |
|---|---|---|---|---|---|
| Cohort 1, MTM1-a | France | 7 | 129Pas | tibialis anterior | HiSeq 4000 |
| Cohort 2, MTM1-b | France | 7 | 50% 129Pas; 50% C57BL/6N | tibialis anterior | HiSeq 4000 |
| Cohort 3, MTM1-c | Switzerland | 7 | 129Pas | tibialis anterior | HiSeq 4000 |
| Cohort 4, MTM1-d | Canada (Maani et al.41) | 5 | C57BL/6J | quadriceps | HiSeq 2500 |
| Cohort DNM2 | France | 7 | C57BL/6N | tibialis anterior | HiSeq 4000 |
| Cohort BIN1 | France | 7 | C57BL/6N | tibialis anterior | HiSeq 4000 |
Figure 2.

MTM1-CNM signature in different species
(A) Venn diagram illustrating the shared dysregulated genes based on the Mtm1−/y versus WT comparison of four mouse cohorts. The percentages of uniquely dysregulated genes and the number of differentially expressed genes in each cohort are indicated in brackets. (B) Gene Ontology (GO) enrichment analysis of differentially expressed genes common to the four Mtm1−/y mouse cohorts. The 20 GO biological process terms with the lowest p values are displayed. (C) Venn diagram illustrating the shared dysregulated genes based on MTM1 versus control comparison in three different species: humans, mice (TA) and dogs (Vastus lateralis). (D) mRNA log2fold change expression of differentially expressed genes common to the three species. (E) GO enrichment analysis of differentially expressed genes common to mice and dogs. The 20 GO biological process terms with the lowest p value are displayed. (F) mRNA log2fold change expression of differentially expressed genes common between mice and dogs determined by RNA-seq (dogs, mice) and qRT-PCR (mice).
All of the common dysregulated genes followed the same trend in the different cohorts. We found that 67 common genes were downregulated and 220 common genes were upregulated. Gene Ontology (GO) analysis revealed an enrichment for muscle development and contraction, cell adhesion, and immune cells (Figure 2B). The most upregulated protein-coding genes were Sox11, Krt18, Mt3, Msln, Hsf2bp, and Fosl1, and the most downregulated genes were Mstn, Cdh4, Edn3, Mtm1, Ighm, Fam19a4, Nt5c1a, and Amd1 (Table 2). We thus report the disease signature for MTM1-CNM that is independent of the genetic and environmental backgrounds in mice.
Table 2.
List of the most dysregulated genes in MTM1-CNM mice
| Gene name | Log2FC cohort MTM1-a | Log2FC cohort MTM1-b | Log2FC cohort MTM1-c | Log2FC cohort MTM1-d |
|---|---|---|---|---|
| Sox11 | 6.16 | 4.70 | 5.80 | 3.97 |
| Krt18 | 5.25 | 3.20 | 7.78 | 5.23 |
| Gm28653 | 5.24 | 2.64 | 5.13 | 3.80 |
| Mt3 | 4.76 | 3.31 | 5.28 | 4.66 |
| Gm13583 | 4.65 | 3.17 | 6.53 | 8.15 |
| Msln | 4.61 | 3.28 | 3.96 | 4.19 |
| Hsf2bp | 4.32 | 3.48 | 4.24 | 4.21 |
| Fosl1 | 4.21 | 2.69 | 4.81 | 6.85 |
| C130080G10Rik | −1.88 | −2.13 | −3.36 | −5.96 |
| Amd1 | −2.04 | −2.66 | −3.17 | −2.72 |
| Nt5c1a | −2.05 | −2.54 | −2.79 | −3.77 |
| Fam19a4 | −2.31 | −3.00 | −2.55 | −3.77 |
| Ighm | −2.33 | −2.89 | −3.34 | −2.55 |
| Mtm1 | −2.43 | −2.12 | −2.10 | −2.55 |
| Edn3 | −2.51 | −3.27 | −2.93 | −1.98 |
| Cdh4 | −2.57 | −3.91 | −3.63 | −2.85 |
| Mstn | −2.59 | −1.79 | −1.89 | −2.72 |
Conserved disease signatures linked to MTM1 mutations among species
We next explored the conservation of the disease signature linked to MTM1 loss in different species. In humans (biceps brachii or quadriceps), Noguchi et al.45 performed microarray analysis on a set of 4,200 genes previously known to be expressed in skeletal muscle from eight patients with different MTM1 mutations. 183 genes were significantly dysregulated compared to unaffected individuals. Of note, MTM1 was not reported as dysregulated in this study. In dogs, Dupont et al.46 used RNA-seq to analyze two hindlimb muscles from Labradors lacking MTM1. They found 824 and 1,122 genes differentially expressed in the biceps femoris and the vastus lateralis, respectively, with 400 genes dysregulated in both muscles. In this study, we used the 632 genes that we identified in the RNA-seq analyses conducted in Mtm1−/y mouse cohorts MTM1-a, MTM1-b, and MTM1-c at 7 weeks (Figure 2A; Table S3).
Interspecies analysis revealed five differentially expressed genes shared by mice, dogs, and humans (Figure 2C). Among them, CHRND and CHRNA1 coding for two subunits of the acetylcholine receptor in the NMJ were upregulated (Figure 2D). MYOG (myogenin), coding for a transcription factor key in muscle differentiation, was upregulated in dogs and mice and downregulated in humans. POPDC3 was upregulated in humans and mice and downregulated in dogs, is also implicated in muscle development, and was found mutated in limb-girdle muscular dystrophy.47 Most of the dysregulated genes were specific to each species; therefore, we analyzed them by GO enrichment and it revealed a few processes that might be species-dependent, such as hemostasis dysregulation in humans or specific impact on kinase pathways in dogs (Figure S1; Table S4).
As the human data were based on microarray analysis of only a subset of genes, additional comparisons were done between dogs and mice and identified 63 additional dysregulated genes (vastus lateralis versus TA) (Figure 2C; Table S5) or 49 genes (biceps femoris versus TA) (Figure S1; Table S6). GO terms related to these genes were highly enriched in muscle development (Figure 2E). Among these genes, Chrna1, Chrnd, and Chrng highlight the NMJ, and Myog and Pax7 highlight the transcriptional regulation of muscle differentiation and regeneration. Other examples confirmed by qRT-PCR included downregulation of Mstn (myostatin), a ligand of transforming growth factor β (TGF-β) receptor involved in muscle growth, and upregulation of Fst (follistatin), coding for an inhibitor of myostatin (Figure 2F). We found upregulation of Igfbp2, coding an insulin-like growth factor-binding protein potentially involved in muscle differentiation and hypertrophy. Cilp was upregulated and codes for a regulator of IGF1 (insulin-like growth factor type 1) and TGF-β signaling. Genes coding for potential regulators of the Rac1-actin pathway, Tiam2 and Arhgap36, were also upregulated.
Overall, defects in muscle development and the NMJ appear conserved in mice, dogs, and humans with MTM1-CNM. The more detailed investigations in mice and dogs highlighted additional pathways of interest such as muscle growth and repair.
Longitudinal molecular profiling of Mtm1−/y mice through disease progression
Several pathways were identified in the MTM1-CNM disease signatures at an age when mice, dogs, and patients are strongly affected. To define the primary molecular causes versus consequences of the disease, we performed longitudinal analyses of the transcriptome and proteome of TA muscles from Mtm1−/y mice at pre-symptomatic age (E18.5) and early (2 weeks) and late (7 weeks) disease stages (Figure 1A). We used the cohort MTM1-a, including untreated Mtm1−/y mice, Mtm1−/y mice rescued by Dnm2 genetic downregulation, treated and healthy Mtm1−/yDnm2+/− mice, and their WT littermates (Table S1).
Principal component analysis (PCA) on transcriptome data showed that age explains most of the variance between the mouse groups as underlined by the first principal component (PC1 58%, PC2 13%, PC3 5% variance; Figure 3A; Figure S2). Separation of the genotypes appeared at PC4 (4% variance). Interestingly, this separation appeared at 2 weeks and increased at 7 weeks. No genes were significantly dysregulated at E18.5, indicating no difference between genotypes at late embryonic stage. A total of 1,175 genes were dysregulated at 2 weeks and 1,981 at 7 weeks (Figure 3B; Table S7). Potential disease causes found at 2 weeks are defects of muscle contraction, sarcomere organization, and cell adhesion (GO term analysis; Figure 3B; Figure S3). Disease consequences found only at 7 weeks highlighted activation of the inflammation pathway, suggesting infiltration of affected muscles by immune cells. Volcano plots display the most dysregulated genes at 2 and 7 weeks (Figures 3C and 3D). As examples, the three most upregulated genes at both ages were Sln (sarcolipin), Krt8, and Krt18 (keratins) (Figures 3C and 3D). Dysregulation of these and other genes was confirmed by qRT-PCR (Figures S4 and S5). The downregulation of Mstn, observed in late disease stage in dogs and mice, was already apparent at the 2-week early stage in mice (Figure 3E). An example for sarcomere organization is Ahnak2, whose protein product (AHNAK2) localizes to Z-line. Tnnt2 (cardiac troponin) and Myl4 (cardiac myosin light chain) are implicated in muscle contraction, expressed in embryonic but not adult skeletal muscle and in adult cardiac muscle under normal conditions, and found upregulated in this myopathy. The cell adhesion was represented for example by Itga3 (integrin). Genes underlying the activation of the inflammation pathway at 7 weeks include Cxcl1 (chemokine) and Tlr2 (Toll-like receptor).
Figure 3.

Longitudinal mRNA profiling of Mtm1−/y mice
(A) Principal component analysis of RNA-seq data. The first and fourth axes are represented. Colored symbols represent genotypes, and shapes represent ages for each mouse. (B) Venn diagram illustrating the shared and specific dysregulated genes based on the Mtm1−/y versus WT comparison at 2 and 7 weeks. The most enriched GO biological processes are represented by dashed boxes. (C and D) Volcano plots representing the differentially expressed genes at (C) 2 weeks and (D) 7 weeks. Upregulated genes are in red, and downregulated genes are in blue. (E) Gene expression data (log-normalized counts) determined by RNA-seq for Mstn (muscle growth), Sln (calcium homeostasis), Ahnak2 (sarcomere organization), Myl4 and Tnnt2 (muscle contraction), Itga3 (cell adhesion), and Cxc3cl1 and Tlr2 (inflammation pathway) across time. Each dot represents an individual mouse; the shaded area represents the confidence interval at 0.95.
In parallel, PCA was also performed on proteome data and showed that the variance between the mouse groups is first explained by age (PC1; 39% variance; Figure 4A) followed by genotypes (PC2; 7%). Similarly to the transcriptome data, the genotype separation appeared at 2 weeks and increased at 7 weeks, indicating no difference at E18.5. Out of 1,462 proteins consistently detected in each of the muscle samples, 168 proteins were dysregulated at 2 weeks and 496 at 7 weeks in the Mtm1−/y mice compared to WT mice (Figure 4B; Table S8). Potential disease causes found at 2 weeks were related mainly to muscle sarcomere and contraction (GO terms analysis; Figure S6). At 7 weeks, defects of the muscle contraction pathway were persistent, and at this late disease stage dysregulation of ribosomal biogenesis (translation) appeared. The transcriptome and proteome data analysis consistently highlighted muscle contraction defects as a main early sign of the disease. The late dysregulation of ribosomal biogenesis may reflect a compensatory mechanism following alteration of protein homeostasis correlated with the strong fiber hypotrophy in MTM1-CNM. Examples of dysregulated proteins implicated in muscle contraction include MYH2 (myosin heavy chain) mutated in a proximal myopathy with ophthalmoplegia (MIM: 606337), and TNNC1 (troponin) mutated in dilated cardiomyopathy (MIM: 611879) (Figures 4C–4E). These proteins strongly correlate pathways and gene families found through the above transcriptome analysis: MYH2 and MYL4 are myosin heavy and light chains, respectively, and TNNT2 and TNNC1 are troponins. All four proteins are implicated in muscle contraction. Pearson correlation analysis did not underline a high correlation between specific genes and proteins at 2 and 7 weeks (Figure S7), as generally reported in the literature.48 In conclusion, while the same dysregulated genes/proteins are not necessarily highlighted by the transcriptome and proteome analyses, the same pathways and functions are consistently defective in early and late disease stages. Dysregulation in muscle contraction appears to be an early defect in the MTM1-CNM pathology in mice.
Figure 4.

Longitudinal protein profiling of Mtm1−/y mice
(A) Principal component analysis of mass spectrometry data. Colored symbols represent genotypes, and shapes represent ages for each mouse. Technical and biological replicates are shown. (B) Venn diagram illustrating the shared and specific dysregulated proteins based on the Mtm1−/y versus WT comparison at 2 and 7 weeks; the most enriched GO biological processes are represented by dashed boxes. (C and D) Volcano plots displaying the differentially expressed proteins at (C) 2 weeks and (D) 7 weeks. Upregulated proteins are in red, and downregulated proteins are in blue. (E) MYH2 and TNNC1 (muscle contraction) and RPL3 (ribosomal biogenesis) expression data obtained by mass spectrometry across time. Each dot represents technical and biological replicates; the shaded areas represent the confidence interval at 0.95.
Disease signature common to several CNM forms
Next, we explored whether a common disease signature can unify the different CNM forms linked to either MTM1, BIN1, or DNM2 mutations. First, we compared the levels of these genes/proteins in the different corresponding models, that is, the Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ mice (Table 3; Figure S8). Apart from the lack of MTM1 and BIN1 proteins in their respective knockout mice, DNM2 was found slightly elevated (2.2-fold; p = 0.057;28) in the Dnm2SL/+ mice. No strong alteration of the level of their RNA was detected in the corresponding RNA-seq data. A slight increase in both BIN1 and DNM2 proteins was found in the Mtm1−/y mice, and DNM2 protein was slightly increased in Bin1mck−/− mice.
Table 3.
RNA and protein levels of Mtm1 (MTM1), Bin1 (BIN1), and Dnm2 (DNM2) in Mtm1−/y, Bin1mck−/−, Dnm2SL/+ and Dnm2RW/+ mice
| Mouse line | RNA/protein | MTM1 | BIN1 | DNM2 |
|---|---|---|---|---|
| Mtm1−/y | protein (WB) | absent | fold = 2 | fold = 2.5 |
| p < 0.05 | p < 0.001 | |||
| age 5 weeks | age 7 weeks | age 5 weeks | ||
| Cowling et al.34 | Lionello et al.39 | Cowling et al.34 | ||
| RNA (qPCR) | fold = 0.5 | N/D | fold = 1.3 | |
| p = 0.0095 | p < 0.05 | |||
| age 7 weeks | age 7 weeks | |||
| RNA (transcriptomic) | fold = 0.2 | fold = 1.4 | fold = 1.02 | |
| p = 1.98E−09 | p = 0.046 | p = 0.95 | ||
| age 7 weeks | age 7 weeks | age 7 weeks | ||
| Bin1mck−/− | protein (WB) | fold = 1.07 | absent | fold = 1.5 |
| p = 0.95 | p = 0.052 | |||
| age 8 weeks | age 8 weeks | |||
| unpublished data | ||||
| RNA (qPCR) | fold = 0.97 | absent | fold = 1.4 | |
| p = 0.28 | p = 0.0091 | p < 0.05 | ||
| age 7 weeks | age 7 weeks | age 7 weeks | ||
| unpublished data | ||||
| RNA (transcriptomic) | fold = 0.93 | fold = 0.10 | fold = 1.1 | |
| p = 0.63 | p = 1.63E−209 | p = 0.42 | ||
| age 7 weeks | age 7 weeks | age 7 weeks | ||
| Dnm2SL/+ | protein (WB) | fold = 0.7 | fold = 0.95 | fold = 2.2 |
| p = 0.016 | p > 0.05 | p = 0.057 | ||
| age 8 weeks | age 8 weeks | age 8 weeks | ||
| Massana Muñoz et al.28 | ||||
| RNA (qPCR) | fold = 1.2 | N/D | fold = 0.9 | |
| p = 0.15 | p > 0.05 | |||
| age 7 weeks | age 7 weeks | |||
| Massana Muñoz et al.28 | ||||
| RNA (transcriptomic) | fold = 0.87 | fold = 1.37 | fold = 1.04 | |
| p = 0.023 | p = 2.34E−09 | p = 0.77 | ||
| age 7 weeks | age 7 weeks | age 7 weeks | ||
| Dnm2RW/+ | protein | fold = 1.2 | fold = 1.8 | fold = 1.05 |
| p = 0.4 | p = 0.07 | p = 0.6 | ||
| age 7 weeks | age 7 weeks | age 7 weeks |
WB, western blot; N/D, not determined.
Then, dysregulated genes were extracted from each individual cohort at 7 weeks—cohorts MTM1-a, MTM1-b, MTM1-c, BIN1 (Bin1mck−/−), and DNM2 (Dnm2SL/+)—and compared to their respective WT littermates. For each cohort, disease models were well separated from the WT controls on PC1 of the PCA (Figures 3A and 5A; Figure S9). The number of dysregulated genes correlated with the severity of the related models at this age: more than 1,200 genes for the most severe Mtm1−/y mice that barely survive beyond 9 weeks, 780 genes for the Dnm2SL/+ mice with strong locomotor deficiency, and 308 for the Bin1mck−/− mice with a conserved locomotor function. Among the 25,494 genes detected in all of the different transcriptomes, 155 common dysregulated genes were identified (Figure 5B; Tables S7, S9, S10, S11, S12, and S13). The main cellular component GO terms highlighted the NMJ, basement membrane, sarcomere, and activation of the inflammation pathway, previously identified as the main pathways dysregulated in the Mtm1−/y cohorts (Figure 5C). Thus, the main disease signature common to the three CNM models underlines defects in sarcomere maturation and function, in NMJ maturation as a main cause of muscle weakness, and alteration in cell adhesion and basement membrane as a potential explanation for the altered fiber shape. As previously noted for the Mtm1−/y cohorts, a potential increase in the inflammation transcriptome supports the infiltration of immune cells in the different models. Several genes associated with inflammation were shared between the different CNM forms as indicated by GO terms linked with macrophages (Figure 5C). Since inflammation was not previously demonstrated in CNM models, we labeled macrophages with an anti-CD68 antibody on TA muscle sections. We found a significant increase in macrophage infiltration in the three CNM mouse models (Figures 5D and 5E).
Figure 5.

CNM disease signature in mice
(A) PCA on RNA-seq data on the 155 genes commonly differentially expressed between the five cohorts; each dot represents a mouse. The three different CNM mouse models are represented by red, orange, and yellow dots and the WT controls are represented by green dots. (B) Venn diagram illustrating the shared and specific dysregulated genes between the five CNM cohorts. (C) GO enrichment analysis for biological processes (BPs), cellular component (CC), and molecular function (MF) of the 155 common differentially expressed genes between the five CNM cohorts. GO terms with highest ratio and lowest q-value are represented. The ratio represents the number of genes dysregulated divided by the total number of genes in the category. The color scale is based on the q-value; dark colors indicate most significantly over-represented terms, while lighter colors indicate the least significant terms. (D) Macrophage localization by immunofluorescence in transverse section from TA muscle at 7 weeks in Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ mice. Nuclei: DAPI (blue), macrophages (red), plasma membrane (WGA, yellow). Arrowheads point to macrophages. Scale bars, 100 μm. (E) Quantification of macrophages. t test: ∗p < 0.05, ∗∗p < 0.01. (F) Transcriptomic expression changes between diseased and WT mice for the five cohorts. The log2fold change expression of Anxa2, Cilp, Fetub, Fgfr4, Fst, Igfbp2, Mstn, Myl4, Runx1, S100a4, Serpinb1a, Sln, Sox11, and Tnnt2 are represented by the bars.
qRT-PCR analyses confirmed the dysregulation of all 14 genes tested in the three different mouse models (Figures S10–S12). In particular, the common CNM disease signature encompassed genes coding for myosin (MYL4) and troponin (TNNT2) for sarcomere organization and contraction, acetylcholine receptor subunits (CHRNA1, CHRNA9, CHRND), the SOX11 and MYOG transcription factors, the calcium regulator sarcolipin (SLN), myostatin, and follistatin, as well as several proteins further studied below (ANXA2, S100A4, CILP, FETUB, SERPINB1A, and IGFBP2) (Figure 5F). Genes specifically dysregulated in each CNM form have been analyzed by GO enrichment and revealed some specific features as hemostasis defects in the BIN1 cohort, or cardiac and lipid metabolism for the DNM2 cohort, which remain to be further explored (Figure S13; Table S14).
Interestingly, orthologs of several genes in the common disease signature were previously associated with neuromuscular diseases: LMNA in Emery-Dreifuss muscular dystrophy, KLHL40 in nemaline myopathy, CHRNA1 and CHRNAD for myasthenic syndrome, and HSPB1 and PDK3 for Charcot-Marie-Tooth peripheral neuropathy. Overall, the main defective pathways found in the MTM1-CNM models are also altered in the BIN1-CNM and DNM2-CNM models, revealing the existence of a pathomechanism common to most CNM forms.
Therapy signature common to several rescuing approaches for different CNM forms
The transcriptome responses to different treatments of these three CNM mouse models was assessed in TA muscle at 7 weeks. Mtm1−/y mice were treated with the drug tamoxifen (cohort MTM1-c), or following genetic crosses with mice either overexpressing human BIN1 (cohort MTM1-b; Mtm1−/yTgBIN1) or with Dnm2 downregulation (cohort MTM1-a; Mtm1−/yDnm2+/−) (Figure 1B). A different methodology to reduce DNM2, systemic injection of ASO Dnm2, was used to treat Bin1mck−/− (cohort BIN1) and Dnm2SL/+ (cohort DNM2) mice. All of these therapies improved the phenotypes of the different disease models (28,34,39,40 and unpublished data for Bin1mck−/−). In each cohort, four groups were studied: treated (=rescued) and untreated CNM disease models and treated and untreated WT controls.
PCA showed that untreated and treated WT controls cluster together, suggesting that treatments had no general effects on WT mice (Figures 3A and 6A; Figure S9). For example, only 112 genes were dysregulated in tamoxifen-treated WT mice, while Dnm2 was indeed found downregulated together with 15 other genes in WT mice treated with ASO Dnm2 (Table S12). The transcriptome of tamoxifen-treated Mtm1−/y mice was similar to that of diseased Mtm1−/y mice, suggesting that tamoxifen did not have a strong transcriptional effect. Genetic downregulation of Dnm2 in Mtm1−/yDnm2+/− led to a partial rescue of the transcriptome of Mtm1−/y mice. When analyzing Mtm1−/yDnm2+/− mice over time, we found a partial rescue of 255 genes of 1,175 genes dysregulated in Mtm1−/y mice at 2 weeks, and 725 genes of 1,981 dysregulated genes at 7 weeks of the disease stage (Figure 6B; Table S7). Myh3, Myh8, and Sln are among the best normalized genes at 2 weeks. Acute downregulation of Dnm2 with ASO Dnm2 in both Bin1mck−/− and Dnm2SL/+ mice also partially rescued their transcriptomes (Figure 6A). Genetic overexpression of BIN1 in Mtm1−/yTgBIN1 normalized the transcriptome to a WT level. As a metric to compare the molecular efficacy of the different therapies, we calculated the ratio of the number of genes dysregulated in the rescued group versus the disease group over the number of genes dysregulated in the disease group versus the WT control; in other words, the ratio of the therapy signature over the disease signature. The percentage of rescued genes was 0.5% for the MTM1-c cohort (tamoxifen), 36% for the MTM1-a cohort, 43% for the DNM2 cohort, 47% for the BIN1 cohort (Dnm2 downregulation), and 96% for the MTM1-b cohort (BIN1 overexpression; 1,680 genes of 1,745). Overall, BIN1 overexpression appears to be the most efficient therapy to normalize the transcriptome defects of CNM in mice.
Figure 6.

Common therapy signature in CNM mice treated with different therapies
(A) PCA on RNA-seq data of the 42 genes commonly rescued in the four cohorts. Each symbol represents a mouse. The three different CNM mouse models are represented by red, orange, and yellow, and the WT controls are represented in green. Treatments are represented by different symbols: squares for the downregulation of Dnm2, either by ASO injection (full square) or by genetic cross (boxed +); the triangle represents the overexpression of human BIN1; and the cross represents the administration of tamoxifen. (B) Venn diagram illustrating the shared and specific dysregulated genes between rescued and diseased mice in each mouse cohort. (C) mRNA expression levels of Anxa2, Cilp, Fetub, and Igfbp2 in CNM mice either diseased or rescued upon therapy, and in untreated and treated controls. Boxplots displaying normalized Ct values. Pairwise significance calculated by a Dunn’s test are represented. (D) Among the 42 genes identified in the therapy signature, 35 have human orthologs and 2 (Scn5a and Sbk3) encode proteins targeted by known drugs.
To determine the common therapy signature, we compared the transcriptome of the rescued mice versus the transcriptome of the diseased mice for all cohorts except the MTM1-c cohort, as tamoxifen treatment had no strong transcriptomic impact. We found 42 genes defining the therapy signature common to all therapies in all CNM forms and that were retrieved in the disease signature for most of them (Figure 6B; Tables S9 and S15). Comparison of the rescued versus WT transcriptomes identified no genes in common that were resistant to all different therapies tested. The expression levels of several of these 42 genes was confirmed by qRT-PCR (Figure 6C; Figures S4, S5, and S10–S12). Depending on the cohorts, expression of these genes was partially or fully rescued upon treatment. For example, Anxa2 expression was not rescued in the MTM1-a cohort (Mtm1−/yDnm2+/−), while Cilp, Fetub, and Igfbp2 were all dysregulated in the different diseased models, and their expression levels were rescued to WT levels following any treatments.
The proteins corresponding to these 42 genes of the common therapy signature could represent novel therapeutic targets. To help pre-selecting the best candidates, we retrieved the 35 corresponding human orthologs and interrogated a drug database (https://drugcentral.org) (Figure 6D). Two proteins (SCN5A, SBK3) appeared directly targeted by several drugs, such as the antiarrhythmic quinidine for the SCN5A sodium channel, or nintedanib, an inhibitor of SBK3 and tyrosine kinases used for pulmonary fibrosis and cancer. Other therapeutic targets that were dysregulated in most cohorts include myosins, troponins, myostatin, or acetylcholine receptor subunits, for which in vivo modulation methods were already validated for other diseases.
Taken together, comparison of the three CNM models and several therapies underlined a common disease signature and a common therapy signature indicating potential therapeutic targets and biomarkers to follow disease severity or progression and therapy efficacy.
Identification of muscle and circulating biomarkers correlating with disease and therapy
To identify potential circulating biomarkers of disease state and therapy efficacy, we compared the list of genes in the disease signature and the therapy signature with proteins detected by mass spectrometry on the serum of WT mice at 8 weeks (Table S16), with public databases listing proteins detected in different fluids in humans and mice (GTEx, BioGPS, Illumina, GXD), and with the literature (Figure 7A). The following proteins were selected: ANXA2, CILP, FETUB, IGFBP2, and MSTN. To identify the best biomarkers, they were further screened by qRT-PCR, western blotting, and an enzyme-linked immunosorbent assay (ELISA) in muscle and in plasma. The levels of all of these RNAs were altered in the disease state and responded to the therapies in the three CNM mouse models, as validated by qRT-PCR in muscle (Figure 6C; Figures S4, S5, and S10–S12). ANXA2, FETUB, and CILP proteins were found dysregulated in muscle from some or all CNM models by western blot (Figure 7B; Figure S14). For example, Anxa2 (ANXA2) was significantly upregulated in muscles from 7-week-old Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ mice at both the RNA and protein levels, and its RNA was found already upregulated, albeit to a lesser extent, at 2 weeks in Mtm1−/y mice (2-fold at 2 weeks and 2.46-fold at 7 weeks).
Figure 7.

Muscle and circulating biomarkers for disease and rescue states
(A) Venn diagram illustrating the strategy to extract biomarkers from the comparison of disease and therapy signatures, proteins detected by mass spectrometry in the sera of WT mice, public databases (GTEx, BioGPS, Illumina, GXD), and literature. (B) Protein levels of ANXA2, FETUB, and CILP in gastrocnemius with standardization by Ponceau red staining in Mtm1−/y (cohort MTM1-a), Bin1mck−/−, and Dnm2SL/+ mouse models at 7 weeks. Protein levels are represented as the fold difference from the average of the WT (4 ≤ n ≤ 9). Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (C) Plasma levels of IGFBP2, ANXA2, CILP, and MSTN (ng/mg or pg/mg protein total) from untreated and treated Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ mouse models and WT controls (3 ≤ n ≤ 12). Student’s t test for untreated cohorts: ∗∗p < 0.01. Tukey’s test for treated cohorts: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Concerning circulating biomarkers, ELISA assays confirmed the presence of ANXA2, CILP, IGFBP2, and MSTN in the plasma from WT mice, as previously detected in serum by mass spectrometry (Figure 7C). Unlike in muscle, CILP levels in plasma were not changed in any CNM mouse models. Interestingly, the plasma level of IGFBP2 was significantly increased in Mtm1−/y mice. The alteration of Igfbp2 RNA levels in the Mtm1−/y muscle increased with age and disease progression, from 6-fold at 2 weeks (early disease stage) to 17-fold at 7 weeks (late disease stage). To assess whether circulating IGFBP2 is a biomarker common to several CNM forms, ELISA assays were performed in Bin1mck−/− and Dnm2SL/+ plasma at 7 weeks. The level of IGFBP2 was changed only in Mtm1−/y mice, suggesting it could be a biomarker specific to the MTM1-CNM form. Similarly, dysregulation of plasma protein content of ANXA2 and MSTN was revealed by ELISA, specifically for the BIN1-CNM or MTM1-CNM models, respectively. In both cases, these biomarkers responded to the therapies. The increase in ANXA2 plasma protein content in Bin1mck−/− mice was normalized upon DNM2 decrease with ASO Dnm2. The strong decrease in MSTN plasma protein content in Mtm1−/y mice was normalized upon BIN1 overexpression.
Overall, this screening strategy, from RNA-seq-based discovery, to qRT-PCR and western blot in muscle, and to ELISA in plasma, discovered ANXA2 as a muscle biomarker for several CNM forms and CILP and FETUB for specific CNM forms. IGFBP2, ANXA2, and MSTN were found as circulating biomarkers for specific CNM forms, and ANXA2 and MSTN plasma levels responded to the therapies.
Discussion
We performed a multi-omics meta-analysis of CNMs through the comparison of mouse models for the three main CNM forms and the comparison of three therapies with different targets. We identified disease signatures for MTM1-CNM conserved in different genetic and environmental backgrounds and in different species (mice, dogs, humans). Longitudinal transcriptome and proteome analysis of Mtm1−/y mice suggested early causal pathomechanisms and late compensatory adjustments. A disease signature common to the three CNM forms was defined, suggesting a common pathomechanism for CNM independent of the mutated genes. Comparison of the molecular effect of the different therapies revealed a correlation between the molecular normalization and the phenotypic rescue. In addition, novel potential therapeutic targets were suggested. Further molecular and biochemical investigations identified several biomarkers for disease state and therapy efficacy in muscle (RNA, proteins) and in plasma.
Pathomechanism of CNMs
We compared the muscle transcriptomes of the Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ mice faithfully reproducing the muscle weakness and histological hallmarks of the three main CNM forms.28,49 The overall transcriptomes easily distinguished the CNM models from their WT littermates.
For MTM1-CNM, to identify the specific disease signature independent from the genetic backgrounds or environment, we increased data heterogeneity by characterizing several Mtm1−/y groups on 129Pas, C57BL/6J, or mixed backgrounds bred in different animal houses, and then focused on the common transcriptome dysregulation. Next, this signature was compared to available transcriptome data from one MTM1 canine model and to a partial microarray analysis (4,200 genes) of patient muscle biopsies.45,46 Our conclusions were supported by previous findings. Dysregulation of NMJ components (CHRNA1, CHRND, CHRNG) were recently reported in the MTM1 dog, and alteration of the NMJ function was suggested in a mtm1 knockdown zebrafish and in Mtm1 mouse models.46,50,51 We found a high increase in Sln expression in the three CNM mouse models, and upregulation of Sln was previously reported following microarray analysis of the Mtm1−/y mouse.49 In addition, longitudinal analyses of the muscle transcriptome and proteome of Mtm1−/y mice at pre-symptomatic (E18.5), early (2 weeks), and late (7 weeks) disease stages highlighted the same dysregulated pathways, although the same dysregulated genes/proteins were not necessarily found (Figures 3 and 4). These ages were chosen to potentially distinguish between early causes of the disease and late consequences or compensatory mechanisms. Further functional investigations are needed to confirm their causality. In addition, the proteome analysis only covered the most abundant proteins. For example, BIN1 and DNM2 were not detected in the muscle proteomes. However, BIN1 and DNM2 proteins were both increased in Mtm1−/y mice using specific antibodies as determined by western blot (Table 3).34,39 Interestingly, we recently showed that the Mtm1−/y mice can be rescued by either decreasing DNM2 or increasing BIN1,37,39 strongly supporting the idea that a DNM2 increase is a disease cause while a BIN1 increase is a compensatory mechanism.
The comparison of the Mtm1−/y, Bin1mck−/−, and Dnm2SL/+ muscle transcriptomes revealed a common disease signature, encompassing sarcomere organization, muscle contraction, muscle development, and cell adhesion. All of these pathways were already the main ones found dysregulated at 2 weeks in Mtm1−/y mice, supporting the hypothesis that their dysregulation represents the main pathomechanism for all CNM forms (Figures 3, 4, and 5). In addition to these pathways, inflammation activation (transcriptome) and ribosomal biogenesis (proteome) were found only at 7 weeks, suggesting that their dysregulation is a consequence or a response to the disease state. Based on these data and on the knowledge that the three CNM proteins regulate membrane remodeling, we propose a model for the pathomechanism of CNM. Alteration of the triad membrane structure would lead to impaired calcium signaling and defective muscle contraction, explaining the strong muscle weakness and hypotonia seen in patients. The myofiber hypotrophy seen in patients and mouse models correlates with alteration of muscle development and regeneration and may be related to dysregulation of IGF modulators as IGFBP2 and/or to the reported decrease in satellite cells in patients.52 The strong myofiber hypotrophy would then trigger a later adaptation on protein homeostasis, as underlined by the increase of the ribosome biogenesis genes found only at 7 weeks. Indeed, defects of protein homeostasis correlate with the alteration of autophagy and the ubiquitin-proteasome pathways found in Mtm1−/y mice.25,53,54 In parallel, a primary defect in cell adhesion would impact the basement membrane and mechanotransduction and may explain defects in muscle contraction and also the altered fiber shape found in patients and mice. This is in agreement with the accumulation of integrins in Mtm1−/y mouse and patient myofibers, and with the proposed link between MTM1 and beta integrin recycling.39,55,56 The observed increased in interfiber space, together with the alteration of muscle regeneration, would cause the late increase in genes implicated in inflammation activation. As we detected a significant increase in RNA markers of inflammatory cells while performing whole-tissue RNA-seq, we conclude there is a significant infiltration of inflammatory cells that was confirmed by immunofluorescence labeling and quantification (Figures 5D and 5E). An inflammatory component was not previously reported for CNM but is common in dystrophies.57 Furthermore, the dysregulated pathways found here were barely underlined previously in the other CNM forms linked to BIN1 or DNM2. Overall, although some of these pathways were previously found altered in MTM1-CNM, these omics analyses allowed us to obtain a more complete and detailed overview of the pathomechanisms and extend it to several other CNMs. The GO term analysis of the common disease signature identified here for different CNM forms mostly reflects general muscle dysfunction and compensatory mechanisms put in place by the myofibers to cope with these defects, and thus highlights pathways also dysregulated in a large number of muscle diseases. However, differences appear when looking at the gene level that may reveal plasticity to impact or compensate the main muscle pathways depending on the sub-class of myopathies and on the primary genetic defect.
To a greater extent, we found several genes mutated in different neuromuscular diseases in the common CNM disease signature. Notably, LMNA is mutated in Emery-Dreifuss muscular dystrophy, and the encoded protein lamin A/C regulates nuclear envelope stability (MIM: 181350 and 616516).58 Of note, lamin A/C and BIN1 both bind the LINC complex that regulates nuclear shape and positioning, and BIN1-CNM patients have an altered nuclear envelope structure.59 KLHL40 is mutated in another congenital myopathy and is a substrate adaptor for the E3-ubiquitin ligase Cullin-3 (MIM: 615348).60,61 Similarly, MTM1 binds the Cullin-3 partner UBQLN2, and MTM1-CNM is linked to defects in the ubiquitin-proteasome pathway.54 Several dysregulated genes in all CNM models are mutated in cardiomyopathy: SCN5A (MIM: 601154), TNNT2 (MIM: 601494), and MYL4 (MIM: 617280). In addition, MYH2 and TNNC1, which are found to be upregulated in the proteome of 2-week Mtm1−/y mice, are also mutated in a proximal myopathy (MIM: 605637) or a cardiomyopathy (MIM: 611879), respectively. Such findings are commonly observed in myopathies where upregulation of genes usually expressed in embryonic muscle or adult cardiac muscle are re-expressed in the affected skeletal muscle. Finally, CHRNA1 (MIM: 601462) and CHRND (MIM: 616322) are mutated in congenital myasthenic syndromes, correlating with the defect in NMJ found in MTM1-CNM models.50,51,62
Common therapeutic targets for CNMs
We compared here three therapies involving three different targets in MTM1-CNM models, BIN1 overexpression, tamoxifen treatment and DNM2 regulation, and two methodologies for the latter target. For the first time, this allows a molecular comparison of the different therapies for CNM. BIN1 overexpression appears to be the most efficient therapy to normalize the molecular defects. The percentage of rescued genes varies greatly with 96% for BIN1 overexpression, 36% for DNM2 downregulation, and 0.5% for tamoxifen in the MTM1-CNM mouse model. In the AAV-MTM1 treated dogs, the percentage of rescued genes was 52% and 43% depending on the muscles analyzed.46 These findings highlight a correlation between the phenotypic and molecular rescue, as modulations of either MTM1, BIN1, or DNM2 improved the lifespan and locomotor and histological phenotypes very efficiently, while tamoxifen treatment resulted in a partial increase in lifespan and a significant amelioration of the histopathology, although to a lesser extent than with genetic crosses.17,31,34,37,39, 40, 41 As a potential explanation for the different rescue efficiency of the transcriptome dysregulation, BIN1 may directly modulate the general transcription program in a disease context, while MTM1 and DNM2 may directly impact the cellular (proteins, membrane) defects. Indeed, BIN1 binds the transcription factor MYC and can shuttle between the cytoplasm and the nucleus in muscle cells.63, 64, 65 Of note, BIN1 overexpression was achieved through genetic cross and is thus chronic from embryogenesis, while AAV-MTM1 and tamoxifen treatments are postnatal. However, transcriptome comparisons between chronic (Dnm2+/− genetic cross for MTM1-a) and acute DNM2 downregulation (ASO Dnm2 injection for DNM2 and BIN1 cohorts) showed similar percentages of rescued genes: 36% in MTM1-a, 43% in DNM2, and 47% in BIN1 cohorts (Figure 6). The rescuing effect of tamoxifen treatment is not based on transcriptome remodeling but might directly involve membrane and protein functions.
We evaluated the toxicity of the different therapies used in this study by comparing the WT treated versus WT mice. Treatment of WT mice did not show a strong impact on the transcriptomes, while it was not reported in AAV-MTM1-treated dogs. For example, injection of ASO Dnm2 decreased Dnm2 expression but had few off-targets. No detectable phenotypic toxicities were observed in the treated WT mice. However, in humans, even if the same gene is targeted (either BIN1 or DNM2), the therapeutic compound and formulation may differ and the delivery method and corresponding dose will change. Here, for BIN1 overexpression we used the human cDNA while for DNM2 downregulation we used ASOs specific to the mouse Dnm2 gene. We also detected the expected overexpression of the human BIN1 gene in the MTM1-b cohort. BIN1 overexpression only changed the expression of three genes in the treated WT mice (Figure S9), while rescuing most transcriptome dysregulation in the Mtm1−/y mice, suggesting this therapy modulates the transcriptome mainly in a disease context (i.e., normalization).
The present data revealed several potential targets that were not directly targeted in the experiments (i.e., not MTM1, BIN1, or DNM2). Genes of interest are expected to be dysregulated in disease and normalized upon efficient therapies, i.e., part of the therapy signature. In addition, known drugs targeting these gene products may be an asset, as this will allow drug repurposing and a faster clinical development. Several genes coding for subunits of the acetylcholine receptor are dysregulated. In particular, acetylcholine esterase inhibitors used in a clinical trial to treat myasthenic syndromes were tested with some success in different CNM forms,62 Scn5a is also found upregulated in the disease signature and normalized in the therapy signature, and it encodes a subunit of the sodium channel that can be inhibited by quinidine, a stereoisomer of quinine. Mstn encoding the myokine myostatin, an inhibitor of muscle growth, is significantly decreased in disease models of all cohorts except the Bin1mck−/− mouse (log2FC = −0.6 while our threshold was −1). Inhibitors of myostatin are being tested in the clinic for other muscle diseases, and one of them (ActRIIB-mFc) showed only a very mild amelioration of the Mtm1RC/y knockin mouse.66 The mild amelioration can be explained by the fact that myostatin is already strongly decreased in the disease state and thus could hardly be better suppressed.67 Sln encoding the calcium regulator sarcolipin is strongly upregulated in disease models of all cohorts except the MTM1-c cohort. Reducing sarcolipin expression through genetic cross or shRNA improved Duchenne muscular dystrophy phenotypes in mice.68 As a last example, myosins and troponins also meet these criteria and are targeted by several pharmacological regulators that may improve the muscle contraction defects of CNM. Indeed, all discussed modulations should be first validated in laboratory models, as it is unclear whether dysregulation of some pathways are disease causing or compensatory.
Potential biomarkers for disease progression and therapy efficacy
We developed a strategy to identify potential biomarkers, combining RNA-seq, qRT-PCR, and western blotting in muscle with mass spectrometry and ELISA assays in blood, supported by database and literature mining. The validity of these biomarkers for monitoring disease progression and therapy efficacy has to be confirmed in human samples. This will require muscle and blood sampling of untreated and treated patients with different CNM forms together with adequate aged-matched controls. We found that the Mstn RNA level is strongly decreased in Mtm1−/y and Dnm2SL/+ mice and to a lesser extent in Bin1mck−/− mice (log2FC = −0.6), and it was normalized upon modulation of MTM1, BIN1, and Dnm2 (Figures S5 and S9–S11).46 In agreement, MSTN was recently found decreased in plasma from MTM1- and DNM2-CNM patients and responded to ASO Dnm2 treatment in Mtm1−/y mice.69 In addition, MSTN plasma level was also normalized upon BIN1 overexpression (Figure 7C). ANXA2 is a calcium-dependent phospholipid-binding protein that has a role in muscle repair,70 and it was validated here as a muscle biomarker for all of the CNM forms that we have tested. ANXA2 was detected in plasma, increased in the Bin1mck−/− mouse model, and normalized upon ASO Dnm2 injection. Moreover, several reports cited ANXA2 to be a valuable biomarker in different cancers.71,72
In particular, these potential biomarkers could be used in clinical trials to monitor the progression/reversion of the disease and/or efficacy of the therapy. Currently, there are two clinical trials ongoing to treat CNM. First, there is the clinical trial (ClinicalTrials.gov: NCT03199469) for X-linked myotubular/CNM in patients under 5 years of age based on MTM1 gene replacement using AAV. Second, a clinical trial based on the decrease/normalization of DNM2 with ASO (DYN101) is ongoing in patients with mutations in MTM1 and DNM2 over 16 years of age (ClinicalTrials.gov: NCT04033159) and is planned for patients between 2 and 17 years of age (ClinicalTrials.gov: NCT04743557).
Conclusions
Herein, we report the first multi-omics analysis of animal models for several CNM forms, and of the effect of different therapies allowing us to reveal a common disease signature and a common therapy signature. We determined the global pathological mechanism and deciphered the molecular impact of therapies. Longitudinal analyses of the treated and untreated MTM1-CNM model highlight potential causes and consequences of the pathology. In addition, we identified several novel biomarkers detectable in muscle and/or plasma through different validated methodologies. These findings and the associated data should be an asset to the community for further investigations. More generally, this study validates the concept of using omics to identify molecular signatures common to different disease forms or to several therapeutic strategies.
Materials and methods
Animals
In this study we used different cohorts of mice. The sample size is given in Table S1. The cohort MTM1-a (WT, Dnm2+/−, Mtm1−/y, and Mtm1−/yDnm2+/−) was previously phenotyped on a 129Pas genetic background.34 The cohort MTM1-b (WT, TgBIN1, Mtm1−/y, and Mtm1−/yTgBIN1) was previously phenotyped on a 50% 129Pas and 50% C57BL/6N genetic background.39 Both cohorts MTM1-a and MTM1-b were bred in an IGBMC animal house in France. The cohort MTM1-c (WT, WT + tamoxifen, Mtm1−/y, and Mtm1−/y + tamoxifen) was previously treated and phenotyped on a 129Pas genetic background.40 Tamoxifen was administered via supplement pellets of diet (30 mg/kg of tamoxifen). This cohort was bred in the animal house of the School of Pharmaceutical Sciences of the University of Geneva (Geneva, Switzerland). The cohort DNM2 (WT + ASO control, WT + ASO Dnm2, Dnm2SL/+ + ASO control, Dnm2SL/+ + ASO Dnm2) was previously treated and phenotyped on a C57BL/6N genetic background.28 The cohort BIN1 (WT + PBS, WT + ASO Dnm2, Bin1mck/− + PBS, Bin1mck−/− + ASO Dnm2) was previously treated and phenotyped on a C57BL/6N genetic background (unpublished data). Both DNM2 and BIN1 cohorts were bred in an IGBMC animal house in France. DNM2 and BIN1 cohorts were treated weekly from 3 to 7 weeks of age with intraperitoneal injections of 25 mg/kg of ASO (IONIS Pharmaceuticals) targeting Dnm2. Only males were analyzed in this study, as only Mtm1−/y males but not Mtm1+/− females are affected. TA muscles were dissected at E18.5, 2 weeks, or 7 weeks and obtained from the previous studies. Data from the MTM1-d cohort (WT, Mtm1−/y) were retrieved from Maani et al.41 This cohort was analyzed on a C57BL/6J genetic background and bred in a University of Toronto animal house in Canada. Quadriceps muscle from 5-week-old animals were considered.
Blood collection
To collect plasma, blood samples were collected on EDTA-coated tubes (Microvette 500 K3E, Sarstedt) by mandibular puncture. Samples were then centrifuged at +4°C during 10 min at 2,000 × g. To collect serum, mandibular puncture was performed on mice. Blood was collected in a sterile empty tube and kept for 30 min. After coagulation, only the supernatant (serum) was kept for further analysis. Plasma and serum samples were stored at −80°C.
RNA extraction and RNA-seq
RNA was extracted from TA muscles using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA). RNA-seq libraries were prepared using the TruSeq stranded mRNA sample preparation kit and poly(A) selection and sequenced on a HiSeq 4000 as single-end 50-bp reads for cohorts MTM1-a, MTM1-b, MTMT1-c, cohort DNM2, and cohort BIN1.
Transcriptome analysis
Reads were preprocessed using cutadapt (version 1.10) in order to remove adaptor, poly(A), and low-quality sequences (Phred quality score below 20). Reads shorter than 40 bases were excluded from further analysis. Reads were mapped to ERCC (External RNA Controls Consortium) spike sequences using Bowtie version 2.2.8, and reads mapping to spike sequences were excluded from further analysis. Reads were mapped onto the mm10 assembly of Mus musculus genome using STAR version 2.5.3. Gene expression quantification was performed from uniquely aligned reads using htseq-count version 0.6.1p1, with annotations from Ensembl version 96 and union mode. Count tables were analyzed by the open-source RStudio environment for R and the Bioconductor software. The DESeq2 package (version 1.16.1) was used to normalize, fit, and compare the data between groups. Cutoff values for differentially expressed gene determinations were as follows: adjusted p value <0.05 and absolute value of log2FC >1. This pipeline was used for cohorts MTM1-a, MTM1-b, MTM1-c, DNM2, and BIN1.
To determine rescued genes, we developed a metric that quantifies the status of a diseased gene after the therapy. A diseased gene is defined as dysregulated in the comparison of disease versus WT (absolute value of log2FC >1 & adjusted p value <0.05). The metric is calculated as the ratio between the log2FC of the two comparisons: rescues versus disease over disease versus WT. We stratified the rescued genes into different categories: excessive rescue (metric > 120), not rescued (0 < metric < 30), partially rescued (30 < metric < 80), rescued (80 < metric < 120), and worsened (metric < 0) (Figure S15).
qRT-PCR
Synthesis of cDNA was performed with SuperScript IV transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). qPCR was done in a LightCycler 480 (Roche Diagnostics, Basel, Switzerland) with SYBR Green master mix I (Roche Diagnostics, Basel, Switzerland), and 0.5 μM forward and reverse primers. Primers were validated by amplicon sequencing and melting curve analysis and are listed in Table S17. Stau1, Rps11, and Rpl27 were used as housekeeping genes to normalize gene expression.
Protein extraction and liquid digestion
TA muscles were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with 1 mM PMSF, 1 mM DTT, and complete mini-EDTA-free protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). A DC protein assay kit (Bio-Rad, Hercules, CA, USA) was used to determine protein concentration. For serum analysis, most abundant serum proteins were depleted with the proteome purify 2 kit (MIDR002-020, R&D Systems) according to the manufacturer instructions before analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Protein mixtures were tricarboxylic acid (TCA) precipitated overnight at 4°C. Samples were then centrifuged at 14,000 rpm for 30 min at 4°C. Pellets were washed twice with 1 mL of cold acetone and centrifuged at 14,000 rpm for 10 min at 4°C. Washed pellets were then urea denatured with 8 M urea in 0.1 mM Tris-HCl, reduced with 5 mM TCEP (tris(2-carboxyethyl)phosphine) for 30 min, and then alkylated with 10 mM iodoacetamide for 30 min in the dark. Both reduction and alkylation were performed at room temperature and under agitation (850 rpm). Double digestion was performed with endoproteinase Lys-C (Wako) at a ratio of 1:100 (enzyme/proteins) in 8 M urea for 4 h, followed by an overnight modified trypsin digestion (Promega) at a ratio of 1:100 (enzyme/proteins) in 2 M urea. Both Lys-C and trypsin digestions were performed at 37°C. Peptide mixtures were then desalted on C18 spin-column and dried on a speed vacuum before LC-MS/MS analysis.
LC-MS/MS analysis
Samples were analyzed using an UltiMate 3000 nano RSLC (Thermo Scientific, San Jose, CA, USA) coupled in line with an LTQ-Orbitrap Elite mass spectrometer via a nano-electrospray ionization source (Thermo Scientific, San Jose, CA, USA). Peptide mixtures were loaded on a C18 Acclaim PepMap 100 trap column (75 μm inner diameter [ID] × 2 cm, 3 μm, 100 Å, Thermo Fisher Scientific) for 3.5 min at 5 μL/min with 2% acetonitrile (ACN)/0.1% formic acid (FA) in H2O and then separated on a C18 Accucore nano-column (75 μm ID × 50 cm, 2.6 μm, 150 Å, Thermo Fisher Scientific) with a 90-min linear gradient from 5% to 35% buffer B (A: 0.1% FA in H2O/B: 99% ACN, 0.1% FA in H2O), then a 20-min linear gradient from 35% to 80% buffer B, followed with 5 min at 99% B and 5 min of regeneration at 5% B. The total duration was set to 120 min at a flow rate of 200 nL/min. The oven temperature was kept constant at 38°C.
The mass spectrometer was operated in positive ionization mode, in data-dependent mode with survey scans from m/z 350 to 1,500 acquired in the Orbitrap at a resolution of 120,000 at m/z 400. The 20 most intense peaks (TOP20) from survey scans were selected for further fragmentation in the linear ion trap with an isolation window of 2.0 Da and were fragmented by collision-induced dissociation (CID) with normalized collision energy of 35%. Unassigned and single charged states were rejected.
The ion target value for the survey scans (in the Orbitrap) and the MS2 mode (in the linear ion trap) were set to 1E6 and 5E3, respectively, and the maximum injection time was set to 100 ms for both scan modes. Dynamic exclusion was used. Exclusion duration was set to 20 s, repeat count was set to 1, and exclusion mass width was ±10 ppm.
Proteome analysis
Proteins were identified by database searching using MaxQuant 1.6.6.0 and Mus musculus database (UniProt proteome database). Oxidation (M) was set as variable modification, and carbamidomethylation (C) was set as a fixed modification. Peptides were filtered with a false discovery rate (FDR) at 1%, and the label-free quantitative values were processed using Perseus 1.6.6.0. 3,521 proteins were identified. Statistical analyses were conducted in R-Bioconductor (R 3.6.3). wrMisc and wrProteo packages were used to normalize and to impute missing data with default parameters. Cutoff values for differentially expressed protein determination were as follows: adjusted p value <0.05 and absolute value log2FC >1.
Western blotting
Denaturation was performed on samples during 5 min at 95°C with 5× lane marker reducing buffer (Thermo Fisher Scientific, Waltham, MA, USA) and loaded on 10% SDS-PAGE gel (161-0173, TGX Fast Cast acrylamide kit, Bio-Rad). Proteins were transferred to a nitrocellulose membrane using a Trans-Blot Turbo RTA transfer kit (Bio-Rad, Hercules, CA, USA). Loading was controlled by Ponceau S (P7170, Sigma-Aldrich) staining and Cy5 dye fluorophore (RPN4000, QuickStain). Membranes were blocked for 1 h with 5% non-fat dry milk in 0.1% TBS with Tween 20 prior to incubations with primary and secondary antibodies. The primary and secondary antibodies used were as follows: ANXA2 (mouse, 1:1,000, Santa-Cruz Biotechnology sc-28385), CILP-1 (rabbit, 1:1,000, Biorbyt, orb182643), FETUB (rabbit, 1:500, Biorbyt,orb252830), MTM1 (2827, 1:700, homemade [34]), BIN1 (R2405, 1:700, homemade [39]), DNM2 (DNM2-R2865, 1:500, homemade [34]), β-actin (mouse, 1:5,000, homemade), peroxidase-coupled goat anti-rabbit (goat, 1:10,000, Jackson ImmunoResearch, 112-036-062), and peroxidase-coupled goat anti-mouse (goat, 1:10,000, Jackson ImmunoResearch, 115-036-068).
ELISA assays
Plasma proteins were quantified by a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Proteins (ANXA2, IGFBP2, CILP, MSTN) were quantified using an ANXA2 ELISA kit (LS-F5798, LSBio), IGFBP2 ELISA kit (ab207615, Abcam), CILP ELISA kit (ABIN5591836, Antibodies-online), and an MSTN ELISA kit (DGDF80, R&D Systems), respectively, according to the manufacturers’ instructions.
Muscle immunofluorescence
Transverse cryosections of TA muscles (8 μm) were permeabilized with 0.5% Triton X-100, blocked in 5% BSA, and incubated overnight at 4°C with anti-CD68 (MCA1957GA, Bio-Rad, 1:100) for identifying macrophages, DAPI for staining nuclei, and wheat germ agglutinin (WGA) conjugated to Alexa Fluor (AF) 647 for labeling the extracellular matrix. Slides were incubated with anti-mouse secondary antibodies for 1 h at room temperature (A-11007, Thermo Fisher Scientific, 1:250), observed, and imaged in a Leica DM 4000 microscope. The images were analyzed using ImageJ software (n = 3 mice per group).
Ortholog retrieval and GO analysis
Orthologs between mice and dogs and humans and mice were retrieved by the bitr function in the clusterProfiler package. GO analyses were performed with the clusterProfiler package (version 3.12.0) using the overrepresentation test and the Benjamini-Hochberg correction for multiple testing. Enrichments with a corrected p value lower than 0.05 were considered significant.7,73
Data representation and statistical analyses
PCA, volcano plot, and qPCR results were generated in R-Bioconductor (R 3.6.3). PCA was generated from the DESeq2 package (version 1.24.0) with variance-stabilizing transformation. All genes were used to generate the PCA from Figures 3A and 4A. Disease signature genes (Table S9) were used to generate the PCA from Figure 5A. Therapy signature genes (Table S15) were used to generate the PCA from Figure 6B. Statistical analyses for qRT-PCR were performed by Dunn’s multiple comparison test. Western blot and ELISA results were analyzed in GraphPad Prism (v9) using a Student’s t test. Venn diagrams were obtained from the InteractiVenn website (http://www.interactivenn.net).74
Data availability
The R script used to process the data has been deposited in GitLab and is freely available at http://git.lbgi.fr/djeddi/Myomics. RNA-seq data were deposited in NCBI GEO: GSE160084. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021725. The mass spectrometry proteomics data from the circulating proteins in serum have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021765.
Acknowledgments
We thank Luc Negroni, Bastien Morlet, Kirsley Chennen, and Matthieu Jung for help in experiments and analyses, Genomeast for RNA-seq, and IGBMC platforms of proteomics and animal housing. This study was supported by institutional funding from the Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique, Université de Strasbourg and by grants from the Agence Nationale de la Recherche (ANR-10-LABX-0030-INRT), a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir (ANR-10-IDEX-0002-02), the Muscular Dystrophy Association (576154), the Fondation pour la Recherche Médicale (201903007992), and by the Association Française contre les Myopathies-Téléthon (no. 20959 to O.M.D. and J.L. and no. 22734 to J.L.).
Author contributions
J.L. designed and supervised the study. S. Djeddi performed most bioinformatics analyses with help from W.R., C. Keime, and J.T. S.F., C. Kretz, and O.M.D. provided mouse samples. D.R., A.M., S.F., and A.-S.S. performed RNA quantification. D.R., S. Djerroud, and X.M.-M. performed western blots. D.R. performed ELISA assays. J.d.C.N. performed immunofluorescence. J.L. and S. Djeddi wrote the manuscript.
Declaration of interests
J.L. is co-founder of Dynacure (Illkirch, France). The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.04.033.
Supplemental information
References
- 1.Karczewski K.J., Snyder M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018;19:299–310. doi: 10.1038/nrg.2018.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subramanian I., Verma S., Kumar S., Jere A., Anamika K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights. 2020;14 doi: 10.1177/1177932219899051. 1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jungbluth H., Treves S., Zorzato F., Sarkozy A., Ochala J., Sewry C., Phadke R., Gautel M., Muntoni F. Congenital myopathies: disorders of excitation-contraction coupling and muscle contraction. Nat. Rev. Neurol. 2018;14:151–167. doi: 10.1038/nrneurol.2017.191. [DOI] [PubMed] [Google Scholar]
- 4.Ravenscroft G., Laing N.G., Bönnemann C.G. Pathophysiological concepts in the congenital myopathies: blurring the boundaries, sharpening the focus. Brain. 2015;138:246–268. doi: 10.1093/brain/awu368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jungbluth H., Wallgren-Pettersson C., Laporte J. Centronuclear (myotubular) myopathy. Orphanet J. Rare Dis. 2008;3:26. doi: 10.1186/1750-1172-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romero N.B. Centronuclear myopathies: A widening concept. Neuromuscul. Disord. 2010;20:223–228. doi: 10.1016/j.nmd.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Laporte J., Hu L.J., Kretz C., Mandel J.L., Kioschis P., Coy J.F., Klauck S.M., Poustka A., Dahl N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996;13:175–182. doi: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- 8.Bitoun M., Maugenre S., Jeannet P.Y., Lacène E., Ferrer X., Laforêt P., Martin J.J., Laporte J., Lochmüller H., Beggs A.H. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 2005;37:1207–1209. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- 9.Bitoun M., Bevilacqua J.A., Prudhon B., Maugenre S., Taratuto A.L., Monges S., Lubieniecki F., Cances C., Uro-Coste E., Mayer M. Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann. Neurol. 2007;62:666–670. doi: 10.1002/ana.21235. [DOI] [PubMed] [Google Scholar]
- 10.Böhm J., Biancalana V., Dechene E.T., Bitoun M., Pierson C.R., Schaefer E., Karasoy H., Dempsey M.A., Klein F., Dondaine N. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum. Mutat. 2012;33:949–959. doi: 10.1002/humu.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Böhm J., Biancalana V., Malfatti E., Dondaine N., Koch C., Vasli N., Kress W., Strittmatter M., Taratuto A.L., Gonorazky H. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. 2014;137:3160–3170. doi: 10.1093/brain/awu272. [DOI] [PubMed] [Google Scholar]
- 12.Nicot A.S., Toussaint A., Tosch V., Kretz C., Wallgren-Pettersson C., Iwarsson E., Kingston H., Garnier J.M., Biancalana V., Oldfors A. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat. Genet. 2007;39:1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 13.Gonorazky H.D., Bönnemann C.G., Dowling J.J. The genetics of congenital myopathies. Handb. Clin. Neurol. 2018;148:549–564. doi: 10.1016/B978-0-444-64076-5.00036-3. [DOI] [PubMed] [Google Scholar]
- 14.Schartner V., Laporte J., Böhm J. Abnormal excitation-contraction coupling and calcium homeostasis in myopathies and cardiomyopathies. J. Neuromuscul. Dis. 2019;6:289–305. doi: 10.3233/JND-180314. [DOI] [PubMed] [Google Scholar]
- 15.Jungbluth H., Gautel M. Pathogenic mechanisms in centronuclear myopathies. Front. Aging Neurosci. 2014;6:339. doi: 10.3389/fnagi.2014.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hnia K., Vaccari I., Bolino A., Laporte J. Myotubularin phosphoinositide phosphatases: Cellular functions and disease pathophysiology. Trends Mol. Med. 2012;18:317–327. doi: 10.1016/j.molmed.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Tasfaout H., Cowling B.S., Laporte J. Centronuclear myopathies under attack: A plethora of therapeutic targets. J. Neuromuscul. Dis. 2018;5:387–406. doi: 10.3233/JND-180309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawlor M.W., Beggs A.H., Buj-Bello A., Childers M.K., Dowling J.J., James E.S., Meng H., Moore S.A., Prasad S., Schoser B., Sewry C.A. Skeletal muscle pathology in X-linked myotubular myopathy: Review with cross-species comparisons. J. Neuropathol. Exp. Neurol. 2016;75:102–110. doi: 10.1093/jnen/nlv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beggs A.H., Böhm J., Snead E., Kozlowski M., Maurer M., Minor K., Childers M.K., Taylor S.M., Hitte C., Mickelson J.R. MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc. Natl. Acad. Sci. USA. 2010;107:14697–14702. doi: 10.1073/pnas.1003677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Böhm J., Vasli N., Maurer M., Cowling B.S., Shelton G.D., Kress W., Toussaint A., Prokic I., Schara U., Anderson T.J. Altered splicing of the BIN1 muscle-specific exon in humans and dogs with highly progressive centronuclear myopathy. PLoS Genet. 2013;9:e1003430. doi: 10.1371/journal.pgen.1003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olby N.J., Friedenberg S., Meurs K., DeProspero D., Guevar J., Lau J., Yost O., Guo L.T., Shelton G.D. A mutation in MTM1 causes X-Linked myotubular myopathy in Boykin spaniels. Neuromuscul. Disord. 2020;30:353–359. doi: 10.1016/j.nmd.2020.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shelton G.D., Rider B.E., Child G., Tzannes S., Guo L.T., Moghadaszadeh B., Troiano E.C., Haase B., Wade C.M., Beggs A.H. X-linked myotubular myopathy in Rottweiler dogs is caused by a missense mutation in exon 11 of the MTM1 gene. Skelet. Muscle. 2015;5:1. doi: 10.1186/s13395-014-0025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buj-Bello A., Laugel V., Messaddeq N., Zahreddine H., Laporte J., Pellissier J.F., Mandel J.L. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc. Natl. Acad. Sci. USA. 2002;99:15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pierson C.R., Dulin-Smith A.N., Durban A.N., Marshall M.L., Marshall J.T., Snyder A.D., Naiyer N., Gladman J.T., Chandler D.S., Lawlor M.W. Modeling the human MTM1 p.R69C mutation in murine Mtm1 results in exon 4 skipping and a less severe myotubular myopathy phenotype. Hum. Mol. Genet. 2012;21:811–825. doi: 10.1093/hmg/ddr512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fetalvero K.M., Yu Y., Goetschkes M., Liang G., Valdez R.A., Gould T., Triantafellow E., Bergling S., Loureiro J., Eash J. Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol. Cell. Biol. 2013;33:98–110. doi: 10.1128/MCB.01075-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X., Gao Y.Q., Zheng Y.Y., Wang W., Wang P., Liang J., Zhao W., Tao T., Sun J., Wei L. The intragenic microRNA miR199A1 in the dynamin 2 gene contributes to the pathology of X-linked centronuclear myopathy. J. Biol. Chem. 2020;295:8656–8667. doi: 10.1074/jbc.RA119.010839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durieux A.C., Vignaud A., Prudhon B., Viou M.T., Beuvin M., Vassilopoulos S., Fraysse B., Ferry A., Lainé J., Romero N.B. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 2010;19:4820–4836. doi: 10.1093/hmg/ddq413. [DOI] [PubMed] [Google Scholar]
- 28.Massana Muñoz X., Kretz C., Silva-Rojas R., Ochala J., Menuet A., Romero N.B., Cowling B.S., Laporte J. Physiological impact and disease reversion for the severe form of centronuclear myopathy linked to dynamin. JCI Insight. 2020;5:e137899. doi: 10.1172/jci.insight.137899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prokic I., Cowling B.S., Kutchukian C., Kretz C., Tasfaout H., Gache V., Hergueux J., Wendling O., Ferry A., Toussaint A. Differential physiological roles for BIN1 isoforms in skeletal muscle development, function and regeneration. Dis. Model. Mech. 2020;13:dmm044354. doi: 10.1242/dmm.044354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller A.J., Baker J.F., DuHadaway J.B., Ge K., Farmer G., Donover P.S., Meade R., Reid C., Grzanna R., Roach A.H. Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol. Cell. Biol. 2003;23:4295–4306. doi: 10.1128/MCB.23.12.4295-4306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Childers M.K., Joubert R., Poulard K., Moal C., Grange R.W., Doering J.A., Lawlor M.W., Rider B.E., Jamet T., Danièle N. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci. Transl. Med. 2014;6:220ra10. doi: 10.1126/scitranslmed.3007523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buj-Bello A., Fougerousse F., Schwab Y., Messaddeq N., Spehner D., Pierson C.R., Durand M., Kretz C., Danos O., Douar A.M. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum. Mol. Genet. 2008;17:2132–2143. doi: 10.1093/hmg/ddn112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raess M.A., Cowling B.S., Bertazzi D.L., Kretz C., Rinaldi B., Xuereb J.M., Kessler P., Romero N.B., Payrastre B., Friant S., Laporte J. Expression of the neuropathy-associated MTMR2 gene rescues MTM1-associated myopathy. Hum. Mol. Genet. 2017;26:3736–3748. doi: 10.1093/hmg/ddx258. [DOI] [PubMed] [Google Scholar]
- 34.Cowling B.S., Chevremont T., Prokic I., Kretz C., Ferry A., Coirault C., Koutsopoulos O., Laugel V., Romero N.B., Laporte J. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J. Clin. Invest. 2014;124:1350–1363. doi: 10.1172/JCI71206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowling B.S., Prokic I., Tasfaout H., Rabai A., Humbert F., Rinaldi B., Nicot A.S., Kretz C., Friant S., Roux A., Laporte J. Amphiphysin (BIN1) negatively regulates dynamin 2 for normal muscle maturation. J. Clin. Invest. 2017;127:4477–4487. doi: 10.1172/JCI90542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buono S., Ross J.A., Tasfaout H., Levy Y., Kretz C., Tayefeh L., Matson J., Guo S., Kessler P., Monia B.P. Reducing dynamin 2 (DNM2) rescues DNM2-related dominant centronuclear myopathy. Proc. Natl. Acad. Sci. USA. 2018;115:11066–11071. doi: 10.1073/pnas.1808170115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tasfaout H., Buono S., Guo S., Kretz C., Messaddeq N., Booten S., Greenlee S., Monia B.P., Cowling B.S., Laporte J. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat. Commun. 2017;8:15661. doi: 10.1038/ncomms15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tasfaout H., Lionello V.M., Kretz C., Koebel P., Messaddeq N., Bitz D., Laporte J., Cowling B.S. Single Intramuscular Injection of AAV-shRNA reduces DNM2 and prevents myotubular myopathy in mice. Mol. Ther. 2018;26:1082–1092. doi: 10.1016/j.ymthe.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lionello V.M., Nicot A.S., Sartori M., Kretz C., Kessler P., Buono S., Djerroud S., Messaddeq N., Koebel P., Prokic I. Amphiphysin 2 modulation rescues myotubular myopathy and prevents focal adhesion defects in mice. Sci. Transl. Med. 2019;11:eaav1866. doi: 10.1126/scitranslmed.aav1866. [DOI] [PubMed] [Google Scholar]
- 40.Gayi E., Neff L.A., Massana Muñoz X., Ismail H.M., Sierra M., Mercier T., Décosterd L.A., Laporte J., Cowling B.S., Dorchies O.M., Scapozza L. Tamoxifen prolongs survival and alleviates symptoms in mice with fatal X-linked myotubular myopathy. Nat. Commun. 2018;9:4848. doi: 10.1038/s41467-018-07058-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maani N., Sabha N., Rezai K., Ramani A., Groom L., Eltayeb N., Mavandadnejad F., Pang A., Russo G., Brudno M. Tamoxifen therapy in a murine model of myotubular myopathy. Nat. Commun. 2018;9:4849. doi: 10.1038/s41467-018-07057-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biressi S., Molinaro M., Cossu G. Cellular heterogeneity during vertebrate skeletal muscle development. Dev. Biol. 2007;308:281–293. doi: 10.1016/j.ydbio.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Egerman M.A., Glass D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014;49:59–68. doi: 10.3109/10409238.2013.857291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bismuth K., Relaix F. Genetic regulation of skeletal muscle development. Exp. Cell Res. 2010;316:3081–3086. doi: 10.1016/j.yexcr.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 45.Noguchi S., Fujita M., Murayama K., Kurokawa R., Nishino I. Gene expression analyses in X-linked myotubular myopathy. Neurology. 2005;65:732–737. doi: 10.1212/01.wnl.0000174625.67484.4d. [DOI] [PubMed] [Google Scholar]
- 46.Dupont J.B., Guo J., Renaud-Gabardos E., Poulard K., Latournerie V., Lawlor M.W., Grange R.W., Gray J.T., Buj-Bello A., Childers M.K., Mack D.L. AAV-mediated gene transfer restores a normal muscle transcriptome in a canine model of X-linked myotubular myopathy. Mol. Ther. 2020;28:382–393. doi: 10.1016/j.ymthe.2019.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vissing J., Johnson K., Töpf A., Nafissi S., Díaz-Manera J., French V.M., Schindler R.F., Sarathchandra P., Løkken N., Rinné S. POPDC3 gene variants associate with a new form of limb girdle muscular dystrophy. Ann. Neurol. 2019;86:832–843. doi: 10.1002/ana.25620. [DOI] [PubMed] [Google Scholar]
- 48.Jiang L., Wang M., Lin S., Jian R., Li X., Chan J., Dong G., Fang H., Robinson A.E., Snyder M.P., GTEx Consortium A quantitative proteome map of the human body. Cell. 2020;183:269–283.e19. doi: 10.1016/j.cell.2020.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Qusairi L., Weiss N., Toussaint A., Berbey C., Messaddeq N., Kretz C., Sanoudou D., Beggs A.H., Allard B., Mandel J.L. T-tubule disorganization and defective excitation-contraction coupling in muscle fibers lacking myotubularin lipid phosphatase. Proc. Natl. Acad. Sci. USA. 2009;106:18763–18768. doi: 10.1073/pnas.0900705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercier L., Böhm J., Fekonja N., Allio G., Lutz Y., Koch M., Goetz J.G., Laporte J. In vivo imaging of skeletal muscle in mice highlights muscle defects in a model of myotubular myopathy. Intravital. 2016;5:e1168553. doi: 10.1080/21659087.2016.1168553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dowling J.J., Joubert R., Low S.E., Durban A.N., Messaddeq N., Li X., Dulin-Smith A.N., Snyder A.D., Marshall M.L., Marshall J.T. Myotubular myopathy and the neuromuscular junction: A novel therapeutic approach from mouse models. Dis. Model. Mech. 2012;5:852–859. doi: 10.1242/dmm.009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shichiji M., Biancalana V., Fardeau M., Hogrel J.Y., Osawa M., Laporte J., Romero N.B. Extensive morphological and immunohistochemical characterization in myotubular myopathy. Brain Behav. 2013;3:476–486. doi: 10.1002/brb3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Al-Qusairi L., Prokic I., Amoasii L., Kretz C., Messaddeq N., Mandel J.L., Laporte J. Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. FASEB J. 2013;27:3384–3394. doi: 10.1096/fj.12-220947. [DOI] [PubMed] [Google Scholar]
- 54.Gavriilidis C., Laredj L., Solinhac R., Messaddeq N., Viaud J., Laporte J., Sumara I., Hnia K. The MTM1-UBQLN2-HSP complex mediates degradation of misfolded intermediate filaments in skeletal muscle. Nat. Cell Biol. 2018;20:198–210. doi: 10.1038/s41556-017-0024-9. [DOI] [PubMed] [Google Scholar]
- 55.Velichkova M., Juan J., Kadandale P., Jean S., Ribeiro I., Raman V., Stefan C., Kiger A.A. Drosophila Mtm and class II PI3K coregulate a PI(3)P pool with cortical and endolysosomal functions. J. Cell Biol. 2010;190:407–425. doi: 10.1083/jcb.200911020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ketel K., Krauss M., Nicot A.S., Puchkov D., Wieffer M., Müller R., Subramanian D., Schultz C., Laporte J., Haucke V. A phosphoinositide conversion mechanism for exit from endosomes. Nature. 2016;529:408–412. doi: 10.1038/nature16516. [DOI] [PubMed] [Google Scholar]
- 57.Chazaud B. Inflammation and skeletal muscle regeneration: Leave it to the macrophages! Trends Immunol. 2020;41:481–492. doi: 10.1016/j.it.2020.04.006. [DOI] [PubMed] [Google Scholar]
- 58.Chatzifrangkeskou M., Bonne G., Muchir A. Nuclear envelope and striated muscle diseases. Curr. Opin. Cell Biol. 2015;32:1–6. doi: 10.1016/j.ceb.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 59.D’Alessandro M., Hnia K., Gache V., Koch C., Gavriilidis C., Rodriguez D., Nicot A.S., Romero N.B., Schwab Y., Gomes E. Amphiphysin 2 orchestrates nucleus positioning and shape by linking the nuclear envelope to the actin and microtubule cytoskeleton. Dev. Cell. 2015;35:186–198. doi: 10.1016/j.devcel.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 60.Garg A., O’Rourke J., Long C., Doering J., Ravenscroft G., Bezprozvannaya S., Nelson B.R., Beetz N., Li L., Chen S. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Invest. 2014;124:3529–3539. doi: 10.1172/JCI74994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ravenscroft G., Miyatake S., Lehtokari V.L., Todd E.J., Vornanen P., Yau K.S., Hayashi Y.K., Miyake N., Tsurusaki Y., Doi H. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy. Am. J. Hum. Genet. 2013;93:6–18. doi: 10.1016/j.ajhg.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robb S.A., Sewry C.A., Dowling J.J., Feng L., Cullup T., Lillis S., Abbs S., Lees M.M., Laporte J., Manzur A.Y. Impaired neuromuscular transmission and response to acetylcholinesterase inhibitors in centronuclear myopathies. Neuromuscul. Disord. 2011;21:379–386. doi: 10.1016/j.nmd.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 63.Cassimere E.K., Pyndiah S., Sakamuro D. The c-MYC-interacting proapoptotic tumor suppressor BIN1 is a transcriptional target for E2F1 in response to DNA damage. Cell Death Differ. 2009;16:1641–1653. doi: 10.1038/cdd.2009.98. [DOI] [PubMed] [Google Scholar]
- 64.Sakamuro D., Elliott K.J., Wechsler-Reya R., Prendergast G.C. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat. Genet. 1996;14:69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 65.Wechsler-Reya R.J., Elliott K.J., Prendergast G.C. A role for the putative tumor suppressor Bin1 in muscle cell differentiation. Mol. Cell. Biol. 1998;18:566–575. doi: 10.1128/mcb.18.1.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lawlor M.W., Read B.P., Edelstein R., Yang N., Pierson C.R., Stein M.J., Wermer-Colan A., Buj-Bello A., Lachey J.L., Seehra J.S., Beggs A.H. Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice. Am. J. Pathol. 2011;178:784–793. doi: 10.1016/j.ajpath.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mariot V., Joubert R., Hourdé C., Féasson L., Hanna M., Muntoni F., Maisonobe T., Servais L., Bogni C., Le Panse R. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017;8:1859. doi: 10.1038/s41467-017-01486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Voit A., Patel V., Pachon R., Shah V., Bakhutma M., Kohlbrenner E., McArdle J.J., Dell’Italia L.J., Mendell J.R., Xie L.H. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017;8:1068. doi: 10.1038/s41467-017-01146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koch C., Buono S., Menuet A., Robé A., Djeddi S., Kretz C., Gomez-Oca R., Depla M., Monseur A., Thielemans L., NatHis-CNM Study Group Myostatin: A circulating biomarker correlating with disease in myotubular myopathy mice and patients. Mol. Ther. Methods Clin. Dev. 2020;17:1178–1189. doi: 10.1016/j.omtm.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bittel D.C., Chandra G., Tirunagri L.M.S., Deora A.B., Medikayala S., Scheffer L., Defour A., Jaiswal J.K. Annexin A2 mediates dysferlin accumulation and muscle cell membrane repair. Cells. 2020;9:1919. doi: 10.3390/cells9091919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tas F., Tilgen Yasasever C., Karabulut S., Tastekin D., Duranyildiz D. Circulating annexin A2 as a biomarker in gastric cancer patients: correlation with clinical variables. Biomed. Pharmacother. 2015;69:237–241. doi: 10.1016/j.biopha.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 72.Sharma M.C. Annexin A2 (ANX A2): An emerging biomarker and potential therapeutic target for aggressive cancers. Int. J. Cancer. 2019;144:2074–2081. doi: 10.1002/ijc.31817. [DOI] [PubMed] [Google Scholar]
- 73.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heberle H., Meirelles G.V., da Silva F.R., Telles G.P., Minghim R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16:169. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R script used to process the data has been deposited in GitLab and is freely available at http://git.lbgi.fr/djeddi/Myomics. RNA-seq data were deposited in NCBI GEO: GSE160084. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021725. The mass spectrometry proteomics data from the circulating proteins in serum have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021765.