

Abstract
Increasing evidence indicates the involvement of myocardial oxidative injury and mitochondrial dysfunction in the pathophysiology of heart failure (HF). Alpha-ketoglutarate (AKG) is an intermediate metabolite of the tricarboxylic acid (TCA) cycle that participates in different cellular metabolic and regulatory pathways. The circulating concentration of AKG was found to decrease with ageing and is elevated after acute exercise and resistance exercise and in HF. Recent studies in experimental models have shown that dietary AKG reduces reactive oxygen species (ROS) production and systemic inflammatory cytokine levels, regulates metabolism, extends lifespan and delays the occurrence of age-related decline. However, the effects of AKG on HF remain unclear. In the present study, we explored the effects of AKG on left ventricular (LV) systolic function, the myocardial ROS content and mitophagy in mice with transverse aortic constriction (TAC). AKG supplementation inhibited pressure overload-induced myocardial hypertrophy and fibrosis and improved cardiac systolic dysfunction; in vitro, AKG decreased the Ang II-induced upregulation of β-MHC and ANP, reduced ROS production and cardiomyocyte apoptosis, and repaired Ang II-mediated injury to the mitochondrial membrane potential (MMP). These benefits of AKG in the TAC mice may have been obtained by enhanced mitophagy, which cleared damaged mitochondria. In summary, our study suggests that AKG improves myocardial hypertrophy remodelling, fibrosis and LV systolic dysfunction in the pressure-overloaded heart by promoting mitophagy to clear damaged mitochondria and reduce ROS production; thus, AKG may have therapeutic potential for HF.
Keywords: Alpha-ketoglutarate, Mitophagy, Transverse aortic constriction, Myocardial hypertrophy, Cardiac insufficiency
Graphical abstract
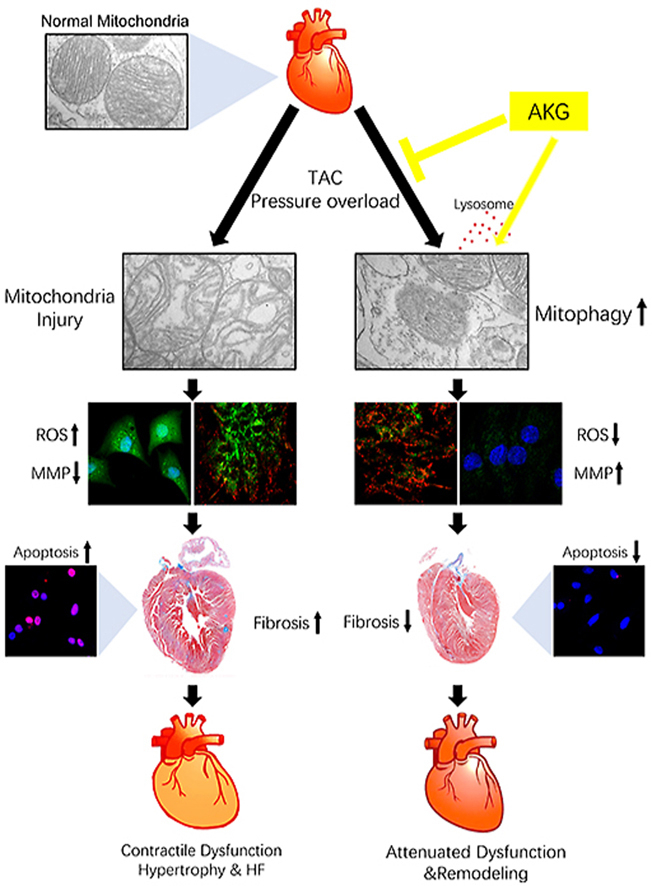
Highlights
-
•
AKG attenuated pressure overload-induced myocardial fibrosis and hypertrophy remodelling.
-
•
AKG improved cardiac function and left ventricular strain in TAC mice.
-
•
AKG increased myocardial mitophagy to clear damaged mitochondria and rescued Ang II-induced MMP impairment in TAC mice.
-
•
AKG reduced intracellular ROS and myocardial apoptosis in the pressure overloaded heart.
1. Introduction
Heart failure (HF) is a complex syndrome with several features, including abnormal myocardial systolic/diastolic function and excessive, continuous neurohormonal activation [1]. Despite the success of neurohormonal interventions, HF remains an enormous health problem. Mitochondrial dysfunction and myocardial metabolic disturbance are pathophysiological processes in HF that cannot be ignored [[2], [3], [4]]. The shift from fatty acids to glucose as a myocardial metabolic substrate for energy production is a major metabolic change in the failing heart and causes changes to intermediate metabolites of the tricarboxylic acid (TCA) cycle. Elevated levels of specific plasma metabolites, including alpha-ketoglutarate (AKG) have been reported as circulating metabolic markers of HF [5,6].
AKG (α-KG), also known as 2-oxoglutarate (2-OG), is an important intermediate in the TCA cycle between succinyl-CoA and isocitrate and regulates adenosine triphosphate (ATP) production as a crucial point in the anaplerotic reaction [7]. The circulating AKG concentration was found to decrease with ageing [8] and is elevated under fasted states [9], after acute exercise [10] and resistance exercise [11], and in HF [5,6]. Clinical studies showed that a high circulating AKG level was associated with HF severity (NYHA classification) and adverse outcome in acute HF (AHF) and chronic HF (AHF) patients [12,13]. As a circulating metabolic marker of HF, AKG may play a special role in risk stratification, prognostic evaluation, and treatment guidance for HF.
In recent decades, the functions of many cell metabolites have been reviewed. AKG has many other important physiological functions beyond its known metabolic roles, such as its role as a signalling molecule, gene expression effector and stress response regulator, and AKG is also a potential antiaging agent and antioxidant [14]. AKG is an essential cofactor and substrate for many biological enzymes, including hypoxia-inducible factor (HIF) prolyl hydroxylases (PHDs) and the histone demethylases JMJDs and ten-eleven translocation hydroxylases (TET1-3), which are involved in DNA demethylation [15], and participates in multiple cellular metabolic and regulatory pathways. Growing evidence suggests that AKG has protective effects, such as its ability to regulate metabolism [16], promote autophagy [17], reduce inflammation [18], delay ageing [19,20] and extend life span [21]. Although previous studies showed the association between a high circulating AKG level with HF severity and short-term adverse outcome in HF patients, there have been fewer relevant studies on the effects of AKG in HF. In the present study, we observed the impact of AKG on the myocardium in mice with HF induced by transverse aortic constriction (TAC) and hypertrophic cardiomyocytes induced by Ang II in vitro.
2. Methods
2.1. Experimental animals
All C57BL/6J mice and new-born Sprague-Dawley (SD) rats used in the study were purchased from the Laboratory Animal Center of Southern Medical University. The 8-week-old male mice received TAC or sham operation as previously described [22]. Mice were ad libitum fed ordinary chow and water with 0% or 2% AKG for 9 weeks under a 12-h light/dark cycle and assigned to four groups. Body weight and echocardiography were monitored at the end of the 6th and 10th weeks after surgery. A diagram showing the mouse treatment timeline is shown in Fig. 1A. The study was conducted according to the recommendations of the Guide for the Care and Use of Laboratory Animals (NIH, 8th edition, 2011). The study was approved by the Ethics Review Committee of Southern Medical University.
Fig. 1.

AKG attenuates cardiac hypertrophy in TAC mice. (A) The time-line diagram of AKG administration in TAC mice. (B) Heart weight-to-lung weight ratio(HW/LW). (C) Heart weight normalized to body weight(HW/BW). (D) Heart weight normalized to tibia length(HW/TL). (E) Circulating AKG concentration of mice. *: P<0.05 vs sham, **: P<0.01 vs sham, ***: P<0.001 vs sham, #: P<0.05 vs TAC, ##: P<0.01 vs TAC, ###: P<0.001 vs TAC. n = 10 each group.
2.2. Isolation and culture of neonatal rat ventricular myocytes (NRVMs)
As previously described [22,23], new-born SD rats were sacrificed via 2% isoflurane inhalation with cervical dislocation. The hearts were removed and digested with 0.2% pancreatin and collagenase II (1 mg/ml). After collection, centrifugation and resuspension, the cells were pre-plated in DMEM:F12 containing 10% foetal bovine serum (FBS), penicillin and streptomycin (100:1, Thermo Fisher) for 90 min for differential adhesion. The cells were placed in culture dishes and cultured in a 5% CO2 incubator at 37°C. After 72 h, the NRVMs were stimulated with 2 μM Ang II (Abcam) and treated with AKG at various concentrations in DMEM:F12 with 10% FBS for 24 h.
2.3. TAC model
The mice were anaesthetized by intraperitoneal injection of a mixture of xylazine (5 mg/kg) and ketamine (100 mg/kg). After anaesthesia, the mice were subjected to TAC as described previously [22]. After insertion of a tracheal tube, the mice were supported by a ventilator (respirator). The mice were dissected through the thorax, and aortic arch banding was performed with a 26-gauge needle. The needle was withdrawn after tight banding. In contrast, sham-operated mice were subjected to the same treatment without banding.
2.4. Conventional echocardiography and speckle-tracking echocardiography (STE)
Echocardiography was conducted in anaesthetized (2% isoflurane) mice using a Vevo 2100 system (VisualSonics, North America). The long and short axes were acquired in B-mode, and measurements were performed in M-mode.
STE analyses of the parasternal long-axis B-mode loops were performed using a VisualSonics Vevo 2100 system as previously described [[24], [25], [26], [27]]. Parasternal long-axis B-mode videos were recorded, and three-dimensional regional endocardial radial strain images were obtained over four consecutive cardiac cycles. Global left ventricular (LV) endocardial longitudinal and radial strain in the long axis and circumferential and radial strain in the short axis (peak strain %) with the maximum opposing wall delay were evaluated (calculated by Vevo Strain software).
2.5. Gravimetric and histological analyses
Ten weeks after TAC surgery, the mice were sacrificed by anaesthesia overdose through intraperitoneal injection of sodium pentobarbital and cervical dislocation. Blood samples were collected by cardiac puncture. Heart weight (HW), lung weight (LW) and tibia length (TL) were measured. For histological analysis, LV tissues were frozen at −80°C for subsequent analysis of frozen sections or fixed with formalin, embedded in paraffin, and sectioned at 5 μm.
2.6. Plasma AKG level measurements
Mouse plasma AKG levels were quantified by hydrophilic interaction chromatography (HILIC)-LC/MS/MS. The HILIC-LC-MS/MS system consisted of an Agilent 6460 Triple Quadrupole LC-MS/MS system with an ESI interface, as described previously [28]. Measurements were carried out as described in a previous study [13].
2.7. Haematoxylin-eosin (HE) staining
Parts of the heart were fixed with 4% paraformaldehyde, embedded in wax and sectioned (5 μm) for HE staining. Photographs of the sections were obtained by microscopy (Olympus, Japan).
2.8. Wheat germ agglutinin (WGA) staining
Cellular cross-sectional areas of the heart tissue were assessed by WGA staining as previously described [22]. The sections were photographed with a Leica SP8 microscope, and images were analysed with ImageJ software (NIH).
2.9. Collagen fibre staining (Masson and Sirius Red staining)
Collagen was quantified by Masson and Sirius Red staining according to the manufacturer's instructions (Servicebio Technology, Wuhan, China). Sections were photographed using a microscope (Olympus, Japan), and the images were analysed with ImageJ software (NIH).
2.10. ROS measurements
Dihydroethidium (DHE) staining of tissue sections and in vitro DCFH-DA staining of cardiomyocytes were used to verify ROS production. As previously described [29], the hearts taken out of the sacrificed mice were mounted in OCT embedding compound and frozen at −80°C. The frozen sections were cut at 5 μm using a cryostat and then thawed. A DHE staining solution (Sigma, USA) was added to the fresh-frozen heart sections after dilution in PBS and incubated at 37°C in the dark for 30 min; the sections were then rinsed twice with cold PBS. Intracellular ROS production was quantified with a 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Beyotime, Jiangsu, China) assay. Cardiomyocytes were incubated with culture medium containing 10 μM DCFH-DA for 30 min at 37°C and washed with PBS in the dark. Photographs were taken with a Leica SP8 microscope. The relative fluorescence density from the ROS was quantified using ImageJ.
2.11. TUNEL assay
Cardiomyocyte apoptosis was examined in vivo and in vitro using the TdT-mediated dUTP nick-end labelling (TUNEL) assay (Beyotime, Jiangsu, China). As previously described [30], after the heart tissues had been embedded and sliced, the sections were dewaxed and stained according to the manufacturer's instructions. The sections were fixed in 4% paraformaldehyde for 30 min and permeabilized in 0.2% Triton X-100 for 5 min. Then, the fixed cells and tissue sections were incubated with TUNEL staining solution at 37°C for 30 min. The cell nuclei were counterstained with DAPI. TUNEL-positive cells were photographed with a Leica SP8 microscope. Bright red fluorescence indicated TUNEL-positive cells, and blue fluorescence indicated nuclei. The images were analysed with ImageJ.
2.12. Detection of mitochondrial and lysosomal colocalization
As previously described [30], mitochondrial and lysosomal colocalization indirectly indicates mitophagy. Cells were co-incubated with LysoTracker Red (50 nM) and MitoTracker Green (100 nM, Molecular Probes, Beyotime, Jiangsu, China) for 30 min and then stained with Hoechst for 10 min. Fluorescence images were obtained with a Leica SP8 microscope. Bright green fluorescence indicated mitochondria, bright red fluorescence indicated lysosomes, and blue fluorescence indicated nuclei. The ratio of LysoTracker-positive foci to MitoTracker-positive foci was used to indirectly indicate mitophagy.
2.13. Measurement of mitochondrial membrane potential (MMP)
JC-1 staining was used to measure MMP according to the manufacturer's instructions. Briefly, as previously described [30], cardiomyocytes were incubated with JC-1 staining solution at 37°C for 20 min. Fluorescence was observed with a Leica SP8 microscope (Germany). Bright red fluorescence indicated J-aggregates, and green fluorescence indicated J-monomers. Images were analysed with ImageJ.
2.14. Transmission electron microscopy (TEM)
Mitochondrial morphology was observed by TEM. As previously described [30], small cardiomyocyte fragments (1 mm3 in volume) were immersed in 2.5% glutaraldehyde in cacodylate buffer (0.1 M, pH 7.4) and fixed for 2 h. The fragments were rinsed 3 times in PBS and then fixed in 1% osmium tetroxide cacodylate buffer (0.1 M, pH 7.4) for 2 h. After rinsing in cacodylate buffer, the fragments were washed three times in PBS. The fragments were then dehydrated in a graded ethanol series (50, 70, 90, and 100%), rinsed with propylene oxide, permeabilized with a 1:1 mixture of propylene oxide and Epon 812, and baked in an oven at 38°C for 3 h. Then, the steps above were repeated using a 1:2 mixture of propylene oxide and Epon 812. Ultrathin sections (80 nm) were cut with an ultramicrotome (UC7, Leica, Germany), after which the sections were stained with uranyl acetate and lead citrate and examined with an electron microscope (JEM-1400, Japan) at 80 kV.
2.15. Western blotting
Proteins were extracted from the cultured cardiomyocytes and heart tissue using RIPA buffer (Fudebio, Hangzhou, China) mixed with protease inhibitor (Fudebio, Hangzhou, China) (1:100) and quantified by BCA assay (Thermo Fisher, USA). The primary antibodies used were anti-β-MHC (1:1000, Abcam, USA), anti-ANP (1:500, Abcam, USA), anti-TGF-β1 (1:1000, Proteintech, USA), anti-PTEN-induced putative kinase-1 (PINK1) (1:1000, Santa Cruz, USA), anti-Parkin (1:1000, Abcam, USA), anti-caspase-3 (1:1000, Cell Signalling Technology, USA), anti-Bcl-2 (1:1000, Proteintech, USA), and anti-β-actin (1:3000, Proteintech, USA). The secondary antibodies used were goat anti-rabbit and anti-mouse IgG-HRP (1:3000, Fudebio, Hangzhou, China). Bands were detected with ECL substrate (Fudebio, Hangzhou, China) and visualized with a GeneGnome imaging system (Syngene Bioimaging). Densitometry was carried out with ImageJ (NIH).
2.16. Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the LV tissues and cardiomyocytes with RNAiso Plus (Takara, Japan) and purified. RNA samples were reverse transcribed to generate cDNA using the reverse transcription reagent PrimeScript™ RT Master Mix (Takara, Japan). Reverse transcription was conducted as follows: 37°C for 15 min, 85°C for 5 s and hold at 4°C. RT-qPCR was performed using TB Green Premix Ex TaqII (Takara, Japan) in a LightCycler 480 system (Roche Diagnostics, Basel, Switzerland). Ten-microlitre reactions were subjected to the following conditions: 95°C for 5 min, 40 cycles of 95°C for 10 s and 55–60°C for 20 s, and 72°C for 32 s. The results were analysed with the 2−△△Ct method and normalized to β-actin gene expression. The primers used are listed in Table 1 (Sangon Biotech Co., Ltd., Shanghai, China).
Table 1.
List of RT-PCR primers.
| Genus | mRNA | Forward | Reverse |
|---|---|---|---|
| mouse | Nppa | AAGAACCTGCTAGACCACCTGGAG | TGCTTCCTCAGTCTGCTCACTCAG |
| mouse | Nppb | GGAAGTCCTAGCCAGTCTCCAGAG | GCCTTGGTCCTTCAAGAGCTGTC |
| mouse | Myh7 | CAGAACACCAGCCTCATCAACCAG | TTCTCCTCTGCGTTCCTACACTCC |
| mouse | TGF-β1 | ACCGCAACAACGCCATCTATGAG | GGCACTGCTTCCCGAATGTCTG |
| mouse | PINK1 | GCAGCAGTCAGCAGCCACTC | AGAGCCTCACACTCCAGGTTAGC |
| mouse | Parkin | ACGACGCTCAACTTGGCTACTC | GCACTCCTCGGCACCATACT |
| mouse | Bcl-2 | CCGTCGTGACTTCGCAGAGATG | ATCCCTGAAGAGTTCCTCCACCAC |
| mouse | Caspase-3 | GAAACTCTTCATCATTCAGGCC | GCGAGTGAGAATGTGCATAAAT |
| mouse | β-actin | TATGCTCTCCCTCACGCCATCC | GTCACGCACGATTTCCCTCTCAG |
| rat | Nppa | GAGCGAGCAGACCGATGAAGC | TCCATCTCTCTGAGACGGGTTGAC |
| rat | Nppb | AGTCTCCAGAACAATCCACGATGC | GCCTTGGTCCTTTGAGAGCTGTC |
| rat | Myh7 | CCAGAACACCAGCCTCATCAACC | CACCGCCTCCTCCACCTCTG |
| rat | PINK1 | GAAGCCACCATGCCCACACTG | CTGCTCCCTTTGAGACGACATCTG |
| rat | Parkin | CACGACGCTCAACTTGGCTACTC | ACGCACTCCTCGGCACCATAC |
| rat | Bcl-2 | TGGAGAGCGTCAACAGGGAGATG | GGTGTGCAGATGCCGGTTCAG |
| rat | Caspase-3 | ACGAACGGACCTGTGGACCTG | GTTTCGGCTTTCCAGTCAGACTCC |
| rat | β-actin | CTGAGAGGGAAATCGTGCGTGAC | AGGAAGAGGATGCGGCAGTGG |
2.17. Statistical analysis
Quantitative data are displayed as the mean ± standard error of the mean (SEM). A normal distribution test was performed to determine whether a parametric or non-parametric test was conducted. Comparisons of two experimental groups were carried out with a two-tailed t-test, while comparisons of more than 3 groups were carried out by ANOVA followed by LSD or Dunnett's T3 post hoc multiple test. All statistical analyses were performed using SPSS 24.0 and GraphPad Prism 8.0 software (San Diego, CA, USA).
3. Results
3.1. AKG supplementation attenuates pressure overload-induced cardiac hypertrophy
Evidence has shown that dietary AKG suppressed chronic inflammation, compressed morbidity and led to health benefits in mice [19]. Here, we examined the effect of dietary AKG supplementation on TAC mice. Ten weeks after surgery, heart weight normalized to tibia length(HW/TL), heart weight normalized to body weight(HW/BW) and the heart weight-to-lung weight ratio(HW/LW) in the TAC mice had increased by 67.48%, 62.26% and 50.14%, respectively, compared with that in the sham mice (P<0.01)(Fig. 1B–D). Pathological heart enlargement and cardiac hypertrophy in the TAC mice were significantly mitigated by AKG supplementation (Fig. 2A–C). In the TAC mice with AKG supplementation, the HW/TL, HW/BW and HW/LW significantly decreased by 42.47% (P<0.01), 24.68% (P<0.05) and 25.05% (P<0.05), respectively (Fig. 1B–D). As shown in Fig. 1E, the circulating AKG concentration was elevated in the TAC mice but the differences were not statistically significant from that in the sham group (63.40 ± 10.27 μM vs 84.65 ± 13.43 μM), and despite being further elevated in TAC mice supplemented with AKG, it was still not statistically significant different from that in the TAC mice (84.65 ± 13.43 μM vs 115.71 ± 8.39 μM).
Fig. 2.

AKG alleviates myocardial hypertrophy induced by pressure overload. Myocardial hypertrophy reflected by (A) Heart photos, (B) HE staining in global and (C) in details, as well as (D) WGA staining(n = 9). (E)Protein expression of ANP and β-MHC in heart tissue(n = 5). (F) mRNA expression of Nppa, Nppb and Myh7 in heart tissue(n = 9); *: P<0.05 vs sham, **: P<0.01 vs sham, ***: P<0.001 vs sham, #: P<0.05 vs TAC, ##: P<0.01 vs TAC, ###: P<0.001 vs TAC.
Evaluation of cardiomyocyte size by WGA staining showed that the mean cross-sectional area of the cardiomyocytes was 91.59% larger in the TAC mice than in the sham mice (P < 0.001) (Fig. 2D). Transcript levels of the myocardial hypertrophy marker genes atrial natriuretic factor (Nppa), brain natriuretic peptide (Nppb) and myosin heavy chain 7 (Myh7), were increased 2.28-fold, 1.14-fold and 1.10-fold, respectively, in the TAC mice (P<0.001)(Fig. 2F). Furthermore, ANP and beta-myosin heavy chain (β-MHC) protein levels in the TAC mice were 1.42-fold and 1.65-fold higher, respectively, than those in the sham group (P<0.001)(Fig. 2E). A 49.11% decrease in average cardiomyocyte cross-sectional area was observed in the TAC mice administered AKG (P < 0.001) (Fig. 2D). Both the mRNA and protein levels of cardiac hypertrophy markers were significantly reduced (Fig. 2E and F).
3.2. AKG supplementation improved cardiac function and LV strain in the TAC mice
TAC leads to an increased cardiac haemodynamic load, resulting in cardiac remodelling and dysfunction. Doppler echocardiography showed that at 6 weeks after TAC surgery, LV fractional shortening (FS) and ejection fraction (EF) in the TAC mice were 35.70% and 31.89% lower than those in the sham mice (23.01 ± 1.02 vs 14.79 ± 0.42 and 46.64 ± 1.74 vs 31.77 ± 0.81, respectively; both P<0.001) (Fig. 3A, H and K), and the LV mass was 51.19% higher (76.29 ± 2.34 mg vs 115.35 ± 5.13 mg,P<0.001)(Fig. 3C). The end-diastolic LV anterior wall (LVAWd) and posterior wall (LVPWd) thicknesses were also increased (0.81 ± 0.02 mm vs 0.97 ± 0.03 mm, P<0.01; 0.63 ± 0.02 mm vs 0.83 ± 0.03 mm, P<0.001)(Fig. 3D and E), indicating LV remodelling, myocardial hypertrophy and cardiac dysfunction induced by pressure overload. Meanwhile, TAC mice supplemented with AKG exhibited a 19.12% decrease in LV mass (115.35 ± 5.13 mg vs 93.29 ± 4.97 mg,P<0.001) and 23.39% and 20.75% increases in LVFS (14.79 ± 0.42 vs 18.26 ± 0.82,P<0.05) and LVEF (31.77 ± 0.81 vs 38.36 ± 1.54,P<0.01), respectively, compared with those in the TAC mice.
Fig. 3.

AKG improves pressure overload-induced LV remodelling and cardiac dysfunction. The echocardiography(ECG) was examined at 6w(A) and 10w(B) after surgery: (C)LV mass, (D)left ventricular anterior wall in diastole(LVAWd), (E)left ventricular posterior wall in diastole(LVPWd), (F)left ventricular internal dimension in diastole(LVIDd), (G) left ventricular internal dimension in systole(LVIDs), (H)fractional shortening(FS), (I)left ventricular diastolic volume(LVdVol), (J) left ventricular systolic volume(LVsVol), (K)ejection fraction(EF); *: P<0.05 vs sham, **: P<0.01 vs sham, ***: P<0.001 vs sham, #: P<0.05 vs TAC, ##: P<0.01 vs TAC, ###: P<0.001 vs TAC. n = 10 each group.
These differences were more pronounced at 10 weeks post-surgery (Fig. 3B–K). At 10 weeks, the LV mass of the TAC mice was 63.17% higher than that of the sham mice (101.26 ± 5.09 mg vs 165.22 ± 13.08 mg,P<0.01), and the thicknesses of the LVAWd and LVPWd were further increased. Additionally, the FS and EF were further impaired (26.28 ± 0.66% vs 12.29 ± 1.12% and 52.09 ± 1.05% vs 26.40 ± 2.27%, respectively; both P<0.001). The LV mass of the TAC mice administered AKG was 39.86% lower than that of the TAC mice without AKG (165.22 ± 13.08 mg vs 99.36 ± 4.47 mg, P<0.01), and the thicknesses of the LVAWd and LVPWd were decreased (1.05 ± 0.05 mm vs 0.91 ± 0.04 mm and 0.99 ± 0.04 mm vs 0.86 ± 0.05 mm, respectively; both P<0.05). Furthermore, the FS (12.29 ± 1.12% vs 23.15 ± 0.96,P<0.001) and EF (26.40 ± 2.27% vs 47.05 ± 1.64,P<0.001) showed further recovery upon AKG supplementation. These results strongly suggest that AKG supplementation protected the TAC mice from pressure overload-induced myocardial hypertrophy, LV remodelling and cardiac dysfunction. Supplementation with 2% AKG had no obvious adverse effects on LV function in the mice.
STE can be used to evaluate ventricular wall strain and activity [24,31](Fig. 4). The results showed that at 10 weeks after surgery, the LV endocardial radial strain (RS) and longitude strain (LS) along the long axis were decreased by 43.90% and 54.18%, respectively, in the AC mice compared with the sham mice (both P<0.001, Fig. 4A and C). The maximum opposing wall delay of the RS and LS in the LV long axis in the TAC mice were 1.95-fold (P<0.001) and 3.66-fold (P<0.05) greater than those in the sham mice, respectively(Fig. 4A and C). The LV endocardial radial strain and circumferential strain (CS) in the short axis in the TAC group were decreased by 54.09% and 65.66% (both P<0.001, Fig. 4B and D), respectively, compared with those in the sham group. Additionally, the maximum opposing wall delay of the RS and CS in the short axis in the TAC mice were 2.83-fold (P<0.01) and 1.44-fold (P<0.001) greater than those of the sham group, respectively(Fig. 4B and D). The STE data indicated a reduction in LV contractility and decreased consistency of ventricular wall motion in the TAC mice. AKG supplementation increased the long axis RS and LS by 50.24% (P<0.01) and 59.23% (P<0.01), respectively, and the short axis RS and CS by 56.10% (P<0.05) and 140.11% (P<0.01), respectively, in the TAC mice, and all the maximum opposing wall delays were improved.
Fig. 4.

AKG improves pressure overload-induced LV strain decline. The LV endocardial strain in long axis(A) and short axis(B) at 10w after surgery measured by Speckle tracking echocardiography: (C) LV endocardial radial strain(RS), longitudinal strain(LS) and maximum opposing wall delay in long axis, (D) LV endocardial radial strain(RS), circumferential strain(CS) and maximum opposing wall delay in short axis; *: P<0.05 vs sham, **: P<0.01 vs sham, ***: P<0.001 vs sham, #: P<0.05 vs TAC, ##: P<0.01 vs TAC, ###: P<0.001 vs TAC. n = 6 each group.
3.3. AKG inhibits myocardial fibrosis under pressure overload
Ventricular fibrosis is an indication of structural remodelling and usually associated with cardiac insufficiency [32]. Myocardial tissue sections were stained with Masson (Fig. 5A and B) and Sirius Red (Fig. 5C) to examine the degree of myocardial fibrosis and observe the impact of AKG on the TAC mice. Compared with that in the sham group, the fibrotic area in the myocardium was significantly increased in the TAC mice (P<0.001), but this increase was dramatically attenuated by AKG supplementation. Fibrosis progression involves the conversion of fibroblasts into active myofibroblasts, and transforming growth factor-β1 (TGF-β1) plays a major role in cardiac pathological fibrogenesis [32]. PCR and Western blot analysis showed that the mRNA and protein levels of TGF-β1 in the myocardium were elevated in the TAC group (P < 0.001, P < 0.05), but AKG supplementation attenuated the overexpression of TGF-β1 in the TAC mice (Fig. 5D and E). This suggests that AKG affects a TGF-β1-related signalling pathway activated by pressure overload to inhibit myocardial fibrosis.
Fig. 5.

AKG inhibits myocardial fibrosis in TAC mice. Collagen quantification was performed on Masson staining (A)in global and (B)in detail(n = 9), and (C)Sirius Red staining(n = 9). (D)Protein(n = 5) and (E)mRNA(n = 9) expression of TGF-β1; *: P<0.05 vs sham, **: P<0.01 vs sham, ***: P<0.001 vs sham, #: P<0.05 vs TAC, ##: P<0.01 vs TAC, ###: P<0.001 vs TAC. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. AKG facilitated myocardial mitophagy, reduced the ROS content and decreased cellular apoptosis in TAC mice
ROS are mainly produced by mitochondria and lead to mitochondrial dysfunction, myocardial injury, and HF [33,34]. Dysfunctional mitochondria in HF produce less ATP and ROS at dangerous quantities, which impairs mitochondrial DNA and respiratory complex proteins, resulting in oxidative damage and eventually cell death [35]. Studies have shown that efficient mitophagy, the selective autophagic removal of abnormal mitochondria in the myocardium, can alleviate oxidative damage and prevent the progression of HF [33,36]. AKG was reported to promote autophagy [17]; therefore, here, we tested the effect of AKG on mitophagy and myocardial apoptosis.
Our results showed the effects of AKG on ROS inhibition and mitophagy activation under pressure overload. Compared with that in the sham mice, the myocardial ROS level was 2.26-fold (P<0.001) higher in the TAC mice, as shown by DHE staining, and AKG supplementation reduced the ROS level by 50.81% (P<0.001) in the TAC mice (Fig. 6A). TUNEL staining showed a significantly increase in the apoptosis rate in the myocardium in the TAC group compared with the sham group (P < 0.001, Fig. 6B), and it generally decreased to the level in the sham group in TAC mice administered with AKG (P < 0.001).
Fig. 6.

Effect of AKG on myocardial ROS content and cellular apoptosis in TAC mice. (A)Dihydroethidium (DHE) staining to reflect myocardial ROS production(n = 9). (B)The apoptosis rate of cardiomyocytes by TUNEL assay(n = 9). (C)Effect of AKG on mRNA expression of PINK1, Parkin, caspase-3 and Bcl-2(n = 9). (D) Relative protein expression of PINK1, Parkin, caspase-3 and Bcl-2(n = 5); *: P<0.05 vs sham, **: P<0.01 vs sham, ***: P<0.001 vs sham, #: P<0.05 vs TAC, ##: P<0.01 vs TAC, ###: P<0.001 vs TAC. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Expression of the mitophagy‐associated markers PINK1 and Parkin and the apoptosis‐associated markers Bcl-2 and caspase-3 was measured by PCR and western blotting. The caspase-3 transcript level in the myocardium was 72.50% higher in the TAC mice than in the sham mice (P<0.001) (Fig. 6C). Additionally, Bcl-2, PINK1, and Parkin transcript levels were 29.95% (P<0.001), 25.51% (P<0.05) and 21.39% (P<0.001) lower, respectively, in the TAC mice(Fig. 6C). Compared with those in the TAC mice, the myocardial transcript level of caspase-3 was 35.93% lower (P<0.01), and the Bcl-2, PINK1-Parkin transcript levels were increased by 25.04% (P<0.05), 181.90% (P<0.01) and 223.42% (P<0.001), respectively, in the TAC mice administered AKG. The changes in protein expression were generally consistent with the changes in transcript levels (Fig. 6D). These results suggest that AKG increased myocardial mitophagy and reduced ROS production and cellular apoptosis under pressure overload.
3.5. AKG alleviates Ang II-induced hypertrophy and injury and reduces cardiomyocyte apoptosis
TAC is a model of pressure overload-induced overactivation of the neuroendocrine system, which leads to elevated circulating Ang II and norepinephrine levels in vivo [22]. In vitro, we used Ang II to induce myocardial hypertrophy and injury and verified the protective effect of AKG on cardiomyocytes. We examined the effects of AKG at two concentrations on mRNA and protein levels in cardiomyocytes. Quantitative RT-PCR (Fig. 7C) showed that after Ang II administration, transcript levels of the Nppa, Nppb and Myh7 genes, markers of myocardial hypertrophy, increased by 39.57% (P<0.001), 56.78% (P<0.05) and 41.48% (P<0.01), respectively, and expression levels of the apoptosis-associated markers caspase-3 and Bcl-2 were increased by 27.23% (P<0.001) and decreased by 32.85% (P<0.001), respectively. The transcript levels of Nppa, Nppb, Myh7 and caspase-3 were decreased, while those of Bcl-2 were increased in Ang II-treated cardiomyocytes supplemented with AKG. Changes in the corresponding proteins detected by Western blot analysis were roughly consistent with changes in the mRNA levels (Fig. 7B). Considering the changes in mRNA and protein expression, we chose to use 2 mM AKG as it was more beneficial to the cardiomyocytes. TUNEL staining showed an increase in cardiomyocyte apoptosis after Ang II treatment (P < 0.001). However, AKG supplementation (2 mM) reduced Ang II-induced apoptosis in the cardiomyocytes (P < 0.05) (Fig. 7A).
Fig. 7.

Effect of AKG on mitophagy, MMP and ROS production in vitro. (A)TUNEL staining(n = 5). (B) Western blotting of β-MHC, PINK1, Parkin, caspase-3, Bcl-2 and ANP(n = 5). (C) mRNA expression levels of Nppa, Nppb, Myh7, Bcl-2, caspase-3, PINK1 and Parkin(n = 9). (D) Colocalization of Mitochondria and Lysosomes(n = 5). (E)TEM showing the effect of 2 mM AKG supplementation on mitochondrial morphology in cardiomyocytes. (F) DCFH-DA staining showing the effect of 2 mM AKG supplementation on ROS level(n = 5). (G)Effect of 2 mM AKG supplementation on MMP(n = 5); *: P<0.05 vs sham, **: P<0.01 vs sham, ***: P<0.001 vs sham, #: P<0.05 vs TAC, ##: P<0.01 vs TAC, ###: P<0.001 vs TAC. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.6. AKG promoted myocardial mitophagy, improved MMP and relieved oxidative damage induced by Ang II in vitro
Next, we examined the impact of AKG on mitophagy in cardiomyocytes. We observed 21.95% (P<0.01) and 25.79% (P<0.001) decreases in the transcript levels of PINK1 and Parkin (Fig. 7C), respectively, in Ang II-treated cardiomyocytes but no significant decrease at the protein level (Fig. 7B), which might be due to the duration of drug treatment and delayed post-transcriptional translation. AKG supplementation increased PINK1 and Parkin expression in Ang II-treated cardiomyocytes, in both mRNA and protein expression level. Fluorescence images showing lysosomal and mitochondrial colocalization revealed that their interaction was increased in Ang II-treated cardiomyocytes administered 2 mM AKG compared to those without AKG supplementation (Fig. 7D).
The TEM results (Fig. 7E) showed that the normal myocardial mitochondria had a regular and complete morphology, but swelling, vacuolization, a reduced density and an incomplete structure were observed in the Ang II-treated myocardial mitochondria, suggesting mitochondrial damage and dysfunction. AKG supplementation (2 mM) reversed Ang II-induced mitochondrial swelling and enhanced the clearance of damaged mitochondria. This suggests that AKG may help rescue mitochondrial morphology and function.
Next, we observed the impact of AKG on changes in MMP and cellular ROS levels induced by Ang II. DCFH-DA staining (Fig. 7F) showed that ROS levels in cardiomyocytes treated with Ang II were significantly increased by approximately 3.94-fold compared to those in the control group (P < 0.001). JC-1 staining (Fig. 7G) showed severe impairment of the MMP in Ang II-treated cardiomyocytes. Supplementation with 2 mM AKG significantly reduced ROS production (P < 0.001) induced by Ang II and partially restored the MMP (P < 0.01). The above in vitro results confirmed that AKG increased mitophagy, improved MMP and relieved oxidative damage induced by Ang II in cardiomyocytes.
4. Discussion
The following are the main findings of the present study: (1) AKG attenuated pressure overload-induced myocardial fibrosis and hypertrophy remodelling. (2) AKG improved cardiac function and LV strain in TAC mice. (3) AKG improved myocardial mitophagy to clear damaged mitochondria in TAC mice and rescued impairment of the MMP induced by Ang II. (4) AKG reduced intracellular ROS and myocardial apoptosis in the pressure overloaded heart (Fig. 8).
Fig. 8.

AKG supplementation promotes mitophagy to clear damaged mitochondria, reduces oxidative damage caused by ROS, attenuates myocardial hypertrophy and fibrosis caused by pressure overload, and improves cardiac insufficiency.
The development of HF is associated with metabolic disturbances in the myocardium, primarily metabolic changes due to mitochondrial dysfunction and a shift from fatty acid to glucose utilization for energy production [37,38]. Changes in specific plasma metabolites, including AKG, in HF patients were shown to act as metabolic biomarkers with clinical applications for diagnostic and prognostic purposes. Previous clinical studies showed circulating AKG levels to be elevated in in AHF and CHF patients [5,6,12,13]. In contrast, in an animal experiment, no significant change in circulating AKG was observed in mice with pressure overload-induced cardiac hypertrophy, but an obvious decrease in circulating AKG was observed in mice with ischaemic HF [37]. Despite the increasing attention to metabolic changes in HF, there have been fewer studies on the effects of AKG on HF. Here, we describe the protective effects of AKG against myocardial hypertrophy remodelling and LV dysfunction in TAC mice, which were determined through the measurement of LV dimensions and strain by echocardiography and STE. As it decreased expression of ANP, β-MHC and TGF-β1, AKG supplementation also improved pathological changes in the hearts of the TAC mice, including changes to cell size and organ weight and fibrosis. Furthermore, our data showed a mild decrease in protein and mRNA levels of ANP in the sham + AKG group compared to the sham group, which may be related to AKG reducing cardiac inflammation in the sham surgery group.
The heart is a high-energy-consuming organ. Cardiac mitochondria produce large quantities of ATP through oxidative phosphorylation to preserve contractile function. However, mitochondria are also the primary source of ROS [34,35], and excessive ROS production pathophysiologically lead to mitochondrial dysfunction, myocardial injury, and HF. Mitophagy, a special type of autophagy in which damaged and dysfunctional mitochondria are selectively cleared by autophagosomes, helps reduce ROS and prevent oxidative damage in the normal heart [35,39]. Failing heart tissues produce more ROS than normal tissues, accompanied by decreased cardiac mitophagy. The development of pressure overload-induced CHF can be prevented by increasing mitophagy [33,36]. Several studies have shown that AKG activates autophagy by inhibiting mammalian target of rapamycin (mTOR) and activating AMP-activated protein kinase (AMPK) [16,17]. Here, we detected the effects of AKG on myocardial mitophagy. Mechanistically, mitophagy in cardiomyocytes is mainly mediated by the cytosolic E3 ubiquitin ligase Parkin and the mitochondrial membrane kinase PINK1 [33]. PINK1, a serine/threonine kinase localized on the mitochondrial surface, is selectively stabilized in mitochondria with diminished electrochemical potentials during mitochondrial depolarization and recruits Parkin to the mitochondria to activate its E3 ubiquitin ligase activity. The Parkin-mediated ubiquitination of mitochondrial outer membrane proteins is a signal for autophagosomal phagocytosis and lysosomal degradation [33,40]. Assessment of mRNA and protein levels in the myocardium showed increased expression of PINK1 and Parkin in the TAC mice treated with AKG, and DHE staining showed a decrease in ROS levels upon AKG supplementation compared to those in the TAC mice, suggesting that AKG increased mitophagy to clear damaged mitochondria and attenuate myocardial oxidative stress under pressure overload.
TAC can be used to generate a model of pressure overload-induced overactivation of the neuroendocrine system, which leads to elevated circulating Ang II and norepinephrine levels [22,41]. Ang II increases ROS production, aggravating mitochondrial dysfunction and leading to myocardial oxidative stress injury and apoptosis [42,43]. A previous study showed a significant decline in ROS levels in AKG-treated bone marrow mesenchymal stromal/stem cells (MSCs) in ageing mice and in MSCs exposed to H2O2 [7]. Our results also confirmed that Ang II-induced ROS levels were decreased in the AKG-treated cardiomyocytes. In our in vitro experiments, we used Ang II to induce cardiomyocyte hypertrophy and injury. DCFH-DA and JC-1 staining showed an increased ROS level and decreased MMP (ΔΨm) in cardiomyocytes exposed to Ang II. Mitochondrial swelling, a reduction in mitochondrial density and vacuolization were also observed by TEM. The decrease of Ang II-induced ROS was likely related to the increase in mitophagy induced by AKG supplementation, which was accompanied by the elimination of damaged mitochondria, restoration of the MMP and decrease in apoptosis. Our in vitro experiments showed that AKG protected cardiomyocytes from Ang II-induced mitochondrial swelling and vacuolization, cellular oxidative injury and apoptosis, which is consistent with the results of in vivo experiments.
The circulating concentration of AKG, which decreases with ageing [8] but increases upon fasting [9], hypoxia [11] and exercise [10,44], reflects changes in metabolic flux. Interestingly, the changes in circulating AKG levels reported in previous studies on HF metabolites are inconsistent. Some clinical studies have shown that an increase in circulating AKG levels in HF patients is associated with HF severity (NYHA classification) and adverse short-term outcomes and that circulating AKG levels thus can be used as a metabolic biomarker and contribute to the diagnosis and prognostic assessment of HF [5,6,12,13]. Another examination of the plasma metabolome in HF patients revealed no significant change in AKG [38]. An animal experiment also found no significant change in circulating AKG concentration in mice with pressure overload-induced cardiac hypertrophy but an obvious decrease in circulating AKG concentration in mice with ischaemic HF [37]. In the present study, we found that the plasma AKG level was elevated in the TAC mice but the differences were not statistically significant from that in the sham group (63.40 ± 10.27 μM vs 84.65 ± 13.43 μM), and the plasma AKG level was further increased in TAC mice supplemented with AKG but still not statistically significant different from that in the TAC mice (84.65 ± 13.43 μM vs 115.71 ± 8.39 μM). It is possible that the number of mice included in the study was relatively small. Meanwhile, as AKG is a TCA cycle metabolite, being transported into cells and consumed could reduce circulating AKG concentration, so the circulating AKG concentrations might not necessarily reflect the intracellular levels, which might also be the reason for no statistically significant differences between the TAC and TAC + AKG groups. As reported in previous studies [19,44,45], we observed no significant adverse effects of AKG administration.
AKG is an important intermediate in the TCA cycle, impacts ATP production as a key point in the anaplerotic pathway [7,15,46,47], involved in the biosynthesis of proteins and amino acids [48] as a substrate of amino acid transferase and glutamate dehydrogenase, and influences epigenetic regulation in ageing [7,19,20]and metabolism [8,49] as an obligate substrate of histone and DNA demethylases [15,50]. Exogenous AKG also affects the cellular energy supply under the effects of various adverse factors [14,[51], [52], [53]], such as bacterial endotoxins and hypoxia. AKG supplementation decreased the levels of ROS [7] and inflammatory cytokines in mice [19], lessening the adverse impact of ageing and oxidative stress. Growing evidence suggests the protective effects of AKG [14], including in cardiomyocytes. Dimethyl α-ketoglutarate(DMKG) has been reported to inhibit maladaptive autophagy in pressure overload-induced cardiomyopathy [54]. Recent studies showed that ablation of oxoglutarate receptor 1(OXGR1, a receptor for AKG, also known as GPR80 or GPR99) in mice resulted in a significant increase in heart hypertrophy following TAC, as well as worse cardiac function compared with wild-type TAC mice, and OXGR1 overexpression in cardiomyocytes reduced the phenylephrine-induced hypertrophy [55]. Another study found that the intracellular levels of AKG were significantly decreased by H2O2 stimulation in rat neonatal cardiomyocytes, while treatment with DMKG decreased intracellular ROS production and suppressed oxidative stress-induced cell death [47].
In the present study, we also observed the cardioprotective effect of AKG under pressure overload, which may occur through multiple mechanisms. On the one hand, myocardial fibrosis, which occurs in the irreversible late stage of HF progression, increases LV stiffness and reduces myocardial contractility and compliance [32]. Cytokines secreted from cardiomyocytes and fibroblasts, including TGF-β1, contribute to the differentiation of fibroblasts into myofibroblasts and remodelling of the extracellular matrix. Our data showed that AKG supplementation helped reduce TGF-β1 transcript and protein expression and lessen cardiac fibrosis in TAC mice, which was also supported by the improvement of LV strain and maximum opposing wall delay measured by STE. On the other hand, decreased mitophagy levels, along with elevated ROS levels and mitochondrial dysfunction, contributes to the progression of HF [33]. Our data suggested that AKG supplementation promoted mitophagy to clear injured mitochondria, reducing myocardial ROS levels and apoptosis in the TAC mice.
4.1. Limitations of the study
Our study has some limitations. As HF is a multifactorial syndrome and there are multiple beneficial mechanisms of AKG, we cannot exclude the notion that AKG utilizes other pathways to protect the heart under pressure overload. More research is needed to determine the mechanism underlying the cardioprotective effects of AKG. In addition, as AKG is an intermediate in the TCA cycle and an important source of glutamate and glutamine involved in the biosynthesis of amino acids and proteins, AKG is produced in skeletal muscle, the myocardium, the liver and other organs. The source of the elevated circulating AKG in HF remains to be clarified. Whether AKG was released from the injured myocardium or produced by other tissues as a protective compensatory adaptation to the HF process is a question that should be resolved.
5. Conclusion
The safety and anti-aging effects of AKG supplementation have been confirmed by many studies, but there have been few studies of the effect of AKG on heart disease. Here, we discovered that AKG supplementation protected the heart from hypertrophy, fibrosis and systolic dysfunction caused by pressure overload and increased mitophagy, alleviated ROS injury and reduced apoptosis induced by Ang II. Because of its generally recognized as safe (GRAS) status and beneficial effect on ageing, an AKG formulation is already sold, and the study of AKG in humans is ongoing [56]. We look forward to more investigations of AKG in heart disease and data from human studies, which may provide new ideas for the treatment of HF.
Declaration of competing interest
The authors declare no competing interests.
Acknowledgments
This project was supported by the Science and Technology Planning Project Foundation of Guangzhou (201707020012), National Natural Science Foundation of China (81970336, 81670367).
Contributor Information
Hao Ren, Email: renhao67@aliyun.com.
Dingli Xu, Email: dinglixu@smu.edu.cn.
References
- 1.Ardehali H., Sabbah H.N., Burke M.A. Targeting myocardial substrate metabolism in heart failure: potential for new therapies[J] Eur. J. Heart Fail. 2012;14(2):120–129. doi: 10.1093/eurjhf/hfr173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lopaschuk G.D., Ussher J.R., Folmes C.D. Myocardial fatty acid metabolism in health and disease[J] Physiol. Rev. 2010;90(1):207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 3.Stanley W.C., Recchia F.A., Lopaschuk G.D. Myocardial substrate metabolism in the normal and failing heart[J] Physiol. Rev. 2005;85(3):1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 4.Huss J.M., Kelly D.P. Mitochondrial energy metabolism in heart failure: a question of balance[J] J. Clin. Invest. 2005;115(3):547–555. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunn W.B., Broadhurst D.I., Deepak S.M. Serum metabolomics reveals many novel metabolic markers of heart failure, including pseudouridine and 2-oxoglutarate[J] Metabolomics. 2007;3(4):413–426. [Google Scholar]
- 6.Lanfear D.E., Gibbs J.J., Li J. Targeted metabolomic profiling of plasma and survival in heart failure patients[J] JACC Heart Fail. 2017;5(11):823–832. doi: 10.1016/j.jchf.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y., Deng P., Liu Y. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations[J] Nat. Commun. 2020;11(1) doi: 10.1038/s41467-020-19360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian Q., Zhao J., Yang Q. Dietary alpha-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice[J] Aging Cell. 2020;19(1) doi: 10.1111/acel.13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw E., Talwadekar M., Rashida Z. Anabolic SIRT4 exerts retrograde control over TORC1 signaling by glutamine sparing in the mitochondria[J] Mol. Cell Biol. 2020;40(2):e00212–e00219. doi: 10.1128/MCB.00212-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nayor M., Shah R.V., Miller P.E. Metabolic architecture of acute exercise response in middle-aged adults in the community[J] Circulation. 2020;142(20):1905–1924. doi: 10.1161/CIRCULATIONAHA.120.050281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oldham W.M., Clish C.B., Yang Y. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress[J] Cell Metabol. 2015;22(2):291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen P.A., Xu Z.H., Huang Y.L. Increased serum 2-oxoglutarate associated with high myocardial energy expenditure and poor prognosis in chronic heart failure patients[J] Biochim. Biophys. Acta. 2014;1842(11):2120–2125. doi: 10.1016/j.bbadis.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Peng Z., Zhan Q., Xie X. Association between admission plasma 2-oxoglutarate levels and short-term outcomes in patients with acute heart failure: a prospective cohort study[J] Mol. Med. 2019;25(1):8. doi: 10.1186/s10020-019-0078-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayliak M.M., Lushchak V.I. Pleiotropic effects of alpha-ketoglutarate as a potential anti-ageing agent[J] Ageing Res. Rev. 2021;66 doi: 10.1016/j.arr.2020.101237. [DOI] [PubMed] [Google Scholar]
- 15.Martínez-Reyes I., Chandel N.S. Mitochondrial TCA cycle metabolites control physiology and disease[J] Nat. Commun. 2020;11(1):102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chin R.M., Fu X., Pai M.Y. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR[J] Nature. 2014;510(7505):397–401. doi: 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chai M., Jiang M., Vergnes L. Stimulation of hair growth by small molecules that activate autophagy[J] Cell Rep. 2019;27(12):3413–3421. doi: 10.1016/j.celrep.2019.05.070. [DOI] [PubMed] [Google Scholar]
- 18.Liu P.S., Wang H., Li X. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming[J] Nat. Immunol. 2017;18(9):985–994. doi: 10.1038/ni.3796. [DOI] [PubMed] [Google Scholar]
- 19.Asadi S.A., Edgar D., Liao C.Y. Alpha-ketoglutarate, an endogenous metabolite, extends lifespan and compresses morbidity in aging mice[J] Cell Metabol. 2020;32(3):447–456. doi: 10.1016/j.cmet.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z., He C., Gao Y. α-ketoglutarate delays age-related fertility decline in mammals[J] Aging Cell. 2021;20(2) doi: 10.1111/acel.13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su Y., Wang T., Wu N. Alpha-ketoglutarate extends Drosophila lifespan by inhibiting mTOR and activating AMPK[J] Aging. 2019;11(12):4183–4197. doi: 10.18632/aging.102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z., Ma Z., Zhang H. Ferulic acid increases intestinal Lactobacillus and improves cardiac function in TAC mice[J] Biomed. Pharmacother. 2019;120 doi: 10.1016/j.biopha.2019.109482. [DOI] [PubMed] [Google Scholar]
- 23.Liu Z., Hua J., Cai W. N-terminal truncated peroxisome proliferator-activated receptor-γ coactivator-1α alleviates phenylephrine-induced mitochondrial dysfunction and decreases lipid droplet accumulation in neonatal rat cardiomyocytes[J] Mol. Med. Rep. 2018;18(2):2142–2152. doi: 10.3892/mmr.2018.9158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Lucia C., Wallner M., Eaton D.M. Echocardiographic strain analysis for the early detection of left ventricular systolic/diastolic dysfunction and dyssynchrony in a mouse model of physiological aging[J] J Gerontol A Biol Sci Med Sci. 2019;74(4):455–461. doi: 10.1093/gerona/gly139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallner M., Duran J.M., Mohsin S. Acute catecholamine exposure causes reversible myocyte injury without cardiac regeneration[J] Circ. Res. 2016;119(7):865–879. doi: 10.1161/CIRCRESAHA.116.308687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shepherd D.L., Nichols C.E., Croston T.L. Early detection of cardiac dysfunction in the type 1 diabetic heart using speckle-tracking based strain imaging[J] J. Mol. Cell. Cardiol. 2016;90:74–83. doi: 10.1016/j.yjmcc.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer M., Cheng S., Jain M. Echocardiographic speckle-tracking based strain imaging for rapid cardiovascular phenotyping in mice[J] Circ. Res. 2011;108(8):908–916. doi: 10.1161/CIRCRESAHA.110.239574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nie J., Xie L., Zhao B.X. Serum trimethylamine N-oxide concentration is positively associated with first stroke in hypertensive patients[J] Stroke. 2018;49(9):2021–2028. doi: 10.1161/STROKEAHA.118.021997. [DOI] [PubMed] [Google Scholar]
- 29.Xiong W., Hua J., Liu Z. PTEN induced putative kinase 1 (PINK1) alleviates angiotensin II-induced cardiac injury by ameliorating mitochondrial dysfunction[J] Int. J. Cardiol. 2018;266:198–205. doi: 10.1016/j.ijcard.2018.03.054. [DOI] [PubMed] [Google Scholar]
- 30.Xiong W., Ma Z., An D. Mitofusin 2 participates in mitophagy and mitochondrial fusion against angiotensin II-induced cardiomyocyte injury[J] Front. Physiol. 2019;10:411. doi: 10.3389/fphys.2019.00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niu P., Li L., Yin Z. Speckle tracking echocardiography could detect the difference of pressure overload-induced myocardial remodelling between young and adult rats[J] J. R. Soc. Interface. 2020;17(163) doi: 10.1098/rsif.2019.0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goh K.Y., He L., Song J. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice[J] Redox Biol. 2019;21 doi: 10.1016/j.redox.2019.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang B., Nie J., Wu L. AMPKα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation[J] Circ. Res. 2018;122(5):712–729. doi: 10.1161/CIRCRESAHA.117.312317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morciano G., Patergnani S., Bonora M. Mitophagy in cardiovascular diseases[J] J. Clin. Med. 2020;9(3):892. doi: 10.3390/jcm9030892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morales P.E., Arias-Durán C., ávalos-Guajardo Y. Emerging role of mitophagy in cardiovascular physiology and pathology[J] Mol. Aspect. Med. 2020;71 doi: 10.1016/j.mam.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Shirakabe A., Zhai P., Ikeda Y. Drp1-Dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure[J] Circulation. 2016;133(13):1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai L., Leone T.C., Keller M.P. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach[J] Circ Heart Fail. 2014;7(6):1022–1031. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng M.L., Wang C.H., Shiao M.S. Metabolic disturbances identified in plasma are associated with outcomes in patients with heart failure: diagnostic and prognostic value of metabolomics[J] J. Am. Coll. Cardiol. 2015;65(15):1509–1520. doi: 10.1016/j.jacc.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 39.Gustafsson Å B., Dorn G.N. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process[J] Physiol. Rev. 2019;99(1):853–892. doi: 10.1152/physrev.00005.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Y., Li T., Li Z. Role of mitophagy in cardiovascular disease[J] Aging Dis. 2020;11(2):419–437. doi: 10.14336/AD.2019.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akers W.S., Cross A., Speth R. Renin-angiotensin system and sympathetic nervous system in cardiac pressure-overload hypertrophy[J] Am. J. Physiol. Heart Circ. Physiol. 2000;279(6):H2797–H2806. doi: 10.1152/ajpheart.2000.279.6.H2797. [DOI] [PubMed] [Google Scholar]
- 42.Hsieh C.C., Li C.Y., Hsu C.H. Mitochondrial protection by simvastatin against angiotensin II-mediated heart failure[J] Br. J. Pharmacol. 2019;176(19):3791–3804. doi: 10.1111/bph.14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao W., Li Y., Jia L. Atg5 deficiency-mediated mitophagy aggravates cardiac inflammation and injury in response to angiotensin II[J] Free Radic. Biol. Med. 2014;69:108–115. doi: 10.1016/j.freeradbiomed.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Yuan Y., Xu P., Jiang Q. Exercise-induced α-ketoglutaric acid stimulates muscle hypertrophy and fat loss through OXGR1-dependent adrenal activation[J] EMBO J. 2020;39(7) doi: 10.15252/embj.2019103304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai X., Yuan Y., Liao Z. α-Ketoglutarate prevents skeletal muscle protein degradation and muscle atrophy through PHD3/ADRB2 pathway[J] Faseb. J. 2018;32(1):488–499. doi: 10.1096/fj.201700670R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zdzisińska B., |urek A., Kandefer-Szerszeń M. Alpha-ketoglutarate as a molecule with pleiotropic activity: well-known and novel possibilities of therapeutic use[J] Arch. Immunol. Ther. Exp. 2017;65(1):21–36. doi: 10.1007/s00005-016-0406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe K., Nagao M., Toh R. Critical role of glutamine metabolism in cardiomyocytes under oxidative stress[J] Biochem. Biophys. Res. Commun. 2021;534:687–693. doi: 10.1016/j.bbrc.2020.11.018. [DOI] [PubMed] [Google Scholar]
- 48.Xiao D., Zeng L., Yao K. The glutamine-alpha-ketoglutarate (AKG) metabolism and its nutritional implications[J] Amino Acids. 2016;48(9):2067–2080. doi: 10.1007/s00726-016-2254-8. [DOI] [PubMed] [Google Scholar]
- 49.Okabe K., Nawaz A., Nishida Y. NAD+ metabolism regulates preadipocyte differentiation by enhancing α-ketoglutarate-mediated histone H3K9 demethylation at the PPARγ promoter[J] Front Cell Dev Biol. 2020;8 doi: 10.3389/fcell.2020.586179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zasłona Z., O'Neill L. Cytokine-like roles for metabolites in immunity[J] Mol. Cell. 2020;78(5):814–823. doi: 10.1016/j.molcel.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Hou Y., Yao K., Wang L. Effects of α-ketoglutarate on energy status in the intestinal mucosa of weaned piglets chronically challenged with lipopolysaccharide[J] Br. J. Nutr. 2011;106(3):357–363. doi: 10.1017/S0007114511000249. [DOI] [PubMed] [Google Scholar]
- 52.Wang L., Yi D., Hou Y. Dietary supplementation with α-ketoglutarate activates mTOR signaling and enhances energy status in skeletal muscle of lipopolysaccharide-challenged piglets[J] J. Nutr. 2016;146(8):1514–1520. doi: 10.3945/jn.116.236000. [DOI] [PubMed] [Google Scholar]
- 53.Guo S., Duan R., Wang L. Dietary α-ketoglutarate supplementation improves hepatic and intestinal energy status and anti-oxidative capacity of Cherry Valley ducks[J] Anim. Sci. J. 2017;88(11):1753–1762. doi: 10.1111/asj.12824. [DOI] [PubMed] [Google Scholar]
- 54.Mariño G., Pietrocola F., Kong Y. Dimethyl α-ketoglutarate inhibits maladaptive autophagy in pressure overload-induced cardiomyopathy[J] Autophagy. 2014;10(5):930–932. doi: 10.4161/auto.28235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Omede A., Zi M., Prehar S. The oxoglutarate receptor 1 (OXGR1) modulates pressure overload-induced cardiac hypertrophy in mice[J] Biochem. Biophys. Res. Commun. 2016;479(4):708–714. doi: 10.1016/j.bbrc.2016.09.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaiser J. Bodybuilding supplement promotes healthy aging and extends life span, at least in mice[J] Science. 2020 doi: 10.1126/science.abe5953. [DOI] [Google Scholar]