

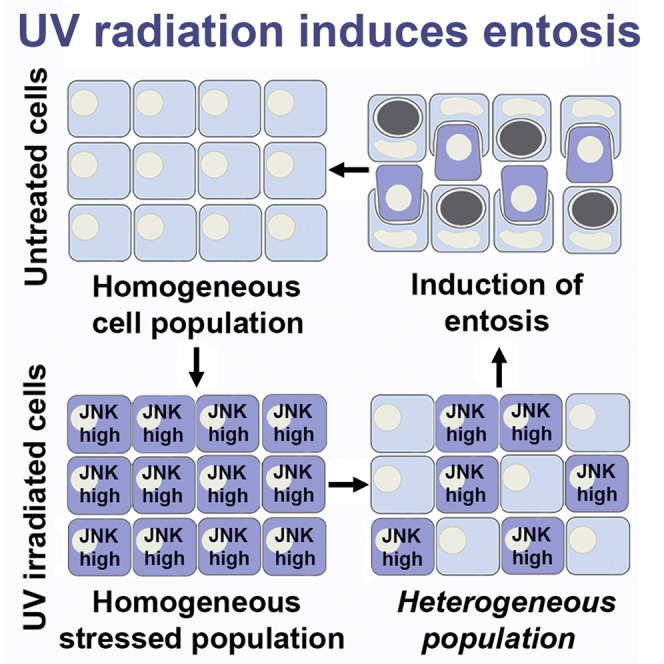
Summary
Entosis is a cell death mechanism that is executed through neighbor cell ingestion and killing that occurs in cancer tissues and during development. Here, we identify JNK and p38 stress-activated kinase signaling as an inducer of entosis in cells exposed to ultraviolet (UV) radiation. Cells with high levels of stress signaling are ingested and killed by those with low levels, a result of heterogeneity arising within cell populations over time.
In stressed cells, entosis occurs as part of mixed-cell death response with parallel induction of apoptosis and necrosis, and we find that inhibition of one form of cell death leads to increased rates of another. Together, these findings identify stress-activated kinase signaling as a new inducer of entosis and demonstrate cross talk between different forms of cell death that can occur in parallel in response to UV radiation.
Subject areas: Cellular physiology, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
UV radiation induces entosis in cancer cells
-
•
JNK and p38 stress kinases regulate entosis induction
-
•
Entosis induction involves heterogeneous inactivation of JNK over time
-
•
Cells ingested by entosis maintain high levels of JNK signaling
Cellular physiology; Molecular biology; Cell biology
Introduction
Tissue homeostasis in multicellular organisms is maintained in part through a balance between proliferation and programmed cell death (PCD) (Kerr et al., 1972). Imbalances in these processes can result in the net loss or accumulation of cells, leading ultimately to developmental defects, degenerative conditions, or the development of diseases such as cancer (Voss and Strasser, 2020). Understanding how PCD is regulated has important implications for understanding both normal physiology and pathological conditions. Numerous PCD mechanisms have been identified in recent years, including regulated forms of necrosis such as necroptosis (Degterev et al., 2005) and ferroptosis (Dixon et al., 2012) and a mechanism called entosis (Overholtzer et al., 2007), each of which can eliminate cells in a regulated manner, in parallel to classical PCD that is executed through apoptosis (Galluzzi et al., 2018). Among these, entosis may be the most unusual because it involves the ingestion and killing of cells by their neighbors, a non-cell-autonomous mechanism that resembles a murder rather than cell suicide (Overholtzer et al., 2007). Entosis has been shown to occur in normal tissues in some contexts, including during embryo implantation (Li et al., 2015) and in Caenorhabditis elegans development (Lee et al., 2019), and is more commonly observed in cancer tissues, where it may promote the development of aggressive disease by driving competition for survival between individual cells (Sun et al., 2014b, 2015). While the mechanisms that control the execution of entosis are well established and involve Rho-kinase (ROCK)- and cadherin-mediated cell ingestion and lysosome-mediated cell killing (Florey et al., 2011; Hinojosa et al., 2017; Overholtzer et al., 2007; Purvanov et al., 2014), it remains less clear what, and how many, upstream signals can induce this process. Entosis occurs in cells that are detached from matrix (Khalkar et al., 2018; Overholtzer et al., 2007) and has also been shown to be induced by nutrient deprivation (Hamann et al., 2017) or as a result of altered regulation of the Rho pathway, myosin and cytoskeletal tension (Liang et al., 2018; Ruan et al., 2018b; Sun et al., 2014b; Wan et al., 2012; Wen et al., 2013; Xia et al., 2014), altered mechanics of mitosis (Durgan et al., 2017; Liang et al., 2021), and changes in intercellular adhesion (Lai et al., 2010; Ruan et al., 2018a; Sun et al., 2014a; Wang et al., 2015). Here, we sought to examine if ultraviolet (UV) radiation, a well-known inducer of other forms of cell death such as apoptosis (Roos and Kaina, 2006), might also induce entosis, and to identify signaling mechanisms that could be involved in entosis induction.
Results
UV radiation induces entosis
To investigate if entosis might be induced by UV radiation, we exposed MCF7 human breast cancer cells, which have been shown to be competent for entosis induction (Hamann et al., 2017; Overholtzer et al., 2007), to increasing doses of UV. UV radiation induced entosis in a dose-dependent and significant manner, as more than 35% of cells were engaged in entosis after treatment with a high dose of UV (100J/m2) (Figures 1A and 1B and see Video S1). Entotic cell structures formed in response to UV were identified by localization of the E-cadherin cell–cell adhesion protein at the interface between the ingested cells and outer, or host cells, and/or by the presence of whole ingested cells inside of lysosomal compartments, as reported (Figure 1B) (Overholtzer et al., 2007). Similar to entosis induced by other stressors, ROCK activity was required for entosis induction in response to UV (Figure 1C) (Overholtzer et al., 2007; Sun et al., 2014b). The knockout of the E-cadherin-encoding gene Cdh1 also inhibited UV-induced entosis, which was restored by E-cadherin reexpression, as shown in cells we previously generated for entosis studies (Figure 1D) (Hamann et al., 2017). Also consistent with entosis induction, internalized entotic cells did not exhibit hallmarks of apoptosis such as condensed or fragmented nuclei, and treatment with an apoptosis inhibitor, the pan-caspase inhibitor ZVAD-FMK, had no effect on entosis induction (Figure S1) (Overholtzer et al., 2007). Finally, while apoptosis was observed to occur in parallel to entosis in UV-irradiated cells, we also did not observe any engulfments of apoptotic cells by neighboring MCF7 cells, out of 50 individual apoptotic cells that were tracked by time-lapse imaging (Figure S1). To examine if UV radiation-induced entosis was unique to MCF7 cells, we also exposed HCT116 human colon cancer cells, BxPC3 human pancreatic cancer cells, and 16HBE human bronchial epithelial cells to UV radiation, in the presence or absence of the ROCK inhibitor Y27632, and examined for entosis induction by confocal microscopy. As shown in Figures 1E and 1F, UV radiation induced entosis in each cell line, in a ROCK-dependent manner, demonstrating that UV radiation induces entosis across different cell types (Figures 1E and 1F).
Figure 1.

UV Radiation induces entosis
(A) UV radiation induces entosis in a dose-dependent manner. Graph shows the percentage of entosis in MCF7 cells after increasing doses of UV radiation, as determined by immunofluorescence 24 h after UV exposure.
(B) Representative images of MCF7 cells after UV radiation. Cell-cell junctions are indicated by E-cadherin immunostaining (green); lysosomal membranes by Lamp1 (red); and nuclei are stained with DAPI (blue). Entotic structures are indicated with arrows.
(C) ROCK inhibition inhibits entosis. Graph shows the quantification of entosis 24 h after exposure to UV radiation with the indicated treatments. Error bars depict mean ± SD, with at least three independent experiments.
(D) Induction of entosis requires E-Cadherin. Graph shows the quantification of entosis in wt MCF7, E-Cadherin (CDH1)-knockout MCF7, and E-Cadherin-knockout with E-Cadherin re-expression (re-exp) cells, 24 h after UV radiation exposure. Error bars depict mean ± SD, with at least three independent experiments.
(E) UV radiation induces entosis in HCT116, BxPC3, and 16HBE cells. Graph shows the quantification of engulfment with the indicated treatment 24 h after UV radiation, as determined by immunofluorescence. Error bars depict mean ± SD, with at least three independent experiments.
(F) Representative images of 16HBE cells 24h after UV radiation. Immunostaining for E-Cadherin (green), and LAMP1 (red) are shown. Cell nuclei are marked by DAPI (blue). Entotic structures are indicated with arrows.
See also Figure S1.
Arrows indicate cells that are ingested; black arrow indicates cell that undergoes entotic cell death, white arrow indicates cell that remains viable for the duration of the movie. Images show DIC and H2B-mCherry fluorescence that marks nuclei (red). Times are indicated as minutes (min).
UV radiation induces entosis through JNK/p38 signaling
We sought to identify what signaling mechanisms might control the induction of entosis in UV-irradiated cells and considered if the induction of p53, a well-known response to DNA damage (Riley et al., 2008) and regulator of entosis in other contexts (Liang et al., 2021; Mackay et al., 2018), might play a role. As shown in Figure S2, the knockdown or knockout of p53 in MCF7 or HCT116 cells had no effect on entosis induction in response to UV, demonstrating that entosis induction in this context does not require p53. We next considered a possible role for the c-Jun N-terminal kinase (JNK) and p38 stress-activated kinases that are known to be induced by UV and contribute to the induction of other forms of cell death (Hotamisligil and Davis, 2016; Karin, 1998; Matsukawa et al., 2004; Sui et al., 2014). The knockdown of JNK1/2 or p38α, or treatment with the JNK inhibitor SP600125 or the p38 inhibitor SB202190, reduced the levels of entosis in UV-irradiated cells, and inhibited the activation of JNK or p38 in response to UV (Figure 2B). Treatment of JNK1/2-knockdown cells with SB202190 further reduced entosis levels, consistent with a role for both JNK and p38 in regulating this process.
Figure 2.

UV-radiation-induced entosis is mediated by the JNK/p38 pathway
(A) UV radiation-induced entosis requires JNK and p38. Graph shows quantification of entosis 24 h after exposure to UV radiation in wt MCF7, JNK1/2 knockdown and p38α knockdown MCF7 cells, with or without the indicated inhibitors, as determined by immunofluorescence. SP600125: JNK inhibitor; SB202190: p38 inhibitor. Error bars depict mean ± SD, with at least three independent experiments. Statistical tests for comparisons of experimental conditions versus control were performed by One-way Anova followed by Dunnett's test; Student's t test was used for the indicated individual comparisons between conditions.
(B) UV radiation activates the JNK/p38 pathway which can be blocked by JNK/p38 inhibition. Western blots show total and phosphorylated (p)-SAPK/JNK (Thr183/Tyr185) (top row) and p-p38 (Thr180/Tyr182) (middle and bottom rows) in MCF7 cells 4 h after exposure to UV radiation, in the presence or absence of JNK and p38 inhibitors or JNK1/2 or p38α knockdowns. Arrowheads indicate protein molecular weight markers.
(C) Anisomycin induces entosis to a similar extent as UV radiation. Graph shows quantification of entosis 24 h after treatment with anisomycin in wt MCF7, JNK1/2 knockdown and p38α knockdown MCF7 cells, with or without the indicated inhibitors, as determined by immunofluorescence. Anisomycin: JNK/p38 agonist; Y27632: ROCK inhibitor. Error bars depict mean ± SD, with at least three independent experiments. Statistical tests for comparisons of experimental conditions versus control were performed by One-way Anova followed by Dunnett's test; Student's t test was used for the indicated individual comparisons between conditions.
(D) Anisomycin activates the JNK/p38 pathway, which is reduced by JNK/p38 inhibition. Western blots show JNK (left blots) and p38 (right blots) activation in response to anisomycin in MCF7 cells after 4 hr of treatment, in the presence or absence of treatment with JNK/p38 inhibitors or JNK1/2 or p38α knockdowns. P-JNK indicates Thr183/Tyr185 phosphorylation; p-p38 indicates Thr180/Tyr182 phosphorylation. Arrowheads indicate protein molecular weight markers.
See also Figure S2.
To further examine JNK and p38, we treated MCF7 cells with anisomycin, a pharmacological activator of these stress-activated kinases. As shown in Figures 2C and 2D, anisomycin induced entosis to a similar extent as UV radiation, and also led to the activation of both JNK and p38. Entosis induced by anisomycin was inhibited by the knockdown of JNK1/2 or p38α, or treatment with the JNK or p38 inhibitors alone or in combination (Figures 2C and 2D), demonstrating altogether that activation of the JNK and p38 stress kinases, through treatment with UV radiation or anisomycin, induces entosis.
UV-radiation-induced entosis results from heterogeneity in JNK signaling
We next sought to determine using an imaging-based approach how the dynamics of JNK activity within individual cells might relate to the induction of entosis within cell populations exposed to UV radiation. A modified version of a fluorescent reporter called JNK kinase translocation reporter (JNK KTR), which localizes to the cytoplasm when JNK is active and nucleus when JNK is inactive, was expressed in MCF7 cells and relative JNK activity was quantified over time by fluorescence microscopy (Figure 3A) (Regot et al., 2014). While all cells initially showed induction of JNK activity after UV exposure, expressed as an increased cytoplasmic-to-nuclear fluorescence ratio of JNK KTR, entotic cell pairs displayed a distinct pattern over time, with internalizing cells maintaining high levels of JNK activity but host cells showing significantly reduced levels, returning to baseline prior to entosis induction (Figures 3B and 3C). These data suggest that JNK may act specifically within internalizing cells to promote uptake into host cells with lower JNK activity. To examine this hypothesis using a genetic approach, we knocked down JNK1/2 expression in MCF7 cells and then labeled this cell population fluorescently by staining with CellTracker Red. After mixing knockdown cells at a 1:1 ratio with control cells stained with CellTracker Green, the coculture was then exposed to UV radiation and the frequency of entosis events with red or green cells on the inside or outside of entotic structures was quantified. As shown in Figure 3D, the knockdown of JNK1/2 inhibited cells from internalizing into their neighbors, leading to an increased frequency of JNK1/2 knockdown cells on the outside of entotic structures, demonstrating that JNK acts within internalizing cells to control entosis in response to UV radiation.
Figure 3.

UV radiation-induced entosis results from heterogeneity in JNK signaling
(A) Representation of JNK KTR in MCF7 cells. Cytoplasmic to nuclear fluorescence ratios of the reporter are quantified for both inner and outer cells of entotic pairs (left), or for control single cell pairs (right). Left schematic depicts inner cell with higher cytoplasmic to nuclear ratio than outer cell, which matches the center cell image (bottom panel: JNK KTR fluorescence in grayscale, C = cytoplasm, N = nucleus; top panel: same entotic cell pair shown with DIC and JNK KTR fluorescence (red)).
(B) JNK activity measurements for control (No UV, black, n = 87 cells), UV-irradiated single cells (red, n = 86 cells) and entotic cell pairs (blue = inner cell, n = 43 cells; green = outer cell, n = 43 cells), made by calculating JNK KTR fluorescence ratios over time. Note inner cells maintain high levels of JNK activity while host cells reduce JNK activity prior to entosis induction (arrow). Graph shows JNK activity 210 min prior to entosis induction to 30 min afterward, as determined by time-lapse imaging. Error bars depict mean ± SD, with at least three independent experiments.
(C) Entotic cell pairs have higher JNK activity ratios compared to neighboring single cells. Graph shows JNK activity ratio (defined in schematic) 210 min prior to entosis induction to 30 min afterward. Black depicts JNK activity ratios of entotic pairs (n = 43 pairs); red line depicts JNK activity ratio for random single cells (n = 43 pairs). Error bars depict mean ± SD, with at least three independent experiments.
(D) JNK1/2 knockdown cells preferentially engulf control cells after exposure to UV radiation. Graph shows the percentage of red labeled-cells that are outer or inner cells when mixed with green-labeled control MCF7 cells and cultured for 24 hr after exposure to UV radiation. Error bars depict mean ± SD. Data are from five independent experiments with at least 50 entotic pairs counted in each experiment.
Engulfment and ingestion of corpses provides survival advantage
We previously reported that entosis can support the survival and proliferation of cells with limited nutrient availability, such as cells starved for amino acids (Krajcovic et al., 2013) or glucose (Hamann et al., 2017), by providing host cells with nutrients derived from digested internalized cells. We have also shown that internalized cells undergo death and are degraded at faster rates during starvation (Hamann et al., 2017), suggesting that stress signaling can upregulate the rate of nutrient scavenging. We quantified the fates of entotic cells that were internalized in UV-irradiated cultures and found that they also underwent death and were degraded at significantly faster rates compared to nonirradiated conditions (Figure 4A). Host cells also died at significantly lower rates in response to UV compared to neighboring nonentotic cells, demonstrating a survival advantage linked to entosis that could result from the ingestion and degradation of internalized cells (Figure 4B). To examine if lysosomal digestion of internalized cells is required for the survival advantage of host cells, lysosome function was inhibited by treatment with the V-ATPase inhibitor concanamycin A (ConA), and the survival rates of host cells and neighboring single cells were quantified by time-lapse imaging. As shown in Figure 4C, treatment with ConA inhibited the degradation of internalized cells and also partially but significantly reduced the survival of host cells in response to UV radiation, consistent with a link between host cell survival and internalized cell degradation. We next asked whether the engulfment and degradation of ingested cells might generally confer a survival advantage in response to UV and examined if cell engulfment through phagocytosis can have a similar effect. Apoptotic cell corpses were fed to UV radiation-exposed J774.1 macrophages, and the overall survival rates of cells that had been fed with corpses were compared to control cells. As shown in Figure 4D, cells that had been fed with apoptotic corpses survived at significantly higher rates than those that were not fed, and this advantage was abrogated by treatment with ConA, demonstrating a general link between the degradation of ingested cells and a survival advantage in response to UV radiation.
Figure 4.

Engulfment and degradation of corpses provides a survival advantage
(A) Entotic death rate of internalized cells increases after exposure to UV radiation. Graph shows internalized cell death rates for MCF7 cells with (red) or without (black) exposure to UV. Death rates are determined over a 10 hr period after the completion of entosis, as determined by a vacuole appearance in the outer cell by DIC microscopy. Error bars depict mean ± SD. Data are from at least three independent experiments with at least 20 entotic pairs counted in each experiment.
(B) Entotic host cells survive at higher rates than neighboring single cells, in a lysosome-dependent manner. Graph shows the quantification of survival rates for entotic host cells and neighboring single cells after exposure to UV radiation with or without ConA treatment. Cell survival rates are determined by morphology using DIC microscopy. Error bars depict mean ± SD, with at least three independent experiments. Entotic host: n = 217; neighboring single cells: n = 312; entotic host with ConA: n = 167; neighboring single cells with conA: n = 435.
(C) Representative images depicting internalized cell degradation in MCF7 cells. The top panel shows internalized cell in UV-irradiated culture that becomes killed and digested over time; bottom panel shows failure of internalized cell degradation in ConA-treated culture, and host cell undergoes cell death (right, arrow). Blue dotted lines depict internalized cells.
(D) Phagocytosis of apoptotic corpses by J774.1 cells provides survival advantage. Graph shows quantification of cell survival in the presence or absence of apoptotic corpses and ConA. Image shows phagocytic ingestion of apoptotic corpse by J774.1 cell; blue dotted line shows corpse inside of phagosome. Error bars depict mean ± SD. Data are from three independent experiments with at least 1000 cells counted in each condition.
(E) Overall cell death rates determined by time-lapse microscopy after exposure to UV radiation in MCF7 and BxPC3 cells, in the presence or absence of Y27632 or Z-VAD-FMK. Y27632: ROCK inhibitor; Z-VAD-FMK: pan-caspase inhibitor. Error bars depict mean ± SD, with at least 350 cells quantified in each condition from three independent experiments. Statistics are obtained using Student's t-test.
(F) Frequencies of individual death types in MCF7 and BxPC3 cells exposed to UV radiation, determined by DIC morphology; representative images of entosis, apoptosis and necrosis shown on right. Note that the inhibition of entosis (with Y27632) results in more necrosis in MCF7 cells, while inhibition of apoptosis (with Z-VAD-FMK) results in more entosis in BxPC3 cells. Graph shows quantification of apoptosis, entosis, and necrosis after exposure to UV radiation, with the indicated treatment, as determined by time-lapse microscopy. Error bars depict mean ± SD, with at least 150 cells quantified in each condition from three independent experiments. Statistical tests were performed using Student's t-test.
Entosis occurs as a part of a mixed death response
Taken together, our data demonstrate that UV radiation and activation of the JNK and p38 stress kinase pathways can induce entosis. While we show that entosis is induced in multiple different cell lines in response to UV, including MCF7, HCT116, BxPC3, and 16HBE, interestingly, the frequency of entosis varies significantly among them, from more than 35% in MCF7 compared to only about 5% in BxPC3 (see Figures 1A and 1E). Although entosis frequencies differed, we found that the total amount of cell death was more similar between cells. For example, ~75% total death was observed in MCF7 cells in response to UV and nearly 60% in BxPC3, due to the induction of other forms of cell death, including apoptosis and necrosis in addition to entosis (Figures 4E and 4F). To investigate the relationship between different forms of cell death in response to UV radiation, we treated MCF7 cells with an inhibitor of entosis, Y27632, and quantified cell death after UV radiation by time-lapse imaging. As shown in Figures 4E and 4F, while treatment with Y27632 nearly completely blocked entosis induction, it had a much smaller effect on the overall amount of cell death, due to a significant increase in the frequency of necrosis. We also treated BxPC3 cells with an inhibitor of apoptosis, the pan-caspase inhibitor Z-VAD-FMK, and quantified UV-induced cell death by time-lapse imaging. While apoptosis was significantly inhibited by treatment with Z-VAD-FMK (by more than 4-fold), the overall rate of cell death was not reduced significantly, as apoptosis-inhibited cells displayed a significant increase in the frequencies of both entosis and necrosis (Figures 4E and 4F). Taken together, these data demonstrate that cells exposed to UV radiation exhibit mixed cell death phenotypes, involving the parallel induction of apoptosis, necrosis, and entosis, and the inhibition of one form of death can lead to compensatory increases in other forms, altering the overall death profile but not necessarily the extent of cell death within the population.
Discussion
Here, we find that entosis is induced by activity of the JNK and p38 stress kinases in response to UV radiation. We find that stress kinase signaling engages entosis by controlling the uptake of cells with high levels of activity into those with reduced activity, an effect that may ultimately clear cells in the population with high levels of stress and select for those with lower stress signaling. We also find that cells with ingested corpses have better overall survival than single cells, suggesting that entosis leads to the clearance of stressed cells in a manner that also provides those with lower stress additional survival advantages.
In previous research, we have shown that nutrient starvation engages entosis in a similar manner, as cells with high levels of stress signaling through the energy-sensing kinase AMP-activated protein kinase (AMPK) are eliminated by those with low AMPK activity (Hamann et al., 2017). The degradation of internalized cells also provides a survival advantage during starvation, an effect that is linked to the scavenging of nutrients from ingested cells (Hamann and Overholtzer, 2017; Hamann et al., 2017; Krajcovic et al., 2013; Krishna et al., 2016). While we show here that UV-irradiated cells also have a survival advantage that is linked to entosis, whether metabolite scavenging underlies the enhanced survival of entotic cells remains to be determined. Significant nucleotide pools are known to be needed for DNA repair following UV radiation, and the sizes of nucleotide pools can be rate limiting for cell survival (Wei et al., 2018), suggesting that the scavenging of nucleotides could plausibly underlie the observed survival advantage of engulfing cells. Alternatively, it is possible that cells with low levels of stress signaling, which are spared entotic death, may have related survival advantages to other forms of cell death as well, and that intrinsic stress resistance contributes to survival. That lysosome inhibition only partially, yet significantly, inhibits the survival advantage of engulfing cells during entosis could be consistent with such a model (Figure 4B).
Our findings demonstrate that entosis occurs in UV-irradiated cell populations as one part of a mixed-cell death response that also involves apoptosis and necrosis. We have shown previously that cell death occurring in glucose-starved cells also involves the parallel induction of these morphologically distinct forms of cell death (Hamann et al., 2017). Similarly, cell death induced by viral infection has been shown to induce apoptosis and necrosis in a mixed, parallel response (Shubina et al., 2020). Our findings here show that the amount of death in different UV-irradiated cell populations is similar despite different frequencies of execution of these different programs (Figures 4E and 4F). We also show that the inhibition of one form of cell death can shift the balance toward a different form of death without significantly affecting the total death amount. While studies over the last decade have elucidated numerous different forms of cell death that can eliminate cells in different contexts (Galluzzi et al., 2018), how mixed death responses are regulated, and how the percentages of different types of death within a mixture may impact tissue physiology or pathophysiology is poorly understood. Our findings identify a new inducer of entosis and reveal cross talk between entosis, apoptosis, and necrosis whose underlying mechanism(s) may be identified in further studies.
Limitations of study
This study focused on identifying JNK and p38 stress-activated kinase signaling as an inducer of entosis. The downstream mechanisms underlying how stress signaling regulates entosis remain to be further elucidated. We also demonstrated cross talk between different forms of cell death, namely entosis, apoptosis, and necrosis, in cells exposed to UV radiation. Future studies may uncover the regulatory mechanisms that control these modes of cross talk.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-E-cadherin | Cell Signaling Technology | Cat#3195; RRID: AB_2291471 |
| Rabbit polyclonal anti-β-Catenin | Sigma | Cat#C2206; RRID: AB_476831 |
| Mouse anti-human LAMP1 | BD Biosciences | Cat#555798; RRID: AB_396132 |
| Goat Alexa Fluor 568 anti-mouse secondary | Life Technologies | Cat#A-11031; RRID: AB_144696 |
| Goat Alexa Fluor 488 anti-rabbit secondary | Life Technologies | Cat#A-11034; RRID: AB_2576217 |
| Mouse monoclonal anti-GAPDH | Santa Cruz | Cat#sc-47724; RRID: AB_627678 |
| Rabbit monoclonal anti-phospho-SAPK/JNK (Thr183/Tyr185) | Cell Signaling Technology | Cat#4668; RRID: AB_823588 |
| Rabbit polyclonal anti-SAPK/JNK | Cell Signaling Technology | Cat#9252; RRID: AB_2250373 |
| Rabbit polyclonal anti-p38 MAPK | Cell Signaling Technology | Cat#9212; RRID: AB_330713 |
| Rabbit polyclonal anti-phospho-p38 MAPK (Thr180/Tyr182) | Cell Signaling Technology | Cat#9211; RRID: AB_331641 |
| Mouse monoclonal anti-p53 | Santa Cruz | Cat#sc-126; RRID: AB_628082 |
| Chemicals, peptides, and recombinant proteins | ||
| Cell Tracker green | Life Technologies | Cat# C7025 |
| Cell Tracker red | Life Technologies | Cat# C34552 |
| Y-27632 | Tocris Bioscience | Cat# 1254 |
| SP600125 | Tocris Bioscience | Cat#1496 |
| SB202190 | Cayman Chemical | Cat#10010399 |
| 4′,6-Diamidino-2-Phenylindole (DAPI) | Life Technologies | Cat#1306 |
| Trihydrochloride (Hoechst) | Thermo Fisher Scientific | Cat#3570 |
| Z-VAD-FMK | Sigma | Cat#V116 |
| Recombinant Murine IFN-γ | Peprotech | Cat#315-05 |
| Concanamycin A | Sigma | Cat#C9705 |
| Critical commercial assays | ||
| Pierce BCA protein assay Kit | Thermo Fisher Scientific | Cat#23225 |
| Experimental models: cell lines | ||
| Human: MCF7 breast cancer cells | Lombardi Cancer Center | NA |
| Human: BxPC3 pancreatic cancer cells | ATCC | Cat#CRL-1687 |
| Human: HCT 116 colorectal cancer cells | CLS | RRID: CVCL_0291 |
| Human: HCT116 p53 KO | CLS | RRID: CVCL_HD97 |
| Mouse: J774.1 macrophages | ATCC | Cat#TIB-67 |
| Human: 16HBE bronchial epithelial cells | Sigma | Cat#SCC150 |
| Human: U-937 monocytes | ATCC | Cat#CRL-1593.2 |
| Human: MCF7 JNKKTR | Overholtzer laboratory | This study |
| Human: MCF7 shp53 | Overholtzer laboratory | This study |
| Recombinant DNA | ||
| Plasmid: pBabe-H2B-mCherry | Overholtzer laboratory | Florey et al. (2011) |
| Plasmid: pLJM1-JNKKTR-mCardinal | Albeck laboratory | This study |
| Plasmid: pLKO.1-shMAPK8 | Sigma | NM_139049 |
| Plasmid: pLKO.1-shMAPK9 | Sigma | NM_139069 |
| Plasmid: pLKO.1-shMAPK14 | MSKCC core facility | NM_001315 |
| Software and algorithms | ||
| NIS Elements software | Nikon | NA |
| Volocity image analysis software | PerkinElmer | NA |
| Prism 7 | GraphPad | NA |
| Other | ||
| 35 mm glass-bottom dishes | MatTek | Cat# P06G-1.5-20-F |
| 12-Well glass-bottom dishes | MatTek | Cat# P12G-1.5-14-F |
| Fetal bovine serum | Sigma | Cat#F2442 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael Overholtzer (overhom1@mskcc.org).
Materials availability
All materials are available from the corresponding author upon reasonable request.
Data and code availability
This study did not generate any unique dataset or code.
Experimental model and subject details
MCF7, J774.1 16HBE, and HCT116, HCT116 p53 knockout cells were cultured in DMEM (11,965-092; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1% penicillin/streptomycin. BxPC3 and U937 cells were cultured in RPMI (11,875-093; Life Technologies) supplemented with 10% FBS and 1% penicillin/streptomycin. Cells with knockdowns were generated as described previously (Florey et al., 2011). The constructs used for JNK1/2 knockdowns were pooled from TRCN0000352709 and TRCN0000196759, obtained from Sigma-Aldrich. The construct used for p38ɑ was TRCN0000000511 obtained from the MSKCC gene editing and screening core facilities. The pBabe-H2B-mCherry construct used in live cell imaging and the generation of H2B-mCherry-expressing cells have been described previously (Florey et al., 2011).
Methods details
Time-lapse microscopy
The time-lapse microscopy was performed in 37°C and 5% CO2 live-cell incubation chambers, as described previously (Hamann et al., 2017). Cells were cultured on glass-bottom dishes. Fluorescence and differential interference contrast (DIC) images were acquired using a Nikon Ti-E inverted microscope attached to a CoolSNAP charge-coupled device camera (Photometrics) and NIS Elements software (Nikon).
Drug treatment
Cells were treated with Y-27632 at 10 μM, SP600126 at 50 μM, SB202190 at 10 μM, Concanamycin A at 100nM, Z-VAD-FMK at 20 μM and Recombinant Murine IFN-γ at 200 U/ml.
Immunofluorescence
The following antibodies were used for immunofluorescence (IF): anti-E-cadherin (1:100; Cell Signaling), anti-β-catenin (1:100; Sigma-Aldrich), anti-Lamp1 (1:100; BD Biosciences), Alexa Fluor 568 goat anti-mouse secondary (1:500; Life Technologies), and Alexa Fluor 488 goat anti-rabbit secondary (1:500; Life Technologies). IF was performed as described previously (Overholtzer et al., 2007). Briefly, cells were cultured on glass-bottom dishes (P35G-1.5-20-C; MatTek) and were fixed in 1:1 methanol/acetone for 5 min at −20°C, followed by three 5-min PBS washes and blocking in 5% BSA, 100 mM glycine in PBS for 1 hr, followed by incubation with primary antibodies at 4°C overnight. Samples were then incubated with secondary antibodies and counterstained with DAPI (1:1,000; Life Technologies). Confocal microscopy was performed with the Ultraview Vox spinning-disk confocal system (PerkinElmer) equipped with a Yokogawa CSU-X1 spinning-disk head and an electron-multiplying charge-coupled device camera (Hamamatsu C9100-13) coupled to a Nikon Ti-E microscope; image analysis was performed using Volocity software (PerkinElmer).
Western blotting
Cells were collected first in lysis buffer consisting of 50mM Tris-Cl pH 6.8, 10% glycerol, and 2% SDS. Then each sample was boiled at 100°C for 5 min, followed by vigorous pipetting to eliminate viscosity. After measuring the protein concentrations using BCA assay (Thermo Fisher Scientific), the samples were prepared with equal protein amounts, and then Western blotting was performed as described (Hamann et al., 2017). The following primary antibodies were used: GAPDH (1:1000; Santa Cruz), p-JNK/SAPK (1:500; Cell Signaling), JNK/SAPK (1:500; Cell Signaling), p38 (1:500; Cell Signaling), p-p38 (1:500; Cell Signaling), p53 (1:1000; Santa Cruz).
Cell death quantification
For UV light exposure, cells were treated with 100J/m2 UV-C (or lower doses as indicated in the figures), using a UV Stratalinker 1800 (Stratagene, La Jolla, CA). To quantify entosis in UV-irradiated or control MCF7, BxPC3, HCT116, and 16HBE cells through immunofluorescence, 400,000 cells were plated on 35 mm glass-bottom dishes in the presence of Y27632 (to inhibit entosis as suspension cells undergo adherence to the plate), and were allowed to adhere overnight. Y27632 was then washed out (PBS x 3) prior to treatment with UV. After the indicated treatment, cells were incubated for the indicated time periods and then fixed and stained as described above. Entotic percentages were determined as described previously (Hamann et al., 2017). Briefly, at least 300 cells were counted in each sample and the percentage of entotic cells were quantified by the number of single cells and cell-in-cell structures; specifically both live and dead cells (defined by LAMP1-positive compartments) were counted as entotic. When one host cell contained two cells, each engulfed cell was counted as an entotic event when they were contained within in separate vacuoles. If two cells were engulfed in a sequential manner (cell-in-cell-in-cell) (Overholtzer et al., 2007), then only one entotic event was counted. For the fate of internalized cells, the death rate was determined as previously described (Hamann et al., 2017). Briefly, entotic events were scored with the appearance of a vacuole within the host cell and cell death was counted by cell morphological changes observed by DIC microscopy. Quantifications of different cell death types (entosis, apoptosis, necrosis) were determined by the morphological appearances of death observed by DIC, as described previously (Hamann et al., 2017) and shown in Figure 4F. For visualization of nuclear morphology in Figure S1, cells were treated with Hoechst stain (5 μg/mL; Thermo Fisher) for 30 min at 37°C. After washing out this nuclear stain with PBS three times, cells were exposed to UV radiation and imaged using time-lapse microscopy to collect both fluorescence images and DIC.
Entosis quantification of outer and inner cells
MCF7 control or JNK1/2 knockdown cells were labeled with 10 μM Cell Tracker dyes (green or red; Life Technologies) for 30 min at 37°C and then plated at a 1:1 ratio at a total cell density of 400,000 cells per 35-mm glass-bottom dish, in the presence of Y27632 to block entosis during plating overnight. Y27632 was then washed out three times with PBS the next day and the cells were exposed to UV radiation. 24 h later, the cells were analyzed by confocal microscopy as described above and previously (Hamann et al., 2017), where heterotypic cell-in-cell structures were counted and the number green-in-red and red-in-green structures were determined.
Survival rate after phagocytosis
For the quantifications of J774.1 survival rate, 15,000 J774.1 cells were plated onto 12-well glass-bottom dishes in the presence of 200 U/ml IFNγ for 2 days, and then were exposed to UV radiation as described above. To prepare apoptotic cells, 600,000 U937 cells were exposed to UV radiation (100J/m2), 16 hr prior to feeding to J774.1 cells, as described (Krishna et al., 2016). J774.1 cells with or without U937 apoptotic corpses were then imaged by time-lapse microscopy at 15 min intervals for 24 hr. Cell death was determined by morphological changes as described above.
JNK KTR activity quantification
The JNK-KTR plasmid construct (pENTR-JNKKTRmRuby2) was obtained from Sergi Regot, and the JNK docking and phosphorylation sequence was subcloned into the pLJM1 lentiviral expression vector to replace mRuby2 with mCardinal. Two-fragment Gibson assembly was performed to fuse the JNK KTR fragment upstream of the mCardinal fluorescent protein on the pLJM1 vector backbone. MCF7 cells expressing JNK KTR were generated by lentiviral transduction as reported previously (Florey et al., 2011). Cells expressing JNK KTR were plated on 35-mm glass-bottom dishes with Y27632 to block entosis from occurring during plating overnight. Y27632 was then washed out three times with PBS the next day and cells were treated with Hoechst stain for 30 min at 37°C. After washing out this nuclear stain with PBS three times, cells were exposed to UV radiation and imaged using time-lapse microscopy to collect both fluorescence images and also DIC, as described above. The fluorescence mean intensities of the cytoplasm and nucleus were determined using NIS Elements software (Nikon). Specifically, the mean intensity in the nucleus was determined using the Draw Object function and manually covering >50% of the nuclear area (defined by Hoechst positivity), followed by automated measurements. The mean intensity of the cytoplasm was determined similarly by drawing >50% of the cytoplasm, as determined by the DIC channel and Hoechst negativity. Background fluorescence was also subtracted prior to determining JNK KTR ratios.
Quantification and statistical analysis
All p values were obtained in Prism 7 using One-Way Anova followed by Dunnett's test, except where Student's t test is indicated (∗∗∗, p < 0.001; ∗∗, p < 0.01; ∗, p < 0.05; n.s., not significant).
Acknowledgments
This research was funded by grants from the National Cancer Institute, RO1CA154649 (MO) and the American Association for Cancer Research/Stand Up To Cancer (SU2C-AACR-IRG-01-16) (JGA). SU2C is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Author contributions
Conceptualization, R.C. and M.O.; Methodology, R.C. and M.O.; Investigation, R.C.; Resources, A.R. and J.G.A; Writing – Original Draft, R.C. and M.O.; Writing – Review & Editing, R.C., A.R., J.G.A and M.O.; Visualization, R.C. and M.O.; Funding Acquisition, J.G.A. and M.O.
Declaration of interests
Memorial Sloan Kettering Cancer Center and one investigator involved in this study (M.O.) have financial interests in Elucida Oncology, Inc.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102902.
Supplemental information
References
- Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G.D., Mitchison T.J., Moskowitz M.A., Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgan J., Tseng Y.Y., Hamann J.C., Domart M.C., Collinson L., Hall A., Overholtzer M., Florey O. Mitosis can drive cell cannibalism through entosis. Elife. 2017;6:e27134. doi: 10.7554/eLife.27134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O., Kim S.E., Sandoval C.P., Haynes C.M., Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011;13:1335–1343. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann J.C., Overholtzer M. Entosis enables a population response to starvation. Oncotarget. 2017;8:57934–57935. doi: 10.18632/oncotarget.20066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann J.C., Surcel A., Chen R., Teragawa C., Albeck J.G., Robinson D.N., Overholtzer M. Entosis is induced by glucose starvation. Cell Rep. 2017;20:201–210. doi: 10.1016/j.celrep.2017.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinojosa L.S., Holst M., Baarlink C., Grosse R. MRTF transcription and Ezrin-dependent plasma membrane blebbing are required for entotic invasion. J. Cell Biol. 2017;216:3087–3095. doi: 10.1083/jcb.201702010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil G.S., Davis R.J. Cell signaling and stress responses. Cold Spring Harb. Perspect. Biol. 2016;8:a006072. doi: 10.1101/cshperspect.a006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann. N Y Acad. Sci. 1998;851:139–146. doi: 10.1111/j.1749-6632.1998.tb08987.x. [DOI] [PubMed] [Google Scholar]
- Kerr J.F., Wyllie A.H., Currie A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalkar P., Diaz-Argelich N., Antonio Palop J., Sanmartin C., Fernandes A.P. Novel methylselenoesters induce programed cell death via entosis in pancreatic cancer cells. Int. J. Mol. Sci. 2018;19:2849. doi: 10.3390/ijms19102849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajcovic M., Krishna S., Akkari L., Joyce J.A., Overholtzer M. mTOR regulates phagosome and entotic vacuole fission. Mol. Biol. Cell. 2013;24:3736–3745. doi: 10.1091/mbc.E13-07-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S., Palm W., Lee Y., Yang W., Bandyopadhyay U., Xu H., Florey O., Thompson C.B., Overholtzer M. PIKfyve regulates vacuole maturation and nutrient recovery following engulfment. Dev. Cell. 2016;38:536–547. doi: 10.1016/j.devcel.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y., Lim D., Tan P.H., Leung T.K., Yip G.W., Bay B.H. Silencing the Metallothionein-2A gene induces entosis in adherent MCF-7 breast cancer cells. Anat. Rec. (Hoboken) 2010;293:1685–1691. doi: 10.1002/ar.21215. [DOI] [PubMed] [Google Scholar]
- Lee Y., Hamann J.C., Pellegrino M., Durgan J., Domart M.C., Collinson L.M., Haynes C.M., Florey O., Overholtzer M. Entosis controls a developmental cell clearance in C. elegans. Cell Rep. 2019;26:3212–3220 e3214. doi: 10.1016/j.celrep.2019.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Sun X., Dey S.K. Entosis allows timely elimination of the luminal epithelial barrier for embryo implantation. Cell Rep. 2015;11:358–365. doi: 10.1016/j.celrep.2015.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Fan J., Wang M., Niu Z., Zhang Z., Yuan L., Tai Y., Chen Z., Song S., Wang X. CDKN2A inhibits formation of homotypic cell-in-cell structures. Oncogenesis. 2018;7:50. doi: 10.1038/s41389-018-0056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J., Niu Z., Zhang B., Yu X., Zheng Y., Wang C., Ren H., Wang M., Ruan B., Qin H. p53-dependent elimination of aneuploid mitotic offspring by entosis. Cell Death Differ. 2021;28:799–813. doi: 10.1038/s41418-020-00645-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay H.L., Moore D., Hall C., Birkbak N.J., Jamal-Hanjani M., Karim S.A., Phatak V.M., Pinon L., Morton J.P., Swanton C. Genomic instability in mutant p53 cancer cells upon entotic engulfment. Nat. Commun. 2018;9:3070. doi: 10.1038/s41467-018-05368-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa J., Matsuzawa A., Takeda K., Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J. Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- Overholtzer M., Mailleux A.A., Mouneimne G., Normand G., Schnitt S.J., King R.W., Cibas E.S., Brugge J.S. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–979. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- Purvanov V., Holst M., Khan J., Baarlink C., Grosse R. G-protein-coupled receptor signaling and polarized actin dynamics drive cell-in-cell invasion. Elife. 2014;3:e02786. doi: 10.7554/eLife.02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S., Hughey J.J., Bajar B.T., Carrasco S., Covert M.W. High-sensitivity measurements of multiple kinase activities in live single cells. Cell. 2014;157:1724–1734. doi: 10.1016/j.cell.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley T., Sontag E., Chen P., Levine A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008;9:402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Roos W.P., Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Ruan B., Wang C., Chen A., Liang J., Niu Z., Zheng Y., Fan J., Gao L., Huang H., Wang X. Expression profiling identified IL-8 as a regulator of homotypic cell-in-cell formation. BMB Rep. 2018;51:412–417. doi: 10.5483/BMBRep.2018.51.8.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan B., Zhang B., Chen A., Yuan L., Liang J., Wang M., Zhang Z., Fan J., Yu X., Zhang X. Cholesterol inhibits entotic cell-in-cell formation and actomyosin contraction. Biochem. Biophys. Res. Commun. 2018;495:1440–1446. doi: 10.1016/j.bbrc.2017.11.197. [DOI] [PubMed] [Google Scholar]
- Shubina M., Tummers B., Boyd D.F., Zhang T., Yin C., Gautam A., Guo X.J., Rodriguez D.A., Kaiser W.J., Vogel P. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J. Exp. Med. 2020;217:e20191259. doi: 10.1084/jem.20191259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui X., Kong N., Ye L., Han W., Zhou J., Zhang Q., He C., Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344:174–179. doi: 10.1016/j.canlet.2013.11.019. [DOI] [PubMed] [Google Scholar]
- Sun Q., Cibas E.S., Huang H., Hodgson L., Overholtzer M. Induction of entosis by epithelial cadherin expression. Cell Res. 2014;24:1288–1298. doi: 10.1038/cr.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Huang H., Overholtzer M. Cell-in-cell structures are involved in the competition between cells in human tumors. Mol. Cell. Oncol. 2015;2:e1002707. doi: 10.1080/23723556.2014.1002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Luo T., Ren Y., Florey O., Shirasawa S., Sasazuki T., Robinson D.N., Overholtzer M. Competition between human cells by entosis. Cell Res. 2014;24:1299–1310. doi: 10.1038/cr.2014.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss A.K., Strasser A. The essentials of developmental apoptosis. F1000Res. 2020;9 doi: 10.12688/f1000research.21571.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q., Liu J., Zheng Z., Zhu H., Chu X., Dong Z., Huang S., Du Q. Regulation of myosin activation during cell-cell contact formation by Par3-Lgl antagonism: entosis without matrix detachment. Mol. Biol. Cell. 2012;23:2076–2091. doi: 10.1091/mbc.E11-11-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Ning X., Chen A., Huang H., Ni C., Zhou C., Yu K., Lan S., Wang Q., Li S. Impaired formation of homotypic cell-in-cell structures in human tumor cells lacking alpha-catenin expression. Sci. Rep. 2015;5:12223. doi: 10.1038/srep12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.W., Lee C.Y., Lee D.J., Chu C.F., Wang J.C., Wang T.C., Jane W.N., Chang Z.F., Leu C.M., Dzhagalov I.L. Equilibrative nucleoside Transporter 3 regulates T cell homeostasis by coordinating lysosomal function with nucleoside availability. Cell Rep. 2018;23:2330–2341. doi: 10.1016/j.celrep.2018.04.077. [DOI] [PubMed] [Google Scholar]
- Wen S., Shang Z., Zhu S., Chang C., Niu Y. Androgen receptor enhances entosis, a non-apoptotic cell death, through modulation of Rho/ROCK pathway in prostate cancer cells. Prostate. 2013;73:1306–1315. doi: 10.1002/pros.22676. [DOI] [PubMed] [Google Scholar]
- Xia P., Zhou J., Song X., Wu B., Liu X., Li D., Zhang S., Wang Z., Yu H., Ward T. Aurora A orchestrates entosis by regulating a dynamic MCAK-TIP150 interaction. J. Mol. Cell. Biol. 2014;6:240–254. doi: 10.1093/jmcb/mju016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Arrows indicate cells that are ingested; black arrow indicates cell that undergoes entotic cell death, white arrow indicates cell that remains viable for the duration of the movie. Images show DIC and H2B-mCherry fluorescence that marks nuclei (red). Times are indicated as minutes (min).
Data Availability Statement
This study did not generate any unique dataset or code.